
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe homogeneous gas-phase formation mechanisms of PCPTs/PCDTs/PCDFs from the radical/radical cross-condensation of 2-CPR and 2-CTPR: a theoretical, mechanistic and kinetics study†
Ying Lia,
Yanan Hana,
Zhuochao Tenga,
Xianwei Zhaoa,
Yanhui Sunb,
Fei Xu *a,
Qingzhu Zhanga and
Wenxing Wanga
*a,
Qingzhu Zhanga and
Wenxing Wanga
aEnvironment Research Institute, Shandong University, Qingdao 266237, P. R. China. E-mail: xufei@sdu.edu.cn; Fax: +86-532-58631992
bCollege of Environment and Safety Engineering, Qingdao University of Science & Technology, Qingdao 266042, P. R. China
First published on 31st March 2021
Abstract
Polychlorinated phenoxathiins (PCPTs) are one group of dioxin-like compounds, which can be considered to be one-oxygen-substituted polychlorinated thianthrene (PCTA) compounds or one-sulfur-substituted polychlorinated dibenzo-p-dioxin (PCDD) compounds. Owing to their high toxicity and wide distribution, clarifying the formation and emission of PCPTs due to combustion and thermal processes can deepen our understanding of the dioxin formation mechanism and allow reduced-emission and dioxin-control strategies to be established. Chlorophenols (CPs) and chlorothiophenols (CTPs) are direct precursors in PCPT formation. In this paper, the homogeneous gas-phase formation mechanisms of PCPTs, as well as polychlorinated dibenzofurans (PCDFs) and polychlorinated dibenzothiophenes (PCDTs), from the cross-condensation of 2-chlorophenoxy radicals (2-CPRs) and 2-chlorothiophenoxy radicals (2-CTPRs) under thermal and combustion conditions were investigated theoretically using a density functional theory (DFT) method. The reaction priorities and effects of water molecules on the formation mechanisms were discussed. The rate constants of crucial elementary steps were calculated from 600–1200 K. The acute and chronic toxicities of the main products were predicted at three trophic levels. This study shows that routes starting with oxygen–carbon condensation are favored over those starting with sulfur–carbon condensation for PCPT formation, and routes ending with Cl loss can occur more easily than those ending with H loss. Water molecules have a negative catalytic effect on CH–S H-transfer steps but a positive catalytic effect on CH–O H-transfer steps.
1. Introduction
Polychlorinated dibenzo-p-dioxins/dibenzofurans (PCDD/Fs) belong to the often-mentioned typical persistent organic pollutant (POP) family because of their carcinogenic, teratogenic, and mutagenic effects and ubiquitous contamination of the global environment via global distillation.1,2 Polychlorinated thianthrenes/dibenzothiophenes (PCTA/DTs) are PCDD/F analogues in which all the oxygen atoms are replaced by sulfur atoms. The replacement of oxygen by sulfur can lower the toxicity of PCDT/TAs; however, the concentrations of PCDT/TAs in some typical regions are higher than those of PCDD/Fs.3,4 Polychlorinated phenoxathiins (PCPTs) are another group of chlorinated tricyclic aromatic heterocycles, which can be considered as one-oxygen-substituted PCTA compounds or one-sulfur-substituted PCDD compounds. Therefore, PCPTs and PCTA/DTs exhibit physicochemical, toxicological, ecotoxicological, and persistent properties similar to those displayed by PCDD/Fs, which can be viewed as dioxin-like compounds.5–10 The structures of PCPT/DD/TAs and PCDF/DTs are depicted in Scheme 1. It is well-established that PCDD/DFs and PCTA/DTs are never intentionally synthesized for commercial aims but are instead generated via a variety of combustion or thermal processes as unwanted byproducts, with examples including municipal and hazardous waste incinerators, industrial incinerators, fly ash, stack gas, and the metal reclamation industry.11–17 The simultaneous occurrence and mutual transformation of PCPTs, PCDD/DFs, and PCTA/DTs18–21 indicate their close correlation and possibly similar formation pathways. However, to our knowledge, there is no information available on the formation of PCPTs. Therefore, obtaining detailed insight into the formation of PCPTs will deepen our understanding of the formation mechanisms of dioxin-like compounds and could lead to emission-reduction and dioxin-control strategies being established.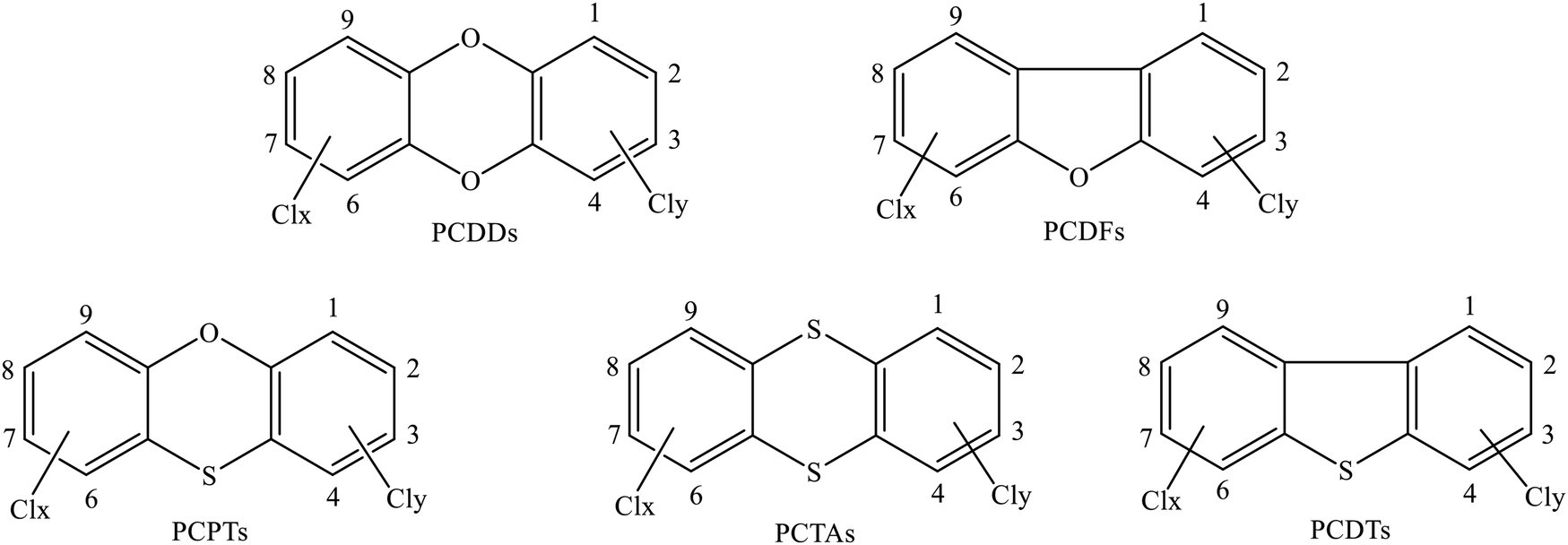 | ||
| Scheme 1 The structures of PCPT/DD/TAs and PCDF/DTs. | ||
The most direct route to the formation of PCDD/Fs and PCTA/DTs is homogeneous gas-phase reactions from chemical precursors. Chlorophenols (CPs) and chlorothiophenols (CTPs) are widely recognized to be the predominant precursors for PCDD/DT and PCTA/DT formation, respectively.3,22–27 CPs mainly arise from direct applications, such as biocides, emissions from operating facilities, bleaching operations, and wood-product leaching.28,29 CTPs have been widely used in various chemical industries, such as in polyvinyl chloride, insecticides, and the manufacturing of dyes, pharmaceuticals, and printing inks.30 Under high-temperature conditions, CPs and CTPs can readily form chlorophenoxy radicals (CPRs) and chlorothiophenoxy radicals (CTPRs) through losing phenoxyl-H and sulfhydryl-H, respectively, via direct H loss or abstraction reactions involving H, OH, Cl, and O(3P). The homogeneous gas-phase formation of PCDD/DFs and PCTA/DTs involves the radical/radical condensation of C(T)PRs and radical/molecule recombination of C(T)PR and C(T)P.3,21,22,27,31 Radical/radical condensation plays a crucial role in PCDD/DF and PCTA/DT formation.3,22–27,31 Based on radical/radical routes, a series of theoretical studies of PCDD/DF and PCTA/DT formation mechanisms from the coupling of 2-C(T)PRs, 2,4,5-TC(T)PRs, 2,4-DC(T)PRs, and 2,4,6-C(T)PRs has been carried out,3,27,31–36 and the consensus has been reached that PCDD and PCTA formation require the condensation of two C(T)PRs with at least one chlorine substituent in the ortho position, while PCDT formation needs two C(T)PRs with at least one available ortho hydrogen.3,27,31–36 For PCDD and PCTA formation, pathways that end with the elimination of Cl are dominant over pathways ending with the elimination of H.3,27,31–36 The chlorine substitution pattern has a significant effect on the self-dimerization of C(T)Ps.3,27,31–36 However, despite there being a lot of studies of the formation mechanisms of PCDD/DFs and PCTA/DTs from the radical/radical self-coupling of CPRs and CTPRs, as far as we know, there is no information available about the formation mechanisms of PCPT/DF/DTs from the cross-coupling of CPRs and CTPRs under high-temperature conditions. Considering the structural resemblance and similar properties of PCPTs and PCTA/DDs, and the coexistence of CTPs and CPs, the study of PCPT formation with both CP and CTP as precursors is significant.
Owing to their ability to form hydrogen bonds, water molecules can participate in many chemical reactions in the environment, such as unimolecular reactions, bimolecular reactions, hydrolysis reactions, and isomerization reactions via increasing or decreasing the energy potentials and catalyzing these reactions.37–41 Water molecules can also form hydrogen-bond clusters in the atmosphere and enhance aerosol nucleation processes.42,43 In real-world waste incineration, the original waste contains an amount of water. Additionally, in some plants, flue gases are quenched with cooling water at the end of furnace operations. Water vapor is abundant and ubiquitous in waste incineration and industrial operations. Therefore, the influence of water on dioxin formation needs to be studied. Several experimental and theoretical studies have been performed to investigate the effects of water on PCDD/F and PCTA/DT formation via de novo synthesis and precursor formation studies36,44–48 but no agreement has been reached. Stieglitz and Ross found that water can enhance the catalytic activity of fly ash and promote the yields of PCDD/Fs.44,45 However, Jay and Briois reported that the presence of water molecules can decrease the PCDD/F formation potential and isomer distribution.46,47 Shi et al. found that water molecules can promote the formation of PCDD/DFs via two routes: water molecules can participate actively in abstraction reactions involving CPs and H/OH radicals via lowering the potential barrier of CPR formation and in the H-shift step for the formation of PDDFs as a positive catalyst through proton transfer reactions via a bridge ring.48 However, Xu et al. indicated that water molecules have a negative catalytic effect on the H-shift step and hinder the formation of PCDTs from CTPs.36
Hence, in this work, we used an overall density functional theory (DFT) approach to study PCPT/DF/DT gas-phase formation with 2-CP and 2-CTP as forerunners under high-temperature pyrolysis or combustion conditions. All possible formation pathways involved in PCPT/DF/DT formation from the cross-condensation of 2-CPR and 2-CTPR were studied. Furthermore, the effects of water molecules on the PCPT/DF/DT formation mechanism are discussed. Some energetically preferred routes with and without water molecules were proposed to explain the isomer patterns of the PCPT/DF/DT products. Moreover, rate constants for the key elementary reactions from 600–1200 K were evaluated. The acute and chronic toxicity properties of the main products were predicted at three trophic levels. The last aim is to compare the formation potentials of PCDD/DF, PCTA/DT, and PCPT/DF/DT products from self- and cross-condensation mechanisms involving 2-CPR and 2-CTPR. The results can be input into dioxin control and prediction models as detailed parameters, which can be used to confirm the formation routes of dioxin-like compounds, reduce dioxin emissions, and establish dioxin-control strategies.
2. Computational methods
All calculations relating to chemical structures, energies, and frequencies of reactants, transition states, and products were performed using the Gaussian 09 program suite,49 along with the hybrid meta function MPWB1K.50 The MPWB1K method is a hybrid DFT model with effective performance in quantum calculations relating to thermochemistry, thermochemical kinetics, hydrogen bonding, and weak interactions.The geometry and vibrational frequency calculations were carried out at the MPWB1K level with a standard 6-31+G(d,p) basis set. The nature of the stationary points, the zero-point energy (ZPE), and the thermal contributions to the free energy of activation can be determined via vibrational frequency calculations. The overall reactions discussed in this paper were verified using intrinsic reaction coordinate (IRC) calculations and the minimum energy path (MEP) analysis of all transition states.51 To calculate the rate constants, 40 non-stationary points near the transition state along the minimum energy path (20 points on the reactant side and 20 points on the product side) were selected for frequency calculations at the MPWB1K/6-31+G(d,p) level. A more flexible basis set, 6-311+G(3df,2p), was used to determine the single-point energies of various species based on the optimized geometries. The profiles of the potential energy surfaces were constructed at the MPWB1K/6-311+G(3df,2p)//MPWB1K/6-31+G(d,p) level, including ZPE correction. All the relative energies quoted and discussed in this paper include ZPE corrections. The rate constants of the key elementary steps involved in this study were calculated over the wide temperature range of 600–1200 K using canonical variational transition-state (CVT) theory with small-curvature tunneling (SCT) correction.52–55 The rate constant calculations were performed using the POLYRATE 9.7 program.56 ECOSAR V2.0 (Ecological Structure Activity Relationships Version 2.0), developed by the US EPA (Environmental Protection Agency), was selected to assess the ecotoxicity properties of PCPT/DF/DTs from the reactions of 2-CPR and 2-CTPR.57
3. Results and discussion
The reliability and accuracy of the MPWB1K6-311+G(3df,2p)//MPWB1K/6-31+G(d,p) level for geometry and energy calculations were studied via comparison with experimental values, as shown in Table S1 of the ESI.† The optimized geometry of phenoxathiin at the MPWB1K/6-31+G(d,p) level is in reasonable accordance with the corresponding experimental values, and the largest discrepancies are within 2.1% and 2.9% for the bond lengths and bond angles, respectively, except for two abnormal values.58 For the reaction C6H5OH + C6H5SH → C12H8OS + 2H2, the reaction enthalpy of 26.32 kcal mol−1 calculated at the MPWB1K/6-311+G(3df,2p) level and at 298.15 K agrees well with the experimental value of 26.12 kcal mol−1, obtained from the measured standard enthalpy of formation (ΔHf,0) values of C6H5OH (−23.02 kcal mol−1), C6H5SH (26.85 kcal mol−1), C12H8OS (30.05 kcal mol−1), and H2 (0.03 kcal mol−1).59–62The formation of 2-C(T)PR from 2-C(T)P is the initial and key step in the formation of PCPT/DF/DTs. In combustion and thermal processes, 2-C(T)PR can be produced through the loss of phenoxyl-H or sulfhydryl-H via the unimolecular cleavage of the O–H or S–H bond or abstraction by active radicals. The potential barrier (ΔE) and reaction heat (ΔH) values of 2-CP and 2-CTP abstraction by H, OH, O(3P), and Cl were calculated at the MPWB1K/6-311+G(3df,2p) level, as shown in Table 1. Data for 2-C(T)P abstraction by H and OH are cited from our previous studies.63–65 The imaginary frequencies, zero-point energies, and total energies for the transition states involved in the formation of PCPT/DF/DTs from the cross-condensation of 2-CPR and 2-CTPR are shown in Table S2 of the ESI.† Cartesian coordinates for the reactants, intermediates, transition states, and products involved in the formation of PCPT/DF/DTs are depicted in Tables S4 and S5 of the ESI.†
| Reaction | ΔE | ΔH | Reference |
|---|---|---|---|
| 2-CP → 2-CPR + H | — | 85.91 | 63 |
| 2-CP + H → 2-CPR + H2 | 13.80 | −12.01 | 63 |
| 2-CP + OH → 2-CPR + H2O | 3.20 | −26.91 | 64 |
| 2-CP + O(3P) → 2-CPR + OH | 7.51 | −11.35 | This paper |
| 2-CP + Cl → 2-CPR + HCl | −2.32 | −14.96 | This paper |
| 2-CTP → 2-CTPR + H | — | 86.24 | This paper |
| 2-CTP + H → 2-CTPR + H2 | 3.42 | −14.43 | 65 |
| 2-CTP + OH → 2-CTPR + H2O | 8.67 | −27.96 | 65 |
| 2-CTP + O(3P) → 2-CTPR + OH | 2.51 | −12.39 | This paper |
| 2-CTP + Cl → 2-CTPR + HCl | −8.30 | −16.00 | This paper |
According to the different positions and numbers of Cl substituents on the benzene ring, PCPT/DF/DTs have different congeners. In this paper, PCPT congeners with zero to two chlorine substitutions are represented by phenoxathiins (PTs), monochlorinated phenoxathiins (MCPTs), and dichlorinated phenoxathiins (DCPTs), respectively. The prefix number represents the position (as given in Scheme 1) of chlorine substitution (e.g., 1-MCPT is the congener of PCPT in which one Cl atom substitutes at the 1 position and 1,6-DCPT is the congener of PCPT in which two Cl atoms substitute at the 1 and 6 positions). Similarly, PCDF/DT congeners with zero to two chlorine atoms are represented by DF/DT, MCDF/MCDT and DCDF/DCDT.
3.1 The formation of PCPTs from the cross-condensation of 2-CPR and 2-CTPR
Fig. 1 and 2 show the homogeneous gas-phase formation of PCPTs from 2-CPR and 2-CTPR, with the potential barrier (ΔE (in kcal mol−1)) and reaction heat (ΔH (in kcal mol−1)) values calculated at the MPWB1K/6-311+G(3df,2p)//MPWB1K/6-31+G(d,p) level. PCPT formation mechanisms involving oxygen–carbon condensation and sulfur–carbon condensation are presented in Fig. 1 and 2, respectively. In Fig. 1, there are two kinds of oxygen–carbon condensation modes: the condensation of chlorophenolic oxygen with the ortho-carbon bonded to the chlorine of the chlorothiophenoxy radical (O/CCl condensation for short); and the condensation of chlorophenolic oxygen with the ortho-carbon bonded to the hydrogen of the chlorothiophenoxy radical (O/CH condensation for short). Eight PCPT formation routes (routes 1–8) and five products (PT, 1-MCPT, 4-MCPT, 1,6-DCPT, and 4,6-DCPT), as illustrated in Fig. 1, are proposed to arise from the oxygen-carbon dimerization of 2-CPR and 2-CTPR. Similarly, there are two kinds of sulfur–carbon condensation modes: the condensation of chlorothiophenolic sulfur with the ortho-carbon bonded to the chlorine of the chlorophenoxy radical (S/CCl condensation for short); and the dimerization of chlorothiophenolic sulfur with the ortho-carbon bonded to the hydrogen of the chlorophenoxy radical (S/CH condensation for short). In Fig. 2, eight PCPT formation routes (routes 9–16) and five products (PT, 1-MCPT, 4-MCPT, 1,6-DCPT, and 1,9-DCPT) arise from sulfur–carbon condensation of 2-CPR and 2-CTPR.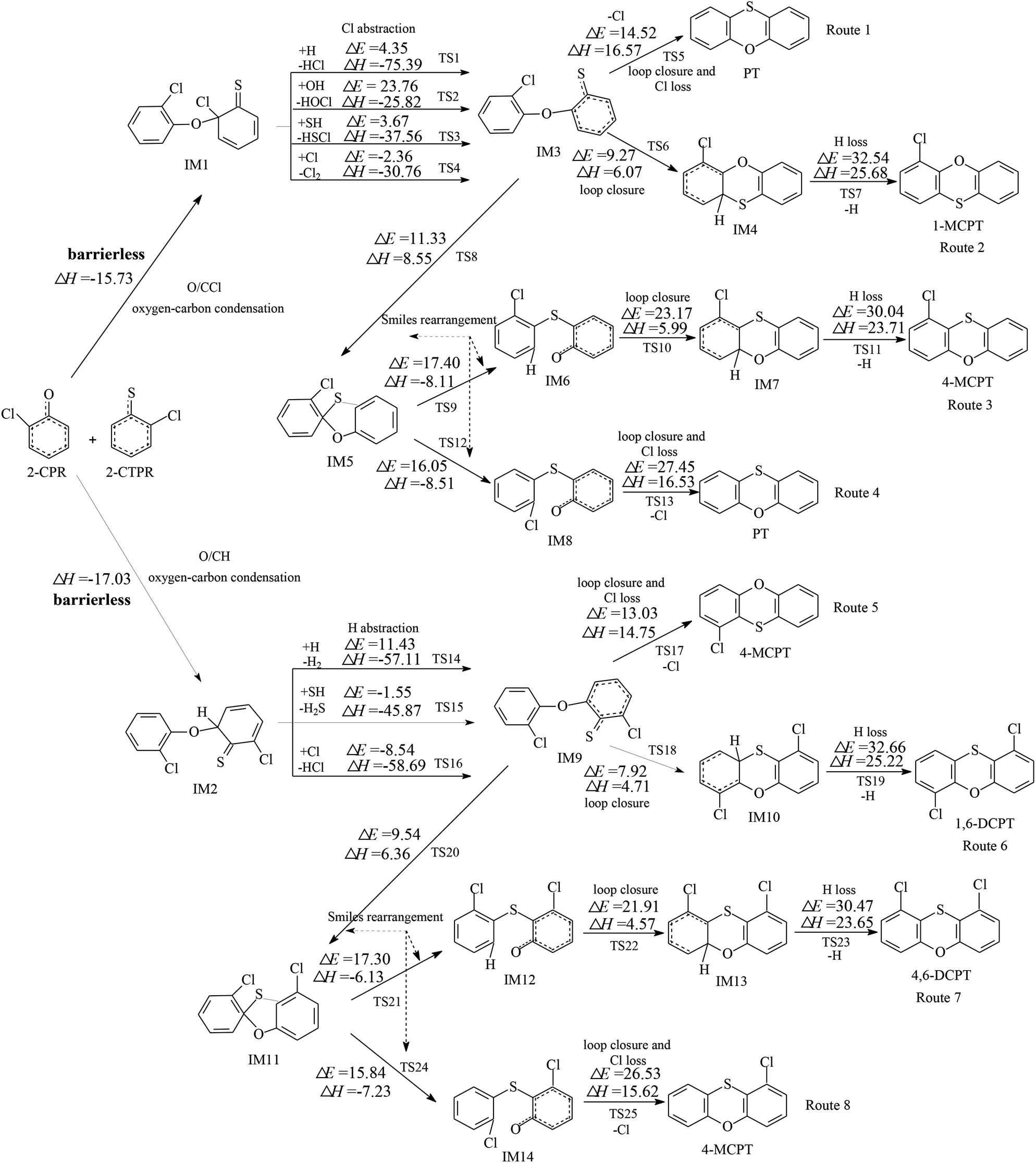 | ||
| Fig. 1 Formation routes of PCPTs including potential barrier (ΔE (in kcal mol−1)) and reaction heat (ΔH (in kcal mol−1)) values via the oxygen–carbon condensation (O/CCl and O/CH) of 2-CPR and 2-CTPR. ΔH is calculated at 0 K. | ||
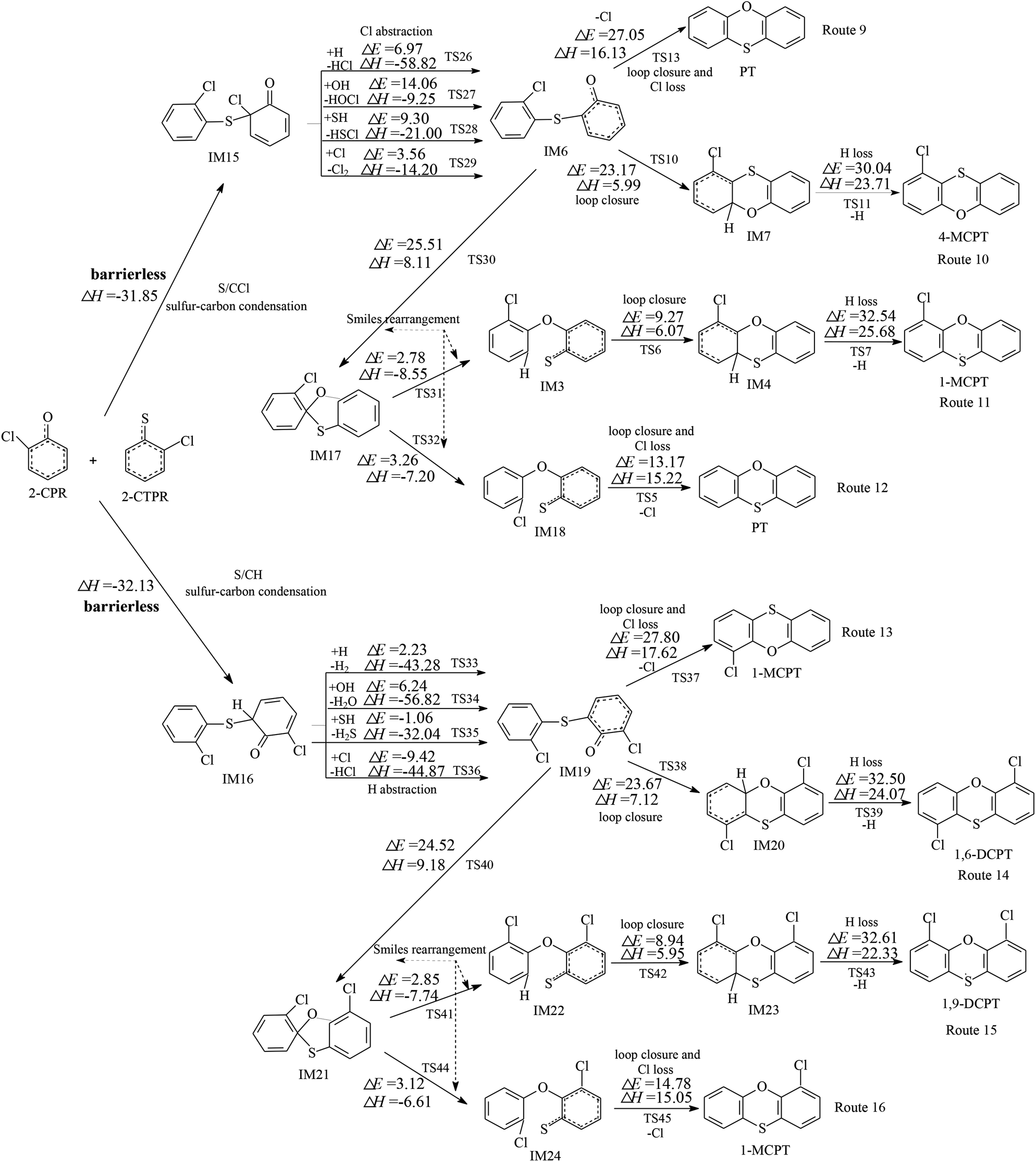 | ||
| Fig. 2 Formation routes of PCPTs including potential barrier (ΔE (in kcal mol−1)) and reaction heat (ΔH (in kcal mol−1)) values via the sulfur–carbon condensation (S/CCl and S/CH) of 2-CPR and 2-CTPR. ΔH is calculated at 0 K. | ||
All four (O/CCl, O/CH, S/CCl, and S/CH) condensation modes are barrierless and strongly exothermic processes, resulting in IM1 (with a reaction heat of −15.73 kcal mol−1), IM2 (with a reaction heat of −17.03 kcal mol−1), IM15 (with a reaction heat of −31.85 kcal mol−1), and IM16 (with a reaction heat of −32.13 kcal mol−1). The sulfur–carbon condensations are more exothermic compared to the oxygen–carbon condensations. This indicates that the sulfur–carbon condensations are favored over the oxygen–carbon condensations. For the oxygen–carbon condensations, O/CH condensation is preferred over O/CCl condensation; for the sulfur–carbon condensations, S/CH dimerization is preferred over S/CCl condensation. This may result from the fact that the steric hindrance effects of Cl atoms are larger than those of H atoms. Of the four intermediates, IM1 and IM15 undergo Cl abstraction, and IM2 and IM16 undergo H abstraction. All the Cl and H abstraction steps involve abstraction by H, OH, SH, and Cl radicals and produce IM3, IM9, IM6, and IM19 with low energy barriers and strong release heats. In routes 1, 5, 9, and 13, the loop closure and Cl-loss steps occur as one-step reactions and are rate-determining steps due to having the highest potential barriers and strongest endothermicities. However, the loop closure and H-loss steps occur separately in routes 2, 6, 10, and 14, and the H-loss steps are the rate-determining steps. Routes 3, 4, 7, 8, 11, 12, 15, and 16 contain two Smiles rearrangement steps after the H or Cl abstraction step. The rate-determining steps are the loop closure and Cl-loss or H-loss steps for routes 3, 4, 7, 8, 11, and 15, but are the first Smiles rearrangement steps for routes 12 and 16.
In Fig. 1, routes 1–4 cover three, four, six, and five elementary steps, respectively, and route 1 has the least elementary steps. Furthermore, the rate-determining step involved in route 1 has the lowest potential barrier (14.52 kcal mol−1) and is the least endoergic (16.57 kcal mol−1) among all four routes. Thus, route 1 is favored over routes 2–4. Similarly, route 5 (with a potential barrier of 13.03 kcal mol−1 and a reaction heat of 14.75 kcal mol−1) is preferred over routes 6–8. Therefore, the thermodynamically favorable PCPT formation routes are route 1 and route 5, resulting in the formation of PT and 4-MCPT. Comparing routes 1 and 5, the initiation step of route 5 is preferred, involving O/CH dimerization, over that of route 1, involving O/CCl dimerization. In addition, the rate-determining step involved in route 5 has a lower potential barrier and less endothermicity than that involved in route 1. Thus, route 5 is preferred over route 1. 4-MCPT is the main product from the oxygen–carbon condensation of 2-CPR and 2-CTPR, as seen in Fig. 1, followed by PT.
The comparison of reaction routes presented in Fig. 2 shows that route 9, ending with a loop-closure and Cl-loss step has one less elementary step than route 10, ending with a H-loss step. Furthermore, the rate-determining step involved in route 9 has a lower potential barrier and is much less endoergic than that involved in route 10. Thus, route 9 is favored over route 10. Route 11 and route 12 involve two more Smiles rearrangement steps compared with route 10 and route 9, respectively. For the same reason, route 12, ending with a loop-closure and Cl-loss step is favored over route 11, ending with a H-loss step. This indicates that routes ending with Cl loss are favored over those ending with H loss, owing to the C–Cl bond being weaker than the C–H bond. Comparing routes 9 and 12, the rate-determining step of route 9 is the loop-closure and Cl-loss step (potential barrier: 27.05 kcal mol−1, reaction heat: 16.13 kcal mol−1), whereas the rate-determining step of route 12 is the first Smiles rearrangement step (potential barrier: 25.51 kcal mol−1, reaction heat: 8.11 kcal mol−1). Although route 9 covers two less elementary steps than route 12, the rate-determining step of route 12 has a lower potential barrier and less endothermicity than route 9. Therefore, route 9 and route 12 should be competitive, resulting in the formation of PT. Similarly, route 13 and route 16 should be competitive, and the dominant product is 1-MCPT. Comparing routes 1 and 5 from Fig. 1 and routes 9, 12, 13, and 16 from Fig. 2, the rate-determining steps of the most favored routes in Fig. 1 contain lower potential barriers (13.03–14.52 kcal mol−1) than those in Fig. 2 (24.52–27.80 kcal mol−1). This means that routes 1 and 5 from Fig. 1 are preferred over routes 9, 12, 13, and 16 in Fig. 2, and the main products from routes starting with the oxygen–carbon and sulfur–carbon condensation of 2-CPR and 2-CTPR are PT and 4-MCPT.
It is interesting to compare PCPT formation from the cross-condensation of 2-CPR and 2-CTPR with PCDD and PCTA formation from the self-condensation of 2-CPR and 2-CTPR.32,36 Our study investigated the PCDD formation routes from the self-coupling of 2-CPR, and the potential barriers of the rate-determining steps are 27.03–27.41 kcal mol−1.32 Fig. S1 in the ESI† shows the PCTA formation routes from the self-coupling of 2-CTPR, and the potential barriers of the loop-closure and Cl-loss steps are 12.28–14.60 kcal mol−1.36 This study shows that the potential barriers of the rate-determining steps from the oxygen–carbon condensation of 2-CPR and 2-CTPR are 13.03–14.52 kcal mol−1. Thus, the PCPT/DD/TA formation potentials from the self- and cross-condensation of 2-CPR and 2-CTPR can be ranked as follows: self-condensation of 2-CTPR (resulting in TA and 1-MCTA) > cross-condensation of 2-CPR and 2-CTPR (resulting in PT and 4-MCPT) > self-condensation of 2-CPR (resulting in DD and 1-MCDD).
3.2 The formation of PCDF/DTs from the cross-condensation of 2-CPR and 2-CTPR
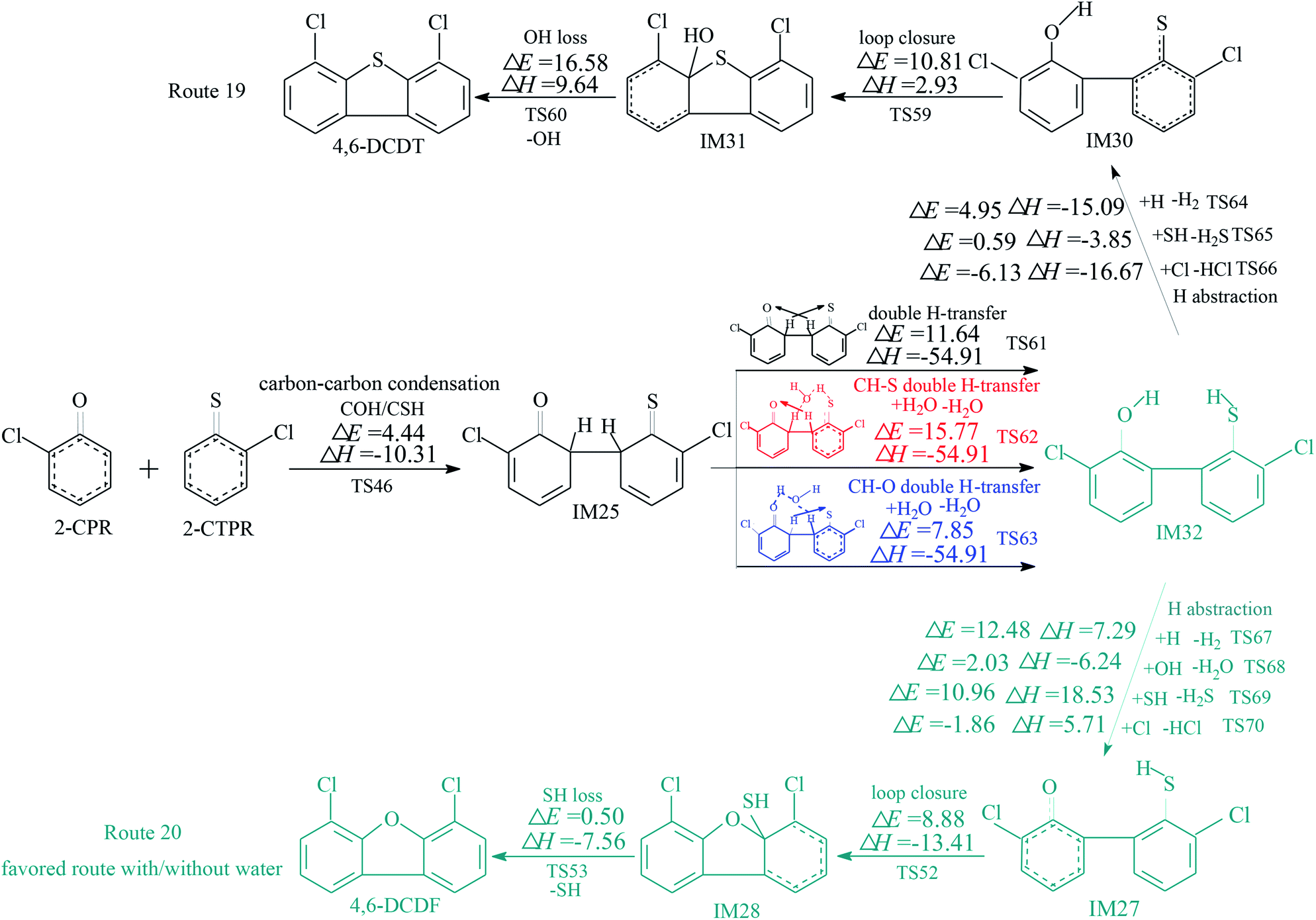
The homogeneous gas-phase formation routes of PCDFs and PCDTs from the cross-condensation of 2-CPR and 2-CTPR with potential barriers (ΔE (in kcal mol−1)) and reaction heats (ΔH (in kcal mol−1)) calculated at the MPWB1K/6-311+G(3df,2p)//MPWB1K/6-31+G(d,p) level are presented in Fig. 3–5. Six PCDF/DT formation routes (routes 17–22) and four products (4,6-DCDF, 4,6-DCDT, 4-MCDF, and 4-MCDT), illustrated in Fig. 3–5, are proposed from the carbon–carbon condensation of 2-CPR and 2-CTPR. Three types of carbon–carbon condensation coupling mechanisms for PCDF/DT formation are presented in Fig. 3–5: the condensation of the ortho-carbon bonded to the hydrogen of the chlorophenoxy radical with the ortho-carbon bonded to the hydrogen of the chlorothiophenoxy radical (COH/CSH condensation for short) in routes 17–20 of Fig. 3 and 4; the condensation of the ortho-carbon bonded to the hydrogen of the chlorophenoxy radical with the ortho-carbon bonded to the chlorine of the chlorothiophenoxy radical (COH/CSCl condensation for short) in route 21 of Fig. 5; and the condensation of the ortho-carbon bonded to the chlorine of the chlorophenoxy radical with the ortho-carbon bonded to the hydrogen of the chlorothiophenoxy radical (COCl/CSH condensation for short) in route 22 of Fig. 5. In Fig. 3 and 5, the routes colored red represent the favored routes without the introduction of water molecules and the blue routes are the favored routes with the introduction of water molecules. The green routes represent the favored routes both with and without the introduction of water molecules. The optimized geometries for selected transition states in the formation processes of PCPT/DT/DFs from the radical/radical cross-condensation of 2-CPR and 2-CTPR are showed in Fig. 6.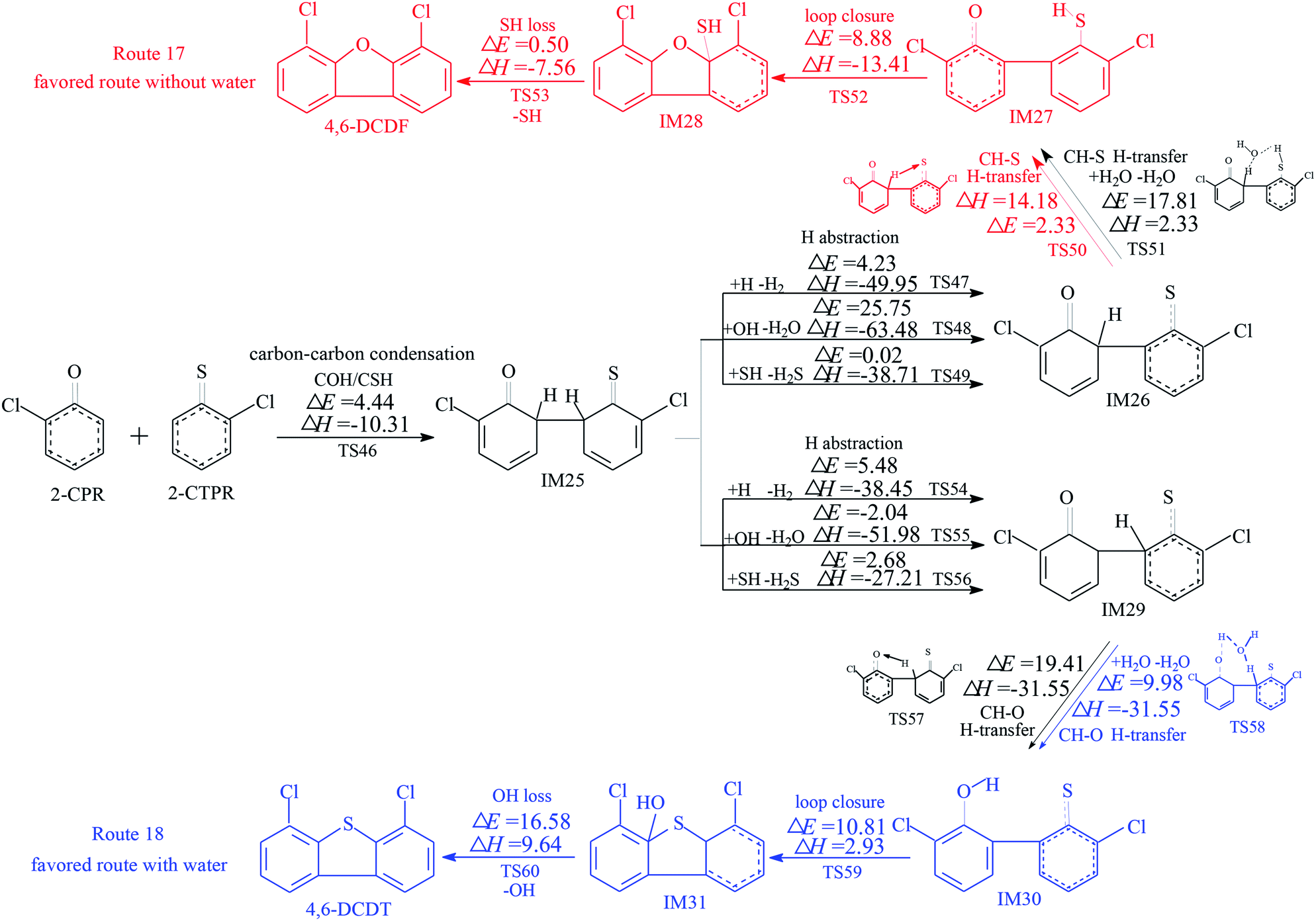 | ||
| Fig. 3 Formation routes of PCDF/DTs including potential barrier (ΔE (in kcal mol−1)) and reaction heat (ΔH (in kcal mol−1)) values via the carbon–carbon condensation (COH/CSH) of 2-CPR and 2-CTPR with H-transfer steps. ΔH is calculated at 0 K. The red route represents the favored route without the introduction of water molecules and the blue route shows the favored route with the introduction of water molecules. | ||
 | ||
| Fig. 4 Formation routes of PCDF/DTs including potential barrier (ΔE (in kcal mol−1)) and reaction heat (ΔH (in kcal mol−1)) values via the carbon–carbon condensation (COH/CSH) of 2-CPR and 2-CTPR with double H transfer steps. ΔH is calculated at 0 K. The green route represents the favored route both with and without the introduction of water molecules. | ||
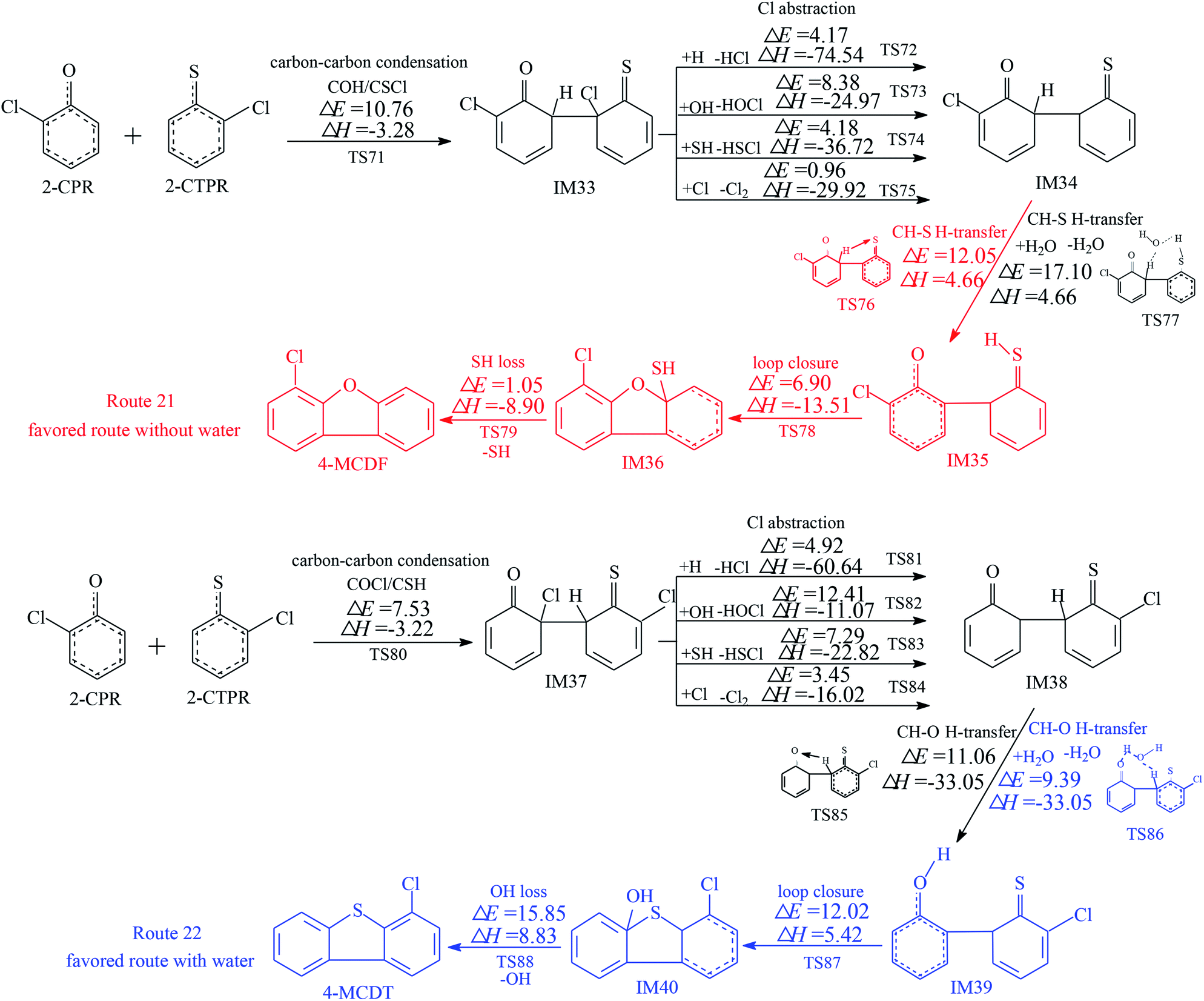 | ||
| Fig. 5 Formation routes of PCDF/DTs including potential barrier (ΔE (in kcal mol−1)) and reaction heat (ΔH (in kcal mol−1)) values via the carbon–carbon condensation (COCl/CSH and COH/CSCl) of 2-CPR and 2-CTPR. ΔH is calculated at 0 K. The red route represents the favored route without the introduction of water molecules, and the blue route shows the favored route with the introduction of water molecules. | ||
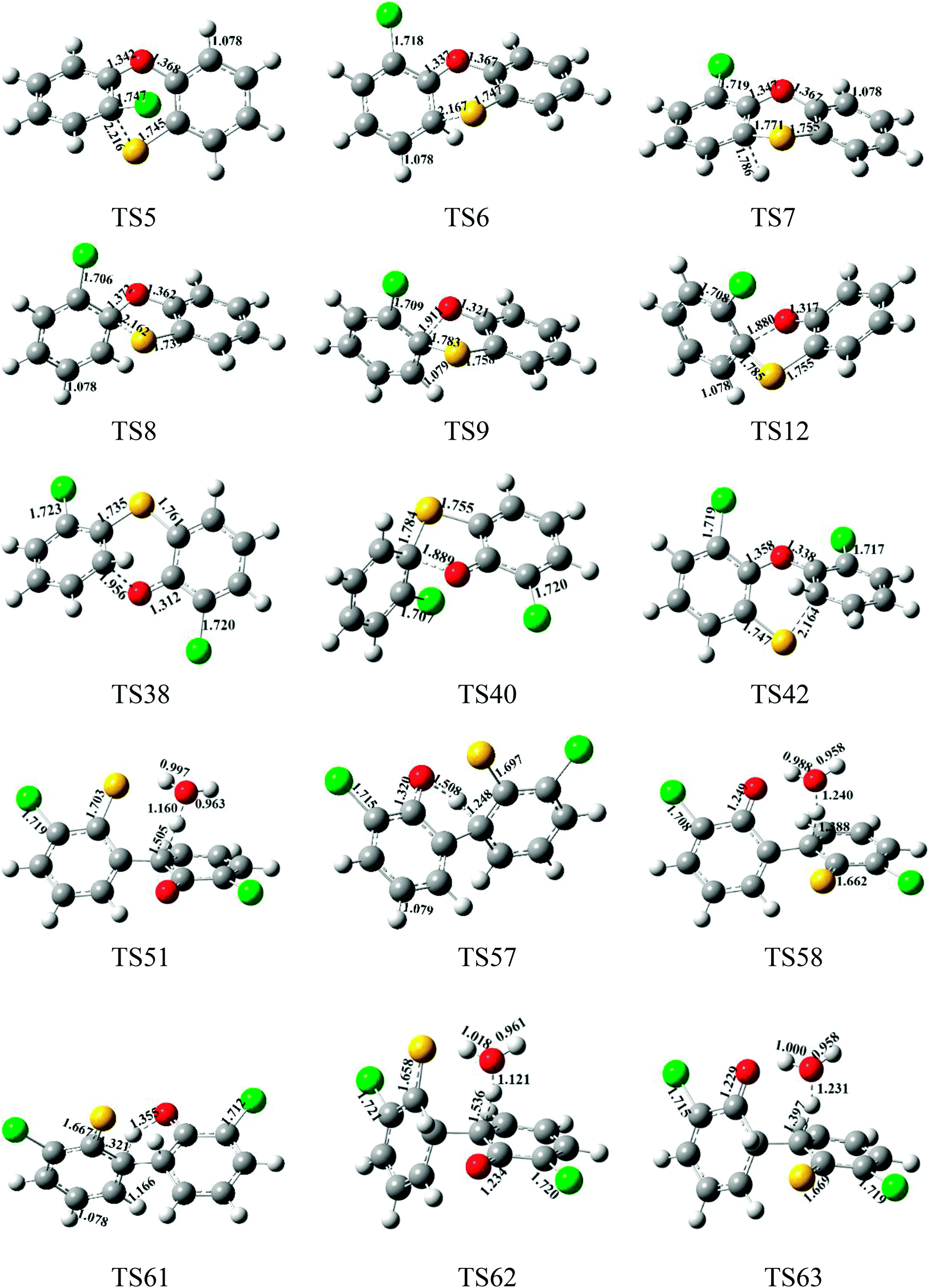 | ||
| Fig. 6 MPWB1K/6-31+G(d,p)-optimized geometries for selected transition states in the formation processes of PCPT/DT/DFs from the radical/radical cross-condensation of 2-CPR and 2-CTPR; distances are in angstroms; gray spheres: C, white spheres: H, red spheres: O, yellow spheres: S, green spheres: Cl. | ||
In Fig. 3, routes 17 and 18 involve the following five elementary steps: (1) COH-CSH condensation; (2) H abstraction; (3) H transfer; (4) loop closure; and (5) SH loss. The H atom shifts from the ortho-carbon of the phenoxy ring to the S atom of the thiophenoxy ring (CH–S H-transfer for short) via TS50 in route 17, whereas the H atom shifts from the ortho-carbon of the thiophenoxy ring to the O atom of the thiophenoxy ring (CH–O H-transfer for short) via TS57 in route 18. Water molecules can participate actively in both the CH–S H-transfer and CH–O H-transfer steps involved in the formation of PCDF/DTs through the formation of hydrogen bonds. As seen in Fig. 3, direct CH–S H-transfer and CH–O H-transfer, via intramolecular isomerization, proceed through a five-membered ring transition state, whereas the H-transfer steps, via a bimolecular reaction with the help of water, proceed through a seven-membered ring transition state. In the seven-membered transition state, the water molecule acts as a bridge, accepting a hydrogen atom from an aromatic ring and simultaneously donating another hydrogen atom to the oxygen keto atom of the other aromatic ring. In route 17, without water molecules, the potential barrier for the CH–S H-transfer step (IM26 → IM27) is 14.18 kcal mol−1. Upon the introduction of water, the potential barrier of the CH–S H-transfer step becomes 17.81 kcal mol−1. The potential barrier of the CH–S H-transfer step with the aid of water molecules is about 4 kcal mol−1 higher than direct CH–S H-transfer without water molecules, which indicates that water molecules have a negative catalytic role in the CH–S H-transfer step and hinder the formation of 4,6-DCDFs. However, for the CH–O H-transfer step in route 18, the potential barrier of the CH–O H-transfer step with the aid of water molecules (9.98 kcal mol−1) is about 10 kcal mol−1 lower than direct CH–O H-transfer without water molecules (19.41 kcal mol−1), which means that water molecules play a positive catalytic role in the CH–O H-transfer step and promote the formation of 4,6-DCDT. Under dry conditions, the rate-determining steps of both routes 17 and 18 are the H-transfer steps. The potential barrier of CH–S H-transfer (14.18 kcal mol−1) is lower than that of CH–O H-transfer (19.41 kcal mol−1). Thus, route 17 is favored over route 18. The main product without water molecules is 4,6-DCDF. With the participation of water molecules, the rate-determining step of route 17 is the bimolecular CH–S H-transfer reaction, but the rate-determining step of route 18 becomes the OH-loss step. The potential barrier of the rate-determining step of route 17 (17.81 kcal mol−1) is higher than that of route 18 (16.58 kcal mol−1). Thus, route 18 was favored over route 17 under wet conditions. The main product with the aid of water molecules is 4,6-DCDT.
In Fig. 4, routes 19 and 20 involve the following five elementary steps: (1) COH/CSH condensation; (2) double H transfer; (3) H abstraction; (4) loop closure; and (5) OH/SH loss. Double H transfer can occur with and without the introduction of water molecules, and it involves a concerted reaction, with two H atoms shifting at the same time. The potential barrier of direct double H transfer via the intramolecular isomerization of TS61 is 11.64 kcal mol−1. Water molecules can participate in CH–S H-transfer with the other H atom directly shifting to the O atom via TS62 (potential barrier: 15.77 kcal mol−1), and they can participate in CH–O H-transfer with the other H atom directly shifting to the S atom via TS63 (potential barrier: 7.85 kcal mol−1). In Fig. 4, the potential barrier of TS62 is higher than that of TS61, whereas the potential barrier of TS63 is lower than that of TS61. This means that water molecules participating in CH–S double H transfer have a negative catalytic effect on PCDF/DT formation, and water molecules participating in CH–O double H transfer have a positive catalytic effect on PCDF/DT formation. Routes 19 and 20 have the same first two steps, and differences occur in the last three elementary steps. For route 19, the OH-loss step involves the highest barrier (16.58 kcal mol−1) and is more endoergic (9.64 kcal mol−1) than any elementary step in the last three elementary steps of route 19, e.g., the OH-loss step is the rate-determining step of route 19. For the same reason, the loop-closure step is the rate-determining step of route 20 (potential barrier: 8.88 kcal mol−1, reaction heat: −13.41 kcal mol−1). Obviously, under conditions both with and without water molecules, the rate-determining step of route 20 can occur via a lower potential barrier than that of route 19, which means that pathway 20 (resulting in 4,6-DCDF) is thermodynamically favored compared with pathway 19 (resulting in 4,6-DCDT). Thus, 4,6-DCDF is the main product from the double H transfer route of the cross-coupling of 2-CPR and 2-CTPR under both dry and wet conditions.
Two reaction routes, shown in Fig. 5, are offered to interpret the formation of 4-MCDF and 4-MCDT. Routes 21 and 22 are similar to routes 17 and 18, involving five elementary processes: (1) COH–CSCl/COCl-CSH condensation; (2) Cl abstraction; (3) H transfer (CH–S H-transfer and CH–O H-transfer with and without water); (4) loop closure; and (5) SH/OH loss. It can be seen from Fig. 3–5 that COH–CSCl/COCl-CSH coupling has higher potential barriers (10.76 and 7.53 kcal mol−1) and is less exothermic (−3.28 and −3.22 kcal mol−1) than COH-CSH coupling (potential barrier: 4.44 kcal mol−1, reaction heat: −10.31 kcal mol−1). These results indicate that COH-CSH condensation is favored over COH-CSCl/COCl-CSH condensation. In route 21, water molecules have an inhibiting effect on CH–S H-transfer and restrain the formation of 4-MCDF, which increases the potential barrier by about 5 kcal mol−1 compared with direct CH–S H-transfer. Direct CH–S H-transfer (potential barrier: 12.05 kcal mol−1, reaction heat: 4.66 kcal mol−1) and CS–H H-transfer with water (potential barrier: 17.10 kcal mol−1, reaction heat: 4.66 kcal mol−1) are the rate-determining steps of route 21 under dry and wet conditions, respectively. However, in route 22, water molecules have a positive catalytic effect on CH–O H-transfer and promote the formation of 4-MCDF, which decreases the potential barrier by about 1.5 kcal mol−1 compared with direct CH–O H-transfer. The rate-determining step of route 22 both with and without water is the OH-loss step, which has the highest potential barrier (15.85 kcal mol−1) and is the most strongly endothermic (8.83 kcal mol−1) in route 22. Under conditions without water, the rate-determining step of route 21 (12.05 kcal mol−1) has a lower potential barrier than that of route 22 (15.85 kcal mol−1), i.e., route 21 is favored over route 22 and 4-MCDF is the main product in a dry incinerator from the CH–CCl coupling of 2-CPR and 2-CTPR. However, under conditions involving water, the rate-determining step of route 21 (17.10 kcal mol−1) requires crossing a higher potential barrier than route 22 (15.85 kcal mol−1), i.e., route 22 is favored over route 21 and 4-MCDT is the dominant product from the CH–CCl coupling of 2-CPR and 2-CTPR in a wet incinerator.
Under dry conditions, without the introduction of water molecules, for PCDF/DT formation from 2-CPR and 2-CTPR, the favored routes are route 17, route 20, and route 21, shown in Fig. 3–5, respectively. The potential barriers of the rate-determining steps of route 17, route 20, and route 21 are 14.18, 8.88, and 12.05 kcal mol−1, which indicates that route 20 can occur more easily than routes 17 and 21, resulting in the formation of 4,6-DCDF. Under wet conditions, with the introduction of water molecules, route 18, route 20, and route 22 are the energetically preferred routes, as shown in Fig. 3–5, and the potential barriers of the rate-determining steps of route 18, route 20, and route 22 are 16.58, 8.88, and 15.85 kcal mol−1, respectively. Thus, under wet conditions, the most favored route is also route 20 for PCDF/DT formation from the cross-condensation of 2-CPR and 2-CTPR.
It is necessary to compare PCDF/DT formation from the cross-condensation of 2-CPR and 2-CTPR with our previous studies of PCDF formation from the self-coupling of 2-CPR and PCDT formation from 2-CTPR (Fig. S1 in the ESI†).32,36 For PCDF formation from the self-coupling of 2-CPR, no matter with or without water molecules, the rate-determining steps are the ring-closure steps, with potential barriers of 28.12 and 29.17 kcal mol−1 and reaction heats of 8.73 and 9.56 kcal mol−1.32 For PCDT formation from the coupling of 2-CTPR, under both dry and wet conditions, the rate-determining step is the ring-closure step, with a potential barrier of 12.37 kcal mol−1 and reaction heat of −0.21 kcal mol−1.36 For PCDF/DT formation from the cross-coupling of 2-CPR and 2-CTPR, the potential barriers of the rate-determining steps are in the range of 8.88–14.18 kcal mol−1 and the reaction heats are in the range of −13.41–4.66 kcal mol−1 under conditions without water molecules; the potential barriers of the rate-determining steps are in the range of 8.88–16.58 kcal mol−1 and the reaction heats are in the range of −13.41–9.64 kcal mol−1 under conditions with water molecules. PCDF/DT formation from the cross-coupling of 2-CPR and 2-CTPR has a lower potential barrier and is more exothermic than from the self-coupling of 2-CPR or 2-CTPR, i.e., PCDF/DT formation from the cross-coupling of 2-CPR and 2-CTPR is more energetically favored than from the self-coupling of 2-CPR or 2-CTPR. The PCDF/DT formation potentials from the self- and cross-condensation of 2-CPR and 2-CTPR can be ranked as follows: cross-coupling of 2-CPR and 2-CTPR (resulting in 4,6-DCDF) > self-coupling of 2-CTPR (resulting in 4,6-DCDT) > self-coupling of 2-CPR (resulting in 4,6-DCDF).
3.3 Rate-constant calculations
In the environmental field, kinetics models are used to account for the potential outcomes regarding contamination of the environment and gaseous routes during production processes. However, for combustion and thermal processes, owing to the strict high-temperature experimental conditions required for dioxin formation and the lack of effective detection methods for short-lifetime intermediates, the availability of kinetics parameters and data, such as pre-exponential factors, activation energies, and rate constants, is limited and insufficient. This causes inaccuracies in dioxin-formation prediction models and difficulties relating to further improving and optimizing dioxin-formation models. Therefore, an alternative method is to use calculated rate constants or other dynamic information directly obtained from quantum calculations of electronic structures, frequencies, and energies.In this study, the rate constants of the crucial elementary reactions for PCPT/DF/DT formation from the cross-condensation of 2-CPR and 2-CTPR were calculated using the CVT/SCT method on the basis of the MPWB1K/6-311+G(3df,2p)//MPWB1K/6-31+G(d,p) energy, as seen in Table S3 of the ESI.† The CVT/SCT values were obtained over a wide temperature range of 600–1200 K, which covers the possible formation temperatures of PCPT/DF/DTs in combustion and thermal processes. Our previous studies of PCDD/DF formation from CPs and PCTA/DT formation with CTPs as precursors have successfully proved the accuracy of the CVT/SCT method for calculating the rate constants for the elementary reactions involved in dioxin-like compound formation.32–36
To be applied more effectively, the CVT/SCT rate constants were fitted, and the Arrhenius formulae are presented in Table 2 for PCPT formation from the oxygen–carbon coupling and sulfur–carbon coupling reactions between 2-CPR and 2-CTPR and in Table 3 for PCDF/DT formation from the carbon–carbon coupling reactions between 2-CPR and 2-CTPR. The pre-exponential factors and activation energies can be obtained from these Arrhenius formulae.
| Reaction | Arrhenius formula |
|---|---|
| IM1 + H → IM3 + HCl via TS1 | k(T) = (7.44 × 10−12) exp (−2921/T) |
| IM1 + SH → IM3 + HSCl via TS3 | k(T) = (2.00 × 10−11) exp (−3289/T) |
| IM3 → PAT + Cl via TS5 | k(T) = (4.87 × 1011) exp (−7520/T) |
| IM3 → IM4 via TS6 | k(T) = (6.69 × 1011) exp (−4742/T) |
| IM4 → 4-MCPAT + H via TS7 | k(T) = (2.79 × 1013) exp (−17407/T) |
| IM3 → IM5 via TS8 | k(T) = (6.91 × 1011) exp (−6087/T) |
| IM5 → IM6 via TS9 | k(T) = (1.49 × 1014) exp (−14986/T) |
| IM6 → IM7 via TS10 | k(T) = (3.71 × 1011) exp (−15672/T) |
| IM7 → 1-MCPAT + H via TS11 | k(T) = (2.44 × 1013) exp (−15995/T) |
| IM5 → IM8 via TS12 | k(T) = (2.27 × 1013) exp (−8498/T) |
| IM8 → PAT + Cl via TS13 | k(T) = (6.88 × 1013) exp (−16356/T) |
| IM2 + H → IM9 + H2 via TS14 | k(T) = (2.58 × 10−7) exp (−1768/T) |
| IM9 → 1-MCPAT + Cl via TS17 | k(T) = (3.79 × 1011) exp (−6743/T) |
| IM9 → IM10 via TS18 | k(T) = (6.84 × 1011) exp (−3962/T) |
| IM10 → 1,6-DCPAT + H via TS19 | k(T) = (3.29 × 1013) exp (−17359/T) |
| IM9 → IM11 via TS20 | k(T) = (2.09 × 1012) exp (−5179/T) |
| IM11 → IM12 via TS21 | k(T) = (3.31 × 1013) exp (−12656/T) |
| IM12 → IM13 via TS22 | k(T) = (2.56 × 1011) exp (−11083/T) |
| IM13 → 1,9-DCPAT + H via TS23 | k(T) = (2.50 × 1013) exp (−16351/T) |
| IM11 → IM14 via TS24 | k(T) = (5.59 × 1013) exp (−8358/T) |
| IM14 → 1-MCPAT + Cl via TS25 | k(T) = (9.34 × 1013) exp (−16363/T) |
| IM15 + H → IM6 + HCl via TS26 | k(T) = (1.06 × 10−10) exp (−4442/T) |
| IM15 + OH → IM6 + HOCl via TS27 | k(T) = (1.60 × 10−11) exp (−8836/T) |
| IM15 + SH → IM6 + HSCl via TS28 | k(T) = (1.49 × 10−11) exp (−6518/T) |
| IM15 + Cl → IM6 + Cl2 via TS29 | k(T) = (1.62 × 10−10) exp (−3299/T) |
| IM6 → IM17 via TS30 | k(T) = (4.28 × 1013) exp (−15588/T) |
| IM17 → IM3 via TS31 | k(T) = (5.27 × 1011) exp (−3271/T) |
| IM17 → IM18 via TS32 | k(T) = (7.99 × 1012) exp (−1931/T) |
| IM16 + H → IM19 + H2 via TS33 | k(T) = (2.06 × 10−11) exp (−1841/T) |
| IM16 + OH → IM19 + H2O via TS34 | k(T) = (1.53 × 10−14) exp (−2044/T) |
| IM19 → 4-MCPAT + Cl via TS37 | k(T) = (4.77 × 1013) exp (−16550/T) |
| IM19 → IM20 via TS38 | k(T) = (2.60 × 1012) exp (−15842/T) |
| IM20 → 4,9-DCPAT + H via TS39 | k(T) = (3.44 × 1013) exp (−17337/T) |
| IM19 → IM21 via TS40 | k(T) = (3.10 × 1014) exp (−17336/T) |
| IM21 → IM22 via TS41 | k(T) = (1.06 × 1013) exp (−1702/T) |
| IM22 → IM23 via TS42 | k(T) = (6.45 × 1011) exp (−4408/T) |
| IM23 → 4,6-DCPAT + H via TS43 | k(T) = (2.44 × 1013) exp (−17412/T) |
| IM21 → IM24 via TS44 | k(T) = (8.86 × 1012) exp (−1828/T) |
| IM24 → 4-MCPAT + Cl via TS45 | k(T) = (1.79 × 1011) exp (−7624/T) |
| Reaction | Arrhenius formula |
|---|---|
| IM25 + H → IM26 + H2 via TS47 | k(T) = (2.09 × 10−12) exp (−2864/T) |
| IM25 + OH → IM26 + H2O via TS48 | k(T) = (1.38 × 10−13) exp (−13998/T) |
| IM25 + SH → IM26 + H2S via TS49 | k(T) = (1.59 × 10−16) exp (−2285/T) |
| IM26 → IM27 via TS50 | k(T) = (6.62 × 1011) exp (−7387/T) |
| IM27 → IM28 via TS52 | k(T) = (1.15 × 1012) exp (−4596/T) |
| IM28 → 4,6-DCDF + SH via TS53 | k(T) = (3.51 × 1012) exp (−460/T) |
| IM25 + H → IM29 + H2 via TS54 | k(T) = (2.38 × 10−11) exp (−3464/T) |
| IM25 + SH → IM29 + H2S via TS56 | k(T) = (1.15 × 10−12) exp (−3779/T) |
| IM29 → IM30 via TS57 | k(T) = (1.04 × 1012) exp (−11786/T) |
| IM30 → IM31 via TS59 | k(T) = (1.45 × 1012) exp (−5780/T) |
| IM31 → 4,6-DCDT + OH via TS60 | k(T) = (5.89 × 1011) exp (−7775/T) |
| IM25 → IM32 via TS61 | k(T) = (1.52 × 1012) exp (−6133/T) |
| IM32 + H → IM30 + H2 via TS64 | k(T) = (2.35 × 10−11) exp (−2932/T) |
| IM32 + SH → IM30 + H2S via TS65 | k(T) = (3.64 × 10−12) exp (−2407/T) |
| IM32 + H → IM27 + H2 via TS67 | k(T) = (3.25 × 10−11) exp (−6646/T) |
| IM32 + OH → IM27 + H2O via TS68 | k(T) = (2.37 × 10−12) exp (−2560/T) |
| IM33 + H → IM34 + HCl via TS72 | k(T) = (1.42 × 10−15) exp (−1011/T) |
| IM33 + OH → IM34 + HOCl via TS73 | k(T) = (8.81 × 10−14) exp (−4175/T) |
| IM33 + SH → IM34 + HSCl via TS74 | k(T) = (1.03 × 10−12) exp (−3705/T) |
| IM33 + Cl → IM34 + Cl2 via TS75 | k(T) = (2.50 × 10−11) exp (−2071/T) |
| IM34 → IM35 via TS76 | k(T) = (2.35 × 1013) exp (−6718/T) |
| IM35 → IM36 via TS78 | k(T) = (1.30 × 1012) exp (−3565/T) |
| IM36 → 4-MCDF + SH via TS79 | k(T) = (4.27 × 1011) exp (−1119/T) |
| IM37 + H → IM38 + HCl via TS81 | k(T) = (3.56 × 10−11) exp (−3261/T) |
| IM37 + OH → IM38 + HOCl via TS82 | k(T) = (4.85 × 10−13) exp (−7399/T) |
| IM37 + SH → IM38 + HSCl via TS83 | k(T) = (1.82 × 10−12) exp (−5469/T) |
| IM37 + Cl → IM38 + Cl2 via TS84 | k(T) = (3.22 × 10−11) exp (−3523/T) |
| IM38 → IM39 via TS85 | k(T) = (3.36 × 1012) exp (−5954/T) |
| IM39 → IM40 via TS87 | k(T) = (9.04 × 1011) exp (−6340/T) |
| IM40 → 4-MCDT + OH via TS88 | k(T) = (2.07 × 1013) exp (−8563/T) |
In order to explore whether the favored routes change with increasing temperature, we fit plots of the CVT/SCT rate constants in the temperature range of 600–1200 K for the rate-determining steps of each PCPT/DT/DF formation route from the cross-condensation of 2-CPR and 2-CTPR, which is shown in Fig. S2 in the ESI.† As can be seen from Fig. S2,† in the range of 600–1200 K, the plot lines have no cross-points, which means that the ranking of CVT/SCT values for the rate-determining steps of the PCPT/DT/DF formation routes from the cross-condensation of 2-CPR and 2-CTPR is consistent. For example, for PCPT formation in routes 1–4, the CVT/SCT values of TS5 in route 1 are consistently larger than those of TS7 in route 2, TS 11 in route 3, and TS 13 in route 4, which agrees well with the thermal analysis that route 1 is favored over routes 2–4. For PCDF/DT formation from the cross-condensation of 2-CPR and 2-CTPR in the range of 600–1200 K via routes 17 and 18, the CVT/SCT values of TS50 in route 17 are consistently larger than those of TS60 in route 18. The favored routes under wet and dry conditions are uniform with increasing temperature in routes 17 and 18. A similar trend occurs for PCDF/DT formation via routes 19 and 20 and routes 21 and 22. Thus, it can be inferred that the favored routes labelled in color in Fig. 3–5 will not change with increasing temperature for PCDF/DT formation.
3.4 Ecotoxicity assessment
Using ECOSAR, the acute and chronic toxicity properties of the main products (PT, 4-MCPT, 1-MCPT, 1,6-DCPT, 4,6-DCPT, 1,9-DCPT, 4-MCDF, 4,6-DCDF, 4-MCDT, and 4,6-DCDT) in this study were predicted at three trophic levels (i.e., green algae, daphnia, and fish). The acute toxicity values were measured using LC50 (50% lethal concentration of the tested compound after daphnia and fish were exposed for 48 and 96 h, respectively) and EC50 (the concentration of the tested chemical at which half of green algae experience growth inhibition after 96 h of exposure) values. The chronic toxicity values are expressed as ChV, representing the repeat dose of a substance that can initiate the development of adverse effects. It can be concluded from Table 4 that the toxicity levels of the products were consistent at the three trophic levels (i.e., green algae, daphnia, and fish) in an aquatic environment, and the toxicity order is 1,6-DCPT/4,6-DCPT/1,9-DCPT > 4,6-DCDT > 4,6-DCDF > 4-MCPT/1-MCPT > 4-MCDT > PT > 4-MCDF. In order to compare the acute and chronic toxicity properties of the products of PCPT formation from the cross-condensation of 2-CPR and 2-CTPR with the acute and chronic toxicity properties of PCDD and PCTA formation from the self-condensation of 2-CPR and 2-CTPR,32,36 we added the toxicity values for DD, 1-MCDD, 2,7-DCDD, TA, 1-MCTA and 2,7-DCTA to Table 4. From Table 4, for the given chlorine substitutions, the toxicities of PCPTs are higher than those of PCDDs and PCTAs. Hence, more attention should be paid to controlling the emission of PCPTs and to the treatment of this pollution.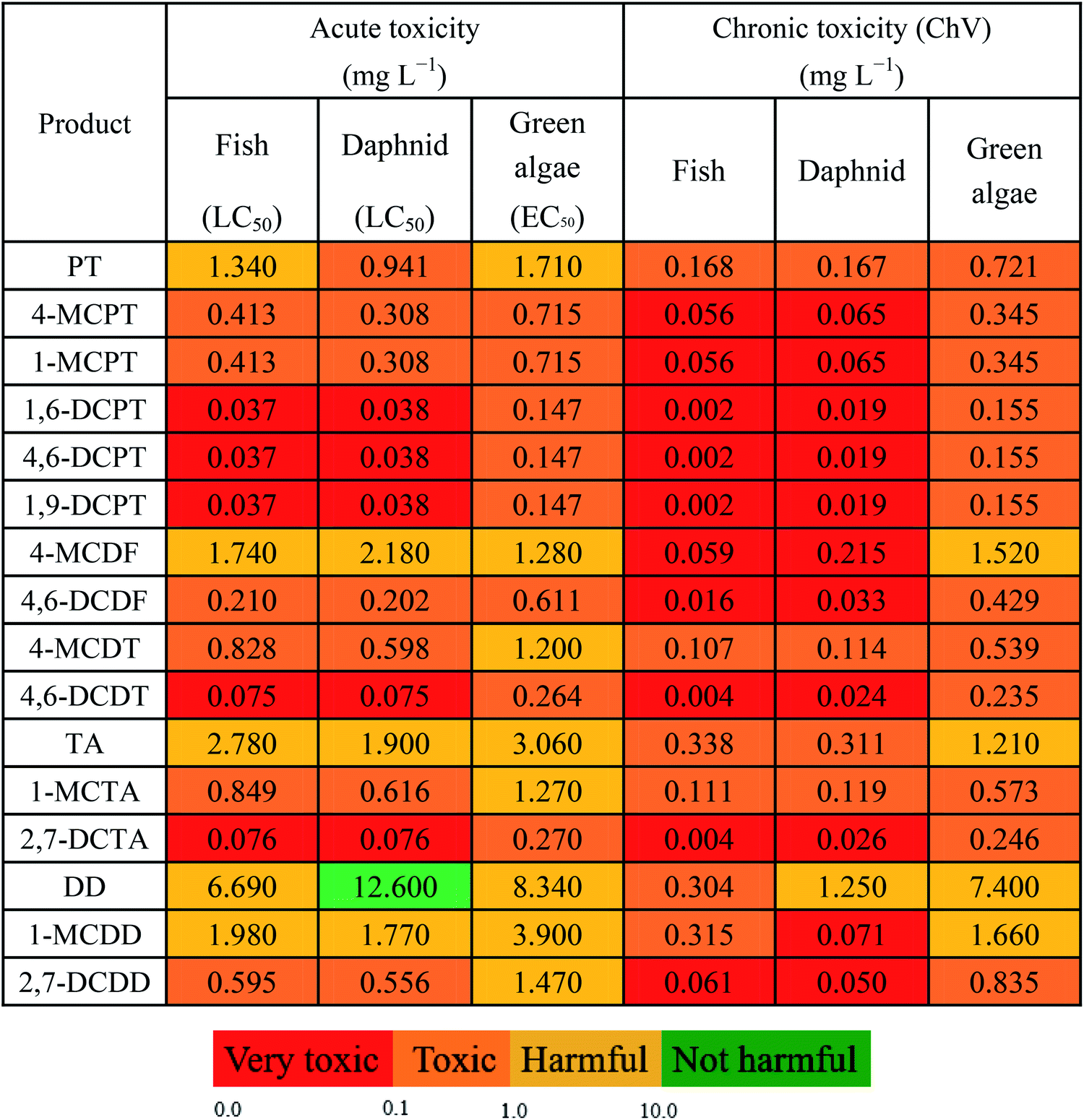 |
4. Conclusions
In this study, the mechanisms of the homogeneous gas-phase formation of PCPT/DF/DTs from the cross-condensation of 2-CPR and 2-CTPR were investigated theoretically using DFT electronic structure theory at the MPWB1K/6-311+G(3df,2p)//MPWB1K/6-31+G(d,p) level. The formation potentials of PCTA/DT products and the roles of water molecules in the mechanisms were discussed. The mechanisms were compared with PCDD/DF formation from the self-coupling of 2-CPR and PCTA/DT formation from 2-CTP. Kinetics calculations were carried out and the rate constants were calculated over the temperature range of 600–1200 K using the CVT/SCT method. The acute and chronic toxicity properties of the main products were predicted at three trophic levels. Four specific conclusions can be drawn, as follows.(1) In PCPT formation from the cross-coupling of 2-CPR and 2-CTPR, routes 1 and 5 are the most favored routes, resulting in the formation of PT and 4-MCPT. Routes starting with oxygen–carbon condensation are favored over those starting with sulfur–carbon condensation, and routes ending with the elimination of Cl are energetically preferred to routes ending with the elimination of H.
(2) In PCDF/DT formation from the cross-coupling of 2-CPR and 2-CTPR, route 20 is the energetically preferred route under both wet and dry conditions, resulting in the formation of 4,6-DCDF. Water molecules participating in CH–S transfer have a negative catalytic effect on PCDF/DT formation and a positive catalytic effect on PCDF/DT formation.
(3) For PCDT/DD/TA formation, the formation potentials for the self- and cross-condensation of 2-CPR and 2-CTPR rank as follows: self-condensation of 2-CTPRs > cross-condensation of 2-CPR and 2-CTPR > self-condensation of 2-CPRs. For PCDF/DT formation, the formation potentials rank as follows: cross-coupling of 2-CPR and 2-CTPR > self-coupling of 2-CTPRs > self-coupling of 2-CPRs.
(4) The order of product toxicity is 1,6-DCPT/4,6-DCPT/1,9-DCPT > 4,6-DCDT > 4,6-DCDF > 4-MCPT/1-MCPT > 4-MCDT > PT > 4-MCDF. The toxicities of PCPTs are higher than those of PCDT/TA/DF/DDs for a given form of chlorine substitution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (project nos. 21876102 and 21976107), the Fundamental Research Funds of Shandong University (No. 2016WLJH51), and the China Postdoctoral Science Foundation funded project (nos. 2017M612277 and 2017T100493). The authors thank Professor Donald G. Truhlar for providing the POLYRATE 9.7 program.References
- A. Schecter, Dioxin and Health, Plenum Press, New York, 1994 Search PubMed.
- H. Hung, P. Blanchard, G. Poole, B. Thibert and C. H. Chiu, Atmos. Environ., 2002, 36, 1041–1050 CrossRef CAS.
- T. Dar, M. Altarawneh and B. Z. Dlugogorski, Environ. Sci. Technol., 2013, 47, 11040–11047 CrossRef CAS PubMed.
- Z. Cai, D. E. Giblin, V. M. S. Ramanujam, M. L. Gross and A. Cristini, Environ. Sci. Technol., 1994, 28, 1535–1538 CrossRef CAS PubMed.
- Y. Wang, X. Zeng, H. Chen and H. Wang, Thermodynamic properties and relative stability of polychlorinated thianthrenes by density functional theory, J. Chem. Eng. Data, 2007, 52, 1442–1448 CrossRef CAS.
- T. Puzyn, P. Rostkowski, A. Swieczkowski, A. Jedrusiak and J. Falandysz, Chemosphere, 2006, 62, 1817–1828 CrossRef CAS PubMed.
- R. Weber, H. Hagenmaier and D. Schrenk, Chemosphere, 1998, 36, 2635–2641 CrossRef CAS.
- S. Nakai, M. P. Espino, Y. Nomura and M. Hosomi, J. Environ. Chem., 2004, 14, 835–844 CrossRef CAS.
- A. Mostrg, T. Puzyn and M. Haranczyk, Environ. Sci. Pollut. Res., 2010, 17, 470–477 CrossRef PubMed.
- Y. Wang and X. L. Zeng, J. Chem. Eng. Data, 2008, 53, 513–519 CrossRef CAS.
- J. C. Harris, P. C. Anderson, B. E. Goodwin and C. E. Rechsteiner, Final Report to ASME, ASME, New York, NY, 1980 Search PubMed.
- R. Addink and E. R. Altwicker, Environ. Sci. Technol., 2004, 38, 5196–5200 CrossRef CAS PubMed.
- A. Yasuhara, T. Katami, T. Okuda, N. Ohno and P. Adriaens, Environ. Sci. Technol., 2001, 35, 1373–1378 CrossRef CAS PubMed.
- H. R. Buser, Chemosphere, 1992, 25, 45–48 CrossRef CAS.
- S. Sinkkonen, J. Paasivirta, J. Koistinen and J. Tarhanen, Chemosphere, 1991, 23, 583–587 CrossRef CAS.
- S. Sinkkonen, E. Kolehmainen, J. Koistinen and M. Lahtipera, J. Chromatogr. A, 1993, 641, 309–317 CrossRef CAS.
- S. Sinkkonen, A. Vattulainen, J. P. Aittola, J. Paasivirta, J. Tarhanen and M. Lahtipera, Chemosphere, 1994, 28, 1279–1288 CrossRef CAS.
- H. R. Buser and C. Rappe, Anal. Chem., 1991, 63, 1210–1217 CrossRef CAS.
- C. Rappe, Chemosphere, 1993, 27, 211–225 CrossRef CAS.
- T. Wiedmann, U. Riehle, J. Kurz and K. Ballschmiter, Fresenius' J. Anal. Chem., 1997, 359, 176–188 CrossRef CAS.
- E. Ferrario, Bull. Soc. Chim. Fr., 1911, 9, 536–537 CAS.
- C. S. Evans and B. Dellinger, Environ. Sci. Technol., 2003, 37, 1325–1330 CrossRef CAS.
- C. S. Evans and B. Dellinger, Environ. Sci. Technol., 2005, 39, 122–127 CrossRef CAS PubMed.
- J. Y. Ryu, J. A. Mulholland, D. H. Kim and M. Takeuchi, Environ. Sci. Technol., 2005, 39, 4398–4406 CrossRef CAS PubMed.
- J. Y. Ryu, J. A. Mulholland and J. E. Oh, Chemosphere, 2004, 55, 1447–1455 CrossRef CAS PubMed.
- M. Altarawneh, B. Z. Dlugogorski, E. M. Kennedy and J. C. Mackie, J. Phys. Chem. A, 2007, 111, 2563–2573 CrossRef CAS PubMed.
- T. Dar, M. Altarawneh and B. Z. Dlugogorski, Organohalogen Compd., 2012, 74, 657–660 Search PubMed.
- R. Buisson, P. Kirk and J. Lester, J. Chromatogr. Sci., 1984, 22, 339–342 CAS.
- R. C. C. Wegman and H. H. Van den Broek, Water Res., 1983, 17, 227–230 CrossRef CAS.
- R. Navarro, K. Bierbrauer, C. Mijangos, E. Goiti and H. Reinecke, Polym. Degrad. Stab., 2008, 93, 585–591 CrossRef CAS.
- T. Dar, K. Shah, B. Moghtaderi and A. J. Page, J. Mol. Model., 2016, 22, 128 CrossRef PubMed.
- Q. Zhang, S. Li, X. Qu, X. Shi and W. Wang, Environ. Sci. Technol., 2008, 42, 7301–7308 CrossRef CAS PubMed.
- X. Qu, H. Wang, Q. Zhang, X. Shi, F. Xu and W. Wang, Environ. Sci. Technol., 2009, 43, 4068–4075 CrossRef CAS PubMed.
- F. Xu, W. Yu, R. Gao, Q. Zhou, Q. Zhang and W. Wang, Environ. Sci. Technol., 2010, 44, 6745–6751 CrossRef CAS PubMed.
- Q. Zhang, W. Yu, R. Zhang, Q. Zhou, R. Gao and W. Wang, Environ. Sci. Technol., 2010, 44, 3395–3403 CrossRef CAS PubMed.
- F. Xu, X. Shi, Y. Li and Q. Zhang, Int. J. Mol. Sci., 2015, 16, 20449–20467 CrossRef CAS PubMed.
- C. Iuga, J. R. Alvarez and A. V. Bunge, J. Phys. Chem. A, 2011, 115, 5138–5146 CrossRef CAS PubMed.
- M. Torrent-Sucarrat, J. S. Francisco and J. M. Anglada, J. Am. Chem. Soc., 2012, 134, 20632–20644 CrossRef CAS PubMed.
- J. Gonzalez, J. M. Anglada, R. J. Buszek and J. S. Francisco, J. Am. Chem. Soc., 2011, 133, 3345–3353 CrossRef CAS PubMed.
- M. K. Hazra and A. Sinha, J. Am. Chem. Soc., 2011, 133, 17444–17453 CrossRef CAS PubMed.
- K. Shao, J. Yan, X. Li, S. Lu, Y. Wei and M. Fu, Chemosphere, 2010, 78, 672–679 CrossRef CAS PubMed.
- Y. Lin, Y. Ji, Y. Li, J. Secrest, W. Xu, F. Xu, Y. Wang, T. An and R. Zhang, Atmos. Chem. Phys., 2019, 19, 8003–8019 CrossRef CAS.
- W. Xu and R. Zhang, J. Chem. Phys., 2013, 139, 1–11 Search PubMed.
- L. Stieglitz, G. Zwick, J. Beck, H. Bautz and W. Ooth, Chemosphere, 1990, 20, 1953–1958 CrossRef CAS.
- B. J. Ross, D. Lacombe and K. P. Naikwadi, Chemosphere, 1990, 20, 1967–1972 CrossRef CAS.
- C. Briois, S. Ryan, D. Tabor, A. Touati and B. Gullett, Environ. Sci. Technol., 2007, 41, 850–856 CrossRef CAS PubMed.
- K. Jay and L. Stieglitz, Chemosphere, 1991, 22, 987–996 CrossRef CAS.
- X. Shi, R. Zhang, H. Zhang, F. Xu, Q. Zhang and W. Wang, Chemosphere, 2015, 137, 142–148 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- M. S. Baldridge, R. Gordon, R. Steckler and D. G. Truhlar, J. Chem. Phys., 1989, 93, 5107–5119 CrossRef.
- A. Gonzalez-Lafont, T. N. Truong and D. G. Truhlar, J. Chem. Phys., 1991, 95, 8875–8894 CrossRef CAS.
- B. C. Garrett and D. G. Truhlar, J. Chem. Phys., 1979, 83, 1052–1079 CrossRef CAS.
- A. Fernandez-Ramos, B. A. Ellingson, B. C. Garret and D. G. Truhlar, in Reviews in Computational Chemistry, Wiley-VCH, Hoboken, NJ, USA, 2007 Search PubMed.
- J. C. Corchado, Y. Y. Chuang, P. L. Fast, J. Villa, W. P. Hu, Y. P. Liu, G. C. Lynch, K. A. Nguyen, C. F. Jackels and V. S. Melissas, POLYRATE version 9.7, University of Minnesota, Minneapolis, Minnesota, MN, USA, 2007 Search PubMed.
- P. Peter, S. Maurizio, D. Martina, W. Dietmar and K. Thomas, Chemosphere, 2008, 71, 1986–1995 CrossRef PubMed.
- V. S. Mastryukov, K. H. Chen, S. H. Slmonsen, N. L. Allinger and J. E. Boggs, J. Mol. Struct., 1997, 413, 1–12 CrossRef.
- J. D. Cox, Pure Appl. Chem., 1961, 2, 125–128 CAS.
- D. W. Scott, J. P. Mccullough and W. N. Hubbard, J. Am. Chem. Soc., 1956, 78, 5463–5468 CrossRef CAS.
- W. V. Steele, R. D. Chirico and S. E. Knipmeyer, J. Chem. Thermodyn., 1993, 25, 965–992 CrossRef CAS.
- J. D. Cox, D. D. Wagman and V. A. Medvedev, CODATA Key Values for Thermodynamics, Hemisphere Publishing Corp., New York, 1984, p. 1 Search PubMed.
- Q. Zhang, X. Qu, H. Wang, F. Xu, X. Shi and W. Wang, Environ. Sci. Technol., 2009, 43, 4105–4112 CrossRef CAS PubMed.
- F. Xu, H. Wang, Q. Zhang, R. Zhang, X. Qu and W. Wang, Environ. Sci. Technol., 2010, 44, 1399–1404 CrossRef CAS PubMed.
- F. Xu, X. Shi, Q. Zhang and W. Wang, Int. J. Mol. Sci., 2015, 16, 18714–18731 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PCTA/DT formation routes from the self-coupling of 2-CTPR; fitted plots of the CVT/SCT rate constants in the temperature range of 600–1200 K for the rate-determining steps in each PCPT/DT/DF formation route; a comparison of the calculated and experimental structural parameters of phenoxathiin; imaginary frequencies, zero-point energies, and total energies for the transition states involved in the formation of PCPT/DF/DTs from the cross-condensation of 2-CPR and 2-CTPR; CVT/SCT rate constants for the formation of PCPT/DT/DFs over the temperature range of 600–1200 K; and Cartesian coordinates for the transition states, reactants, intermediates, and products involved in the formation of PCPT/DT/DFs. See DOI: 10.1039/d1ra00599e |
| This journal is © The Royal Society of Chemistry 2021 |