DOI:
10.1039/D1RA00514F
(Paper)
RSC Adv., 2021,
11, 7511-7520
Aerobically stable and substitutionally labile α-diimine rhenium dicarbonyl complexes†
Received
20th January 2021
, Accepted 5th February 2021
First published on 15th February 2021
Abstract
New synthetic routes to aerobically stable and substitutionally labile α-diimine rhenium(I) dicarbonyl complexes are described. The molecules are prepared in high yield from the cis–cis–trans-[Re(CO)2(tBu2bpy)Br2]− anion (2, where tBu2bpy is 4,4′-di-tert-butyl-2,2′-bipyridine), which can be isolated from the one electron reduction of the corresponding 17-electron complex (1). Compound 2 is stable in the solid state, but in solution it is oxidized by molecular oxygen back to 1. Replacement of a single bromide of 2 by σ-donor monodentate ligands (Ls) yields stable neutral 18-electron cis–cis–trans-[Re(CO)2(tBu2bpy)Br(L)] species. In coordinating solvents like methanol the halide is replaced giving the corresponding solvated cations. [Re(CO)2(tBu2bpy)Br(L)] species can be further reacted with Ls to prepare stable cis–cis–trans-[Re(CO)2(tBu2bpy)(L)2]+ complexes in good yield. Ligand substitution of Re(I) complexes proceeds via pentacoordinate intermediates capable of Berry pseudorotation. In addition to the cis–cis–trans-complexes, cis–cis–cis- (all cis) isomers are also formed. In particular, cis–cis–trans-[Re(CO)2(tBu2bpy)(L)2]+ complexes establish an equilibrium with all cis isomers in solution. The solid state crystal structure of nearly all molecules presented could be elucidated. The molecules adopt a slightly distorted octahedral geometry. In comparison to similar fac-[Re(CO)3]+complexes, Re(I) diacarbonyl species are characterized by a bend (ca. 7°) of the axial ligands towards the α-diimine unit. [Re(CO)2(tBu2bpy)Br2]− and [Re(CO)2(tBu2bpy)Br(L)] complexes may be considered as synthons for the preparation of a variety of new stable diamagnetic dicarbonyl rhenium cis-[Re(CO)2]+ complexes, offering a convenient entry in the chemistry of the core.
Introduction
The chemistry of metal complexes of the bidentate diimine Re(I) tricarbonyl core (fac-[ReI(CO)3]+) is widely investigated due to the rich photochemistry and photophysics of the molecules,1–3 their potential applications as diagnostic and anticancer agents,4,5 electro- and photoinduced-catalysts,6,7 artificial photosynthetic8 and supramolecular materials.9 In comparison, the chemistry of the Re(I) dicarbonyl core (cis-[ReI(CO)2]+) is rare. In general the chemistry of Re(I) dicarbonyl complexes is dominated by π-acids ligands which effectively replace electronically the role of the “missing” carbonyl and stabilize the d6 core via π-back bonding. In most cases, such species are substitutionally inert, thereby limiting our understanding of the core. To our knowledge, there are no examples of [2 + 1] complexes of cis-[ReI(CO)2] species (where 2 + 1 indicates the combination of a bidentate and a monodentate ligand) in which one of the two is not a π-acid ligand of the PR3 (phosphine and phosphites), NCR or CNR (nitrile or isonitrile), CN− or NO+ type.
Chemically the majority of Re(I) dicarbonyl species is prepared in one of three ways: by trimethylamine N-oxide (Me3NO), photolytic or thermal decarbonylation. Trimethylamine N-oxide (Me3NO) reacts selectively and irreversibly with metal bound CO following a second-order rate law consistent with a bimolecular mechanism.10 The reagent has been used successfully for the decarbonylation of different Re tricarbonyl complexes.10–17 Kurtz et al., e.g., prepared a series of compounds of the type cis-[Re(diimine)(CO)2(L)Cl] (where L = P(OEt)3, PMe3) by exploiting the trans-labilizing effect of phosphorus ligands to facilitate carbonyl replacement.12,17 Similarly the group of Wilson synthesized and structurally characterized analogous complexes as the active photoactivated species in a study that reported the in vitro anticancer activity of rhenium(I) tricarbonyl complexes bearing water-soluble phosphines.13
Nitrile and isonitrile ligands (together with other π-acids such as PR3, vide infra) dominate the coordination chemistry of dicarbonyl Re(I) complexes. In the early 2000's Kromer and Alberto introduced and studied the substitution chemistry of cis–trans-[Re(CO)2(CH3CN)2Br2]− species.18,19 The two acetonitriles are strongly bound and are not substituted by mono- and bidentate ligands. In the last decade the groups of Ko and Ishitani have introduced tunable isocyano and acetonitrile rhenium(I) diimine luminophores of general formula cis,cis-[Re(CO)2(N⁀N)(CNR)2]+ and cis,cis- and cis–trans-[Re(CO)2(L′/′′)2(N⁀N)]+ (with L′ = CH3CN and L′′ = CH3CN, py, PR3 or halide).7,20–25 Complexes are prepared by photo-substitutions/decarbonylation of fac-[Re(CO)3(CNR)2Br] in the presence of diimine ligands (N⁀N) with broadband or UV excitation. N⁀N isonitrile analogues of the Kromer complexes were similarly obtained by photolytic decarbonylation of fac-[Re(CO)3(N⁀N)(CNR)]+ in the presence of CNR. We have described and structurally characterized cis–fac-[Re(CO)2(CNR)3Br] obtained from the reaction of cis-[Re(CO)2Br4]−/2− with CNR in a study aimed at the preparation of cardiolite-inspired carbon monoxide releasing molecules.26 More recently Triantis reported the synthesis and characterization of the dicarbonyl mixed ligand cis-[Re(CO)2(quin)(CNR)(PPh3)] by refluxing a toluene solution of fac-[Re(CO)3(quin)(CNR)] with triphenylphosphine (quin = quinaldic acid).27
The vast majority of complexes of the cis-[ReI(CO)2] core are those with PR3 ligands (mono-, bi- or tridentate chelates) occupying coordination sites of the metal. Species of formula cis–trans-[ReI(CO)2(PR3)2(N⁀N)]+ have been known for fifty years,28 but it was in the last two decades that their luminescent properties have been studied in details, particularly since the realization of linear and closed multicomponent systems bridged by R2P⁀PR2 chelates. The groups of Sullivan and Meyer28–30 and Ishitani31–41 have pioneered this chemistry and several examples of luminescent monomeric35,42 or R2P⁀PR2 bridged linear and ring shaped polymeric systems have been reported.43,44
Our group has been interested in the chemistry of 18- and 17-electron fac-[ReI(CO)3]+ and cis-[ReII(CO)2]2+ species for their potential use in medicinal chemistry, particularly as anticancer45–48 and antibacterial agents26,49,50 and CO-releasing molecules respectively.51–54 While abundant literature is available for the biological effects of Re(I) tricarbonyl complexes, very little is known for corresponding Re(I) dicarbonyl species. We, therefore, set out to establish synthetic procedures that would allow accessing the latter core. Of interest to us was the realization of stable and substitutionally labile molecules that could allow preparation of a variety of molecules, not limited to π-acid ligands. In this contribution we describe our efforts and we introduce new synthetic routes to aerobically stable α-diimine cis-[ReI(CO)2]+ complexes with σ- and π-donor only ligands. The species may be considered as cis-[Re(CO)2]+ synthons, offering a convenient entry in the chemistry of the core. The synthesis, spectroscopic and structural properties of molecules are described.
Results and discussion
Synthesis
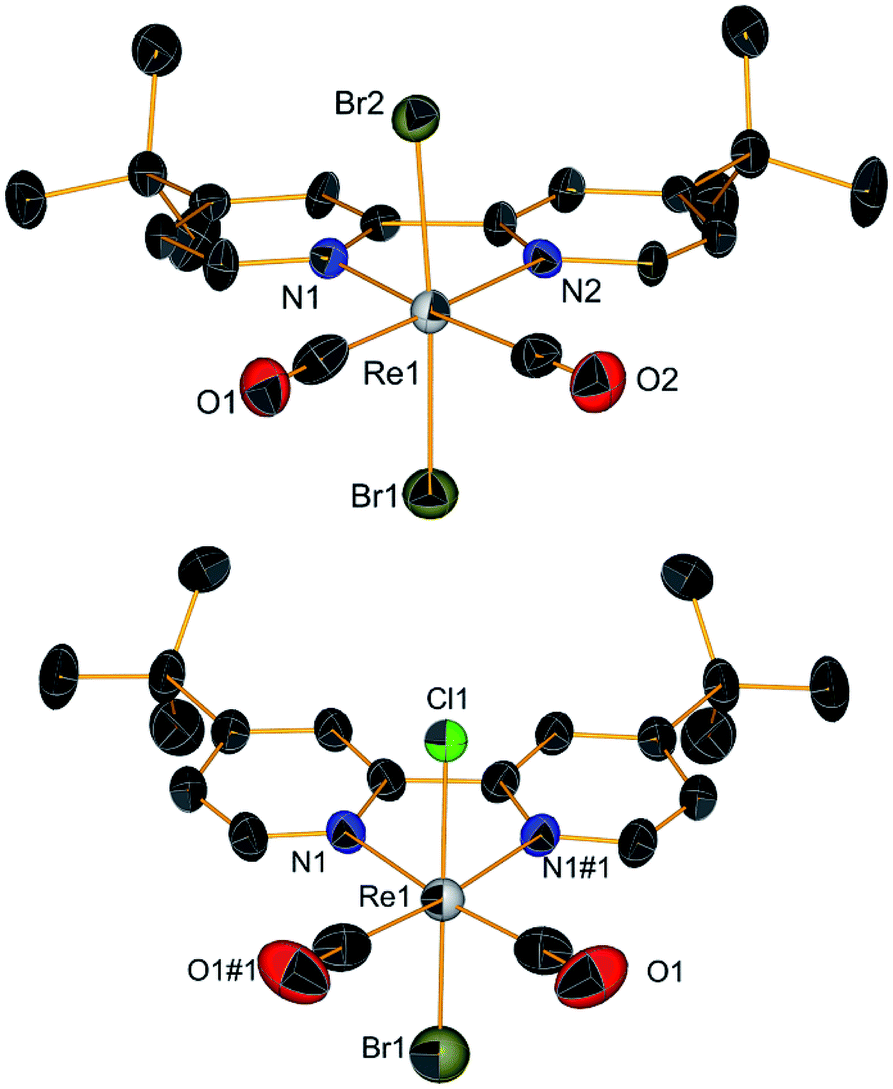
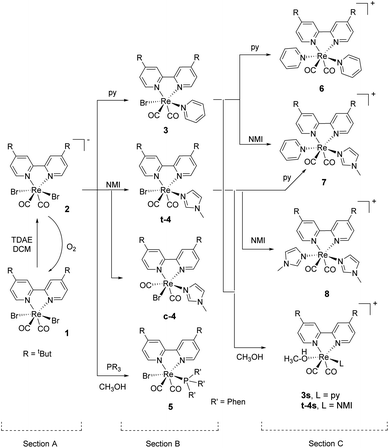
Scheme 1 summarizes the synthetic knowledge we were able to acquire in this study. The starting point of our synthetic strategy entailed the preparation of the [ReII(CO)2(tBu2bpy)Br2] complex (1, Scheme 1, section A) via reaction of the diimine ligand (tBu2bpy) with the (Et4N)2[ReII(CO)2Br4] salt (ReII-a), as previously described.55 Alternatively, we found that disproportionation/reduction of (Et4N)[ReIII(CO)2Br4] (ReIII-a) in the presence of the bipyridine ligand yields the same species. This new procedure, while lower yielding, may be useful to avoid the established one electron reduction step of ReIII-a to ReII-a, carried out under inert atmosphere in a glove box. In this second procedure, addition of tBu2bpy to ReIII-a occurs at room temperature and the reaction is allowed to proceed for 3 hours in DCM. A purification by flash column chromatography on silica separates compound 1 (first red fraction) from the fac-[ReI(CO)3(tBu2bpy)Br] side product (second fraction, yellow). Measurable crystals of 1 were grown from pentane layered on a DCM solution of the complex (Fig. 1, vide infra for crystallographic details). Carbon monoxide stretching vibrations measured by IR (frequencies at 1993 and 1853 cm−1) are in agreement with the +2 oxidation state of rhenium. Reduction of complex 1 took place under inert conditions due to the oxygen sensitivity of the tetrakis(dimethylamino)ethylene (TDAE) reductant. The addition of 0.5 equivalent of TDAE to a DCM solution of compound 1 leads immediately to precipitation of a dark fine powder identified as the (TDAE)[ReI(CO)2(tBu2bpy)Br2]2 salt (2). This compound is air sensitive in solution (it oxidizes back to Re(II) giving 1) but stable in the solid state. C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching vibrations (frequencies at 1864 and 1774 cm−1) clearly show a red shift compared to complex 1. The shift confirms the reduction to rhenium(I) as it is due to increase electronic density on the Re ion and consequently to stronger π-backbonding to the CO's (vide infra Table 1). Under similar reaction conditions, reduction of complex 1 was also achieved with cobaltocene and yielded (CoCp2)[Re(CO)2(tBu2bpy)Br2]. Multiple attempts of crystallization of 2 (inert atmosphere, glove box), or the equivalent CoCp2+ salt, failed. We only succeeded in isolating the [ReII(CO)2(tBu2bpy)BrCl] complex (Cl-1) from layering pentane on a chloroform solution of 2 (Fig. 1). Even though crystals grew under inert conditions, the X-ray structure revealed the oxidized Re(II) product. Moreover, a surprising exchange of bromide for chloride, abstracted from chloroform, was observed. We did not study this reaction further and made no attempts to elucidate its possible mechanism.
O stretching vibrations (frequencies at 1864 and 1774 cm−1) clearly show a red shift compared to complex 1. The shift confirms the reduction to rhenium(I) as it is due to increase electronic density on the Re ion and consequently to stronger π-backbonding to the CO's (vide infra Table 1). Under similar reaction conditions, reduction of complex 1 was also achieved with cobaltocene and yielded (CoCp2)[Re(CO)2(tBu2bpy)Br2]. Multiple attempts of crystallization of 2 (inert atmosphere, glove box), or the equivalent CoCp2+ salt, failed. We only succeeded in isolating the [ReII(CO)2(tBu2bpy)BrCl] complex (Cl-1) from layering pentane on a chloroform solution of 2 (Fig. 1). Even though crystals grew under inert conditions, the X-ray structure revealed the oxidized Re(II) product. Moreover, a surprising exchange of bromide for chloride, abstracted from chloroform, was observed. We did not study this reaction further and made no attempts to elucidate its possible mechanism.
 |
| | Scheme 1 General synthetic scheme to [Re(CO)2(tBu2bpy)Br(L)] and [Re(CO)2(tBu2bpy)(L)2]+ compounds. | |
 |
| | Fig. 1 Crystal structures of compound 1 (top) and the [Re(CO)2(tBu2bpy)BrCl] complex (Cl-1) obtained from the attempted crystallization of 2 in CHCl3 under inert atmosphere. Thermal ellipsoids are at 30% probability. | |
Table 1 Physical properties for complexes 2, 3, t-4, 6, 7 and 8
| Complex |
ν(CO) [cm−1] |
Predicted ν(CO) [cm−1] |
λmax [nm (M−1 cm−1)] |
| 1 |
1993, 1853 |
1995 |
301(12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 993), 426 (4753) 993), 426 (4753) |
| 2 |
1864, 1774 |
1864 |
295 (20331), 306 (14436), 418 (3444), 567 (546) |
| 3 |
1885, 1807 |
1882 |
298 (16616), 306 (18852), 384 (7188), 518 (2922) |
| t-4 |
1876, 1789 |
1872 |
297 (17723), 306 (20141), 382 (3154), 518 (2705) |
| 6 |
1899, 1818 |
1900 |
301 (19479), 359 (10961), 471 (3104), |
| 7 |
1893, 1810 |
1890 |
304 (12397), 358 (4644), 461 (1963) |
| 8 |
1886, 1800 |
1880 |
304 (14826), 356 (2702), 483 (2194), |
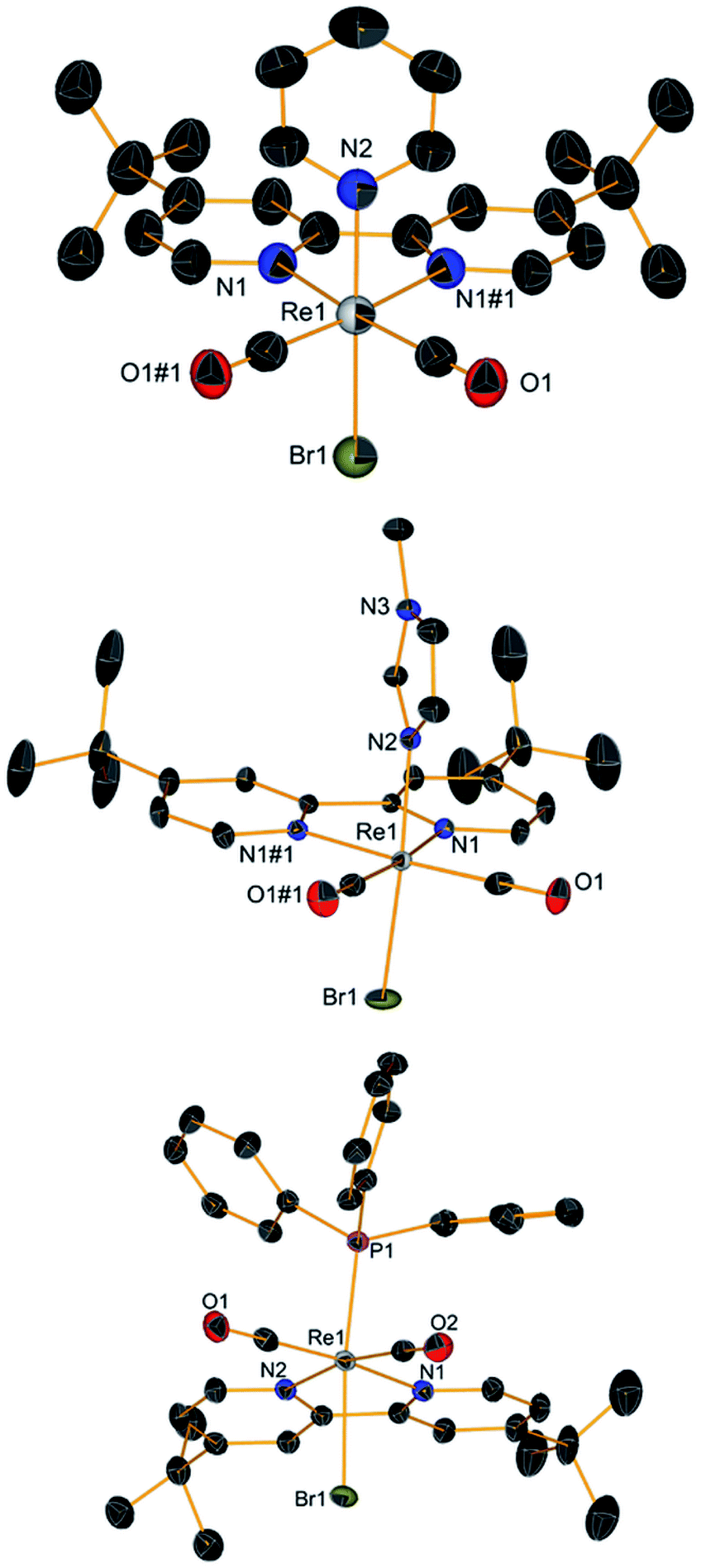
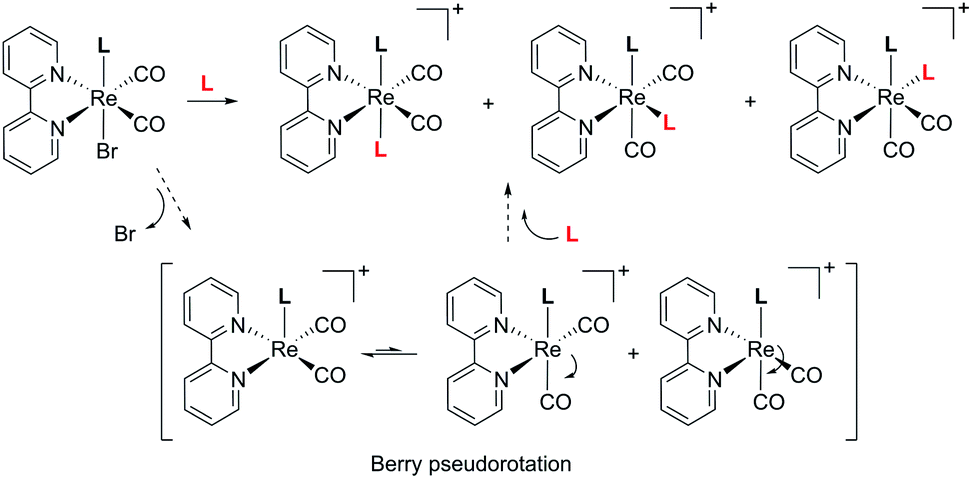
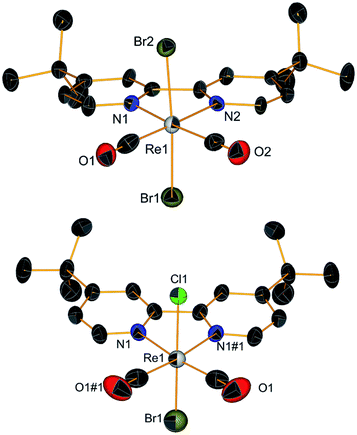
With complex 2 in our hands, we moved to explore the substitution chemistry of the species (Scheme 1, section B). Substitution reactions of one bromide ion were studied under inert conditions with σ-donor ligands L, such as pyridine (py) and N-methylimidazole (NMI). Reactions were initially attempted with increasing equivalents of L in different solvents like methanol. However, due to low yields and back oxidation of 2 to 1, we ultimately decided to use py and NMI as solvents for the same reactions. This procedure gave excellent results with yields for the desired [ReI(CO)2(tBu2bpy)Br(L)] products (3 for L = py and t-4 with L = NMI, see Scheme 1) > 85%. Substitution of one Br− ligand for py or NMI imparts aerobic stability to [ReI(CO)2(tBu2bpy)Br(L)] species, both in solid state and in solution. The presence of two coordinated π-donor bromide ions stabilizes the 17-electron complexes. The effect is lost when a bromide is exchanged by a σ-donor, and the resulting 18-electron complexes are stable. Complexes [Re(CO)2(tBu2bpy)Br(py)] (3) and [Re(CO)2(tBu2bpy)Br(NMI)] (t-4) are stable enough to be purified via flash column chromatography. We found that aluminum oxide is a better stationary phase than silica for these rhenium(I) dicarbonyl species. During the purification of the complexes, we noticed a faint green-blue band eluting first on alumina (3 and t-4 are isolated as brick red powders). In the case of the NMI reaction we were able to isolate enough of the product and we identified it as the all cis-[Re(CO)2(tBu2bpy)Br(NMI)] isomers (c-4, see Scheme 1). This green-blue side product was also crystalized along 3 and t-4 from layering pentane on DCM solutions of the complexes (Fig. 2 and 3). The presence of c-4 is interesting as it suggests a dissociative type of substitution mechanism, whereby replacement of Br in 2 may proceed via a pentacoordinate intermediate capable of Berry pseudorotation. Reaction of 2 with a strong π-acid ligand such as triphenylphosphine (PR3) yields the equivalent [Re(CO)2(tBu2bpy)Br(PR3)] species (5) in low yield. This type of complexes are well known and other synthetic procedures (e.g. Me3NO or photolytic decarbonylation of the fac-[Re(CO)3]+ core) appear superior. The solid-state structure of 5 is also given in Fig. 2.
 |
| | Fig. 2 Crystal structures (top to bottom) of compounds 3, t-4 and 5. Thermal ellipsoids are at 30% probability. | |
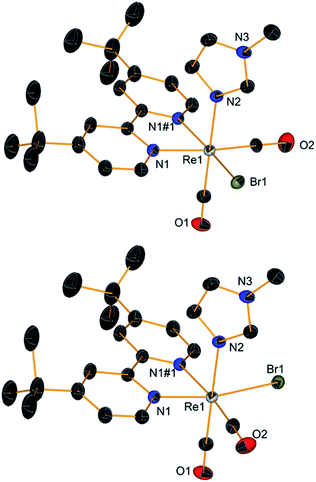 |
| | Fig. 3 Crystal structures of all cis isomers c-4. Thermal ellipsoids are at 30% probability. | |
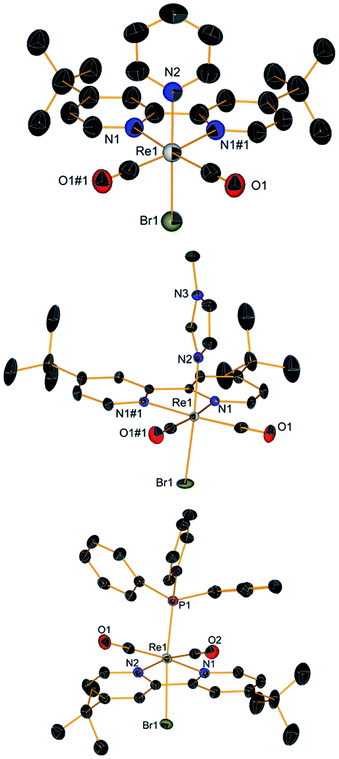
As mentioned above, 3 and t-4 are aerobically stable in solution. When dissolved in methanol, HPLC-MS analysis revealed the formation of the solvated [Re(CO)2(tBu2bpy)(L)(CH3OH)]+ ion in nearly quantitative yield (3s and t-4s in Scheme 1, section C). Indeed, we found no differences in the HPLC traces of the species whether treated with AgPF6 (1.1 eq.) or not. The presence of the solvated ion clearly indicated the possibility of replacing the second coordinated Br. Therefore, we attempted the preparation of [Re(CO)2(tBu2bpy)(L)(L′)]+ complexes with the same σ-donor ligands L. Addition of py or NMI to 3 or t-4, gives the corresponding [Re(CO)2(tBu2bpy)(L)2]+ or [Re(CO)2(tBu2bpy)(L)(L′)]+ species in good yield (6–8, Scheme 1, section C). Complexes [Re(CO)2(tBu2bpy)(py)2](PF6) (6), [Re(CO)2(tBu2bpy)(py)(NMI)](PF6) (7) and [Re(CO)2(tBu2bpy)(NMI)2](PF6) (8) were isolated as PF6− salts following treatment of aqueous solutions of the corresponding bromide salts with KPF6. We note that 7 can be obtained from either reaction of 3 with NMI or t-4 with py. X-ray quality single crystals of compounds 6–8 were obtained by layering pentane on a DCM solution of the species. Their molecular structures are shown in Fig. 4. These molecules are also aerobically stable both in solid state and in solution. Complexes are soluble in DCM, acetonitrile and methanol and they are all photolitically stable.
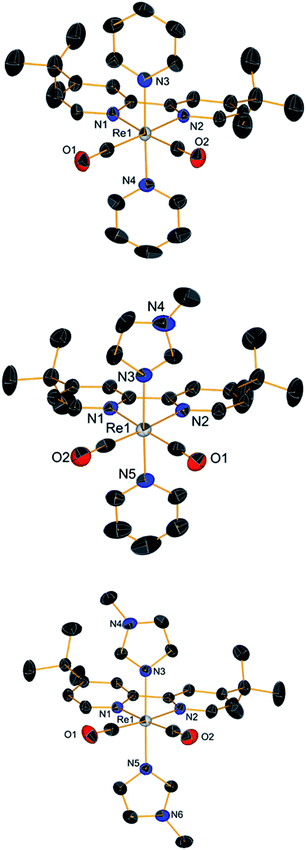 |
| | Fig. 4 Crystal structures (top to bottom) of compounds 6, 7 and 8. Thermal ellipsoids are at 30% probability. | |
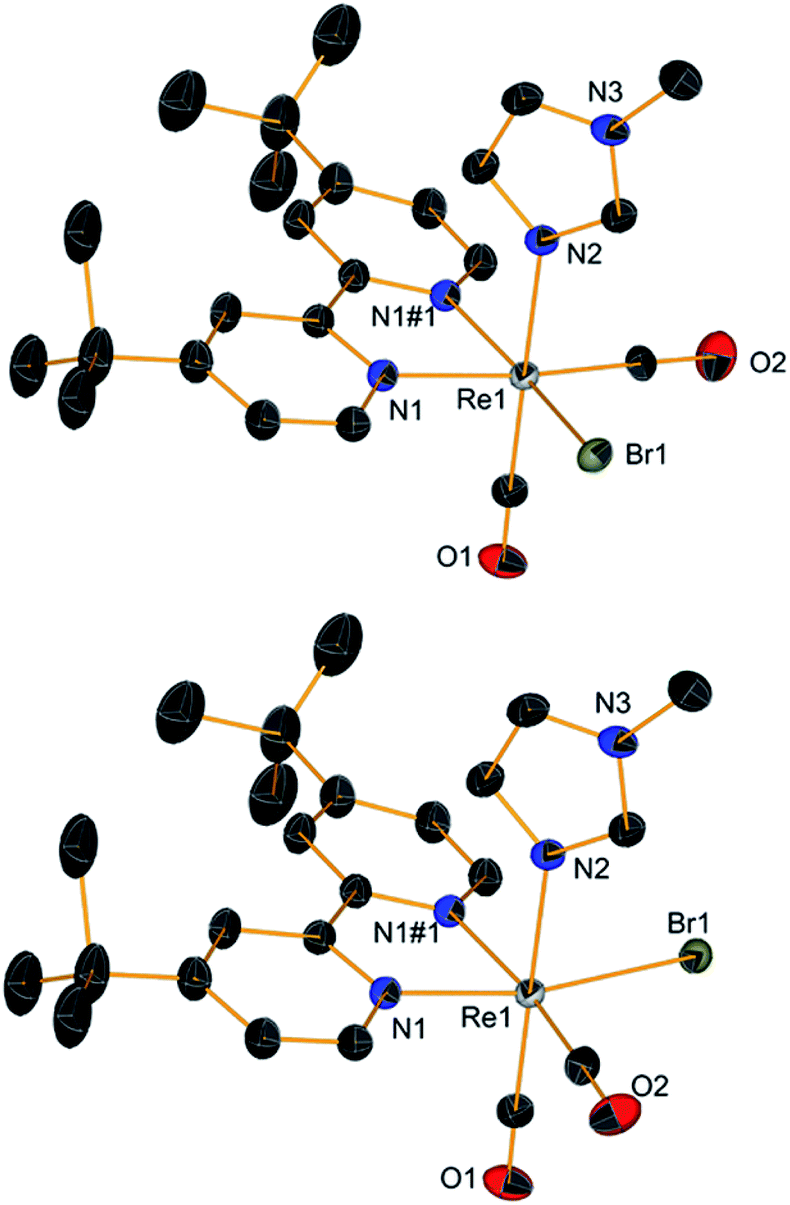
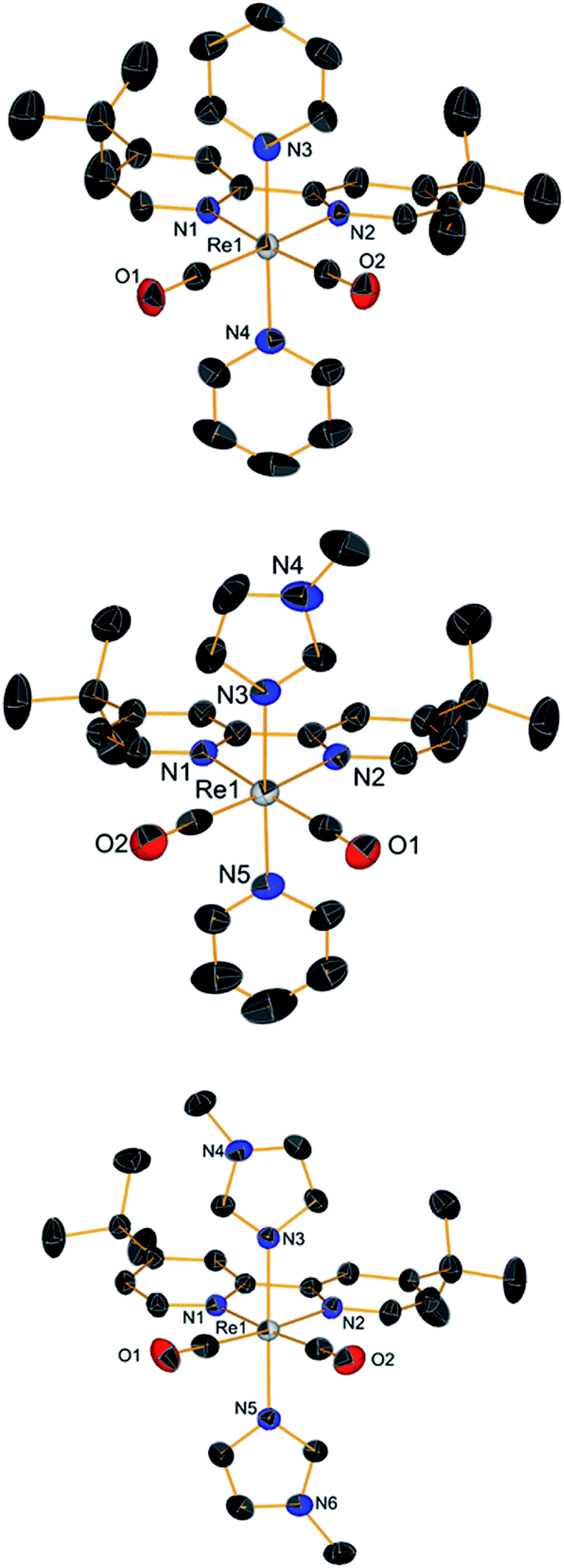
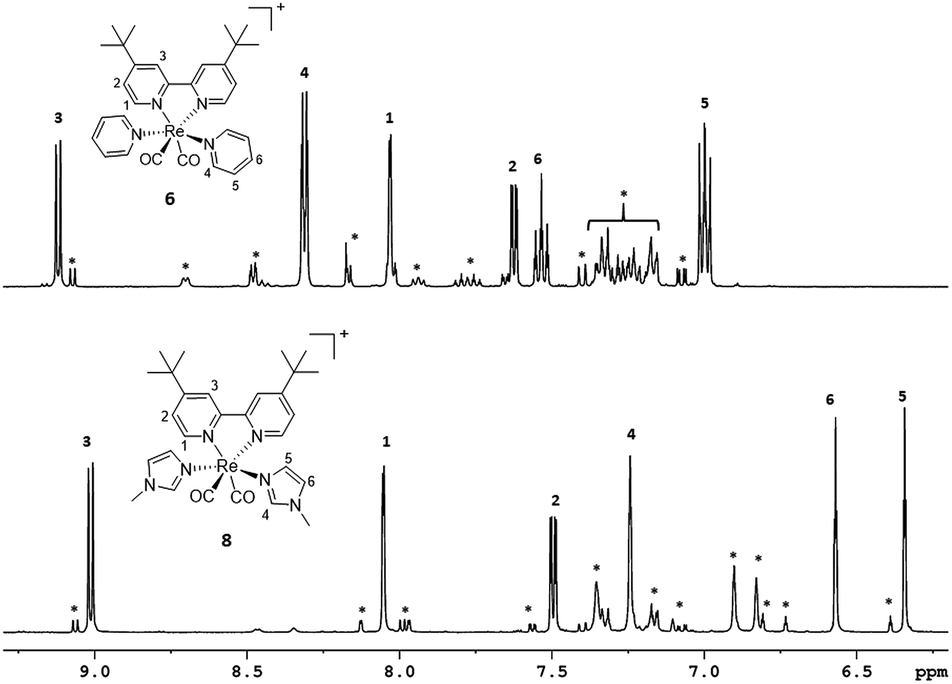
When NMR spectra of [Re(CO)2(tBu2bpy)(L)(L′)]+ species were analyzed, we realized that all cis isomers also formed (Scheme 2 and Fig. 5). As it is the case for the first substitution reaction of 2, further ligand replacement of [Re(CO)2(tBu2bpy)Br(L)] complexes occurs according to a dissociative type of substitution mechanism proceeding via a pentacoordinate intermediate capable of Berry pseudorotation. Attempts to isolate the major trans stereoisomers in pure form (6–8, Scheme 1, section C) did not succeed. Although the trans and all cis isomers are well separated on alumina (ethyl acetate/pentane mobile phase) NMR spectra of purified 6–8 still revealed the presence of the other geometrical isomers, pointing to a rapid equilibrium of the species in solution. Similar results were obtained when single crystals of 6–8 were analyzed by the technique. Trans and cis isomers ratios were determined to be respectively 70.9–29.1% after purification and 79.5–20.5% from crystals for 6; 85.5–14.5% and 85.6–15.4% for 7; 86.6–13.4% and 88.9–11.1% for 8. The relative cis enantiomers ratio was closely distributed as 1:1 for all complexes.
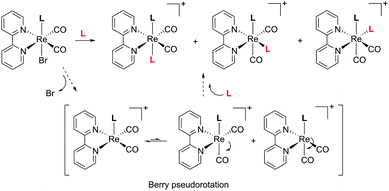 |
| | Scheme 2 Major stereoisomers formed in the reaction of [Re(CO)2(tBu2bpy)Br(L)] complexes with a second ligand L, and most probable substitution mechanism. | |
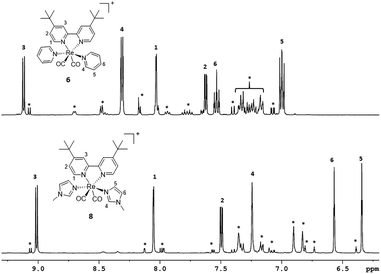 |
| | Fig. 5 Aromatic region of 1H-NMR spectra of purified complexes 6 and 8. Asterisks indicate signals of all cis isomers formed in solution. | |
X-ray crystallography
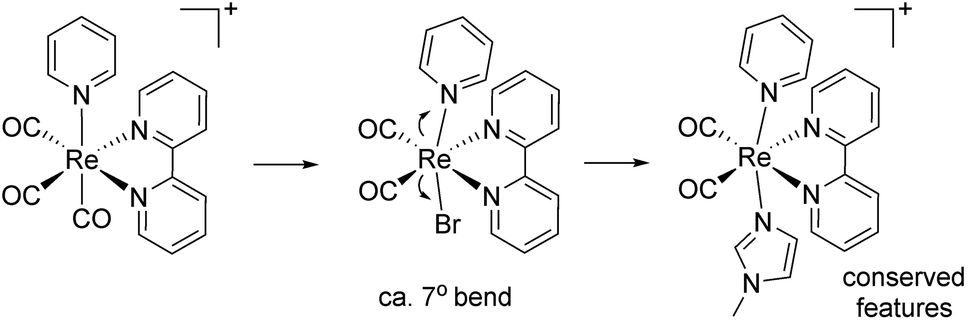
Crystallographic details, selected bond lengths and angles of complexes 1–8 (Fig. 1–4), are reported in ESI. As mentioned in the introduction, cis-[Re(CO)2]+ complexes lacking at least a π-acid ligands are not known. Structural analysis of the species and comparison to related fac-[Re(CO)3]+ species, revealed some interesting characteristics. All cis-[Re(CO)2]+ complexes show a distorted octahedral geometry around the Re ion. Perhaps the most striking feature is represented by the bending (ca. 7°) of the trans axial ligands (tLs) towards the α-diimine ligand (Scheme 3). In [Re(CO)2(tBu2bpy)(L)2]n complexes, the angle formed by the tL-Re-tL unit is on average 172°, with t-4 showing the lowest value (169.38(10)°). The tL-Re-tL angle is significantly smaller than the corresponding fac-[Re(CO)3]+ analogs (on average by ca. 7°). A conquest search of the CCDC indicates that other crystallographic parameters around the rhenium ion in cis-[Re(CO)2]+ species (bond lengths and angles) are not significantly dissimilar than the ones reported for comparable fac-[Re(CO)3(α-diimine)(L)]+ species (L = py or NMI).
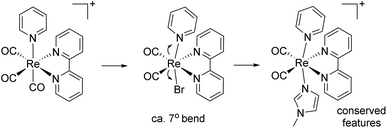 |
| | Scheme 3 Main structural differences between related diimine cis-[Re(CO)2]+ and fac-[Re(CO)3]+ species. | |
Spectroscopic characterization
Physical properties of cis-[Re(CO)2]+ complexes are summarized in Table 1. The IR spectra of the compounds show the typical pattern expected for the dicarbonyl moiety with cis geometry, with bands in the range of 1900 to 1800 cm−1 (Fig. 6). As expected, the carbonyl stretching frequencies are significantly lower than the corresponding fac-[Re(CO)3]+ tricarbonyl species. The comparison of the symmetric CO stretching vibration frequencies between complexes can be correlated to the electron donating ability of each ligand bound to the metal center. Thus, e.g., the stepwise substitution of the π-donor halide with the σ-donor py induced a stepwise hypsochromic shift of the CO stretching vibration (ca. 15 cm−1, see Fig. 6) due to decrease π-backbonding from the rhenium ion. Previously, we have shown that the symmetric CO stretching vibration frequency can be predicted by empirical calculations based on individual ligand contribution.56–58 This method provides precise estimates of frequencies for the cis-[Re(CO)2]+ cores, among others. This assessment is established by the formula
with SR = 2.00 and IR = −2115.6. The ∑[IRp(L)] term is the sum of the contribution for each ligand to the frequency. SR and IR are constants, which depend on the metal center, its oxidation state and the number of coordinated CO. The measured frequencies are in good agreement with values predicted from this empirical parametrization (Fig. 6).
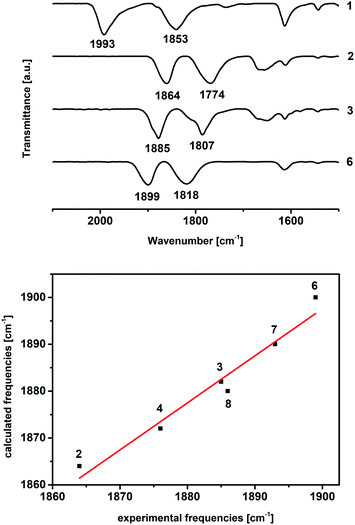 |
| | Fig. 6 IR spectra of 1, 2, 3, 6 (top). Measured versus predicted symmetric stretching vibration of CO bond for complex 2, 3, t-4, 6, 7, 8 (bottom). | |
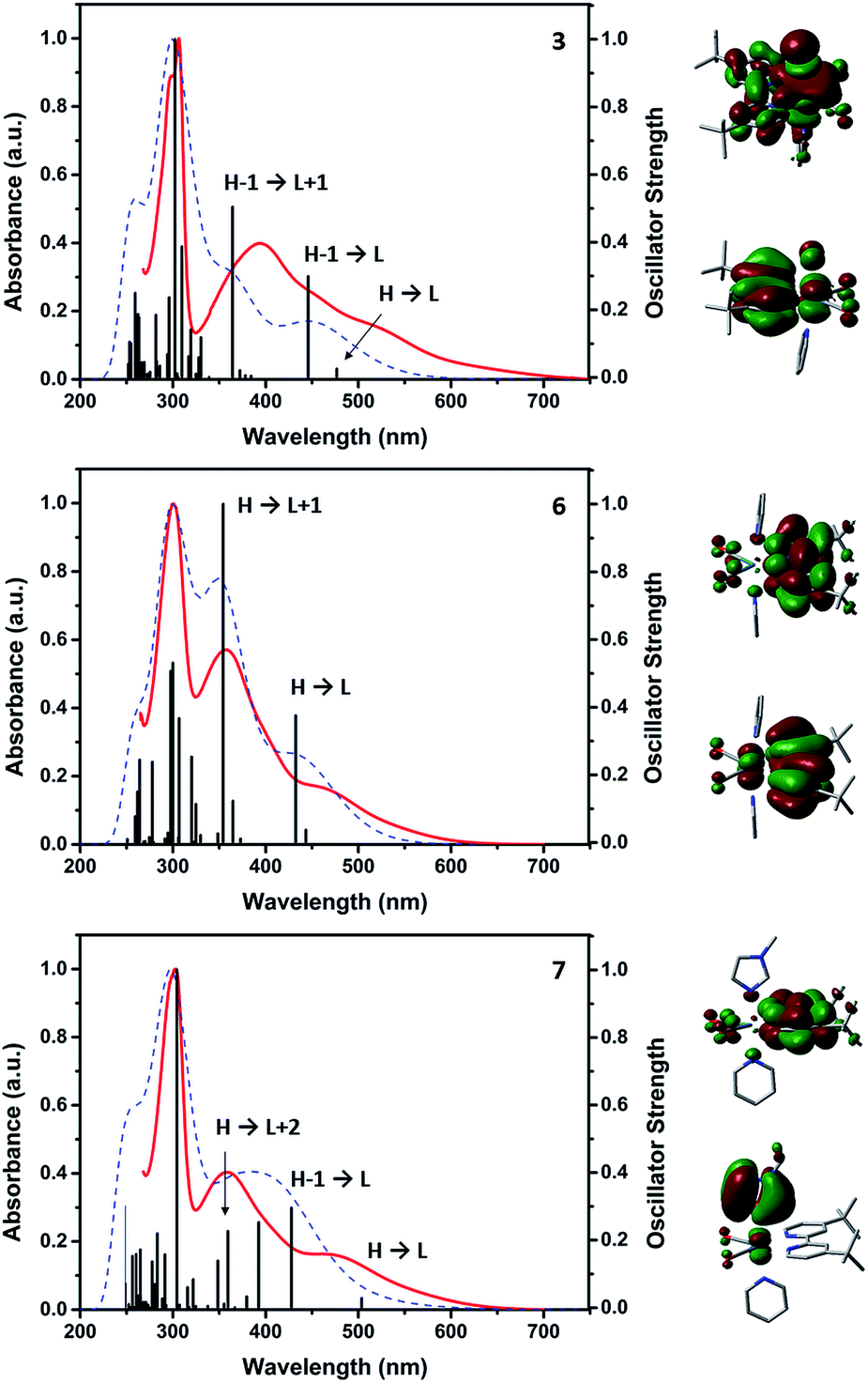
The UV-Vis spectra of the compounds are generally characterized by three main absorptions. All complexes show similar π → π* intra-ligand transitions (LLCT) as sharp bands in the 300 nm region attributed to the diimine-ligand system. In addition, the spectra show two lower lying less intense absorption in the 350–400 and 460–520 nm regions respectively. We initially hypothesized that the latter could be attributed to metal-to-ligand charge transfer transitions (MLCT), but TDDFT and orbital analysis revealed that these absorptions can at best be described as metal–ligand-to-ligand charge transfer transitions (MLLCT) of substantial (>60%) ligand character. These MLLCTs involve frontier, HOMO−1 and LUMO+1 orbital transitions. In the case of 2 and the [ReI(CO)2(tBu2bpy)Br(L)] or [ReI(CO)2(tBu2bpy)(L)2]+ species (where L = NMI, i.e. t-4, c-4 and 8, see ESI†) the absorptions are well separated and show similar extinction coefficients. In complexes where L = py, the MLLCT in the 350–400 nm region is more intense and the two absorptions are not as well-separated (Fig. 7).
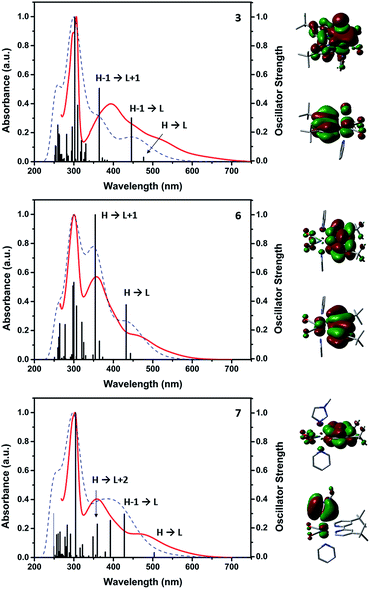 |
| | Fig. 7 Measured (red) and calculated (blue) UV-Vis spectra (all in DMF) of selected complexes (3, 6 and 7) together with calculated oscillator strength (black). The HOMO and LUMO of each corresponding molecule are shown to the right of the graphs. | |
In relative terms, substitution of the halide by L in [ReI(CO)2(tBu2bpy)Br(L)] complexes, induces an hypsochromic shift of both MLLCT maxima, which are centered respectively around 380 and 520 nm in [ReI(CO)2(tBu2bpy)Br(L)] species and 360 and 470 nm in [ReI(CO)2(tBu2bpy)(L)2]+ cations (Table 1). With the exception of NMI complexes, the shift is mainly attributed to the destabilization of LUMO orbitals energies (see ESI†). With the exception of 5, in contrast to fac-[ReI(CO)3]+ complexes, the significant ligand character of the MLLTCs of cis-[ReI(CO)2]+ species renders the same nonemissive, and none of the complexes show photoluminescent properties.
Conclusions
We prepared and investigated a new series of stable rhenium(I) dicarbonyl species, obtained from the cis–cis–trans-[Re(CO)2(tBu2bpy)Br2]− anion (2) by the reduction of the corresponding rhenium(II) complex (1). Substitution of a single bromide in 2 by monodentate ligands yielded 18-electron cis–cis–trans-[Re(CO)2(tBu2bpy)Br(L)] compounds. The second bromide could be easily substituted in coordinating solvents, such as methanol and then replaced by a second σ-donor monodentate ligand giving cis–cis–trans-[Re(CO)2(tBu2bpy)(L)2]+ cations. We observed the formation of cis–cis–cis-[Re(CO)2(tBu2bpy)(L)2]+ isomers, induced by a Berry pseudorotation involved in the ligand substitution mechanism. Crystallography analysis showed a slightly distorted octahedral geometry for all species, in which trans axial monodentate ligands are bent (ca. 7°) towards the α-diimine. In the IR spectrum, these complexes are characterized by a typical pattern expected for the dicarbonyl moiety with cis geometry, with bands in the range of 1900 to 1800 cm−1. Symmetric stretching vibration of CO bond were analyzed and compared with empirical calculations, showing a good correlation with predictive theoretical models. TDDFT calculations allowed assignment of UV-Vis electronic transitions and orbitals involved in the same. The complexes are characterized by metal–ligand-to-ligand charge transfer transitions (MLLCT) of substantial ligand character. [Re(CO)2(tBu2bpy)Br2]− and [Re(CO)2(tBu2bpy)Br(L)] complexes may be considered as synthons for the preparation of a variety of new stable diamagnetic dicarbonyl rhenium cis-[Re(CO)2]+ complexes, offering a convenient entry in the chemistry of the core.
Experimental section
Reagents and chemicals
All reagent and solvents were purchased from standard sources and used without further purification. Complexes [Re(CO)5Br],59 (Et4N)2[Re(CO)3Br3]60 and (Et4N)[Re(CO)2Br4]61 were synthesized according to published procedures. Unless otherwise noted, solvents used in the preparation of all molecules were dry and O2-free.
Instruments and analysis
NMR spectra were measured on a Bruker Advance III 500 or 400 MHz. The corresponding 1H chemical shifts are reported relative to residual solvent protons. Mass analyses were performed either using ESI-MS on a Bruker FTMS 4.7-T Apex II in positive mode or MALDI with a Bruker UltrafleXtreme MALDI-TOF. UV-Vis spectra were measured on a Jasco V730 spectrophotometer. IR spectra were recorded on a Bruker TENSOR II with the following parameters: 16 scans for background, 32 scans for sample with a resolution of 4 cm−1 in the 4000 to 600 cm−1 region. Analytical HPLCs were performed with a Merck Hitachi L-7000 system, which comprises a pump L-7100 and a UV-detector L-7400. For preparative HPLC, a column Macherey-Nagel Nucleodur C18 HTec (5 μm particle size, 110 Å pore size, 250 × 21 mm) was used. Aqueous trifluoroacetic acid 0.1% solution (A) and pure methanol (B) were respectively used as solvents. The compounds were analyzed using the following gradient: 0–5 min (75% A), 5–35 (75% A → 0% A), 35–45 min (100% B) or 0–5 min (50% A), 5–30 (50% A → 0% A), 30–45 min (100% B), the flow rate set to 5 mL min−1 and the compounds detected at 260 nm. Single crystal diffraction data collection was performed on a Stoe IPDS2 diffractometer (CuKα1 (λ = 1.5406 Å)) equipped with a cryostat from Oxford Cryosystems. The structure were solved with the ShelXT structure solution program using Intrinsic Phasing and refined with the ShelXL refinement package using Least Squares minimisation. All crystal structures are deposited at the Cambridge Crystallographic Data Centre. CCDC numbers 2056997–2057005 contain the supplementary crystallographic data for this paper.
DFT and TDDFT calculations
All computations were performed with the Gaussian 09 programs. Geometry optimizations as well as frequency calculations were performed in the gas phase. The hybrid meta-GGA functional wB97XD,62–66 designed to account for dispersion, was used in combination with the standard SDD basis sets.67 For the spin state of the complexes (singlet state in all cases expect 1), the default spin formalism was followed in the calculations and default Gaussian 09 values were adopted for the numerical integration grids, self-consistent-field (SCF) and geometry optimization convergence criteria. Geometries were optimized without symmetry restrictions. The nature of the stationary points was checked by computing vibrational frequencies in order to verify true minima. No imaginary frequencies were observed for the reported values. Electronic transition energies and oscillator strengths were then calculated at their wB97XD -optimized geometries using TDDFT. For these calculations the 40 lowest energy electronic excitations were calculated for each compound, and solvent effects were added via a solvent continuum dielectric model using DMF as the solvent. The calculated molecular orbitals were visualized using GaussView.
Synthetic procedures
[Re(CO)2(tBu2bpy)Br2] (1). Complex (Et4N)[Re(CO)2Br4] (20 mg, 0.029 mmol) and 4,4′-di-tert-butyl-2,2′-bipyridine (8 mg, 0.029 mmol) were dissolved in DCM (7 mL). The mixture was stirred at room temperature for 3 h. The solvent was evaporated, and the product was purified on silica by flash column chromatography (eluent:EtOAc/pentane 1:8). Compound 1 was isolated as a red solid. Yield: 13 mg, 0.019 mmol, 67%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CH2Cl2 solution of the compound giving dark-red crystals. IR (cm−1), νCO: 1993, 1853. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 426 (4753), 301 (12993). NMR: not available because of paramagnetic compound. ESI-MS (MeOH): m/z, 693.7 [M + Na]+.
(TDAE)[Re(CO)2(tBu2bpy)Br2]2 (2). Complex 1 (58 mg, 0.086 mmol) was dissolved in DCM (20 mL) in a schlenk flask in the glove box. TDAE (10 μL, 0.043 mmol, 0.5 eq.) was dissolved in DCM (1 mL) and slowly added to the stirring solution of 1. A dark powder began to precipitate immediately. The mixture was stirred for 15 min, and then carefully filtered to collect a purple solid, which was then dried in vacuo. Yield: 62 mg, 0.04 mmol, 94%. IR (cm−1), νCO: 1864, 1774. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 567 (546), 418 (3444), 306 (14436), 295 (20331). 1H-NMR (400 MHz, CD2Cl2, ppm), δ: 8.56 (d, J = 0.61, 5.26 Hz, 2H, H4 (bpy)), 8.44 (d, 2H, H1 (bpy)), 7.32 (dd, J = 1.96, 5.14 Hz, 2H, H2 (bpy)), 2.95 (s, 2H, H(TDAE)), 2.93 (s, 2H, H(TDAE)), 2.76 (s, 8H, H(TDAE)), 1.38 (s, 18H, H7 (bpy)). 13C-NMR (101 MHz, CD2Cl2, ppm), δ: 161.44 (2C, C3 (bpy)), 156.93 (2C, C5 (bpy)), 149.50 (2C, C1 (bpy)), 121.28 (2C, C4 (bpy)), 118.50 (2C, C2 (bpy)), 38.93 (2C, C(TDAE)), 35.41 (2C, C6 (bpy)), 30.91 (6C, C7 (bpy)). ESI-MS (MeOH): m/z, 670.6 [M]+.
[Re(CO)2(tBu2bpy)Br(py)] (3). To a solid sample of complex 2 (51 mg, 0.0332 mmol), degassed in a flask, anhydrous pyridine (5 mL, excess) was added. The suspension was heated to 100 °C for 60 min. The solvent was then evaporated and the black residue was washed with water to remove the TDAEBr2. The product was purified on aluminum oxide by flash column chromatography (eluent:EtOAc/pentane 1:1, increased to 2:1). Compound 3 was isolated as a brown solid. Yield: 38.2 mg, 0.0570 mmol, 85%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CDCl3 solution of the compound giving dark-orange crystals. IR (cm−1), νCO: 1885, 1807. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 518 (2922), 384 (7188), 306 (18852), 298 (16616). 1H-NMR (400 MHz, CD2Cl2, ppm), δ: 9.10 (d, J = 5.87 Hz, 2H, H4 (bpy)), 8.46 (d, 2H, H8 (py)), 8.10 (d, J = 1.83 Hz, 2H, H1 (bpy)), 7.52 (dd, J = 1.96, 5.87 Hz, 2H, H2 (bpy)), 7.48 (t, J = 7.64 Hz, 1H, H10 (py)), 6.92 (t, 2H, H9 (py)), 1.43 (s, 18H, H7 (bpy)). 13C-NMR (101 MHz, CD2Cl2, ppm), δ: 163.59 (2C, C3 (bpy)), 156.73 (2C, C5 (bpy)), 155.97 (2C, C8 (py)), 152.42 (2C, C1 (bpy)), 135.62 (1C, C10 (py)), 125.68 (2C, C9 (py)), 124.98 (2C, C4 (bpy)), 120.21 (2C, C2 (bpy)), 35.97 (2C, C6 (bpy)), 30.67 (6C, C7 (bpy)). ESI-MS (MeOH), m/z: 691.8 [M + Na]+.
[Re(CO)2(tBu2bpy)Br(NMI)] (t-4). To a solid sample of complex 2 (104 mg, 0.0675 mmol), degassed in a flask, anhydrous N-methylimidazole (5 mL, excess) was added. The suspension was heated to 110 °C for 60 min. The solvent was then evaporated and the black residue was washed with water to remove the TDAEBr2. The product was purified on aluminum oxide by flash column chromatography (eluent:EtOAc/pentane 1:1, increased to 3:1). The first fraction was dried as a green solid, complex c-4. Yield: 2 mg, 0.00297 mmol, 2%. Compound t-4 was isolated from the second fraction as a brown solid. Yield: 77.6 mg, 0.1154 mmol, 86%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CDCl3 solution of t-4 giving dark-violet crystals. IR (cm−1), νCO: 1876, 1789. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 518 (2705), 382 (3154), 306 (20141), 297 (17723). 1H NMR (400 MHz, CD2Cl2, ppm) δ: 9.01 (d, J = 5.50 Hz, 2H, H4 (bpy)), 8.08 (s, 2H, H1 (bpy)), 7.47 (dd, J = 1.83, 5.75 Hz, 2H, H2 (bpy)), 7.30 (broadened, 1H, H8 (NMI)), 6.56 (s, 1H, H10 (NMI)), 6.38 (s, 1H, H9 (NMI)), 3.60 (s, 3H, H11 (NMI)), 1.43 (s, 18H, H7 (bpy)). 13C-NMR (101 MHz, CD2Cl2, ppm), δ: 163.67 (2C, C3 (bpy)), 156.99 (2C, C5 (bpy)), 152.62 (2C, C1 (bpy)), 124.37 (2C, C4 (bpy)), 121.57 (2C, C2 (bpy)), 119.64 (1C, C9 (NMI)), 35.83 (2C, C6 (bpy)), 34.43 (1C, C11 (NMI)), 30.79 (6C, C7 (bpy)). ESI-MS (MeOH): m/z, 694.8 [M + Na]+.
[Re(CO)2(tBu2bpy)Br(PPh3)] (5). To a solid sample of complex 2 (15 mg, 0.0194 mmol in Re) and triphenylphosphine (52 mg, 0.194 mmol, 10 eq.), degassed in a flask, anhydrous DME (11 mL) was added. The mixture was stirred at reflux (80 °C) during 1 h. The solvent was then evaporated and the black solid was washed with H2O to remove TDAE. The product was purified on silica by flash column chromatography (eluent:EtOAc/pentane 1:4, increased to 1:1). Compound 5 was isolated as a red solid. Yield: 2 mg, 0.0023, 12%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CH2Cl2 solution of the compound giving dark-red crystals. IR (cm−1), νCO: 1917, 1839. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 447, 303. 1H NMR (400 MHz, CD2Cl2, ppm), δ: 8.33 (d, J = 5.87 Hz, 2H, H4 (bpy)), 7.93 (s, 2H, H1 (bpy)), 7.21 (m, 15H, H(PPh3)), 7.04 (m, 2H, H2 (bpy)), 1.37 (d, J = 0.98 Hz, 18H, H7 (bpy)). ESI-MS (MeOH): m/z, 874.9 [M + Na]+.
[Re(CO)2(tBu2bpy)(py)2](PF6) (6). Complex 3 (38 mg, 0.0570 mmol) was dissolved in MeOH (8 mL) and pyridine (0.5 mL). The solution was heated to 75 °C overnight. The solvent was evaporated and the solid was dissolved in water, then filtered to remove possible impurities. KPF6 (15 mg, 0.0855 mmol, 1.5 eq.) dissolved in water was added and the precipitate was centrifugated. Compound 6 was filtered as a red solid. Yield: 27.3 mg, 0.0335 mmol, 59%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CH2Cl2 solution of the compound giving brown crystals. IR (cm−1), νCO: 1899, 1818. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 471 (3104), 359 (10961), 301 (19479). 1H NMR (400 MHz, CD2Cl2, ppm) δ: 9.20 (d, J = 5.87 Hz, 2H, H4 (bpy)), 8.40 (m, 4H, H8 (py)), 8.11 (d, J = 1.83 Hz, 2H, H1 (bpy)), 7.71 (dd, 2H, H2 (bpy)), 7.62 (tt, 2H, H10 (py)), 7.08 (m, 4H, H9 (py)), 1.43 (s, 18H, H7 (bpy)). 13C-NMR (101 MHz, CD2Cl2, ppm), δ: 206.33 (2C, C (CO)), 166.20 (2C, C3 (bpy)), 157.08 (2C, C5 (bpy)), 155.70 (4C, C8 (py)), 152.49 (2C, C1 (bpy)), 137.66 (2C, C10 (py)), 127.58 (4C, C9 (py)), 126.65 (2C, C4 (bpy)), 121.73 (2C, C2 (bpy)), 36.63 (2C, C6 (bpy)), 30.72 (6C, C7 (bpy)). ESI-MS (MeOH): m/z, 668.9 [M]+.
[Re(CO)2(tBu2bpy)(py)(NMI)](PF6) (7). Complex 3 (11 mg, 0.0164 mmol) was dissolved in MeOH (7 mL) and N-methylimidazole (1 mL). The solution was heated to 80 °C for 3 h and 60 °C overnight. The solvent was evaporated and the solid was dissolved in water, then filtered to remove possible impurities. KPF6 dissolved in water was added and the precipitate was centrifuged. Compound 7 was isolated as a red solid. Yield: 10 mg, 0.0122 mmol, 75%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CH2Cl2 solution of the compound giving dark-orange crystals. IR (cm−1), νCO: 1893, 1810. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 461 (1963), 358 (4644), 304 (12397). 1H NMR (400 MHz, CD2Cl2, ppm) δ: 9.15 (d, J = 5.87 Hz, 2H, H4 (bpy)), 8.43 (d, J = 1.47 Hz, 2H, H8 (py)), 8.13 (d, J = 1.71 Hz, 2H, H1 (bpy)), 7.64 (dd, J = 1.96, 5.87 Hz, 2H, H2 (bpy)), 7.59 (m, 1H, H10 (py)), 7.30 (s, 1H, H11 (NMI)), 7.04 (t, 2H, H9 (py)), 6.67 (s, 1H, H13 (NMI)), 6.44 (s, 1H, H12 (NMI)), 3.54 (s, 3H, H14 (NMI)), 1.43 (s, 18H, H7 (bpy)). 13C-NMR (101 MHz, CD2Cl2, ppm), δ: 206.88 (2C, C(CO)), 165.55 (2C, C3 (bpy)), 156.57 (2C, C5 (bpy)), 155.36 (2C, C8 (py)), 152.25 (2C, C1 (bpy)), 141.51 (1C, C11 (NMI)), 136.90 (1C, C10 (py)), 132.06 (1C, C13 (NMI)), 126.21 (2C, C9 (py)), 126.14 (2C, C4 (bpy)), 122.37 (2C, C2 (bpy)), 121.20 (1C, C12 (NMI)), 36.29 (2C, C6 (bpy)), 34.78 (1C, C14 (NMI)), 30.49 (6C, C7 (bpy)). ESI-MS (MeOH): m/z, 671.9 [M]+.
[Re(CO)2(tBu2bpy)(NMI)2](PF6) (8). Complex t-4 (15 mg, 0.0223 mmol) was suspended in MeOH (6 mL) and N-methylimidazole (0.5 mL). The solution was heated to 80 °C for 2 h and 60 °C overnight, the solvent was then evaporated. The solid was dissolved in water, presolved KPF6 was added. The solution was stirred for 10 minutes and then centrifugated. Compound 8 was isolated as a red solid. Yield: 7 mg, 0.00854 mmol, 38%. Single crystals suitable for X-ray diffraction were grown by layering pentane on a CH2Cl2 solution of the compound giving brown crystals. IR (cm−1), νCO: 1886, 1800. UV-Vis (DMF), λmax (ε) [nm (M−1 cm−1)]: 483 (2194), 356 (2702), 304 (14826). 1H NMR (400 MHz, CD2Cl2, ppm) δ: 9.10 (d, J = 5.87 Hz, 2H, H4 (bpy)), 8.14 (d, J = 1.71 Hz, 2H, H1 (bpy)), 7.58 (dd, J = 1.96, 5.87 Hz, 2H, H2 (bpy)), 7.33 (s, 2H, H8 (NMI)), 6.66 (m, 2H, H10 (NMI)), 6.43 (t, J = 1.41 Hz, 2H, H9 (NMI)), 3.54 (s, 6H, H11 (NMI)), 1.44 (s, 18H, H7 (bpy)). 13C-NMR (101 MHz, CD2Cl2, ppm), δ: 207.73 (2C, C(CO)), 165.17 (2C, C3 (bpy)), 156.62 (2C, C5 (bpy)), 152.26 (2C, C1 (bpy)), 141.46 (2C, C8 (NMI)), 131.95 (2C, C10 (NMI)), 125.78 (2C, C4 (bpy)), 122.16 (2C, C2 (bpy)), 120.96 (2C, C9 (NMI)), 36.23 (2C, C6 (bpy)), 34.72 (2C, C11 (NMI)), 30.53 (6C, C7 (bpy)). ESI-MS (MeOH): m/z, 674.9 [M]+.
Author contributions
K. S. investigation, formal analysis, data curation, methodology, writing – original draft; A. C. crystallography; F. Z. conceptualization, supervision, validation, writing – review & editing, funding acquisition, resources, project administration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the Swiss National Science Foundation (Project# 200021_196967) is gratefully acknowledged.
Notes and references
- A. J. Lees and S. S. Sun, in CCC II, ed. J. A. McCleverty and T. J. Meyer, Pergamon, Oxford, 2003, pp. 731–742, DOI:10.1016/B0-08-043748-6/01077-X.
- S. Sato and O. Ishitani, Coord. Chem. Rev., 2015, 282–283, 50–59 CrossRef CAS.
- D. J. Stufkens, Comments Inorg. Chem., 1992, 13, 359–385 CrossRef CAS.
- A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS.
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS.
- G. F. Manbeck, J. T. Muckerman, D. J. Szalda, Y. Himeda and E. Fujita, J. Phys. Chem. B, 2015, 119, 7457–7466 CrossRef CAS.
- J. Agarwal, E. Fujita, H. F. Schaefer and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 Search PubMed.
- K. Oppelt, R. Fernández-Terán, R. Pfister and P. Hamm, J. Phys. Chem. C, 2019, 123, 19952–19961 CrossRef CAS.
- D. L. Reger, K. J. Brown and M. D. Smith, J. Organomet. Chem., 2002, 658, 50–61 CrossRef CAS.
- I. M. Lorkovic, M. S. Wrighton and W. M. Davis, J. Am. Chem. Soc., 1994, 116, 6220–6228 CrossRef CAS.
- W. L. Ingham and N. J. Coville, Organometallics, 1992, 11, 2551–2558 CrossRef CAS.
- D. A. Kurtz, B. Dhakal, E. S. Donovan, G. S. Nichol and G. A. N. Felton, Inorg. Chem. Commun., 2015, 59, 80–83 CrossRef CAS.
- S. C. Marker, S. N. MacMillan, W. R. Zipfel, Z. Li, P. C. Ford and J. J. Wilson, Inorg. Chem., 2018, 57, 1311–1331 CrossRef CAS.
- O. Kaufhold, A. Stasch, T. Pape, A. Hepp, P. G. Edwards, P. D. Newman and F. E. Hahn, J. Am. Chem. Soc., 2009, 131, 306–317 CrossRef CAS.
- S. Du, J. A. Kautz, T. D. McGrath and F. G. A. Stone, Dalton Trans., 2005, 3672–3680, 10.1039/B510026G.
- M. S. Jana, A. K. Pramanik, D. Sarkar, S. Biswas and T. K. Mondal, J. Mol. Struct., 2013, 1047, 73–79 CrossRef CAS.
- D. A. Kurtz, K. R. Brereton, K. P. Ruoff, H. M. Tang, G. A. N. Felton, A. J. M. Miller and J. L. Dempsey, Inorg. Chem., 2018, 57, 5389–5399 Search PubMed.
- L. Kromer, B. Spingler and R. Alberto, J. Organomet. Chem., 2007, 692, 1372–1376 CrossRef CAS.
- L. Kromer, B. Spingler and R. Alberto, Dalton Trans., 2008, 5800–5806, 10.1039/B805410J.
- C.-C. Ko, L. T.-L. Lo, C.-O. Ng and S.-M. Yiu, Chem. - Eur. J., 2010, 16, 13773–13782 CrossRef CAS.
- A. W.-Y. Cheung, L. T.-L. Lo, C.-C. Ko and S.-M. Yiu, Inorg. Chem., 2011, 50, 4798–4810 CrossRef CAS.
- S. Sato, A. Sekine, Y. Ohashi, O. Ishitani, A. M. Blanco-Rodríguez, A. Vlček, T. Unno and K. Koike, Inorg. Chem., 2007, 46, 3531–3540 CrossRef CAS.
- Y. Xiao, A. W.-Y. Cheung, S.-W. Lai, S.-C. Cheng, S.-M. Yiu, C.-F. Leung and C.-C. Ko, Inorg. Chem., 2019, 58, 6696–6705 CrossRef CAS.
- C.-O. Ng, S.-C. Cheng, W.-K. Chu, K.-M. Tang, S.-M. Yiu and C.-C. Ko, Inorg. Chem., 2016, 55, 7969–7979 CrossRef CAS.
- W.-K. Chu, X.-G. Wei, S.-M. Yiu, C.-C. Ko and K.-C. Lau, Chem. - Eur. J., 2015, 21, 2603–2612 CrossRef CAS.
- E. Kottelat, V. Chabert, A. Crochet, K. M. Fromm and F. Zobi, Eur. J. Inorg. Chem., 2015, 5628–5638, DOI:10.1002/ejic.201500756.
- C. Triantis, A. Shegani, C. Kiritsis, M. Ischyropoulou, I. Roupa, V. Psycharis, C. Raptopoulou, P. Kyprianidou, M. Pelecanou, I. Pirmettis and M. S. Papadopoulos, Inorg. Chem., 2018, 57, 8354–8363 CrossRef CAS.
- J. V. Caspar, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1984, 23, 2104–2109 CrossRef CAS.
- E. Schutte, J. B. Helms, S. M. Woessner, J. Bowen and B. P. Sullivan, Inorg. Chem., 1998, 37, 2618–2619 CrossRef CAS.
- A. S. DelNegro, S. M. Woessner, B. P. Sullivan, D. M. Dattelbaum and J. R. Schoonover, Inorg. Chem., 2001, 40, 5056–5057 CrossRef CAS.
- O. Ishitani, K. Kanai, Y. Yamada and K. Sakamoto, Chem. Commun., 2001, 1514–1515, 10.1039/B104220N.
- H. Tsubaki, A. Sekine, Y. Ohashi, K. Koike, H. Takeda and O. Ishitani, J. Am. Chem. Soc., 2005, 127, 15544–15555 CrossRef CAS.
- Y. Yamamoto, S. Sawa, Y. Funada, T. Morimoto, M. Falkenström, H. Miyasaka, S. Shishido, T. Ozeki, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 14659–14674 CrossRef CAS.
- Y. Yamamoto, Y. Tamaki, T. Yui, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2010, 132, 11743–11752 CrossRef CAS.
- T. Morimoto, M. Ito, K. Koike, T. Kojima, T. Ozeki and O. Ishitani, Chem. - Eur. J., 2012, 18, 3292–3304 CrossRef CAS.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266–13269 CrossRef CAS.
- T. Asatani, Y. Nakagawa, Y. Funada, S. Sawa, H. Takeda, T. Morimoto, K. Koike and O. Ishitani, Inorg. Chem., 2014, 53, 7170–7180 CrossRef CAS.
- J. Rohacova, A. Sekine, T. Kawano, S. Tamari and O. Ishitani, Inorg. Chem., 2015, 54, 8769–8777 CrossRef CAS.
- T. Morimoto and O. Ishitani, Acc. Chem. Res., 2017, 50, 2673–2683 CrossRef CAS.
- Y. Yamazaki, J. Rohacova, H. Ohtsu, M. Kawano and O. Ishitani, Inorg. Chem., 2018, 57, 15158–15171 CrossRef CAS.
- Y. Yamazaki and O. Ishitani, Inorg. Chem., 2019, 58, 12905–12910 CrossRef CAS.
- C.-O. Ng, S.-M. Yiu and C.-C. Ko, Inorg. Chem., 2014, 53, 3022–3031 CrossRef CAS.
- S. E. Kabir, J. Alam, S. Ghosh, K. Kundu, G. Hogarth, D. A. Tocher, G. M. G. Hossain and H. W. Roesky, Dalton Trans., 2009, 4458–4467, 10.1039/B815337J.
- B. Laramée-Milette, F. Nastasi, F. Puntoriero, S. Campagna and G. S. Hanan, Chem. - Eur. J., 2017, 23, 16497–16504 CrossRef.
- J. Delasoie, A. Pavic, N. Voutier, S. Vojnovic, A. Crochet, J. Nikodinovic-Runic and F. Zobi, Eur. J. Med. Chem., 2020, 204, 112583 CrossRef CAS.
- J. Delasoie, P. Schiel, S. Vojnovic, J. Nikodinovic-Runic and F. Zobi, Pharmaceutics, 2020, 12, 480 CrossRef CAS.
- G. Santoro, T. Zlateva, A. Ruggi, L. Quaroni and F. Zobi, Dalton Trans., 2015, 44, 6999–7008 RSC.
- J. Rossier, D. Hauser, E. Kottelat, B. Rothen-Rutishauser and F. Zobi, Dalton Trans., 2017, 46, 2159–2164 RSC.
- S. Nasiri Sovari and F. Zobi, Chemistry, 2020, 2, 418–452 CrossRef.
- S. N. Sovari, S. Vojnovic, S. S. Bogojevic, A. Crochet, A. Pavic, J. Nikodinovic-Runic and F. Zobi, Eur. J. Med. Chem., 2020, 205, 112533 CrossRef CAS.
- J. Delasoie, N. Radakovic, A. Pavic and F. Zobi, Appl. Sci., 2020, 10, 7380 Search PubMed.
- G. Santoro, R. Beltrami, E. Kottelat, O. Blacque, A. Y. Bogdanova and F. Zobi, Dalton Trans., 2016, 45, 1504–1513 RSC.
- L. Prieto, J. Rossier, K. Derszniak, J. Dybas, R. M. Oetterli, E. Kottelat, S. Chlopicki, F. Zelder and F. Zobi, Chem. Commun., 2017, 53, 6840–6843 RSC.
- H. B. Suliman, F. Zobi and C. A. Piantadosi, Antioxid. Redox Signaling, 2016, 24, 345–360 CrossRef CAS.
- F. Zobi, A. Degonda, M. C. Schaub and A. Y. Bogdanova, Inorg. Chem., 2010, 49, 7313–7322 CrossRef CAS.
- F. Zobi, Inorg. Chem., 2009, 48, 10845–10855 CrossRef CAS.
- F. Zobi and O. Blacque, Dalton Trans., 2011, 40, 4994–5001 RSC.
- F. Zobi, Inorg. Chem., 2010, 49, 10370–10377 CrossRef CAS.
- S. P. Schmidt, W. C. Trogler, F. Basolo, M. A. Urbancic and J. R. Shapley, Inorg. Synth., 1990, 160–165, DOI:10.1002/9780470132593.ch42.
- R. Alberto, A. Egli, U. Abram, K. Hegetschweiler, V. Gramlich and P. A. Schubiger, Dalton Trans., 1994, 2815–2820, 10.1039/DT9940002815.
- U. Abram, R. Hübener, R. Alberto and R. Schibli, Z. Anorg. Allg. Chem., 1996, 622, 813–818 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- Q. Wu and W. Yang, J. Chem. Phys., 2002, 116, 515–524 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2056997–2057005. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00514f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Aurélien Crochet
,
Aurélien Crochet