DOI:
10.1039/D0RA10425F
(Paper)
RSC Adv., 2021,
11, 10385-10392
Discovery and characterization of naturally occurring potent inhibitors of catechol-O-methyltransferase from herbal medicines†
Received
11th December 2020
, Accepted 28th February 2021
First published on 11th March 2021
Abstract
Human catechol-O-methyltransferase (hCOMT) is considered a therapeutic target due to its crucial roles in the metabolic inactivation of endogenous neurotransmitters and xenobiotic drugs. There are nevertheless few safe and effective COMT inhibitors and there lacks a diversity in structure. To discover novel safe and effective hCOMT inhibitors from herbal products, in this study, 53 herbal products were collected and their inhibitory effects against hCOMT were investigated. Among them, Scutellariae radix (SR) displayed the most potent inhibitory effect on hCOMT with an IC50 value of 0.75 μg mL−1. To further determine specific chemicals as COMT inhibitors, an affinity ultrafiltration coupled with liquid chromatography-mass spectrometry method was developed and successfully applied to identify COMT inhibitors from SR extract. The results demonstrated that scutellarein 2, baicalein 9 and oroxylin A 12 were potent COMT inhibitors, showing a high binding index (>3) and very low IC50 values (32.9 ± 3.43 nM, 37.3 ± 4.32 nM and 18.3 ± 2.96 nM). The results of inhibition kinetics assays and docking simulations showed that compounds 2, 9 and 12 were potent competitive inhibitors against COMT-mediated 3-BTD methylation, and they could stably bind to the active site of COMT. These findings suggested that affinity ultrafiltration allows a rapid identification of natural COMT inhibitors from a complex plant extract matrix. Furthermore, scutellarein 2, baicalein 9 and oroxylin A 12 are potent inhibitors of hCOMT in SR, which could be used as promising lead compounds to develop more efficacious non-nitrocatechol COMT inhibitors for biomedical applications.
Introduction
Human catechol-O-methyltransferase (COMT, EC 2.1.1.6), an important phase II conjugative enzyme, transfers the methyl of S-adenosyl-L-methionine (SAM) to one of the hydroxyl groups of the catechol substrate in the presence of magnesium ions (Mg2+).1,2 COMT is widely distributed in both the central nervous system (CNS) and peripheral tissues such as liver, kidney and intestines, and plays a central role in the metabolic inactivation of endogenous neurotransmitters and xenobiotic drugs and hormones incorporating catecholic structures.3–5 In genetic analysis, COMT polymorphisms are related to memory, pain, hypertension and cancer, all of which are involved in the catechol metabolism.6 Since the metabolism of catecholamine neurotransmitters and estrogen is affected by COMT, inhibition of COMT may serve as a promising adjunct therapy for Parkinson's disease, Alzheimer's disease, schizophrenia and depression.7,8 Notably, different COMT inhibitors are widely used for the treatment of Parkinson's disease, and effectively improve the pharmacokinetic behavior of exogenously administered L-DOPA, extend the half-life, and increase bioavailability.9–12
Due to the important role of COMT in the pathophysiology of different human diseases, they have undertaken extensive efforts to identify COMT inhibitors and several types of COMT inhibitors with different structures were developed.13,14 First-generation COMT inhibitors are characterized by a catechol skeleton, such as pyrogallol and caffeic acid.1,14 These compounds with poor pharmacokinetics, low selectivity and high toxicity with less medicinal value as pharmacological agents.15 The second-generation COMT inhibitors with nitrocatechol structure characteristics displayed highly potent COMT inhibitory activity.16 Among them, three COMT inhibitors including tolcapone, entacapone and opicapone have been used for the symptomatic treatment of Parkinson's disease. However, tolcapone was withdrawn in many countries due to severe hepatotoxicity.17 Despite opicapone being employed with rather high cost, more investigations still needed to test its efficacy in more detail.18 Considering the function of COMT in the detoxification or metabolic inactivation of catechol drugs as well as the defects of current COMT inhibitors, more potent COMT inhibitors with low toxicity and improved safety profiles are warranted.
Historically, many Chinese proprietary medicines have been used in clinical practice mainly for anti-oxidant and anti-inflammatory activity, and neuroprotection, and a great mass of biologically active compounds have been obtained from herbal medicines, which inspired the discovery of novel COMT inhibitors from the herbal products.19–21 We first evaluated the inhibition potency against COMT of 53 Chinese herbal products with anti-inflammatory and anti-oxidant effects on the basis of the microplate reader-based fluorescence assay. Among them, an extract of Scutellariae radix (Chin.: Huang–Qin) (SR) displayed the strongest inhibitory effect on COMT with IC50 values of 0.75 μg mL−1. This finding encouraged us to further investigate specific components of SR against COMT via affinity ultrafiltration procedure.22,23 After affinity selection, scutellarein 2, baicalein 9 and oroxylin A 12 were discovered with binding indexes (BI) over 3. Subsequently, the inhibitory potentials of flavones discovered from SR on human COMT were investigated. Besides, the inhibition behaviors and inhibition mechanisms of novel potent COMT inhibitors were characterized by in vitro kinetic assays and docking simulations. All studies demonstrated that affinity ultrafiltration allows for a rapid identification of natural COMT inhibitors from complex plant extract matrices, as well as some flavones that are potent inhibitors of hCOMT in SR, while these constituents can be used as promising lead compounds to develop more efficacious COMT inhibitors to combat Parkinson's disease, Alzheimer's disease and related disorders.
Experimental
Chemical and reagents
The 53 Chinese medicine products used in this study were obtained from Jiangyin Tianjiang Pharmaceutical Co., Ltd. (Jiangyin, China). 3-(Benzo[d]thiazol-2-yl)-7,8-dihydroxy-2H-chromen-2-one (3-BTD), was used as the specific substrate of COMT. Previous studies reported the synthesis of 3-BTD and its methylated product 3-(benzo[d]thiazol-2-yl)-7-hydroxy-8-methoxy-2H-chromen-2-one (3-BTMD).24 Recombinant human S-COMT (1.4 mg mL−1) was prepared in phosphate buffer (pH 7.4) and stored at −80 °C. DTT (dithiothreitol), SAM (S-adenosyl-L-methionine), entacapone were purchased from Sigma (Louis, USA). Magnesium chloride hexahydrate (MgCl2·6H2O) was analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Baicalein, scutellarein, oroxylin A, wogonin, scutellarin, oroxin A and wogonoside were all purchased from Chengdu Purfield Biotechnology Co., Ltd. (Chengdu, China), and baicalin was prepared in our laboratory. The purity of the above eight flavones is higher than 98%. A stock solution (100 mM) of each compound was prepared in DMSO and stored at 4 °C. Tris–HCl buffer: Tris buffer (50 mM, pH 8.3) was prepared with Millipore water, adjusted to pH 7.4 with 0.1 M HCl, and stored at 4 °C for further use. Three stock solutions of MgCl2 (50 mM), DTT (4 mM) and SAM (2 mM) were prepared in Millipore water and stored at −20 °C. Millipore water (Millipore, Bedford, USA), LC grade DMSO (Tedia, USA) were used throughout.
Affinity ultrafiltration mass spectrometry
For centrifugal ultrafiltration procedure, 2 μL of 40.0 mg mL−1 SR sample solution was incubated in 10 mM PBS (pH 7.4) buffer in a total volume of 200 μL for 0.5 hours at 37 °C with 80 μL of COMT (1.4 mg mL−1). After incubation, each mixture was filtered through a Microcon (Millipore, Bedford, MA, USA) YM-10 centrifugal filter, which contains a regenerated cellulose ultrafiltration membrane with a retention capacity of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MW cutoff by centrifugation at 10000 × g for 20 min at 4 °C. The enzyme–ligand complex solution was washed 4 times with ice-cold 150 mM ammonium acetate (pH 7.5) to remove unbound compounds.22,25 Subsequently, 50% methanol (300 μL, 3 times) was added to the ultrafilter, containing the compound initially bound to the receptor was collected. The eluted samples were quickly evaporated under vacuum and reconstituted in 50% methanol before LC-MS analysis. For comparison, the control experiments with denatured enzymes were performed before each screening experiment.
000 MW cutoff by centrifugation at 10000 × g for 20 min at 4 °C. The enzyme–ligand complex solution was washed 4 times with ice-cold 150 mM ammonium acetate (pH 7.5) to remove unbound compounds.22,25 Subsequently, 50% methanol (300 μL, 3 times) was added to the ultrafilter, containing the compound initially bound to the receptor was collected. The eluted samples were quickly evaporated under vacuum and reconstituted in 50% methanol before LC-MS analysis. For comparison, the control experiments with denatured enzymes were performed before each screening experiment.
SR samples were analyzed on a Shimazu L20A UPLC system (Shimazu) coupled with a SCIEX Triple TOF 5600 mass spectrometer (AB SCIEX) operating in the negative ion mode, and the composition was determined using mixed standards. The chromatographic separations were carried out using a C18 column (250 mm × 2.0 mm, 4.6 μm, Shimadzu) at a flow rate of 0.5 mL min−1 and maintained at 40 °C. The mobile phase was consisted acetonitrile (A) and water/0.1% formic acid (B) with a gradient of 0–2.0 min, 85% B; 2.0–10.0 min, 85–75% B; 10.0–25.0 min, 7–70% B; 25.0–35.0 min, 70–65% B; 35.0–50.0 min, 65–55% B; 50–50.5 min, 55–10% B; 50.5–55 min, 10% B for washing the pillar, 55–55.5 min, 10–85% B and 55.5–60.0 min, balance to 85% for 5 min. The detection wavelength was set at 280 nm. Full-scan mass range spectra was set from 100–2000 m/z with major ESI source settings: ion spray voltage +5000 kV/−4500 kV; probe heater temperature 450 °C, curtain gas 35 psi; ion source gas 1 and gas 250 psi. The declustering potential and collision energy were −80 V and −35 V, respectively.
COMT inhibition assay
3-BTD, a highly selective optical probe substrate for human catechol-O-methyltransferase (hCOMT), was used for sensing the residual activities of hCOMT with or without inhibitors. In brief, the incubation mixture with a total volume of 200 μL consisted of 50 mM Tris–HCl (pH 7.4), 50 mM MgCl2, 40 mM DTT, 0.2 μM 3-BTD, recombinant human S-COMT (1.75 μg mL−1, final concentration) and various inhibitors. After 5 min preincubation at 37 °C, the O-methylation reaction was initiated by adding 2 mM SAM. Then incubation at 37 °C for another 6 min, 0.2 mL of 0.1% formic acid in ice-cooled acetonitrile (4 °C) was added to terminate the enzyme reaction. The incubation mixtures were subsequently centrifuged at 20000 × g, 4 °C for 20 min and an aliquot of the supernatant was analysed by a multi-mode microplate reader (SpectraMaxs iD3®, Molecular Devices, Austria) to analyse its product (3-BTMD). 3-BTMD (the 8-O-methyl metabolite of 3-BTD) was excited at 390 nm, and the emission wavelength is set at 510 nm. A positive control (entacapone) was also carried out under the same conditions. Control experiments were performed without substrate, or COMT enzyme source. The residual activity of hCOMT was calculated by the following formula: residual activity (%) = (fluorescence intensity of 3-BTMD in the presence of inhibitor)/fluorescence intensity of 3-BTMD in the negative control (DMSO only) × 100%.
Inhibition kinetic analyses
The inhibition mode and corresponding inhibition constant (Ki) of scutellarein, baicalein and oroxylin A against COMT were studied by using the specific fluorescent substrate 3-BTD of COMT. In the presence of multiple concentrations of inhibitors, different concentrations of substrate were used to determine the reaction rate. After that, a Lineweaver–Burk plot was drawn to determine the inhibition pattern of the tested inhibitor, while the second plot of the slopes from the Lineweaver–Burk plot was used to calculate the corresponding inhibition constant (Ki) value. The following equations are competitive inhibition (1), non-competitive inhibition (2) or mixed inhibition (3) for fitting data respectively.| | |
V = VmaxS/[(Km + I/Ki) + S]
| (1) |
| | |
V = VmaxS/[(Km + S)(1 + I/Ki)]
| (2) |
| | |
V = VmaxS/[(Km + S)(1 + I/αKi)]
| (3) |
where V is the reaction rate; Ki is the inhibition constant, describing the affinity of the inhibitor to the enzyme; S and I are the concentrations of the substrate and the inhibitor, respectively; Vmax is the maximum velocity; (substance concentration at 0.5Vmax). Km is the Michaelis constant (substrate concentration at 0.5Vmax). The goodness of fit parameter is used to determine the most appropriate type of inhibition kinetics.
Molecular docking simulations
The crystal structure of COMT (PDB ID: 3BWY) was retrieved from a Protein Data Bank (http://www.rcsb.org/). Before performing all docking experiments via AutoDock Vina, the accuracy of the docking procedure was verified by redocking the original ligand 3,5-dinitrophenol into the COMT crystal structure and RMSD was calculated.26 Subsequently, taking the following steps to process the receptor coordinate file for docking: previous to adding the hydrogens and merging non-polar hydrogens, both water molecules and irrelevant heteroatoms were removed, and charges were added using the Kollman method. On the other hand, the ligand compounds, including oroxylin A, scutellarein, and baicalein, were imported into AutoDock Tools, accompanied with the Torsion Tree root detected. The searching conformation space that was represented by a grid box with a spacing of 0.375 Å and dimensions of (80 × 80 × 80) points and the grid center XYZ set at −5.308, −11.975, and −15.176 respectively, was sufficiently large to wrap the active pocket. Discovery Studio (BIOVIA Discovery Studio 2020, Dassault Systèmes, SanDiego, USA) visualizer version was eventually employed to further analyze docking poses with the lowest binding affinity.
Statistical analysis
All kinetic and inhibition experiments were carried out in triplicate and the data obtained from experiments are expressed as mean ± SD. The IC50 values (the concentration of inhibitor that reduces enzyme activity by 50%) were calculated by nonlinear regression using GraphPad Prism 6.0 software (GraphPad Software, Inc., La Jolla, USA).
Results
Discovery of herbal product(s) with hCOMT inhibition activity
To discover novel safe and effective hCOMT inhibitors from herbal products, 53 common types of herbal products with anti-inflammatory, anti-oxidant or neuroprotective activities were collected, and their inhibitory effects towards hCOMT were investigated using 3-BTD as the substrate.19 Extracts of Scutellariae radix (SR), Glabrous greenbrier, Peppermint, Arcece peel, Asiatic pennywort and India madder root displayed moderate to strong inhibition against hCOMT at the concentration of 10 μg mL−1 (Fig. 1). Due to high inhibitory potency of SR, the dose-dependent inhibition of the SR was further examined to quantify the inhibitory effects against hCOMT (Fig. S2†). The IC50 value of SR is as low as 0.75 μg mL−1, which indicates that it's potential to offer potent COMT inhibitors.
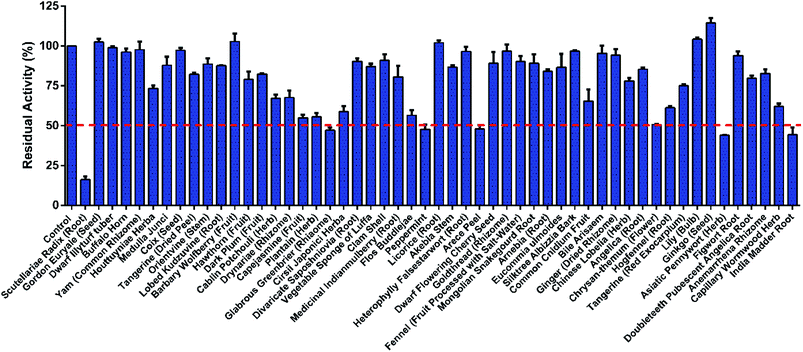 |
| | Fig. 1 The residual activities of all tested herbal products (10 μg mL−1, final concentration) on the catalytic activities of COMT-mediated 8-O-methylation. The data were shown as mean ± SD. | |
Identification of COMT inhibitors from SR by affinity ultrafiltration mass spectrometry
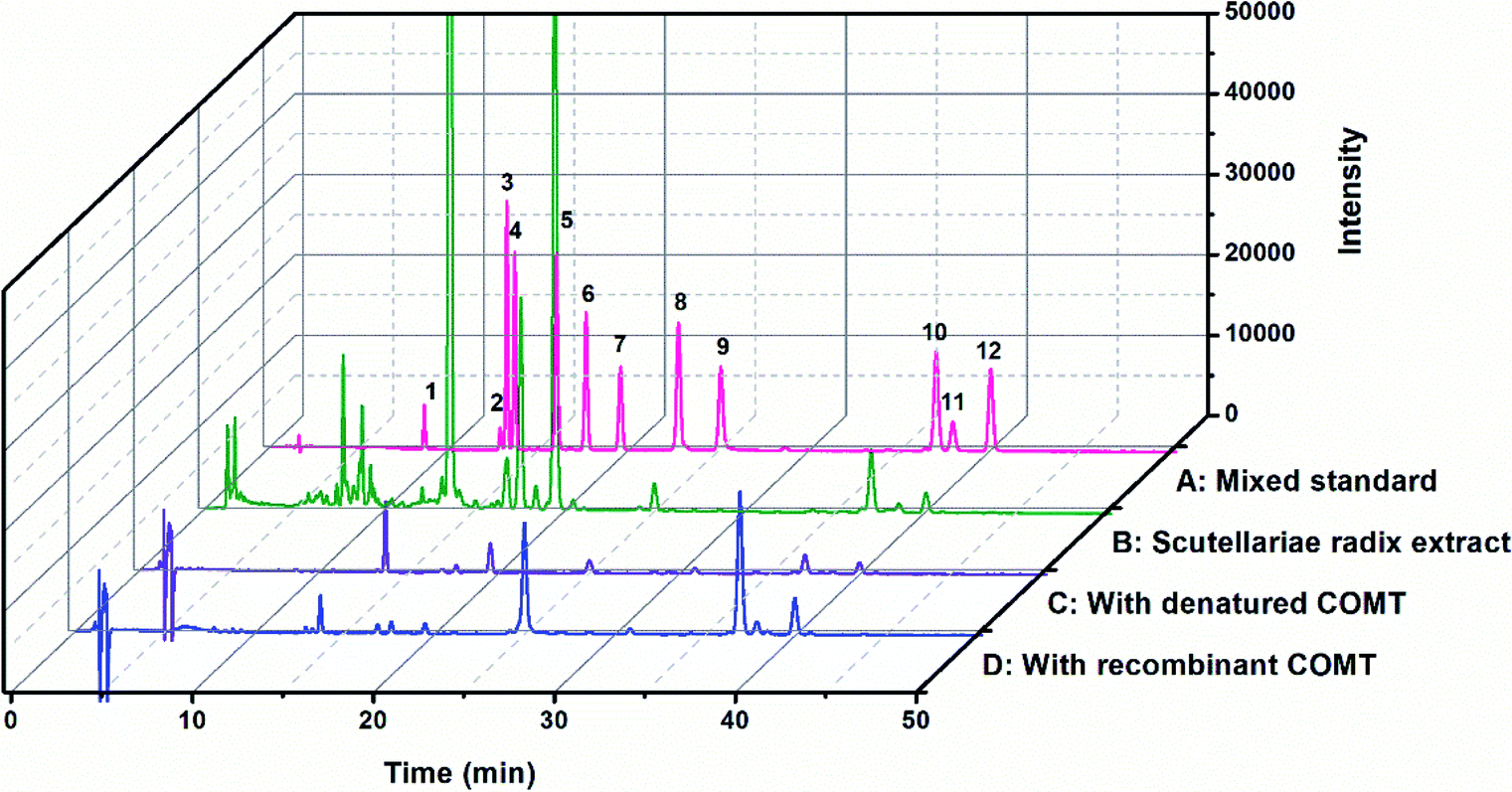
The chemical constituents of SR have been investigated, notably flavones like baicalin 4, wogonoside 7, baicalein 9 and wogonin 10, which are critical for the quality control of SR.27,28 To explore the inhibitory potentials of the major constituents of SR on hCOMT, twelve flavones were selected as criteria to optimize chromatographic conditions for analysis the chemical constituents of SR (Fig. S7†). The baseline separations of flavones standards and SR extracts are shown in Fig. 2. The biological affinity screening for SR toward hCOMT was performed using affinity ultrafiltration procedure. To assess the specific association of a given compound with the receptor, we calculated a binding index (BI) defined as the ratio of UV response of the compound detected in the target COMT versus the control incubations. Four trapped ligands (2, 9, 10 and 12) from SR extract showed specific binding to COMT with BI over 3 (Fig. 2 and Table S1†). Compounds 3, 4, 6 and 7 displayed non-specific binding to COMT compared to the denatured COMT with BI below 2. All identified ligands above were identified by authentic standards and LC-TOF-MS/MS, and the structures shown in Fig. 3.
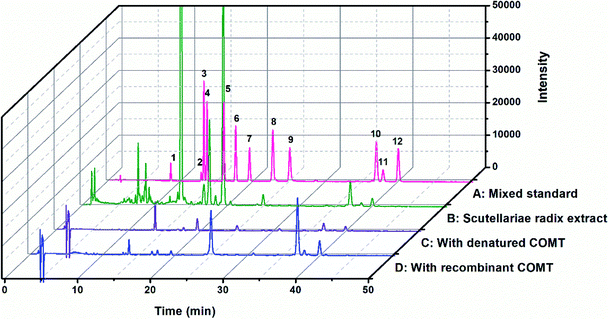 |
| | Fig. 2 SR extract incubated with hCOMT after performing ultrafiltration ((A) pink lines: mixed standard; (B) green lines: chromatogram of SR extract; (C) purple lines: control experiments incubated with denatured hCOMT; (D) blue lines: experiments incubated with active hCOMT). | |
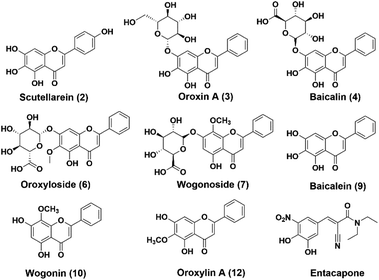 |
| | Fig. 3 Chemical structures of identified COMT inhibitors in SR and entacapone. | |
hCOMT inhibitory activities of the identified bioactive constituents
The inhibitory effects of eight trapped compounds on hCOMT were conducted using three inhibitor concentrations (1.0 μM, 10 μM and 100 μM). Three compounds (scutellarein 2, baicalein 9 and oroxylin A 12) with BI values over 3 could inhibit the catalytic activity of hCOMT and lead to the residual activity of hCOMT lower than 30% at the concentration of 1.0 μM (Fig. S3† and Table 1). To quantify the inhibitory effects of these compounds against hCOMT, dose-dependent inhibition curves were plotted using different inhibitor concentrations. Scutellarein 2, baicalein 9 and oroxylin A 12 showed strong inhibitory effects on hCOMT with the IC50 values of 0.032 ± 0.003 μM, 0.037 ± 0.004 μM and 0.018 ± 0.002 μM, respectively, which were comparable with the positive drug entacapone (IC50 = 0.012 ± 0.001 μM) (Table 1 and Fig. S4†). These findings demonstrated that the inhibitory potency of these three bioactive ingredients towards hCOMT were the same order of magnitude as that of entacapone. Additionally, glycoside forms of these ingredients with BI below 2 (scutellarin 1, baicalin 4 and oroxin A 3) showed weaker inhibitory activity than that of aglycones with the IC50 values 0.221 ± 0.032 μM, 0.432 ± 0.056 μM and 38.130 ± 8.68 μM, respectively (Table 1). These results indicated that the ligands from SR which were identified by the centrifugal ultrafiltration method were COMT inhibitors, and the inhibitory activity of these flavones against hCOMT corresponded to the results from the affinity ultrafiltration assay.
Table 1 COMT inhibitory activities of the identified bioactive constituents in SR
| Peak |
Constituents |
Inhibitiona (%) |
IC50 (μM) |
| Inhibition by 1.0 μmol L−1 flavonoids. |
| 2 |
Scutellarein |
16.27 ± 1.41 |
0.032 ± 0.003 |
| 3 |
Oroxin A |
80.46 ± 2.27 |
38.130 ± 8.680 |
| 4 |
Baicalin |
42.66 ± 4.72 |
0.432 ± 0.056 |
| 6 |
Oroxyloside |
40.49 ± 2.59 |
0.171 ± 0.025 |
| 7 |
Wogonoside |
87.58 ± 1.87 |
>100 |
| 9 |
Baicalein |
21.32 ± 0.41 |
0.037 ± 0.004 |
| 10 |
Wogonin |
83.61 ± 4.49 |
24.090 ± 4.960 |
| 12 |
Oroxylin A |
27.43 ± 2.35 |
0.018 ± 0.002 |
Inhibitory behaviors of potent flavone-type COMT inhibitors
The strongly inhibitory potency of scutellarein 2, baicalein 9, and oroxylin A 12 against hCOMT promoted us to further investigate the inhibition modes and to obtain the corresponding inhibition constants by using varying concentrations of 3-BTD as the substrate in the presence or absence of the tested inhibitor. The Lineweaver–Burk and the corresponding second plots demonstrated that these three natural ingredients against hCOMT displayed competitive inhibition behavior toward 3-BTD methylation catalyzed by hCOMT (Fig. 4). The Ki values of scutellarein 2, baicalein 9, oroxylin A 12 were evaluated as 14.09 ± 3.73 nM 14.51 ± 4.58 nM and 8.82 ± 2.33 nM, respectively (Table 2). Notably, scutellarein 2 and baicalein 9 possess the structure of catechol and are likely to be involved as a substrate reaction for the COMT, while there are no catechol groups in oroxylin A 12, but IC50 and Ki values of oroxylin A 12 were better than those of scutellarein 2 and baicalein 9 with catechol groups. These finding led us to further study the inhibitory mechanism of these three similar COMT inhibitors, which will be helpful to develop novel non-catechol COMT inhibitors.
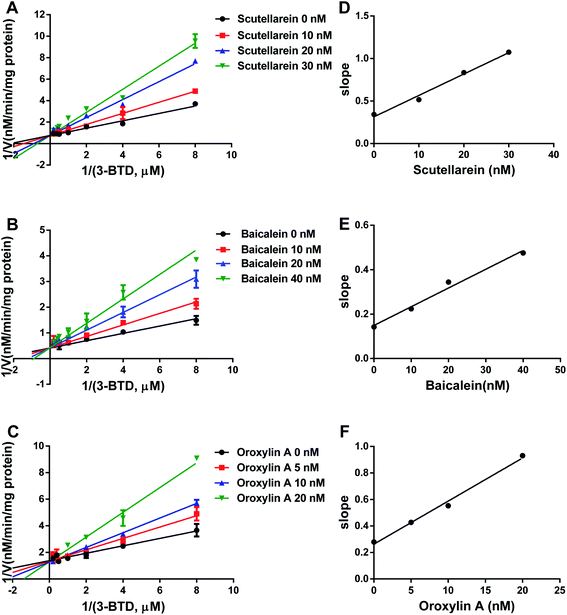 |
| | Fig. 4 (Left) The Lineweaver–Burk plots of scutellarein 2 (A), baicalein 9 (B) and oroxylin A 12 (C) against COMT-mediated 3-BTD-O-methylation. (Right) The second plots of slopes from the Lineweaver–Burk plot for COMT inhibition by scutellarein 2 (D), baicalein 9 (E) and oroxylin A 12 (F). All data are shown as mean ± SD. | |
Molecular docking simulations
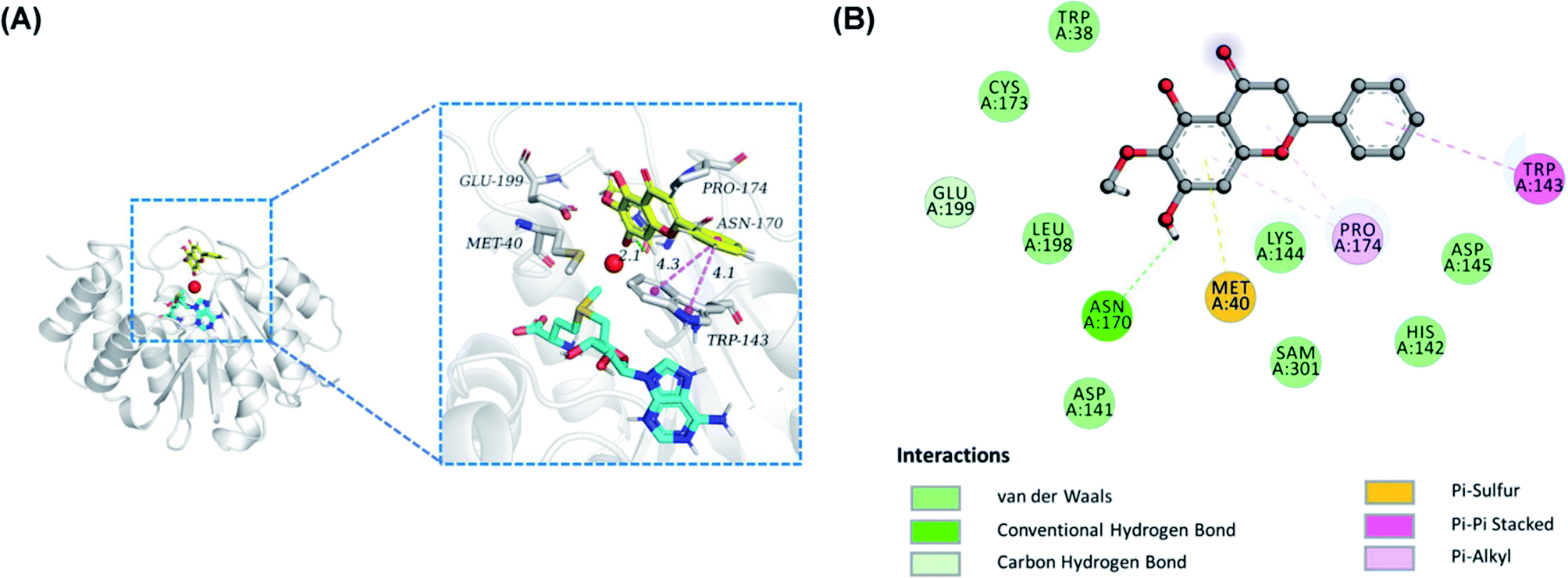
To insight into the geometrically stable conformation of the potent flavones against COMT-mediated 3-BTD-O-methylation, molecular docking simulations were performed by employing a previously reported S-COMT crystal structure (PDB ID: 3BWY).26 Notably, the conformation of redocking original ligand 3,5-dinitrophenol well coincided with that of crystallographic structure, with a RMSD value of 0.56 Å, implying that a desirable accuracy of the docking procedure. Based on Fig. S3,† these compounds including oroxylin A, scutellarein and baicalein could bound at COMT in a similar binding mode with slightly different binding interaction, and the corresponding binding affinities were −6.5, −7.3 and −7.3 kcal mol−1, respectively, which indicated that these compounds had a good affinity with COMT. In the predicted conformation of oroxylin A 12 (Fig. 5), the inhibitory effect may be generated by it strongly interact with Asn-170 via hydrogen bond (colored by green) and with TRP-143 via pi–pi stacked interaction (colored by violet), as well as with Met-40 via pi-sulfur. Furthermore, oroxylin A 12 were in contact with TRP-38, ASP-141, HIS-142, LYS-144, ASP-145, CYS-173, and LEU-198 by van der Waals interactions. Remarkably, according to previous reports, the substrate-binding site of COMT is a shallow pocket defined by some hydrophobic residues including Met40, Leu198, Trp143, and “gatekeeper” residues Trp38 and Pro174.29 These results indicated that oroxylin A 12 could be well trapped by the catechol pocket of COMT against 3-BTD-8-O-methylation in a competitive inhibition manner.
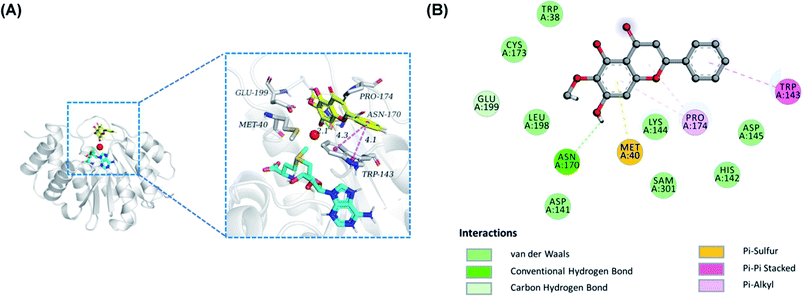 |
| | Fig. 5 (A) The binding poses of Mg2+ (colored by red), SAM (colored by cyan), oroxylin A (colored by yellow) into S-COMT in panorama and part. (B) 2D representation of the key interactions between oroxylin A and amino acids around the catechol-pocket of hCOMT. | |
Discussion
It is well-known that COMT is a therapeutic target for the treatment of Parkinson's disease and various central and peripheral nervous system disorders.30,31 Although three COMT inhibitors have been used in clinical practice, they still have many deficiencies such as poor pharmacokinetics, poor selectivity and high toxicity.32,33 Therefore, it makes sense to develop more potent COMT inhibitors with low toxicity, especially those that discover new skeleton structures from natural products. Studies have shown that a wide range of natural compounds (such as chlorogenic acid, caffeic acid, (−)-epigallocatechin-3-O-gallate, EVOO phenolic oleacein, quercetin) isolated from herbs or medicinal plants are effective inhibitors of COMT.34–37 Encouraged by these findings, we endeavored to find novel natural COMT inhibitors from medicinal plants or edible herbs.
Following screening 53 herbal products, SR displayed the strongest inhibitory effect on hCOMT with an IC50 value of 0.75 μg mL−1. Further study showed that three baicalein analogs showed strong inhibitory effects on hCOMT, and glycoside forms of these ingredients showed weaker inhibitory activity than that of aglycones. Notably, these flavone glycosides could be hydrolyzed to form the corresponding aglycones in the intestine when SR was ingested. SR has been widely employed as a Chinese medicinal plant for the treatment of various inflammatory, hepatitis, tumors and diarrhea for thousands of years.38,39 Modern pharmacological studies revealed that SR has neuroprotective effects and is regarded as a potential drug for the therapy of AD and PD.40,41 These results shows that strong COMT inhibitory activity may be another mechanism for the pharmaceutical effects of SR in addition to its anti-inflammatory and anti-oxidant activities, as well as medicinal plants rich in flavonoids might assist in the discovery of new treatments for neurological diseases such as Parkinson's disease through COMT inhibition.
Table 2 Inhibition kinetic parameters of scutellarein 2, baicalein 9 and oroxylin A 12 against hCOMT
| Constituents |
Ki (nM) |
Inhibition mode |
Goodness of fit (R2) |
| Oroxylin A |
8.82 ± 2.33 |
Competitive |
0.98 |
| Scutellarein |
14.09 ± 3.73 |
Competitive |
0.98 |
| Baicalein |
14.51 ± 4.58 |
Competitive |
0.99 |
Furthermore, to identify COMT inhibitors in a multicomponent SR extract, an affinity ultrafiltration mass spectrometry method was developed based on the affinity between hCOMT and its ligand. This method doesn't need to label the receptor so that the native conformation of the protein can be retained during the interaction.42,43 The denatured COMT group was introduced as a control to efficiently find specifical ligand towards hCOMT, which significantly reduced false positive results. The results demonstrated that scutellarein 2, baicalein 9 and oroxylin A 12 with a binding index (BI) over 3 were found to be naturally occurring potent inhibitors of COMT, which are comparable to the marketed entacapone. It is worth mentioning that Zhang et al. has reported that baicalein could be metabolized by COMT at high concentration with the Km value of 1967 ± 389 μM.44 However, our results showed that the Ki of baicalein inhibiting COMT was 14.51 ± 4.58 nM, which indicated that baicalein was mainly act as an inhibitor at low concentration, rather than primarily metabolized by COMT.
Taking characteristic natural products as the leading compounds or candidate drugs is an important avenue of new drug development.45 As we all know, the content of flavonoids (baicalein, scutellarein and oroxylin A) in the roots of this medicinal plant are very high.46 It is facile to separate these natural compounds from medicinal plants and then semi-synthesize various flavonoid derivatives following chemical modification. From the perspective of a medicinal chemist, oroxylin A 12 has several functional groups (such as hydroxyl groups), which could be easily modified to produce a series of derivatives. This greatly facilitated medicinal chemists to design new flavonoid derivatives as potent COMT inhibitors. Lamentably, this study only evaluated the identified individuals, and thereby was deficient in the structure–activity relationships of flavonoids. In the near future, it is necessary to obtain a variety of flavonoids with diverse structures for detailed structure–activity relationship study, which will greatly facilitate medicinal chemists to design more diversified flavonoid derivatives as potent COMT inhibitors.
Conclusions
In this study, 53 Chinese herbal products with anti-inflammatory, anti-oxidant or neuroprotective effects were collected, and their inhibitory effects towards hCOMT investigated by a microplate reader-based fluorescence assay. Among them, SR displayed the strongest inhibitory effect on hCOMT with an IC50 value of 0.75 μg mL−1. To discover and identify COMT inhibitors from SR extract, an affinity ultrafiltration coupled with liquid chromatography-mass spectrometry method was developed and successfully applied to assess COMT inhibitors in a multicomponent SR extract. This procedure provided a better tool for rapid screening and identifying COMT inhibitors from complex plant extract matrices, compared with the commonly used bioassay-guided column separation technique. The results demonstrated that scutellarein 2, baicalein 9 and oroxylin A 12 with binding index (BI) over 3 were found to be naturally occurring potent inhibitors of COMT, comparable to the marketed drug entacapone, with the IC50 values were 32.93 ± 3.43 nM, 37.34 ± 4.32 nM and 18.28 ± 2.96 nM, respectively. Further investigations on inhibition kinetics showed that these three natural ingredients displayed competitive inhibition behavior against COMT mediated 3-BTD methylation, with the Ki values of 14.09 ± 3.73 nM, 14.51 ± 4.58 nM and 8.82 ± 2.33 nM for scutellarein 2, baicalein 9 and oroxylin A 12, respectively. Molecular docking simulations demonstrated that the potent flavones could simultaneously bind on the catalytic site. In summary, these findings suggested that affinity ultrafiltration allows a rapid identification of natural COMT inhibitors from complex plant extract matrix. Baicalein, scutellarein and oroxylin A are potent inhibitors of hCOMT in SR, while these constituents can be used as promising lead compounds to develop novel efficacious non-nitrocatechol COMT inhibitors.
Author contributions
PW, Y-LX and G-BG conceived and designed the study. D-FZ, Y-FF, F-YW and F-BH conducted the study and drafted the manuscript. PW and G-BG supervised the study and revised the manuscript. F-JG, S-YL and LY provided the technical support and advices for the study. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by National Key Research and Development Program of China (2017YFC1700200, 2017YFC1702000), National Natural Science Foundation of China (81973393, 81773810), Natural Science Foundation of Shanghai (18ZR1436500), and the Fundamental Research Funds for the Central Universities (DUT20RC(4)013).
Notes and references
- H. C. Guldberg and C. A. Marsden, Pharmacol. Rev., 1975, 27, 135–206 CAS.
- I. J. Kopin, Pharmacol. Rev., 1985, 37, 333–364 CAS.
- P. T. Männistö and S. Kaakkola, Pharmacol. Rev., 1999, 51, 593–628 Search PubMed.
- L. E. Kiss and P. Soares-da-Silva, J. Med. Chem., 2014, 57, 8692–8717 CrossRef CAS PubMed.
- P. N. Palma, M. J. Bonifácio, A. I. Loureiro, L. C. Wright, D. A. Learmonth and P. Soares-da-Silva, Drug Metab. Dispos., 2003, 31, 250–258 CrossRef CAS PubMed.
- M. Ikeda, H. Iijima, I. Shinoda, H. Iwamoto and Y. Takeda, Food Funct., 2018, 9, 2865–2871 RSC.
- D. Scheggia, S. Sannino, M. L. Scattoni and F. Papaleo, CNS Neurol. Disord.: Drug Targets, 2012, 11, 209–221 CrossRef CAS PubMed.
- M. N. Perkovic, D. S. Strac, L. Tudor, M. Konjevod, G. N. Erjavec and N. Pivac, Curr. Alzheimer Res., 2018, 15, 408–419 CrossRef CAS PubMed.
- I. Katsaiti and J. Nixon, J. Parkinson's Dis., 2018, 8, 217–231 CAS.
- I. Buchler, D. Akuma, V. Au, G. Carr, P. de León, M. DePasquale, G. Ernst, Y. Huang, M. Kimos, A. Kolobova, M. Poslusney, H. Wei, D. Swinnen, F. Montel, F. Moureau, E. Jigorel, M. E. D. Schulze, M. Wood and J. C. Barrow, J. Med. Chem., 2018, 61, 9647–9665 CrossRef CAS PubMed.
- R. G. Robinson, S. M. Smith, S. E. Wolkenberg, M. Kandebo, L. Yao, C. R. Gibson, S. T. Harrison, S. Polsky-Fisher, J. C. Barrow, P. J. Manley, J. J. Mulhearn, K. K. Nanda, J. W. Schubert, B. W. Trotter, Z. Zhao, J. M. Sanders, R. F. Smith, D. McLoughlin, S. Sharma, D. L. Hall, T. L. Walker, J. L. Kershner, N. Bhandari, P. H. Hutson and N. A. Sachs, ACS Chem. Neurosci., 2012, 3, 129–140 CrossRef CAS PubMed.
- C. N. Patel, J. J. Georrge, K. M. Modi, M. B. Narechania, D. P. Patel, F. J. Gonzalez and H. A. Pandya, J. Biomol. Struct. Dyn., 2018, 36, 3938–3957 CrossRef CAS PubMed.
- P. T. Männistö, I. Ulmanen, K. Lundström, J. Taskinen, J. Tenhunen, C. Tilgmann and S. Kaakkola, Prog. Drug Res., 1992, 39, 291–350 Search PubMed.
- N. Jatana, N. Apoorva, S. Malik, A. Sharma and N. Latha, Cent. Nerv. Syst. Agents Med. Chem., 2013, 13, 166–194 CrossRef CAS PubMed.
- A. D. Ericsson, J. Neurol. Sci., 1971, 14, 193–197 CrossRef CAS PubMed.
- M. J. Bonifácio, P. N. Palma, L. Almeida and P. Soares-da-Silva, CNS Drug Rev., 2007, 13, 352–379 CrossRef PubMed.
- C. W. Olanow, Arch. Neurol., 2000, 57, 263–267 CrossRef CAS PubMed.
- M. Fabbri, J. J. Ferreira, A. Lees, F. Stocchi, W. Poewe, E. Tolosa and O. Rascol, Mov. Disord., 2018, 33, 1528–1539 CrossRef PubMed.
- B. Y. K. Law, A. G. Wu, M. J. Wang and Y. Z. Zhu, J. Alzheimer's Dis., 2017, 60, S151–s160 Search PubMed.
- K. Gasiorowski, E. Lamer-Zarawska, J. Leszek, K. Parvathaneni, B. B. Yendluri, Z. Błach-Olszewska and G. Aliev, CNS Neurol. Disord.: Drug Targets, 2011, 10, 184–191 CrossRef CAS PubMed.
- B. Y. Zeng, Int. Rev. Neurobiol., 2017, 135, 57–76 CrossRef PubMed.
- S. Qin, M. Meng, D. Yang, W. Bai, Y. Lu, Y. Peng, G. Song, Y. Wu, Q. Zhou, S. Zhao, X. Huang, J. D. McCorvy, X. Cai, A. Dai, B. L. Roth, M. A. Hanson, Z. J. Liu, M. W. Wang, R. C. Stevens and W. Shui, Chem. Sci., 2018, 9, 3192–3199 RSC.
- J. R. Yang, J. G. Luo and L. Y. Kong, Chin. J. Nat. Med., 2015, 13, 208–214 CAS.
- P. Wang, Y. L. Xia, L. W. Zou, X. K. Qian, T. Y. Dou, Q. Jin, S. Y. Li, Y. Yu, D. D. Wang, Q. Luo, G. B. Ge and L. Yang, Chem.–Eur. J., 2017, 23, 10800–10807 CrossRef CAS PubMed.
- Z. W. Ning, L. X. Zhai, T. Huang, J. Peng, D. Hu, H. T. Xiao, B. Wen, C. Y. Lin, L. Zhao and Z. X. Bian, Food Funct., 2019, 10, 1893–1902 RSC.
- N. N. Zulkipli, R. Zakaria, I. Long, S. F. Abdullah, E. F. Muhammad, H. A. Wahab and T. H. Sasongko, Molecules, Basel, Switzerland, 2020, vol. 25 Search PubMed.
- H. B. Li, Y. Jiang and F. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 812, 277–290 CrossRef CAS.
- H. Bochoráková, H. Paulová, J. Slanina, P. Musil and E. Táborská, Phytother. Res., 2003, 17, 640–644 CrossRef PubMed.
- K. Rutherford, I. Le Trong, R. E. Stenkamp and W. W. Parson, J. Mol. Biol., 2008, 380, 120–130 CrossRef CAS PubMed.
- A. Serretti and P. Olgiati, CNS Neurol. Disord.: Drug Targets, 2012, 11, 299–305 CrossRef CAS PubMed.
- A. Tammimäki and P. T. Männistö, Pharmacogenet. Genomics, 2012, 22, 673–691 CrossRef PubMed.
- S. Z. Marsala, M. Gioulis, R. Ceravolo and M. Tinazzi, Clin. Neuropharmacol., 2012, 35, 185–190 CrossRef CAS PubMed.
- T. Müller, Drugs, 2015, 75, 157–174 CrossRef PubMed.
- B. T. Zhu, P. Wang, M. Nagai, Y. Wen and H. W. Bai, J. Steroid Biochem. Mol. Biol., 2009, 113, 65–74 CrossRef CAS PubMed.
- O. Weinreb, S. Mandel, T. Amit and M. B. Youdim, J. Nutr. Biochem., 2004, 15, 506–516 CrossRef CAS PubMed.
- E. Cuyàs, S. Verdura, J. Lozano-Sánchez, I. Viciano, L. Llorach-Parés, A. Nonell-Canals, J. Bosch-Barrera, J. Brunet, A. Segura-Carretero, M. Sanchez-Martinez, J. A. Encinar and J. A. Menendez, Food Chem. Toxicol., 2019, 128, 35–45 CrossRef PubMed.
- Y. Cao, Z. J. Chen, H. D. Jiang and J. Z. Chen, J. Phys. Chem. B, 2014, 118, 470–481 CrossRef CAS PubMed.
- Q. Zhao, X. Y. Chen and C. Martin, Sci. Bull., 2016, 61, 1391–1398 CrossRef CAS PubMed.
- T. Zhao, H. Tang, L. Xie, Y. Zheng, Z. Ma, Q. Sun and X. Li, J. Pharm. Pharmacol., 2019, 71, 1353–1369 CrossRef CAS PubMed.
- J. Choi, C. C. Conrad, C. A. Malakowsky, J. M. Talent, C. S. Yuan and R. W. Gracy, Biochim. Biophys. Acta, 2002, 1571, 201–210 CrossRef CAS.
- J. W. Song, J. Y. Long, L. Xie, L. L. Zhang, Q. X. Xie, H. J. Chen, M. Deng and X. F. Li, Chin. Med., 2020, 15, 102 CrossRef CAS PubMed.
- B. Zhang, S. Zhao, D. Yang, Y. Wu, Y. Xin, H. Cao, X. P. Huang, X. Cai, W. Sun, N. Ye, Y. Xu, Y. Peng, S. Zhao, Z. J. Liu, G. Zhong, M. W. Wang and W. Shui, Chin. Med., 2020, 6, 213–225 CAS.
- L. Xie, D. Y. Lee, Y. Shang, X. Cao, S. Wang, J. Liao, T. Zhang and R. Dai, Phytomedicine, 2020, 77, 153284 CrossRef CAS PubMed.
- R. Y. Zhang, Y. L. Cui, Y. Wang, X. G. Tian, L. Zheng, H. J. Cong, B. Wu, X. K. Huo, C. Wang, B. J. Zhang, X. B. Wang and Z. H. Yu, Eur. J. Drug Metab. Pharmacokinet., 2017, 42, 981–992 CrossRef CAS PubMed.
- J. A. Beutler, Curr. Protoc. Pharmacol., 2009, 46, 9.11.1–9.11.21 Search PubMed.
- R. X. Liu, G. H. Song, P. G. Wu, X. W. Zhang, H. J. Hu, J. Liu, X. S. Miao, Z. Y. Hou, W. Q. Wang and S. L. Wei, Chin. J. Nat. Med., 2017, 15, 152–160 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10425f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac,
Yang-Liu Xia
*ac,
Yang-Liu Xia