
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sesquiterpenoids of diverse types from the rhizomes of Acorus calamus†
Zhi-You Hao *ab,
Yan-Fei Liub,
Yan-Gang Caoa,
Dong Liangb,
Huan Luob,
Chun-Lei Zhangb,
Yan Wangb,
Ruo-Yun Chenb and
De-Quan Yu*b
*ab,
Yan-Fei Liub,
Yan-Gang Caoa,
Dong Liangb,
Huan Luob,
Chun-Lei Zhangb,
Yan Wangb,
Ruo-Yun Chenb and
De-Quan Yu*b
aSchool of Pharmacy, Henan University of Chinese Medicine, Zhengzhou 450046, P. R. China. E-mail: hzy@hactcm.edu.cn
bState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, P. R. China. E-mail: dqyu@imm.ac.cn
First published on 15th April 2021
Abstract
Six new sesquiterpenoids (1–6), named calamusins L–Q, together with fourteen known ones were isolated from the ethanol extract of the rhizomes of Acorus calamus. The new compounds and their absolute configurations were determined based on extensive spectroscopic analyses and computational methods. All of the new compounds were evaluated for their neuroprotective effect against serum withdrawal, rotenone, and OGD-induced PC12 cell injury, and it was revealed that compounds 1 and 6 increased the cell survival rate of the OGD-treated PC12 cells moderately at 10 μM.
Introduction
Acorus calamus Linn. (Chinese name is “Shui Chang Pu”) is a perennial aquatic plant of the family Araceae, and widely distributed in China.1 The rhizomes of Acorus calamus were commonly used in traditional Chinese medicine to treat amnesia, phlegm syncope, epilepsy, rheumatism, and various mental illnesses. Previous bioactive studies displayed the plant possesses antitumor,2–4 antivirus,5,6 antibacterial,7,8 neuroprotective,9–11 antidiabetic12 and anti-inflammatory13 activities. Especially, extensive clinical evidence proved it was effective to treat neurological disorders, such as Alzheimer's disease, Parkinson's disease, senile memory impairment, and schizophrenia.14,15 Probably, one reason A. calamus could treat these diseases was its neuroprotective bioactivities. Chemical investigation indicated it contains wide variety of sesquiterpenoids,16–19 phenylpropanoids, lignans, alkaloids,20 flavonoids, steroids, triterpenoid saponins, and volatiles.21–23 Among them, β-asarone, a phenylpropanoid, was reported mostly and regarded as the main neuroprotective constituent of A. calamus.24–27 However, toxicological studies suggested it possess carcinogenicity and genetic toxicity.28,29 Apparently, in this sense, it's not reasonable to view β-asarone as the representative neuroprotective constituent of A. calamus. Therefore, it is necessary to evaluate the neuroprotective bioactivities of other types of compounds.Our previous study on the 95% aqueous ethanol extract of A. calamus reported nine new sesquiterpenoids (calamusins A–I) and their bioactivities.16 During the continued investigation, six new (1–6) and fourteen known sesquiterpenoids of diverse types were characterized. The isolation and structure elucidation of these compounds are described herein, as well as the in vitro neuroprotective assays of the new compounds (Fig .1).
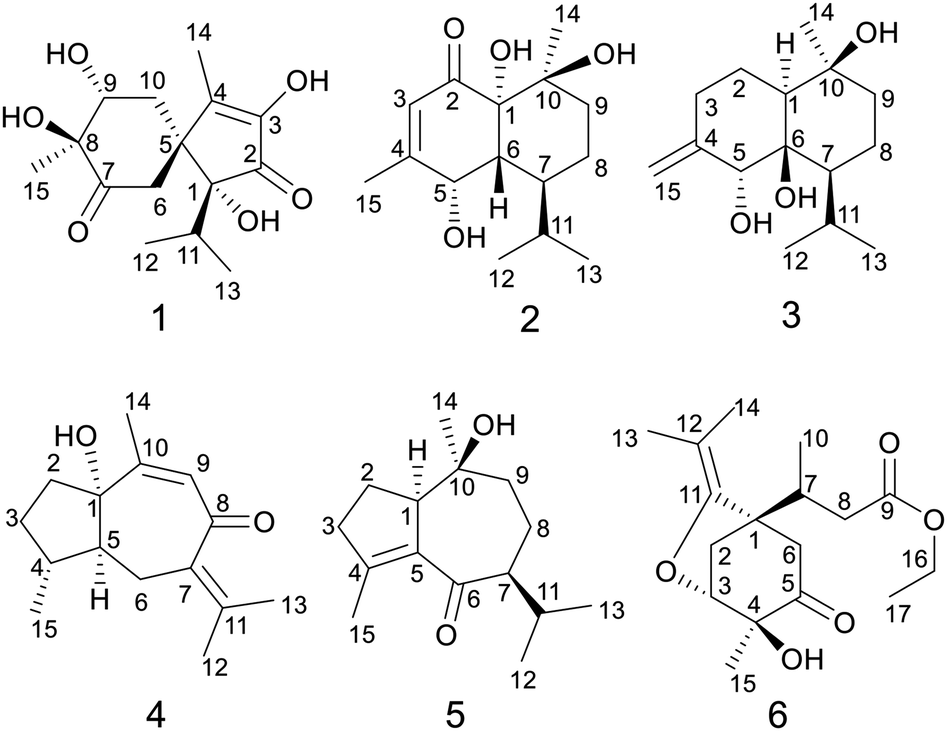 | ||
| Fig. 1 Chemical structures of compounds 1–6. | ||
Results and discussion
Calamusin L (1) was assigned the molecular formula C15H22O6, with 5 hydrogen deficiencies, as deduced from HRESIMS (m/z 299.1490 [M + H]+, m/z 321.1313 [M + Na]+) and NMR data. The IR spectrum showed the absorptions of hydroxy (3390 cm−1) and carbonyl (1708, 1651 cm−1) groups. The 1H NMR spectrum of 1 (Table 1) showed two secondary methyl groups [δH 0.68 (d, J = 6.8 Hz), Me-12; 1.01 (d, J = 6.6 Hz), Me-13], two tertiary methyl groups [δH 1.84 (s), Me-14; 1.29 (s), Me-15], an oxygenated methine group [δH 3.91 (m), H-9], and other five aliphatic protons. The 13C NMR (Table 3) and DEPT spectra of 1 exhibited 15 carbon resonances corresponding to four methyls, two methylenes, two methines (one aliphatic, δC 35.3; one oxygenated, δC 74.7), and seven quaternary carbons (two oxygenated ones at δC 85.2 and 80.1; an aliphatic one at δC 51.9; two olefinic ones at δC 149.2 and 146.1; an α,β-unsaturated keto carbonyl group at δC 200.8; an saturated keto carbonyl group at δC 213.7). Analysis of 1H–1H COSY spectrum revealed the presence of two discrete spin systems (Fig. 2) corresponding to a –CH–CH2– unit (C9–C10) and a CH3–CH–CH3 moiety (C12–C11–C13, an isopropyl group). The HMBC correlations (Fig. 2) were displayed from H-14/10/6 to C-5/4, Me-15 to C-7/8/9, and Me-13/Me-12 to C-1, allowing the establishment of an acorane-type sesquiterpene skeleton, which exists extensively in genus Acorus.16,30 Furthermore, NOESY correlations (Fig. 2) between H-11/12/13 and H-6, Me-14/10b (δH 1.71, β) and H-9 (β), H-10a (δH 2.28, α) and H-15 (α) determined the relative configuration of 1. The absolute configuration of 1 was obtained by analyses of CD spectrum. According to the octant rule of saturated cyclohexanones,31 the negative Cotton effect that occurred at 273 nm (Δε −1.29) based on the n → π* transition of cyclohexanone indicated that the configuration of compound 1 is 1R,5S,8S,9R.| No. | 1 | 2 | 3 |
|---|---|---|---|
| a Measured at 600 MHz. | |||
| 1 | 1.84 (dd, 12.8/3.4) | ||
| 2 | 1.91 (m) | ||
| 1.37 (m) | |||
| 3 | 5.72 (brs) | 2.35 (m) | |
| 2.10 (m) | |||
| 5 | 4.29 (m) | 4.02 (s) | |
| 6 | 2.80 (overlap) | 2.18 (dd, 12.0/3.6) | |
| 7 | 1.99 (m) | 1.71 (m) | |
| 8 | 1.48 (overlap) | 1.52 (m) | |
| 1.38 (m) | 1.45 (overlap) | ||
| 9 | 3.91 (m) | 1.73 (m) | 1.75 (m) |
| 1.48 (overlap) | 1.45 (overlap) | ||
| 10 | 2.28 (m) | ||
| 1.71 (m) | |||
| 11 | 2.04 (m) | 2.13 (m) | 2.16 (m) |
| 12 | 0.68 (d, 6.8) | 0.96 (d, 7.2) | 0.91 (d, 6.9) |
| 13 | 1.01 (d, 6.6) | 0.76 (d, 7.2) | 0.88 (d, 6.9) |
| 14 | 1.84 (s) | 1.42 (s) | 1.14 (s) |
| 15 | 1.29 (s) | 2.05 (d, 1.2) | 4.84 (s) |
| OH | 5.08 (s, 1-OH) | ||
| OH | 4.96 (d, 7.2, 5-OH) | ||
| OH | 2.92 (brs, 10-OH) | ||
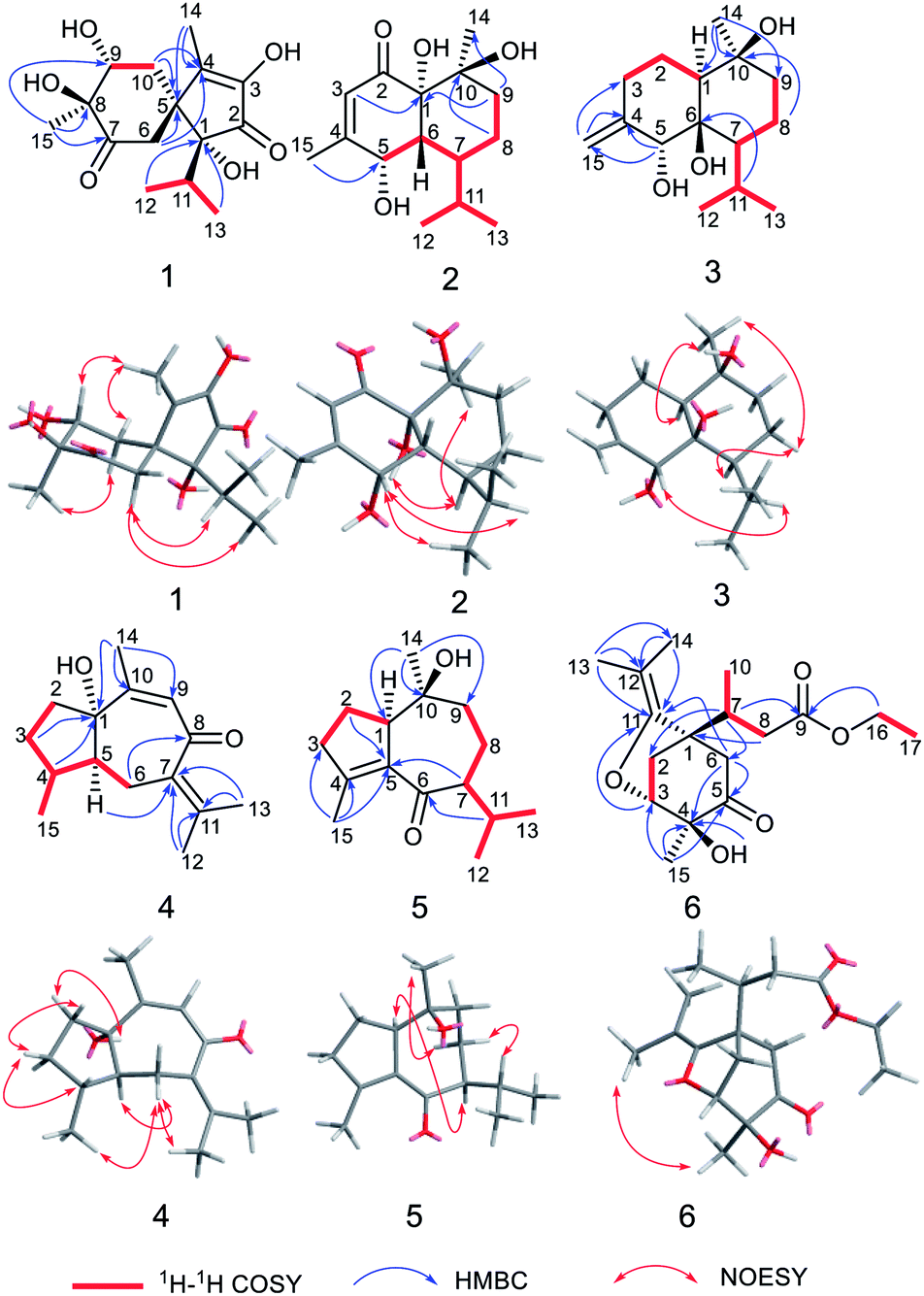 | ||
| Fig. 2 1H–1H COSY, HMBC, and NOESY correlations of compounds 1–6. | ||
The molecular formula of calamusin M (2) was determined to be C15H24O4, with four hydrogen deficiencies, on the basis of HRESIMS (m/z 291.1575 [M + Na]+). The UV spectrum showed the absorption of an α,β-unsaturated carbonyl at 226 nm. The IR spectrum showed absorption bands corresponding to hydroxy (3384 cm−1) and carbonyl (1670 cm−1) groups. The 1H NMR spectrum (Table 1) showed signals corresponding to the protons of two secondary methyl groups [δH 0.96 (d, J = 7.2 Hz), Me-12; 0.76 (d, J = 7.2 Hz), Me-13], an olefinic tertiary methyl group [δH 2.05 (d, J = 1.2 Hz), Me-15], an aliphatic tertiary methyl group [δH 1.42 (s), Me-14], one olefinic methine [δH 5.72 (brs), H-3], and an oxygen-bearing methine [δH 4.29 (m), H-5]. 13C NMR (Table 3) and DEPT-135/90 spectra showed 15 signals corresponding to four methyl groups, two methylene groups, five methines (an oxygenated one at δC 68.0; three aliphatic ones at δC 43.1, 38.0, and 26.2; an olefinic one at δC 125.7), four quaternary carbons [an olefinic quaternary carbon at δC 159.1; two oxygenated quaternary carbons at δC 77.9 and 72.4; an α,β-unsaturated keto carbonyl group at δC 201.4]. The cross peaks of 1H–1H COSY spectrum suggested the presence of C9–C8–C7–C6–C5 and C12–C11(C7)–C13 moieties (Fig. 2). Additionally, coupling constant of Me-15 (1.2 Hz) in the 1H NMR spectrum and the weak cross signal of 1H–1H COSY spectrum between Me-15 and H-3 suggested an allylic system of C15–C4–C3 is present in the structure. The HMBC correlations (Fig. 2) from Me-15 to C-5, H-8 to C-10, H-9 to C-1/14, and H-3 to C-1 suggested C-5 is linked with C-4, C-10 is linked with C-9, C-10/Me-14 are linked with C-10, and C-1 is linked with C-2. The above data, similar to those of tatarinowin C,32 suggested that compound 2 possessed a cadinane-type sesquiterpene carbon skeleton. Analysis of the chemical shifts and degrees of unsaturation indicated that C-1, C-5, and C-10 are all linked with hydroxy groups. The combined analysis of coupling constant and NOESY spectrum allowed the determination of the relative configuration of 2. The rather small coupling constant (3.5 Hz) between H-5 and H-6 indicated that the two protons are cis-oriented (β-oriented). Furthermore, the NOESY correlations (Fig. 2) of H-5 with H-11/Me-13 suggested the isopropyl group is β-oriented, and H-7 is α-oriented. In addition, the NOESY correlations of H-7 with 1-OH/Me-14 suggested 1-OH and Me-14 are both α-oriented. According to the octant rule of n → π* transition of α,β-unsaturated cyclohexanone unit, the absolute configuration of 2 was determined to be 1S,5S,6R,7S,10S on the basis of the positive Cotton effect observed in the CD spectrum at 343 nm (Δε +1.03).33
Calamusin N (3) was assigned the molecular formula C15H26O3, with 3 hydrogen deficiencies, as deduced from HRESIMS (m/z 277.1777 [M + Na]+) and NMR data. The IR spectrum showed bands corresponding to hydroxy groups (3372 and 3288 cm−1). 1H NMR spectrum (Table 1) showed proton signals corresponding to two secondary methyl groups [δH 0.91 (d, J = 6.9 Hz), Me-12; 0.88 (d, J = 6.9 Hz), Me-13], one tertiary methyl group [δH 1.14 (s), Me-14], an oxygen-bearing methine group [δH 4.02 (s), H-5], and two olefinic protons [δH 4.84 (2H, s), H-15]. 13C NMR (Table 3) and DEPT-135/90 spectra showed 15 carbon signals corresponding to three methyl groups, five methylene groups (four aliphatic ones at δC 22.9, 30.5, 19.6, and 44.1; an olefinic one at δC 112.4), three methines (an oxygenated one at δC 76.2), as well as three quaternary carbons (two oxygenated ones at δC 77.7 and 72.1; an olefinic one at δC 150.4). The cross signals of 1H–1H COSY spectrum (Fig. 2) of 3 suggested the presence of C9–C8–C7–C11(C12)–C13 and C1–C2–C3 structural moieties. Additionally, the observed HMBC (Fig. 2) correlations between H-15 and C-4/C-3, H-5 and C-4/C-15, H-11 and C-6, H-8 and C-10, and Me-14 and C-1/9/10 suggested C-4 is linked with C-3/5/15, C-6 is linked with C-7, C-10 is linked with C-9, and C-10 is attached to C-1 and C-14, allowing the establishment of a cadinene-type sesquiterpene carbon skeleton. The NOESY spectrum showed correlations (Fig. 2) of H-5 and H-11, H-7 and H-8a (δ 1.52), H-8a and Me-14, Me-14 and H-1, which indicated 5-OH/Me-14/H-1 are α-oriented, the isopropyl group at position 7 and 10-OH are β-oriented. As the structural analysis, H-5 and H-7 should be closely spatially if 6-OH was α-oriented, but the correlation was not appeared between the above two in the NOESY spectrum, indicating 6-OH is β-oriented.
Determination of the absolute configuration of 3 was implemented by comparison of the experimental and calculated specific optical rotation (SOR) of the two stereoisomers. The SOR of each configuration of 3 was calculated using DFT theory method at the pbe1pbe/aug-cc-pVTZ level. Finally, the calculated SOR of (1S,5R,6R,7S,10S)-3 ([α]D +41.9) had the same trend as the experimental vale ([α]D +23.4, MeOH), while the calculated data of (1R,5S,6S,7R,10R)-3 ([α]D −42.8) had the opposite sign to the experimental value, indicating the absolute configuration of 3 was 1S,5R,6R,7S,10S.
The molecular formula of calamusin O (4) was determined to be C15H22O2 by HRESIMS (m/z 235.1688 [M + H]+) and NMR spectra. The 1H NMR spectrum (Table 2) showed signals corresponding to the protons of three tertiary methyl groups [δH 1.92 (s), Me-12; 1.82 (s), Me-13; 1.95 (d, J = 1.2 Hz), Me-14], one secondary methyl groups [δH 1.08 (d, J = 7.0 Hz), Me-15], and an olefinic proton [δH 5.67 (q, J = 1.2 Hz), H-9]. 13C NMR (Table 3) and DEPT-135/90 spectra displayed 15 signals corresponding to four methyl groups, three methylenes, three methines (two aliphatic ones at δC 36.8 and 56.0; an olefinic one at δC 128.1), five quaternary carbons (three olefinic ones at δC 135.7, 153.8, and 141.2; an oxygenated aliphatic one at δC 86.0; an α,β-unsaturated keto carbonyl group at δC 194.6). The 1H–1H COSY correlations (Fig. 2) between H-5 and H-4/H-6, H-4 and H-3/H-5/Me-15, and H-2 and H-3 allowed the formation of C2–C3–C4(C15)–C5–C6 moiety. A rather weak correlation was displayed between H-9 and Me-14 in the 1H–1H COSY spectrum, and a rather small coupling constant (4J9,14 = 1.2 Hz) was also observed in 1H NMR, indicating H-9 and Me-14 are in an allylic system. In addition, the observed HMBC correlation from Me-14 to C-10/C-9 suggested the aforementioned allylic system is C14–C10–C9. Furthermore, the HMBC correlations (Fig. 2) from H-14 to C-1, H-3 to C-1, H-4 to C-1, H-5 to C-7, Me-12/Me-13 to C-11/C-7, and H-6 to C-8 indicated C-1 is linked with C-10, C-2 is attached to C-1, C-5 is attached to C-1, C-7 is attached to C-6, Me-12/Me-13 are linked with C-11, and C-8 is linked with C-8. The above data determined the planar structure of 4, which is a guaiane-type sesquiterpene. The NOESY spectrum of 4 displayed correlations (Fig. 2) between H-6a (δH 2.62) and H-5/Me-15/Me-12, H-3b (δH 1.25) and H-4/H-2a (δH 1.99), and H-2b (δH 1.92) and 1-OH, which suggest H-5, Me-15, and 1-OH are all on the same side of the cyclic system and are specified as α-oriented. The absolute configuration of 4 was elucidated by the quantum chemistry calculations on ECD spectra. Firstly, the ECD calculations were conducted using TDDFT method at the ωb97xd/def2tzvp level in methanol. The overall calculated ECD spectrum of each configuration was then generated according to the Boltzmann weighting of the conformers with Boltzmann-population of over 1%. The tendency of the experimental ECD spectrum of 4 was similar to that of the calculated (1S,4R,5S)-4 (Fig. 3), confirming the absolute configuration of 4 to be 1S,4R,5S.
| No. | 4 | 5 | 6 |
|---|---|---|---|
| a Measured at 600 MHz. | |||
| 1 | 3.23 (m) | ||
| 2 | 1.99 (m) | 1.94 (m) | 2.29 (d, 12.0) |
| 1.92 (overlap) | 2.08 (overlap) | ||
| 3 | 1.84 (overlap) | 2.41 (m) | 4.14 (d, 6.0) |
| 1.25 (m) | 2.32 (m) | ||
| 4 | 2.74 (m) | ||
| 5 | 1.85 (m) | ||
| 6 | 2.62 (d, 12.6) | 2.94 (d, 15.6) | |
| 1.83 (overlap) | 2.28 (dd, 15.6/3.0) | ||
| 7 | 2.29 (ddd, 11.4/6.0/1.8) | 2.57 (m) | |
| 8 | 1.82 (m) | 2.66 (dd, 15.0/3.6) | |
| 1.17 (m) | 2.10 (dd, 15.0/10.2) | ||
| 9 | 5.67 (q, 1.2) | 1.95 (overlap) | |
| 1.76 (td, 13.2/3.6) | |||
| 10 | 0.96 (d, 7.2) | ||
| 11 | 2.05 (m) | ||
| 12 | 1.92 (s) | 0.88 (d, 6.6) | |
| 13 | 1.82 (s) | 0.89 (d, 5.4) | 1.56 (s) |
| 14 | 1.95 (d, 1.2) | 0.90 (s) | 1.57 (s) |
| 15 | 1.08 (d, 7.0) | 1.99 (m) | 1.27 (s) |
| 16 | 4.12 (q, 7.2) | ||
| 17 | 1.22 (t, 7.2) | ||
| OH | 3.80 (s, 6-OH) | 4.71 (s) | |
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a Overlapped by solvent reagents. | ||||||
| 1 | 85.2 | 77.9 | 48.6 | 86.0 | 57.0 | 49.8 |
| 2 | 200.8 | 201.4 | 22.9 | 38.1 | 24.3 | 33.6 |
| 3 | 149.2 | 125.7 | 30.5 | 29.8a | 39.2 | 81.1 |
| 4 | 146.1 | 159.1 | 150.4 | 36.8 | 154.8 | 74.7 |
| 5 | 51.9 | 68.0 | 76.2 | 56.0 | 138.1 | 207.7 |
| 6 | 39.7 | 43.1 | 77.7 | 24.9 | 203.5 | 49.2 |
| 7 | 213.7 | 38.0 | 46.2 | 135.7 | 58.5 | 34.6 |
| 8 | 80.1 | 18.7 | 19.6 | 194.6 | 25.0 | 38.6 |
| 9 | 74.7 | 37.3 | 44.1 | 128.1 | 47.5 | 173.1 |
| 10 | 43.1 | 72.4 | 72.1 | 153.8 | 74.1 | 15.0 |
| 11 | 35.3 | 26.2 | 25.1 | 141.2 | 28.4 | 151.1 |
| 12 | 21.2 | 21.9 | 24.2 | 23.0 | 21.4 | 101.5 |
| 13 | 17.4 | 15.3 | 18.9 | 22.3 | 19.1 | 18.8 |
| 14 | 11.7 | 25.6 | 23.8 | 22.7 | 20.8 | 16.8 |
| 15 | 20.1 | 21.6 | 112.4 | 16.0 | 16.7 | 20.6 |
| 16 | 60.7 | |||||
| 17 | 14.5 | |||||
Calamusin P (5) was assigned the molecular formula C15H24O2 on the basis of HRESIMS (m/z 237.1849 [M + H]+) and NMR spectra. 1H NMR spectrum (Table 2) displayed proton signals corresponding to two secondary methyl groups [δH 0.88 (d, J = 6.6 Hz), Me-12; 0.89 (d, J = 5.4 Hz), Me-13], two tertiary methyl groups [δH 0.90 (s), Me-14; 1.99 (m), Me-15]. 13C NMR (Table 3) and DEPT 135/90 spectra showed 15 carbon signals corresponding to four methyl groups, four methylenes, three methines, and four quaternary carbons (an oxygenated aliphatic one at δC 74.1; two olefinic ones at δC 154.8 and 138.1; an α,β-unsaturated keto carbonyl group at 203.5). The 1H–1H COSY spectrum showed correlations (Fig. 2) corresponding to C1–C2–C3 and C9–C8–C7–C11(C12)–C13 moieties. In the HMBC spectrum, correlations (Fig. 2) from H-11 to C-6, H-7 to C-5, Me-15 to C-3/C-4/C-5, H-2 to C-5, and Me-14 to C-1/C-10/C-9 suggested C-6 is linked with C-7, C-5 is linked with C-6, C-4 is attached to C-3 and C-15, C-5 is linked with C-1, and C-10 is linked with Me-14/C-1/C-9, indicating it possesses a carbon skeleton of guaiane-type sesquiterpene. In the NOESY spectrum of 5, cross signals (Fig. 2) of H-11 with H-8a (δH 1.82) and H-8b (δH 1.17) with Me-14 indicated the isopropyl group at position 7, 10-OH, and H-8a are located on the same side (specified to be β-oriented) of the ring system, and H-8b and Me-14 (α-oriented) are on the other side of the ring system. Additionally, the NOESY correlation between H-1 and H-7 indicated H-1 is α-oriented. The absolute configuration of 5 was determined with the same method as compound 4. The tendency of the experimental ECD spectrum of 5 was similar to that of the calculated (1R,7S,10S)-5 (Fig. 3), confirming the absolute configuration of 5 to be 1R,7S,10S.
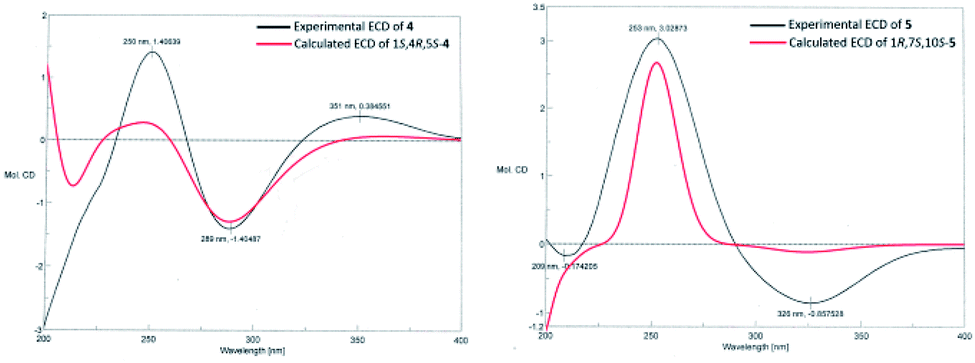 | ||
| Fig. 3 Calculated and experimental ECD spectra of compounds 4 and 5. | ||
Calamusin Q (6) was deduced to have the molecular formula C17H26O5 by combined analysis of HRESIMS (m/z 311.1859 [M + H]+) and NMR spectra. The IR spectrum showed the absorptive bands corresponding to hydroxyl (3439 cm−1) and carbonyl (1724 cm−1) groups. 1H NMR spectrum (Table 2) showed proton signals corresponding to an ethoxyl group [δH 4.12(q, J = 7.2 Hz), H-16; 1.22 (t, J = 7.2 Hz), Me-17], one secondary methyl group [δH 0.96 (d, J = 7.2 Hz), Me-10], and three tertiary methyl groups [δH 1.56 (s), Me-13; 1.57 (s), Me-14; 1.27 (s), Me-15]. A resonance corresponding to an oxygen-bearing methine was also observed [δH 4.14 (m), H-3] in 1H NMR spectrum. In addition, it showed resonance signals assignable to several aliphatic methylenes and methines between δH 2.05 and 3.00. 13C NMR (Table 3) and DEPT-135/90 spectra showed 17 carbon signals corresponding to five methyl groups, four methylenes (an oxygenated one at δC 60.7), two methines (an oxygenated one at δC 81.1), and six quaternary carbons (a ketone carbonyl group at δC 207.7; an ester carbonyl group at δC 173.1; a double band at δC 151.1 and 101.5; an oxygenated one at δC 74.7). Additionally, the olefinic carbon C-11 must be linked with an oxygen atom according to its chemical shift (δc-11 151.1). The cross signals observed in 1H–1H COSY spectrum indicated the presence of structural units of C16–C17, C8–C7–C10, and C2–C3. The HMBC spectrum displayed cross peaks of Me-13 and C-14/12/11, H-16 and C-9, H-8 and C-1, H-7 and C-2/6, Me-15 and C-3/4/5, –OH and C-4, H-6 and C-4/C-5, and H-6 and C-11 indicating Me-13/Me-14 is attached to C-12, the ethoxyl group is linked with C-9, C-1 is linked with C-7/C-2/C-6, C-4 is linked with C-3/5/15, the hydroxyl group is linked with C-4, C-5 is linked with C-6, and C-11 is attached to C-1. Furthermore, the HMBC correlation from H-3 to C-11 and the chemical shifts of C-3 (δC 81.1) and C-11(δC 151.1) indicated that an oxygen bridge connected C-3 and C-11.
The NOESY correlation between the protons of Me-13 and Me-15 suggested Me-15 and the oxygen bridge are on the same side of the cyclohexanone unit, then the hydroxyl group and C-7 are on the other side. A positive Cotton effect occurred at 302 nm (Δε +0.65) in CD spectrum indicated the absolute configurations of the chiral centers at the cyclohexanone unit are 1S,3R,4S, according to the n → π* transition of saturated cyclopentanone.31
According to the empirical isoprene rule, compound 6 can't be considered as a sesquiterpene. However, as our conjecture, the biogenic precursors of 6 is a previously reported acorane type sesquiterpenoid, calamusin D,16 in which a series of oxidation, dehydration, oxidative cleavage, and ethylation reactions occurred to yield compound 6 (Fig. 4). As this conjecture, the absolute configuration of C-7 in compound 6 should be S, the same as C-4 in compound calamusin D. This was supported by NMR chemical shifts calculations of both the possible isomers 6a (1S,3R,4S,7S) and 6b (1S,3R,4S,7R) using the GIAO method at the mPW1PW91/6-311G(d,p) level in acetone. The calculated 1H and 13C NMR chemical shifts of the two possible isomers were compared with the experimental values of 6 by utilizing DP4+ probability analysis, which showed that 6a was predicted to be correct with probabilities of 100% for the 1H NMR data, 93.4% for the 13C NMR data, and 100% for their combination. The absolute configuration of 6 was thus proposed to be 1S,3R,4S,7S.
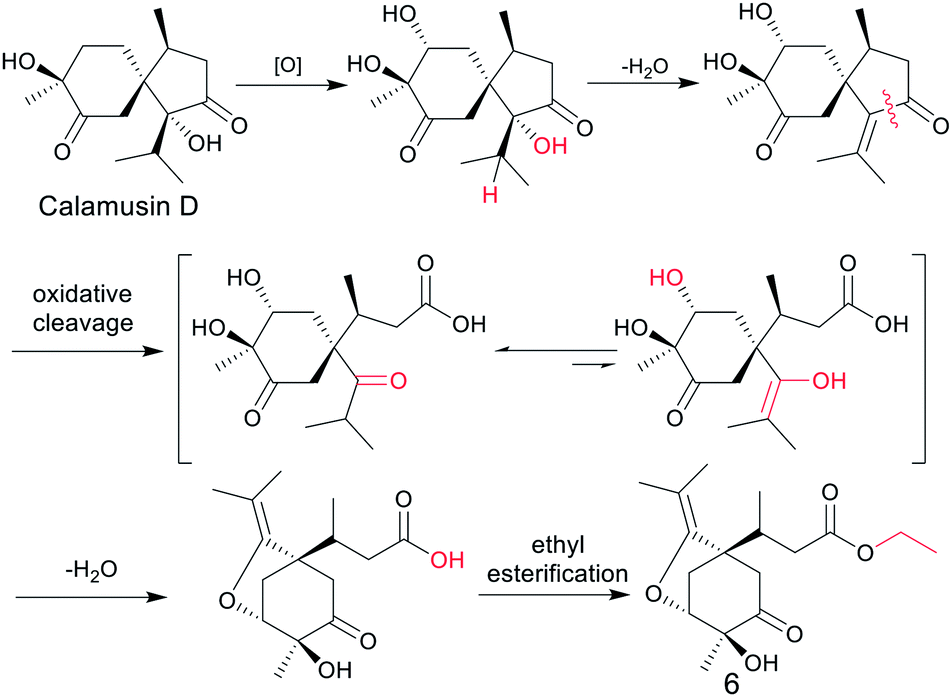 | ||
| Fig. 4 Plausible biosynthetic pathway of compound 6. | ||
The known compounds were identified by comparison of their spectroscopic data with reported data. They were hedytriol (7),34, (−)-1β,4β,7α-trihydroxyeudesmane (8),35 oplodiol (9),36 6-eudesmene-1β,4β-diol (10),37 4β,5α,10β-trihydroxycadinan (11),38 tatarinowin A (12),25 bullatantriol (13),39 homalomenol A (14),40 4′-dihydrophaseic acid (15),41 blumenol C (16),42, (6R,9S)-9-hydroxy-4-megastigmen-3-one (17),42, (+)-dehydrovomifoliol (18),43 1,10-seco-4ξ-hydroxy-muurol-5-ene-1,10-diketone (19),44 and 2-hydroxyacorenone (20).45
Compounds 1–6 were evaluated for their neuroprotective effect on PC12 cells induced by serum withdrawal, rotenone, and OGD (Oxygen and Glucose Deprivation) in vitro with MTT method. As shown in Table 4, compounds 1 and 6 increased the cell survival rate of the OGD-treated group moderately, while compounds 3 and 5 decreased the cell survival rate of the serum withdrawal-treated PC12 cells moderately, and none of the compounds showed activity in the rotenone-treated models at 10 μM. Few investigations have been taken previously about neuroprotective activity and the corresponding mechanisms of sesquiterpenes from A. calamus. However, a study by Li et al.19 indicated the sesquiterpenes, calamusin D (with a same carbon skeleton of compound 1) and acoric acid (with a same carbon skeleton of compound 6), showed significant cell proliferation activity on the human neuroblastoma cell line SK-N-BE (2), suggesting the both possessed neuroprotective activity. Therefore, more sesquiterpenes of diverse types, such as acorane- and 1,2-secoacorane-types, should be evaluated for their neuroprotective activity, and what's more, the action mechanisms and structure–activity relationships should be investigated in the future to discover the drug leads to treat neurodegenerative diseases.
| Compounds | Serum withdrawal | Rotenone | OGD |
|---|---|---|---|
| a ##p < 0.01 vs. control; *p < 0.05, **p < 0.01 vs. model. | |||
| Control | 100.0 ± 7.2 | 100.0 ± 5.9 | 100.0 ± 6.2 |
| Model | 51.0 ± 5.6### | 71.0 ± 6.1### | 58.2 ± 7.7### |
| 1 | 47.2 ± 4.5 | 71.3 ± 4.6 | 74.3 ± 8.2** |
| 2 | 46.1 ± 4.1 | 73.7 ± 6.1 | 60.2 ± 8.5 |
| 3 | 41.6 ± 5.8** | 68.2 ± 4.8 | 63.3 ± 9.0 |
| 4 | 47.7 ± 3.7 | 69.9 ± 5.2 | 63.4 ± 8.6 |
| 5 | 43.2 ± 5.6** | 68.6 ± 4.7 | 60.7 ± 8.4 |
| 6 | 46.5 ± 6.7 | 72.4 ± 6.8 | 68.9 ± 7.6* |
Experimental
General experimental procedures
Optical rotations were measured with a JASCO P-2000 polarimeter. UV spectra were recorded on a JASCO V-650 spectrophotometer. IR spectra were recorded on a Nicolet 5700 spectrometer using FT-IR microscope transmission method. NMR spectra were recorded with Varian INOVA-600 and Bruker AV-600 spectrometers. HRESIMS were obtained on an Agilent 1100 series LC/MSD ion trap mass spectrometer. CD spectra were recorded on a JASCO J-815 spectropolarimeter. Analyticl HPLC was carried out on an Agilent 1200 INFINITY system with an Agilent ZORBAX SB-C18 column (5 μm, 4.6 × 150 mm). Semi-preparative HPLC was carried out on a Shimadzu LC-6AD pump with a Shimadzu SPD-M20A detector, an Agilent ZORBAX SB-C18 column (9.4 × 250 mm, 5 μm), and an YMC-PACK SIL-06 column (20 mm × 250 mm, 5 μm). Colum chromatography was performed on silica gel (200–300 mesh, Qingdao Marine Chemical Factory, China), Sephadex LH-20 (GE) and ODS (50 μm, YMC, Japan), respectively. TLC was carried out with GF254 plates (Qingdao Marine Chemical Factory, China).Plant material
The rhizomes of A. calamus were purchased from a Chinese herb store in Anguo county, Hebei Province, China, and authenticated by Professor Lin Ma, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College. A voucher specimen (no. ID-S-2281) has been deposited at the Herbarium of the Institute of Material Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, China.Extraction and isolation
Dried rhizomes of A. calamus (19.8 kg) were smashed and extracted with 95% ethanol (3 × 2 h). The concentrated extract (3.1 kg) was suspended in water (3 L) and partitioned with petroleum ether, ethyl acetate and n-butanol (3 L × 3), successively. The EtOAc portion (310 g) was subjected to silica gel column (10.5 × 100 cm) eluted with CH2Cl2/MeOH (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 20:1, 15:1, 10:1, 8:1, 6:1, 5:1, 3:1, 1:1, 0:1; v/v) to give ten fractions (F1–F10). F6 (21 g) was chromatographed with silica gel (200–300 mesh, 10 × 60 cm) eluting with a gradient of CH2Cl2:MeOH (10:1, 7:1, 5:1, 3:1, 0:1) to give five fractions, named F6a–F6e. F6a (2.3 g) was chromatographed on a reversed phase C18 silica gel column (4.5 × 42 cm) with a gradient MeOH–H2O (30:70, 50:50, 70:30, 100:0) to give six fractions, named F6a1–F6a6. F6a2 was purified by normal-phase silica gel semi-preparative HPLC (87% n-hexane/2-propanol, 4 mL min−1) to yield 1 (5.5 mg, tR = 50.5 min). F6a3 was purified by C18 ODS semi-preparative HPLC (25% MeCN/H2O, 2.5 mL min−1) to yield 2 (12.4 mg, tR = 45.6 min) and 3 (5.5 mg, tR = 39.2 min). F6a5 was purified by C18 ODS semi-preparative HPLC (37.5% MeCN/H2O, 2.5 mL min−1) to yield 12 (2 mg, tR = 42.4 min), 4 (5.8 mg, tR = 44.1 min), 20 (21.5 mg, tR = 49.2 min), and 6 (8.2 mg, tR = 59.1 min). F6a6 was purified by C18 ODS semi-preparative HPLC (54% MeCN:MeOH(1:1)/H2O, 2.5 mL min−1) to yield compound 5 (4.7 mg, tR = 28.5 min). F6b (5.7 g) was chromatographed on a reversed phase C18 silica gel column (4.5 × 42 cm) with a gradient MeOH–H2O (30:70, 50:50, 70:30, 100:0) to give eight fractions, named F6b1–F6b8. F6b2 was chromatographed on Sephadex LH-20 column (MeOH) to give 7 (20.8 mg) and 8 (5.5 mg). F6b3 was purified by C18 ODS preparative HPLC (35% MeCN:MeOH/H2O, 5 mL min−1) to yield compound 9 (60.2 mg, tR = 112 min). F6b5 was purified by C18 ODS semi-preparative HPLC (26% MeCN:MeOH/H2O, 2.5 mL min−1) to yield compound 10 (16.8 mg, tR = 118 min) and 14 (5.1 mg, tR = 125 min). F6b4 was chromatographed over silica gel (PE:acetone, 3:1) to give 11 (10.3 mg). F6c was chromatographed on a reversed phase C18 silica gel column (4.5 × 42 cm) with a gradient MeOH–H2O (30:70, 50:50, 70:30, 100:0) to give 7 fractions, named F6c1–F6c7. F6c1 was chromatographed over Sephadex LH-20 (MeOH), followed by recrystallization in acetone, to give 13 (15.2 mg). F6c2 was purified by C18 ODS semi-preparative HPLC (8% MeCN:MeOH/H2O, 2.5 mL min−1) to yield compound 15 (6.0 mg, tR = 59.6 min). F6c3 was purified by C18 ODS semi-preparative HPLC (35% MeOH/H2O, 2.5 mL min−1) to yield compound 16 (3.6 mg, tR = 110 min) and 17 (4.6 mg, tR = 115 min). F6c4 was purified by C18 ODS semi-preparative HPLC (12% MeCN/H2O, 2.5 mL min−1) to yield compound 18 (2.5 mg, tR = 61.7 min). F6c6 was purified by C18 ODS semi-preparative HPLC (41% MeCN/H2O, 2.5 mL min−1) to yield compound 19 (4.3 mg, tR = 10.0 min).
ε) 269 nm (4.69); CD (MeOH) λmax (Δε): 315 nm (−1.29), 273 nm (−1.29), 224 nm (+0.78); IR νmax 3390, 1708, 1411, 1372, 1077 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 1 and 3; HRESIMS m/z: 299.1490 [M + H]+ (calcd for C15H23O6, 299.1495), 321.1313 [M + Na]+ (calcd for C15H22O6Na, 321.1314).ε) 226 nm (3.94); CD (MeOH) λmax (Δε) 343 nm (1.03), 242 nm (−5.11), 219 nm (−0.31); IR νmax 3384, 2957, 1670, 1437, 1375, 1259, 1041 cm−1; For 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 1 and 3; HRESIMS: m/z 291.1575 [M + Na]+ (calcd for C15H24O4Na, 291.1572).ε) 251 nm (3.72), 202 nm (3.81); CD (MeOH) λmax (Δε): 351 nm (0.38), 289 nm (−1.40), 250 nm (+1.41); IR νmax 3413, 2957, 1710, 1642, 1449, 1375, 1299 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 2 and 3; HRESIMS: m/z 235.1688 [M + H]+ (calcd 235.1698).ε) 253 nm (3.80); CD (MeOH) λmax (Δε): 326 nm (−0.86), 253 nm (+3.03); IR νmax 3422, 2958, 1670, 1609, 1462, 1374, 1194, 1112 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 2 and 3; HRESIMS: m/z 237.1849 [M + H]+ (calcd for C15H25O2, 237.1855).ε) 202 nm (4.01); CD (MeOH) λmax (Δε): 348 nm (−0.04), 302 nm (+0.65), 249 nm (−0.18), 222 nm (+0.74); IR νmax 3439, 2980, 1724, 1457, 1374, 1303, 1177, 1100 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 2 and 3; HRESIMS: m/z 311.1859 [M + H]+ (calcd for C17H27O5, 311.1858), 333.1682 [M + Na]+ (calcd for C17H26O5Na, 333.1678).
:1, 20:1, 15:1, 10:1, 8:1, 6:1, 5:1, 3:1, 1:1, 0:1; v/v) to give ten fractions (F1–F10). F6 (21 g) was chromatographed with silica gel (200–300 mesh, 10 × 60 cm) eluting with a gradient of CH2Cl2:MeOH (10:1, 7:1, 5:1, 3:1, 0:1) to give five fractions, named F6a–F6e. F6a (2.3 g) was chromatographed on a reversed phase C18 silica gel column (4.5 × 42 cm) with a gradient MeOH–H2O (30:70, 50:50, 70:30, 100:0) to give six fractions, named F6a1–F6a6. F6a2 was purified by normal-phase silica gel semi-preparative HPLC (87% n-hexane/2-propanol, 4 mL min−1) to yield 1 (5.5 mg, tR = 50.5 min). F6a3 was purified by C18 ODS semi-preparative HPLC (25% MeCN/H2O, 2.5 mL min−1) to yield 2 (12.4 mg, tR = 45.6 min) and 3 (5.5 mg, tR = 39.2 min). F6a5 was purified by C18 ODS semi-preparative HPLC (37.5% MeCN/H2O, 2.5 mL min−1) to yield 12 (2 mg, tR = 42.4 min), 4 (5.8 mg, tR = 44.1 min), 20 (21.5 mg, tR = 49.2 min), and 6 (8.2 mg, tR = 59.1 min). F6a6 was purified by C18 ODS semi-preparative HPLC (54% MeCN:MeOH(1:1)/H2O, 2.5 mL min−1) to yield compound 5 (4.7 mg, tR = 28.5 min). F6b (5.7 g) was chromatographed on a reversed phase C18 silica gel column (4.5 × 42 cm) with a gradient MeOH–H2O (30:70, 50:50, 70:30, 100:0) to give eight fractions, named F6b1–F6b8. F6b2 was chromatographed on Sephadex LH-20 column (MeOH) to give 7 (20.8 mg) and 8 (5.5 mg). F6b3 was purified by C18 ODS preparative HPLC (35% MeCN:MeOH/H2O, 5 mL min−1) to yield compound 9 (60.2 mg, tR = 112 min). F6b5 was purified by C18 ODS semi-preparative HPLC (26% MeCN:MeOH/H2O, 2.5 mL min−1) to yield compound 10 (16.8 mg, tR = 118 min) and 14 (5.1 mg, tR = 125 min). F6b4 was chromatographed over silica gel (PE:acetone, 3:1) to give 11 (10.3 mg). F6c was chromatographed on a reversed phase C18 silica gel column (4.5 × 42 cm) with a gradient MeOH–H2O (30:70, 50:50, 70:30, 100:0) to give 7 fractions, named F6c1–F6c7. F6c1 was chromatographed over Sephadex LH-20 (MeOH), followed by recrystallization in acetone, to give 13 (15.2 mg). F6c2 was purified by C18 ODS semi-preparative HPLC (8% MeCN:MeOH/H2O, 2.5 mL min−1) to yield compound 15 (6.0 mg, tR = 59.6 min). F6c3 was purified by C18 ODS semi-preparative HPLC (35% MeOH/H2O, 2.5 mL min−1) to yield compound 16 (3.6 mg, tR = 110 min) and 17 (4.6 mg, tR = 115 min). F6c4 was purified by C18 ODS semi-preparative HPLC (12% MeCN/H2O, 2.5 mL min−1) to yield compound 18 (2.5 mg, tR = 61.7 min). F6c6 was purified by C18 ODS semi-preparative HPLC (41% MeCN/H2O, 2.5 mL min−1) to yield compound 19 (4.3 mg, tR = 10.0 min).
ε) 269 nm (4.69); CD (MeOH) λmax (Δε): 315 nm (−1.29), 273 nm (−1.29), 224 nm (+0.78); IR νmax 3390, 1708, 1411, 1372, 1077 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 1 and 3; HRESIMS m/z: 299.1490 [M + H]+ (calcd for C15H23O6, 299.1495), 321.1313 [M + Na]+ (calcd for C15H22O6Na, 321.1314).ε) 226 nm (3.94); CD (MeOH) λmax (Δε) 343 nm (1.03), 242 nm (−5.11), 219 nm (−0.31); IR νmax 3384, 2957, 1670, 1437, 1375, 1259, 1041 cm−1; For 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 1 and 3; HRESIMS: m/z 291.1575 [M + Na]+ (calcd for C15H24O4Na, 291.1572).ε) 251 nm (3.72), 202 nm (3.81); CD (MeOH) λmax (Δε): 351 nm (0.38), 289 nm (−1.40), 250 nm (+1.41); IR νmax 3413, 2957, 1710, 1642, 1449, 1375, 1299 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 2 and 3; HRESIMS: m/z 235.1688 [M + H]+ (calcd 235.1698).ε) 253 nm (3.80); CD (MeOH) λmax (Δε): 326 nm (−0.86), 253 nm (+3.03); IR νmax 3422, 2958, 1670, 1609, 1462, 1374, 1194, 1112 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 2 and 3; HRESIMS: m/z 237.1849 [M + H]+ (calcd for C15H25O2, 237.1855).ε) 202 nm (4.01); CD (MeOH) λmax (Δε): 348 nm (−0.04), 302 nm (+0.65), 249 nm (−0.18), 222 nm (+0.74); IR νmax 3439, 2980, 1724, 1457, 1374, 1303, 1177, 1100 cm−1; for 1H (600 MHz, acetone-d6) and 13C NMR (150 MHz, acetone-d6) data, see Tables 2 and 3; HRESIMS: m/z 311.1859 [M + H]+ (calcd for C17H27O5, 311.1858), 333.1682 [M + Na]+ (calcd for C17H26O5Na, 333.1678).Specific optical rotation, ECD, and NMR calculations
Conformational analyses were carried out via random searching in the GMMX software using the MMFF94 force field. Subsequently, the conformers were re-optimized using DFT at the B97D/TZVP or ωb97xd/def2tzvp levels in MeOH by the GAUSSIAN 16 program.46 The SORs of conformers of 3 were calculate at the pbe1pbe/aug-cc-pVTZ level. The ECD spectra of conformers of compounds 4 and 5 were calculated using the TDDFT methodology at the ωb97xd/def2tzvp level. The calculated SORs and ECD spectra of the conformers were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy (ΔG).Geometrically optimized conformers for the possible diastereomers of 6a/6b proceeded to GIAO magnetic shielding constants at the mPW1PW91/6-311G(d,p) level. The NMR chemical shifts of the isomers were obtained by Boltzmann averaging of the 1H and 13C NMR chemical shifts of the stable conformers.47 The calculated NMR properties of the optimized structures were averaged as described above, and DP4+ probability analysis was facilitated using an Excel sheet (DP4+) provided by Grimblat et al.48
Neuroprotective assays
The in vitro neuroprotective activity was tested by the method described previously.49,50Conclusions
Six new sesquiterpenes, named calamusins L–Q (1–6), were isolated from the rhizomes of A. calamus, together with fourteen known ones. The new compounds and their absolute configurations were determined based on extensive spectroscopic analyses and computational methods. It was hypothesized that compound 6 was a secosesquiterpene, which was formed from calamusin D during the biosynthesis after a series of oxidation, dehydration, oxidative cleavage, and ethylation reactions. Furthermore, compounds 1–6 were tested for their neuroprotective effects, and it was observed that compounds 1 and 6 increased the cell survival rate of the OGD-treated PC12 cells moderately, while compounds 3 and 5 decreased the cell survival rate of the serum withdrawal-treated PC12 cells moderately.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was financially supported by Chinese Academy of Medical Sciences Initiative for Innovative Medicine [CAMS-12M-1-010], the State Key Laboratory of Bioactive Substance and Function of Natural Medicines (GTZA201803), and the Drug Innovation Major Project (2018ZX09711001-008-009).Notes and references
- Chinese Material Medica Editorial Board of Chinese Material Medica, Chinese Material Medica, Shanghai Scientific & Technical Publishers, Shanghai, 1999 Search PubMed.
- S. R. Haghighi, M. H. Asadi, H. Akrami and A. Baghizadeh, Avicenna J. Phytomed., 2017, 7, 145–156 CAS.
- K. A. Lopatina, E. A. Safonova, K. V. Nevskaya, M. N. Stakheeva, A. M. Gur'ev, E. P. Zueva, T. G. Razina, E. N. Amosova, S. G. Krylov and M. V. Belousov, Bull. Exp. Biol. Med., 2017, 164, 102–105 CrossRef CAS PubMed.
- B. K. Das, A. H. M. V. Swamy, B. C. Koti and P. C. Gadad, Heliyon, 2019, 5, e01585 CrossRef PubMed.
- S. Reddy, G. Rao, B. Shetty and G. Hn, Turk. Neurosurg., 2015, 25, 425–431 Search PubMed.
- X. Yao, Y. Ling, S. Guo, W. Wu, S. He, Q. Zhang, M. Zou, K. S. Nandakumar, X. Chen and S. Liu, Phytomedicine, 2018, 42, 258–267 CrossRef CAS PubMed.
- A. Sagar and S. Rana, J. Pure and Appl. Microbio., 2012, 6, 1455–1460 Search PubMed.
- R. K. Joshi, World J. Microbiol. Biotechnol., 2016, 32, 164 CrossRef PubMed.
- V. Chinappan, M. A. Prabha and G. Sivakumar, Faseb J, 2011, 25, 677.12 Search PubMed.
- A. Muthuraman and N. Singh, J. Ethnopharmacol., 2012, 142, 723–731 CrossRef CAS PubMed.
- S. Subamalani, A. Sasikumar, S. Manikandan and C. Ramaswamy, Int. J. Pharm. Sci. Res., 2018, 9, 4832–4841 CAS.
- Y. X. Liu, M. M. Si, W. Lu, L. X. Zhang, C. X. Zhou, S. L. Deng and H. S. Wu, J. Ethnopharmacol., 2015, 166, 168–175 CrossRef CAS PubMed.
- S. Ahmed, S. Gul, M. Zia-Ul-Haq and M. S. Stankovic, Bol. Latinoam. Caribe Plant. Med. Aromat., 2014, 13, 38–46 Search PubMed.
- V. Sharma, R. Sharma, D. S. Gautam, K. Kuca, E. Nepovimova and N. Martins, J. Clin. Med., 2020, 9, 1176 CrossRef CAS PubMed.
- R. Tundis, M. Bonesi, F. Menichini and M. R. Loizzo, Mini-Rev. Med. Chem., 2016, 16, 605–618 CrossRef CAS PubMed.
- Z.-Y. Hao, D. Liang, H. Luo, Y.-F. Liu, G. Ni, Q.-J. Zhang, L. Li, Y.-K. Si, H. Sun, R.-Y. Chen and D.-Q. Yu, J. Nat. Prod., 2012, 75, 1083–1089 CrossRef CAS PubMed.
- C. X. Zhou, D. Qiao, Y. Y. Yan, H. S. Wu, J. X. Mo and L. S. Gan, Chin. Chem. Lett., 2012, 23, 1165–1168 CrossRef CAS.
- J. Li, J. P. Zhao, S. X. Li, B. Li, Y. W. Ou and Q. R. Liu, Chem. Nat. Compd., 2015, 51, 1099–1102 CrossRef CAS.
- J. Li, J. Zhao, W. Wang, L. Li, L. Zhang, X. F. Zhao, Q. R. Liu, F. Liu, M. Yang, I. A. Khan and S. X. Li, Molecules, 2017, 22, 529 CrossRef PubMed.
- J. Li, Z. X. Li, J. P. Zhao, W. Wang, X. F. Zhao, B. Xu, L. Li, L. Zhang, J. Ren, I. A. Khan and S. X. Li, Chem. Biodiversity, 2017, 14, e1700201 CrossRef PubMed.
- R. Kumar, S. Sharma, S. Sharma and N. Kumar, J. Appl. Res. Med. Aroma., 2016, 3, 136–141 Search PubMed.
- R. Shukla, P. Singh, B. Prakash and N. K. Dubey, J. Essent. Oil Bear. Pl., 2016, 19, 542–552 CrossRef CAS.
- S. Suzgec-Selcuk, G. Ozek, A. H. Mericli, K. H. C. Baser, Y. Haliloglu and T. Ozek, J. Essent. Oil Bear. Pl., 2017, 20, 646–661 CrossRef CAS.
- X. He, Q. Cai, J. Li and W. Guo, Neurosci. Lett., 2018, 666, 78–84 CrossRef CAS PubMed.
- X.-G. Tong, G.-S. Wu, C.-G. Huang, Q. Lu, Y.-H. Wang, C.-L. Long, H.-R. Luo, H.-J. Zhu and Y.-X. Cheng, J. Nat. Prod., 2010, 73, 1160–1163 CrossRef CAS PubMed.
- Y. Yang, L. Xuan, H. Chen, S. Dai, L. Ji, Y. Bao and C. Li, Evid. Based Complement. Alternat. Med., 2017, 2017, 8516518 Search PubMed.
- K. Y. C. Lam, P. Yao, H. Y. Wang, R. Duan, T. T. X. Dong and K. W. K. Tsim, Plos One, 2017, 12, e0179077 CrossRef PubMed.
- R. W. Wiseman, E. C. Miller, J. A. Miller and A. Liem, Cancer Res., 1987, 47, 2275–2283 CAS.
- T. Uebel, L. Hermes, S. Haupenthal, L. Muller and M. Esselen, J. Appl. Toxicol., 2020, 1–14 Search PubMed.
- G. Ni, G. R. Shi, D. Zhang, N. J. Fu, H. Z. Yang, X. G. Chen and D. Q. Yu, Planta Med., 2016, 82, 632–638 CrossRef CAS PubMed.
- H. B. Kagan, Determination of Configurations by Dipole Moments, Determination of Configurations by Dipole Moments, CD or ORD, Thieme Publishers, Stuttgart, 1977 Search PubMed.
- X. L. Feng, Y. Yu, H. Gao, Z. Q. Mu, X. R. Cheng, W. X. Zhou and X. S. Yao, RSC Adv, 2014, 4, 42071–42077 RSC.
- G. Snatzke, Tetrahedron, 1965, 21, 421–438 CrossRef CAS.
- W. M. Zhu, Q. Zhao, S. L. Li and X. J. Hao, J. Asian Nat. Prod. Res., 2007, 9, 277–283 CrossRef CAS PubMed.
- J. E. de Menezes, F. E. Machado, T. L. Lemos, E. R. Silveira, R. Braz Filho and O. D. Pessoa, Z. Naturforsch., C: J. Biosci., 2004, 59, 19–22 CrossRef CAS PubMed.
- M. Ono, M. Yamashita, K. Mori, C. Masuoka, M. Eto, J. Kinjo, T. Ikeda, H. Yoshimitsu and T. Nohara, Food Sci. Tech. Res., 2008, 14, 499–508 CrossRef CAS.
- W. C. Su, J. M. Fang and Y. S. Cheng, Phytochemistry, 1995, 39, 603–607 CrossRef CAS.
- M. Andersson, O. Bergendorff, R. Shan, P. Zygmunt and O. Sterner, Planta Med., 1997, 63, 251–254 CrossRef CAS PubMed.
- T. V. Sung, B. Steffan, W. Steglich, G. Klebe and G. Adam, Phytochemistry, 1992, 31, 3515–3520 CrossRef CAS PubMed.
- K.-Y. Jung, D.-S. Kim, S.-R. Oh, I.-S. Lee, J.-J. Lee, H.-K. Lee, D.-H. Shin, E.-H. Kim and C.-J. Cheong, Arch. Pharm Res., 1997, 20, 363–367 CrossRef CAS PubMed.
- Z. Zhang, W. Zhang, Y. P. Ji, Y. Zhao, C. G. Wang and J. F. Hu, Phytochemistry, 2010, 71, 693–700 CrossRef CAS PubMed.
- B. D'Abrosca, M. DellaGreca, A. Fiorentino, P. Monaco, P. Oriano and F. Temussi, Phytochemistry, 2004, 65, 497–505 CrossRef PubMed.
- H. Kai, M. Baba and T. Okuyama, Chem. Pharm. Bull., 2007, 55, 133–136 CrossRef CAS PubMed.
- K. S. Ngo, W. T. Wong and G. D. Brown, J. Nat. Prod., 1999, 62, 549–553 CrossRef CAS PubMed.
- K. Nawamaki and M. Kuroyanagi, Phytochemistry, 1996, 43, 1175–1182 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03; Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. C. Carter, C. L. Petersen, K. L. Wendt, S. K. Helff, A. L. Risinger, S. L. Mooberry and R. H. Cichewicz, J. Nat. Prod., 2019, 82, 886–894 CrossRef CAS PubMed.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS PubMed.
- H. J. Ji, D. M. Wang, J. F. Hu, M. N. Sun, G. Li, Z. P. Li, D. H. Wu, G. Liu and N. H. Chen, Eur. J. Pharmacol., 2014, 723, 259–266 CrossRef CAS PubMed.
- S. Y. Shao, Z. M. Feng, Y. N. Yang, J. S. Jiang and P. C. Zhang, Org. Biomol. Chem., 2017, 15, 7034–7039 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: UV, IR, ECD, MS, 1D and 2D NMR spectra for compounds 1–6. See DOI: 10.1039/d1ra00350j |
| This journal is © The Royal Society of Chemistry 2021 |