
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2,3-Dimethoxy-2,3-dimethyl-1,4-dioxane as a useful precursor to 2,3-dimethylene-1,4-dioxane for [4+2] cycloaddition reaction†
Makoto Shimizu *ab,
Toshihiro Yamamotob,
Hiroaki Shindob,
Isao Mizotab and
Yusong Zhua
*ab,
Toshihiro Yamamotob,
Hiroaki Shindob,
Isao Mizotab and
Yusong Zhua
aSchool of Energy Science and Engineering, Nanjing Tech University, Nanjing 211816, Jiangsu Province, China
bDepartment of Chemistry for Materials, Graduate School of Engineering, Mie University, Tsu, Mie 514-8507, Japan. E-mail: mshimizu@chem.mie-u.ac.jp
First published on 18th February 2021
Abstract
2,3-Dimethoxy-2,3-dimethyl-1,4-dioxane readily prepared from biacetyl serves as a stable precursor to 2,3-dimethylene-1,4-dioxane which undergoes a [4+2] cycloaddition reaction with dienophiles to give functionalized cyclohexene derivatives. The cycloaddition adducts obtained by the present procedure are transformed into potentially useful intermediates for biologically important materials.
1 Introduction
In conjunction with the synthetic study of biologically important 1,2-dihydroxycyclohexanes and their 1,2-hydroxylamine analogues such as alpha-1 adrenoceptor antagonist,1 sulfonamide CA inhibitor,2 and fliposome (DDS)3 (Scheme 1), we needed a simple and reliable method for the synthesis of these particular structures. Regarding the construction of six-membered carbocycles, the power of the Diels–Alder reaction means it has been recognized as one of the most reliable and straightforward approaches to these molecules, and several functionalized 1,3-dienes have been devised.4 Among the dienes explored, 2,3-dialkoxy-1,3-butadienes5 have received considerable attention since the cycloaddition products with dienophiles are latent 2-hydroxycyclohexanones which can be transformed into a variety of cyclohexanol derivatives. We have been interested in the synthesis and use of 2,3-dimethylene-1,4-dioxane for constructing thiophene derivatives,6 which include 3,4-ethylenedioxythiophene (EDOT) commonly used as a starting material for poly(3,4-ethylenedioxythiophene) (PEDOT), one of the most useful electron-conducting polymers.7 During these investigations, Diels–Alder cycloaddition using this particular diene intrigued us. A literature search into such diene has revealed that some derivatives are known, but many of them are unstable at room temperature.8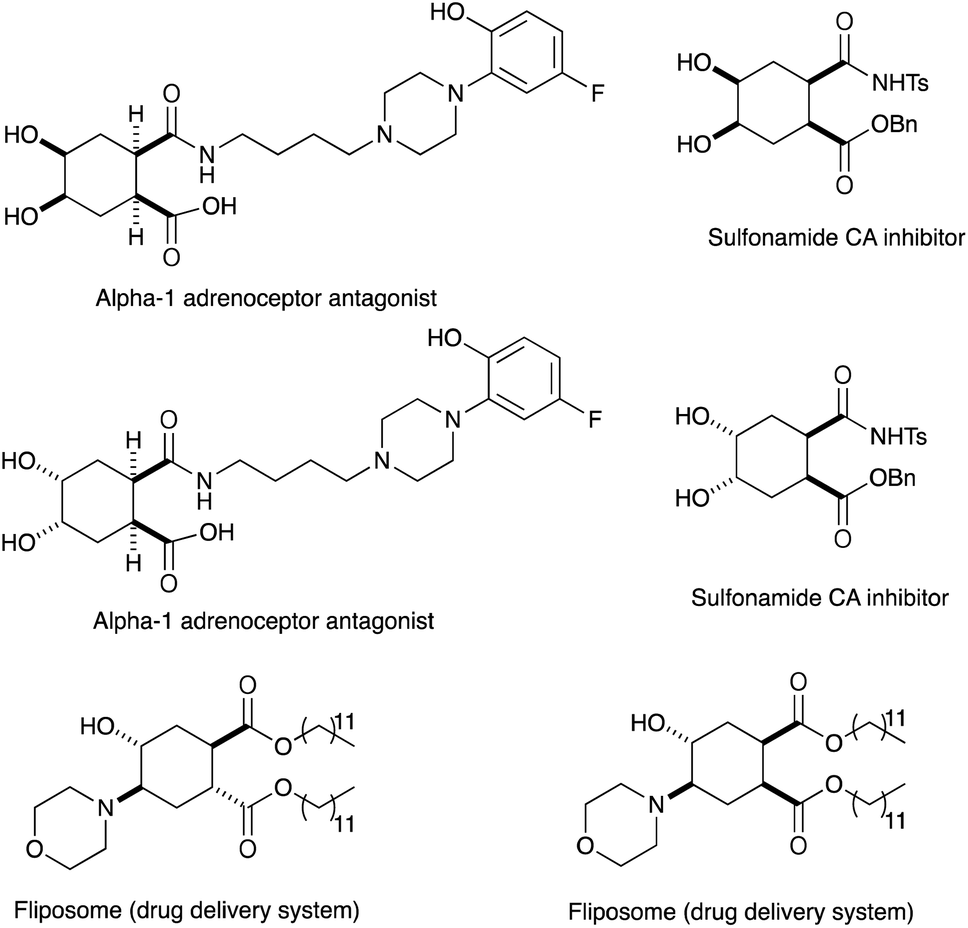 | ||
| Scheme 1 Biologically important materials containing a cyclohexanol moiety. | ||
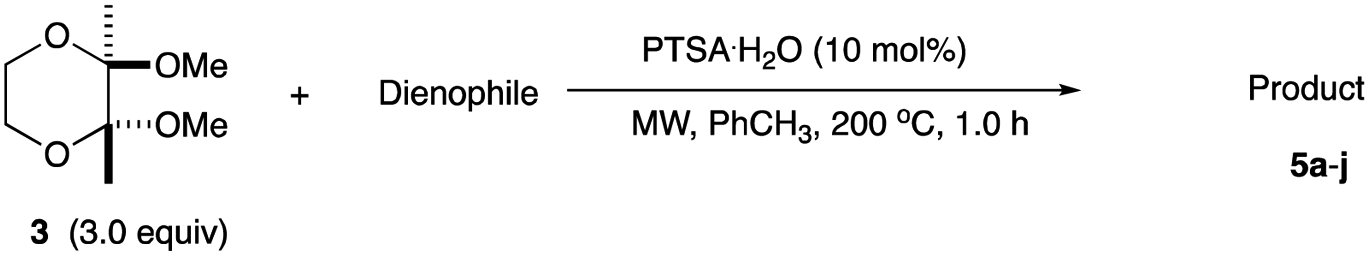
In an effort to find a stable precursor to this diene, we screened several compounds, and among the derivatives examined 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 shows a reasonable stability and can work as a good precursor to 2,3-dimethylene-1,4-dioxane 2. This paper describes [4+2] cycloaddition reactions using 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 with dienophiles (Scheme 2, eqn (1)) and subsequent transformations of the cycloaddition products.
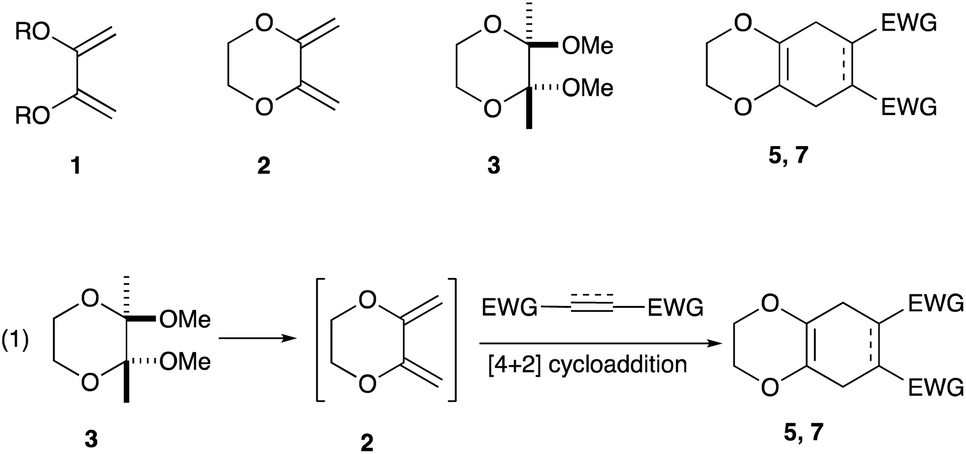 | ||
| Scheme 2 Particular dienes, a precursor, and adducts. | ||
2 Results and discussion
An initial examination was carried out to find good precursors to the diene 2. The investigation involves the use of readily available starting materials to synthesize a relatively large amount of the desired product. Among the derivatives examined 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 show promising results (Scheme 3).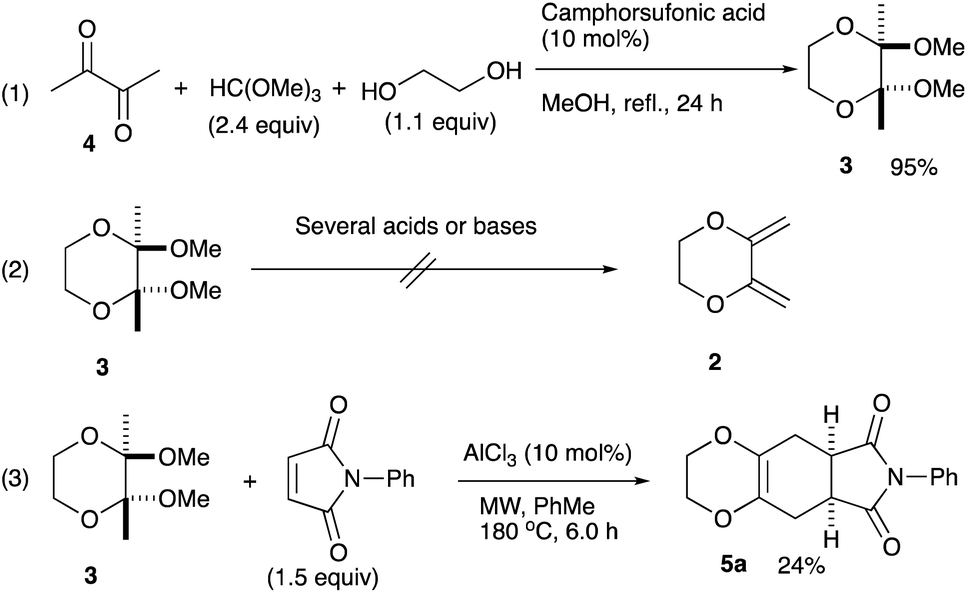 | ||
| Scheme 3 Preparation of 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 and its reaction. | ||
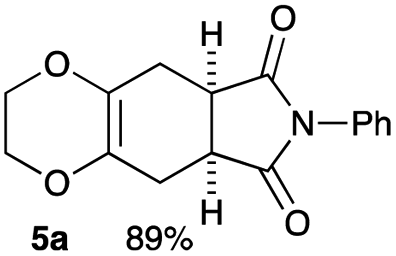
2,3-Dimethoxy-2,3-dimethyl-1,4-dioxane 3 was readily prepared from butane-2,4-dione 4 under camphorsulfonic acid-catalyzed acetalization conditions in 95% yield (Scheme 3, eqn (1)). Several attempts to convert 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 into 2,3-dimethylene-1,4-dioxane 2 under either acidic or basic conditions met with disappointing results, where only a trace amount of the desired diene 2 was obtained (Scheme 3, eqn (2)). However, the treatment of 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 with N-phenylmaleimide in the presence of a catalytic amount of aluminum chloride under microwave irradiation at 180 °C produced the [4+2] cycloaddition product 5a in 24% yield (Scheme 3, eqn (3)), indicating that the in situ generation of the diene 2 could be used for the cycloaddition process. We next examined the best cycloaddition reaction conditions using N-phenylmaleimide as a dienophile, and Table 1 summarizes the results.
|
|||||
|---|---|---|---|---|---|
| Entry | 3: equiv. | Solv. | Acid | Temp. (°C) | Yielda (%) |
| a Isolated yield.b p-Toluenesulfonic acid·H2O.c Camphorsulfonic acid.d 1,2-Dichloroethane. | |||||
| 1 | 2.0 | PhCH3 | AlCl3 | 180 | 51 |
| 2 | 2.0 | PhCH3 | AlCl3 | 200 | 68 |
| 3 | 2.0 | PhCH3 | AlCl3 | 220 | 44 |
| 4 | 3.0 | PhCH3 | AlCl3 | 200 | 73 |
| 5 | 5.0 | PhCH3 | AlCl3 | 200 | 59 |
| 6 | 3.0 | PhCH3 | Et2AlCl | 200 | 75 |
| 7 | 3.0 | PhCH3 | ZrCl4 | 200 | 80 |
| 8 | 3.0 | PhCH3 | ZnCl2 | 200 | 32 |
| 9 | 3.0 | PhCH3 | PTSA·H2Ob | 200 | 89 |
| 10 | 3.0 | PhCH3 | CSAc | 200 | 82 |
| 11 | 3.0 | PhCH3 | AcOH | 200 | 0 |
| 12 | 3.0 | nHex | PTSA·H2O | 200 | 0 |
| 13 | 3.0 | DCEd | PTSA·H2O | 200 | 73 |
| 14 | 3.0 | EtCN | PTSA·H2O | 200 | 34 |
| 15 | 3.0 | THF | PTSA·H2O | 200 | 19 |
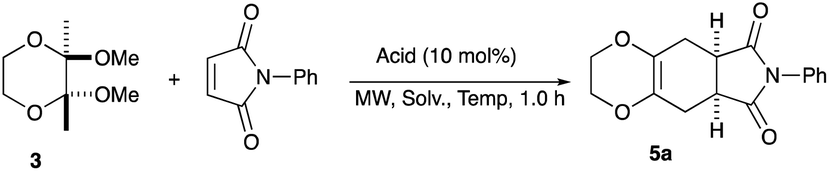
Due to the instability of the intermediary 2,3-dimethylene-1,4-dioxane 2, excess amounts of 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 were used throughout the present study and the yields were determined on the basis of the dienophile, N-phenylmaleimide. The use of 2 equivalents of 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 in the presence of AlCl3 increased the product yield up to 51% (entry 1). A better yield was obtained when the reaction was carried out at 200 °C, whereas the yield decreased at 220 °C (entries 2 and 3). We next examined the amount of 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3. The use of 3.0 equivalents of 3 recorded a better yield of the desired product 5a, although a large amount of 3 did not improve its formation (entries 4 and 5). Further examination into the use of other Lewis acids revealed that Et2AlCl and ZrCl4 could be used with comparable efficiency, although ZnCl2 was less effective (entries 6–8). The use of Brønsted acids recorded better results. Among the Brønsted acids examined the presence of p-toluenesulfonic acid·H2O recorded the best result, while camphorsulfonic acid could be used (entries 9 and 10). However, simple carboxylic acids (acetic acid) appear to be too weak to promote the 2,3-dimethylene-1,4-dioxane 2 formation (entry 11). The reactions in other solvents were also examined briefly. Although nhexane, EtCN, and THF were not suitable, DCE may be used with a small decrease in the product yield (entries 12–15). Thus, the optimized reaction conditions (entry 9) were found and used for further [4+2] cycloaddition reactions with 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3. Table 2 summarizes [4+2] cycloaddition reactions using various dienophiles.
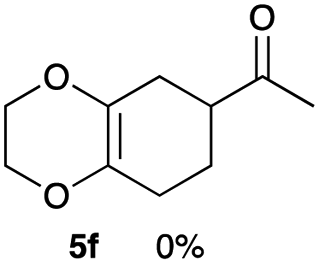
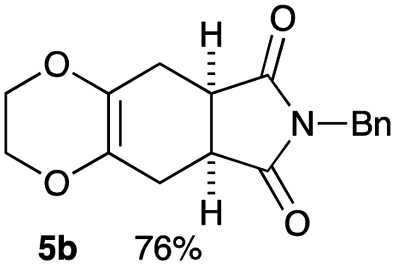
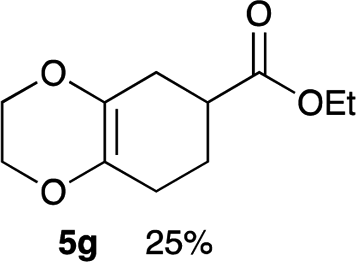
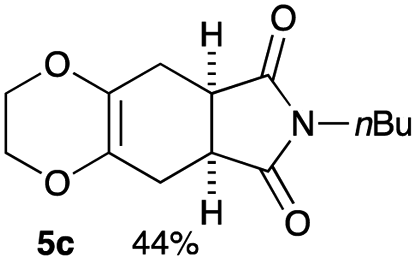
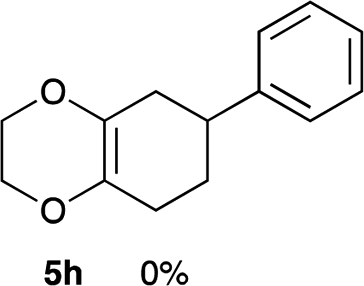
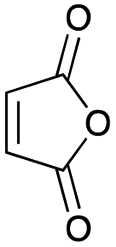
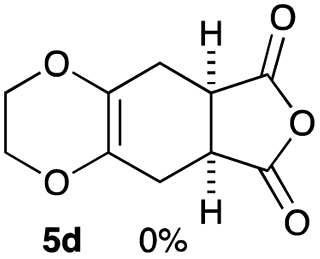
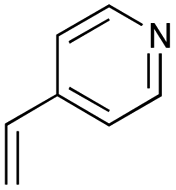
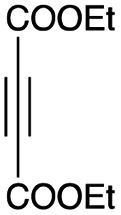
As shown in Table 2, among N-substituted maleimides examined the reaction using N-phenyl maleimide gave the addition product in the best yield of 89%, while its benzyl and nbutyl analogues recorded decreased yields of 76 and 44%, respectively (entries 1–3). However, maleic anhydride could not be used as the dienophile for the present cycloaddition (entry 4). The use of diethyl acetylenedicarboxylate gave the addition product in 58% yield (entry 5). Regarding the mono-substituted olefins, although MVK did not give the addition product 5f, ethyl acrylate afforded the cycloaddition the adduct 5g in 25% yield (entries 6 and 7). While simple styrene did not give the cycloaddition product due to the polystyrene formation, 4- and 2-vinylpyridines gave the desired products in 17 and 25%, respectively (entries 8–10). These results indicate that the present procedure suffers from the side reaction of polymerization of the intermediate 2,3-dimethylene-1,4-dioxane 2 due to the high reaction temperature necessary for the formation of this particular diene 2 from the precursor 3. Therefore, an excess amount of the diene precursor 3 is required for the optimum results and relatively stable dienophiles at high temperatures appear to be applicable. Regarding the reactivity and stability of 2,3-dimethylene-1,4-dioxane derivatives, several substituted derivatives were examined, and Table 3 summarizes the results.
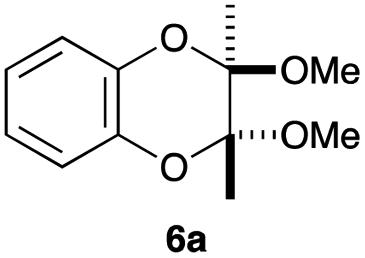
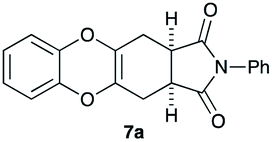
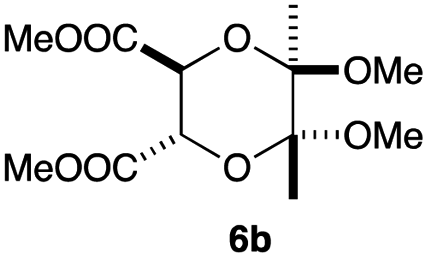
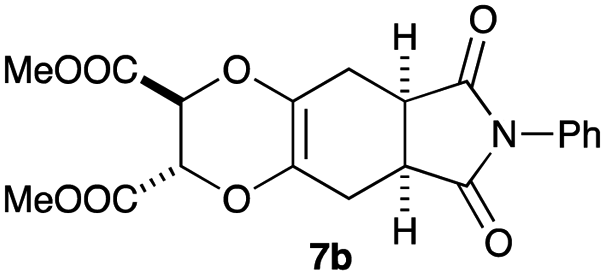
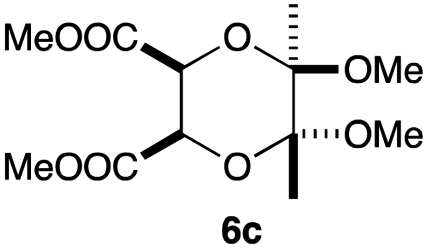
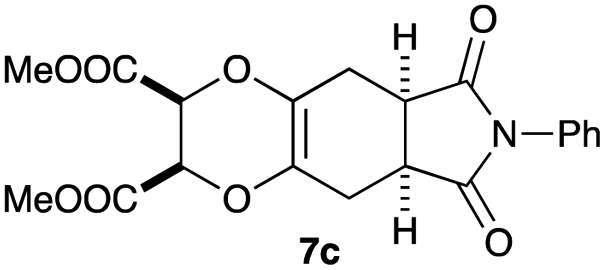
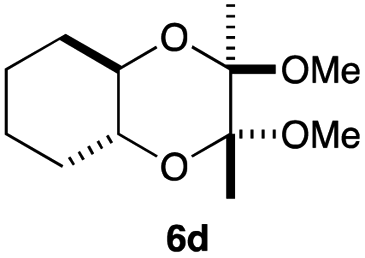
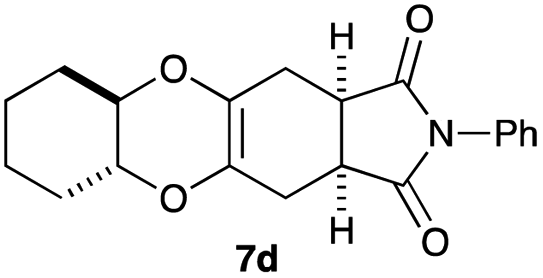
As can be seen from Table 3, the use of other substituted precursor 6a–d did not record better results than that of 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3. The benzo derivatives 6a gave a small amount of the desired product (entry 1). 5,6-Dimethoxycarbonyl derivatives 6b, c may be used, in which the cis-isomer 6c was preferred (entries 2 and 3). Cyclohexane derivative 6d could be used as a diene precursor for the present cycloadditions, giving the adduct 7d in good yield (entry 4). In the cases of the low yields of the desired products, undesired polymerization of the intermediate diene was a major side reaction, and several attempts (addition of radical scavengers, reactions at lower temperatures, etc.) did not improve the product yields. Regarding the pathways of the present cycloaddition, there exist two representative mechanisms; non-polar and polar ones.9 Considering the nature of the diene and dienophiles used in the present study, the present [4+2] cycloaddition reaction would proceed via a polar mechanism. We next examined the transformation of the Diels–Alder adduct 5a to useful intermediates to biologically important materials. Scheme 4 summarizes the results.
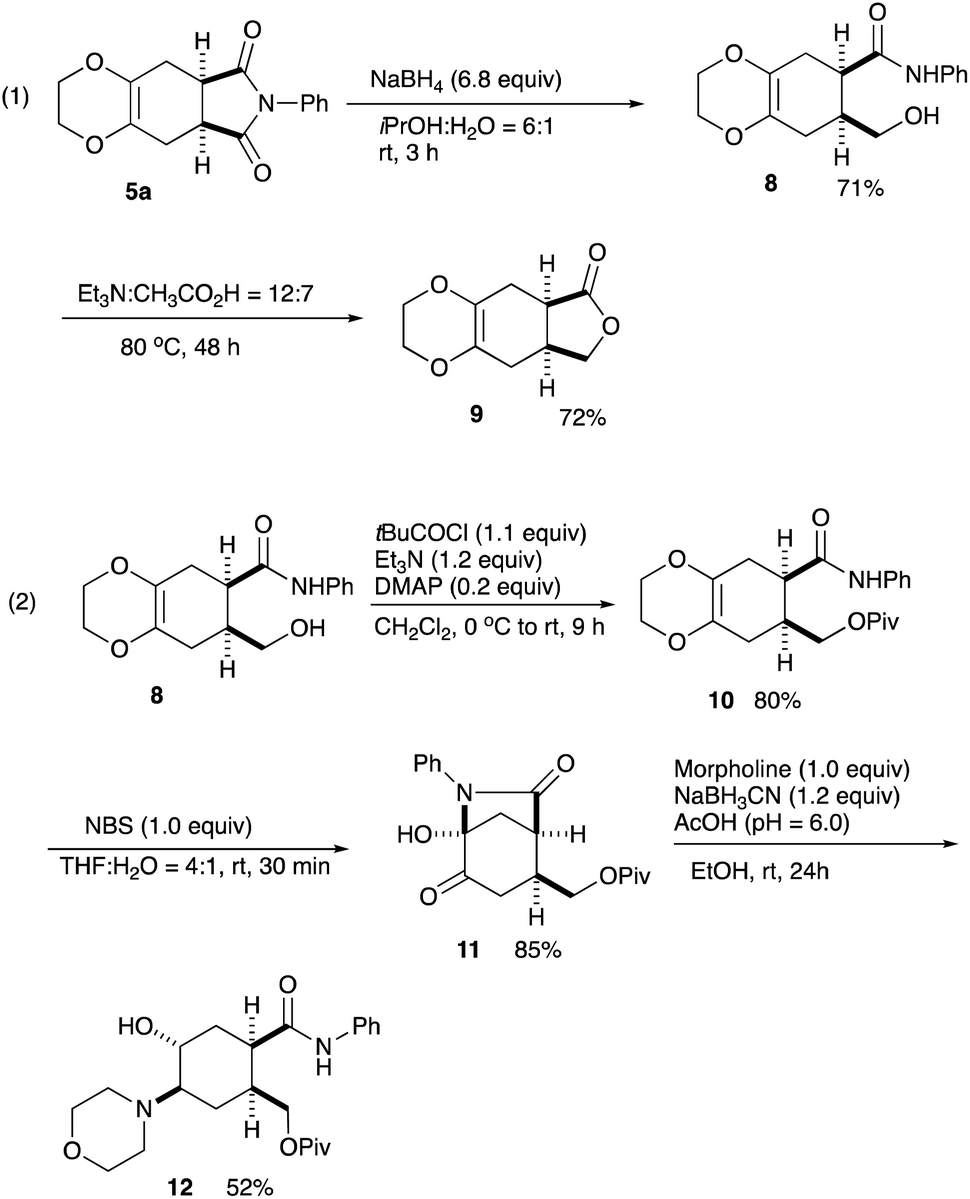 | ||
| Scheme 4 Useful transformations of the Diels–Alder adduct 5a. | ||
Regarding the imide moiety, NaBH4 reduction readily opened up the five-membered ring to give the hydroxy amide 8 in 71% yield. The subsequent lactonization was conducted with Et3N in AcOH at 80 °C to give the γ-lactone 9 in 72% yield (Scheme 4, eqn (1)).10 Removal of the dioxane ring was next examined. Several oxidative transformations were attempted, in which the primary hydroxy group often participated in the oxidation to give many byproducts, and therefore, this hydroxy function was protected as the pivalate 10. Treatment of the alcohol 8 with pivaloyl chloride in the presence of DMAP/Et3N in CH2Cl2 at 0 °C to rt for 9 h gave the pivalate 10 in 80% yield. The pivalate 10 was then treated with NBS in THF–H2O at rt for 30 min cleanly cleaved the dioxane ring to give the cyclized bicyclic product 11 in 85% yield. Toward the synthesis of important compounds for the drug delivery system (see, Scheme 1), a morpholino moiety was introduced via reductive amination. The lactam 11 was reduced with NaBH3CN in the presence of morpholine in EtOH to give the amino alcohol 12 in 52% yield.11 This amino alcohol 12 is a potential intermediate for the synthesis of a series of fliposomes (DDS) (Scheme 4, eqn (2)). We next focussed on the symmetrical features of the Diels–Alder adduct 5a.
Desymmetrization12 reaction of the Diels–Alder adducts 5a was examined to obtain chiral molecules. Several years ago, we reported the synthesis of deoxybiotine using the reductive desymmetrization reaction of imides as a crucial step (Scheme 5, eqn (1)).13 The same procedure was applied to the present Diels–Alder adducts (Scheme 5, eqn (2)), and Table 4 summarizes the results.
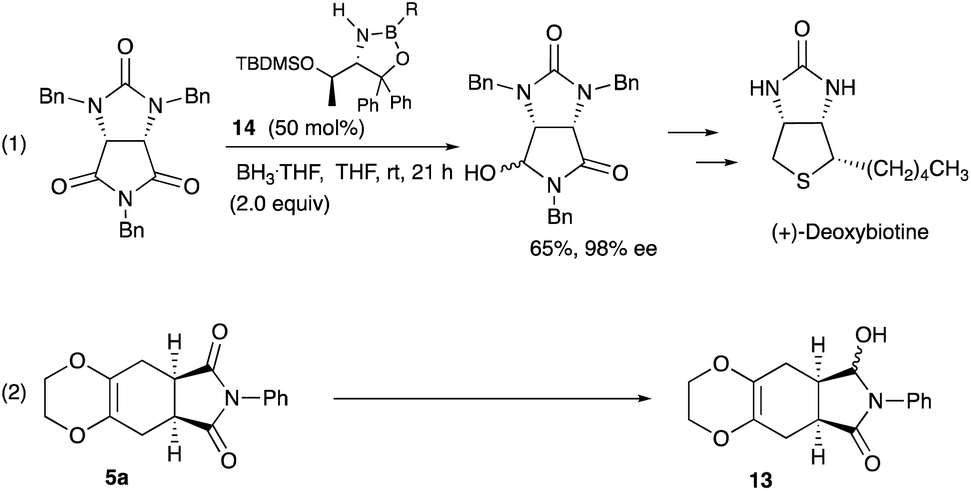 | ||
| Scheme 5 Desymmetrization reaction using the oxazaborolidine catalyst 14. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | R | BH3·THF (equiv.) | Solvent | Yielda (%) | eeb (%) |
| a Isolated yield.b Determined by HPLC using a chiral stationary column (Daicel IB) after transformation into the ethoxy derivative 15 (Scheme 6).c Oxazaborolidine (50 mol%) was used.d Reaction was carried out at 0 °C.e Reaction was carried out for 0.5 h. | |||||
| 1 | Hc | 2.0 | THF | 58 | 98 |
| 2 | H | 2.0 | THF | 51 | 89 |
| 3 | OMe | 2.0 | THF | 61 | 98 |
| 4 | OMed | 2.0 | THF | 11 | 77 |
| 5 | OMee | 2.0 | THF | 55 | 93 |
| 6 | OMe | 1.0 | THF | 11 | 80 |
| 7 | OMe | 2.5 | THF | 62 | 91 |
| 8 | OEt | 2.5 | THF | 37 | 81 |
| 9 | OMe | 3.0 | THF | 45 | 99 |
| 10 | OMe | 2.0 | PhCH3 | 32 | 99 |
| 11 | OMe | 2.0 | CH2Cl2 | 51 | 99 |
| 12 | OMe | 2.0 | Et2O | 55 | 97 |
| 13 | OMe | 2.0 | Dioxane | 52 | 89 |
| 14 | OMe | 2.0 | DME | 55 | 99 |
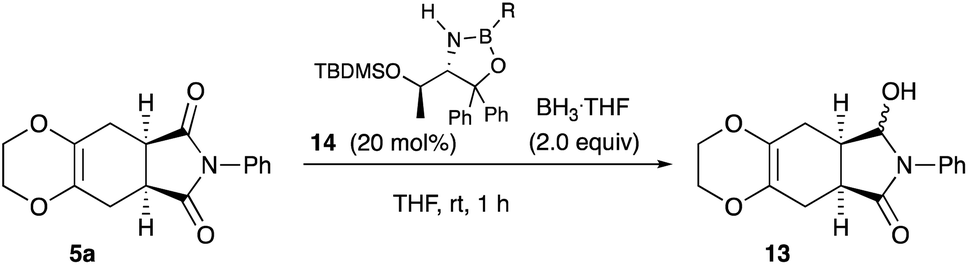
As shown in Table 4, the oxazaborolidine 14 (R = H, 50 mol%) used for the synthesis of (+)-deoxybiotine worked well to give the hydroxy lactone 13 with a high ee of 98% (entry 1) which was determined after the transformation into the ethoxy derivative 15 (Scheme 6).14 However, decreasing the amount of the catalyst to 20 mol% decreased the ee value to 89% (entry 2). Introducing an alkoxy group onto the boron atom sometimes improves the discrimination ability. Actually, the use of the B-methoxy derivative 14 (R = OMe, 20 mol%) recorded an excellent ee of 98% (entry 3). Lowering the reaction temperature did not improve the result (entry 4). A decreased reaction time decreased both the yield and the ee (entry 5). The amount of the reducing reagent BH3·THF was next examined. As you can see, the best result was obtained when the reaction was carried out with 2 equivalents of BH3·THF (entries 3, 6, 7, and 9). The use of the B-ethoxy derivative 14 (R = OEt, 20 mol%) did not afford a satisfactory result (entry 8). Regarding the solvents, CH2Cl2, Et2O, and DME may be used besides THF (entries 11, 12, and 14). Thus, the Diels–Alder adducts obtained from the present reaction served as good substrates for further functional group transformations.
 | ||
| Scheme 6 Transformation of the hydroxy lactam 13 into the ethoxy lactam 15. | ||
3 Conclusions
In conclusion, 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 readily prepared from biacetyl in good yield works as a convenient precursor to 2,3-dimethylene-1,4-dioxane 2 which can construct functionalized cyclohexene derivatives via a [4+2] cycloaddition reaction with dienophiles. The Diels–Alder adducts obtained by the present procedure are transformed into potentially useful intermediates for biologically important materials. As mentioned in the total synthesis of (+)-biotine and other examples,12,13 the desymmetrization of the Diels–Alder adducts also serves as a straightforward method for the preparation of multi-functionalized chiral compounds in a single operation.4 Experimental
4.1 General aspects
Reactions under microwave irradiation were carried out using a Biotage Initiator. Analytical techniques: melting points (mp) were determined using a YAMATO MP-21 instrument and are uncorrected. (Solid compounds were not recrystallized.) Infrared spectra were determined on a JASCO FT/IR-460 plus spectrometer. 1H NMR and 13C NMR spectra were recorded with a JEOL ECX-400P, or a JEOL A-500 spectrometer using tetramethylsilane as an internal standard. Mass spectra were recorded on a JEOL MS-700D spectrometer. High-performance liquid chromatography (HPLC) was carried out on a Hitachi L-4200 detector and a Hitachi L-6200 pump using a chiral stationary column (Daicel IB). Materials: toluene (PhCH3), dichloroethane (DCE), dichloromethane (CH2Cl2), and nhexane were dried over CaH2, distilled, and stored over molecular sieves 4 Å. Propionitrile (EtCN) was distilled from phosphorus pentaoxide and then from calcium hydride, and stored over Molecular Sieves 4A. 1,2-Dimethoxyethane (DME) and 1,4-dioxane were distilled from calcium hydride and then cupper(I) chloride, and stored over sodium. Methanol was distilled from Mg and stored over molecular sieves 3 Å. Ethanol was distilled from EtONa and stored over molecular sieves 4 Å. Isopropyl alcohol was distilled from iPrONa and stored over molecular sieves 4 Å. Triethylamine was distilled from CaH2. Tetrahydrofuran (THF) and diethyl ether (Et2O) were purified by the Glass Contour Organic Solvent Purification System of Nikko Hansen & Co., Ltd. Purification of products was performed by column chromatography on silica gel (Kanto Silica Gel 60N) and/or preparative TLC on silica gel (Merck Kiesel Gel GF254 or Wako Gel B-5F). The staring materials 6a–d were prepared as reported.154.2 Synthesis of 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3
To a solution of ethylene glycol (7.88 g, 127 mmol) in MeOH (63 mL) was added 2,3-butanedione (10.0 g, 116 mmol), trimethyl orthoformate (29.6 g, 279 mmol), and camphorsulfonic acid (2.93 g, 12.6 mmol) at room temperature under an argon atmosphere. The mixture was stirred at reflux for 24 h. Triethylamine (1.9 mL) was added to the reaction mixture to quench the reaction. The mixture was diluted with Et2O and washed with water, sat. aqueous NaHCO3, and brine, dried over anhydrous Na2SO4, and filtered. The solvents were evaporated in vacuo and then the residue was purified by chromatography on silica gel (nHex![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 1:1, as an eluent) to give 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 (19.4 g, 95%).
:ethyl acetate = 1:1, as an eluent) to give 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 (19.4 g, 95%).
Yield 95% (19.4 g); a yellow oil; 1H NMR (500 MHz, CDCl3) δ 3.95 (dd, J = 12.2, 19.5 Hz, 2H), 3.43 (dd, J = 12.2, 19.5 Hz, 2H), 3.31 (s, 6H), 1.29 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 98.5, 58.9, 48.1, 17.8; IR (neat) 2954, 2831, 1449, 1373, 1308, 1264, 1269, 1141, 1084, 1037, 941, 918, 871, 829 cm−1; HRMS (EI) calcd for C8H16O4 (M)+ 176.1049, found 176.1050.
4.3 Synthesis of (5aR*,8aS*)-7-phenyl-2,3,5,5a,8a,9-hexahydro-6H-[1,4]dioxino[2,3-f]isoindole-6,8(7H)-dione 5a (general procedure for the [4+2] cycloaddition)
To a microwave vial were added N-phenylmaleimide (17.3 mg, 0.1 mmol), 2,3-dimethoxy-2,3-dimethyl-1,4-dioxane 3 (52.9 mg, 0.3 mmol), p-toluenesulfonic acid·H2O (1.9 mg, 0.01 mmol), and toluene (1 mL). The vial was sealed with a cap, placed in the microwave cavity, and heated under the following conditions. Absorption level: low, temperature: 200 °C, vial type: 0.5–2.0 mL, pre-stirring: 30 s, reaction time: 1 h, initial power: 0, dynamic deflector optimization: off. After the reaction, a solution of sat. aqueous NaHCO3 was added to the mixture, which was extracted with EtOAc (10 mL × 3). The combined extracts were washed with sat. aqueous NaCl and dried over Na2SO4. After filtration and concentration, the mixture was purified by silica gel column chromatography (nhexane:ethyl acetate = 2:1) to give (5aR*,8aS*)-7-phenyl-2,3,5,5a,8a,9-hexahydro-6H-[1,4]dioxino[2,3-f]isoindole-6,8(7H)-dione 5a.
Yield 89% (25.3 mg); orange solid; mp = 120–124 °C; Rf = 0.40 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 2.57–2.71 (m, 4H), 3.23–3.30 (m, 2H), 3.99–4.09 (m, 4H), 7.26–7.29 (m, 2H), 7.36–7.41 (m, 1H), 7.44–7.49 (m, 2H); 13C NMR (126 MHz, CDCl3): δ 25.8, 39.5, 64.9, 126.2, 127.7, 128.5, 129.1, 131.9, 178.3; IR (neat): 2926, 1711, 1598, 1500, 1386, 1229, 1195, 1039, 917, 732, 693 cm−1; HRMS (EI) calcd for C16H15NO4 (M)+ 285.1001, found 285.1001.
4.4 (5aR*,8aS*)-7-benzyl-2,3,5,5a,8a,9-hexahydro-6H-[1,4]dioxino[2,3-f]isoindole-6,8(7H)-dione 5b
Yield 76% (22.8 mg); orange oil; Rf = 0.50 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 2.47–2.59 (m, 4H), 3.07–3.12 (m, 2H), 3.75–3.80 (m, 2H), 3.91–3.97 (m, 2H), 4.66 (s, 2H), 7.22–7.35 (m, 5H); 13C NMR (126 MHz, CDCl3): δ 25.9, 39.6, 42.6, 64.8, 127.6, 127.8, 128.4, 128.6, 135.9, 179.0; IR (neat): 2928, 2872, 1700, 1431, 1399, 1346, 1277, 1227, 1173, 1033, 951, 734, 702 cm−1; HRMS (EI) calcd for C10H10NO4 (M − C7H7)+ 208.0610, found 208.0610.
4.5 (5aR*,8aS*)-7-Butyl-2,3,5,5a,8a,9-hexahydro-6H-[1,4]dioxino[2,3-f]isoindole-6,8(7H)-dione 5c
Yield 44% (11.6 mg); orange oil; Rf = 0.50 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 0.92 (t, J = 7.3 Hz, 3H), 1.25–1.32 (m, 2H), 1.51–1.57 (m, 2H), 2.50–2.59 (m, 4H), 3.07–3.08 (m, 2H), 3.52 (t, J = 7.0 Hz, 2H), 3.95–4.05 (m, 4H).; 13C NMR (126 MHz, CDCl3): δ 13.6, 19.9, 25.8, 25.9, 29.7, 38.8, 64.9, 127.7, 179.4; IR (neat): 2933, 2873, 1775, 1700, 1439, 1402, 1356, 1227, 1197, 1113, 1042, 946, 890 cm−1; HRMS (EI) calcd for C10H10NO4 (M − C4H9)+ 208.0610, found 208.0611.
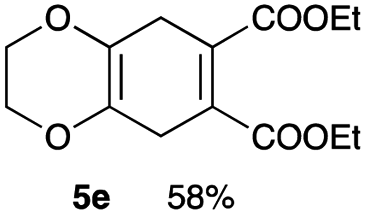
4.6 Diethyl 2,3,5,8-tetrahydrobenzo[b][1,4]dioxine-6,7-dicarboxylate 5e
Yield 58% (16.4 mg); yellow oil; Rf = 0.60 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 1.30 (t, J = 7.1 Hz, 6H), 3.13 (s, 4H), 4.10 (s, 4H), 4.23 (q, J = 7.2 Hz, 4H); 13C NMR (126 MHz, CDCl3): δ 14.0, 29.9, 61.3, 65.9, 125.7, 131.7, 167.0; IR (neat): 2979, 2360, 1720, 1459, 1262, 1214, 1133, 1060, 917, 889, 763 cm−1; HRMS (EI) calcd for C14H18O6 (M)+ 282.1103, found 282.1101.
4.7 Ethyl 2,3,5,6,7,8-hexahydrobenzo[b][1,4]dioxine-6-carboxylate 5g
Yield 24% (5.0 mg); colorless oil; Rf = 0.30 (nhexane:ethyl acetate = 15:1); 1H NMR (400 MHz, CDCl3): δ 1.26 (t, J = 7.1 Hz, 3H), 1.73–1.80 (m, 1H), 1.98–2.03 (m, 1H), 2.14–2.15 (m, 2H), 2.27–2.39 (m, 1H), 2.36–2.45 (m, 1H), 4.01–4.35 (m, 6H); 13C NMR (126 MHz, CDCl3): δ 13.8, 22.7, 25.6, 30.1, 38.3, 40.0, 64.6, 64.7, 128.1, 128.4, 176.0; IR (neat): 3448, 2977, 2933, 2873, 1731, 1456, 1381, 1348, 1313, 1277, 1131, 1042, 891, 876 cm−1. HRMS (EI) calcd for C11H16O4+ 212.1049, found 212.1055.
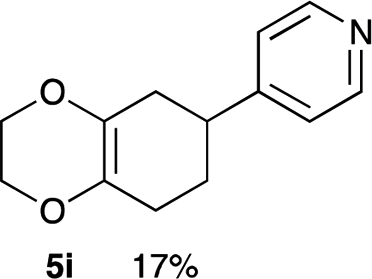
4.8 4-(2,3,5,6,7,8-Hexahydrobenzo[b][1,4]dioxin-6-yl)pyridine 5i
Yield 17% (3.6 mg); dark orange oil; Rf = 0.20 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3): δ 1.74–1.97 (m, 2H), 2.13–2.37 (m, 4H), 2.88–2.93 (m, 1H), 4.04–4.13 (m, 4H), 7.16–7.17 (m, 2H), 8.52–8.53 (m, 2H); 13C NMR (126 MHz, CDCl3): δ 25.7, 29.0, 32.7, 39.5, 64.6, 64.7, 122.3, 128.9, 129.9, 149.9, 154.2, 162.1; IR (neat): 2921, 2871, 1718, 1598, 1415, 1201, 1125, 1002, 908, 870 cm−1; HRMS (EI) calcd for C8H11O2 (M − C5H4N)+ 139.0759, found 139.0761.
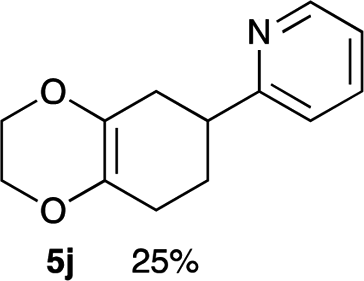
4.9 2-(2,3,5,6,7,8-Hexahydrobenzo[b][1,4]dioxin-6-yl)pyridine 5j
Yield 25% (5.5 mg); dark orange oil; Rf = 0.20 (nhexane:ethyl acetate = 4:1); 1H NMR (400 MHz, CDCl3): δ 1.85–2.51 (m, 6H), 3.04–3.10 (m, 1H), 3.98–4.21 (m, 4H), 7.12–7.14 (m, 1H), 7.19–7.21 (m, 1H), 7.61–7.64 (m, 1H), 8.55–8.56 (m, 1H); 13C NMR (126 MHz, CDCl3): δ 26.0, 28.7, 32.0, 42.4, 64.6, 64.7, 121.2, 121.4, 129.3, 129.8, 136.4, 149.2, 164.2; IR (neat): 2923, 2364, 1712, 1590, 1472, 1434, 1275, 1200, 1126, 1028, 885, 868 cm−1; HRMS (EI) calcd for C8H11O2 (M − C5H4N)+ 139.0759, found 139.0761.
4.10 (3aR*,11aS*)-2-Phenyl-3a,4,11,11a-tetrahydro-1H-benzo[5,6][1,4]dioxino[2,3-f]isoindole-1,3(2H)-dione 7a
Yield 20% (6.5 mg); orange solid; mp = 172–177 °C; Rf = 0.20 (nhexane:ethyl acetate = 5:1); 1H NMR (400 MHz, CDCl3): δ 2.63–2.72 (m, 4H), 3.33–3.35 (m, 2H), 6.64–6.67 (m, 2H), 6.80–6.86 (m, 2H), 7.30–7.48 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 24.3, 38.7, 116.1, 124.0, 126.3, 127.6, 128.7, 129.2, 131.8, 142.6, 177.7; IR (neat): 2914, 1705, 1598, 1493, 1384, 1262, 1194, 1169, 925, 850, 747 cm−1; HRMS (EI) calcd for C14H10NO4 (M − C6H5)+ 256.0610, found 256.0597.
4.11 Dimethyl (2S,3S*,5aR,8aS*)-6,8-dioxo-7-phenyl-2,3,5a,6,7,8,8a,9-octahydro-5H-[1,4]dioxino[2,3-f]isoindole-2,3-dicarboxylate 7b
Yield 34% (13.7 mg); orange oil; Rf = 0.10 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 2.65–2.81 (m, 4H), 3.26–3.32 (m, 2H), 3.66 (s, 3H), 3.81 (s, 3H), 5.01 (s, 1H), 5.03 (s, 1H), 7.36–7.38 (m, 5H); 13C NMR (126 MHz, CDCl3): δ 25.2, 25.7, 52.8, 52.9, 72.7, 72.8, 126.5, 127.5, 128.5, 128.9, 132.1, 167.4, 167.9, 177.9, 178.1; IR (neat): 2957, 2856, 1761, 1712, 1498, 1435, 1388, 1217, 1083, 911, 731, 695 cm−1; HRMS (EI) calcd for C20H19NO8 (M)+ 401.1111, found 401.1117.
4.12 Dimethyl (2R*,3S*,5aR*,8aS*)-6,8-dioxo-7-phenyl-2,3,5a,6,7,8,8a,9-octahydro-5H-[1,4]dioxino[2,3-f]isoindole-2,3-dicarboxylate 7c
Yield 50% (19.9 mg); orange oil; Rf = 0.10 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 2.71–2.88 (m, 4H), 3.31–3.33 (m, 2H), 3.80 (s, 6H), 4.83 (s, 2H), 7.26–7.48 (m, 5H); 13C NMR (126 MHz, CDCl3): δ 25.2, 39.1, 52.8, 73.2, 125.9, 126.1, 126.7, 127.8, 128.6, 129.1, 166.7, 171.0, 172.2; IR (neat): 2956, 1761, 1710, 1500, 1440, 1388, 1187, 1048, 913, 726 cm−1; HRMS (EI) calcd for C18H16NO6 (M − C2H3O2)+ 342.0978, found 342.0988.
4.13 (3aR*,5aR*,9aR*,11aS*)-2-Phenyl-3a,4,5a,6,7,8,9,9a,11,11a-decahydro-1H-benzo[5,6][1,4]dioxino[2,3-f]isoindole-1,3(2H)-dione 7d
Yield 71% (24.0 mg); orange oil; Rf = 0.20 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 1.30–1.36 (m, 4H), 1.76 (brs, 2H), 2.08 (brs, 2H), 2.54–2.76 (m, 4H), 3.23–3.30 (m, 2H), 3.41–3.53 (m, 2H), 7.26–7.48 (m, 5H); 13C NMR (126 MHz, CDCl3): δ 23.8, 23.9, 25.4, 26.1, 29.9, 39.4, 39.6, 60.2, 77.0, 126.2, 127.3, 128.5, 129.1, 132.0, 178.3, 178.4; IR (neat): 2939, 2863, 1704, 1500, 1392, 1228, 1196, 1037, 925, 769 cm−1; HRMS (EI) calcd for C20H21NO4 (M)+ 339.1471, found 339.1488.
4.14 Synthesis of (6R*,7S*)-7-(hydroxymethyl)-N-phenyl-2,3,5,6,7,8-hexahydrobenzo[b][1,4]dioxine-6-carboxamide 8
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed NaBH4 (25.7 mg, 0.68 mmol). To it was added a solution of (5aR*,8aS*)-7-phenyl-2,3,5,5a,8a,9-hexahydro-6H-[1,4]dioxino[2,3-f]isoindole-6,8(7H)-dione 5a (28.5 mg 0.10 mmol) in iso-PrOH (1.8 mL)–H2O (0.3 mL) at rt, and the mixture was stirred at rt for 3 h. The reaction was quenched with sat. aqueous NaHCO3, and the whole mixture was extracted with CH2Cl2 (30 mL × 3). The combined extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was purified by silica gel column chromatography (dichloromethane:isopropyl alcohol = 20:1) to give the title compound.
Yield 71% (20.5 mg); yellow solid; mp = 258–262 °C; Rf = 0.30 (nhexane:ethyl acetate = 1:1); 1H NMR (400 MHz, CDCl3): δ 2.05–2.33 (m, 5H), 2.82–2.86 (m, 1H), 3.27–3.44 (m, 2H), 3.92–4.04 (m, 4H), 4.53 (t, J = 5.2 Hz, 1H), 7.00–7.03 (m, 1H), 7.27–7.30 (m, 2H), 7.58–7.59 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 25.3, 27.2, 38.2, 41.7, 59.4, 64.0, 119.1, 123.0, 127.5, 127.7, 128.6, 139.3, 171.6; IR (neat): 3308, 2963, 2933, 1718, 1652, 1598, 1525, 1442, 1331, 1204, 1047, 983, 856 cm−1; HRMS (EI) calcd for C16H19NO4 (M)+ 289.1314, found 289. 1312.
4.15 Synthesis of (5aR*,8aS*)-2,3,5,8,8a,9-hexahydro-[1,4]dioxino[2,3-f]isobenzofuran-6(5aH)-one 9
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed (6R*,7S*)-7-(hydroxymethyl)-N-phenyl-2,3,5,6,7,8-hexahydrobenzo[b][1,4]dioxine-6-carboxamide 8 (42.2 mg, 0.15 mmol). To it was added triethylamine (2.4 mL) and acetic acid (1.4 mL) at rt, and the mixture was stirred at 80 °C for 48 h. The reaction was cooled to 0 °C and quenched with sat. aqueous NaHCO3. The whole mixture was extracted with CH2Cl2 (30 mL × 3). The combined extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was purified by silica gel column chromatography (nhexane:ethyl acetate = 1:1) to give the title compound.
Yield 72% (20.6 mg); colorless oil; Rf = 0.70 (nhexane:ethyl acetate = 1:1); 1H NMR (400 MHz, CDCl3): δ 2.09–2.83 (m, 6H), 4.00–4.09 (m, 5H), 4.26–4.30 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 23.2, 26.8, 33.1, 38.1, 64.6, 72.0, 127.1, 127.6, 177.8; IR (neat): 2975, 2915, 2878, 1773, 1716, 1451, 1375, 1276, 1208, 1043, 971, 890 cm−1; HRMS (EI) calcd for C10H12O4 (M)+ 196.0736, found 196. 0743.
4.16 Synthesis of ((6S*,7R*)-7-(phenylcarbamoyl)-2,3,5,6,7,8-hexahydrobenzo[b][1,4]dioxin-6-yl)methyl pivalate 10
In a 100 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed (6R*,7S*)-7-(hydroxymethyl)-N-phenyl-2,3,5,6,7,8-hexahydrobenzo[b][1,4]dioxine-6-carboxamide 8 (867.9 mg, 3.0 mmol). To it was added triethylamine (0.50 mL, 3.6 mmol), DMAP (73.3 mg, 0.60 mmol), and dichloromethane (20 mL) at rt. The mixture was cooled to 0 °C and to it was slowly added pivaloyl chloride (0.40 mL, 3.3 mmol). The mixture was allowed to stand at rt for 9 h. The reaction was quenched with HCl (2 M, 20 mL). The whole mixture was extracted with CH2Cl2 (20 mL × 3). The combined extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was purified by silica gel column chromatography (nhexane:ethyl acetate = 2:1) to give the title compound.
Yield 80% (901.3 mg); yellow solid; mp = 154–162 °C; Rf = 0.40 (nhexane:ethyl acetate = 2:1); 1H NMR (400 MHz, CDCl3): δ 1.19 (s, 1H), 2.17–2.23 (m, 1H), 2.31–2.36 (m, 1H), 2.41–2.54 (m, 3H), 2.84–2.89 (m, 1H), 4.08–4.13 (m, 5H), 4.26–4.31 (m, 1H), 7.00–7.13 (m, 1H), 7.30–7.34 (m, 2H), 7.50–7.57 (m, 2H), 7.85 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 26.7, 27.1, 27.6, 34.9, 38.8, 42.8, 64.6, 64.7, 64.7, 120.0, 124.3, 127.9, 128.4, 128.9, 137.8, 137.8, 170.2, 178.7; IR (neat): 3319, 2972, 2928, 2873, 1720, 1664, 1600, 1540, 1442, 1283, 1199, 1160, 1041, 982, 887 cm−1; HRMS (EI) calcd for C21H27NO5 (M)+ 373.1889, found 373.1876.
4.17 Synthesis of [(1R*,2S*,5R*)-5-hydroxy-4,7-dioxo-6-phenyl-6-azabicyclo[3.2.1]octan-2-yl]methyl pivalate 11
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed [(6S*,7R*)-7-(phenylcarbamoyl)-2,3,5,6,7,8-hexahydrobenzo[b][1,4]dioxin-6-yl]methyl pivalate (75.0 mg, 0.20 mmol). To it was added THF (8.0 mL), NBS (35.6 mg, 0.20 mmol), and H2O (2.0 mL) successively at rt. The mixture was stirred at rt for 30 min. The reaction was quenched with sat. aqueous NaHSO3. The whole mixture was extracted with Et2O (10 mL × 3). The combined extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was purified on silica gel TLC (nhexane:ethyl acetate = 1:1) to give the title compound.
Yield 85% (58.6 mg); yellow powder; mp = 62–64 °C; Rf = 0.70 (nhexane:ethyl acetate = 1:1); 1H NMR (400 MHz, CDCl3): δ 1.24 (s, 9H), 2.04–2.12 (m, 1H), 2.48–2.55 (m, 1H), 2.59–2.68 (m, 1H), 2.77–2.81 (m, 1H), 2.88–2.94 (m, 1H), 3.03–3.04 (m, 1H), 4.09–4.17 (m, 1H), 4.25–4.29 (m, 1H), 4.41 (s, 1H), 7.08–7.14 (m, 2H), 7.24–7.28 (m, 1H) 7.35–7.38 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 27.1, 37.1, 38.6, 38.8, 40.7, 42.5, 64.8, 90.5, 124.9, 127.2, 127.2, 129.1, 129.1, 133.5, 171.3, 178.0, 204.8; IR (neat): 3404, 2973, 2908, 2875, 1719, 1598, 1542, 1501, 1367, 1285, 1159, 1035 cm−1;. HRMS (EI) calcd for C19H23O5 (M)+ 345.1576, found 345.1572.
4.18 Synthesis of [(1S*,2R*,4R*,5R*)-4-hydroxy-5-morpholino-2-(phenylcarbamoyl)cyclohexyl]methyl pivalate 12
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed [(1R*,2S*,5R*)-5-hydroxy-4,7-dioxo-6-phenyl-6-azabicyclo[3.2.1]octan-2-yl]methyl pivalate 11 (34.5 mg, 0.10 mmol). To it was added EtOH (4.0 mL), morpholine (8.7 μL, 0.10 mmol), and NaBH3CN (7.5 mg, 0.12 mmol) successively at rt. To the mixture was added AcOH to reach pH 6.0, and it was stirred at rt for 24 h. The reaction was quenched with sat. aqueous NaHCO3. The whole mixture was extracted with CH2Cl2 (10 mL × 3). The combined extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was purified by silica gel column chromatography (dichloromethane:methanol = 10:1) to give the title compound.
Yield 52% (21.7 mg); colorless oil; Rf = 0.30 (dichloromethane:methanol = 10:1); 1H NMR (400 MHz, CDCl3): δ 1.09, (s, 9H)1.53–1.57 (m, 1H), 1.74–1.84 (m, 1H), 2.01–2.04 (m, 1H), 2.22–2.25 (m, 1H), 2.31–2.35 (m, 1H), 2.46–2.47 (m, 1H)2.52–2.54 (m, 1H), 2.57–2.74 (m, 4H), 3.71–3.74 (m, 4H), 3.75–3.76 (m, 1H) 4.05 (s, 1H) 4.29–4.34 (m, 1H), 4.43–4.48 (m, 1H), 7.07–7.10 (m, 1H), 7.28–7.32 (m, 2H), 7.57–7.59 (m, 2H), 7.83 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 21.0, 27.0, 31.7, 34.1, 38.6, 45.6, 50.2, 64.5, 64.5, 67.2, 119.8, 124.2, 128.9, 138.0, 171.0, 179.1; IR (neat): 3310, 2961, 1722, 1600, 1544, 1500, 1442, 1286, 1161, 1119, 755, 517, 504 cm−1; HRMS (EI) calcd for C23H34N2O5 (M)+ 418.2468, found 418.2477.
4.19 Synthesis of (5aS,8aR)-8-hydroxy-7-phenyl-2,3,5,5a,7,8,8a,9-octahydro-6H-[1,4]dioxino[2,3-f]isoindol-6-one 13
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed (2S,3R)-2-amino-3-(tert-butyldimethylsilyloxy)-1,1-diphenylbutan-1-ol16 (7.4 mg, 0.02 mmol). To it was added THF (1.0 mL) and trimethyl borate (1.0 M, 0.02 mL, 0.02 mmol), and the mixture was stirred at rt for 1 h. To the mixture were slowly added solutions of (5aR*,8aS*)-7-phenyl-2,3,5,5a,8a,9-hexahydro-6H-[1,4]dioxino[2,3-f]isoindole-6,8(7H)-dione 5a (28.5 mg 0.1 mmol) in THF (2 mL) and BH3·THF (1.0 M, 0.20 mL, 0.20 mmol). After stirring at rt for 1 h, the reaction was quenched with sat. aqueous NaHCO3. The whole mixture was extracted with AcOEt (10 mL × 3). The combined extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was purified on silica gel TLC (nhexane:ethyl acetate = 2:1) to give the title compound.
Yield 61% (17.5 mg); colorless oil; Rf = 0.10 (nhexane:ethyl acetate = 1:1); [α]14D −42.3 (c 0.012, CHCl3); 1H NMR (400 MHz, CDCl3): δ 2.30–2.65 (m, 4H), 2.76–2.90 (m, 3H), 4.02–4.14 (m, 4H), 5.55–5.57 (m, 1H), 7.20–7.54 (m, 5H); 13C NMR (100 MHz, CDCl3): δ 22.9, 26.1, 37.9, 38.6, 64.6, 64.7, 88.5, 122.5, 126.0, 126.6, 128.3, 129.2, 137.7, 174.8; IR (neat): 3425, 2920, 1682, 1497, 1404, 1278, 1207, 1128, 1039, 890, 758 cm−1; HRMS (EI) calcd for C16H16NO3+ (M − HO)+ 270.1130, found 270.1142.
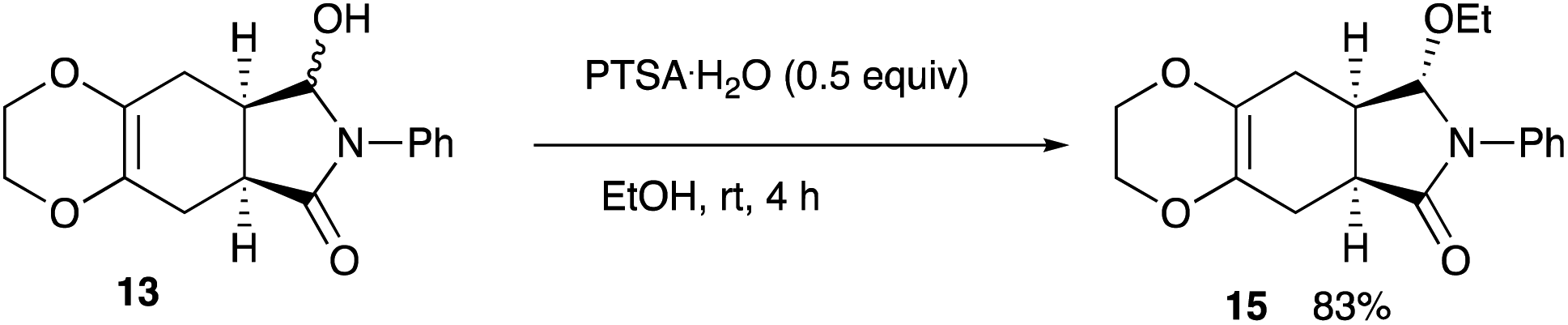
4.20 Determination of the enantiomeric purity of the reduction product via the ethoxy derivative 15
In a 30 mL two-necked round-bottomed flask equipped with a magnetic stirring bar, a rubber septum, and an argon balloon was placed (5aS,8aR)-8-hydroxy-7-phenyl-2,3,5,5a,7,8,8a,9-octahydro-6H-[1,4]dioxino[2,3-f]isoindol-6-one 13 (8.1 mg, 0.028 mmol). To it was added PTSA·H2O (2.7 mg, 0.014 mmol) and EtOH (4.0 mL), and the mixture was stirred at rt for 4 h. The reaction was quenched with sat. aqueous NaHCO3. The whole mixture was extracted with CH2Cl2 (5 mL × 3). The combined extracts were washed with brine (15 mL), dried (Na2SO4), and concentrated in vacuo. The crude product was purified on silica gel TLC (nhexane:ethyl acetate = 1:1) to give (5aS,8S,8aR)-8-ethoxy-7-phenyl-2,3,5,5a,7,8,8a,9-octahydro-6H-[1,4]dioxino[2,3-f]isoindol-6-one 15.
Yield 83% (7.4 mg); colorless oil; Rf = 0.50 (nhexane:ethyl acetate = 1:1); [α]25D −46.8 (c 0.089, CHCl3); 1H NMR (400 MHz, CDCl3): δ 1.18 (dd, J = 6.7, 7.3 Hz, 3H), 1.95–2.05 (m, 1H), 2.28–2.45 (m, 2H), 2.65–2.76 (m, 2H), 3.19–3.22 (m, 1H), 3.49–3.53 (m, 2H), 3.95–4.12 (m, 5H), 4.88 (s, 1H), 7.16–7.19 (m, 1H), 7.33–7.36 (m, 2H), 7.47–7.49 (m, 2H); 13C NMR (126 MHz, CDCl3): δ 15.3, 22.8, 26.2, 35.9, 38.0, 63.9, 64.6, 94.6, 122.6, 125.7, 126.5, 128.5, 128.9, 138.7, 174.8; IR (neat): 2974, 2911, 1714, 1598, 1497, 1395, 1280, 1200, 1071, 920, 890, 859 cm−1; HRMS (EI) calcd for C16H16NO4 (M − C2H5)+ 286.1079, found 286.1071.
The enantiomeric purity of the above ethoxide 15 was determined by HPLC using a chiral stationary column (Daicel IB). Flow rate 1.0 mL min−1, nhexane:iPrOH = 9:1, detection at 254 nm, set temperature 35 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) and on Innovative Areas “Organic Synthesis Based on Reaction Integration. Development of New Methods and Creation of New Substances” from JSPS and MEXT.Notes and references
- (a) P. K. S. Savithru, P. Gupta, V. Palle, M. S. Rao, R. S. Kombu, K. Rauthan, and V. K. Ramanathan, WO 2007/039809 A1, 2007; (b) D. B. Bylund, in Encyclopedia of Biological Chemistry, ed. W. J. Lennarz and M. D. Lane, Elsevier Inc., Amsterdam, 2nd edn, 2013, pp. 57–60 Search PubMed.
- A. Yıldırım, U. Atmaca, A. Keskin, M. Topal, M. Çelik, I. Gülçin and C. T. Supuran, Bioorg. Med. Chem., 2015, 23, 2598–2605 CrossRef.
- (a) N. M. Samoshina, X. Liu, B. Brazdova, A. H. Franz, V. V. Samoshin and X. Guo, Pharmaceutics, 2011, 3, 379–405 CrossRef CAS; (b) X. Liu, Y. Zheng, N. M. Samoshina, A. H. Franz, X. Guo and V. V. Samoshin, J. Liposome Res., 2012, 22, 319–328 CrossRef CAS; (c) J. Lou, X. Zhang and M. D. Best, Chem.–Eur. J., 2019, 25, 20–25 CrossRef CAS.
- For reviews, see: (a) U. Pindur, G. Lutz and C. Otto, Chem. Rev., 1999, 93, 741–761 CrossRef; (b) A. Kumar, Chem. Rev., 2001, 101, 1–20 CrossRef CAS; (c) S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359–5406 CrossRef CAS; (d) P. Wessig and G. Müller, Chem. Rev., 2008, 108, 2051–2063 CrossRef CAS; (e) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983–2992 RSC; (f) V. Nair, R. S. Menon, A. T. Biju and K. G. Abhilash, Chem. Soc. Rev., 2012, 41, 1050–1059 RSC.
- For 2,3-dialkoxy-1,3-butadienes, see: (a) R. K. Summerbell and G. J. Lestina, J. Am. Chem. Soc., 1957, 79, 3878–3884 CrossRef CAS; (b) D. R. Anderson and T. H. Koch, J. Org. Chem., 1978, 43, 2726–2728 CrossRef CAS; (c) H.-D. Scharf, H. Plum, J. Fleischhauer and W. Schleker, Chem. Ber., 1979, 112, 862–882 CrossRef CAS; (d) E. McDonald, A. Suksamrarn and R. D. Wylie, J. Chem. Soc., Perkin Trans. 1, 1979, 1893–1900 RSC; (e) N. Ruiz, M. D. Pujol, G. Guillaumet and G. Coudert, Tetrahedron Lett., 1992, 33, 2965–2968 CrossRef CAS; (f) G. Torres-García and J. Mattay, Tetrahedron, 1996, 52, 5421–5426 CrossRef; (g) F. von Kieseritzky, F. Allared, E. Dahlstedt and J. Hellberg, Tetrahedron Lett., 2004, 45, 6049–6050 CrossRef CAS; (h) Y. Kasano, A. Okada, D. Hiratsuka, Y. Oderaotoshi, S. Minakata and M. Komatsu, Tetrahedron, 2006, 62, 537–542 CrossRef CAS; (i) B. J. Compton, D. S. Larsen, L. Larsen and R. T. Weavers, Tetrahedron Lett., 2008, 49, 219–221 CrossRef CAS; (j) B. Linclau, P. J. Clarke and M. E. Light, Tetrahedron Lett., 2009, 50, 7144–7147 CrossRef CAS.
- I. Hachiya, T. Yamamoto, T. Inagaki, T. Matsumoto, A. Takahashi, I. Mizota and M. Shimizu, Heterocycles, 2014, 88, 607–612 CrossRef CAS.
- For a review, see: L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS and references therein.
- Regarding 2,3-dimethylene-1,4-dioxane 2, it was reported that this particular diene polymerized noticeably within 5 min at 25 °C. At the end of 21 h a white, glass-like polymer was obtained which did not melt at 240 °C. See, ref. 5a.
- For non-polar mechanisms, see for example: (a) D. A. Singleton, B. E. Schulmeier, C. Hang, A. A. Thomas, S.-W. Leung and S. R. Merrigan, Tetrahedron, 2001, 57, 5149–5160 CrossRef CAS; (b) R. Jasiński, React. Kinet., Mech. Catal., 2016, 119, 49–57 CrossRef. For polar mechanisms, see for example: (c) R. Jasiński, Monatsh. Chem., 2016, 147, 1207–1213 CrossRef; (d) R. Jasiński, J. Mol. Graphics Modell., 2017, 75, 55–61 CrossRef; (e) R. Jasiński, J. Fluorine Chem., 2018, 206, 1–7 CrossRef.
- M. D. Barker, R. A. Dixon, S. Jones and B. J. Marsh, Tetrahedron, 2006, 62, 11663–11669 CrossRef CAS.
- E. Podyacheva, O. I. Afanasyev, A. A. Tsygankov, M. Makarova and D. Chusov, Synthesis, 2019, 51, 2667–2677 CrossRef CAS and references therein.
- For reviews, see: (a) M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765–1784 RSC; (b) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS; (c) T. Suzuki, Tetrahedron Lett., 2017, 58, 4731–4739 CrossRef CAS.
- M. Shimizu, Y. Nishigaki and A. Wakabayashi, Tetrahedron Lett., 1999, 40, 8873–8876 CrossRef CAS.
- M. Ostendorf, R. Romagnoli, I. C. Pereiro, E. C. Roos, M. J. Moolenaar, W. N. Speckamp and H. Hiemstra, Tetrahedron: Asymmetry, 1997, 8, 1773–1789 CrossRef CAS.
- (a) C. Ramarao, R. Nandipati, R. Navakoti and R. Kottamasu, Tetrahedron Lett., 2012, 53, 637–640 CrossRef CAS; (b) D. E. A. Brittain, C. M. Griffiths-Jones, M. R. Linder, M. D. Smith, C. McCusker, J. S. Barlow, R. Akiyama, K. Yasuda and S. V. Ley, Angew. Chem., Int. Ed., 2005, 44, 2732–2737 CrossRef CAS; (c) N. Mariet, H. Pellissier, M. Ibrahim-Ouali and M. Santelli, Eur. J. Org. Chem., 2004, 2679–2691 CrossRef CAS.
- (a) M. Shimizu, M. Kamei and T. Fujisawa, Tetrahedron Lett., 1995, 36, 8607–8610 CrossRef CAS; (b) M. Shimizu, K. Tsukamoto and T. Fujisawa, Tetrahedron Lett., 1997, 38, 5193–5196 CrossRef CAS; (c) M. Shimizu, K. Tsukamoto, T. Matsutani and T. Fujisawa, Tetrahedron, 1998, 54, 10265–10274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00329a |
| This journal is © The Royal Society of Chemistry 2021 |