
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium(III) catalyzed olefination and deuteration of tetrahydrocarbazole†
Wan Peng,
Qiaohong Liu,
Fucheng Yin,
Cunjian Shi,
Limei Ji,
Lailiang Qu,
Cheng Wang,
Heng Luo,
Lingyi Kong * and
Xiaobing Wang*
* and
Xiaobing Wang*
Jiangsu Key Laboratory of Bioactive Natural Product Research, State Key Laboratory of Natural Medicines, School of Traditional Chinese Pharmacy, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, People's Republic of China. E-mail: xbwang@cpu.edu.cn; cpu_lykong@126.com; Fax: +86-25-83271405; Tel: +86-25-83271405
First published on 23rd February 2021
Abstract
The rhodium-catalyzed olefination and deuteration of tetrahydrocarbazoles in water with the aid of an N,N-dimethylcarbamoyl-protected group is presented. This olefination method features a broad substrate scope, good functional-group tolerance, and high efficiency in water. Practical applications of the protocol are illustrated by the synthesis of various evodiamine derivatives. As such, this environmentally friendly approach to directly modify natural products will attract much attention in academic and industrial research.
Introduction
C–H functionalization strategies, which emerged as an increasingly powerful synthetic method, can functionalize the inactivated C–H bond or C–C bond directly, but provide high reaction efficiency and atom economy.1 Rhodium catalysts have become quite popular in this field due to their high efficiency, good regioselectivity, and wide functional group tolerance.2 The tetrahydrocarbazole framework is a common subunit in many bioactive compounds, and serves as the core to a wide variety of natural products, bioactive molecules and pharmaceuticals (Scheme 1a).3 Tetrahydrocarbazole is one type of indole in which the C2 and C3 position were fused with a six-membered ring. Few studies on the C–H functionalization of tetrahydrocarbazole were reported, such as N,N-dimethylcarbamoyl-protected tetrahydrocarbazoles undergo directly oxidative to exclusively provide products from C(sp3)–H oxidation (2014).4 However, indoles and indolines mediated by organocatalysts and transition metals have made fruitful and important progress.5 For instance, alkenylation,6 arylation,7 alkylation8 and others9 of indoles and indolines at the C7 position have been described. The Oestreich group reported the palladium(II)-catalyzed arylation of 2,3-substituted indolines (Scheme 1b).10 These results led us to explore whether this C8-functionalization strategy can be applied in tetrahydrocarbazoles.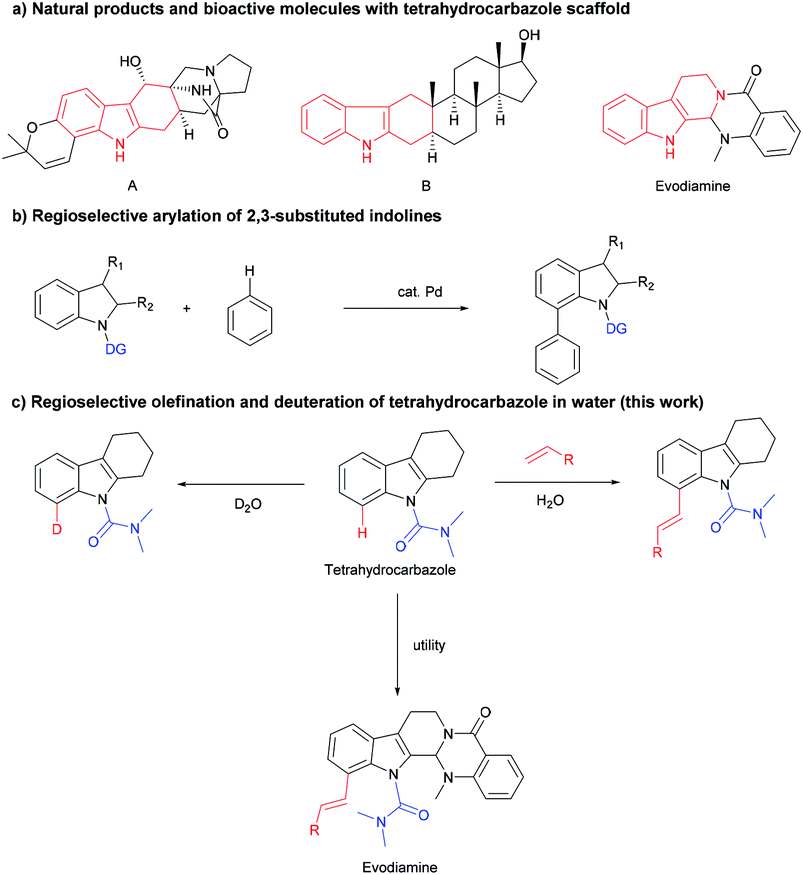 | ||
| Scheme 1 Outline of the research. | ||
The regioselective modification of natural products via the metal-catalyzed C–H bond activation strategy is difficult,11 we demonstrated an efficient rhodium-catalyzed synthetic route for regioselective olefination of tetrahydrocarbazole in water. The present protocol using water as the solvent was compatible with a wide range of substituents on the tetrahydrocarbazole substrate. Furthermore, olefinated products could also be generated in a moderate yield at a lower reaction temperature in the developed system. It was also found that regioselective alkenylation of tetrahydrocarbazole derivatives followed by one-pot subsequent deprotection provides the desired product in good yield. In addition, synthetic applications of this novel alkenylation method were further demonstrated in an efficient modification of evodiamine.
On the other hand, isotopic labelling is of high significance for their utility, e.g. for investigating reaction mechanisms,12 exploring the ADMET properties of existing drug candidates,13 or in nuclear magnetic resonance spectroscopy14 and mass spectrometry.15 Since the FDA approved deutetrabenazine, which is the first deuterated drug in 2017,16 deuterium has received unprecedented widespread attention. Versatile applications of deuterium-labelled compounds demand effective synthetic methods to regioselectively introduce deuterium atoms at specific positions. Hydrogen isotope exchange (HIE) assisted by metal-catalyzed C–H activation has become a widely utilized and elegant method for synthesizing isotopically labelled compounds, allowing direct and selective late-stage incorporation of deuterium or tritium (2H or 3H).17 Inspired by our above developed rhodium-catalyzed regio-olefination, we have also successfully achieved deuteration labelling of tetrahydrocarbazole with D2O, which serves as an economical deuterium source and solvent in this reaction.
Results and discussion
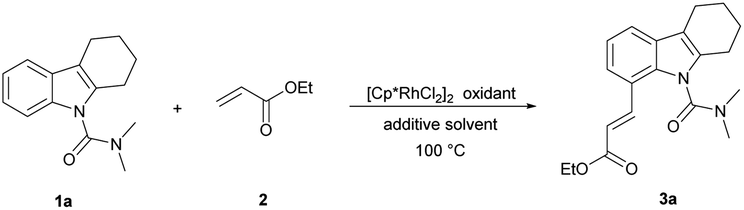
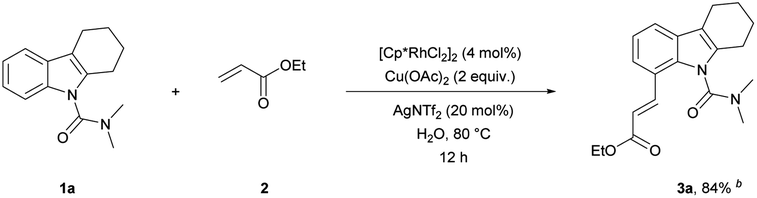
Initially, we started this idea using N,N-dimethylcarbamoyl-protected18a tetrahydrocarbazole 1a and ethyl acrylate 2 as the template substrates. To our delight, the desired olefinated product 3a was obtained in good yield (75%) under the conditions of 5 mol% [Cp*RhCl2]2, 20 mol% AgSbF6, and 3 equiv. of Cu(OAc)2 in DCE at 100 °C for 12 h (Table 1, entry 1). Then, through evaluating different types of oxidants, Cu(OAc)2 was found to be superior to the rest (entries 2–5).18a Thereafter, further screening of reaction solvents revealed that DCE showed the best performance in this reaction using AgSbF6 as an additive (entries 6–9). Addition of AgNTf2 in place of AgSbF6 could improve the yield of 3a (entry 10).18b Further shortening or extending the reaction time offered no improvement to the yield (Table S1 in the ESI†). Moreover, decreasing the reaction temperature to 80 °C was beneficial to the olefination, and the product 3a was obtained in 93% yield (entry 11 and Table S2†). To our delight, we accidentally found that the reaction proceeded smoothly in water without obvious decrease in the yield (entry 12). Considering the cost and environmental factor, we decided to employ the reaction using water as the solvent. Lowering the amount of catalyst, oxidant, additive and ethyl acrylate (Tables S3–S5†), we got the final optimized reaction conditions (entry 13). In addition, we also found that this reaction could proceed and provided the desired alkenylated product 3a in moderate yield (78%) when the temperature was decreased to 60 °C (entry 14). 3a was observed with a yield of 10% when the reaction was carried out without an oxidant (entry 15). The reaction hardly happened without an additive (entry 16). These results showed that the oxidant and additive had crucial roles in this olefination.
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Solvent | Additive | Yieldb (%) |
| a 1a (0.1 mmol, 1 equiv.), 2 (5 equiv.), [Cp*RhCl2]2 (5 mol%), AgSbF6 (20 mol%), Cu(OAc)2 (3 equiv.), solvent (1.0 mL), 100 °C, 12 h.b Data were obtained by 1H NMR analysis with CH2Br2 as reference.c 80 °C.d 1a (0.1 mmol, 1 equiv.), 2 (2 equiv.), [Cp*RhCl2]2 (4 mol%), AgSbF6 (20 mol%), Cu(OAc)2 (2 equiv.), H2O (1.0 mL), 80 °C, 12 h.e 60 °C. DCE = dichloroethane, BQ = benzoquinone, PhCF3 = benzotrifluoride, CH3CN = acetonitrile, t-AmOH = 2-methyl-2-butanol. | ||||
| 1 | Cu(OAc)2 | DCE | AgSbF6 | 75 |
| 2 | AgOAc | DCE | AgSbF6 | Trace |
| 3 | BQ | DCE | AgSbF6 | Trace |
| 4 | PhI(OAc)2 | DCE | AgSbF6 | — |
| 5 | Ag2CO3 | DCE | AgSbF6 | 28 |
| 6 | Cu(OAc)2 | Dioxane | AgSbF6 | 40 |
| 7 | Cu(OAc)2 | PhCF3 | AgSbF6 | 61 |
| 8 | Cu(OAc)2 | CH3CN | AgSbF6 | — |
| 9 | Cu(OAc)2 | t-AmOH | AgSbF6 | 17 |
| 10 | Cu(OAc)2 | DCE | AgNTf2 | 86 |
| 11c | Cu(OAc)2 | DCE | AgNTf2 | 93 |
| 12c | Cu(OAc)2 | H2O | AgNTf2 | 90 |
| 13d | Cu(OAc)2 | H2O | AgNTf2 | 86 |
| 14d,e | Cu(OAc)2 | H2O | AgNTf2 | 78 |
| 15 | — | H2O | AgNTf2 | 10 |
| 16 | Cu(OAc)2 | H2O | — | Trace |
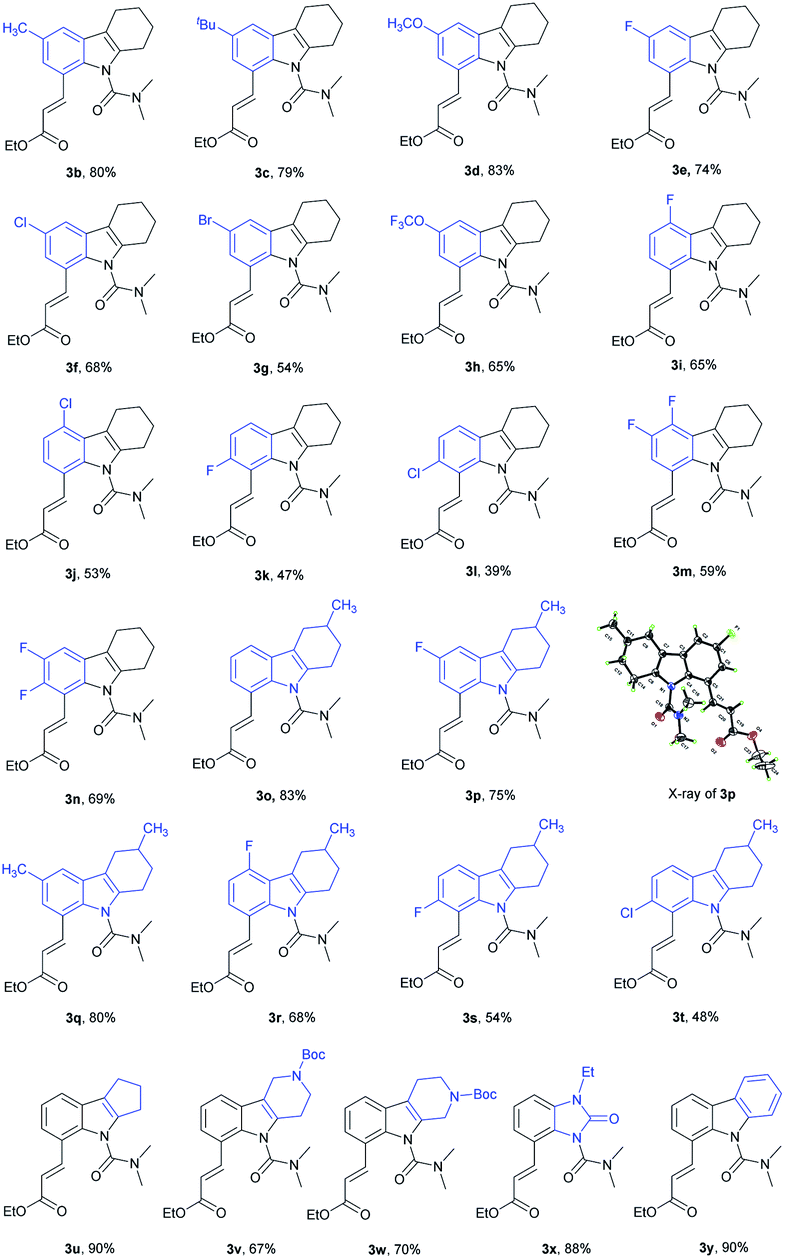
With the optimal reaction conditions established, we next explored olefination with a variety of substituted tetrahydrocarbazoles (Table 2). For C6-substituted substrates, moderate to good yields were obtained with both electron-donating (3b, 3c) and electron-withdrawing groups (3d–3h) compared to C5-substituted substrates (3i, 3j) and C7-substituted substrates (3k–3m). It is noteworthy that the halogen groups such as F (3e, 3n) and Cl (3f) performed well in C8-olefination. In comparison with substrates bearing electron-donating groups (–CH3, –C(CH3)3), those bearing electron-withdrawing groups (–F, –Cl and –Br) decreased the overall yields, which might imply an electrophilic activation pattern. We were pleased to find that tetrahydrocarbazoles containing a methyl group at the six-membered ring were olefinated in good yields, with good selectivity (3o–3t) and did not affect the efficiency of this process. The size of the aliphatic ring was well tolerated (3u). The presence of an additional piperidine ring was also tolerated (3v, 3w), providing a moderate yield (67%). Notably, a fluzoxazone analogue was embedded in drug candidates in excellent yield (3x). Interestingly, medicinally useful carbazole was alkenylated to give the mono-olefinated product 3y in good yield.
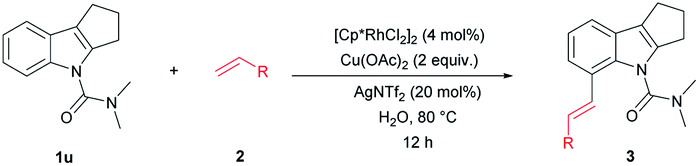
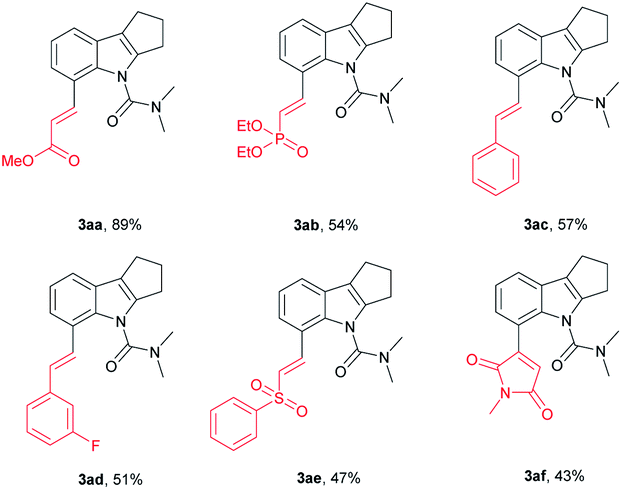
We further investigated whether the protocol could be applied to a wide range of olefin coupling partners (Table 3). Methyl acrylate (2a), phosphonate (2b), styrene (2c), 3-fluorostyrene (2d) and phenyl vinyl sulfone (2e) were effectively coupled with 1u to furnish the corresponding products (3aa, 3ab, 3ac, 3ad and 3ae) in moderate to good yields (47–89%), respectively. In addition, maleimide was also successfully coupled in moderate yield (3af).
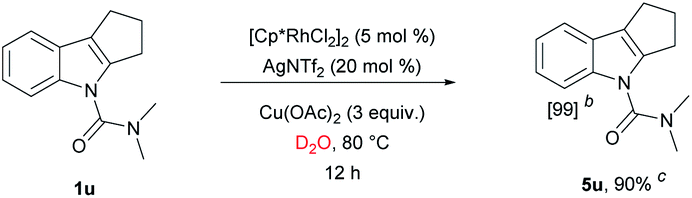
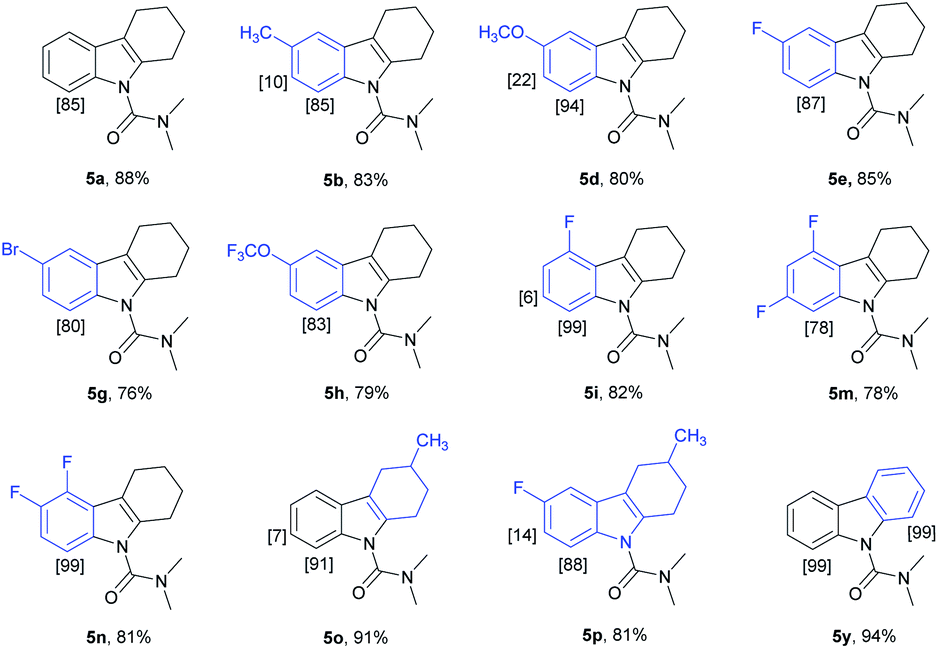
We have established a method for the olefination of tetrahydrocarbazole in water. This approach led us to explore whether this functionalization strategy could be applied for deuteration. We studied the selective deuteration process of tetrahydrocarbazole with substrate 1u as the initial substrate. On the basis of olefination conditions, different deuterium-containing solvents (D2O, [D4]-methanol, [D1]-TFA, CDCl3) were tested (Table S6 in the ESI†).19 To our delight, 1u was proceeded with 5 mol% [Cp*RhCl2]2, 20 mol% AgNTf2, and 3 equiv. Of Cu(OAc)2, providing deuterated product 5u in 90% yield. This result was very meaningful as D2O acted as not only a deuterium provider, but the solvent of the reaction, and encouraged us to explore the utility of this method. Gratifyingly, most substrates were compatible, which greatly enhanced the scope of the method (Table 4). Under the reaction condition, the CH3–, CH3O–, F–, Cl– and Br–substituted at the C6-position (5b, 5d–5e, 5g) could be smoothly deuterated. The F-substituent at C-5 (5i, 5n) had little influence on the results for the deuteration rates and gave more than 99% D incorporation. Carbazole was also shown to exhibit high levels of reactivity.
Next, the practicality of this protocol was examined. As shown in Scheme 2a, the reaction was scalable in normal reaction equipment and gave the olefinated products (52% and 80%) using water and DCE as solvent, respectively. Compounds 3u and 5y were further deprotected under basic conditions to provide the corresponding products in 75% and 89% yield (Scheme 2b). In order to improve the synthetic utility of this protocol and avoid the tedious purification, we next pursued the direct synthesis of 3u′ by a one-pot method. Following rhodium-catalyzed olefination, the product was filtered out through a celite pad without additional purification. Then, the directing group was removed, and the desired product 3u′ was obtained in 71% yield (Scheme 2c), which further demonstrated the applicability of the method.
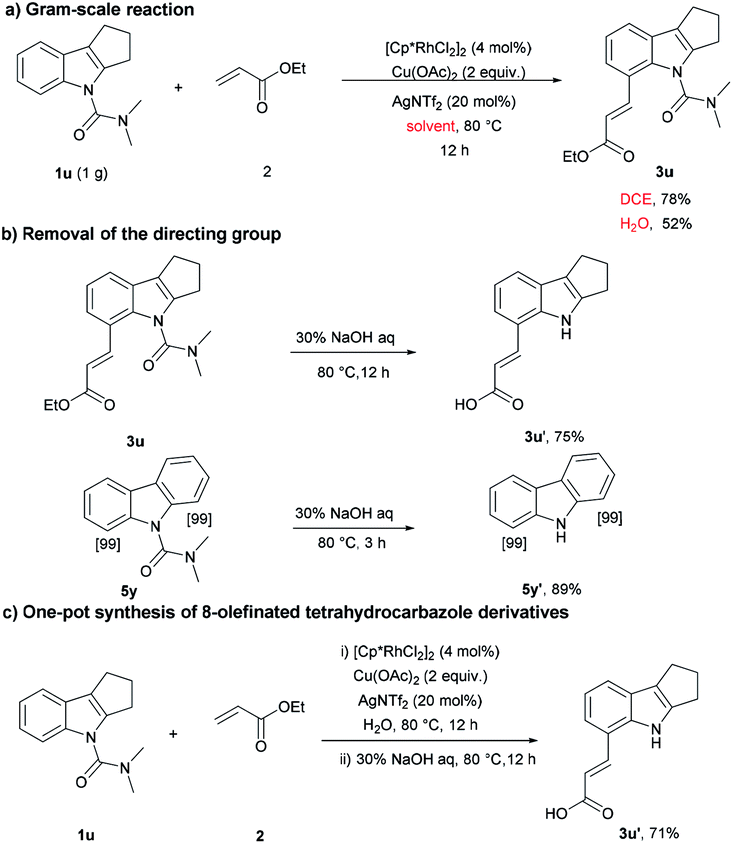 | ||
| Scheme 2 The utility of this method. | ||
An application of this novel methodology on the olefination of a more complex natural product evodiamine was tested (Scheme 3). The method was used to directly modify evodiamine and obtain evodiamine derivatives (4a–4d) with moderate yields.
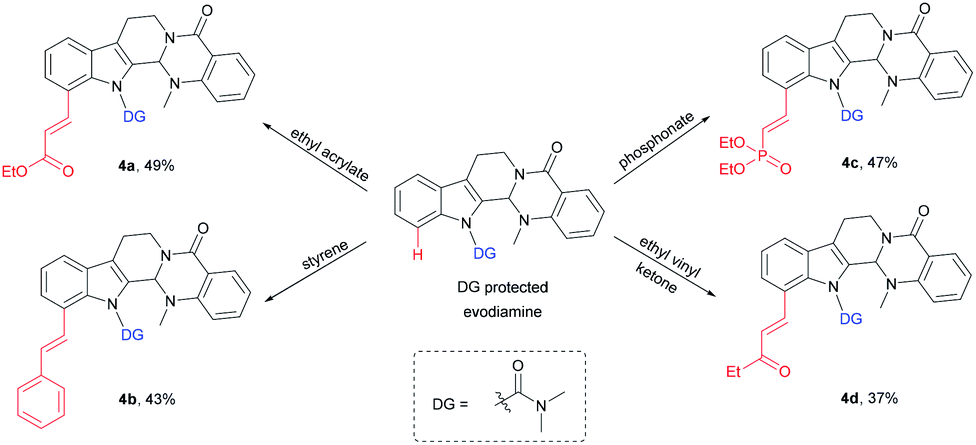 | ||
| Scheme 3 Modification of natural product evodiamine. | ||
To perceive the reaction mechanism, the following experiments were performed (Scheme 4).20 First, N,N-dimethyl-carbamoyl-protected tetrahydrocarbazole 1a under the reaction condition with 10 equiv. of D2O gave a C8-deuterated compound in 60%, indicating the reversibility of the C–H activation step. Next, an intermolecular competition experiment between electronically diverse tetrahydrocarbazoles led to the conclusion that an electron-rich substrate tended to react at a higher rate. Subsequently, radical trapping experiments were explored. The yield of the desired compound decreased remarkably with the addition of the radical scavengers. Adding different proportions of free radical scavenger had almost no obvious change, and the yield was similar to the variable without adding oxidant. These results suggest that a radical should not be involved in the reaction and the added radical scavengers may destroy the properties of the oxidant copper acetate.
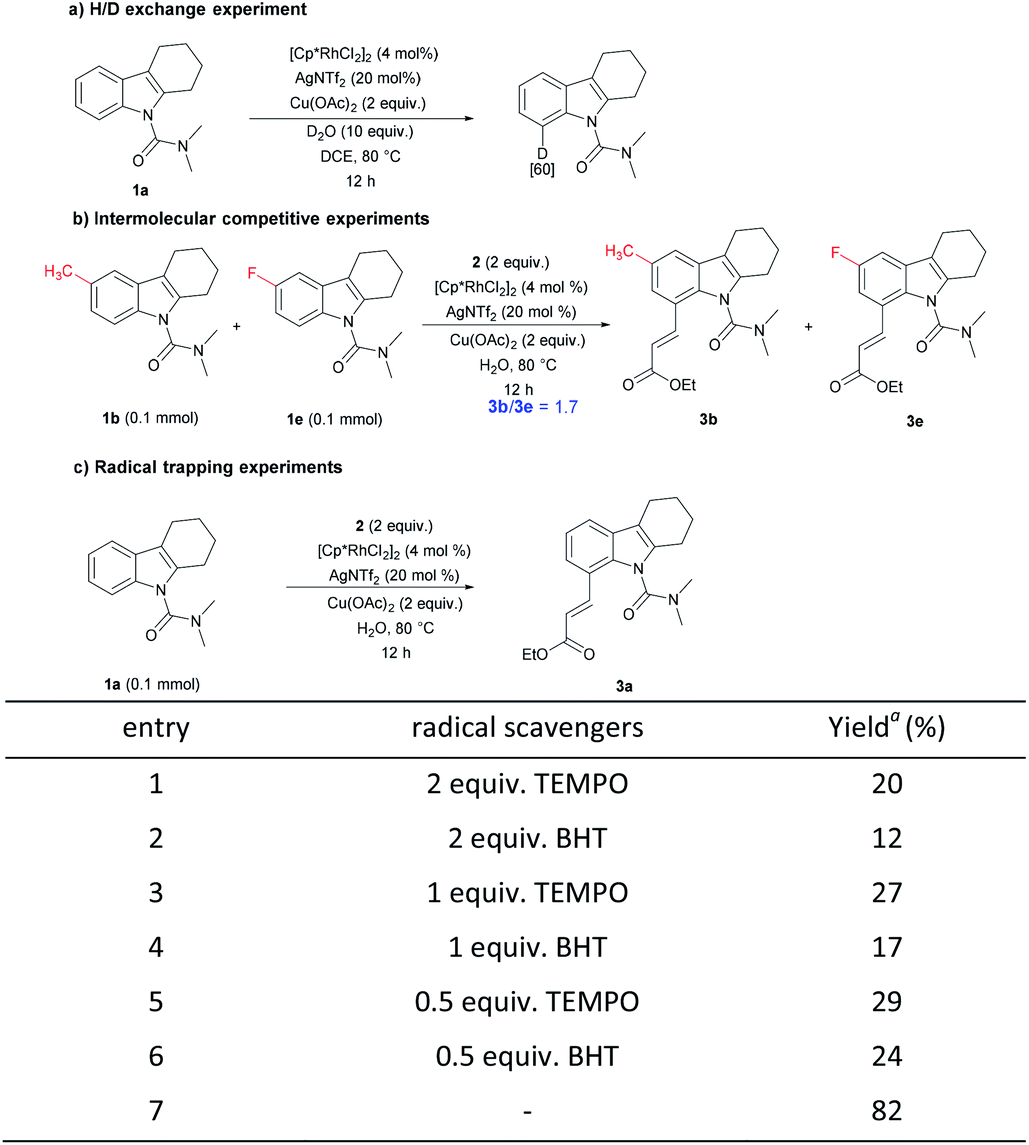 | ||
| Scheme 4 Mechanistic studies. a Data were obtained by 1H NMR analysis with CH2Br2 as reference. BHT = 2,6-di-tert-butyl-4-methylphenol, TEMPO = 2,2,6,6-tetramethylpiperidinooxy. | ||
To develop a better understanding of the catalyst reaction state, on-line 1H NMR experiments were carried out (Fig. 1). There was a slight change in the peak position of the substrate after adding the catalyst [Cp*RhCl2]2. After adding sodium acetate, the peak shape of the substrate changed significantly.21a,b We speculate that the -OAc group promotes the weak coupling between the catalyst and the substrate, resulting in the change of the peak, producing the intermediate I′. These data indicated that intermediate I′ may be the reaction state of the catalyst in the system. Considering that C8-olefination could occur in DCE using Ag2CO3 as the oxidant, the reaction of 1e in CDCl3 was monitored with 1H NMR spectroscopy.21c,d The 1H NMR analysis experiments were carried out in the same clean NMR tube in a continuous sequence. As shown in Fig. 2, the signals of 1e and 2 disappeared while that of product 3e increased with time. The reaction rate slowed down after 3.5 h. The contrast spectra of 3e (top trace of Fig. 2) indicated nearly no byproduct formation.
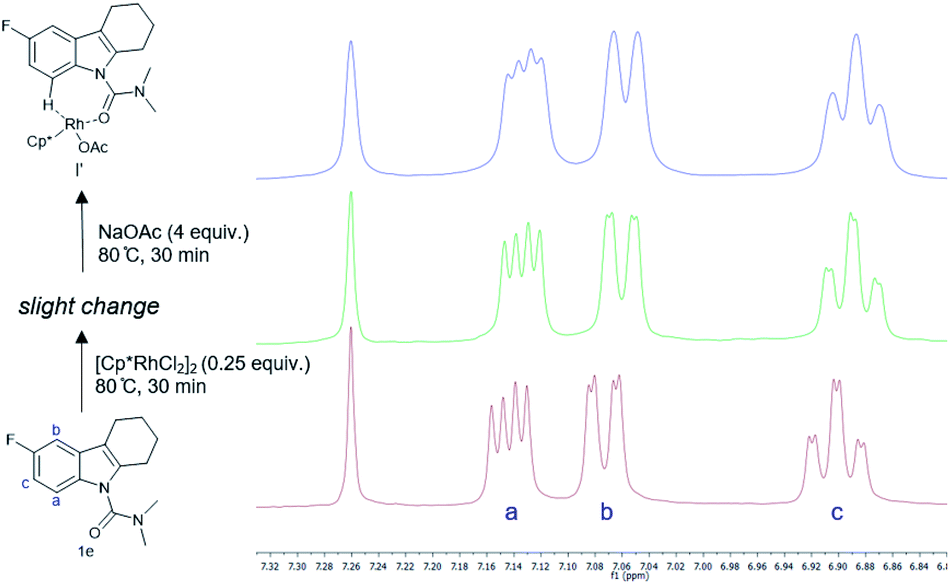 | ||
| Fig. 1 Real-time on-line 1H NMR monitoring of substrate-Rh-NaOAc interaction. | ||
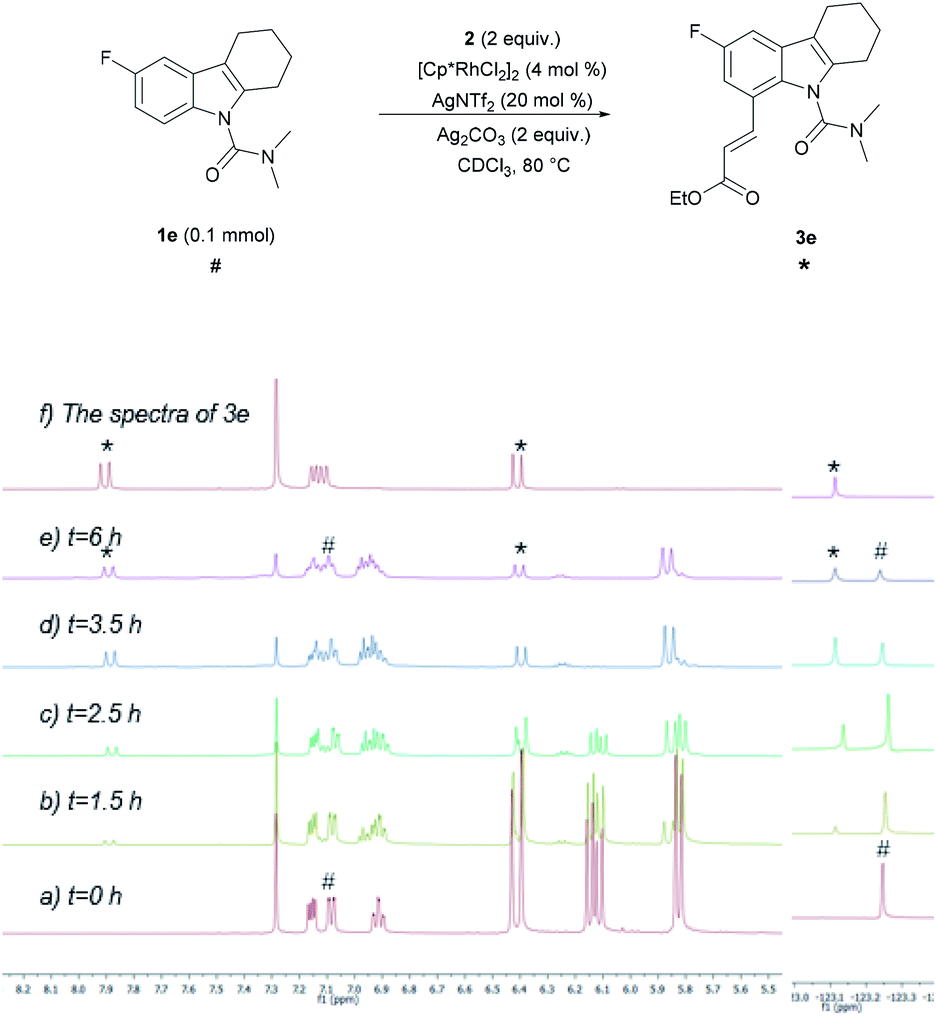 | ||
| Fig. 2 C–H olefination monitored by 1H NMR (left) and 19F NMR (right) spectroscopy. The spectrum (a) was acquired prior to heating, the spectrum (b) after heating for 1.5 h, the spectrum (c) after heating for 2.5 h, the spectrum (d) after heating for 3.5 h and the spectrum (e) after heating for 6 h. *3e (product) peaks. #1e (reactant) peaks. | ||
Based on the above mechanistic studies and precedent literature,22 we have proposed a tentative catalytic cycle for this reaction (Scheme 5). We presume that, initially, a more active cationic rhodium complex I is formed with AgNTf2 and Cu(OAc)2, followed by the C–H functionalization of substrate 1u to generate the six membered rhodacycle II. A subsequent olefin insertion step provides intermediate III. Then, rhodacycle III provides the eight-membered IV, which undergoes β-hydride elimination to continue the catalytic cycle and produces the desired olefinated product. Meanwhile, the RhIII complex regenerates RhI by reoxidation, thereby completing the mechanistic cycle. Besides, the presence of intermediates (II and III or IV) was also detected by ESI-MS supporting the plausibility of our reaction hypothesis (see the ESI†).
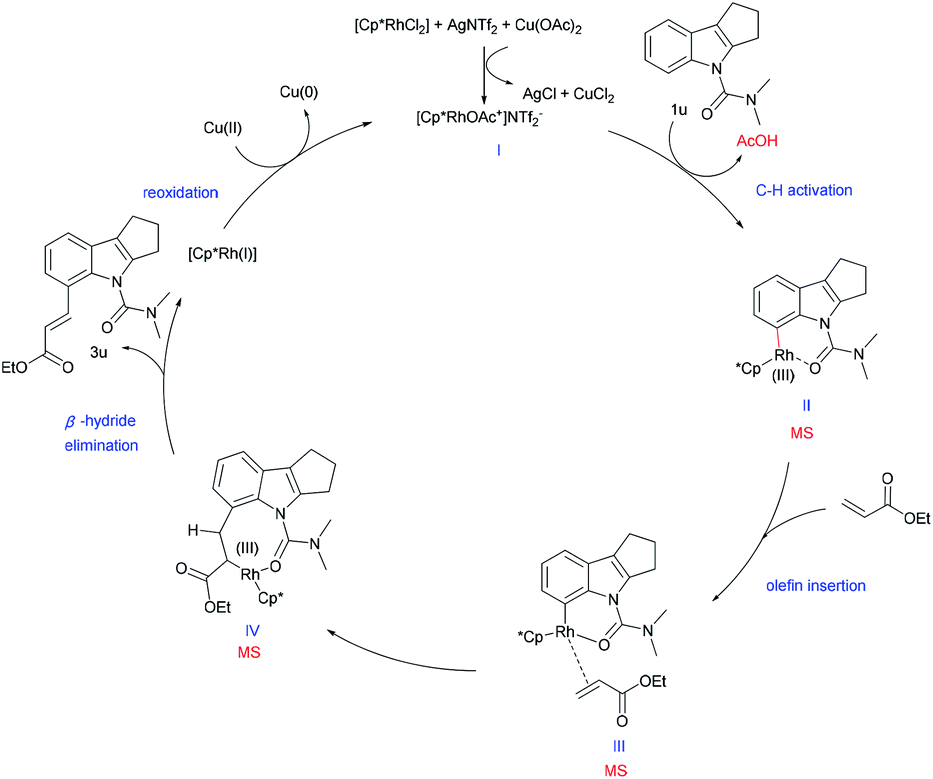 | ||
| Scheme 5 A plausible mechanistic cycle. | ||
Conclusion
In summary, we have reported an efficient, selective, direct olefination and deuteration of tetrahydrocarbazoles attached to the N,N-dimethylcarbamoyl-protected group by rhodium catalysis. Substitutions at various positions of the tetrahydrocarbazoles were tolerated. Easy removal of the template and gram–scale reaction demonstrate the extensive utility of this protocol. This method has been successfully applied to the natural product evodiamine. In this olefination reaction, water is an efficient solvent. The deuterium reaction uses D2O, which is desirable and atom economical, as the deuterium source. Exclusive site selectivity, functional group tolerance, and late-stage modifications are the important practical features. We believe that the new facile and green approach for modifying natural products could pave a new way in drug discovery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the support from the Natural Science Foundation of Jiangsu Province (Grants No BK20201332), the Six Talent Peaks Project of Jiangsu Province (Wang, X. B. SWYY-107), Jiangsu Province ‘333’ Project (Wang, X. B.), 111 Center from Ministry of Education of China and the State Administration of Foreign Experts Affairs of China (No: B18056), and the establishment and application of key technology system for obtaining and identifying effective/active small molecules of natural medicine (2017ZX09101003-001-007).Notes and references
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS; (b) S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601 RSC; (c) J. Chen, J. Xu, Y. Zhou, S. Xie, F. Gao, X. Xu, X. Xu and Z. Jin, Org. Lett., 2019, 21, 7928 CrossRef CAS; (d) B. Wang, Y. Zhou, N. Xu, X. Xu, X. Xu and Z. Jin, Org. Lett., 2019, 21, 1885 CrossRef CAS; (e) M. Shang, J. Z. Chan, M. Cao, Y. Chang, Q. Wang, B. Cook, S. Torker and M. Wasa, J. Am. Chem. Soc., 2018, 140, 10593 CrossRef CAS; (f) M. Shang, M. Cao, Q. Wang and M. Wasa, Angew. Chem., Int. Ed., 2017, 56, 13338 CrossRef CAS; (g) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- (a) C. Gao, B. Li, X. Geng, Q. Zhou, X. Zhang and X. Fan, Green Chem., 2019, 21, 5113 RSC; (b) S. Guo, Y. Liu, L. Zhao, X. Zhang and X. Fan, Org. Lett., 2019, 21, 6437 CrossRef CAS; (c) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS; (d) H. Zhang, Z. Yang, J. Liu, X. Yu, Q. Wang and Y. Wu, Org. Chem. Front., 2019, 6, 967 RSC.
- (a) J. S. Cannon and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 4288 CrossRef CAS; (b) G. Dong, C. Sheng, S. Wang, Z. Miao, J. Yao and W. Zhang, J. Med. Chem., 2010, 53, 7521 CrossRef CAS; (c) S. K. Venkatesan and V. K. Dubey, Sci. World J., 2012, 963658 Search PubMed; (d) X. Wang, J. You, J. B. King, D. R. Powell and R. H. Cichewicz, J. Nat. Prod., 2012, 75, 707 CrossRef CAS.
- Y. Nakano and D. W. Lupton, Chem. Commun., 2014, 50, 1757 RSC.
- (a) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef CAS; (b) T. A. Shah, P. B. De, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2019, 55, 572 RSC; (c) Q. Wang, C. L. Zhi, P. P. Lu, S. Liu, X. Zhu, X. Q. Hao and M. P. Song, Adv. Synth. Catal., 2019, 361, 1253 CrossRef CAS; (d) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403 CrossRef CAS.
- (a) L. Xu, C. Zhang, Y. He, L. Tan and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 321 CrossRef CAS; (b) L. Xu, L. Tan and D. Ma, Synlett, 2017, 28, 2839 CrossRef CAS; (c) B. Urones, R. G. Arrayas and J. C. Carretero, Org. Lett., 2013, 15, 1120 CrossRef CAS; (d) X.-F. Yang, X.-H. Hu, C. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 2532 RSC; (e) L. Guo, Y. Chen, R. Zhang, Q. Peng, L. Xu and X. Pan, Chem.–Asian J., 2017, 12, 289 CrossRef CAS.
- (a) X. Qiu, P. Wang, D. Wang, M. Wang, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504 CrossRef CAS; (b) Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495 CrossRef CAS; (c) Y. Yang and Z. Shi, Chem. Commun., 2018, 54, 1676 RSC.
- (a) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062 CrossRef CAS; (b) X. Zhou, S. Yu, Z. Qi, L. Kong and X. Li, J. Org. Chem., 2016, 81, 4869 CrossRef CAS.
- (a) J. Lv, X. Chen, X.-S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W.-Y. Sun and K. N. Houk anorylation, Nature, 2019, 575, 336 CrossRef CAS; (b) G. Chen, X. Zhang, R. Jia, B. Li and X. Fan, Adv. Synth. Catal., 2018, 360, 3781 CrossRef CAS; (c) S. Guo, Y. Liu, X. Zhang and X. Fan, Adv. Synth. Catal., 2020, 362, 3011 CrossRef CAS; (d) M. Chen, Y. Luo, C. Zhang, L. Guo, Q. Wang and Y. Wu, Org. Chem. Front., 2019, 6, 116 RSC; (e) Y. Kim, J. Park and S. Chang, Org. Lett., 2016, 18, 1892 CrossRef CAS.
- (a) L.-Y. Jiao, P. Smirnov and M. Oestreich, Org. Lett., 2014, 16, 6020 CrossRef CAS; (b) L.-Y. Jiao and M. Oestreich, Chem.-Eur. J., 2013, 19, 10845 CrossRef CAS.
- (a) S. Fang, X. Wang, F. Yin, P. Cai, H. Yang and L. Kong, Org. Lett., 2019, 21, 1841 CrossRef CAS; (b) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489 CrossRef CAS; (c) D. S. Peters, F. E. Romesberg and P. S. Baran, J. Am. Chem. Soc., 2018, 140, 2072 CrossRef CAS; (d) T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751 CrossRef CAS.
- (a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS; (b) V. Soulard, G. Villa, D. P. Vollmar and P. Renaud, J. Am. Chem. Soc., 2017, 140, 155 CrossRef.
- (a) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182 CrossRef CAS; (b) Y. Zhang, M. D. Tortorella, Y. Wang, J. Liu, Z. Tu, X. Liu, Y. Bai, D. Wen, X. Lu, Y. Lu and J. J. Talley, ACS Med. Chem. Lett., 2014, 5, 1162 CrossRef CAS; (c) Y. Zhu, J. Zhou and B. Jiao, ACS Med. Chem. Lett., 2013, 4, 349 CrossRef CAS.
- K. Inomata, A. Ohno, H. Tochio, S. Isogai, T. Tenno, I. Nakase, T. Takeuchi, S. Futaki, Y. Ito, H. Hiroaki and M. Shirakawa, Nature, 2009, 458, 106 CrossRef CAS.
- (a) K. Voronin, A. J. Allentoff, S. J. Bonacorsi Jr, C. Mapelli, S. X. Gong, V. Lee, D. Riexinger, N. Sanghvi, H. Jiang and J. Zeng, J. Labelled Compd. Radiopharm., 2014, 57, 579 CrossRef CAS; (b) G. N. Nilsson and W. J. Kerr, J. Labelled Compd. Radiopharm., 2010, 53, 662 CrossRef CAS; (c) J. Atzrodt and V. Derdau, J. Labelled Compd. Radiopharm., 2010, 53, 674 CrossRef CAS.
- C. Schmidt, Nat. Biotechnol., 2017, 35, 493 CrossRef CAS.
- (a) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744 CrossRef CAS; (b) H. Jiang, S. Gao, J. Xu, X. Wu, A. Lin and H. Yao, Adv. Synth. Catal., 2016, 358, 188 CrossRef CAS; (c) J. J. Tan, B. Liu and S. S. Su, Org. Chem. Front., 2018, 5, 3093 RSC; (d) M. S. Ledovskaya, K. S. Rodygin and V. P. Ananikov, Org. Chem. Front., 2018, 5, 226 RSC.
- (a) Z. Song, R. Samanta and A. P. Antonchick, Org. Lett., 2013, 15, 5662 CrossRef CAS; (b) H. Jiang, S. Gao, J. Xu, X. Wu, A. Lin and H. Yao, Adv. Synth. Catal., 2016, 358, 188 CrossRef CAS.
- S. Ma, G. Villa, P. S. Thuy-Boun, A. Homs and J. Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 734 CrossRef CAS.
- (a) Z. Shi, M. Boultadakis-Arapinis and F. Glorius, Chem. Commun., 2013, 49, 6489 RSC; (b) Y. Qiu, W. J. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5828 CrossRef CAS.
- (a) X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799 CrossRef CAS; (b) A. I. VanderWeide, W. W. Brennessel and W. D. Jones, J. Org. Chem., 2019, 84, 12960 CrossRef CAS; (c) A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109 CrossRef CAS; (d) M. Zhang, A. Luo, Y. Shi, R. Su, Y. Yang and J. You, ACS Catal., 2019, 9, 11802 CrossRef CAS.
- (a) S. Chen, M. Zhang, R. Su, X. Chen, B. Feng, Y. Yang and J. You, ACS Catal., 2019, 9, 6372 CrossRef CAS; (b) S. K. Banjare, R. Chebolu and P. C. Ravikumar, Org. Lett., 2019, 21, 4049 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization, and copies of 1H and 13C NMR spectra. CCDC 2025858. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00236h |
| This journal is © The Royal Society of Chemistry 2021 |