
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of 3′-[18F]fluorothymidine-5′-squaryl as a bioisostere of 3′-[18F]fluorothymidine-5′-monophosphate†
D. Brickute‡
a,
A. Beckley‡a,
L. Allott a,
M. Bragaa,
C. Barnesa,
K. J. Thorleyb and
E. O. Aboagye*a
a,
M. Bragaa,
C. Barnesa,
K. J. Thorleyb and
E. O. Aboagye*a
aComprehensive Cancer Imaging Centre, Faculty of Medicine, Imperial College London, Hammersmith Hospital, W12 0NN, London, UK. E-mail: eric.aboagye@imperial.ac.uk
bUniversity of Kentucky, Department of Chemistry, Lexington, KY 40506, USA
First published on 29th March 2021
Abstract
The squaryl moiety has emerged as an important phosphate bioisostere with reportedly greater cell permeability. It has been used in the synthesis of several therapeutic drug molecules including nucleoside and nucleotide analogues but is yet to be evaluated in the context of positron emission tomography (PET) imaging. We have designed, synthesised and evaluated 3′-[18F]fluorothymidine-5′-squaryl ([18F]SqFLT) as a bioisostere to 3′-[18F]fluorothymidine-5′-monophosphate ([18F]FLTMP) for imaging thymidylate kinase (TMPK) activity. The overall radiochemical yield (RCY) was 6.7 ± 2.5% and radiochemical purity (RCP) was >90%. Biological evaluation in vitro showed low tracer uptake (<0.3% ID mg−1) but significantly discriminated between wildtype HCT116 and CRISPR/Cas9 generated TMPK knockdown HCT116shTMPK−. Evaluation of [18F]SqFLT in HCT116 and HCT116shTMPK− xenograft mouse models showed statistically significant differences in tumour uptake, but lacked an effective tissue retention mechanism, making the radiotracer in its current form unsuitable for PET imaging of proliferation.
Introduction
Bioisosteres improve the biological or physical properties of a molecule (e.g. toxicity, bioavailability, metabolism) by replacing problematic functional groups without making significant change to the structure.1 Problematic moieties can hinder the progression of otherwise promising lead candidates from entering clinical trials and therefore, their bioisosteric replacement is more cost-effective than implementing an alternative pharmacophore. Acidic functional groups are important in drug discovery with >450 marketed drugs containing a carboxylic acid; however, poor membrane permeability and metabolic instability support their bioisosteric replacement.2The squaryl moiety, derived from squaric acid (3,4-dihydroxycyclobut-3-ene-1,2-dione), has been investigated as an acidic bioisostere in a number of compounds. The aromatic structure mimics the electrostatic properties of the phosphate group by forming resonance structures which result in two negatively charged carbonyls. The synthesis of novel oligodeoxynucleotide analogues containing the squaryl moiety as a phosphate mimic was reported;3–6 work by Sekine et al. has shown that base-recognition was maintained, opening up a potential for use in a variety of applications where precise hybridisation is required.4 Xie et al. (2003) developed protein tyrosine phosphatase (PTPase) inhibitors using the squaryl moiety as a non-hydrolysable bioisostere of phosphotyrosine with a reduced negative charge to improve bioavailability;7 squaric acid bound to the active site PTPase mimicked natural phosphate substrates. Potent squaryl containing analogues of γ-amino-butyric acid and L-glutamate were synthesised and investigated as neurochemically interesting molecules.8 Despite potential use in therapeutic molecules for many biological applications, squaryl (and phosphate bioisosteres more generally) has not been evaluated in the context of molecular imaging. An understanding of the relationship between squarate and phosphate in terms of biological properties (i.e. recognition by kinases), pharmacokinetics (PK) and metabolism may result in useful diagnostic imaging agents.
Positron emission tomography (PET) is a powerful and highly sensitive molecular imaging technique for probing living systems using sub-pharmacological doses of radiolabelled compounds for minimal biological perturbation.9 Phosphate groups play an important role in the uptake and retention of some PET radiopharmaceuticals inside the cell, including 3′-[18F]fluorothymidine ([18F]FLT), used to image rapidly proliferating cancer cells (Fig. 1). The in vivo metabolism of [18F]FLT is well understood.10–12 [18F]FLT follows the salvage pathway of DNA synthesis and undergoes three intracellular 5′-phosphorylations. Thymidine kinase-1 (TK1) converts [18F]FLT into [18F]FLT-monophosphate ([18F]FLTMP), which is subsequently phosphorylated into [18F]FLT-di/triphosphate ([18F]FLTDP/[18F]FLTTP) by thymidylate kinase (thymidine monophosphate kinase, TMPK; EC 2.7.4.9) and nucleotide diphosphate kinase (NDPK) respectively; preceding DNA incorporation. However, [18F]FLT uptake is largely dependent upon TK1 and therefore is unable to trace tumours that rely primarily on de novo thymidine monophosphate synthesis. We envisaged that imaging downstream of TK1, at the level of TMPK activity, where the de novo and salvage pathways converge would eliminate the dependence on TK1 activity. To achieve this, a radiotracer that mimics [18F]FLTMP would be required, as it is not a substrate for the TK1 enzyme but can be further phosphorylated by TMPK and NDPK. Despite [18F]FLTMP appearing to exhibit ideal characteristics for imaging TMPK activity, the radiosynthesis would be challenging and the radiotracer would be charged under physiological conditions (monophosphate pKa: 1.6, 6.6) preventing cell membrane permeability.13,14
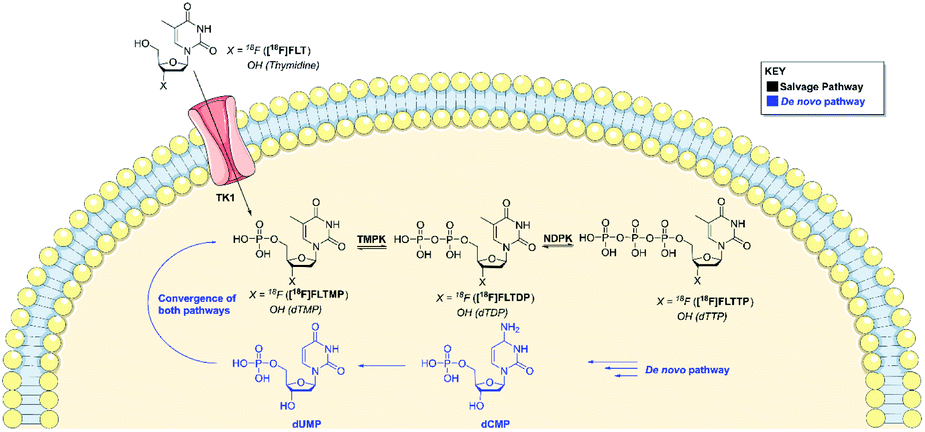 | ||
| Fig. 1 A schematic showing the de novo and salvage pathways of DNA synthesis. Both pathways converge at the formation of dTMP/[18F]FLTMP. | ||
Nucleotide derivatives with a masked monophosphate group (i.e. prodrugs) have several advantages over their unmasked counterparts such as overcoming reliance on nucleoside transporters for cellular uptake and phosphorylation by rate limiting kinases. The use of phosphoramidate pro-nucleotides (ProTides), which are clinically approved prodrugs exploited in nucleoside therapy, presents a potential solution.15,16 The ProTide moiety masks the charged phosphate for improved cellular membrane permeability yet releases the charged phosphate group once inside the cell. Cavaliere et al. (2020) developed a radiosynthetic route to access 3′-[18F]FLT ProTide; in vitro and in vivo biological evaluation is reportedly ongoing.17 The 3′-[18F]FLT ProTide is likely to passively diffuse across the cell membrane, however the PET readout may be rate-limited by the enzymatic cleavage of the ProTide moiety.
We proposed that the use a squaryl group as a phosphate bioisostere, substituted at the 5′-position of [18F]FLT, could be an alternative strategy. In order for a 5′-squaryl derivative of [18F]FLT to image proliferation downstream of TK1, it must not be transported into the cell by nucleoside transporters, e.g. equilibrative nucleoside transporter 1/2 (ENT1/2) and instead passively diffuse across the cell membrane. Experimental data on the membrane permeability of the squaryl group is limited; however, Lassalas et al. (2016) studied the structure activity relationship between organic acids and bioisosteres by experimentally determining and compiling data on lipophilicity (Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D7.5), acidity (pKa) and membrane permeability from parallel artificial membrane permeability assays (PAMPA).2 It was concluded that a simple squaryl substituted molecule may be significantly more cell membrane permeable than a similar phosphate despite comparable acidity and lipophilicity.2 This work was of particular interest for our application and encouraged the design, synthesis and biological evaluation of 3′-[18F]fluorothymidine-5′-squaryl ([18F]SqFLT) (Fig. 2). We hypothesised that the reduced negative charge and the potential increase in membrane permeability of the squaryl group compared to phosphate, may allow sufficient membrane permeability for PET imaging of proliferating cancer cells due to the high sensitivity of PET imaging.2,7 A high contrast PET image is not only dependent upon radiotracer uptake in target tissue, but rapid clearance from blood and non-target tissues (e.g. muscle).
D7.5), acidity (pKa) and membrane permeability from parallel artificial membrane permeability assays (PAMPA).2 It was concluded that a simple squaryl substituted molecule may be significantly more cell membrane permeable than a similar phosphate despite comparable acidity and lipophilicity.2 This work was of particular interest for our application and encouraged the design, synthesis and biological evaluation of 3′-[18F]fluorothymidine-5′-squaryl ([18F]SqFLT) (Fig. 2). We hypothesised that the reduced negative charge and the potential increase in membrane permeability of the squaryl group compared to phosphate, may allow sufficient membrane permeability for PET imaging of proliferating cancer cells due to the high sensitivity of PET imaging.2,7 A high contrast PET image is not only dependent upon radiotracer uptake in target tissue, but rapid clearance from blood and non-target tissues (e.g. muscle).
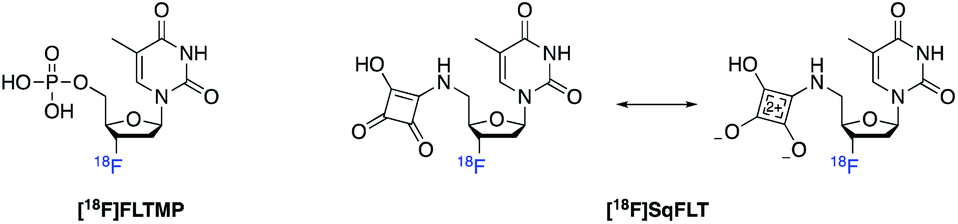 | ||
| Fig. 2 [18F]FLTMP and the proposed [18F]SqFLT phosphate mimic with resonance structure. | ||
The purpose of this study was two-fold: firstly, to evaluate if squaryl moiety was an effective bioisostere for the phosphate group in the context of a radiolabelled nucleoside ([18F]FLT); secondly, evaluate if [18F]SqFLT could image the DNA synthesis. This work represents the first radiosynthesis, in vitro and in vivo imaging and biodistribution of a radiolabelled squarylamide-thymidine derivative and may aid the development of therapeutic agents.
Results and discussion
[18F]FLT has been studied extensively as a proliferation radiotracer and exhibits selective uptake via the thymidine salvage pathway, mediated by TK1. [18F]FLT is unable to image tumours that utilise the de novo pathway primarily, and therefore the development of a radiotracer that bypasses TK1 is clinically relevant.18 In this study, we proposed that [18F]SqFLT bearing a 5′-squaryl moiety as a phosphate bioisostere may result in a membrane permeable analogue of [18F]FLTMP. A requirement for successful PET imaging of TMPK activity is the phosphorylation of [18F]SqFLT, mimicking the formation of [18F]FLTDP, the diphosphate species of [18F]FLT. Molecular in silico modelling was used to determine if [18F]SqFLT could occupy the binding pocket of TMPK in a similar way to TMP and [18F]FLT; a first step towards phosphorylation if it acts as a substrate (as opposed to an enzyme antagonist).Molecular modelling
Using Gaussian 09 and UCSF Chimera,19,20 TMP with adenosine diphosphate (ADP), and SqFLT with ADP were docked into human TMPK to study the interactions between the ligands and the enzyme; the structures were geometrically optimized using the oniom method.21 The enzyme was treated using a molecular mechanics UFF force field, while the substrates (TMP and SqFLT) were modelled with density functional theory (B3LYP/6-31G*). Encouragingly, SqFLT occupied the catalytic site of TMPK, comparable to TMP (Fig. 3). Moreover, the hydroxyl group of the squaryl moiety was in close proximity to the phosphate of ADP, which could promote phosphotransfer in the presence of ADP and Mg2+. These preliminary data suggested a close relationship between SqFLT and bound TMP, by visual inspection of the image and the calculated free energy, encouraging further investigation.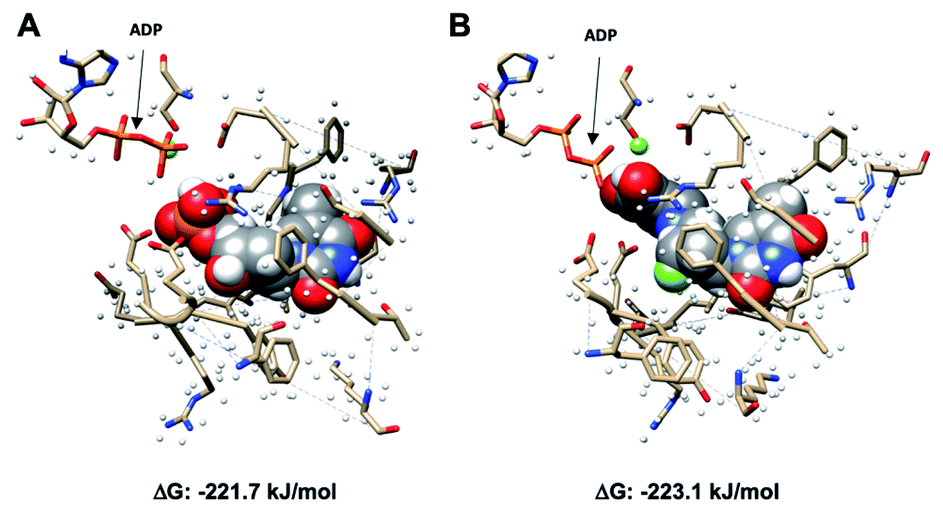 | ||
| Fig. 3 Molecular modeling of SqFLT and TMP (space-filled model) into the human ADP-bound dTYMK. (A) dTMP bound to the active site of TMPK (P-loop) in the presence of ADP and Mg2+; (B) SqFLT bound to the active site of TMPK (P-loop) in the presence of ADP and Mg2+; ΔG = free energies (kJ mol−1) of predicted binding interactions; color code: carbon: black; hydrogen: white; oxygen: red; nitrogen: blue; phosphorus: orange; fluorine: large green; magnesium: small green. | ||
Chemical synthesis
A radiosynthetic route to access [18F]SqFLT was established using precursor 6 (Scheme 1A). Compound 1 was synthesized following a literature procedure to ensure the 3′-OH was in the R-configuration so that the desired S-configuration was produced by inversion of stereochemistry upon nucleophilic substitution with [18F]fluoride (Scheme S1†). In brief, the 4,4′-dimethoxytrityl protecting group (DMTr) of 1 was removed from the 5′-OH moiety resulting in 2 in good yield (80%). The 5′-OH was converted into 5′-NH2 through a tetrachlorophthalic anhydride intermediate (3) giving compound 4 in a combined yield of 51%. The 5′-NH2 was boc-protected (5) and the nosylate leaving group installed on the 3′-OH resulting in radiochemical precursor 6. A non-radioactive standard for [18F]SqFLT ([18F]11) was synthesized, along with standards for key radiolabelled intermediates (Scheme 1B). Commercially available 3′-fluorothymidine ([19F]FLT) was modified at the 5′-OH position to bear a 5′-NH2 (9) as described above. The squaryl group was installed by reacting 3,4-diethoxy-3-cyclobutene-1,2-dione with the 5′-NH2 of 9 to produce 10 in a yield of 55%; subsequent hydrolysis of 10 produced [19F]SqFLT (11). All compounds were characterised by NMR and HRMS (spectra shown in the ESI, Fig. S1–S26†).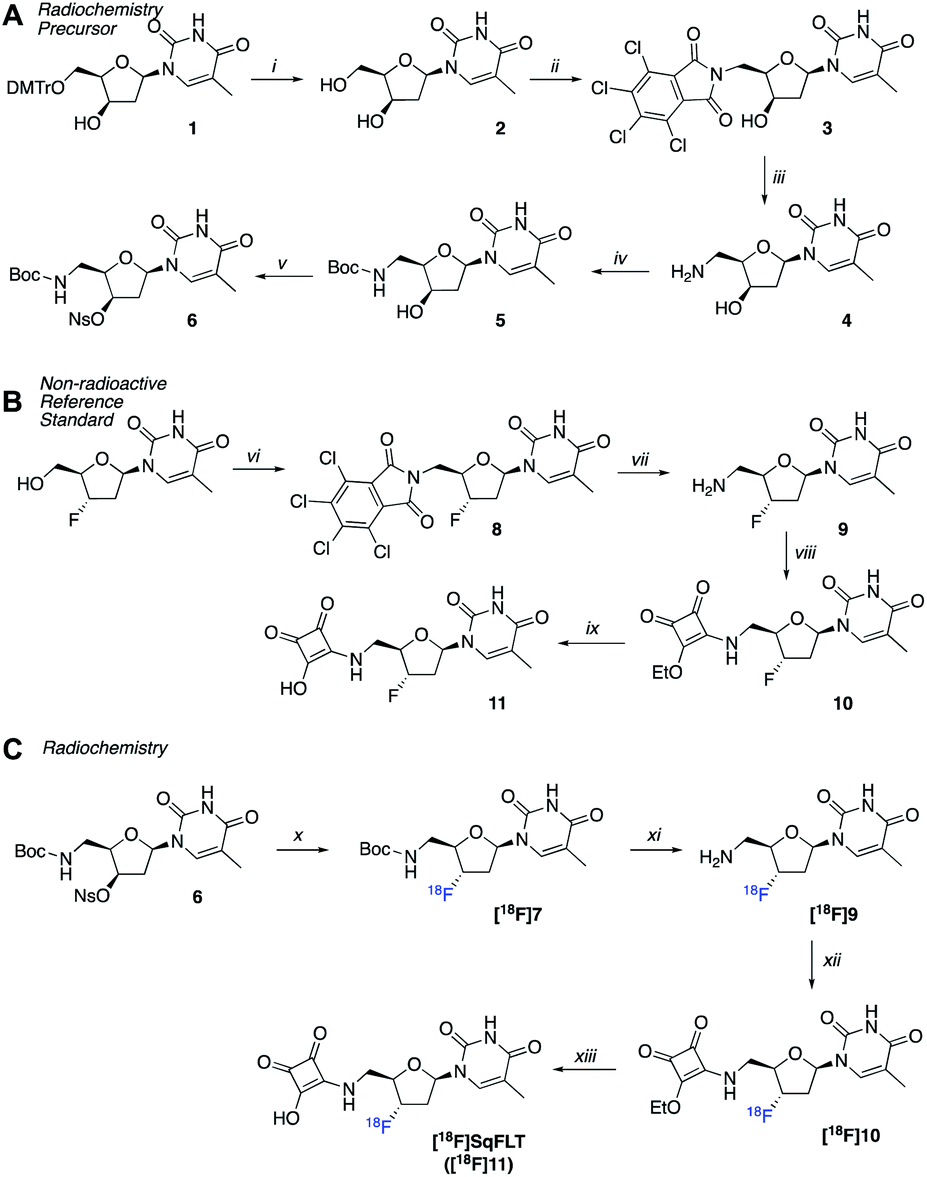 | ||
| Scheme 1 (A) Synthesis of precursor 6; (B) synthesis of reference compound 11; (C) radiosynthesis of [18F]SqFLT. Reaction conditions: (i) TFA, DCM, RT, 2 h; (ii) triphenylphosphine, tetrachlorophtalimide, DIAD, dry THF, RT, 4 days; (iii) ethylenediamine, MeCN, THF, EtOH, 60 °C, 1 h, then 45 °C, 2 h; (iv) Boc2O, NaHCO3, THF, H2O, 0 °C, 1 h, then RT, 2 h; (v) NsCl, pyridine, silver trifluoromethanesulfonate, RT, 2 h; (vi) triphenylphosphine, tetrachlorophtalimide, DIAD, dry THF, RT, 4 days; (vii) ethylenediamine, MeCN, THF, EtOH, RT, 2 h; (viii) 3,4-diethoxy-3-cyclobutene-1,2-dione, EtOH, RT, 1.5 h; (ix) HCl (4 M), acetone, RT, 16 h; (x) automated: [18F]TBAF, 2-methyl-2-butanol, MeCN, 125 °C, 30 min; (xi) phosphoric acid (2.29 M), 75 °C, 25 min; (xii) NH3 in MeOH, 3,4-diethoxy-3-cyclobutene-1,2-dione, pH 7, 50 °C, 40 min; (xiii) HCl (4N), EtOH, 85 °C, 40 min. | ||
Radiochemistry
Precursor 6 was radiolabelled using [18F]TBAF by SN2 displacement of the nosylate leaving group producing [18F]7. The nucleophilicity of the [18F]fluoride ion was modulated by the addition of 2-methyl-2-butanol in MeCN (1:1 v/v) as a polar and protic co-solvent.22 The absence of 2-methyl-2-butanol resulted in poor radiolabelling and the formation of unwanted radioactive products, suspected to be [18F]fluoronosylate. The 18F-fluorination of precursor 6 was automated using the GE FASTLab™ platform (Fig. S27†) and the resulting compound [18F]7 was purified by HLB-SPE in a 24.7 ± 7.4% RCY. Subsequent reactions were performed manually in Wheaton vials. The boc-group of [18F]7 was hydrolysed under acidic conditions to give [18F]9 and the reaction mixture was neutralized to pH 7. The free 5′-NH2 reacted with 3,4-diethoxy-3-cyclobutene-1,2-dione to give [18F]10 which was purified by semi-preparative radio-HPLC and formulated into EtOH for the final hydrolysis to [18F]SqFLT ([18F]11). Once neutralized, [18F]SqFLT was suitable for in vitro and in vivo evaluation. The overall radiochemical yield (RCY) was 6.7 ± 2.5% (decay corrected to start of synthesis) and the radiochemical purity (RCP) was >90%; the molar activity was 4.38 GBq μmol−1. The radiotracer was co-injected with [19F]SqFLT reference material which confirmed the identity of the product (Fig. S30†); the identity of intermediate product [18F]10 was also confirmed by co-injecting a reference standard (Fig. S29†). The overall 4 ± 1 h radiosynthesis was not fully optimized but produced [18F]SqFLT in sufficient quantity and quality for initial in vitro and in vivo evaluation. Attempts to simplify the radiochemistry by forming [18F]10 from a single 18F-fluorination reaction with an appropriate precursor proved unsuccessful and formed multiple radioactive products (data not shown).
The hydrophilicity of [18F]SqFLT (>90% RCP) was determined experimentally by its LogD7.5 using the shake flask method. The radiotracer was very hydrophilic with a LogD7.5 value of −2.90 ± 0.24 which was 2.4-fold lower than calculated LogP values for the tracer and [18F]FLTMP (Table S1†). The simple model squaryl compound (3-(benzylamino)-4-hydroxycyclobut-3-ene-1,2-dione) reported by Lassalas et al. (2016) exhibited significantly higher permeability compared to a phosphate analogue, however it was considerably less polar (LogD7.4 −1.4) than [18F]SqFLT, owed to the highly hydrophilic structure of nucleosides.2 Being a highly hydrophilic molecule, we were sceptical that [18F]SqFLT would be cell permeable and aimed to determine this experimentally in radioactive cell uptake experiments.
In vitro evaluation
A CRISPR/Cas9 generated knockdown of TMPK in HCT116 cells (HCT116shTMPK−) was developed for use in radioactive uptake experiments alongside wildtype (WT) HCT116 cells, to determine if the uptake of [18F]SqFLT paralleled TMPK expression. Western blot analysis and quantification of HCT116shTMPK− and WT HCT116 cell lines showed a 2.9-fold reduction of TMPK expression in the mutant cell line (33 ± 2%) (Fig. 4A and B) . The uptake of [18F]SqFLT in mutant HCT116shTMPK− was significantly lower (0.14 ± 0.04% ID g−1) compared to WT HCT116 (0.24 ± 0.04% ID mg−1), despite low absolute uptake in both cell lines attributed to rate limiting permeability of [18F]SqFLT (Fig. 4C). The in vitro uptake kinetic profile was determined in WT HCT116 cells over 120 min (Fig. 4D). The peak uptake of [18F]SqFLT occurred between 0–15 min followed by a drop of 2-fold between 15–120 min; the efflux was not the result of an affinity of [18F]SqFLT to multidrug resistance proteins (Fig. S34†) and suggests that a permanently phosphorylated [18F]SqFLT species was not formed, which would have enhanced retention inside the cell. The uptake of [18F]SqFLT was confirmed to be independent of TK1 and nucleoside transporters (ENT1/2) by incubating pharmacological doses (1–10 μg mL−1) of [19F]SqFLT with [18F]FLT; thymidine was used as a positive control. An increase in thymidine concentration inversely correlated with [18F]FLT uptake in HCT116 cells, suggesting saturation of TK1 and ENT1/2; the same increase of [19F]SqFLT concentration did not inhibit [18F]FLT uptake (Fig. S33†). Encouraged by the in vitro uptake data, [18F]SqFLT was evaluated in vivo.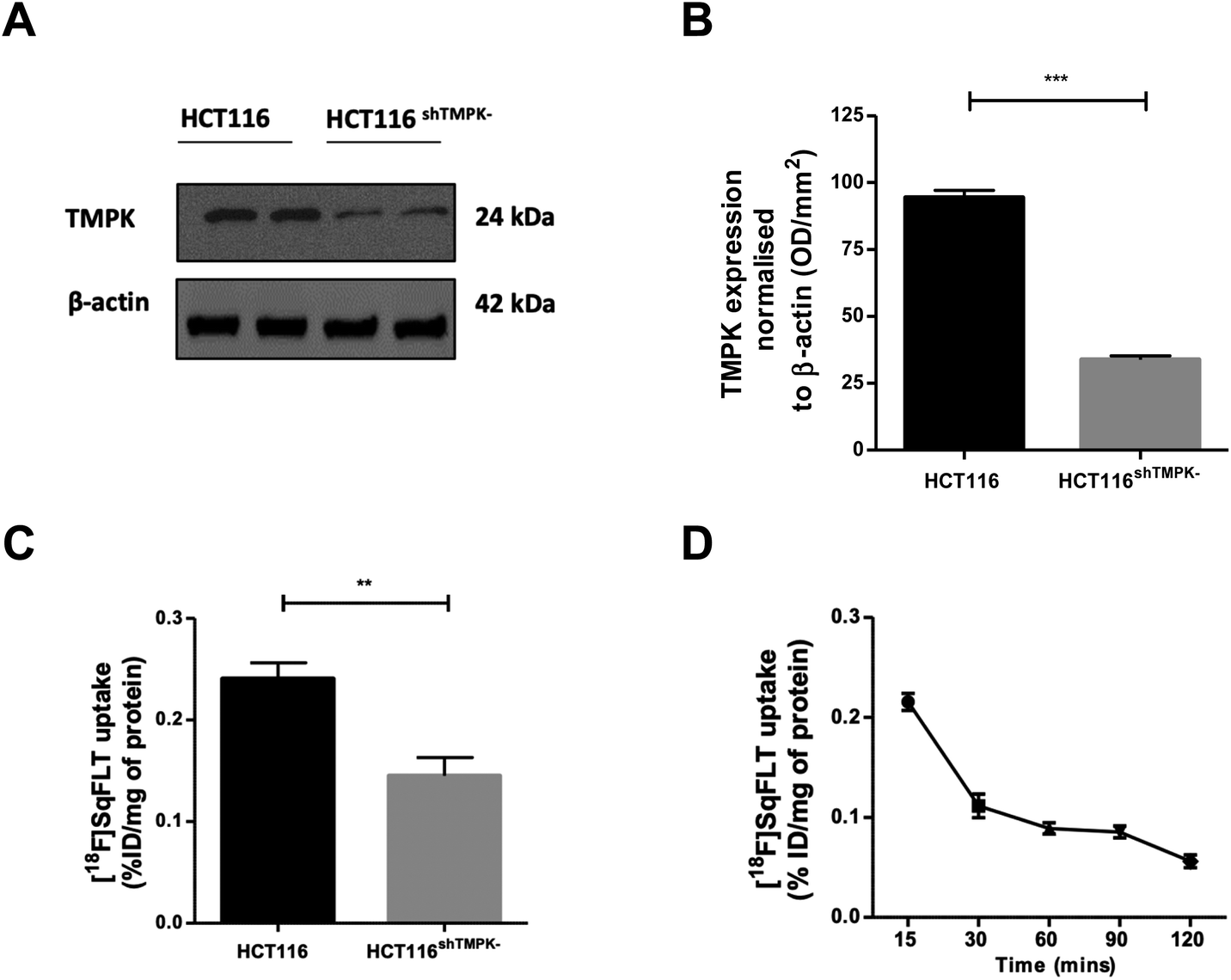 | ||
| Fig. 4 (A) Western blot analysis of TMPK expression in WT HCT116 and HCT116shTMPK− cell lines; (B) quantification of TMPK expression normalized to β actin (n = 3; mean ± SEM); (C) uptake of [18F]SqFLT (0.74 MBq) in WT HCT116 and HCT116shTMPK− cell lines following 60 min incubation (n = 6; mean ± SEM); (D) uptake of [18F]SqFLT (0.74 MBq) at 5 time points over 120 min (n = 3; mean ± SEM). Unpaired two-tailed T-test significance where p < 0.05 is indicated by **p <0.01 and ***p <0.001. | ||
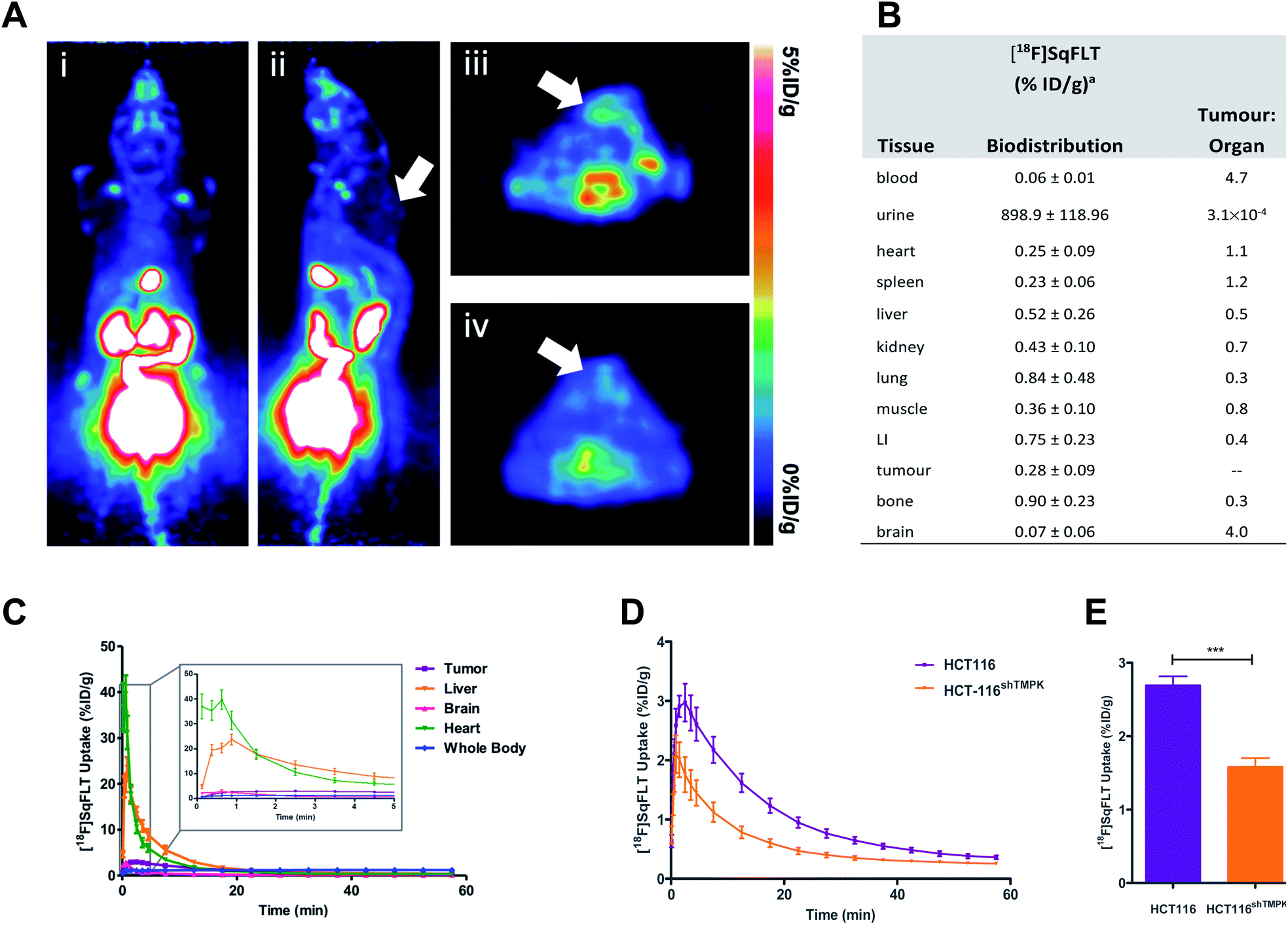 | ||
| Fig. 5 In vivo evaluation of [18F]SqFLT in female BALB/c athymic nude mice (6–8 weeks old) injected subcutaneously with either WT HCT116 or HCT116shTMPK− cells (5 × 106 cells per mouse). (A) Representative (i) coronal and (ii) sagittal PET-derived maximum intensity projection images of 60 min dynamic scans in athymic nude mice bearing HCT116 tumours (indicated by white arrow) injected with 0.74 MBq of [18F]SqFLT; (iii & iv) axial PET images of summed 60 min dynamic scans showing HCT116 tumours (indicated by white arrows). (B) Biodistribution analysis at 60 min p.i. (n = 5), LI = large intestine; (C) average time-activity curves (TACs) derived from region-of-interest analysis of PET imaging data (including detail of uptake kinetics between 0–5 min of [18F]SqFLT injection in HCT116 tumors, liver, brain, heart, whole body). Average TAC for bladder, kidneys and gallbladder are shown in the ESI (Fig. S35).† Data represent mean ± SEM (n >5) and are expressed as % of injected dose per gram (%ID g−1). (D) Time-activity curve of HCT116 and tumours derived from region-of-interest analysis of PET imaging data acquired over 60 min p.i.; (E) quantification of tumour uptake between 2–10 min post tracer injection. | ||
In vivo evaluation
The uptake of [18F]SqFLT was evaluated by dynamic PET imaging in female BALB/c athymic nude mice, used previously in our work with [18F]FLT-PET,23–25 inoculated with mutant HCT116shTMPK− and WT HCT116 cells (Fig. 5A). The overall uptake into the tumours was low (Fig. 5B), however pharmacokinetic analysis (time–activity curve) of the 60 min scan showed significant differences between mutant and wildtype xenografts (Fig. 5D and E); peak radiotracer uptake occurred at 2–10 min post-injection, with 2.69 ± 0.12 vs. 1.58 ± 0.12% ID g−1 (WT and mutant, respectively). This suggests some specificity towards TMPK, but a lack of sensitivity to depict tumour proliferative activity. [18F]SqFLT was eliminated through the kidney, liver, and bladder (Fig. 5C); partial biliary elimination was inferred from the high radioactive accumulation in the gall bladder (Fig. S35†). Efflux of [18F]SqFLT through ABCB1 transporter was discounted as a mechanism for the rapid tumoral elimination by treating HCT116 cells with verapamil (3 μM), which did not change radiotracer uptake (Fig. S34†); this neither explains potential efflux by other ABC transporters nor nucleophilic attack on squaryl by cytoplasmic thiol containing compounds. Insights from the in silico modelling demonstrated that [18F]SqFLT may fit well into the binding pocket but unfortunately cannot prove [18F]SqFLT is a substrate for phosphorylation, or a selective antagonist. If [18F]SqFLT is a substrate for phosphorylation, to explain the in vitro and in vivo data, [18F]SqFLT-PO4 must degrade back to [18F]SqFLT and diffuses out of the cell. This hypothesis needs to be further tested, however there are limited conflicting reports on the use of squaric acid phosphates as DNA precursors.3,4 While we see selective uptake in paired TMPK-high and TMPK-low cells, the evidence from this work suggests that [18F]SqFLT behaves more in line with an antagonist than a substrate.26–28 Future studies of [18F]SqFLT or analogues should consider how the sex of a mouse cancer model influences tumour uptake and tissue biodistribution, as previously reported for [18F]FLT;29 given the poor in vivo performance of [18F]SqFLT, this was not further considered as part of this study. Incubation of [18F]SqFLT with human liver microsomes (HLM) showed no metabolism over 60 min.Conclusion
The aim of this study was to evaluate the use of the squaryl moiety as a bioisostere for the phosphate group in the context of fluorine-18 radiolabelled nucleosides. We synthesised [18F]SqFLT, a nucleoside derivative of [18F]FLTMP and investigated the radiotracer in vitro and in vivo. Promising in silico data showed that [18F]SqFLT may occupy the active site of TMPK in a favourable orientation for phosphorylation or antagonism of TMPK, which encouraged the synthesis and in vitro evaluation of radiotracer. Uptake was low, but statistically significant differences between wildtype and a CRISPR/Cas9 generated TMPK knockdown cell line was observed. The in vivo PET imaging of [18F]SqFLT in wildtype and TMPK mutant xenografts showed a similar statistically significant difference in uptake, but tumour washout was rapid as a result of poor cellular retention of the tracer. Low uptake in vitro and in vivo was likely due to poor passive diffusion across the cell membrane resulting from high hydrophilicity of tracer; further in vitro studies confirmed that [18F]SqFLT was not a substrate for nucleoside transporters TK1 or ENT1/2. Taken together, the data suggest that [18F]SqFLT is not an optimal radiotracer for imaging cellular proliferation, despite it's favourable properties for targeting TMPK activity.This work presents synthetic and radiosynthetic routes to 5′-squaryl nucleosides that may be modified for other applications (i.e. synthesis of radiolabelled nucleoside dimers). In addition, the in vivo pharmacokinetics of squaryl-containing nucleosides are reported for the first time. We demonstrate the benefits and limitations of the squaryl group as a nucleoside phosphate bioisostere; while it may be considered a good mimic of the phosphate group in terms of physical properties (i.e. charge at physiological pH), it does not mimic the biological properties of the phosphate group and in this regard, it is a poor phosphate mimic.3 Further modulation of the acidity is required for [18F]SqFLT to cross the cell membrane (i.e. prodrug strategies). It is noteworthy that nucleoside derivatives are themselves hydrophilic (cLogP of [18F]FLT is −0.74) and therefore combining a charged squaryl moiety was detrimental to cell permeability in this context; this may not be the case when combined with lipophilic substrates as demonstrated in the literature.2
Experimental
Materials and methods
All reagents and solvents were purchased from commercial sources and were used without further purification unless otherwise stated. HPLC grade acetonitrile and trifluoroacetic acid (TFA), dimethyl sulfoxide (DMSO), dichloromethane (DCM), ethyl acetate (EtOAc), ethanol (EtOH) and hexane were purchased from Sigma Aldrich (Gillingham, Dorset, UK). [18F]Fluoride was produced by a GE PETrace cyclotron by 16 MeV irradiation of enriched [18O]H2O target, supplied by Alliance Medical Radiopharmacy Ltd (Warwick, UK). Automated radiosynthesis were performed using the GE FASTlab™ automated synthesis module (GE Healthcare Life Sciences, Amersham, UK). Solid phase extraction (SPE) cartridges were purchased from Waters and used according to the manufacturers recommended guidelines. Tetrabutylammonium hydrogen carbonate (TBAHC) 0.7 M in H2O was purchased from ABX GmbH (Radeberg, Germany). FLT was purchased from Carbosynth (Compton, UK). All RCY are decay-corrected to the start of synthesis, according to nomenclature guidelines.30 Radioactive semi-preparative HPLC was performed using a Shimadzu LC20-AT pump attached to a custom-built system, equipped with an Agilent Eclipse XDB-C18, 5 μm (250 × 9.4 mm) column. The mobile phase was 15% EtOH/85% H2O at a flow rate of 3 mL min−1. A detailed description of the analytical HPLC systems used in this work is presented in the ESI.† The radiosynthesis of [18F]FLT was performed on the GE FASTlab™ using commercially available synthesis cassettes (GE Healthcare, UK). Typically, [18F]fluoride (ca. 4 GBq, 2 mL), was used in the synthesis, which resulted in ca 440 MBq of [18F]FLT in >98% RCP and 10.9 ± 3.6% RCY.Molecular modelling
Gaussian 09 and UCSF Chimera were used to dock, TMP with adenosine diphosphate (ADP), and SqFLT with ADP into human TMPK.20 The structures were geometrically optimized using the oniom method. The enzyme was treated using a molecular mechanics UFF force field, while the substrates (TMP and SqFLT) were modelled with density functional theory (B3LYP/6-31G*).Chemical synthesis
:3, v/v) to give 1 as a white solid (2.2 g, 65%). 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H, NH), 7.60 (m, 1H), 7.47–7.38 (m, 2H), 7.35–7.18 (m, 7H), 6.95–6.81 (m, 4H), 6.11 (dd, J = 8.1, 2.3 Hz, 1H, H-1′), 5.20 (d, J = 3.5 Hz, 1H, 3′-OH), 4.24–4.05 (m, 2H, H-3′, H-4′), 3.73 (2× s, 6H, OCH3), 3.38 (dd, J = 10.4, 8.0 Hz, 1H, H-5′), 3.18 (dd, J = 10.3, 2.9 Hz, 1H, H-5′), 2.59–2.51 (m, 1H, H-2′), 1.86 (dd, J = 14.4, 2.5 Hz, 1H, H-2′), 1.64 (d, J = 1.1 Hz, 3H, 5-CH3). 13C NMR (101 MHz, DMSO-d6) δ 164.28 (CO-4), 158.49 (CHPh3), 150.97 (CO-2), 145.40 (CPh3), 137.25 (CH-6), 136.19 (CPh3), 135.98 (CPh3), 130.24 (CHPh3), 130.20 (CHPh3), 128.25 (CHPh3), 128.21 (CHPh3), 127.08 (CHPh3), 113.60 (CHPh3), 108.73 (C-5), 85.98 (CPh3), 84.63 (CH-4′), 83.81 (CH-1′), 69.42 (CH-3′), 63.29 (CH2-5′), 55.48 (CH3O), 41.28 (CH2-2′), 12.91 (5-CH3). HRMS (ESI) = 567.2110 (M + Na)+. Calc for C31H32N2O7Na: 567.2107.:MeOH, 97:3, v/v) to afford the product (560 mg, 80%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.21 (s, 1H, NH), 7.80 (q, J = 1.1 Hz, 1H, H-6), 6.06 (dd, J = 8.4, 2.6 Hz, 1H, H-1′), 5.23 (d, J = 3.5 Hz, 1H, 3′-OH), 4.67 (t, J = 5.7 Hz, 1H, 5′-OH), 4.31–4.17 (m, 1H, H-3′), 3.88–3.55 (m, 3H, H-4′, H-5′, H-5′′), 2.62–2.50 (m, 1H, H-2′), 1.85 (ddd, J = 14.7, 2.7, 1.0 Hz, 1H, H-2′′), 1.76 (d, J = 1.2 Hz, 3H, 5-CH3). 13C NMR (101 MHz, DMSO-d6) δ 163.81 (CO-4), 150.56 (CO-2), 137.17 (CH-6), 108.64 (C-5), 84.77 (CH-4′), 83.44 (CH-1′), 68.64 (CH-3′), 59.57 (CH2-5′), 40.67 (CH2-2′), 12.46 (5-CH3). HRMS (ESI) = 243.0977 (M + H)+. Calc. for C10H15N2O5: 243.0981.:1:1 v/v, 40 mL) was added dropwise ethylenediamine (159 μL, 2.4 mmol). The reaction mixture was stirred first at 60 °C for 1 h, then at 45 °C for 2 h, after which it was concentrated in vacuo. The resulting material was purified by silica gel column chromatography (EtOAc:MeOH:TEA, 7:3:0.1) to provide the product (126 mg, 98%) as a pale yellow solid. 1H NMR (400 MHz, deuterium oxide) δ 7.79 (q, J = 1.2 Hz, 1H, H-6), 6.17 (dd, J = 8.4, 2.9 Hz, 1H, H-1′), 4.52 (ddd, J = 5.6, 3.3, 1.0 Hz, 1H, H-3′), 4.08 (td, J = 6.1, 3.3 Hz, 1H, H-4′), 3.17 (d, J = 6.1 Hz, 2H, H-5′, H-5′′), 2.78 (ddd, J = 15.4, 8.4, 5.6 Hz, 1H, H-2′), 2.14 (ddd, J = 15.4, 3.0, 1.0 Hz, 1H, H-2′′), 1.91 (d, J = 1.2 Hz, 3H, 5-CH3). 13C NMR (101 MHz, deuterium oxide) δ 168.67 (CO-4), 153.34 (CO-2), 138.19 (CH-6), 111.05 (C-5), 85.20 (CH-4′), 83.20 (CH-1′), 69.74 (CH-3′), 39.98 (CH2-5′), 39.33 (CH2-2′), 11.83 (5-CH3). HRMS (ESI) = 242.1140 (M + H)+. Calc. for C10H16N3O4: 242.1141.:1 mixture of THF/H2O (1.5 mL) NaHCO3 (93.3 mg, 1.11 mmol) and Boc2O (96.9 mg, 0.44 mmol) were added consecutively at 0 °C. After 1 h at 0 °C, the solution was stirred at RT for 2 h. After which time it was filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography with 100% EtOAc to provide the product (80 mg, 64%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.22 (s, 1H, NH), 7.79 (m, 1H, H-6), 6.89 (t, J = 6.0 Hz, 1H, NH-Boc), 6.03 (dd, J = 8.4, 2.4 Hz, 1H, H-1′), 5.26 (d, J = 3.3 Hz, 1H, 3′-OH), 4.17 (dt, J = 6.2, 3.2 Hz, 1H, H-3′), 3.79 (td, J = 6.4, 3.0 Hz, 1H, H-4′), 3.25 (t, J = 5.9 Hz, 2H, H-5′, H-5′′), 2.57 (ddd, J = 14.2, 8.5, 5.4 Hz, 1H, H-2′), 1.87 (dd, J = 14.8, 2.5 Hz, 1H, H-2′′), 1.77 (d, J = 1.2 Hz, 3H, 5-CH3), 1.38 (s, 9H, boc). 13C NMR (101 MHz, DMSO-d6) δ 163.81 (CO-4), 155.95 (CO-Boc), 150.53 (CO-2), 137.12 (CH-6), 108.68 (C-5), 83.53 (CH-4′), 82.49 (CH-1′), 77.97 (C-Boc), 68.55 (CH-3′), 40.31 (CH2-5′), 39.20 (CH2-2′), 28.21 (CH3-Boc), 12.44 (5-CH3). HRMS (ESI) = 342.1671 (M + H)+. Calc. for C15H24N3O6: 342.1665.13C NMR (101 MHz, DMSO) δ 163.61 (CO), 163.47 (CO), 150.33 (CO-2), 138.24 (TCP-C), 136.44 (CH-6), 128.26 (TCP-C), 128.13 (TCP-C), 109.68 (C-5), 94.04 (d, J = 176.1 Hz, CH-3′), 84.60 (CH-1′), 80.76 (d, J = 25.5 Hz, CH-4′), 40.21 (d, J = 10.3 Hz, CH2-5′), 35.77 (d, J = 20.2 Hz, CH2-2′), 11.89 (5-CH3). HRMS (ESI) = 509.9588 (M + H)+. Calc. for C18H13N3O5CI4F: 509.9593.
:1:1, 20 mL) was added dropwise ethylenediamine (293 μL, 4.4 mmol). The reaction mixture was stirred at RT for 2 h, after which time it was concentrated in vacuo. The resulting material was purified by silica gel column chromatography (EtOAc:MeOH:TEA, 7:3:1%) to provide the product (158 mg, 66%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (q, J = 1.3 Hz, 1H, H-6), 6.16 (dd, J = 8.3, 6.6 Hz, 1H, H-1′), 5.43–5.20 (m, 1H, H-3′), 4.01 (dt, J = 28.3, 5.2 Hz, 1H, H-4′), 3.01–2.57 (m, 2H, H-5′, H-5′′), 2.46–2.25 (m, 2H, H-2′, H-2′′), 1.79 (d, J = 1.2 Hz, 3H, CH3-5). 19F NMR (376 MHz, DMSO-d6) δ −173.92. 13C NMR (101 MHz, DMSO-d6) δ 163.64 (CO-4), 150.48 (CO-2), 136.03 (CH-6), 109.78 (C-5), 94.67 (d, J = 173.6 Hz, CH-3′), 85.54 (d, J = 22.5 Hz, CH-4′), 83.61 (CH-1′), 42.96 (d, J = 10.0 Hz, CH2-5′), 36.18 (d, J = 20.3 Hz, CH2-2′), 12.13 (CH3-5). HRMS (ESI) = 244.1097 (M + H)+. Calc. for C10H15N3O3F: 244.1097.:MeOH, 95:5) to provide the product (116 mg, 55%) as a white solid. 1H NMR (400 MHz, DMSO-d6) presence of rotamers is observed: δ 11.38 (s, 1H, NH-3), 8.93 (s, 0.5H, NH), 8.75 (s, 0.5H, NH), 7.40 (m, 1H, H-6), 6.16 (q, J = 7.0 Hz, 1H, H-1′), 5.61–5.02 (m, 1H, H-3′), 4.65 (2× q, 2H, sqCH2), 4.27–4.01 (m, 1H, H-4′), 3.93–3.55 (m, 2H, H-5′, H-5′′), 2.49–2.20 (m, 2H, H-2′, H-2′′), 1.77 (, 3H, CH3-5), 1.35 (2× t, 3H, 2× sqCH3). 19F NMR (376 MHz, DMSO-d6) δ −175.01 and −175.50. 13C NMR (101 MHz, DMSO-d6) δ 189.26, 188.93 (Sq-C), 182.56, 182.26 (Sq-C), 177.42, 176.95 (Sq-C), 173.17, 172.41 (Sq-C), 163.56 (CO-4), 150.37(CO-2), 136.05, 135.90 (CH-6), 109.89 (C-5), 93.47 (d, J = 175.7 Hz, CH-3′), 84.06 (CH-1′), 82.65 (m, CH-4′), 68.94 (CH2-sq), 44.85 (m, CH2-5′), 35.57 (m, CH2-2′), 15.56 (CH3-sq), 12.04, 11.98 (CH3-5). HRMS (ESI) = 368.1249 (M + H)+. Calc. for C16H19N3O6F: 368.1258.:1 v/v) was added to the reactor and heated at 125 °C for 30 min. Once cooled, the reaction mixture was diluted in water (40 mL) and passed through an Oasis Plus HLB cartridge [pre-conditioned with EtOH (5 mL) and water (10 mL)]. Compound [18F]7 was retained on the cartridge and eluted manually into a clean Wheaton vial with EtOH (500 μL). Phosphoric acid (1 mL, 2.29 M) was added and the reaction was heated at 75 °C for 25 min to hydrolyse [18F]7 into [18F]9. Once cooled, the reaction mixture was neutralized by the addition of NH3 in MeOH (350 μL), followed by the addition of 3,4-diethoxy-3-cyclobutene-1,2-dione (15 μL) to give [18F]10. The resulting mixture was heated to 50 °C for 40 min and diluted into water (10 mL) for semi-preparative radio-HPLC purification. [18F]10 was collected into water (10 mL) and passed through an Oasis (30 mg) HLB cartridge [pre-conditioned with EtOH (5 mL) and water (10 mL)]. The cartridge was eluted in a minimum volume of EtOH (300 μL) into a clean Wheaton vial containing HCl (4 M, 200 μL). The reaction mixture was heated to 85 °C for 40 min, cooled and neutralized using NaOH (4 M) to give [18F]SqFLT (59.3 ± 15.6 MBq) for further evaluation.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) D7.5 determination. To a 1.5 mL microcentrifuge tube was added PBS pH 7.5 (500 μL) and octanol (500 μL). The tube was shaken for 15 min, followed by centrifugation (13000g, 5 min). [18F]SqFLT (3 MBq) in PBS (<5% EtOH) was added and the samples shaken for 30 min, followed by centrifugation (13000g, 5 min). Aliquots from the octanol layer and PBS layer were removed (100 μL) and placed in counting tubes. Radioactivity was measured using a γ-counter and the partition coefficient was calculated using LogD7.5 = log10[Oct/PBS]. The LogD7.5 value was reported as mean ± SD (n = 3, triplicate analysis).000 cells per well or 1500000 cells (per 15 cm petri dish for HPLC) for the stated cell lines. All cells were grown for 24 h in a volume of 2 mL per well or 10 mL per petri dish unless stated otherwise. All cells were maintained at 60–70% confluence prior to experiments, passaged in our laboratory for fewer than 6 months on receipt and were tested mycoplasma free. Experiments were performed using cells under 10 passages.000g, 3 min), and the supernatant removed and filtered (0.2 μm). The supernatant was diluted (1:9) with H2O + 0.1% TFA and injected onto a HPLC instrument bearing a 1 mL injection loop and PosiRam (LabLogic, Sheffield, UK) metabolite detector. Samples of a known volume were taken from the supernatant and the entire protein pellet were counted using an auto-gamma counter (Perkin Elmer, London, UK) to determine the extraction efficiency. All experiments were performed in triplicate (n = 3).
D7.5 determination. To a 1.5 mL microcentrifuge tube was added PBS pH 7.5 (500 μL) and octanol (500 μL). The tube was shaken for 15 min, followed by centrifugation (13000g, 5 min). [18F]SqFLT (3 MBq) in PBS (<5% EtOH) was added and the samples shaken for 30 min, followed by centrifugation (13000g, 5 min). Aliquots from the octanol layer and PBS layer were removed (100 μL) and placed in counting tubes. Radioactivity was measured using a γ-counter and the partition coefficient was calculated using LogD7.5 = log10[Oct/PBS]. The LogD7.5 value was reported as mean ± SD (n = 3, triplicate analysis).000 cells per well or 1500000 cells (per 15 cm petri dish for HPLC) for the stated cell lines. All cells were grown for 24 h in a volume of 2 mL per well or 10 mL per petri dish unless stated otherwise. All cells were maintained at 60–70% confluence prior to experiments, passaged in our laboratory for fewer than 6 months on receipt and were tested mycoplasma free. Experiments were performed using cells under 10 passages.000g, 3 min), and the supernatant removed and filtered (0.2 μm). The supernatant was diluted (1:9) with H2O + 0.1% TFA and injected onto a HPLC instrument bearing a 1 mL injection loop and PosiRam (LabLogic, Sheffield, UK) metabolite detector. Samples of a known volume were taken from the supernatant and the entire protein pellet were counted using an auto-gamma counter (Perkin Elmer, London, UK) to determine the extraction efficiency. All experiments were performed in triplicate (n = 3).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Imperial College NIHR Biomedical Research Centre award (WSCC_P62585), Cancer Research UK grant (C2536/A16584), Medical Research Council grant (MC-A652-5PY80), and Experimental Cancer Medicine Centres grant (C37/A7283). We would like to thank “The Friends of Hammersmith Hospital” (Registered Charity No: 209182) for the funding the purchase of our PosiRAM radio-metabolite detector. Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. We thank Ning Wang for contributing towards compound characterisation.Notes and references
- N. A. Meanwell, Top. Med. Chem., 2014, 9, 283–381 CrossRef.
- P. Lassalas, B. Gay, C. Lasfargeas, M. J. James, V. Tran, K. G. Vijayendran, K. R. Brunden, M. C. Kozlowski, C. J. Thomas, A. B. Smith, D. M. Huryn and C. Ballatore, J. Med. Chem., 2016, 59, 3183–3203 CrossRef CAS.
- E. M. Dürr, W. Doherty, S. Y. Lee, A. H. El-Sagheer, A. Shivalingam, P. J. McHugh, T. Brown and J. F. McGouran, ChemistrySelect, 2018, 3, 12824–12829 CrossRef PubMed.
- K. Sato, K. Seio and M. Sekine, J. Am. Chem. Soc., 2002, 124, 12715–12724 CrossRef CAS PubMed.
- K. Seio, T. Miyashita, K. Sato and M. Sekine, Eur. J. Org. Chem., 2005, 24, 5163–5170 CrossRef.
- M. Lu, Q. Bin Lu and J. F. Honek, Bioorg. Med. Chem. Lett., 2017, 27, 282–287 CrossRef CAS PubMed.
- J. Xie, A. B. Comeau and C. T. Seto, Org. Lett., 2004, 6, 83–86 CrossRef CAS PubMed.
- P. C. M. Chan, R. J. Roon, J. F. Koerner, N. J. Taylor and J. F. Honek, J. Med. Chem., 1995, 38, 4433–4438 CrossRef CAS PubMed.
- L. Allott and E. O. Aboagye, Mol. Pharm., 2020, 17, 2245–2259 CrossRef CAS PubMed.
- H. Barthel, M. C. Cleij, D. R. Collingridge, O. C. Hutchinson, S. Osman, Q. He, S. K. Luthra, F. Brady, P. M. Price and E. O. Aboagye, Cancer Res., 2003, 63, 3791–3798 CAS.
- A. F. Shields, J. R. Grierson, O. Muzik, J. C. Stayanoff, J. M. Lawhorn-Crews, J. E. Obradovich and T. J. Mangner, Mol. Imaging Biol., 2002, 4, 83–89 CrossRef.
- L. M. Kenny, D. M. Vigushin, A. Al-Nahhas, S. Osman, S. K. Luthra, S. Shousha, R. C. Coombes and E. O. Aboagye, Cancer Res., 2005, 65, 10104–10112 CrossRef CAS PubMed.
- V. Vanheusden, H. Munier-Lehmann, S. Pochet, P. Herdewijn and S. Van Calenbergh, Bioorg. Med. Chem. Lett., 2002, 12, 2695–2698 CrossRef CAS PubMed.
- B. Roy, A. Depaix, C. Périgaud and S. Peyrottes, Chem. Rev., 2016, 116, 7854–7897 CrossRef CAS PubMed.
- Y. Mehellou, H. S. Rattan and J. Balzarini, J. Med. Chem., 2018, 61, 2211–2226 CrossRef CAS PubMed.
- C. McGuigan, R. N. Pathirana, N. Mahmood, K. G. Devine and A. J. Hay, Antiviral Res., 1992, 17, 311–321 CrossRef CAS.
- A. Cavaliere, K. C. Probst, S. J. Paisey, C. Marshall, A. K. H. Dheere, F. Aigbirhio, C. McGuigan and A. D. Westwell, Molecules, 2020, 25, 704 CrossRef CAS PubMed.
- M. A. Moroz, T. Kochetkov, S. Cai, J. Wu, M. Shamis, J. Nair, E. De Stanchina, I. Serganova, G. K. Schwartz, D. Banerjee, J. R. Bertino and R. G. Blasberg, Clin. Cancer Res., 2011, 17, 1099–1110 CrossRef CAS PubMed.
- D. J. Frisch, M. J. Trucks, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Scalmani, G. Barone, V. Mennucci, B. Petersson, G. A. Nakatsuji, H. Caricato, M. Li, X. Hratchian, H. P. Izmaylov, A. F. Bloino, J. Zheng and G. Sonnenber, Gaussian, Inc., Wallingford CT, 2009.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H. B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- D. W. Kim, D. S. Ahn, Y. H. Oh, S. Lee, H. S. Kil, S. J. Oh, S. J. Lee, J. S. Kim, J. S. Ryu, D. H. Moon and D. Y. Chi, J. Am. Chem. Soc., 2006, 128, 16394–16397 CrossRef CAS PubMed.
- J. Leyton, G. Smith, M. Lees, M. Perumal, Q. De Nguyen, F. I. Aigbirhio, O. Golovko, Q. He, P. Workman and E. O. Aboagye, Mol. Cancer Ther., 2008, 7, 3112–3121 CrossRef CAS PubMed.
- J. Leyton, J. P. Alao, M. Da Costa, A. V. Stavropoulou, J. R. Latigo, M. Perumal, R. Pillai, Q. He, P. Atadja, E. W. F. Lam, P. Workman, D. M. Vigushin and E. O. Aboagye, Cancer Res., 2006, 66, 7621–7629 CrossRef CAS PubMed.
- K. Heinzmann, Q. D. Nguyen, D. Honess, D. M. Smith, S. Stribbling, D. Brickute, C. Barnes, J. Griffiths and E. Aboagye, J. Nucl. Med., 2018, 62, 1558–1565 CrossRef PubMed.
- S. he Huang, A. Tang, B. Drisco, S. Q. Zhang, R. Seeger, C. Li and A. Jong, DNA Cell Biol., 1994, 13, 461–471 CrossRef PubMed.
- P. Y. Ke, Y. Y. Kuo, C. M. Hu and Z. F. Chang, Genes Dev., 2005, 19, 1920–1933 CrossRef CAS PubMed.
- C. M. Hu, M. T. Yeh, N. Tsao, C. W. Chen, Q. Z. Gao, C. Y. Chang, M. H. Lee, J. M. Fang, S. Y. Sheu, C. J. Lin, M. C. Tseng, Y. J. Chen and Z. F. Chang, Cancer Cell, 2012, 22, 36–50 CrossRef CAS PubMed.
- S. R. Chan, K. Salem, J. Jeffery, G. L. Powers, Y. Yan, K. I. Shoghi, A. M. Mahajan and A. M. Fowler, J. Nucl. Med., 2018, 59, 833–838 CrossRef CAS PubMed.
- H. H. Coenen, A. D. Gee, M. Adam, G. Antoni, C. S. Cutler, Y. Fujibayashi, J. M. Jeong, R. H. Mach, T. L. Mindt, V. W. Pike and A. D. Windhorst, Nucl. Med. Biol., 2017, 55, 5–11 CrossRef PubMed.
- M. Yun, S. J. Oh, H. J. Ha, J. S. Ryu and D. H. Moon, Nucl. Med. Biol., 2003, 30, 151–157 CrossRef CAS PubMed.
- Z. J. Jia, S. Kelberlau, L. Olsson, G. Anilkumar and B. Fraser-Reid, Synlett, 1999, 5, 565–566 CrossRef.
- P. D. Hsu, E. S. Lander and F. Zhang, Cell, 2014, 157, 1262–1278 CrossRef CAS PubMed.
- S. Stella, P. Alcon and G. Montoya, Nat. Struct. Mol. Biol., 2017, 24, 882 CrossRef CAS PubMed.
- L. Allott, C. Miranda, A. Hayes, F. Raynaud, C. Cawthorne and G. Smith, EJNMMI Radiopharm. Chem., 2019, 4, 1–20 CrossRef PubMed.
- P. Workman, E. O. Aboagye, F. Balkwill, A. Balmain, G. Bruder, D. J. Chaplin, J. A. Double, J. Everitt, D. A. H. Farningham, M. J. Glennie, L. R. Kelland, V. Robinson, I. J. Stratford, G. M. Tozer, S. Watson, S. R. Wedge, S. A. Eccles, V. Navaratnam and S. Ryder, Br. J. Cancer, 2010, 102, 1555–1577 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00205h |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |