DOI:
10.1039/D1RA00237F
(Paper)
RSC Adv., 2021,
11, 9849-9855
Preparation and anti-coking application of sol–gel SiO2 coating in a delayed coking furnace
Received
11th January 2021
, Accepted 10th February 2021
First published on 8th March 2021
Abstract
To isolate iron sulphide and reduce coke adhesion reactions that occur on the surface of a delayed coking furnace, a SiO2 coating was developed on Cr9Mo alloy by employing the sol–gel method. The coating was characterised through Fourier transformation infrared spectroscopy, X-ray diffraction, scanning electron microscopy, energy dispersive spectroscopy, potentiodynamic scanning, InfiniteFocus optical 3D surface metrology, thermal shock and coking experiments. After heat treatment at 550 °C, the silicon methyl groups were oxidised and the coating exhibited a Si–O–Si connected crosslinked network structure. The coating surface was uniformly dense with a roughness and thickness of 0.2 and approximately 4 μm, respectively. The coating still adhered to the substrate tightly after 20 cycles of thermal shock treatment. Compared with an uncoated sample, the coating effectively improved the corrosion resistance of the substrate, suppressed the iron sulphide reaction, and reduced coke adhesion onto the sample. The coating had a better inhibition effect on coke fouling in a delayed coking furnace.
1 Introduction
As one of the most widely used residue conversion processes, delayed coking is employed to thermally crack residues into light components and value-added products.1–3 The residue is rapidly heated to the temperature required for coking (∼500 °C) using a heating furnace, and then it undergoes cracking and condensation in a coke drum. In this entire process, the coking reaction is delayed into a coke tower; hence, the process is called ‘delayed coking’.3,4 However, in actual production, coke becomes deposited on the surface of a furnace tube,1,4–8 which leads to a decrease in the heat transfer efficiency for high wall thermal resistance, an increase in the medium flow resistance in the tube, and furnace tube blowout.9,10 Therefore, the problem of coking inhibition in a delayed coking furnace has received increasing attention.
Currently, several approaches are used to suppress coking, described as follows. (i) Improving feedstock quality: the critical decomposition temperature of oil products is increased by adding aromatic hydrocarbons to the residue to delay the coking reaction into the coke tower.7,8,11 (ii) Adding inhibitors to raw materials: an inhibitor with specific functions can slow down the coking process because inhibitors can change free radical reactions to inhibit or stop free radical formation.9,12–14 (iii) Optimising heating furnace operations: by changing the flow field distributions in the furnace tube, the average flow velocity of a boundary layer medium is increased to the highest possible value to reduce the boundary layer thickness.15,16 Simultaneously, the selection of appropriate temperature and pressure is important.2,3 (iv) Surface treatment: a layer of inert coating is applied to the furnace tube surface, by using different methods to prevent direct contact between the oil and inner walls of the tube to stop vulcanisation and metal catalytic coking.17–26
Coking mainly results from formation of iron sulphide on the furnace tube surface.4,7,8,10,27–29 Dickakia found a visible layer of iron disulphide between a tube wall and the first layer of coke.30 The iron disulphide behaved like Velcro to trap bitumen molecules and attach them to the inner wall of the furnace tube.30 Inert coatings can cover metal active sites, prevent these sites from interacting with coke precursors, reduce metal surface roughness and surface energy, and change the thickness of furnace tube boundary layers to suppress the coking of the inner walls of furnace tubes. Efforts have been taken to resolve the problem of coke deposition on the furnace tube surface by using coating techniques. Coatings of Mn and Cr oxides on the metal surface manufactured through element diffusion and selective oxidation can reduce the occurrence of catalytic coking and prevent carburisation.17,18 A protective layer of SiO2 forms on the wall of halogen-treated pyrolysis pipes containing silane, ethylsilane, and siloxane. Moreover, this SiO2 layer can be used to isolate pipe walls from the gas present in the pipe to inhibit coking.19 TiO2,20 SiO2,21 Al2O3,22 TiN,23,24 and SiO2/S25 coatings prepared through chemical vapour deposition can prevent direct contact between hydrocarbons and metal tube walls for coking inhibition. However, these coatings are not suitable for use in a delayed coking furnace. Metal-infused surface treatment is adopted for solving the problem of delayed coking in a tube coking furnace. In this method, a protective nanofilm layer is formed. Nanofilms of 50–100 nm isolate the furnace tube from the residue, eliminate chemical foundations for coke adherence, and highly reduce the sticking effect of mesophase wetting.26
In this study, a uniformly dense SiO2 coating was prepared on the Cr9Mo alloy, using methyl triethoxysilane (MTES) and alkaline silica sol and employing the sol–gel method. The molecular structure and residual phase composition of the gel solution were analysed and identified through Fourier-transform infrared spectroscopy (FTIR) and X-ray diffraction (XRD). The surface morphology, surface element composition, and sectional element distribution of the SiO2 coating were characterised through scanning electron microscopy (SEM), InfiniteFocus optical 3D surface metrology, and energy dispersive spectroscopy (EDS). The service resistibility of the coating against peeling off was studied using thermal shock experiments. The decay resistance of the coating was evaluated through potentiodynamic scanning (PDS). A delayed coking furnace simulation device was used to evaluate the anti-coking performance of the SiO2 coating, and coke was analysed and compared through XRD.
2 Experimental details
2.1 Chemical materials
MTES (SiC7H18O3, Aladdin, 98%) and alkaline silica sol (mSiO2·nH2O, Alfa Aesar, 30%) were used as SiO2 precursors. Acetic acid (CH3COOH, Aladdin, 99.5%) and deionised water (H2O, ECUST) were used as hydrolysis accelerators and isopropanol (C3H8O, Aladdin, 99.5%) was used as a component compatibiliser.
2.2 Materials pre-treatment and preparation
The sample was shaped into a sheet of 10 mm × 10 mm × 3 mm through electrical discharge machining, and a small hole with a diameter of 1 mm was drilled at the top of the sample for subsequent testing. 400–2000 grit sandpapers were used to remove oxides from the Cr9Mo alloy surface. Subsequently, the sample was placed in absolute ethanol for ultrasonic cleaning for approximately 5 min and was dried with hot air for further use.
2.3 Preparation of the coatings
First, 20 ml of MTES was mixed with 0.5 ml of acetic acid. After 30 min hydrolysis at 20 °C, the silica sol was slowly added to the mixed liquor. Specific amounts of deionised water and isopropanol were added, and the resulting mixture was placed on a magnetic stirrer at 20 °C. After stirring for 24 h, a gel was obtained. Subsequently, the pretreated Cr9Mo alloy sample was fixed on a computer-controlled vertical elevator (PTL-MMB02-200, Hefei tactic material technology) and immersed into the gel solution. After 30 s, the sample was removed at 15 mm min−1 rate, and then the sample was heated in an oven at 60 °C for 5 min. Finally, the sample was placed in a muffle furnace at 550 °C for 2 h. Fig. 1 illustrates the coating preparation process.
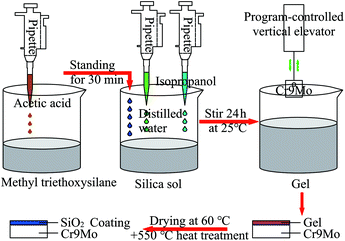 |
| | Fig. 1 The coating preparation process. | |
2.4 Characterisation of the coatings
The molecular structure and residual phase composition of the gel solution were analysed and identified through XRD (18KW/D/max2550VB/PC, Japan) and FTIR (7800–350/cm 0.01/cm/6700, USA). The surface morphology, surface element composition, and sectional element distribution of the SiO2 coating were characterised using SEM (Hitachi-S-3400N, Japan), InfiniteFocus optical 3D surface metrology (InfiniteFocus G4, Austria) and EDS (133eV/Falcon 60S, USA). The decay resistance of the coating was evaluated through PDS (PARSTAT-2273, USA).
2.5 Anti-coking tests of SiO2 coatings
To evaluate the anti-coking performance of the SiO2 coating, Saudi Arabian light crude vacuum residues were added to the high-pressure reactor with 1/3 of total volume (Table 1). The uncoated and SiO2 coated samples (10 mm × 10 mm × 3 mm) were suspended in the tube to simulate furnace tube coking. Nitrogen was passed into the reactor at a 150 ml min−1 rate for 10 min to replace the air. The reaction preservation time was 30, 60, and 120 min at 500 °C, and the rotation speed of the stirrer was adjusted to 120 rpm to make the residue flow in the heating vessel. After heat preservation, the test samples were removed and placed in acetone to dissolve the residual material present on the surface. Then, the samples were dried for analyses. Fig. 2 presents the experimental schematic diagram.
Table 1 Composition of vacuum residue
| Component |
Saturates/% |
17.3 |
| Aromatic/% |
52.7 |
| Resins/% |
23.9 |
| Asphaltenes/% |
6.1 |
| Element |
C/% |
85.19 |
| H/% |
10.42 |
| S/% |
4.06 |
| N/% |
0.32 |
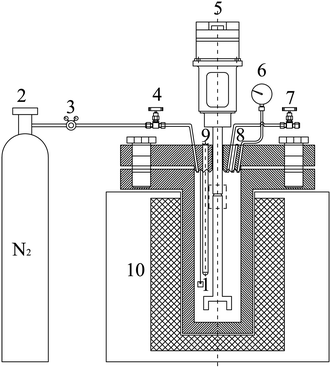 |
| | Fig. 2 Schematic diagram of the experimental set-up: (1) sample; (2) nitrogen gas bottle; (3) relief valve; (4) gas inlet valve; (5) stirring motor; (6) pressure gauge; (7) pressure relief valve; (8) bursting disc; (9) temperature measuring instrument; (10) heating furnace. | |
3 Results and discussion
3.1 FT-IR and XRD analyses
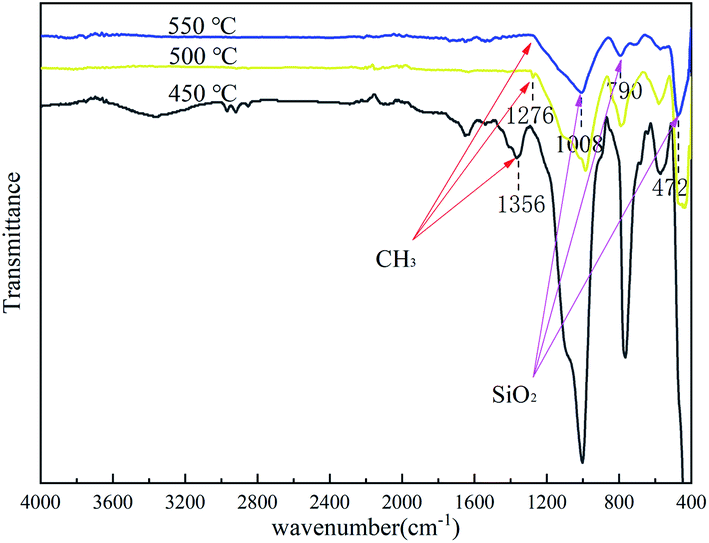
To determine the characteristics of the coating formed on the Cr9Mo alloy surface, FTIR was used to characterise the coating after each heat treatment at 450, 500 and 550 °C for 2 h (Fig. 3). With an increase in temperature, the variable-angle vibration peak of the methyl group appearing near 1276 cm−1 weakened and eventually disappeared, indicating that the methyl group underwent complete oxidative decomposition at 550 °C. The Si–O–Si stretching vibration, and characteristic SiO2, and Si–O–Si swing vibration peaks observed near 1008, 790 and 472 cm−1, respectively, indicated that the coating was composed of a Si–O–Si connected crosslinked network structure.31,32 To further determine the composition of the coating, the gel solution was analysed through XRD after heat treatment at 550 °C for 2 h (Fig. 4). The result showed only one peak at 2θ = 22°, which confirmed that the coating contained amorphous silica.33,34
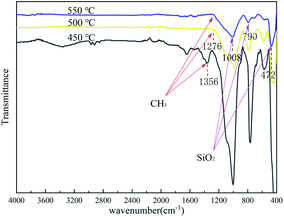 |
| | Fig. 3 FTIR of coated sample after heat-treatment at 450 °C, 500 °C and 550 °C for 2 h. | |
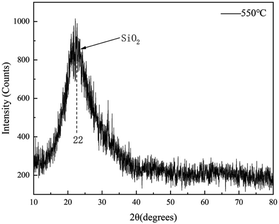 |
| | Fig. 4 XRD of coating after heat-treatment at 550 °C. | |
3.2 SEM and EDS analyses
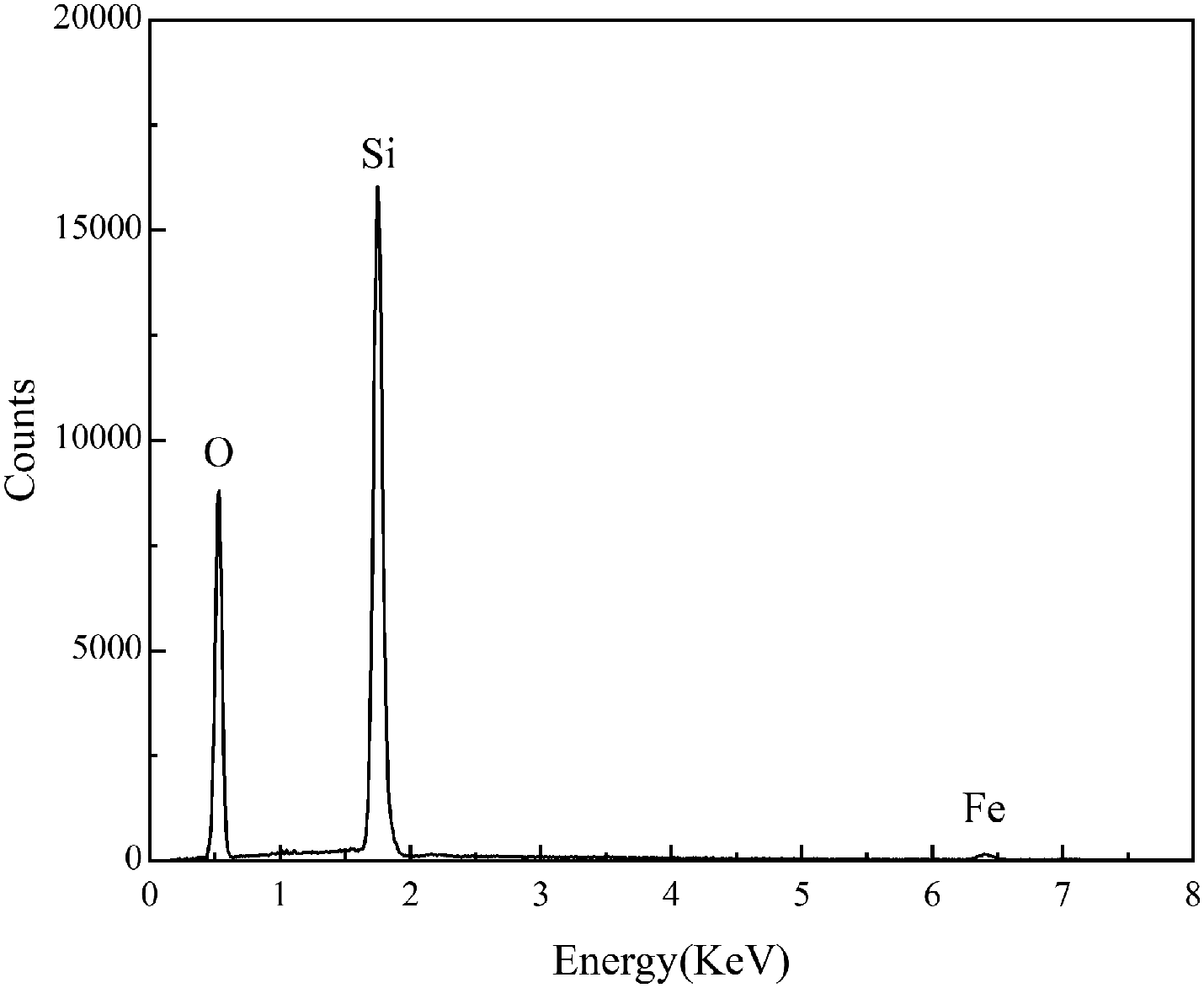
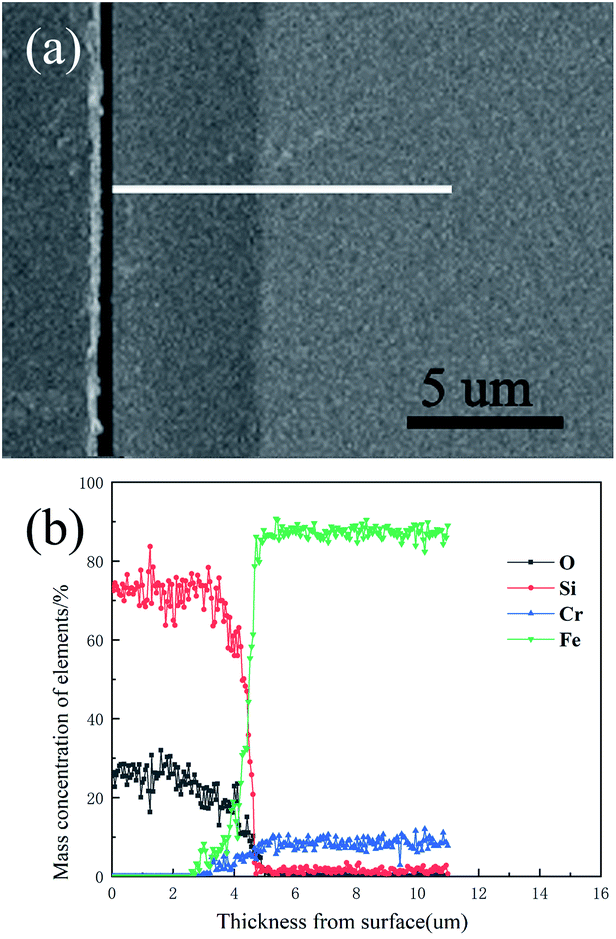
The surface morphology of the pre-treated and 550 °C heat-treated coated samples was compared using SEM (Fig. 5a–d). The coated surface was uniformly dense without cavities or cracks, and the coating covered the uneven area resulting from grinding with the 2000 grit sandpaper. The EDS results (Fig. 6) revealed the main chemical components of the coating as O and Si. The Fe signal originated from the substrate. Fig. 7 shows the SEM morphology and EDS maps of the coating surface. Si and O were uniformly distributed in the coating. The coating was tightly bonded to the substrate, and the measured coating thickness was approximately 4 μm (Fig. 8a). Along the direction perpendicular to the coating, the coating surface was free of Fe and Cr, and the surface silicon and oxygen content was relatively higher, indicating that the substrate surface was completely covered with the coating (Fig. 8b). As the depth increased, the Fe and Cr content gradually increased and a transition layer appeared. According to the mechanism of coating formation, the gradual increase in Fe and Cr content may be caused by the formation of oxide and Si–O–M covalent bonds at the junction of the coating and substrate.35–39 The transition layer tightly combined the coating and metal substrate, which can effectively prevent the coating from being peeled off during use.
 |
| | Fig. 5 SEM morphology of the uncoated sample and coated sample: (a and b) uncoated sample; (c and d) 550 °C heat-treated coated sample. | |
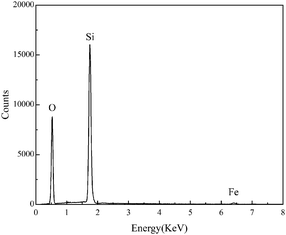 |
| | Fig. 6 EDS analysis of the surface of the 550 °C heat-treated coated sample. | |
 |
| | Fig. 7 SEM morphology and EDS maps of the surface of the 550 °C heat-treated coated sample. | |
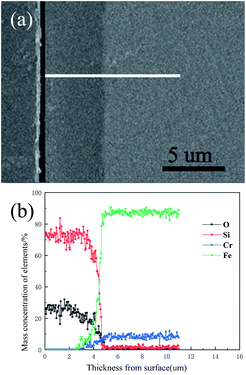 |
| | Fig. 8 Cross-sectional morphology and elemental distribution of the 550 °C heat-treated coated sample: (a) cross-sectional morphology; (b) elemental distribution. | |
3.3 Surface roughness analyses
On the inner surface of the heating furnace tubes, the surface roughness an important factor that affects the transportation, adhesion, and removal of coke.40,41 The surface roughness of the uncoated sample after pre-treatment with 400–2000 grit sandpaper (Fig. 9a) and that of the coated sample after heat treatment at 550 °C (Fig. 9b) were analysed using InfiniteFocus optical 3D surface metrology. The microscope can capture lateral and longitudinal resolutions of 400 and 10 nm, respectively. After the substrate was coated, its roughness reduced from 0.417 to 0.199 μm (Table 2), and only small protrusions and depressions were present on the coating surface because of the evaporation and condensation of the gel liquid during the heat treatment.42 The lower the roughness is, the smoother the surface is. Hence, coke is difficult to grow and deposit on the surface, which results in a thin flow boundary layer, and coke is easily carried away by the fluid shear stress.
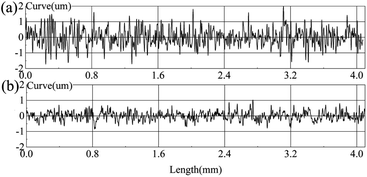 |
| | Fig. 9 Surface roughness of the uncoated sample and coated sample: (a) uncoated sample after treatment with 400–2000 grit sandpaper; (b) coated sample after heat treatment at 550 °C. | |
Table 2 Composition of the vacuum residue
| Status |
Ra/μm |
Rq/μm |
Rz/μm |
| Substrate |
0.417 |
0.533 |
3.230 |
| Coating |
0.199 |
0.255 |
1.447 |
3.4 Service resistibility analysis
The service resistibility of coatings depends on their structural quality. When the furnace tube is subjected to a temperature change, such as start-up and stop-down, stresses are produced that may cause coatings to rupture. Fig. 10 shows the SEM morphology of the coated samples after 20 cycles of thermal shock from 500 to 25 °C. In this process, the sample was repeatedly heated in a muffle furnace for 30 min and taken out to cool down to room temperature at about 25 °C. After 20 cycles of thermal shock experiment, the coated sample was still uniformly dense without any obvious cracks on the surface (Fig. 10a), and the coating remained tightly adhered to the substrate (Fig. 10b). The coating can withstand the temperature changes in the delayed coking production process.
 |
| | Fig. 10 SEM morphology of the coated sample after 20 cycles of thermal shock circulation: (a) surface of the coated sample; (b) cross-section of the coated sample. | |
3.5 Electrochemical performance analysis
The presence of sulphides in the residue is likely to result in the corrosion of the walls of the heating furnace tube, thereby causing coke to deposit on the furnace tube surface and making it difficult to peel off the coke.4 Therefore, the corrosion potential and current were obtained using polarisation curves to analyse the electrochemical performance of the coating. A Princeton electrochemical test system (PARSTAT-2273) was used to determine the corrosion resistance of the samples (Fig. 11). The experiment adopted a three-electrode system, and the coated or uncoated sample was used as the working electrode. A platinum electrode was used as the auxiliary electrode, and a saturated calomel battery (SCE) was used as the reference electrode. The area exposed to the test solution was 1 cm2. The measured potential was relative to the SCE potential. The test medium was a mixed solution of 0.35% (NH4)2SO4 and 0.05% NaCl. The scanning range and speed were −0.8 to 0 V and 1 mV s−1, respectively. A comparison between the results showed that the corrosion potential of the coated substrate substantially moved in the positive direction. The self-corrosion current density and polarisation resistance of the coated substrate decreased and increased, respectively, indicating that electron transfer on the electrode surface and alloy dissolution corrosion were difficult to achieve. The coating effectively improved the corrosion resistance of the substrate and provided a better protection effect to a certain extent for the substrate in the residue environment.
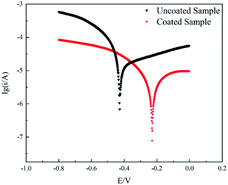 |
| | Fig. 11 Polarization curves of uncoated and coated samples. | |
3.6 Anti-coking performance of SiO2 coatings
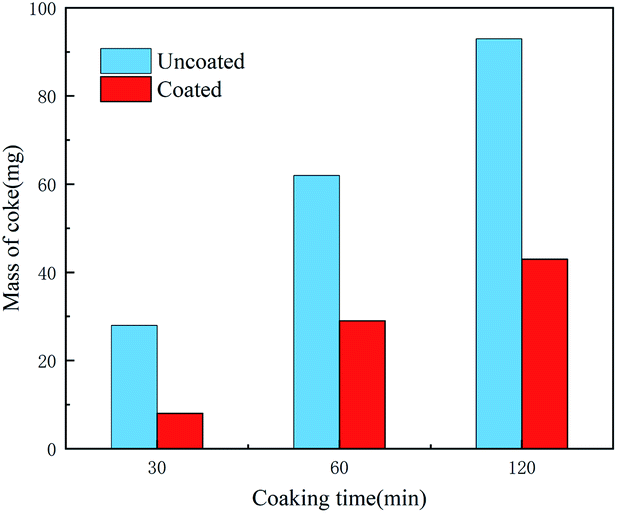
Coking experiments were performed on both the coated and uncoated samples. Fig. 12 shows the SEM morphology of the samples after different coking times and Fig. 13 shows the coke amount deposited on the samples. For the coking time of 30 min, the coke amount present on the coated sample surface is considerably lower than that on the uncoated sample surface. The uncoated sample was severely coked with a large amount of coke. Coke present on the coated sample (Fig. 12b) fell off and that on the uncoated sample (Fig. 12a) remained firmly adhered to the substrate. This may be because the sulphide present in the residue formed a sulphide corrosion layer on the metal surface and provided a basis for coke to adhere to the pipe walls.30 In contrast, the coated samples revealed a bare silica coating. Lesser coke adhered onto the surface of the coated samples, indicating that the coating eliminated metal sulphuration and coke adhesion. Therefore, the coating surface cannot form a sulphide corrosion layer, and coke can be easily peeled off with a flow of residual oil. With an increase in the coking time, coke gradually accumulated on the uncoated sample surface and became hard (Fig. 12c and e). The removal of this coke from the base surface was difficult. The coke amount of the coated sample also increased, but the coke layer was thin and cracked (Fig. 12d and f). Peeling began to occur due to the internal stress caused by the temperature difference, which further confirmed that the coating can substantially reduce coke adhesion. A comparison of the XRD results of the uncoated and coated samples (Fig. 14) revealed that the coke of the uncoated sample contained iron sulphide and that of the coated sample did not.4 This finding indicated that the iron sulphide reaction was difficult on the coated sample. It was difficult for coke to adhere to the surface and coke was easily removed along with the flowing oil.30 Thus, the SiO2 coating has a good coking inhibition ability.
 |
| | Fig. 12 SEM morphology of the uncoated sample coking for: (a) 30 min; (c) 60 min; (e) 120 min. SEM morphology of the coated sample coking for: (b) 30 min; (d) 60 min; (f) 120 min. | |
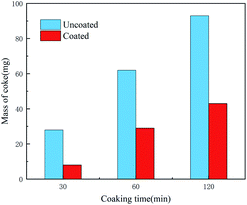 |
| | Fig. 13 Coking amount of coated and uncoated sample after deferent coking time. | |
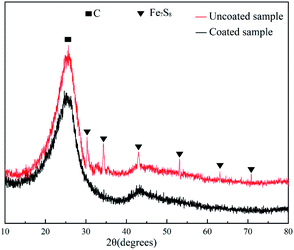 |
| | Fig. 14 XRD of coking on the surface of the uncoated sample and the coated sample at 500 °C for 120 min. | |
4 Conclusions
Metal sulphide and coke adhesion reactions can lead to carbon deposit formation in delayed coking furnaces. In this work, a uniformly dense SiO2 coating was prepared on Cr9Mo alloy using the sol–gel method. This coating effectively inhibited coke fouling in a delayed coking furnace.
The structural results (FTIR and XRD) evidenced that the silicon methyl groups of the coating oxidised at 450 to 550 °C and that the coating was composed of a Si–O–Si connected crosslinked network structure after heat treatment at 550 °C. Morphological tests using SEM, EDS and InfiniteFocus optical 3D surface metrology showed that the 4 μm coating was uniformly dense and smooth and was firmly bound to the substrate. The coating was composed of silicon and oxygen and isolated the substrate from the residue. The coating withstood 20 cycles of thermal shock treatment from 500 °C to 25 °C and was still uniformly dense and tightly adhered to the substrate. According to the PDS results, compared with the uncoated sample, the corrosion potential of the coated samples increased from −0.4 to −0.23 V. The coating effectively improved the corrosion resistance of the substrate.
Coking experiments were performed on both coated and uncoated samples at 500 °C. The coke present on the surface of the uncoated samples was hard, and this coke contained iron sulphide. The coke present on the surface of the coated sample was brittle and was easily peeled off from the coating. The SiO2 coating has better coking suppression performance in delayed coking heating furnaces.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by Engineering Center of Efficient Green Process Equipment and Energy Conservation: no. 2011007411009, Ministry of Education and the Project of China Petroleum & Chemical Corporation: no. 116060.
Notes and references
- H. P. Halim, J. S. Im and C. W. Lee, Carbon Lett., 2013, 14, 152–161 CrossRef.
- M. S. Rana, V. Sámano, J. Ancheyta and J. A. I. Diaz, Fuel, 2007, 86, 1216–1231 CrossRef CAS.
- A. N. Sawarkar, A. B. Pandit, S. D. Samant and J. B. Joshi, Can. J. Chem. Eng., 2010, 85, 1–24 CrossRef.
- H. Bisht, V. S. Balachandran, M. Patel, G. D. Sharma, A. H. Yadav, D. D. Biswas, S. Pacharu, S. Mandal and A. K. Das, Fuel Process. Technol., 2018, 172, 133–141 CrossRef CAS.
- S. Rahmani, W. Mccaffrey and M. R. Gray, Energy Fuels, 2001, 16, 177–182 Search PubMed.
- A. H. Alshareef, A. Scherer, X. Tan, K. Azyat, J. M. Stryker, R. R. Tykwinski and M. R. Gray, Energy Fuels, 2011, 25, 2130–2136 CrossRef CAS.
- H. K. Lemke and W. K. Stephenson, Pet. Sci. Technol., 1998, 16, 335–360 CrossRef CAS.
- M. Derakhshesh, P. Eaton, B. Newman, A. Hoff, D. Mitlin and M. R. Gray, Energy Fuels, 2013, 27, 1856–1864 CrossRef CAS.
- K. Fujimoto, M. Sato, M. Yamada, R. Yamashit and K. Shibat, Carbon, 1986, 24, 397–401 CrossRef CAS.
- M. Hazelton, T. Stephenson, J. Lepore, V. Subramani and D. Mitlin, Fuel, 2015, 160, 479–489 CrossRef CAS.
- N. P. Zambrano, L. J. Duarte, J. C. Poveda-Jaramillo, H. J. Picón, F. M. Ortega and M. E. Niño-Gómez, Energy Fuels, 2017, 32, 2722–2732 CrossRef.
- A. Niaei, D. Salari, J. Towfighi, P. Panahi and R. Nabavi, Pet. Sci. Technol., 2008, 26, 2170–2181 CrossRef CAS.
- M. Alhreez and D. S. Wen, RSC Adv., 2019, 9, 19560–19570 RSC.
- J. D. Wang, M. F. Reyniers, K. M. V. Geem and B. Marin, Ind. Eng. Chem. Res., 2008, 47, 1468–1482 CrossRef CAS.
- C. L. Su, H. Y. Shi, P. Li and J. T. Cao, Meas. Control, 2015, 48, 54–59 CrossRef.
- F. Rodríguez-Reinoso, P. Santana, E. R. Palazon, M. A. Diez and H. Marsha, Carbon, 1998, 36, 105–116 CrossRef.
- B. B. Bao, J. L. Liu, H. Xu, Z. Y. Wang and K. Zhang, Braz. J. Chem. Eng., 2018, 35, 721–730 CrossRef CAS.
- B. Wang, S. Wang, B. Liu, Q. Zhu and X. Y. Li, Surf. Coat. Technol., 2019, 378, 124952 CrossRef CAS.
- S. Kukes, R. J. Harris, A. R. Porter and R. E. Larry, US Pat., 4410418, 1983 Search PubMed.
- Y. Zhang, S. Zhang, T. Zhang, T. X. Zou, Q. Zhu, J. L. Wang and X. Y. Li, Surf. Coat. Technol., 2016, 296, 108–116 CrossRef CAS.
- L. Ma, W. J. Lv, Q. Yang and Y. Fan, Mater. Res. Innovations, 2014, 18, 527–531 Search PubMed.
- C. H. Yang, G. Z. Liu, X. Q. Wang, R. P. Jiang, L. Wang and X. W. Zhang, Ind. Eng. Chem. Res., 2012, 51, 1256–1263 CrossRef CAS.
- S. Y. Tang, S. Gao, S. X. Wang, J. L. Wang, Q. Zhu, Y. Q. Chen and X. Y Li, Surf. Coat. Technol., 2014, 258, 1060–1067 CrossRef CAS.
- S. Y. Tang, S. Gao, S. W. Hu, J. L. Wang, Q. Zhu, Y. Q. Chen and X. Y. Li, Ind. Eng. Chem. Res., 2014, 53, 5432–5442 CrossRef CAS.
- J. X. Zhou, H. Xu, J. L. Liu and Z. M. Jiang, Mater. Lett., 2007, 61, 5087–5090 CrossRef CAS.
- R. Pelham, M. Spears and M. Pozvonkov, NPRA Annu. Meeting, Phoenix, 2010, pp. 1–30 Search PubMed.
- T. Stephenson, A. Kubis, M. Derakhshesh, M. Hazelton, C. Holt, P. Eaton, B. Newman, A. Hoff, M. Gray and D. Mitlin, Energy Fuels, 2011, 25, 4540–4551 CrossRef CAS.
- W. Wang and A. P. Watkinson, Heat Transfer Eng., 2015, 36, 623–631 CrossRef CAS.
- W. Wang and A. P. Watkinson, Proc Int. Conf. on Heat Exch. Fouling and Clean. IX, Crete Island, 2011, pp. 23–30 Search PubMed.
- I. A. Wiehe, Process Chemistry of Petroleum Macromolecules, CRC Press, 2008 Search PubMed.
- A. Fidalgo and L. M. Ilharco, J. Non-Cryst. Solids, 2004, 347, 128–137 CrossRef CAS.
- D. Balgude and A. Sabnis, J. Sol-Gel Sci. Technol., 2012, 64, 124–134 CrossRef CAS.
- M. Momeni, H. Saghafian, F. Golestani-Fard, N. Barati and A. Khanahmadi, Appl. Surf. Sci., 2017, 392, 80–87 CrossRef CAS.
- K. Kato, J. Mater. Sci., 1992, 27, 1445–1448 CrossRef CAS.
- D. Xiong, J. Cheng, H. Li, W. Deng and K. Ye, Microelectron. Eng., 2010, 87, 1741–1746 CrossRef CAS.
- D. Balgude and A. Sabnis, J. Sol-Gel Sci. Technol., 2012, 64, 124–134 CrossRef CAS.
- D. C. L. Vasconcelos, J. A. N. Carvalho, M. Mantel and W. L. Vasconcelos, J. Non-Cryst. Solids, 2000, 273, 135–139 CrossRef CAS.
- S. H. Zaferani, M. Peikari, D. Zaarei, I. Danaee, J. M. Fakhraei and M. Mohammadi, Corrosion, 2013, 69, 372–387 CrossRef CAS.
- E. Owczarek, Acta Phys. Pol., A, 2019, 135, 147–152 CrossRef CAS.
- J. C. Song, M. Y. Liu, X. X. Sun, J. S. Wang and J. L. Zhu, Appl. Therm. Eng., 2018, 132, 740–759 CrossRef CAS.
- S. Y. Tang, X. Y. Luo, C. Cai, J. L. Wang and A. J. Tang, Energy Fuels, 2018, 32, 1223–1229 CrossRef CAS.
- T. Y. Liu, L. G. Teoh, C. K. Huang, C. K. Huang and Y. C, Ind. Eng. Chem. Res., 1995, 34, 773–785 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Xiangxiang Wang,
Hao Wang,
Hong Xu and
Jiangfeng Yang
*,
Xiangxiang Wang,
Hao Wang,
Hong Xu and
Jiangfeng Yang