
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of the cofactor specificity of wild-type phosphite dehydrogenase and its mutant using molecular dynamics simulations†
Kunlu Liu‡
a,
Min Wang‡a,
Yubo Zhoub,
Hongxiang Wangb,
Yudong Liub,
Lu Han*a and
Weiwei Han *a
*a
aKey Laboratory for Molecular Enzymology and Engineering of Ministry of Education, School of Life Science, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: luhan@jlu.edu.cn; weiweihan@jlu.edu.cn
bHigh School Attached to Northeast Normal University, 506 Ziyou Road, Changchun 130012, China
First published on 19th April 2021
Abstract
Phosphite dehydrogenase (Pdh) catalyzes the NAD-dependent oxidation of phosphite to phosphate with the formation of NADH. It can be used in several bioorthogonal systems for metabolic control and related applications, for example, bioelectricity. At present, NAD has poor stability at high concentrations and costs are expensive. Implementation of a non-natural cofactor alternative to the ubiquitous redox cofactor nicotinamide adenosine dinucleotide (NAD) is of great scientific and biotechnological interest. Several Pdhs have been engineered to favor a smaller-sized NAD analogue with a cheaper price and better thermal stability, namely, nicotinamide cytosine dinucleotide (NCD). However, the conformational changes of two cofactors binding to Pdh remain unknown. In this study, five molecular dynamics (MD) simulations were performed to exploit the different cofactors binding to wild-type (WT) Pdh and mutant-type (MT) Pdh (I151R/P176E/M207A). The results were as follows: First, compared with WT Pdh, the cofactor-binding pocket of mutant Pdh became smaller, which may favor a smaller-sized NCD. Second, secondary structure analysis showed that the alpha helices in residues 151–207 partly disappeared in mutant Pdh binding to NAD or NCD. Our theoretical results may provide a basis for further studies on the Pdh family.
1. Introduction
Phosphite dehydrogenase (Pdh) catalyzes the NAD-dependent oxidation of phosphite to phosphate with the formation of NADH.1–11 This kind of enzyme regeneration module is combined with a coenzyme-consuming reaction module to achieve the ideal coenzyme balance in multi-enzyme molecular machines. The multi-enzyme reaction sometimes takes place at high temperature, NAD and NAD(P)H may be decomposed, which results in the decrease of coenzyme concentration during the operation of the multi-enzyme molecular machine in vitro, thereby affecting the catalytic efficiency of Pdh. Moreover, NAD is expensive and less specific. Implementation of a non-natural cofactor alternative to the ubiquitous redox cofactor nicotinamide adenosine dinucleotide (NAD) is of great scientific and biotechnological interest. Thus, redox enzymes with a more stringent preference to NAD analogues (e.g., NCD) needed to be developed for cheaper price and better thermal stability. They can be used in several biorthogonal systems for metabolic control and related applications, for example, bioelectricity.12–17According to sequence comparisons of Pdh in the NAD+-dependent D-2-hydroxyacid dehydrogenase family,18,19 Pdh shares 56% identity with other members of this family. Three conserved catalytic residues have been reported to be useful for catalysis by Pdh.19,20 A conserved histidine residue (His293 in Pdh) is proposed to act as a general base for the nucleophilic water molecule attacking on phosphite, Glu264 orients and modulates the pKa of the active site His293, and Arg237 is near the substrate, phosphite, prior to nucleophilic attack.19,21 The site-specific mutants of residues demonstrated that the mutations were at His293, Arg237, and Glu264 severely compromises catalysis relative to the WT.19,22
In 2019, Zhao et al. made a semi-rational approach to characterize several mutant Pdh responsible for NCD-binding capability by X-ray crystallography.1 They obtained mutants with substantially improved NCD preference.1 Although more and more synthetic systems using non-natural cofactors can be treated as new tools for widespread applications by chemical and synthetic biologists, some issues remain to be elucidated. For example, the conformational changes of two cofactors binding to Pdh remain unknown.
In this study, five MD simulations were performed to explore the different cofactors binding to WT Pdh and MT Pdh. This study will provide useful information to address challenging problems for chemical and synthetic biologists.
2. Materials and methods
2.1. Preparation of the protein structures
Five different systems were used to identify the binding mechanism with Pdh: (1) free Pdh, (2) WT Pdh_NCD, (3) MT Pdh_NCD, (4) WTPdh_NAD, and (5) MT Pdh_NAD. The initial coordination of Pdh with NCD was obtained from the Protein Data Bank (PDB) (PDB code 6IH3).1 The 3D structure of NAD was obtained from PDB (PDB code 4E5N). The MT Pdh were generated using Pymol. Protonation states were conducted at physiological pH by using the H++ server (http://biophysics.cs.vt.edu/index.php). All residues were assigned in their standard protonation (pH = 7), and all missing hydrogen atoms were generated using Discovery Studio 4.0 client software. NCD and NAD were then optimized according to the density functional theory at the B3LYP/6-31G* level23 by using Gaussian 09 software.242.2. Molecular docking
NAD was docked to Pdh with AutoDock Vina software.25,26 AutoDockTools were used for docking. We adjusted the box to the centre of the primary and secondary binding sites. The three-dimensional grids were set as 46 × 40 × 36 points with a grid spacing of 1 Å. NAD was docked to Pdh in the binding pocket, and the corresponding energy evaluations were also generated. The most popular and stable docking conformations were selected as the initial binding modes for the subsequent MD simulations.2.3. MD simulation
Gromos 53A6 force field27,28 was applied to describe proteins (WT and MT) and ligands (NAD and NCD). The parameterization of the two ligands was produced by the PRODRG2.5 server. All of the systems were subjected to MD simulations by using a SPC water model in a periodic box.29 Chloride and sodium ions were added to a random location as a replacement for the water molecules in the simulation box to neutralize the systems. Energy minimization was performed using the steepest descent method to equilibrate the initial structure. Afterward, 100 ps NVT (constant number of particles, volume, and temperature) and 100 ps NPT were adopted to stabilize the environment of the systems at 300 K and 1 bar. The coupling constant was set to 0.1 and 2.0 ps for temperature and pressure. The MD simulations were performed using the GROMACS 4.5.2 software package30,31 on the basis of the predicted complexes generated by AutoDock Vina.25,26 During the 200 ns simulation, the LINCS algorithm32 was employed to fix all of the bonds, and the long-range electrostatics was computed by the particle mesh Ewald method33 with a grid spacing of 0.16 nm.2.4. Cross-correlation analysis and principal component analysis (PCA)
Bio3D version 2.3.0 was used to perform cross-correlation analysis. The coordinate axis scales indicated the atomic number in the covariance matrix map. PCA can provide a detailed picture of biomolecular motion to identify large-scale motions and the correlated movements of biological protein systems. In this study, PCA was visualized by VMD version 1.9.3. The energy of macromolecular conformations such as the first two principal components (PC1, PC2) were characterized in free energy landscape (FEL). FEL is used to describe the energies of sets of macromolecular conformations.34,353. Results and discussion
3.1. Quantum chemical calculations and structural stability of the five systems
Pdh has drawn attention because it can be used as a cofactor regeneration system for the transfer of NAD+ to NADH in cofactor recycling. Moreover, due to their relatively inexpensive substrate (i.e., phosphite) and high catalytic efficiency, Pdhs are attractive candidates in the enzymatic regeneration of NADH from NAD+.36–41X-ray crystallographic analysis revealed that Pdh has two catalytic domains in Fig. 1. His293, Asp261, Val262, Ala235, Gly295, Val262, Ala235, Gly295, Thr104, Ser296, Leu100, Lys76, Val156, Thr101, Gly157, Ala155, Gly154, Met153, Ile177, Asp175, Gly152, Pro176, Thr214, Val208, Pro209, Lys188, Met207, and Cys236 (important residues for NCD binding) were shown in Fig. S1.† NAD was docked to WT Pdh and MT Pdh (Fig. 1d and 2).
 | ||
| Fig. 1 Structure of the complex. (a) 2D structure topology, (b) 3D structure of Pdh (PDB code 6IH3), (c) structure of NCD, (d) structure of NAD. | ||
 | ||
| Fig. 2 The docked pose of Pdh_NAD. (a) The docked pose of WT Pdh_NAD, (b) the docked pose of MT Pdh_NAD. | ||
The most popular and stable docking conformations were selected. It was well known that molecular docking was determined not only by the highest score conformation but also by the orientation. See Fig. S2,† it can be seen that the lowest energy docked complexes (NAD) were similar to the reference (NCD). Fig. 2a showed that Gly295, His293, Ala235, Met207, Cys236, Pro209, Val208, Pro209, Val262, Arg237, Val156, Ala155, Gly154, Cys174, Ile177, Cys174, Asp175, and Pro176 were important residues for NAD binding to WT Pdh. In MT Pdh_NAD, Arg151, Cys174, Asp175, Glu176, Ile177, Gly152, Met153, Gly154, Pro209, Ala207, Asn208, Asn234, Ile218, Thr104, Tyr258, and Leu232 were in the NAD binding pocket. The active residues for NAD and NCD binding to Pdh were different. The reason may be lay in the different size of NAD and NCD.
Quantum chemical calculation was used on two ligands by using Gaussian 09(ref. 24) to optimize the structures. The differences in energy between these HOMO and LUMO frontier orbitals can be used to predict the strength and stability of electron transfer.42 Fig. 3 showed the HOMO and LUMO orbits drawn by the Multiwfn program.43 Egap is the energy differences between HOMO and LUMO orbits. In terms of Egap, the cofactors NCD had smaller energy than that of NAD, indicating that electron transfer might occur more easily in NCD than in NAD. Therefore, NCD had more interactions than NAD, including H bonds, salt bridges, and Van der Waals' force (VDW) interactions.
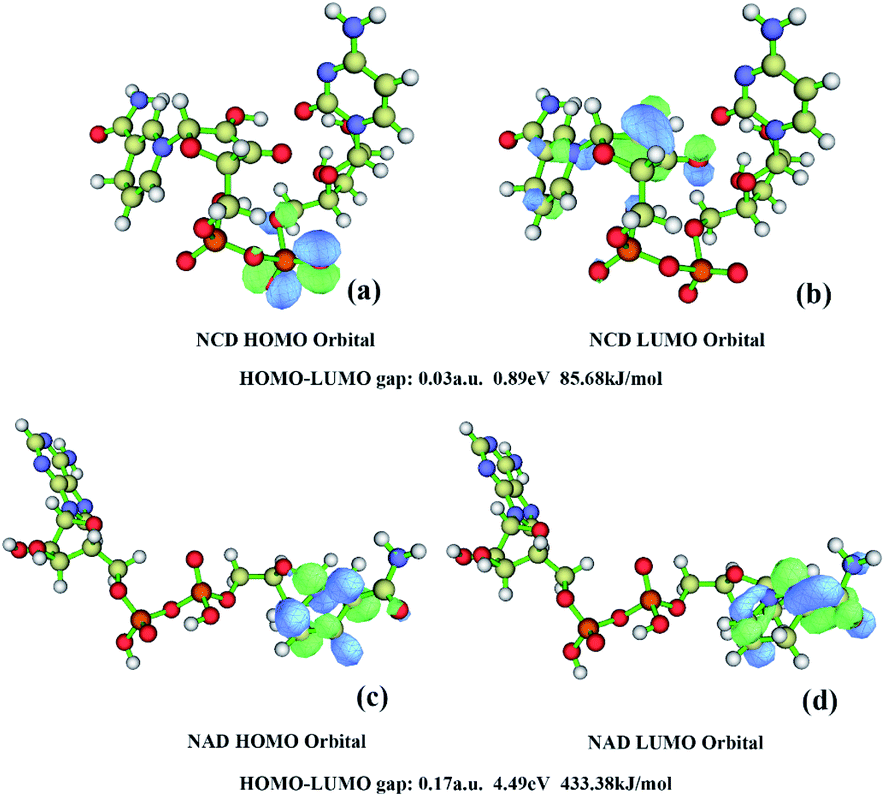 | ||
| Fig. 3 Quantum chemical calculations. (a) HOMO orbital of NCD, (b) LUMO orbital of NCD, (c) HOMO orbital of NAD, (d) LUMO orbital of NAD. | ||
After 200 ns simulations, the root-mean-square deviations (RMSDs) of the Cα atom backbone of the five systems were calculated during simulations in Fig. 4. As shown in Fig. 4a, the RMSDs of free Pdh and WT Pdh_NAD could be stabilized at 6 Å, and that of WT Pdh_NCD could be stabilized at 7.5 Å, indicating that the structures of the three systems already reached a state of relative equilibrium. Meanwhile, the RMSDs of MT Pdh_NAD and MT Pdh_NCD could be stabilized at approximately 9 Å after 20 ns, suggesting that the structures of the two systems reached a state of relative equilibrium.
 | ||
| Fig. 4 Structural stability of the five systems during 200 ns simulations. (a) RMSD plot, (b) SASA plot, (c) Rg plot. | ||
The solvent-accessible surface area (SASA) of the five systems was calculated by the VMD program. The SASA of residues to find the points on a sphere that were exposed to solvent. In Fig. 4b, the WT Pdh_NCD complex became larger than WT Pdh_NAD. In MT Pdh, MT Pdh_NAD became larger than MT Pdh_NCD, suggesting that the SASA of MT Pdh_NCD was smaller.
The radius gyration (Rg) of the five systems was calculated by the VMD program. In Fig. 4c, the Rg values of WT Pdh_NAD could be stabilized at 20 Å, and those of WT Pdh_NCD could be stabilized at 22 Å after 20 ns. As shown in Fig. 4c, the Rg values of MT Pdh_NAD and free Pdh can be stabilized at 22 Å, and those of MT Pdh_NCD can be stabilized at 21 Å after 20 ns. This finding may facilitate the conformational rearrangement of the NAD binding domain in the MT Pdh to move to the catalytic domain in the MT Pdh_NCD complex.
3.2. Conformational changes caused by two cofactor binding to WT and MT Pdh
The conformational changes caused by the five systems were investigated and compared. To investigate the conformational changes, we obtained the differences in the secondary structure (DSSP) by using the do_dssp command. The DSSP of residues 151–207 in MT Pdh differed from that in free Pdh and WT Pdh significantly (Fig. 5). As shown in Fig. 5c and e, the alpha helices in residues 151–207 partly disappeared in MT Pdh binding to NAD or NCD. The alpha helix probability (%) of residues 154–163 of the five systems during MD simulations were shown in Table 1. Residues 154–163 all contained a helix in free Pdh and WT Pdh binding to NAD or NCD. However, the helix of MT Pdh partly disappeared. The reason may be due to the partly disappeared alpha helix, wherein the smaller cofactor, NCD, binds to MT Pdh.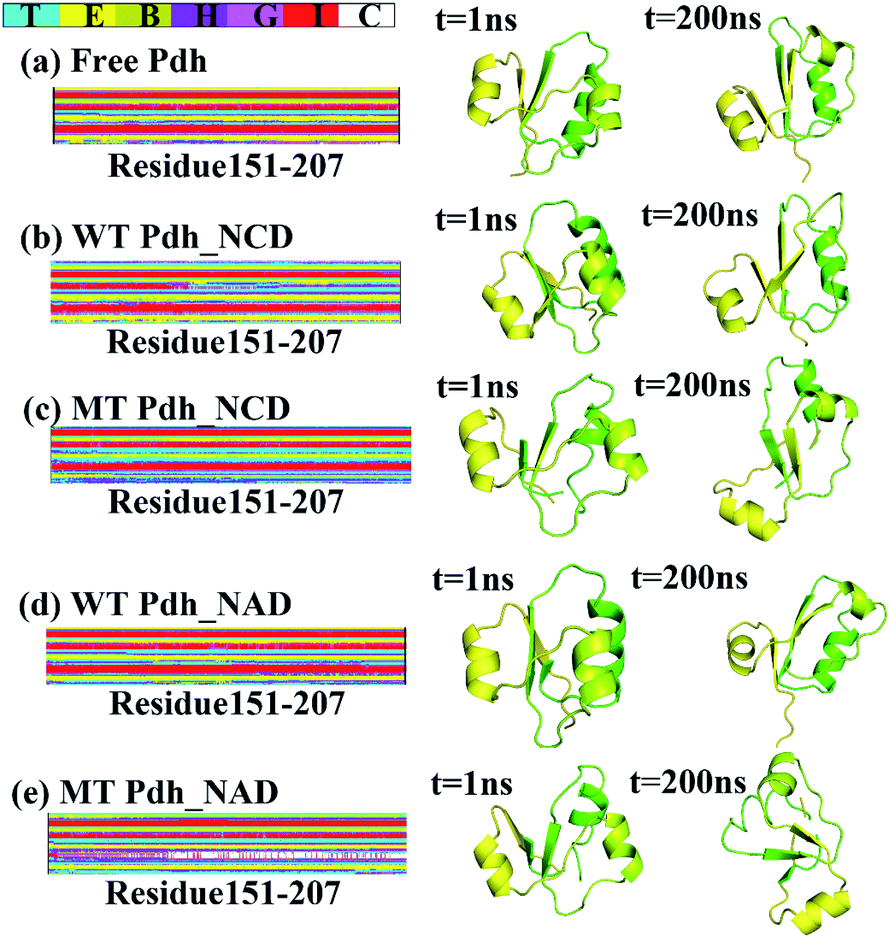 | ||
| Fig. 5 Secondary structure analysis. (a) Free Pdh, (b) WT Pdh_NCD, (c) MT Pdh_NCD, (d) WT Pdh_NAD, (e) MT Pdh_NAD. T: turn, E: extended conformation, B: isolated bridge, H: alpha helix, G: 3-10 helix, I: pi-helix, C: coil. | ||
| G154 | A155 | V156 | G157 | R158 | A159 | I160 | A161 | Q162 | R163 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Free Pdh | 1.85 | 96.19 | 99.04 | 99.99 | 99.99 | 100 | 99.98 | 99.81 | 95.64 | 90.01 |
| WT Pdh_NCD | 0.12 | 15.77 | 18.81 | 82.88 | 99.73 | 99.97 | 99.98 | 99.93 | 98.47 | 83.31 |
| MT Pdh_NCD | 0 | 0 | 0.65 | 98.84 | 99.39 | 99.76 | 99.88 | 98.67 | 93.26 | 66.51 |
| WTPdh_NAD | 0.67 | 88.50 | 99.49 | 99.99 | 100 | 100 | 99.81 | 99.89 | 97.41 | 83.15 |
| MT Pdh_NAD | 0 | 0 | 0 | 0.42 | 2.75 | 4.57 | 10.29 | 10.19 | 8.62 | 6.88 |
3.3. Residue cross-correlation and FEL calculations
Correlation matrix analysis can promote the understanding of protein regions that have intense relevant conformational changes, which can shed light on the dynamic motion of Pdh induced by cofactor binding. In this study, covariance matrix maps were constructed on the basis of the first two eigenvectors to depict the linear correlations of each pair of Cα atoms during the MD simulations. The matrix maps of the five systems indicated that significant motions, correlated or noncorrelated, mainly occurred among the regions of residues 150–200 and residues 200–300. Particularly, the motion in residues 151–207 deserved more attention (Fig. 6), because it occurred near the cofactor active site. | ||
| Fig. 6 Residue cross correlation analysis. (a) Free Pdh, (b) WT Pdh_NCD, (c) MT Pdh_NCD, (d) WT Pdh_NAD, (e) MT Pdh_NAD. | ||
The probabilities of PC1 and PC2 of the five systems were shown in Table 2. The two most stable conformations of the Pdh structure in the five systems were shown in the left and right panels in Fig. 7. Residues 154–163 all contained a helix in free Pdh, WT Pdh_NAD, and WT Pdh_NCD. However, in MT Pdh_NAD and MT Pdh_NCD, the alpha helix partly disappeared.
| PC1 | PC2 | |
|---|---|---|
| Free Pdh | 32.26 | 18.72 |
| WT Pdh_NCD | 34.12 | 18.35 |
| MT Pdh_NCD | 52.35 | 14.46 |
| WT Pdh_NAD | 63.16 | 9.12 |
| MT Pdh_NAD | 27.81 | 19.34 |
 | ||
| Fig. 7 Principal component analysis. (a) Free Pdh, (b) WT Pdh_NCD, (c) MT Pdh_NCD, (d) WT Pdh_NAD, (e) MT Pdh_NAD. | ||
PCA was used to confirm whether conformational changes were stable or not. The FEL calculations were shown in Fig. 7. In the FEL of the five systems, the different energies of the corresponding structures were shown in different colours. The conformations found in the blue area were more stable and had lower energy states than those found in the red area.
3.4. Structural stability of the cofactor-binding pocket
The RMSD, SASA, Rg, and relative frequency of residues 151–207 were also calculated in Fig. 8. The changes of the data indicated that the structure in residues 151–207 in WT Pdh with NAD or NCD was different compared with that in MT Pdh with the cofactors. | ||
| Fig. 8 Structural stability of the cofactor-binding pocket during 200 ns simulations. (a) RMSD plot, (b) SASA plot, (c) Rg plot. | ||
POCASA44 (http://altair.sci.hokudai.ac.jp/g6/service/pocasa/) was used to calculate the binding pocket size during MD simulations (Parameters were listed as follow: The radius of probe sphere value was 2 Å. Single Point Flag value was 16, Protein Depth Flag value was 18. The number of results to show was 5. The size of unit grid was 1 Å. The type of atom was all).
The binding pocket conformations of the four systems at 0, 50, and 200 ns were shown in Fig. 9. The volume of the pockets in WT Pdh_NAD was smaller than that in WT Pdh_NCD (Fig. 9a and b), the volume of the pockets in MT Pdh_NAD was larger than that in MT Pdh_NCD (Fig. 9c and d). A small cofactor-binding pocket will be useful for NCD, a small-sized cofactor, to bind to MT Pdh.
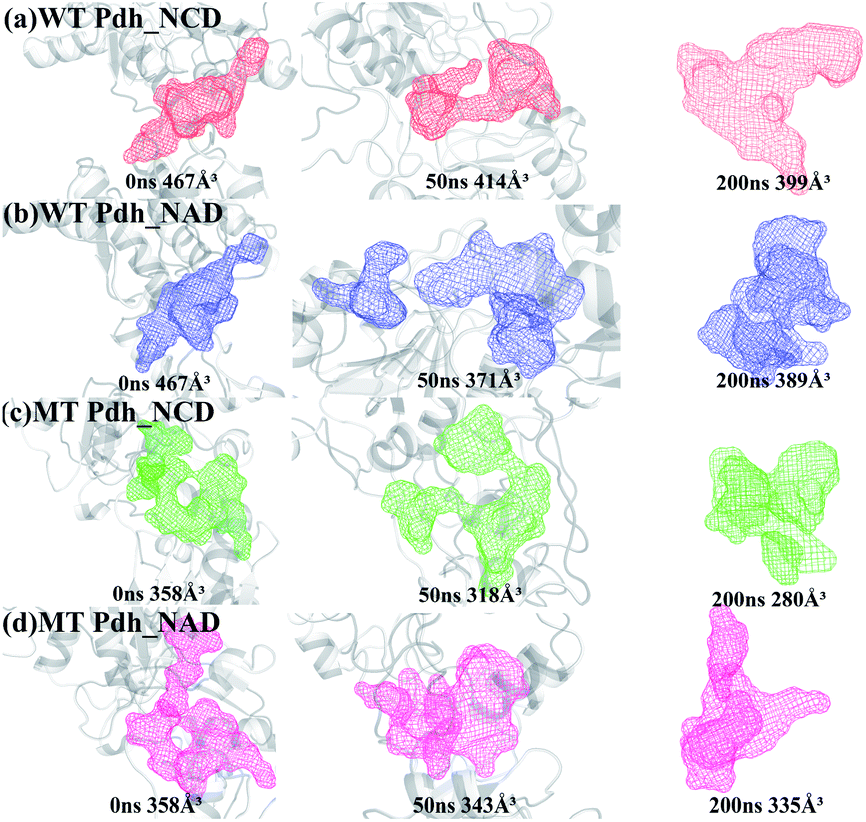 | ||
| Fig. 9 The active pocket size of the four systems at 0 ns,50 ns and 200 ns MD simulations. (a) WT Pdh_NCD, (b) WT Pdh_NAD, (c) MT Pdh_NCD, (d) MT Pdh_NAD. | ||
The distances between Glu264N and Asn234OD1 of the three systems including free Pdh, MT Pdh_NAD, and MT Pdh_NCD were calculated in Fig. 10. As shown in Fig. 10a, only the distance between Glu264N and Asn234OD1 in the MT Pdh_NCD complex was stabilized within 4 Å after 50 ns. Thus, the introduction of new hydrophobic interactions or hydrogen bonds in MT Pdh_NCD can be used to improve its catalytic efficiency.
 | ||
| Fig. 10 The distance between Glu264N and Asn234OD1. (a) The distance between Glu264N and Asn234OD1 of Free Pdh, MT Pdh_NAD and MT Pdh_NCD, (b) relative frequency of Free Pdh, MT Pdh_NAD and MT Pdh_NCD, (c) 3D structure of the distance. | ||
4. Conclusions
Several Pdhs have been engineered to favor a smaller-sized NAD analogue (i.e., NCD). However, the conformational changes of two cofactors binding to Pdh remain unknown. In this study, five MD simulations were performed to exploit the different cofactors binding to WT Pdh and MT Pdh. The results were as follows: First, compared with WT Pdh, the cofactor-binding pocket of MT Pdh became smaller, which may favour a smaller-sized NCD. Second, secondary structure analysis showed that the alpha helices in residues 151–207 partly disappeared in MT Pdh binding to NAD or NCD. The MT Pdh moved inward the cofactor-binding pocket, which presents a more restricted region and a compact cavity, indicating an increased NCD preference. These structural data reveal that the cofactor-binding pocket of Pdh became more crowded upon the introduction of large and polar residues such as Arg or Glu. Moreover, these mutations also led to the reorientation of skeletal amino acid residues inward to narrow the binding cleft. However, these charged side chains provided additional interactions with NCD. Together, these structural changes created a compacted cofactor-binding pocket and molecular interactions beneficial to NCD residence. Our theoretical results may provide a basis for further studies on Pdh.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [31870201] and the Overseas Cooperation Project of Jilin Province [20200101068JC]. This work was performed at the High Performance Computing Center of Jilin University.Notes and references
- Y. Liu, Y. Feng, L. Wang, X. Guo, W. Liu, Q. Li, X. Wang, S. Xue and Z. K. Zhao, ACS Catal., 2019, 9, 1883–1887 CrossRef CAS.
- V. Schreiber, F. Dantzer, J. C. Ame and G. De Murcia, Nat. Rev. Mol. Cell Biol., 2006, 7, 517–528 CrossRef CAS PubMed.
- M. Pittelli, L. Formentini, G. Faraco, A. Lapucci, E. Rapizzi, F. Cialdai, G. Romano and G. Moneti, J. Biol. Chem., 2010, 285, 34106–34114 CrossRef CAS PubMed.
- M. S. Bonkowski and D. A. Sinclair, Nat. Rev. Mol. Cell Biol., 2016, 17, 679–690 CrossRef CAS PubMed.
- W. Ying, Antioxid. Redox Signaling, 2008, 10, 179–206 CrossRef CAS PubMed.
- K. W. Ryu, T. Nandu, J. Kim, S. Challa, R. J. Deberardinis and W. L. Kraus, Science, 2018, 360, 618 CrossRef CAS PubMed.
- M. Wang, B. Chen, Y. Fang and T. Tan, Biotechnol. Adv., 2017, 35, 1032–1039 CrossRef CAS PubMed.
- D. V. Titov, V. Cracan, R. P. Goodman, J. Peng, Z. Grabarek and V. K. Mootha, Science, 2016, 352, 231–235 CrossRef CAS PubMed.
- C. Del Nagro, Y. Xiao, L. Rangell, M. Reichelt and T. O'Brien, J. Biol. Chem., 2014, 289, 35182–35192 CrossRef CAS PubMed.
- A. K. Holm, L. M. Blank, M. Oldiges, A. Schmid, C. Solem, P. R. Jensen and G. M. Vemuri, J. Biol. Chem., 2010, 285, 17498–17506 CrossRef CAS PubMed.
- M. J. A. van Hoek and R. M. H. Merks, BMC Syst. Biol., 2012, 6, 22 CrossRef PubMed.
- A. R. Rovira, A. Fin and Y. Tor, J. Am. Chem. Soc., 2017, 139, 15556–15559 CrossRef CAS PubMed.
- F. Hallé, A. Fin, A. R. Rovira and Y. Tor, Angew. Chem., Int. Ed., 2018, 57, 1087–1090 CrossRef PubMed.
- T. Knaus, C. E. Paul, C. W. Levy, S. de Vries, F. G. Mutti, F. Hollmann and N. S. Scrutton, J. Am. Chem. Soc., 2016, 138, 1033–1039 CrossRef CAS PubMed.
- D. Ji, L. Wang, S. Hou, W. Liu, J. Wang, Q. Wang and Z. K. Zhao, J. Am. Chem. Soc., 2011, 133, 20857–20862 CrossRef CAS PubMed.
- L. Wang, D. Ji, Y. Liu, Q. Wang, X. Wang, Y. J. Zhou, Y. Zhang, W. Liu and Z. K. Zhao, ACS Catal., 2017, 7, 1977–1983 CrossRef CAS.
- D. Ji, L. Wang, Y. Zhou, W. Yang, Q. Wang and Z. K. Zhao, Cuihua Xuebao, 2012, 33, 530–535 CrossRef CAS.
- G. A. Grant, Biochem. Biophys. Res. Commun., 1989, 165, 1371–1374 CrossRef CAS PubMed.
- Y. Zou, H. Zhang, J. S. Brunzelle, T. W. Johannes, R. Woodyer, J. E. Hung, N. Nair, W. A. van der Donk, H. Zhao and S. K. Nair, Biochemistry, 2012, 51, 4263–4270 CrossRef CAS PubMed.
- A. M. G. Costas, A. K. White and W. W. Metcalf, J. Biol. Chem., 2001, 276, 17429–17436 CrossRef CAS PubMed.
- H. A. Relyea and W. A. van der Donk, Bioorg. Chem., 2005, 33, 171–189 CrossRef CAS PubMed.
- R. Woodyer, J. L. Wheatley, H. A. Relyea, S. Rimkus and W. A. van der Donk, Biochemistry, 2005, 44, 4765–4774 CrossRef CAS PubMed.
- I. L. Rogers and K. J. Naidoo, J. Comput. Chem., 2017, 38, 1789–1798 CrossRef CAS PubMed.
- J. Z. Vilseck, J. Kostal, J. Tirado-Rives and W. L. Jorgensen, J. Comput. Chem., 2015, 36, 2064–2074 CrossRef CAS PubMed.
- N. T. Nguyen, T. H. Nguyen, T. N. H. Pham, N. T. Huy, M. V. Bay, M. Q. Pham, P. C. Nam, V. V. Vu and S. T. Ngo, J. Chem. Inf. Model., 2020, 60, 204–211 CrossRef CAS PubMed.
- T. Gaillard, J. Chem. Inf. Model., 2018, 58, 1697–1706 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- O. Guvench and A. D. MacKerell, Methods Mol. Biol., 2008, 443, 63–88 CrossRef CAS PubMed.
- N. W. Ockwig, R. T. Cygan, L. J. Criscenti and T. M. Nenoff, Phys. Chem. Chem. Phys., 2008, 10, 800–807 RSC.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel and B. Hess, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- M. J. Abraham and J. E. Gready, J. Comput. Chem., 2011, 32, 2031–2040 CrossRef CAS PubMed.
- R. Kumar, R. Maurya and S. Saran, J. Biomol. Struct. Dyn., 2019, 37, 781–795 CrossRef CAS PubMed.
- R. E. Isele-Holder, W. Mitchell and A. E. Ismail, J. Chem. Phys., 2012, 137, 174107 CrossRef PubMed.
- P. O'Driscoll, E. Merenyi, C. Karmonik, R. Grossman, Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 2014, pp. 734–737 Search PubMed.
- J. X. Zhu, Y. Li, J. Z. Wang, Z. F. Yu, Y. Liu, Y. Tong and W. W. Han, Front. Chem., 2018, 6, 437 CrossRef CAS PubMed.
- J. M. Vrtis, A. K. White, W. W. Metcalf and W. A. van der Donk, Angew. Chem., Int. Ed., 2002, 41, 3257–3259 CrossRef CAS PubMed.
- H. K. Chenault and G. M. Whitesides, Appl. Biochem. Biotechnol., 1987, 14, 147–197 CrossRef CAS PubMed.
- W. A. van der Donk and H. Zhao, Curr. Opin. Biotechnol., 2003, 14, 421–426 CrossRef CAS PubMed.
- H. Zhao and W. A. van der Donk, Curr. Opin. Biotechnol., 2003, 14, 583–589 CrossRef CAS PubMed.
- T. W. Johannes, R. D. Woodyer and H. Zhao, Biotechnol. Bioeng., 2007, 96, 18–26 CrossRef CAS PubMed.
- R. Woodyer, W. A. van der Donk and H. Zhao, Comb. Chem. High Throughput Screening, 2006, 9, 237–245 CrossRef CAS PubMed.
- J. S. Griffith and L. E. Orgel, Q. Rev., Chem. Soc., 1957, 11, 381–393 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Yu, Y. Zhou, I. Tanaka and M. Yao, Bioinformatics, 2010, 26, 46–52 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00221j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |