
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of ultra-high molecular weight poly(ethylene)-co-(1-hexene) copolymers through high-throughput catalyst screening†
Thomas J. Williams a,
Jessica V. Lamba,
Jean-Charles Buffeta,
Tossapol Khamnaenb and
Dermot O'Hare*a
a,
Jessica V. Lamba,
Jean-Charles Buffeta,
Tossapol Khamnaenb and
Dermot O'Hare*a
aChemistry Research Laboratory, Department of Chemistry, University Oxford, 12 Mansfield Road, OX1 3TA, Oxford, UK. E-mail: dermot.ohare@chem.ox.ac.uk
bSCG Chemicals Co., Ltd, 1 Siam Cement Rd, Bangkok 10800, Thailand
First published on 2nd February 2021
Abstract
A family of permethylindenyl titanium constrained geometry complexes, Me2SB(R′N,3-RI*)TiX2 ((3-R-η5-C9Me5)Me2Si(R′TiX2)), supported on solid polymethylaluminoxane (sMAO) are investigated as slurry-phase catalysts for ethylene/H2 homopolymerisation and ethylene/1-hexene copolymerisation by high-throughput catalyst screening. Me2SB(tBuN,I*)TiCl2 supported on sMAO [sMAO-Me2SB(tBuN,I*)TiCl2] is responsive to small quantities of H2 (<1.6%), maintaining high polymerisation activities (up to 4900 kgPE molTi−1 h−1 bar−1) and yielding polyethylenes with significantly decreased molecular weight (Mw) (from 2700 to 41 kDa with 1.6% H2). In slurry-phase ethylene/1-hexene copolymerisation studies, a decrease in polymerisation activity and polymer molecular weights compared to ethylene homopolymerisation is observed. Compared to many solid supported system, these complexes all display high 1-hexene incorporation levels up to a maximum incorporation of 14.2 mol% for sMAO-Me2SB(iPrN,I*)TiCl2). We observe a proportionate increase in 1-hexene incorporation with concentration, highlighting the ability of these catalysts to controllably tune the amount of 1-hexene incorporated into the polymer chain to produce linear low-density polyethylene (LLDPE) materials.
Introduction
The incorporation of longer chain α-olefin monomers into polyethylene chains increases the degree of polymer branching, which lowers the melting point, crystallinity, and density of the polymers.1 This can lead to significant increases in polymer flexibility, which gives the resultant polymers applications in packaging, foams, elastic fibers, and adhesives.2Metallocene catalysts containing two η5-cyclopentadienyl (C5H5, Cp) ligands and two σ-type ligands (Cp2MX2) have similar reactivities with both ethylene and longer chain α-olefins;3 allowing them to incorporate much larger percentages of higher α-olefins than traditional Ziegler–Natta catalysts.4 Unlike the latter, copolymerisation using metallocene catalysts often results in regular comonomer distributions and forms high strength, high clarity polymers.5,6
Constrained geometry complexes (CGCs), bridged half-metallocenes containing amide ligands, such as the Dow Chemical Co. complexes {(3-tBu-η5-C5H3)Me2Si(tBuN)}TiMe2 (Me2SB(tBuN,Cp3-tBu)TiMe2), Me2SB(tBuN,Cp*)TiMe2, Me2SB(tBuN,I)TiMe2, and Me2SB(tBuN,3-OMeI)TiMe2,7 have been shown to be highly efficient ethylene/olefin copolymerisation catalysts, with high levels of olefin incorporated into the polymer chains.5,8,9 For example, in the solution phase, α-olefin incorporations of 25.3 mol% have been observed for ethylene/1-octene copolymerisation using Me2SB(tBuN,Cp*)TiMe2/[HNMe(C18H37)2][B(C6F5)4] (20 bar ethylene and 300 g 1-octene),7 and incorporations of 69.9% for ethylene/1-hexene copolymerisation using Me2SB(tBuN,Cp*)Ti(CH2Ph)2/MAO (1 bar ethylene and 44.5 mmol 1-hexene).10,11 These CGCs are of industrial interest due to their enhanced ability to copolymerise ethylene and longer chain α-olefins when compared to Cp2MX2 metallocene catalysts.7,8,11,12 This has been attributed to the less crowded coordination sphere, decreased tendency to undergo chain transfer reactions, and smaller bite angle (Cpcent–M–N angle) of CGCs compared to metallocenes (Cpcent–M–Cpcent) (by approximately 25–30°).13
CGCs are highly tuneable, and variation of the complex components can dramatically influence polymerisation activities.13 It has been found that for CGCs containing a substituted indenyl fragment, the addition of electron-donating substituents leads to both increased copolymerisation activity and polymer molecular weights.14 One advantage of CGCs is their ability to produce polyethylenes with very ultra-high molecular weights, with Mw often in excess of 1000 kDa.10,15–17 The long polymer chains transfer pressure more effectively to the polymer backbone, resulting in very tough materials with the highest impact strength of any thermoplastic currently produced.17 The extremely low moisture absorption, very low friction coefficient, biological inertness, and self-lubricating nature of UHMWPE have led to their use in fishing lines, joint replacements, and impact-resistant materials in the military.17–19
We recently reported the synthesis and characterisation of a new family of CGCs based on the permethylindenyl ligand (C9Me7, Ind*, I*): {(3-R-η5-C9Me5)Me2Si(R′N)}TiX2 (Me2SB(R′N,3-RI*)TiX2; R = H and Et; R′ = iPr, tBu, and nBu; X = Cl, Me, CH2Ph, and CH2SiMe3) (Chart 1).20,21
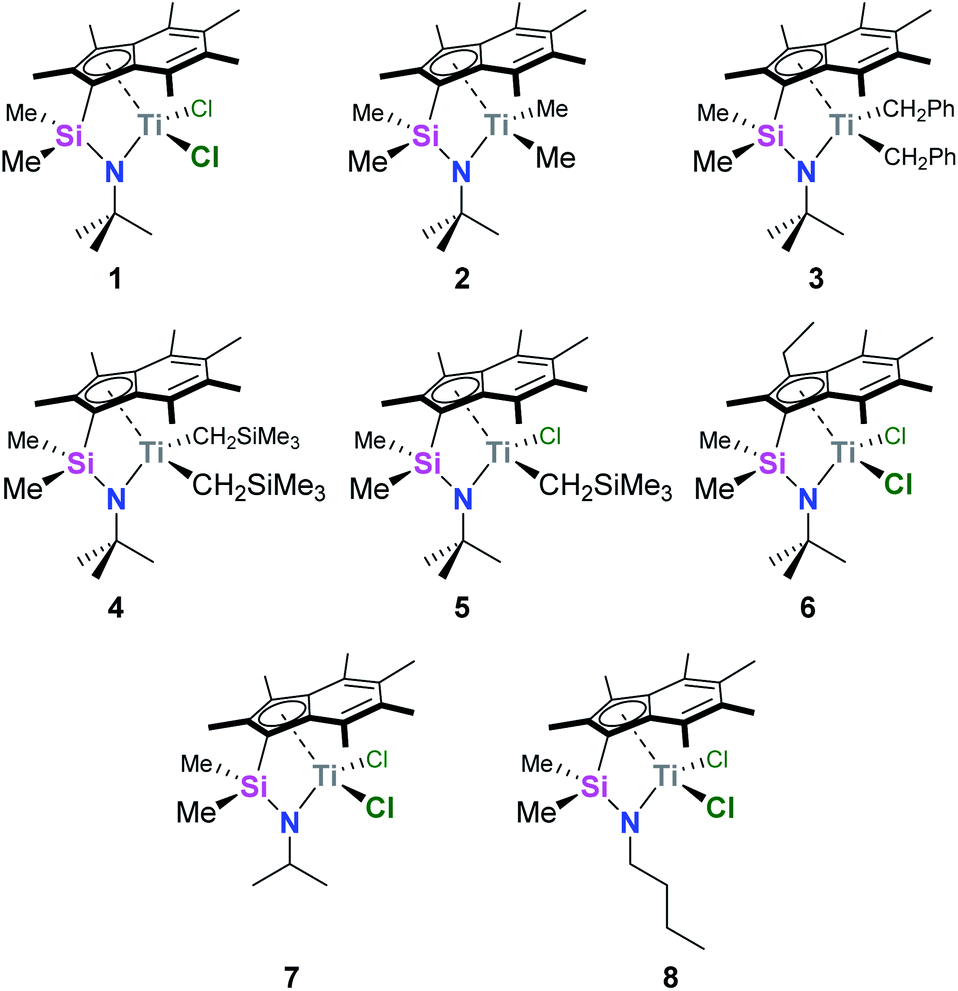 | ||
| Chart 1 Permethylindenyl CGCs: Me2SB(tBuN,I*)TiCl2 (1),20 Me2SB(tBuN,I*)TiMe2 (2),21 Me2SB(tBuN,I*)Ti(CH2Ph)2 (3),21 Me2SB(tBuN,I*)Ti(CH2SiMe3)2 (4),21 Me2SB(tBuN,I*)Ti(Cl)CH2SiMe3 (5),21 Me2SB(tBuN,3-EtI*)TiCl2 (6),21 Me2SB(iPrN,I*)TiCl2 (7),20 and Me2SB(nBuN,I*)TiCl2 (8).20 | ||
When immobilised on solid polymethylaluminoxane (sMAO),22 an insoluble form of oligomeric MAO, the CGCs were found to be very active catalysts for slurry-phase ethylene polymerisation, ethylene/1-hexene copolymerisation, and ethylene/styrene copolymerisation with activities up to 7048, 4248, and 2036 kgPE molTi−1 h−1 bar−1 respectively.21 The catalysts showed low levels of 1-hexene and styrene incorporation (1.9–2.4 mol% and 1.6–2.5 mol% respectively) with 1-hexene incorporation levels found to increase with increasing copolymerisation temperature.21
Herein, we report a systematic investigation of the polymerisation performance of sMAO supported permethylindenyl titanium constrained geometry complexes for ethylene and ethylene/1-hexene copolymerisation using a high-throughput catalyst screening methodology.
Results and discussion
The CGCs in Chart 1 were immobilised on solid polymethylaluminoxane (sMAO) with an initial aluminium to titanium catalyst loading ([AlsMAO]0/[Ti]0) of 200, using a procedure described in previous work.20 The catalysts were studied under high-throughput conditions for ethylene homopolymerisation with or without dihydrogen (H2), and ethylene/1-hexene copolymerisation. The high-throughput system allowed a large number of parallel experiments to be run simultaneously, enabling the screening of different conditions in a shorter time period.23Ethylene/H2 homopolymerisation
sMAO supported Me2SB(tBuN,I*)TiCl2 (1sMAO), Me2SB(tBuN,I*)TiMe2 (2sMAO), Me2SB(tBuN,I*)Ti(CH2Ph)2 (3sMAO), Me2SB(tBuN,I*)Ti(CH2SiMe3)2 (4sMAO), Me2SB(tBuN,I*)Ti(Cl)CH2SiMe3 (5sMAO), and Me2SB(tBuN,3-EtI*)TiCl2 (6sMAO) were studied for ethylene homopolymerisation and H2 response. High-throughput polymerisation studies were conducted in a parallel pressure reactor (PPR) at 80 °C with 8.3 bar ethylene, 0.8% (0.07 bar) or 1.6% (0.13 bar) H2 supplied by a mixed H2/N2 feed, 5 mL heptane, 10 μmol triisobutylaluminium (TiBA, Al(CH2CH(CH3)2)3) scavenger, and 0.075–0.40 mg pre-catalyst ([AlsMAO]0/[Ti]0 = 200) for 1 hour or until 8.3 bar of ethylene uptake was reached.Polymerisation activities decreased with the addition of H2, however, the catalysts remained very active; activities of 6700, 5700, and 4800 kgPE molTi−1 h−1 bar−1 for 1sMAO with 0, 0.8, and 1.6% H2 respectively (Fig. 1 and Table 1). The decrease in polymerisation activity with increasing H2 pressure was found to be greater for the alkylated catalysts (2sMAO, 3sMAO, and 4sMAO) than the dichloride (1sMAO and 6sMAO) and mono-chloride (5sMAO) catalysts; with 1.6% H2, activity decreased by 28, 30, and 42% for 1sMAO, 5sMAO, and 6sMAO when compared to ethylene homopolymerisation, and by 43, 54 and 65% for 4sMAO, 2sMAO, and 3sMAO. The differences in the relative changes in activities and the absolute activities of sMAO-Me2SB(tBuN,I*)TiX2 (1sMAO–4sMAO) catalysts also suggests that the initiator groups remain coordinated to the surface of the support and influence the nature of the active species through a secondary coordination effect.24 Chlorides initiating group could also block the active sites.
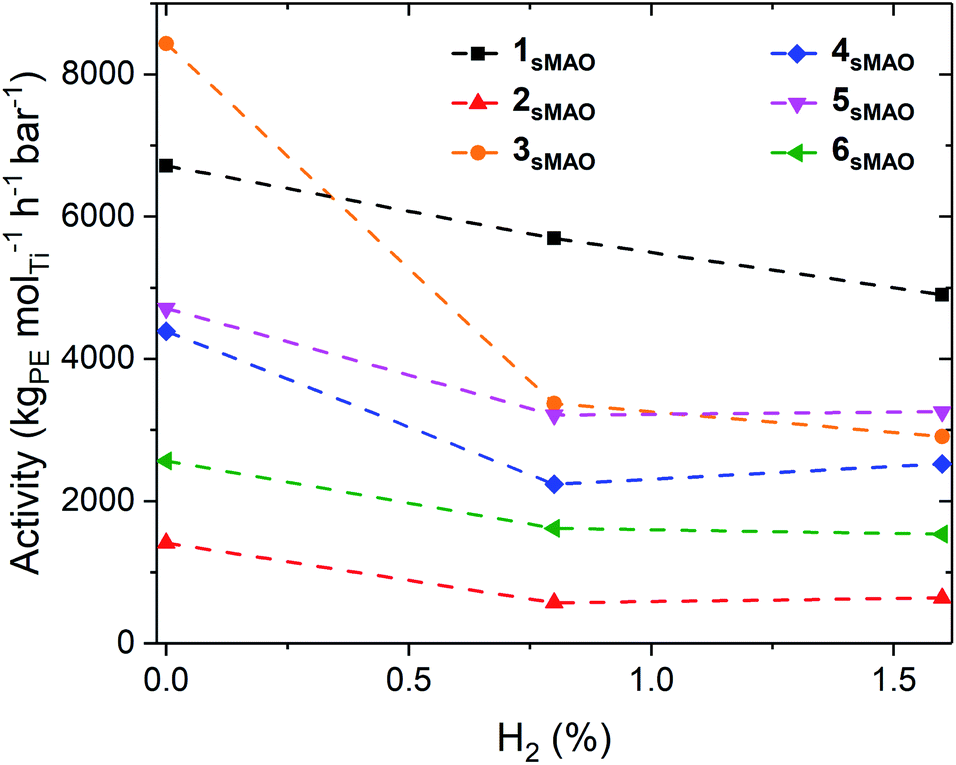 | ||
| Fig. 1 Slurry-phase ethylene polymerisation activity as a function of H2 (%) using sMAO supported Me2SB(tBuN,I*)TiCl2 (1sMAO) (black square), Me2SB(tBuN,I*)TiMe2 (2sMAO) (red up triangle), Me2SB(tBuN,I*)Ti(CH2Ph)2 (3sMAO) (orange circle), Me2SB(tBuN,I*)Ti(CH2SiMe3)2 (4sMAO) (blue diamond), Me2SB(tBuN,I*)Ti(Cl)CH2SiMe3 (5sMAO) (pink down triangle), and Me2SB(tBuN,3-EtI*)TiCl2 (6sMAO) (green left triangle) with 0, 0.8, and 1.6% H2. Polymerisation conditions: 8.3 bar ethylene, 0.075–0.40 mg pre-catalyst ([AlsMAO]0/[Ti]0 = 200), 5.0 mL heptane, 10 μmol TiBA, and 80 °C. Reactions quenched at 8.3 bar ethylene uptake or after 60 minutes. | ||
| Catalyst | H2b | Activityc | Mwd | Mw/Mn |
|---|---|---|---|---|
| a Polymerisation conditions: 8.3 bar ethylene, 0.075–0.40 mg pre-catalyst ([AlsMAO]0/[Ti]0 = 200), 5.0 mL heptane, 10 μmol TiBA, and 80 °C. Reactions quenched at 8.3 bar ethylene uptake or after 60 minutes.b %.c kgPE molTi−1 h−1 bar−1, reported to 2 significant figures.d kDa, reported to 2 significant figures. | ||||
| 1sMAO | 0 | 6700 | 2700 | 3.2 |
| 0.8 | 5700 | 80 | 2.4 | |
| 1.6 | 4900 | 41 | 2.9 | |
| 2sMAO | 0 | 1400 | 1400 | 3.4 |
| 0.8 | 570 | 85 | 2.7 | |
| 1.6 | 640 | 45 | 2.7 | |
| 3sMAO | 0 | 8400 | 1200 | 3.4 |
| 0.8 | 3400 | 84 | 2.8 | |
| 1.6 | 2900 | 47 | 2.7 | |
| 4sMAO | 0 | 4400 | 1800 | 3.5 |
| 0.8 | 2200 | 82 | 2.6 | |
| 1.6 | 2500 | 43 | 2.6 | |
| 5sMAO | 0 | 4700 | 1500 | 3.8 |
| 0.8 | 3200 | 73 | 2.7 | |
| 1.6 | 3300 | 42 | 2.7 | |
| 6sMAO | 0 | 2600 | 1400 | 3.4 |
| 0.8 | 1600 | 80 | 2.8 | |
| 1.6 | 1500 | 42 | 2.7 | |
Over the course of the polymerisation runs, the in situ ethylene uptake rate profiles show lower uptake rates for ethylene polymerisation with H2 compared to without H2 (Fig. 2 and S1–S3†). The lower activities and ethylene uptake rates for ethylene/H2 polymerisation are attributable to the formation of a metal hydride species from chain transfer to H2, which requires reactivation by propagation.25–27 The lower polymerisation activities may also be due to the formation of dormant bimetallic resting states with a bridging hydride, as has been proposed in the solution phase, that require reactivation to form the cationic methyl species.28,29 For 1sMAO, ethylene polymerisation with 0.8% H2 initially shows a higher ethylene uptake rate than for polymerisation without H2; H2 may activate an alternative site for a short period,27 which then becomes deactivated as polymerisation progresses (Fig. 2).
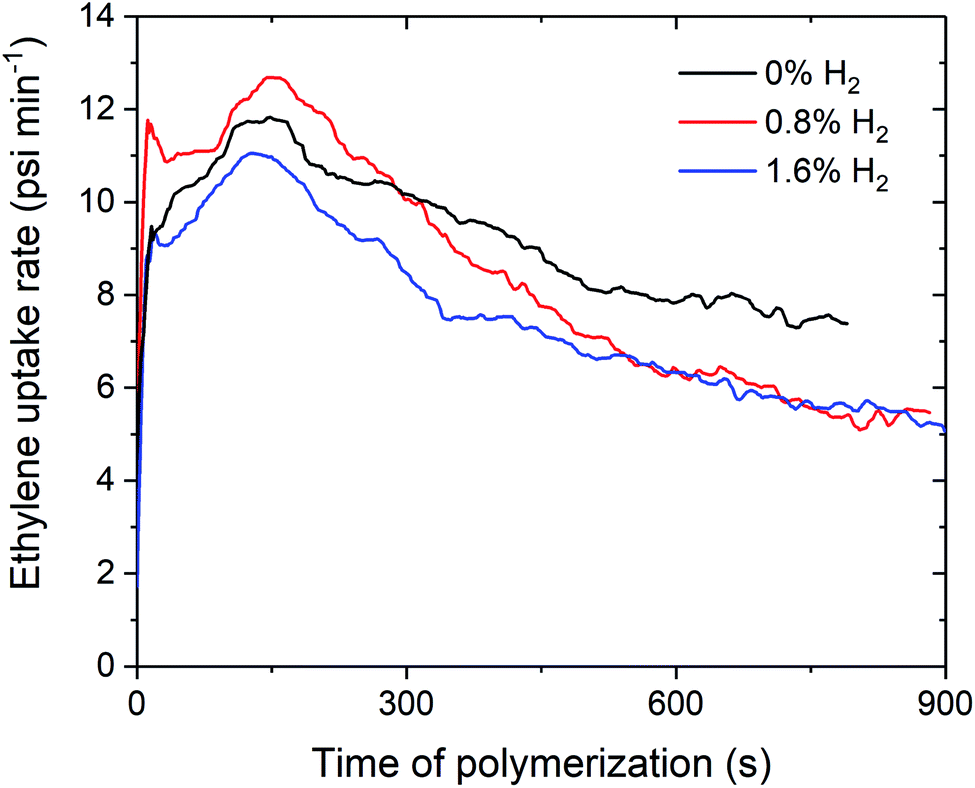 | ||
| Fig. 2 Slurry-phase polymerisation ethylene uptake rate as a function of time of polymerisation using sMAO supported Me2SB(tBuN,I*)TiCl2 (1sMAO) with 0 (black), 0.8 (red), and 1.6% H2 (blue). Polymerisation conditions: 8.3 bar ethylene, 0.20 mg pre-catalyst ([AlsMAO]0/[Ti]0 = 200), 5.0 mL heptane, 10 μmol TiBA, and 80 °C. Reactions quenched at 8.3 bar ethylene uptake or after 60 minutes. | ||
Polymer molecular weights (Mw) decreased with increased addition of H2; Mw of ∼80 and ∼45 kDa with 0.8 and 1.6% H2 respectively for all catalysts (Table 1, Fig. S9 and S11–S13†). The narrowing of the molecular weight distributions with increased addition of H2, (Mw/Mn of 3.8 and 2.7 for 5sMAO with 0 and 1.6% H2) suggests increased control in the reaction.30 Crystallisation-elution fractionation (CEF) showed that the maximum elution temperature (Tel,max) of the polymers decreased slightly in the presence of H2 (Tel,max of 113.3, 112.1, and 111.8 °C with 0, 0.8, and 1.6% H2 respectively for 2sMAO), indicating a slight decrease in melting point and crystallinity (Table S1 and Fig. S18–S20†). The amorphous fraction (AF) increased in the presence of H2; AF of 0.2, 0.5 and 0.7 with 0, 0.8, and 1.6% H2 respectively for 2sMAO.
Ethylene/1-hexene copolymerisation
sMAO supported Me2SB(tBuN,I*)TiCl2 (1sMAO), Me2SB(tBuN,I*)TiMe2 (2sMAO), Me2SB(tBuN,I*)Ti(CH2Ph)2 (3sMAO), Me2SB(tBuN,I*)Ti(CH2SiMe3)2 (4sMAO), Me2SB(tBuN,I*)Ti(Cl)CH2SiMe3 (5sMAO), Me2SB(tBuN,3-EtI*)TiCl2 (6sMAO), Me2SB(iPrN,I*)TiCl2 (7sMAO), and Me2SB(nBuN,I*)TiCl2 (8sMAO) (Chart 1) were studied as catalysts for ethylene/1-hexene copolymerisation.Large reductions in activity were observed for ethylene/1-hexene copolymerisation compared to ethylene homopolymerisation (6700 and 3600 kgPE molTi−1 h−1 bar−1 for 1sMAO with 0 and 250 μL 1-hexene respectively), indicating that the negative comonomer effects outweigh the positive effects (Table 2, Fig. 3 and S8†).21,31,32 A large decrease in ethylene polymerisation activity is observed with increasing volumes of 1-hexene. For example, a decrease from 4700 to 1500 kgPE molTi−1 h−1 bar−1 for 5sMAO with 0 and 250 μL 1-hexene respectively.
| Catalyst | 1-Hexeneb | Activityc | Mwd | Mw/Mn | Incorporatione | Tel,maxf |
|---|---|---|---|---|---|---|
| a Polymerisation conditions: 8.3 bar ethylene, 0.075–0.40 mg pre-catalyst ([AlsMAO]0/[Ti]0 = 200), 5.0 mL heptane, 10 μmol TiBA, and 80 °C. Reactions quenched at 5.5 bar ethylene uptake or after 60 minutes.b μL.c kgPE molTi−1 h−1 bar−1, reported to 2 significant figures.d kDa, reported to 2 significant figures.e Mol%.f °C. | ||||||
| 1sMAO | 0 | 6700 | 2700 | 3.2 | — | 112.1 |
| 125 | 5200 | 270 | 3.0 | 5.6 | 85.1 | |
| 250 | 3600 | 330 | 2.5 | 6.6 | 73.5 | |
| 2sMAO | 0 | 1400 | 1400 | 3.4 | — | 113.3 |
| 125 | 380 | 390 | 2.8 | 3.4 | 89.0 | |
| 250 | 250 | 300 | 2.6 | 6.3 | 70.5 | |
| 3sMAO | 0 | 8400 | 1200 | 3.4 | — | 111.4 |
| 125 | 2700 | 490 | 2.7 | 3.1 | 89.8 | |
| 250 | 3000 | 380 | 2.4 | 6.3 | 71.6 | |
| 4sMAO | 0 | 4400 | 1800 | 3.5 | — | 113.3 |
| 125 | 1100 | 390 | 2.8 | 3.5 | 84.8 | |
| 250 | 280 | 200 | 2.3 | 7.1 | 69.5 | |
| 5sMAO | 0 | 4700 | 1500 | 3.8 | — | 113.3 |
| 125 | 2200 | 360 | 2.5 | 3.6 | 88.0 | |
| 250 | 1500 | 270 | 2.5 | 7.4 | 70.9 | |
| 6sMAO | 0 | 2600 | 1400 | 4.0 | — | 113.4 |
| 125 | 1700 | 250 | 2.9 | 5.6 | 81.9 | |
| 250 | 1100 | 220 | 2.6 | 8.4 | 66.0 | |
| 7sMAO | 0 | 1200 | 1700 | 2.5 | — | 113.9 |
| 125 | 380 | 250 | 2.7 | 6.3 | 98.8 | |
| 250 | 390 | 260 | 2.4 | 14.2 | 80.5 | |
| 8sMAO | 0 | 490 | 1700 | 6.5 | — | — |
| 125 | 220 | 210 | 4.3 | 1.6 | — | |
| 250 | 190 | 360 | 3.9 | 4.7 | — | |
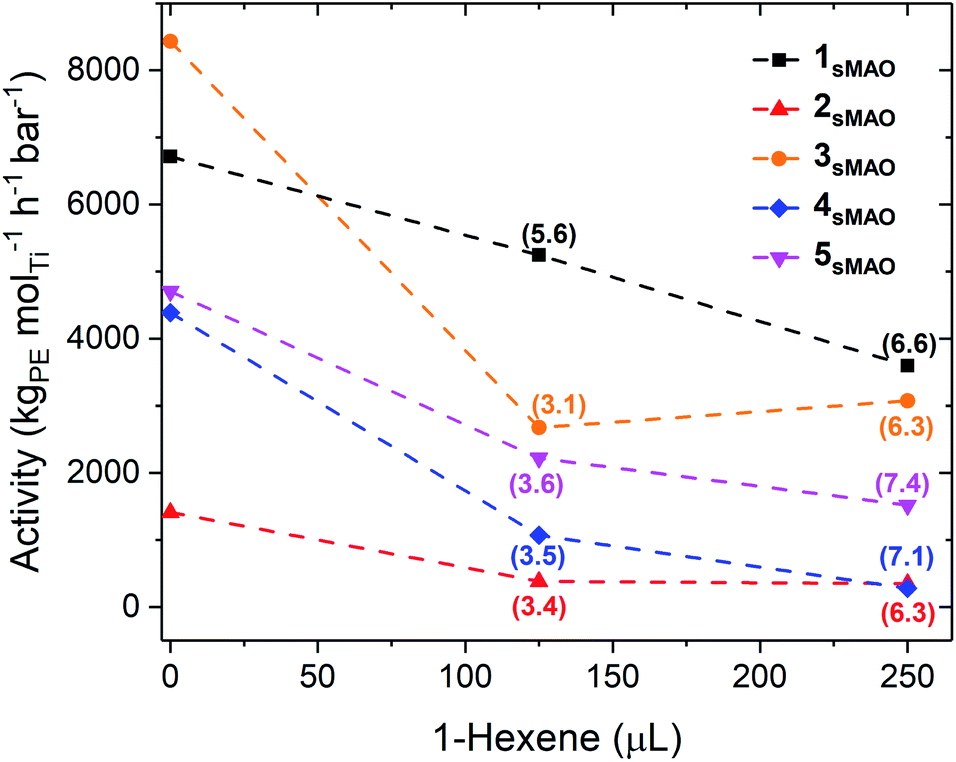 | ||
| Fig. 3 Slurry-phase ethylene polymerisation activity as a function of 1-hexene (μL) using sMAO supported Me2SB(tBuN,I*)TiCl2 (1sMAO) (black square), Me2SB(tBuN,I*)TiMe2 (2sMAO) (red up triangle), Me2SB(tBuN,I*)Ti(CH2Ph)2 (3sMAO) (orange circle), Me2SB(tBuN,I*)Ti(CH2SiMe3)2 (4sMAO) (blue diamond), and Me2SB(tBuN,I*)Ti(Cl)CH2SiMe3 (5sMAO) (pink down triangle) with 0, 125, and 250 μL 1-hexene. 1-Hexene incorporation (mol%) shown in parenthesis. Polymerisation conditions: 8.3 bar ethylene, 0.075–0.40 mg pre-catalyst ([AlsMAO]0/[Ti]0 = 200), 5.0 mL heptane, 10 μmol TiBA, and 80 °C. Reactions quenched at 5.5 bar ethylene uptake or after 60 minutes. | ||
Many theories have been proposed for the positive comonomer effect, including fracturing of catalyst particles exposing new sites, the formation of new active species by coordination of α-olefins, and activation of dormant active sites; however, many of these have been refuted for molecular catalyst systems.33 Studies have also shown that the addition of 1-hexene to an alkane reaction mixture leads to a 7–10% increase in ethylene solubility between 70–90 °C,34 as well as improved diffusion of ethylene close to the catalytic site, which improves polymerisation activity.35 The negative effects of comonomer addition are proposed to be due to competitive binding between ethylene and α-olefins and, if the rate of migratory insertion of the α-olefin is slower than that of ethylene, the rate of chain propagation will decrease leading to a decrease in polymerisation activity.33 The negative effects of comonomers on ethylene polymerisation activity may also be due to slower rates of insertion; the increased steric bulk of α-olefin comonomers in the polymer chain can lead to reduced rates of ethylene insertion.36
Through monitoring changes in temperature during polymerisation, an exothermic temperature spike to approximately 85 °C was observed at the start of the copolymerisation experiments. As the alkyl catalysts (2sMAO, 3sMAO, and 4sMAO) are much more sensitive to polymerisation temperature than the dichloride catalysts (1sMAO, 6sMAO, 7sMAO, and 8sMAO),21 this thermal spike caused more substantial decreases in polymerisation activities for these catalysts; activity decreases from 6700 to 3600 kgPE molTi−1 h−1 bar−1 for 1sMAO and from 4400 to 280 kgPE molTi−1 h−1 bar−1 for 4sMAO with 0 and 250 μL 1-hexene respectively.
The decreases in polymerisation activity with increasing volumes of 1-hexene are highlighted by the in situ ethylene uptake rate profiles, where sharp decreases in uptake rates with 125 and 250 μL 1-hexene are observed when compared to ethylene homopolymerisation (Fig. 4 and S4–S7†). Polymerisation activity was observed to increase with increasing electron-donating ability of the amido fragment (tBu > iPr > nBu; 1sMAO > 7sMAO > 8sMAO) (Fig. S8†), as observed in previous work.20 Kamigaito et al. and Nomura et al. have also observed similar effects when using Me2SB(RN,Cp*)TiCl2/MAO (R = tBu, Ph, and C6F5;37 R = tBu and Cy)38,39 catalysts for solution-phase ethylene/styrene copolymerisation.
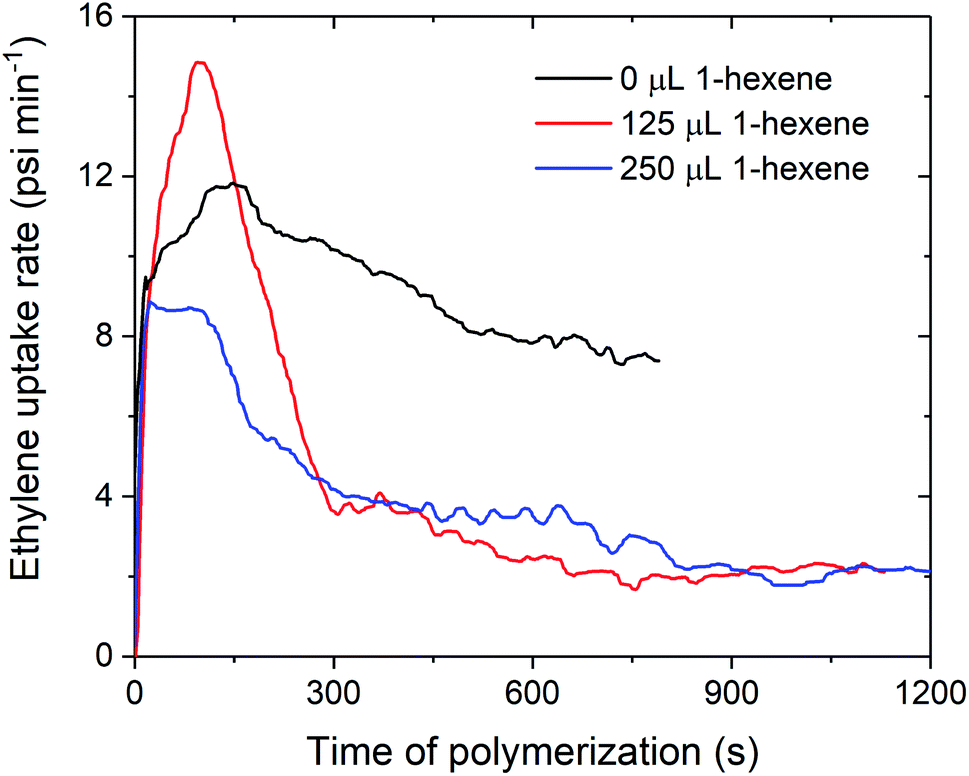 | ||
| Fig. 4 Slurry-phase polymerisation ethylene uptake rate as a function of time of polymerisation using sMAO supported Me2SB(tBuN,I*)TiCl2 (1sMAO) with 0 (black), 125 (red), and 250 μL 1-hexene (blue). Polymerisation conditions: 8.3 bar ethylene, 0.20 mg pre-catalyst ([AlsMAO]0/[Ti]0 = 200), 5.0 mL heptane, 10 μmol TiBA, and 80 °C. Reactions quenched at 5.5 bar ethylene uptake or after 60 minutes. | ||
Klosin et al. have previously reported the effects of variation of the indenyl moiety on ethylene/1-octene copolymerisation, finding that increased electron-donating ability led to higher activities and polymer molecular weights.14 The opposite effect is observed for these systems, where 6sMAO shows a lower ethylene polymerisation activity than 1sMAO, attributed to its decreased thermal stability; activities of 1100 and 3600 kgPE molTi−1 h−1 bar−1 respectively with 250 μL 1-hexene. The lower polymerisation activity of 3-ethylpentamethylindenyl supported catalysts relative to the permethylindenyl analogs has been observed previously for ethylene polymerisation using 1sMAO and 6sMAO with 2 bar ethylene and 50 mL solvent at temperatures above 70 °C,21 and when using sMAO-Me2SB(2,7-tBuFlu,3-RI*)ZrCl2 catalysts.40 6sMAO also shows greater decreases in activities for ethylene/1-hexene copolymerisation compared to ethylene homopolymerisation (35 and 58% decreases for 125 and 250 μL 1-hexene respectively) than 1sMAO (22 and 46% decreases respectively). Similar to alkylated catalysts (2sMAO, 3sMAO, and 4sMAO), this may be due to the exothermic temperature spike at the beginning of the copolymerisation experiment and the lower thermal stability of 6sMAO compared to 1sMAO.
The catalysts produced polymers with very high levels of 1-hexene incorporation for supported systems (up to 14.2 mol% for 7sMAO), confirming the production of ethylene/1-hexene copolymers. This is a trait commonly observed for CGCs that is attributed to the open metal centre resulting from the strain-inducing ansa-bridge (Table 2).8,12,13
The incorporation levels observed for these catalysts are lower than for solution-phase ethylene/1-hexene copolymerisation using Me2SB(tBuN,Cp*)Ti(CH2Ph)2 with an MAO cocatalyst (65–70% 1-hexene incorporation).10 However, supported catalysts typically give lower incorporation levels than homogeneous catalysts due to mass transfer effects, where both the support and the propagating polymer chain cause diffusional resistance of the comonomer towards the active sites.41,42 The active sites of supported catalysts may also become blocked with polymer more quickly than the same catalysts in solution and therefore become inaccessible.42
It was found that 1sMAO, 6sMAO, and 7sMAO produced polymers with similar incorporation levels with 125 μL 1-hexene (5.6–6.3 mol%). However, 7sMAO produced polymers with much higher incorporation levels than 6sMAO and 1sMAO with 250 μL 1-hexene (14.2, 8.4, and 6.6 mol% respectively). This suggests that higher levels of 1-hexene incorporation accompany reduced steric bulk in the amido substituent, likely due to easier coordination of 1-hexene to the metal centre. Catalysts containing at least one alkyl ligand (2sMAO, 3sMAO, 4sMAO, and 5sMAO) produced polymers with similar incorporation levels; 3.1–3.6 and 3.3–7.4 mol% with 125 and 250 μL 1-hexene respectively.
A similar effect was also observed by Chen and Marks for solution-phase ethylene/1-hexene copolymerisation using Me2SB(tBuN,Cp*)TiMe2/(BC6F5)3 and Me2SB(tBuN,Cp*)Ti(CH2Ph)2/MAO where both alkyl ligand containing catalysts produced polymers with ∼70% 1-hexene incorporation.10 8sMAO consistently produced polymers with lower incorporation levels (1.6 and 4.7 mol% with 125 and 250 μL 1-hexene respectively), which may be due to the reduced electron donating ability of nBu. A proportionate increase in 1-hexene incorporation was observed for ethylene/1-hexene copolymerisation using 2sMAO–7sMAO (the amount of 1-hexene incorporated into the polyethylene chain approximately doubled when the amount of 1-hexene in the system was doubled), which gives great potential for these catalysts to controllably tune the amount of 1-hexene incorporated into the polymer chain.
Gel permeation chromatography (GPC) showed that as the amount of 1-hexene added to the system increased, the molecular weights (Mw) of the polymers significantly decreased; the polymers produced using 1sMAO showed an eight-fold decrease in polymer molecular weights on the addition of 250 μL 1-hexene (Mw of 2700 and 330 kDa with 0 and 250 μL 1-hexene respectively) (Table 2, Fig. S10 and S14–S17†). The decrease in polymer molecular weights likely results from frequent chain termination following 1-hexene insertion and chain transfer to 1-hexene monomers, coupled with a decrease in the rate of chain propagation.31,43 This effect has been observed and studied for ethylene/α-olefin polymerisation using other CGC systems, such as Me2SB(tBuN,Cp*)TiMe2, Me2SB(tBuN,2-RI)TiMe2, and Me2SB(tBuN,3-RI)TiMe2, with work having been undertaken in an attempt to negate the molecular weights decrease by adding heteroatom substituents in the 2- and 3-positions on the indenyl moiety.7,14
The catalysts produced polymers with relatively narrow molecular weight distributions (Mw/Mn), which became narrower with increasing volumes of 1-hexene; Mw/Mn of 3.2, 3.0, and 2.7 for 1sMAO with 0, 125, and 250 μL 1-hexeneThe polymers produced using 8sMAO showed wider molecular weight distributions than the polymers produced using the other catalysts (Mw/Mn of 6.5, 4.4, and 4.0 with 0, 125, and 250 μL 1-hexene respectively), suggesting the potential for more than one active site (Fig. S17†).
CEF showed that the maximum elution temperatures (Tel,max) of the polymers dramatically decreased with increasing volumes of 1-hexene, indicative of higher levels of 1-hexene incorporation; Tel,max of 112.1, 85.1, and 73.5 °C for 1sMAO with 0, 125, and 250 μL 1-hexene respectively (Table 2 and Fig. S21–S23†).
The decreases in Tel,max are attributable to the weakening of intramolecular forces between the polymer chains with increasing incorporation of 1-hexene and decreasing molecular weights of the polymers.44 The amorphous fraction (AF) also increased with increasing 1-hexene concentration; AF of 0.2, 0.7, and 27.2 wt% for 4sMAO with 0, 125, and 250 μL 1-hexene respectively (Table S2†). This corroborates with the high temperature 13C{1H} NMR spectra (Fig. S24–S27†).
Conclusions
A series of eight permethylindenyl constrained geometry titanium complexes (Me2SB(R′N,3-RI*)TiX2) supported on solid polymethylaluminoxane (sMAO) have been studied for ethylene homopolymerisation, H2 response, and ethylene/1-hexene copolymerisation in a high-throughput catalyst screening system.The catalysts displayed very high ethylene homopolymerisation activities; maximum activity of 8400 kgPE molTi−1 h−1 bar−1 for sMAO-Me2SB(tBuN,I*)Ti(CH2Ph)2. sMAO-Me2SB(tBuN,I*)TiCl2 displayed the best H2 response, displaying modest decreases in activity (6700 and 4900 kgPE molTi−1 h−1 bar−1 with 0 and 1.6% H2 respectively), large decreases in polymer molecular weights (Mw of 2700 and 41 kDa with 0 and 1.6% H2 respectively), and narrow molecular weight distributions (Mw/Mn of 2.4–3.2).
The addition of 1-hexene to the system caused a decrease in polymerisation activity and polymer molecular weights (activities of 6700 and 3600 kgPE molTi−1 h−1 bar−1 and Mw of 2700 and 330 kDa for sMAO-Me2SB(tBuN,I*)TiCl2 with 0 and 250 μL 1-hexene respectively), highlighting a negative comonomer effect.
The catalysts displayed high 1-hexene incorporation levels for supported systems with a maximum incorporation of 14.2 mol% for sMAO-Me2SB(iPrN,I*)TiCl2, demonstrating the formation of ethylene/1-hexene copolymers. A proportionate increase in 1-hexene incorporation with 1-hexene concentration was observed, demonstrating the potential capacity of these catalysts to controllably tune the amount of 1-hexene incorporated into the polymer chain to produce industrially relevant linear low-density polyethylene (LLDPE) materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. J. W., J. V. L. and J.-C. B. would like to thank SCG Chemicals Co., Ltd. (Thailand) for financial support; T. J. W. additionally thanks the Engineering and Physical Sciences Research Council (U.K.) for financial support (CASE studentship). We also thank Prof. Vincenzo Busico for the use of HTExplore (University of Naples), and Mr Alessio Mingione and Prof. Roberta Cipullo (HTExplore) for running the high-throughput polymerisation experiments and polymer analysis.Notes and references
- B. Paredes, R. van Grieken, A. Carrero, I. Suarez and J. B. P. Soares, Macromol. Chem. Phys., 2011, 212, 1590–1599 CrossRef CAS.
- W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907–3945 CrossRef CAS.
- W. Kaminsky, A. Funck and H. Haehnsen, Dalton Trans., 2009, 8803–8810 RSC.
- K. Nomura and J. Liu, in Organometallic Reactions and Polymerization, ed. K. Osakada, Springer-Verlag Berlin, Berlin, 2014, vol. 85, pp. 51–88 Search PubMed.
- A. Shamiri, M. H. Chakrabarti, S. Jahan, M. A. Hussain, W. Kaminsky, P. V. Aravind and W. A. Yehye, Materials, 2014, 7, 5069–5108 CrossRef.
- J. Imuta, A. Todo, T. Tsutsui, T. Hachimori and N. Kashiwa, Bull. Chem. Soc. Jpn., 2004, 77, 607–615 CrossRef CAS.
- J. Klosin, P. P. Fontaine and R. Figueroa, Acc. Chem. Res., 2015, 48, 2004–2016 CrossRef CAS.
- J. C. Stevens, F. J. Timmers, D. R. Wilson, G. F. Schmidt, P. N. Nickias, R. K. Rosen, G. W. Knight and S. Y. Lai, EP416815A2, 1991.
- Y. Nakayama, Y. Sogo, Z. G. Cai and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1223–1229 CrossRef CAS.
- Y.-X. Chen and T. J. Marks, Organometallics, 1997, 16, 3649–3657 CrossRef CAS.
- J. Suhm, M. J. Schneider and R. Mülhaupt, J. Mol. Catal. A: Chem., 1998, 128, 215–227 CrossRef CAS.
- J. A. M. Canich, EP420436A1, 1991.
- H. Braunschweig and F. M. Breitling, Coord. Chem. Rev., 2006, 250, 2691–2720 CrossRef CAS.
- J. Klosin, W. J. Kruper, P. N. Nickias, G. R. Roof, P. De Waele and K. A. Abboud, Organometallics, 2001, 20, 2663–2665 CrossRef CAS.
- H. G. Alt, A. Weis, A. Reb and R. Ernst, Inorg. Chim. Acta, 2003, 343, 253–274 CrossRef CAS.
- J.-C. Buffet, Z. R. Turner, R. T. Cooper and D. O'Hare, Polym. Chem., 2015, 6, 2493–2503 RSC.
- S.-J. Park and M.-K. Seo, Interface Science and Composite, 1st edn, Elsevier, 2011, vol. 8, pp. 431–499 Search PubMed.
- A. Wang, D. C. Sun, C. Stark and J. H. Dumbleton, Wear, 1995, 181–183, 241–249 CrossRef CAS.
- M. C. Sobieraj and C. M. Rimnac, J. Mech. Behav. Biomed. Mater., 2009, 2, 433–443 CrossRef CAS.
- T. J. Williams, J.-C. Buffet, Z. R. Turner and D. O'Hare, Catal. Sci. Technol., 2018, 8, 5454–5461 RSC.
- T. J. Williams, A. D. H. Smith, J.-C. Buffet, Z. R. Turner and D. O'Hare, Mol. Catal., 2020, 486, 110872 CrossRef CAS.
- E. Kaji and E. Yoshioka, US8404880, 2013.
- V. Busico, R. Pellecchia, F. Cutillo and R. Cipullo, Macromol. Rapid Commun., 2009, 30, 1697–1708 CrossRef CAS.
- C. M. R. Wright, Z. R. Turner, J.-C. Buffet and D. O'Hare, Chem. Commun., 2016, 52, 2850–2853 RSC.
- A. T. Rappe, W. M. Skiff and C. J. Casewit, Chem. Rev., 2000, 100, 1435–1456 CrossRef CAS.
- W. Kaminsky and H. Lüker, Macromol. Rapid Commun., 1984, 5, 225–228 CrossRef CAS.
- K. J. Chu, J. B. P. Soares and A. Penlidis, Macromol. Chem. Phys., 2000, 201, 552–557 CrossRef CAS.
- F. Q. Song, R. D. Cannon and M. Bochmann, J. Am. Chem. Soc., 2003, 125, 7641–7653 CrossRef CAS.
- M. Bochmann, Organometallics, 2010, 29, 4711–4740 CrossRef CAS.
- D. Mathis, F. Rouholahnejad and P. Chen, Helv. Chim. Acta, 2010, 93, 212–219 CrossRef CAS.
- R. Quijada, G. B. Galland and R. S. Mauler, Macromol. Chem. Phys., 1996, 197, 3091–3098 CrossRef CAS.
- R. Quijada, J. Dupont, M. S. L. Miranda, R. B. Scipioni and G. B. Galland, Macromol. Chem. Phys., 1995, 196, 3991–4000 CrossRef CAS.
- J. C. W. Chien and T. Nozaki, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 227–237 CrossRef CAS.
- B. J. Banasazak, D. Lo, T. Widya, W. H. Ray, J. J. de Pablo, A. Novak and J. Kosek, Macromolecules, 2004, 37, 9139–9150 CrossRef.
- A. Novak, M. Bobak, J. Kosek, B. J. Banaszak, D. Lo, T. Widya, W. H. Ray and J. J. de Pablo, J. Appl. Polym. Sci., 2006, 100, 1124–1136 CrossRef CAS.
- G. Fink, R. Mülhaupt and H. H. Brintzinger, Ziegler Catalysts: Recent Scientific Innovations and Technological Improvements, Springer Berlin Heidelberg, 2012 Search PubMed.
- M. Kamigaito, T. K. Lal and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4649–4660 CrossRef CAS.
- K. Nomura, H. Okumura, T. Komatsu and N. Naga, Macromolecules, 2002, 35, 5388–5395 CrossRef CAS.
- K. Nomura, H. Okumura, T. Komatsu, N. Naga and Y. Imanishi, J. Mol. Catal. A: Chem., 2002, 190, 225–234 CrossRef CAS.
- J.-C. Buffet, Z. R. Turner and D. O'Hare, Chem. Commun., 2018, 54, 10970–10973 RSC.
- L. A. Williams and T. J. Marks, Organometallics, 2009, 28, 2053–2061 CrossRef CAS.
- K. Czaja, L. A. Novokshonova and N. J. Kovaleva, Macromol. Chem. Phys., 1999, 200, 983–988 CrossRef CAS.
- N. Herfert, P. Montag and G. Fink, Makromol. Chem., 1993, 194, 3167–3182 CrossRef CAS.
- M. D. F. V. Marques and R. B. Tiosso, Journal of Materials Science and Engineering with Advanced Technology, 2011, 4, 149–173 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectroscopy, gel permeation chromatography, crystallisation elution fractionation, ethylene uptake profile. See DOI: 10.1039/d1ra00446h |
| This journal is © The Royal Society of Chemistry 2021 |