
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-catalyzed synthesis of spirooxindoles
P. V. Saranya
a,
Mohan Neetha
a,
Thaipparambil Aneeja
a and
Gopinathan Anilkumar
 *abc
*abc
aSchool of Chemical Sciences, Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala 686560, India. E-mail: anilgi1@yahoo.com; anil@mgu.ac.in
bAdvanced Molecular Materials Research Centre (AMMRC), Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala 686560, India
cInstitute for Integrated Programmes and Research in Basic Sciences (IIRBS), Mahatma Gandhi University, Priyadarsini Hills P O, Kottayam, Kerala 686560, India
First published on 10th February 2021
Abstract
Spirooxindole is a principal bioactive agent and is observed in several natural products including alkaloids. They are broadly studied in the pharmaceutical field and have a significant role in the evolution of drugs such as anti-viral, anti-cancer, anti-microbial etc. In organic chemistry, an indispensable role is presented by transition metal catalysts. An effective synthetic perspective to spirooxindoles is the use of transition metals as the catalyst. This review discusses the synthesis of spirooxindoles catalyzed by transition metals and covers literature up to 2020.
 P. V. Saranya | P. V. Saranya was born in 1997 in Kerala, India. She received her B. Sc. degree from Kannur University (Payyanur College, Payyanur) and M. Sc. degree from the Department of Applied Chemistry, Cochin University of Science and Technology. She qualified in the CSIR-UGC National Eligibility Test 2019 with a research fellowship. Currently she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Mohan Neetha | Mohan Neetha was born in 1996 in Ernakulam, Kerala, India. She pursued her B.Sc. Degree from St. Albert's College, Ernakulam in 2016 and her M.Sc. Degree from Sacred Heart College, Thevara in 2018. She acquired First rank in B.Sc. Chemistry Examination held in 2016 from Mahatma Gandhi University, Kottayam. She qualified KSCSTE Fellowship-2018 as well as CSIR-UGC National Eligibility Test-2019 and is presently doing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Thaipparambil Aneeja | Thaipparambil Aneeja was born in Chittariparamba, Kannur, Kerala, India. She obtained her B.Sc. degree from Kannur University (Nirmalagiri College, Kuthuparamba) in 2016 and her M.Sc. degree from the Department of Applied Chemistry, Cochin University of Science and Technology in 2018. She passed the CSIR-UGC National Eligibility Test 2019 for a research fellowship. Currently she is pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his Ph. D in 1996 from Regional Research Laboratory (renamed as National Institute for Interdisciplinary Science and Technology NIIST-CSIR), Trivandrum with Dr Vijay Nair. He did postdoctoral studies at University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 110 papers in peer-reviewed journals, 7 patents, eight book chapters and edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, in press). He has received Dr S. Vasudev Award from Govt. of Kerala, India for best research (2016) and Evonik research proposal competition award (second prize 2016). His h-index is 29. |
1 Introduction
Compounds having at least two molecular rings with one common atom are called spiro compounds. They can be heterocyclic or fully carbocyclic. The spiro atom is the one which connects the two or three rings. These compounds are useful in the synthesis of various novel therapeutic agents.Spirooxindole is a type of spiro compound which has a prominent role in synthetic and pharmaceutical chemistry. Several natural products such as horsfiline,1 alantrypinone,2 elacomine and isoelacomine3 contain spirooxindole framework in their structure. Spirooxindoles have wide applications as anti-cancer,4,5 anti-inflammatory,6 anti-microbial,7,8 anti-oxidant,9,10 anti-viral,11 and anti-malarial12 agents (Fig. 1).
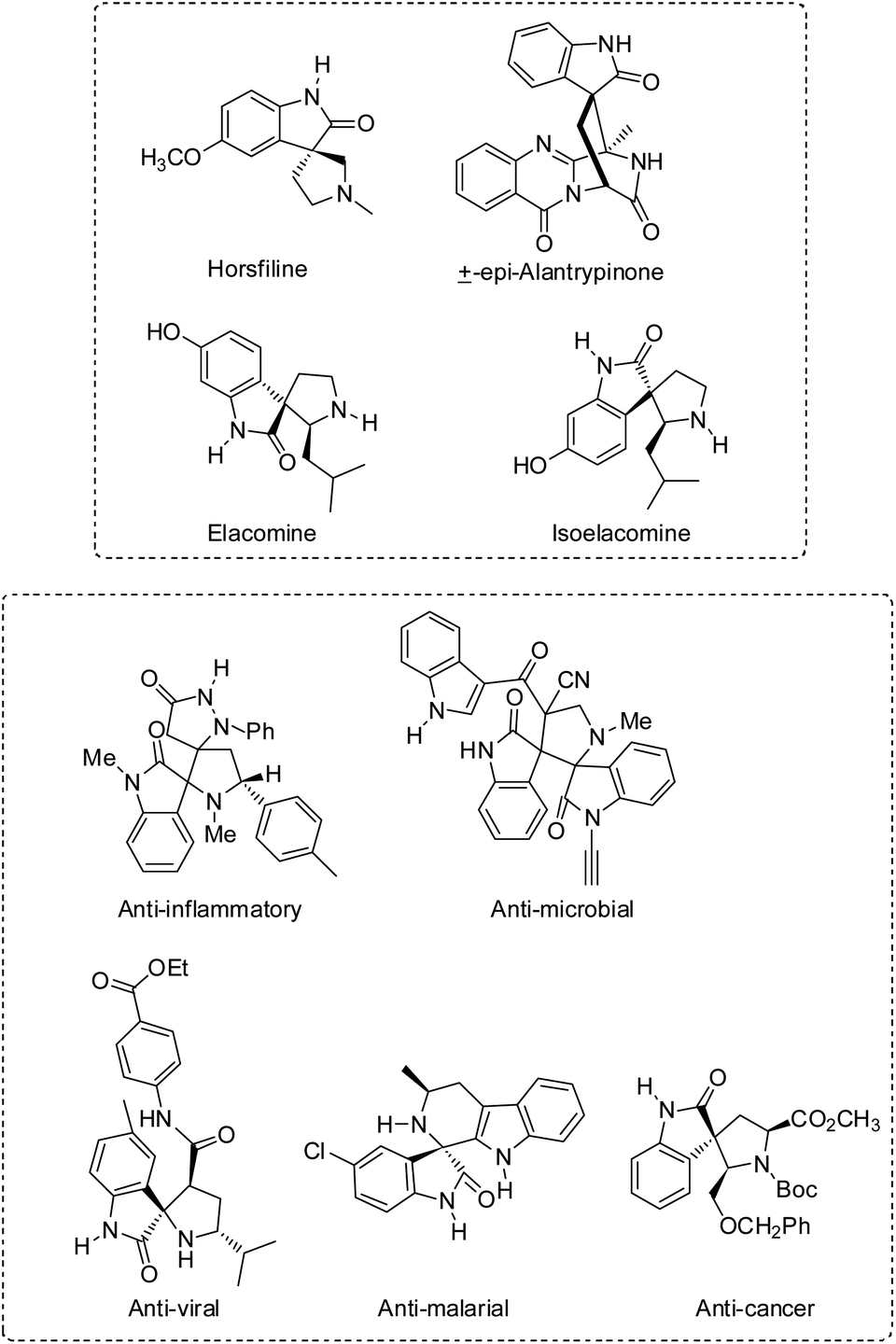 | ||
| Fig. 1 Examples of natural products and bioactive molecules containing a spirooxindole core. | ||
Spirooxindole synthesis is a rapidly developing research area wherein the strategies towards enantioselective synthesis is undergoing large-scale investigations.13–15 Methyleneindolinones,16 isatin derivatives17 etc. are widely employed as the starting materials for spirooxindole synthesis. Expeditious development occurred in the field of spirooxindole synthesis from 2012 to 2020.18–20
The synthesis of spirooxindoles catalyzed by different reagents has been reported which includes iodine/H2O2,21 amine,22 β-cyclodextrin,23 L-proline,24 ethylenediaminediacetate,25 imidazole,26 citric acid27 and many others. Transition metals are efficient catalysts, as they are facile in losing and gaining of electrons, and most of them are malleable, ductile and easily available. In organic synthesis, transition metal-catalyzed reactions have advantages like gentle reaction conditions and are compatible with an extensive span of functionalities. Consequently, transition metal-catalyzed approaches are deliberated most assiduously. Ligand exchange, elimination, coordination etc. are the different modes through which the transition metal catalyst can stimulate the starting materials. Ligand alteration is a suitable pathway for changing the selectivities of these catalysts. Recently, investigations in the transition metal-catalyzed synthesis of spirooxindoles are advancing rapidly.
Previously, reviews were reported on the synthesis of spirooxindole via green protocols.28 In addition, several other reviews are available on the catalytic asymmetric synthesis of spirooxindole.29–31 In the present review, we highlight the transition metal-catalyzed synthesis of spirooxindoles up to 2020. For better conception, the review is categorized based on the transition metal catalyst used and subcategorized according to the starting materials.
2 Ag-catalyzed spirooxindole synthesis
Silver has helped in the development of novel reactions at a much lower cost compared to platinum and gold. The construction of both intramolecular and intermolecular bonds can be intermediated by Ag catalysts.32 Silver can manifest Ag(I)/Ag(III) redox chemistry33 and can function as one-electron oxidant. Several of the silver catalyst can effectively activate C–H bond present in the substrate to generate various useful organic scaffolds.Spirooxindoles containing pyrrolidine and nitrile were synthesized by utilizing various silver catalysts from 2011 to 2018. Among the various silver catalysts used, Ag nanoparticle-catalyzed reactions are economic because the catalyst can be separated effortlessly and there is no necessity for any ligands. Large surface area is an assisting factor in their activity and selectivity. The other catalysts employed include – AgOAc, AgF and bimetallic catalyst of Ag(I) with Pd(0).
2.1 Reaction involving oxoindoline derivatives
Wang et al. proposed a method for the building up of spirooxindole-pyrrolidines 3 carrying four vicinal stereogenic centers from azomethine ylides 1 and 2-oxoindolin-3-ylidene 2 which are N-unprotected, through 1,3-dipolar cycloaddition catalyzed by AgOAc/TF-BiphamPhos complexes.34 They could attain finer diastereo-, enantio- and regioselectivity with the catalyst. The reaction involved 15 mol% of triethylamine, 5 mol% of AgOAc/(S)-TF-BiphamPhos (L1) in dichloromethane at room temperature (Scheme 1). High to excellent yields of adducts with enantioselectivities 50–71% and diastereoselectivities >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :<2 were obtained via the reaction between 2-oxoindolin-3-benzylidene and imino esters gained from aromatic aldehydes. The enantioselectivities were not significantly affected by the electronic and positional properties of aromatic ring substituents. 92% of adduct with 50% ee were afforded by imino esters derived from aliphatic cyclohexane carbaldehyde. Good yields and diastereo- and enantioselectivities were attained with electron-deficient and electron-rich substituents on the benzene ring of 2-oxoindolin-3-ylidenes. Moderate enantioselectivity and high diastereo- and regioselectivity were afforded by the adduct from methyl oxoindolylidene acetate derived from Wittig reagent and isatin.
:<2 were obtained via the reaction between 2-oxoindolin-3-benzylidene and imino esters gained from aromatic aldehydes. The enantioselectivities were not significantly affected by the electronic and positional properties of aromatic ring substituents. 92% of adduct with 50% ee were afforded by imino esters derived from aliphatic cyclohexane carbaldehyde. Good yields and diastereo- and enantioselectivities were attained with electron-deficient and electron-rich substituents on the benzene ring of 2-oxoindolin-3-ylidenes. Moderate enantioselectivity and high diastereo- and regioselectivity were afforded by the adduct from methyl oxoindolylidene acetate derived from Wittig reagent and isatin.
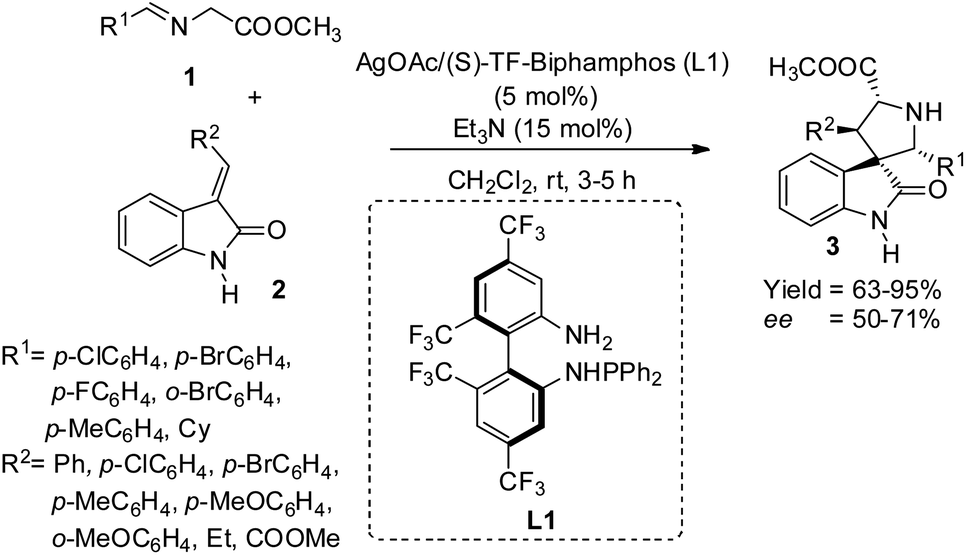 | ||
| Scheme 1 Synthesis of spirooxindole-pyrrolidines from azomethine ylides and derivatives of N-unprotected 2-oxoindoline-3-ylidene. | ||
Wide substrate scope was a major highlight of the reaction involving oxoindoline derivatives.
2.2 Reaction involving alkene derivatives
A novel method for the synthesis of nitrile-containing spirooxindoles 5 mediated by AgF was developed by Liu and co-worker.35 Activated alkenes 4 were dialkylated with acetonitrile as solvent, 4 equiv. of AgF as catalyst in an atmosphere of nitrogen at 110 °C in which the C–H bond of acetonitrile was activated by AgF (Scheme 2). Moderate yield with diastereoselectivities 1.1–1.2:1 were obtained when indoles having alkyl groups on nitrogen were used as the substrates. The corresponding products were not yielded by substrates with electron-deficient indole or with simple indole. Moderate yield was obtained with substrate in which the linked nitrogen bears a tBu group. The yield of the product was not significantly affected by the aromatic ring substituents. When tBu was on alkene moiety the product was not obtained, but phenyl group gave 51% of product. The diastereomeric ratio could be increased to 4–4.9:1 when 0.2 mmol K2CO3 was added at a lowered reaction temperature of 80 °C (Scheme 3).
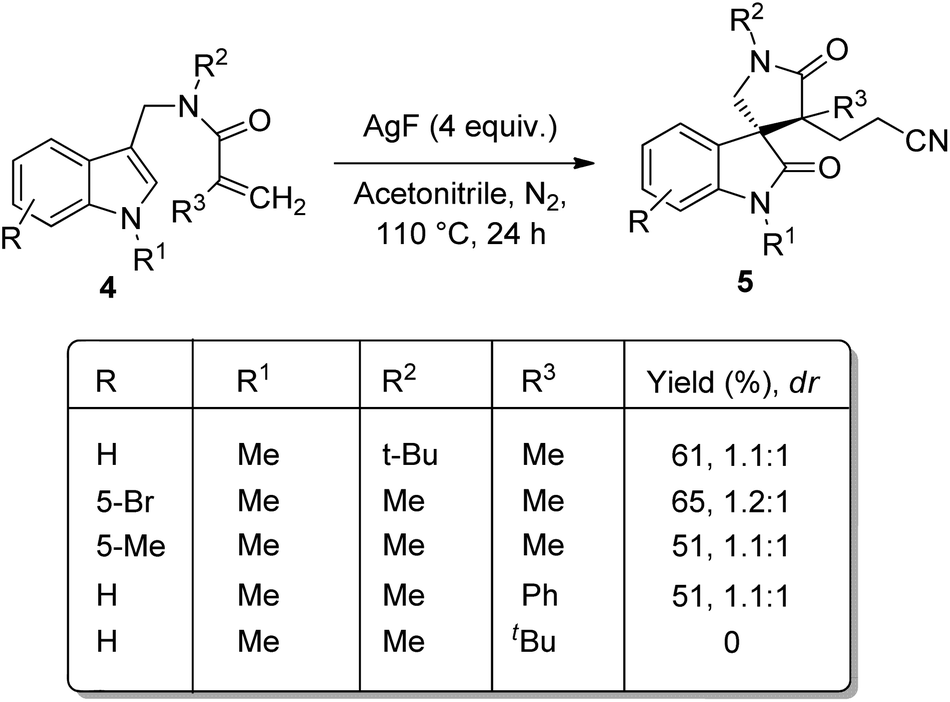 | ||
| Scheme 2 Yield and diastereomeric ratio for the synthesis of nitrile-containing spirooxindoles through AgF mediated dialkylation. | ||
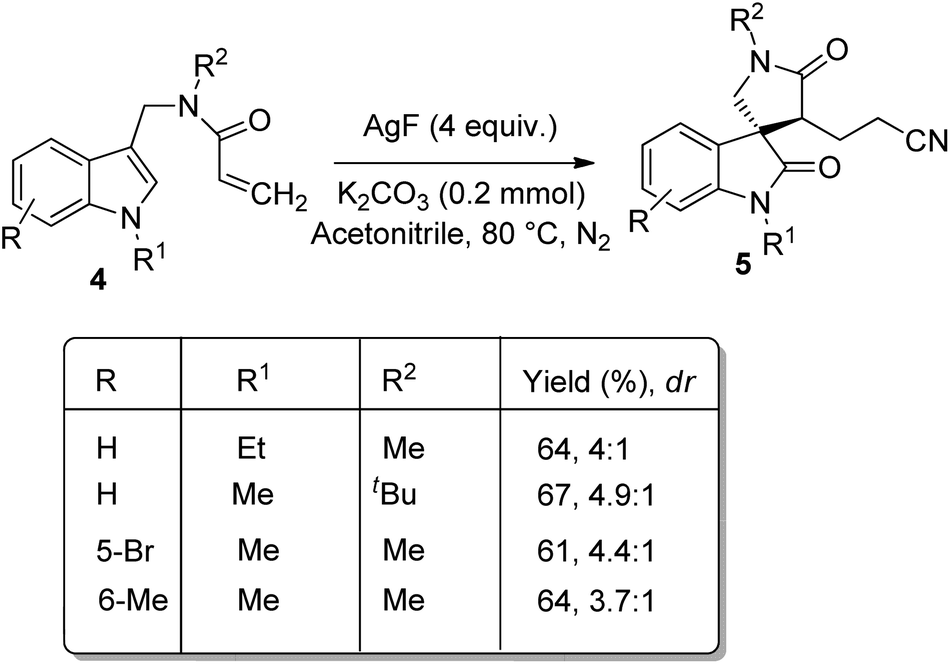 | ||
| Scheme 3 Yield and diastereomeric ratio for the synthesis of nitrile-containing spirooxindoles with the addition of K2CO3 at 80 °C. | ||
Further explorations in this field of spirooxindole synthesis using alkene as starting materials did not expand much after 2014.
2.3 Reactions involving isatin derivatives
A method for the synthesis of spirooxindoles catalyzed by silver nanoparticles was established.36 It is a green approach in which the aqueous extract of the leaves of Ferula latisecta was used to make up Ag nanoparticles. A series of spirooxindoles 9 were synthesised using the condensation reaction between isatin 6, β-diketone 7 and enamines 8, catalyzed by Ag nanoparticles in water as solvent at 90 °C (Scheme 4). Spirooxindoles were obtained in a reaction time of 15–30 minutes in high to excellent yields.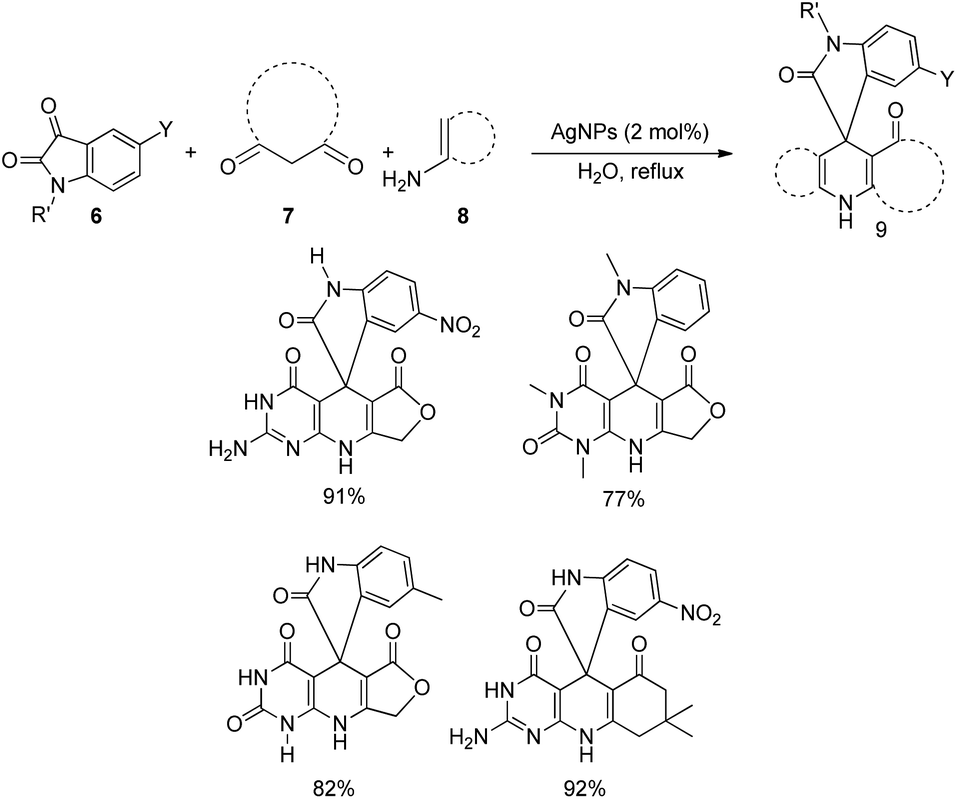 | ||
| Scheme 4 Synthesis of spirooxindoles catalyzed by silver nanoparticles. | ||
Millington and co-workers reported a bimetallic Pd(0)/Ag(I)-catalyzed synthesis of spirooxindoles, epi-spirotryprostatin A and its analogues.37 There are two approaches in which the first one, produces spirooxindoles via Peterson olefination followed by 1,3-dipolarcycloaddition. Here initially, a β-hydroxysilane 12 was obtained by the reaction between trimethylsilylmethyl magnesium chloride 10 and N-methyl isatin 6 followed by the addition of NH4Cl solution 11. In the second step, the β-hydroxysilane 12 was treated with potassium hydride 13 and triethylamine 14, in the presence of 10 mol% AgO to form an intermediate 15. Mixture of stereoisomers of spirooxindoles (17 and 18) were obtained in a 1:2 ratio by the reaction between the intermediate and the imine 16. The cycloaddition was catalyzed by Ag(I)oxide in Et3N, KH and toluene at 0–20 °C.
In the second approach, reaction between acryloyl chloride 20 and o-iodoaniline 19 was succeeded by an N-methylation to give precursor 21, which then underwent intra-molecular Heck reaction using Pd(OAc)2/PPh3. Spirooxindoles 22 were obtained with endo-selectivity with isomer ratios 4.2:1 to >9:1 through cycloaddition cascade process between the intermediate 15 and imine 16, catalyzed by Ag(I). Both the approaches are portrayed in Scheme 5.
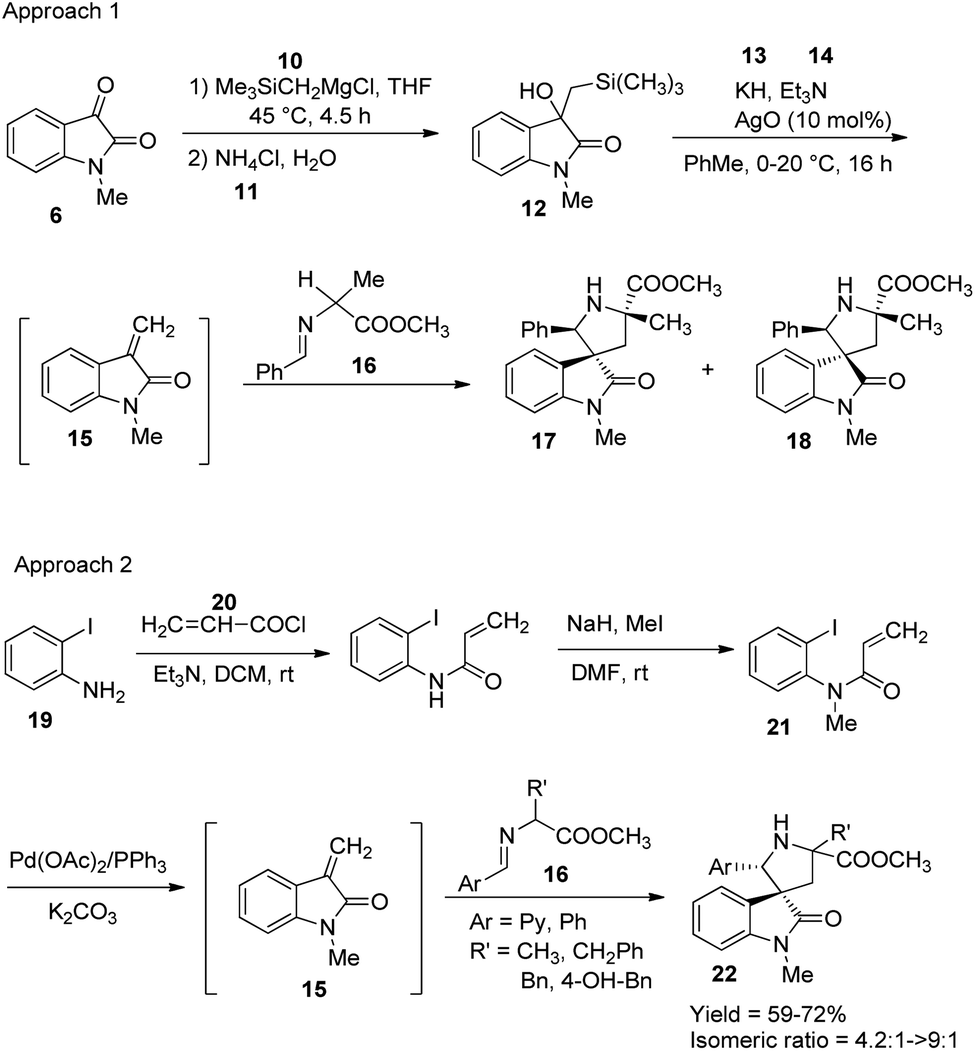 | ||
| Scheme 5 Synthesis of spirooxindole via Peterson olefination/1,3-dipolar cycloaddition and intramolecular Heck/1,3-dipolar cycloaddition. | ||
The group also synthesised epi-spirotryprostatin A and its analogues using the second approach. The products were obtained as a stereoisomeric mixture in which the product of cycloaddition of syn-dipole led to the minor isomer (Scheme 6).
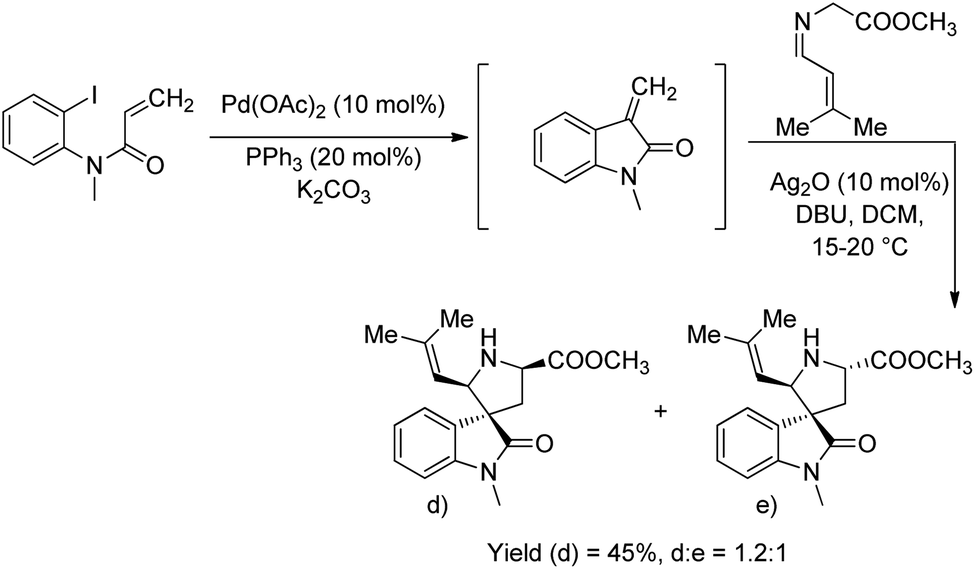 | ||
| Scheme 6 Synthesis of epi-spirotryprostatin A and its analogues. | ||
3 Mn-catalyzed spirooxindole synthesis
Manganese is an excellent candidate as a catalyst due to its earth-abundance, cheapness and non-toxicity. In organic reactions, catalyst derived from manganese could gain a ubiquitous role because of the higher activity and cost-effectiveness. The electrophilic trait in starting materials, can be raised by some Mn catalysts because of their Lewis acidic nature.38 Immense electron mobility could be observed in many of the manganese derived catalysts.Various reports for the synthesis of spirooxindoles from isatin derivatives, catalyzed by manganese were published in 2013 and 2014. In all these cases, the catalyst used was manganese ferrite nanoparticles. The importance of this catalyst lies in its uncomplicated recoverability and reusability up to many cycles of reaction.
3.1 Reactions involving isatin derivatives
In 2013, Naeimi and co-workers demonstrated an environmentally benign method for the construction of novel spirooxindoles 25 by means of a one-pot procedure.38 The reaction was effected by the condensation between anilinolactone 23, isatin 6 and dimedone 24 with 5 mol% of MnFe2O4 NPs as catalyst, water as solvent at 90 °C (Scheme 7). The most important advantage of this procedure is that, the catalyst is green, can be recovered magnetically and can be reused. The reaction time was high in the case of anilinolactones having electron-deficient substituents on the aryl ring compared to electron-rich ones. The yield was not significantly affected by the nature of substituents on isatin.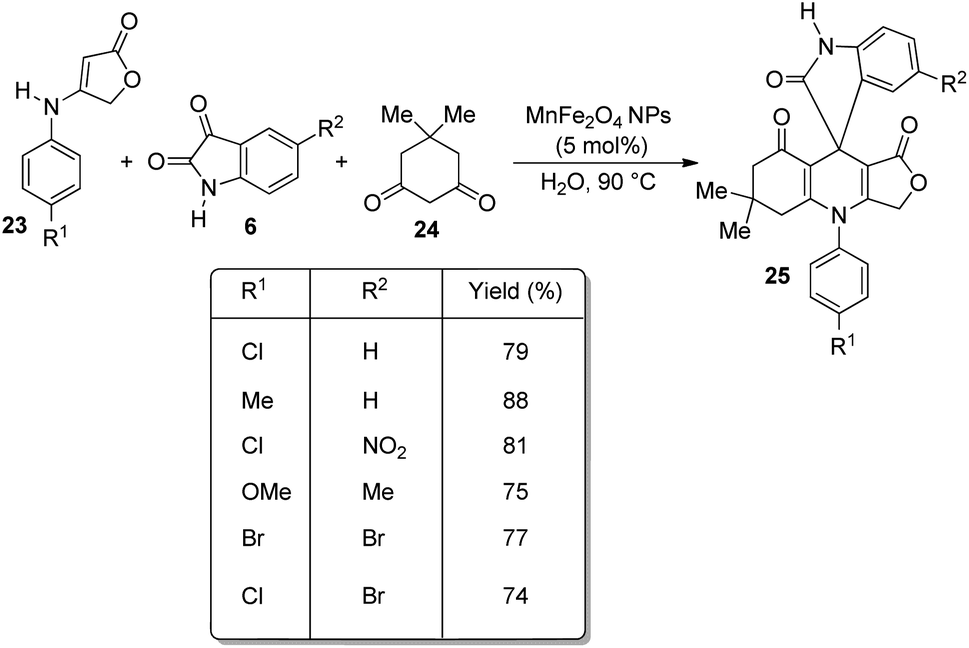 | ||
| Scheme 7 Scope of the condensation reaction between anilinolactone, isatin and dimedone. | ||
Two mechanistic pathways were suggested for the reaction (Scheme 8). In the first approach Lewis acid sites present in nano MnFe2O4 caused activation of isatin 6 and further nucleophilic addition occurred between activated isatin and dimedone 24. The intermediate 26 so obtained reacted with anilinolactone 23 and subsequent cyclization gave the required product 25. The second step was also catalyzed by MnFe2O4. According to the second pathway, anilinolactone 23 reacted with isatin 6 generating an intermediate 27. Next step was the nucleophilic addition between dimedone 24 and the intermediate 27. The spirooxindole product 25 was obtained via intramolecular ring closing step. In this approach also all the steps were catalyzed by nano MnFe2O4.
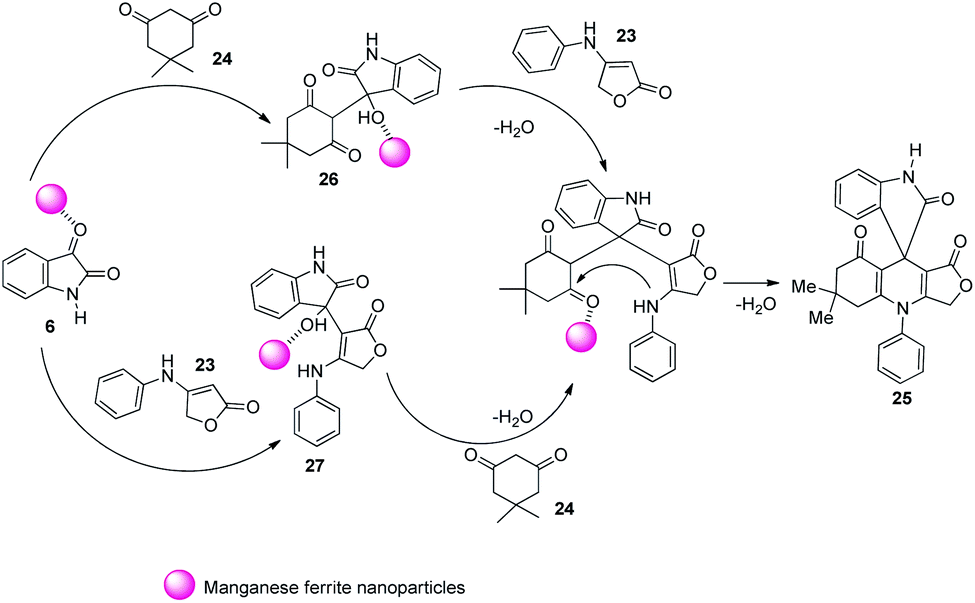 | ||
| Scheme 8 Plausible mechanism for spirooxindole synthesis by condensation between anilinolactone, isatin and dimedone [reproduced with permission from ref. 38]. | ||
This method was further explored and was reported by the same group in the next year.39 Here, anilinolactone 23, isatin 6 and dicyanomethane 28 undergo one-pot reaction in PEG-400 as solvent, catalyzed by 5 mol% of MnFe2O4 nanoparticles at 90 °C (Scheme 9). The activity of the catalyst was not diminished even after five cycles of reaction. Recyclability of the catalyst and PEG-400 and the green conditions are the significances of this method.
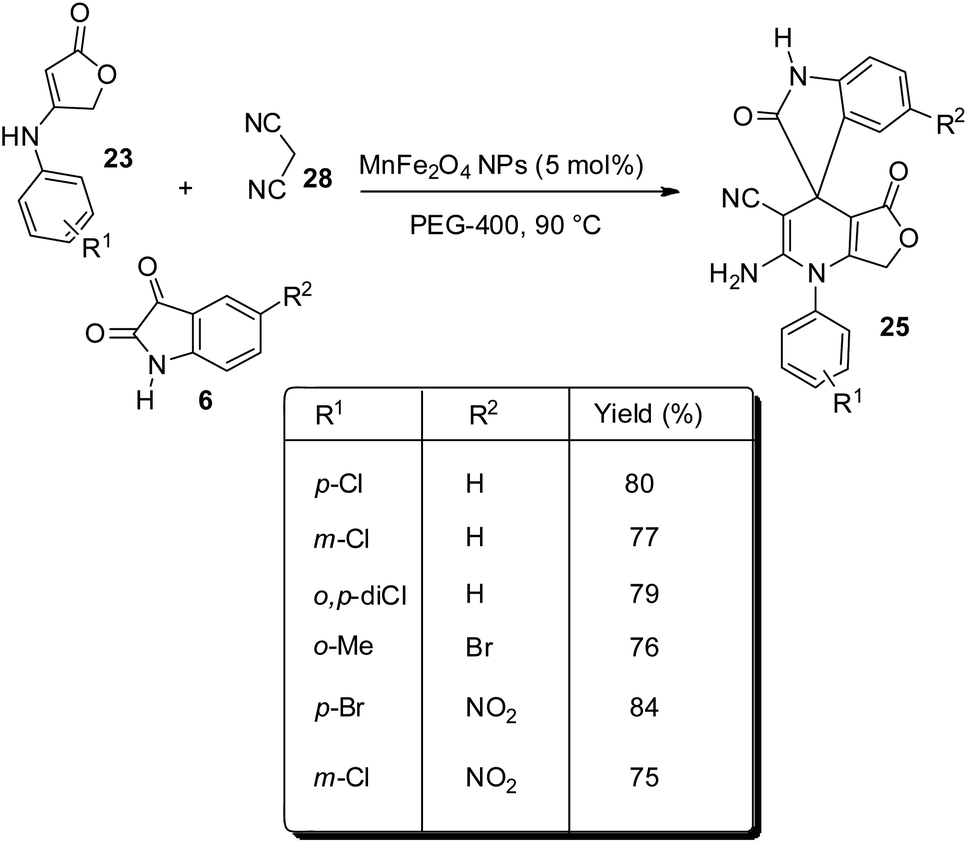 | ||
| Scheme 9 Synthesis of spirooxindole by condensation between anilinolactone, isatin and dicyanomethane in PEG-400. | ||
An eco-friendly methodology for the production of spirooxindoles catalyzed by MnFe2O4@NH2@2AB-Ni was established.40 It is a nanocatalyst, the surface of which was functionalised with amino group and then a nickel complex was immobilized on it. Under the optimized conditions, derivatives of spirooxindole 32, 33 were produced by the condensation reaction between barbituric acid 30 or 2,6-diaminopyrimidine-4(3H)-one 31, cyclic 1,3-diketone 29 and different isatins 6 in the presence of water at 90 °C, and the reaction was catalyzed by 10 mg of MnFe2O4@NH2@2AB-Ni (Scheme 10).
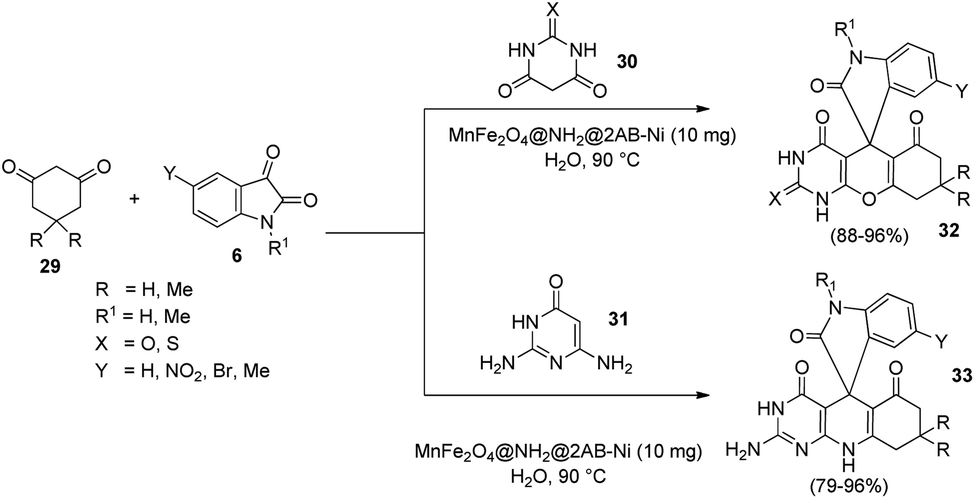 | ||
| Scheme 10 Synthesis of spirooxindole catalyzed by MnFe2O4@NH2@2AB-Ni. | ||
4 Zn-catalyzed spirooxindole synthesis
The biological relevance, abundance and unique capabilities of Zn aided in the application of it as a substitute for metals like Rh, Ir etc. Numerous reports are accessible on organic transformations attained by employing catalytic quantities of Zn.41,42 A pair of electrons can be accepted by Zn2+ by acting as Lewis acid but it is deficient in redox properties due to the filled d10 configuration. The zinc ion can function as redox-stable Lewis acid catalyst.Diverse Zn catalysts like zinc sulphide nanoparticles, zinc triflates etc. were applied for the synthesis of spirooxindoles bearing frameworks such as dihydrofuran, tetrahydrofuran and so on. Metal triflates are having Lewis acidic properties and are generally eco-friendly. Typical chemical and physical characteristics are exhibited by ZnS nanoparticles in comparison with ZnS in bulk and this property adds to their catalytic efficiency.
4.1 Reactions involving isatin derivatives
In an interesting work put forth by Dandia and co-workers, spirooxindole derivatives were synthesized using ZnS NPs as catalyst.43 The reaction follows a green procedure through which derivatives of spiro[chromene-4,3′-indoline] 37 and spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] 36 were generated by means of Knoevenagel condensation followed by Michael addition. The optimized reaction conditions include water as solvent, 10 mol% of ZnS NPs as catalyst under ultrasonic irradiation. Under the optimized reaction conditions, 3-methyl-1-phenyl-2-pyrazolidin-5-one 34, activated methylene compound 35 and different isatins 6 were reacted to afford derivatives of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] 36. Derivatives of spiro[chromene-4,3′-indoline] 37 were produced by utilizing dimedone 24, activated methylene compound 35 and isatins 6 (Scheme 11). The reaction components bearing different substituents underwent the reaction evenly and rendered excellent yields.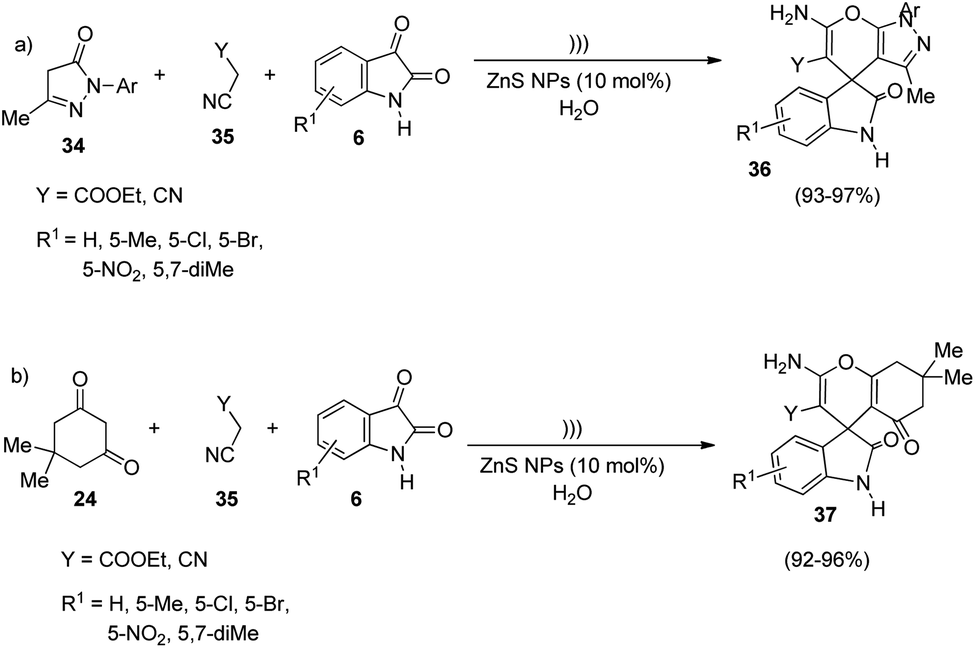 | ||
| Scheme 11 ZnS nanoparticles-catalyzed synthesis of (a) spiro[indoline-3,4′-pyrano[2,3-c]pyrazole]; (b) spiro[chromene-4,3′-indoline]. | ||
Wang and co-workers developed a novel procedure for the synthesis of 3,3′-dihydrofuran spirooxindoles 39 with excellent yields and diastereo- and enantioselectivities, utilizing a dinuclear zinc catalyst, in 2019.44 The reaction proceeds through Knoevenagel/Michael/Pinner/Isomerization path, via condensation between α-hydroxy ketones 38, substituted isatins 6 and dicyanomethane 28 catalyzed by dinuclear zinc generated in situ by reaction between 2 mol% of ligand L2 and 4 mol% of diethyl zinc (Scheme 12). The optimized reaction conditions include DCM as solvent at a temperature of 25 °C (Scheme 13).
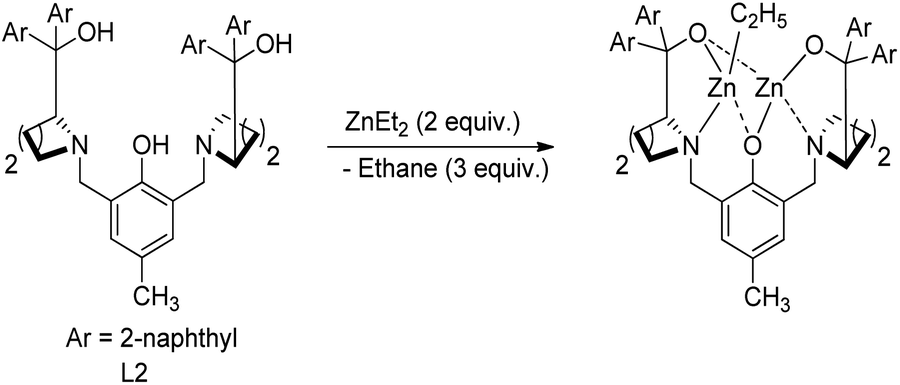 | ||
| Scheme 12 Generation of dinuclear zinc catalyst. | ||
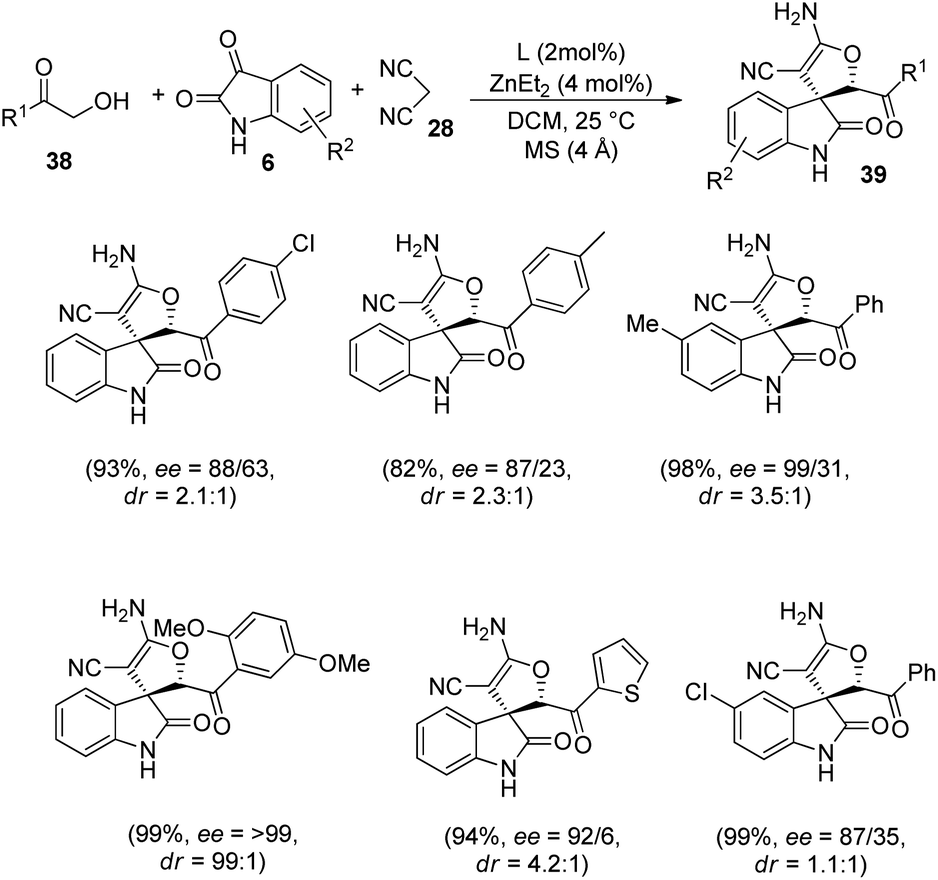 | ||
| Scheme 13 Synthesis of 3,3′-dihydrofuran spirooxindoles using dinuclear zinc catalyst. | ||
In the case of α-hydroxy ketones 38, substituents R′ such as benzene rings which are o-, m- and p-substituted, α-heteroaromatic ring and β-naphthyl ring afforded good to excellent yields and enantioselectivities. The ee values of 29% and >99% were obtained by using 2-hydroxy acetone and α-hydroxy-2,5-dimethoxy acetophenone respectively. Excellent yields and ee values were observed in the case of isatin bearing electron-releasing as well as electron-deficient substituents. Relatively higher enantioselectivities were afforded by isatins having electron-rich groups compared to electron-poor ones. A gram scale reaction between the three components was also performed via the protocol, and they could regain the yield and stereoselectivity.
4.2 Reactions involving isothiocyanatooxindole derivatives
Alkenes with electron-deficient groups and carbonyl compounds were excellent cyclization partners for isothiocyanatooxindole derivatives which were adaptable under the conditions of transition metal-catalyzed synthesis of spirooxindoles. In the case of reaction with the electrophiles, the isothiocyanatooxindoles performed as excellent donors.Xiao et al. demonstrated a method for the emergence of polycyclic spirooxindoles 42 via Michael addition/cyclization cascade utilizing Zn(OTf)2/bis(oxazoline) complex, where the ligand is having a chiral center.45 The reaction between 3-isothiocyanato oxindole 40 and 3-nitro-2H-chromene 41 was optimized by using 11 mol% of ligand (S,S)-L5 and 10 mol% of Zn(OTf)2 in toluene at room temperature (Scheme 14). The substrate scope investigations were carried out by using differently substituted 3-isothiocyanato oxindoles 40 and 3-nitro-2H-chromenes 41. Great enantio- and high diastereoselectivities and good to efficient yields were obtained with different 3-nitro-2H-chromenes 41 irrespective of the electronic nature and position of the substituents. Excellent stereoselectivity and yield were afforded by 3-nitro-2H-benzo[h]chromene, but the enantioselectivity was less with reactive alkyl substrates. Excellent stereoselectivity and yield were also obtained when the methylene group was substituted for 3-nitro-2H-chromene oxygen atom. In the case of 3-isothiocyanato oxindoles 40, very good stereoselectivities and yields were gained when the 5th position bears substituents OMe, F and Me. The reaction exhibited tolerance towards substrates which are benzyl protected. β-Nitro styrene and β-methyl-β-nitrostyrene instead of 3-nitro-2H-chromenes underwent the reaction giving respective yields of 29% and 82%.
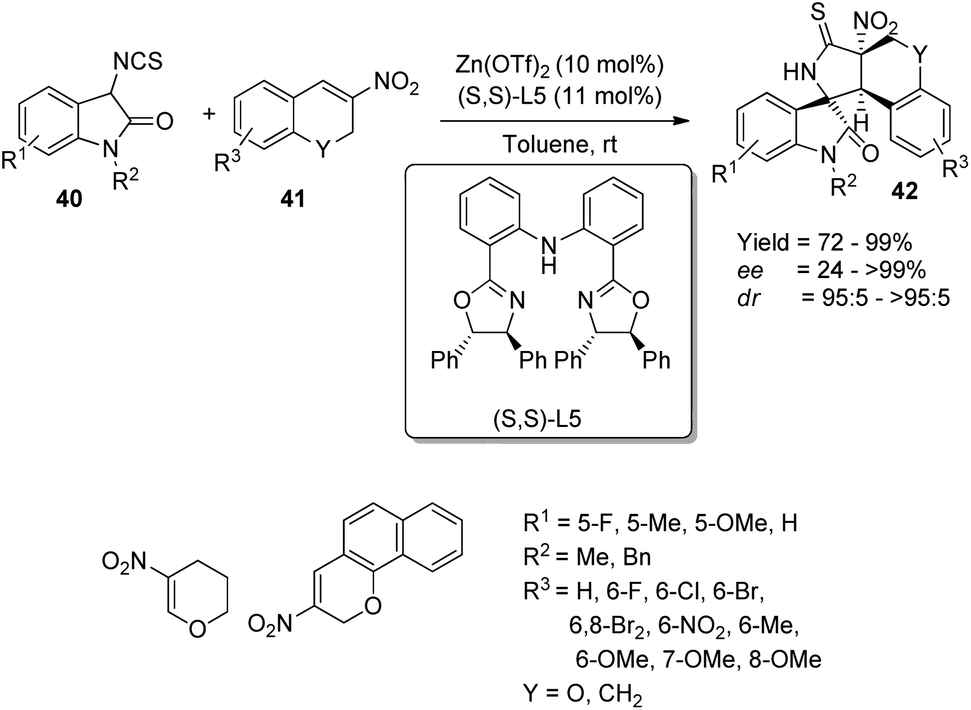 | ||
| Scheme 14 Synthesis of spirooxindole derivatives catalyzed by Zn(OTf)2/bis(oxazoline) complex. | ||
A method for the formation of derivatives of polycyclic spirooxindoles 44 employing Zn(OTf)2/diphenylamine linked bis(oxazoline) complex, which is a chiral catalyst was proposed by Yuan and co-workers.46 The Michael/cyclization reaction between 3-nitroindoles 43 and 3-isothiocyanato oxindoles 40 was catalyzed by 10 mol% Zn(OTf)2 with 11 mol% ligand L4 in presence of toluene at 50 °C (Scheme 15). 95–99% of the products were provided by 3-nitroindoles with different substituents on the benzene ring irrespective of their electronic nature. Various N1-substituted 3-nitroindoles also underwent the reaction except the one bearing methyl. The outcomes were high to excellent with different substituents at the benzene ring and N1-position of 3-isothiocyanato oxindoles. They also compared the metal catalyst with the organocatalyst in their previous work and could demonstrate the superiority of metal catalyst.
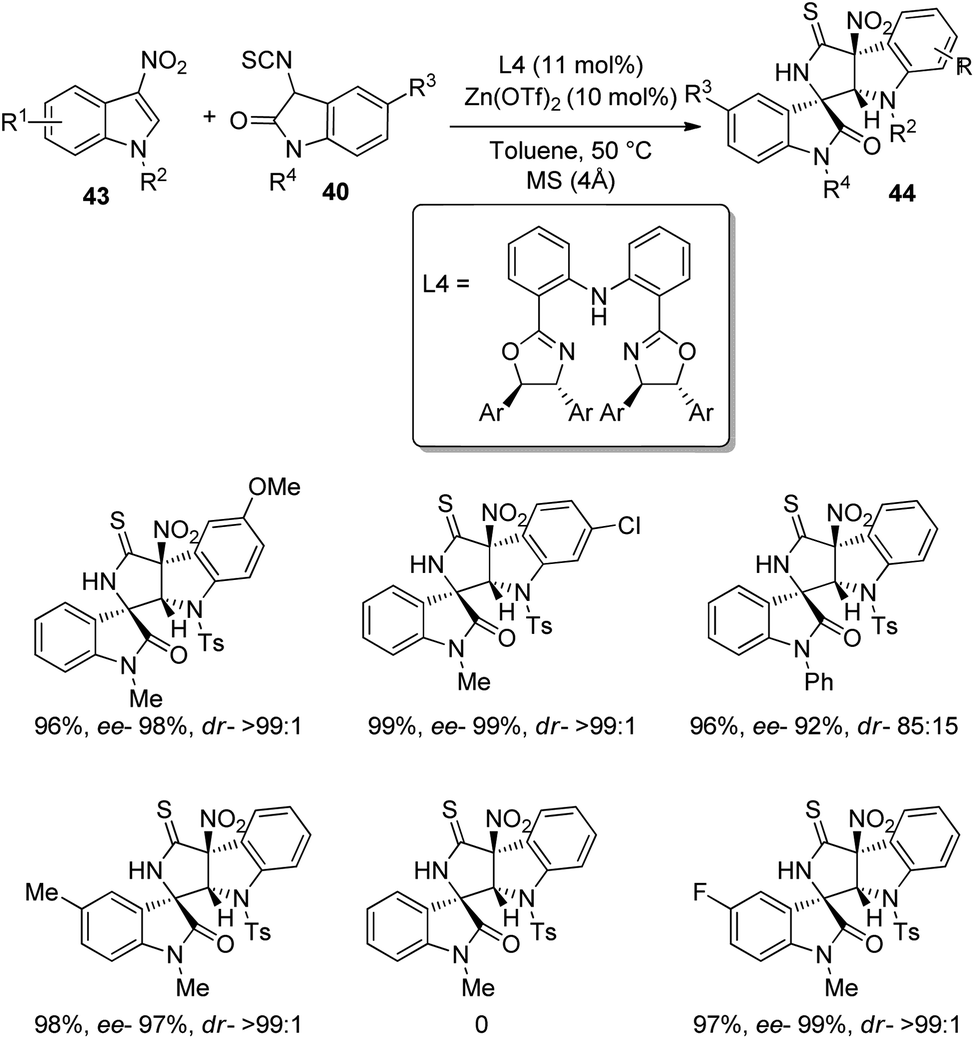 | ||
| Scheme 15 Synthesis of polycyclic spirooxindoles catalyzed by Zn(OTf)2/diphenylamine linked bis(oxazoline) complex. | ||
4.3 Reaction involving β,γ-unsaturated-α-ketoamide derivatives
In 2020, Wang and team produced chiral tetrahydrofuran spirooxindoles 47 by using a dinuclear zinc catalyst.47 Here, it is a two-step reaction in which Michael/Hemiketalization is followed by Friedel–Crafts reaction. In the first step, α-hydroxyaryl ketone (derivative of 38) is reacted with β,γ-unsaturated-α-ketoamide 45 in CH2Cl2 solvent at 20 °C catalyzed by dinuclear zinc which was formed in situ by the reaction between 20 mol% of diethyl zinc and 10 mol% of ligand L3. In the second step, trifluoroacetic acid 46 was added and the reaction was carried out at room temperature for 60 min.For the substrate scope assessment, α-hydroxyacetophenone was reacted with a series of β,γ-unsaturated-α-ketoamides 45 (Scheme 16). It afforded 80–85% of products with 8:1 to 13:1 dr values when the N-aromatic ring at the p-position bears either electron-rich or electron-poor groups. Substrates with benzyl, ethyl and methyl as the N-substituents were tolerated. In the case of Ar2 groups at the p-position, good dr values and yields were observed and the ee value was relatively higher for Cl. Excellent ee was obtained when o- or m-position of Ar2 is having Br and the p-position is having electron-releasing substituents. The products were also afforded by fused and heterocyclic ring substrates.
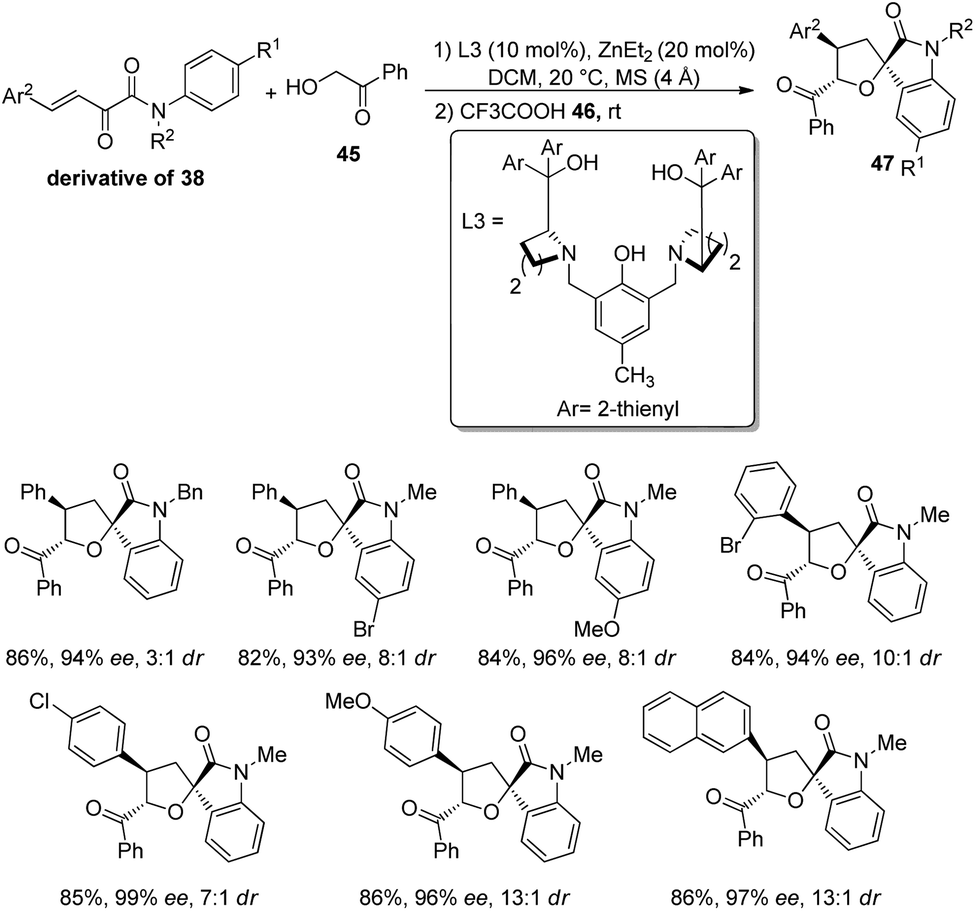 | ||
| Scheme 16 Synthesis of tetrahydrofuran-spirooxindoles with α-hydroxy acetophenone and β,γ-unsaturated-α-ketoamides. | ||
Further the influence of substituents on α-hydroxyaryl ketones (derivative of 38) were also assessed (Scheme 17). Electron-releasing as well as electron-deficient groups on the aryl group provided good stereoselectivities for the corresponding spirooxindoles. 2-Methoxy substituted and 4-methoxy substituted α-hydroxyaryl ketones afforded the products with ee values 75% and 96% respectively. Then, 76% of the tetrahydrofuran spirooxindole 49 with 91% ee and 4:1 dr value was gained by the implementation of substrate 48 which has an N-aromatic ring ortho-substituted by OMe group (Scheme 18). Wang and co-workers could also synthesise tetrahydrofuran spirooxindoles by using this protocol on gram scale.
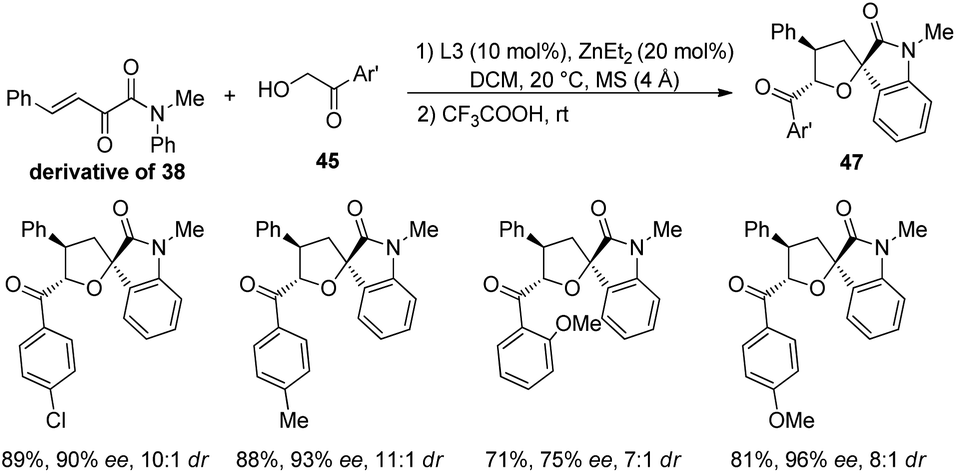 | ||
| Scheme 17 Synthesis of tetrahydrofuran-spirooxindoles with β,γ-unsaturated-α-ketoamide and a series of α-hydroxy aryl ketones. | ||
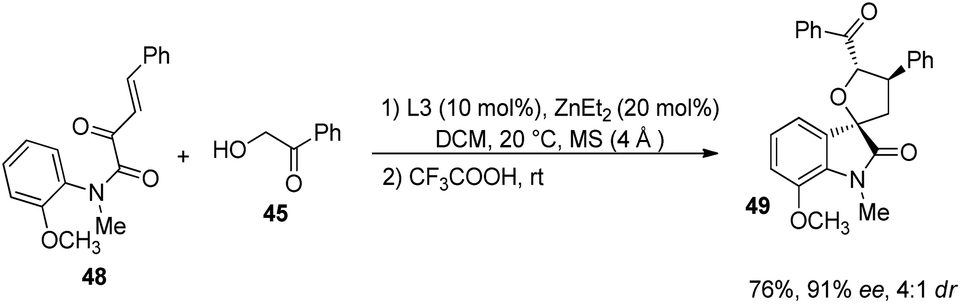 | ||
| Scheme 18 Synthesis of tetrahydrofuran-spirooxindole corresponding to substrate having N-aromatic ring ortho-substituted by OMe group. | ||
Efficient enantioselectivities and good diastereoselectivities were afforded by β,γ-unsaturated-α-ketoamide derivatives in the synthesis of spirooxindole.
5 Au-catalyzed spirooxindole synthesis
Gold-catalyzed organic transformations developed notably in the past years. Gold shows greater resistance towards oxidation. High selectivities and activities are exhibited by most of the Au catalysts. Gold(I) catalysts being more stable are widely employed in reactions than gold(III).48 But, gold(III) catalysts also play function like activating unsaturated bonds through electrophilic π-activation.49Gold-catalyzed spirooxindole synthesis was not well explored and only two works-one in 2016 and another in 2018 were reported. The strategies employed sodium tetrachloroaurate and JohnphosAu(CH3CN)SbF6 as the catalyst respectively.
5.1 Reaction involving isatin derivatives
Praveen and co-workers devised a procedure for the construction of spirooxindoles catalyzed by gold(III) and studied the cytotoxic and antimicrobial characters.50 A series of spirooxindoles 51 were produced by reacting 4-hydroxycoumarin 50 and isatins 6 which were N-substituted. The reaction was catalyzed by 2 mol% NaAuCl4·2H2O in ethanol solvent at 40 °C under microwave (Scheme 19). The yield of the desired product was good to excellent with different substituents on the isatin. Based on this strategy, they could build bis-spirooxindole system also.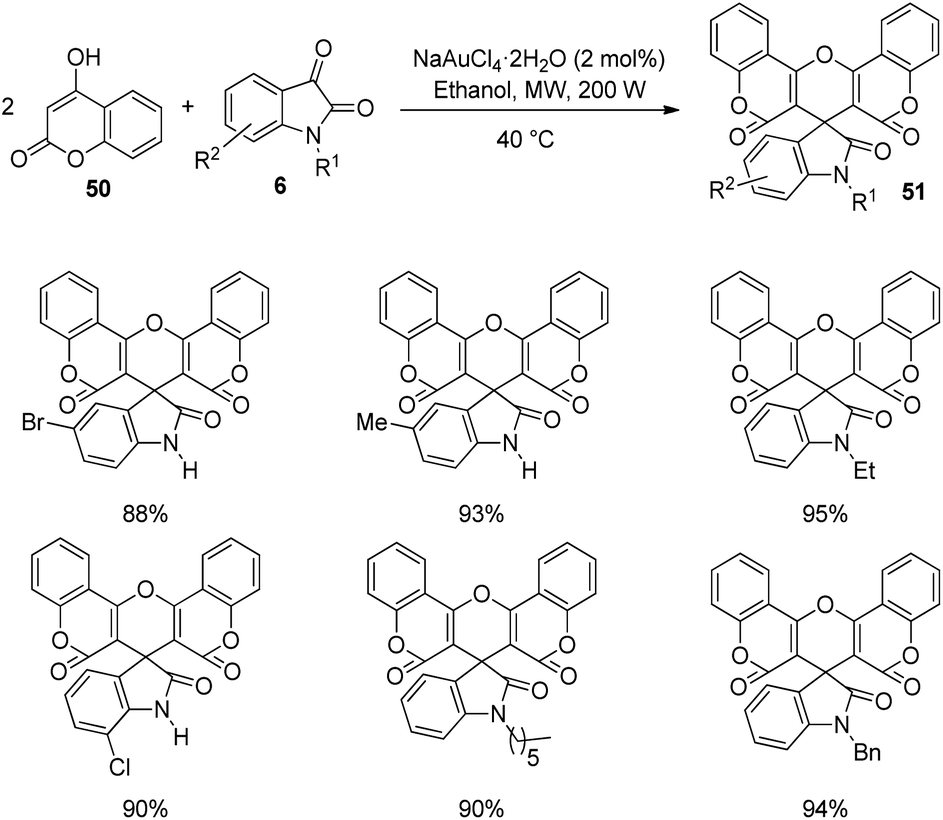 | ||
| Scheme 19 Substrate scope for the gold-catalyzed spirooxindole synthesis. | ||
Here, isatin 6 was activated by gold catalyst which acted as Lewis acid. An intermediate 52 was produced via the nucleophilic addition between isatin 6 and 4-hydroxycoumarin 50. The catalyst coordinates with carbonyl oxygen of second intermediate 53 which was formed through elimination of water molecule from first intermediate 52. Third intermediate 54 was generated through the addition of 4-hydroxycoumarin 50 to second intermediate 53. The desired spirooxindole product 51 was achieved by means of cyclization followed by dehydration. The scheme furnishes the possible mechanism for the reaction (Scheme 20).
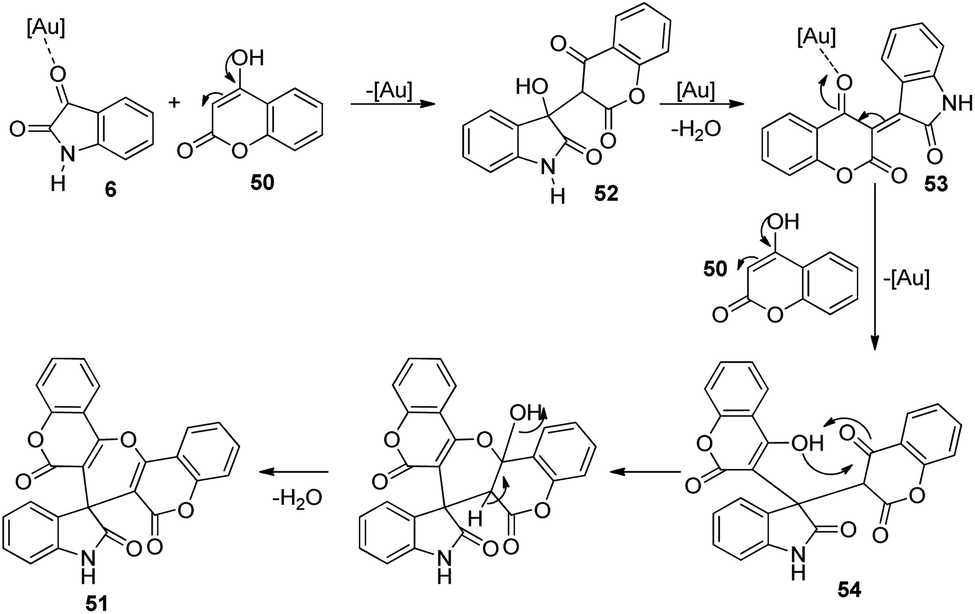 | ||
| Scheme 20 Proposed mechanism for the construction of spirooxindoles [reproduced with permission from ref. 50]. | ||
5.2 Reaction involving diaryl alkyne derivatives
A strategy for the development of derivatives of spirooxindoles using gold catalyst via diaryl alkyne bicyclization was reported by Xu et al.51 In the first step, fused indoles 56 were formed from diaryl alkyne 55 by making use of gold catalyst JohnphosAu(CH3CN)SbF6 in 1,2-dichloroethane at 60 °C for 12 h (Scheme 21). Electron-deficient and electron-releasing groups on the diaryl alkynes 55 rendered fused indoles 56 without significant electronic and steric effects. Substrate containing naphthyl group afforded two isomers of the corresponding product. The yield was 75% with substrate bearing an alkynyl substituent. In the second step the substrates 56 thus obtained were protected with (Boc)2O 57 and converted into spirooxindoles 59 via oxidation with PCC 58 in dichloromethane (Scheme 22).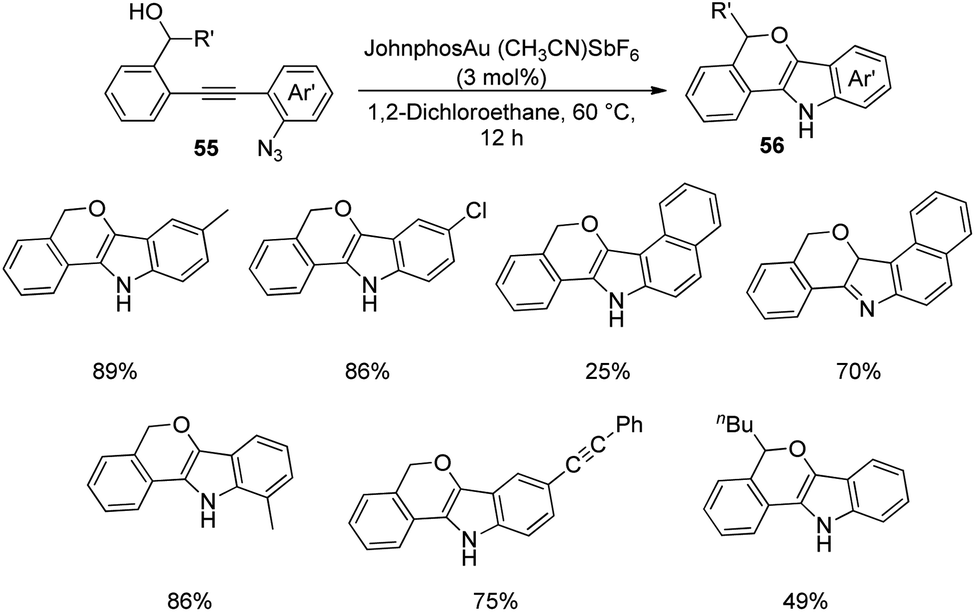 | ||
| Scheme 21 Formation of fused indoles from diaryl alkenes catalyzed by gold. | ||
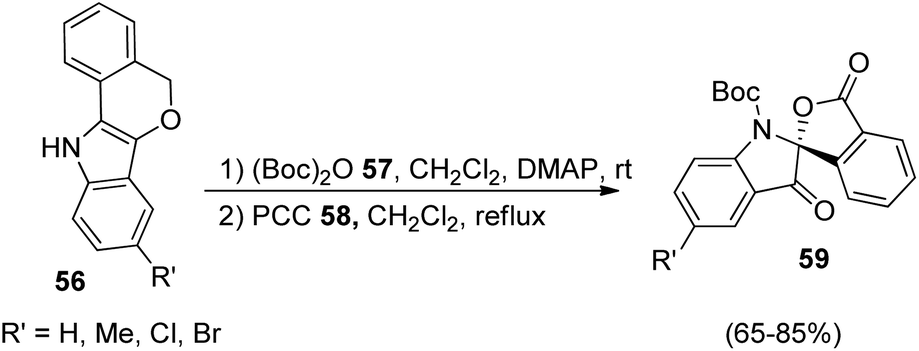 | ||
| Scheme 22 PCC oxidation of Boc-protected fused indoles to produce spirooxindoles. | ||
This protocol is found to be highly atom-economic with an added advantage of gentle reaction conditions.
6 Ni-catalyzed spirooxindole synthesis
Nickel is admirable in terms of catalyst cost compared to metals like Pt, Rh and so on. Great variability of oxidation states are exhibited by nickel catalysts. Reactants can be activated and the conversion to products can be achieved effortlessly. Most of the nickel catalysts exhibit great affinity to unsaturated molecules. Hence they can easily coordinate to alkynes and alkenes and can activate them.52The generation of spirooxindoles by means of nickel-catalyzed approaches were reported from 2011 to 2020, most of them based upon isatin derivatives. Pyrazolophthalazinyl-, pyrazolopyrridine-, thiochromanyl-spirooxindoles etc. were built by availing catalysts including nickel chloride, nickel oxide and nickel(II)acetate. In most of these protocols, the mostly employed catalyst was NiO nanoparticle which is superior in terms of catalytic activities and environmental benignity.
6.1 Reactions involving isatin derivatives
Zhang and co-workers established a plan for the evolution of pyrazolophthalazinyl spirooxindoles 61 by applying nickel chloride as catalyst.53 The reaction between phthalhydrazide 60, isatin 6 and activated methylene compound 35 was optimized in PEG 600 as the solvent, NiCl2 as catalyst at 100 °C (Scheme 23). The product yield was slightly higher for dicyanomethane compared with cyanoacetic esters. Both electron-deficient and electron-rich group substituted isatins afforded the products in excellent yields (80–95%). A large scale reaction offered 88% yield of the product.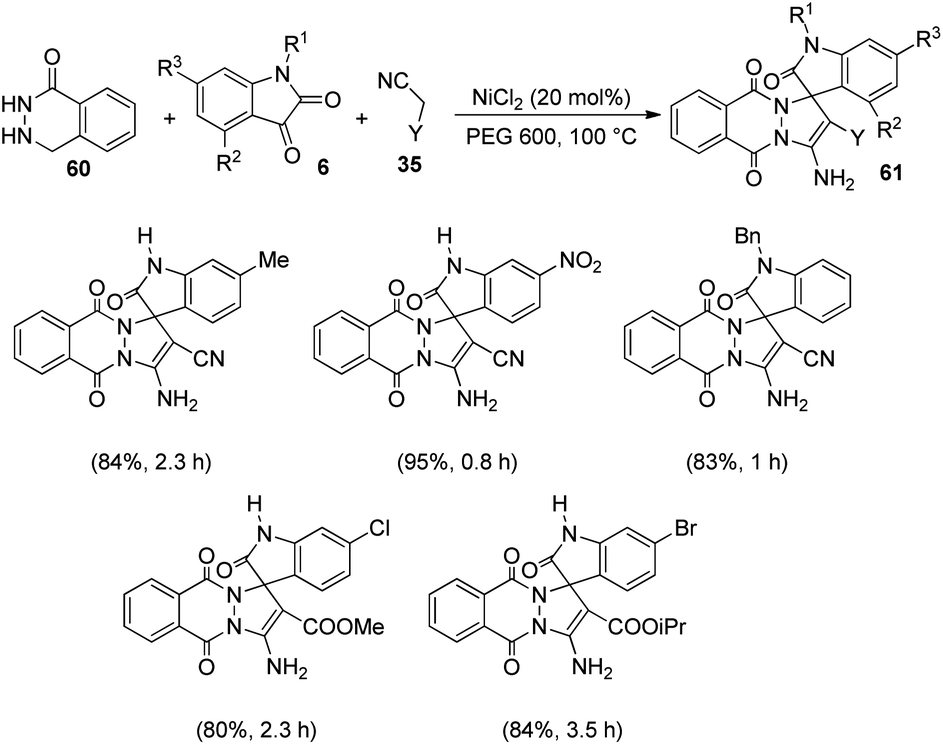 | ||
| Scheme 23 NiCl2 catalyzed synthesis of pyrazolophthalazinyl spirooxindoles. | ||
The reaction follows a Knoevenagel condensation/Michael addition/cycloaddition/isomerization sequence in which the conversion of cyano group into amine was activated by NiCl2. In the first step an adduct 62 was formed by the Knoevenagel condensation of activated methylene compound 35 with isatin 6. Then there occurs a Michael addition between the carbon–carbon double bond of the adduct 62 and phthalhydrazide 60. The required product 61 was achieved in the next step which involves cycloaddition and isomerization. The suggested mechanism is depicted in Scheme 24.
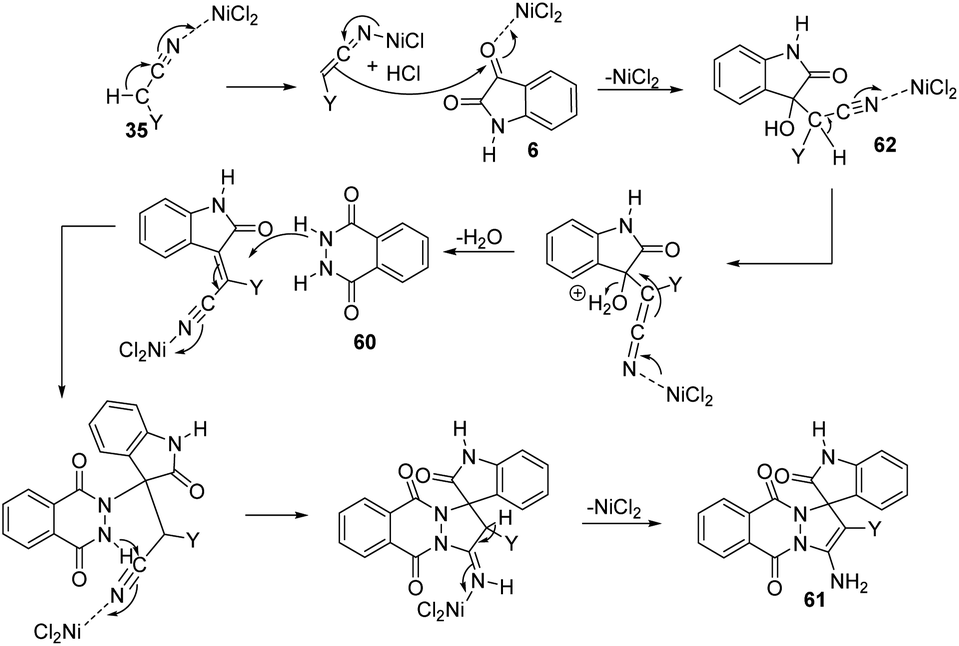 | ||
| Scheme 24 The suggested mechanism for the synthesis of pyrazolophthalazinyl spirooxindole utilizing NiCl2 as the catalyst [reproduced with permission from ref. 53]. | ||
An environmentally-benign strategy for the synthesis of spirooxindoles by employing NiO NPs was demonstrated by Nasseri and co-workers.54 The optimized reaction condition includes 0.0007 g of nano-NiO in aqueous medium at room temperature in which dicyanomethane 28, cyclic 1,3-diketone 29 and isatins 6 reacted to formulate derivatives of spirooxindole 63. High to excellent yields were given by different cyclic 1,3-diketones and isatins. Due to the presence of electron-releasing Me groups, dimedone reacted rapidly relative to 1,3-cyclohexadione. Further, the authors could also extend the reaction by employing 4-hydroxycoumarine 50 as an alternative to 1,3-cyclohexadione and obtained the corresponding spirooxindole 64 (Scheme 25).
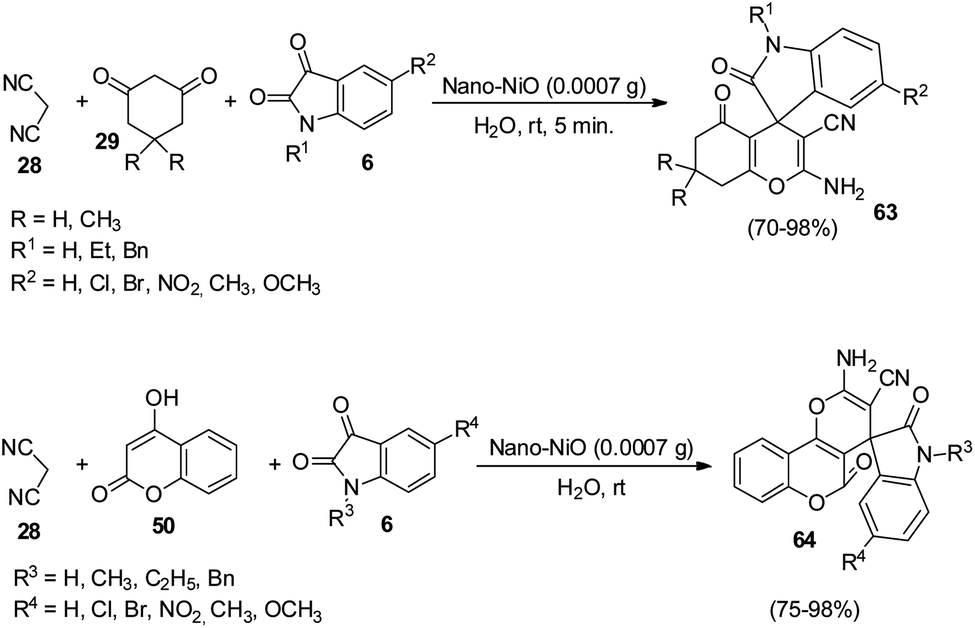 | ||
| Scheme 25 Synthesis of spirooxindoles using dicyanomethane, dicarbonyl compounds/4-hydroxycoumarine and isatins. | ||
A strategy for the synthesis of spirooxindole-fused pyrazolo pyridine derivatives 66 utilizing silica supported NiO nanoparticles as catalyst was put forth by Yagnum et al. in which they also studied the anti-microbial activities of the novel compounds.55 The one-pot reaction between three components-dicyanomethane 28, isatin 6 and 3-methyl-1-phenyl-1H-pyrazole-5-amine 65 was advanced in ethanol, 6.5 mol% of NiO–SiO2 at reflux temperature (Scheme 26). Isatins with electron-withdrawing and electron-releasing groups were tolerated in the reaction. The yield was a little higher for 5-substituted and 7-substituted isatins relative to 4,7-disubstituted ones. There was no prominent role for steric and electronic factors. According to the suggested mechanism, the reaction follows a Knoevenagel condensation/Michael addition/cyclization/isomerization sequence. Anti-fungal and anti-bacterial properties were displayed by most of the spirooxindole derivatives so-obtained.
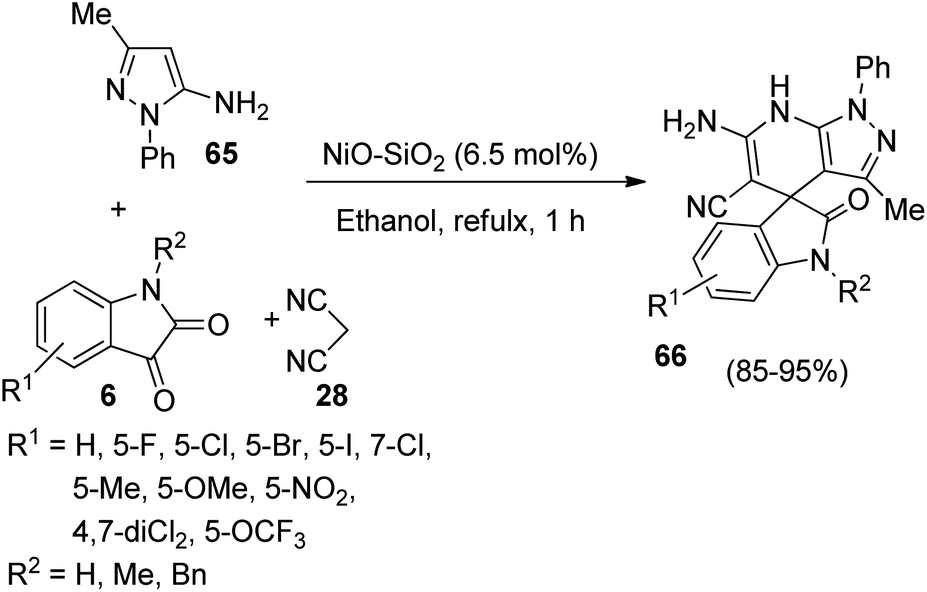 | ||
| Scheme 26 Synthesis of spirooxindole fused pyrazolo pyrrolidine using NiO–SiO2 catalyst. | ||
A green methodology for the building up of derivatives of spirooxindoles 68, 63, 64 by applying another NiO based catalyst was designed by Moqadam et al.56 Here the catalyst was NiO@g-C3N4, in which nanosheets of graphitic carbon nitride was used to carry NiO nanoparticles, through which the catalytic activity was enhanced by decreasing the nanoparticle aggregation. The reaction between dicyanomethane 28, isatin 6 and 4-hydroxycoumarin 50 (or 1,3-diketoester 67 or cyclic 1,3-diketone 29) was catalyzed by 50 mg of NiO@g-C3N4 in ethanol at 80 °C under reflux (Scheme 27). The starting materials with diverse substituents were used to explore the scope of this reaction. The reactivity and outcomes were best for dimedone relative to 4-hydroxycoumarin, 1,3-diketoesters and 1,3-cyclohexadione. In the case of isatins, better upshots were obtained with electron-withdrawing substituents on the aromatic ring. The development of products was diminished with electron-releasing groups on the N atom of isatin.
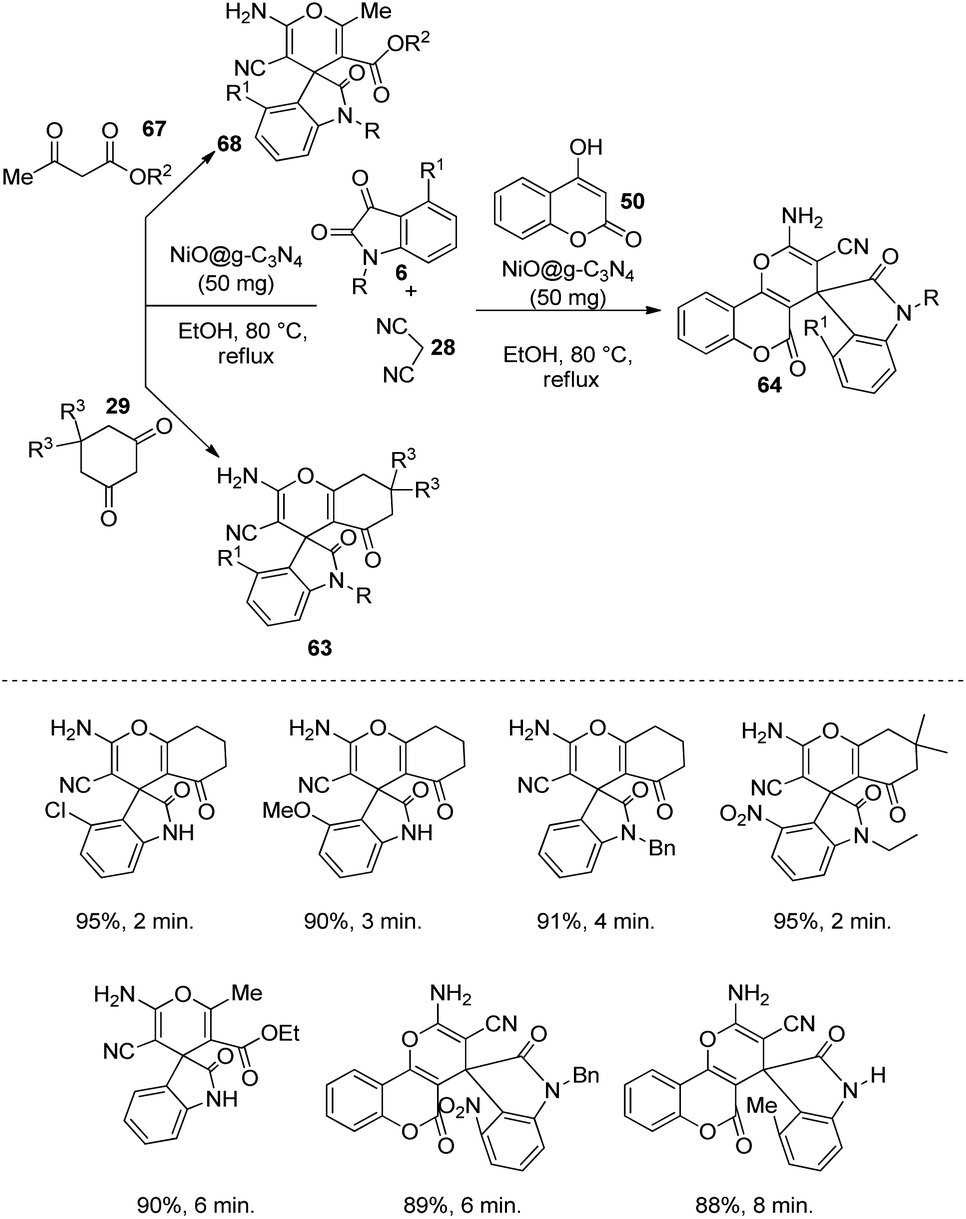 | ||
| Scheme 27 NiO@g-C3N4-catalyzed synthesis of spirooxindoles. | ||
6.2 Reaction involving isothiocyanatooxindole derivatives
A scheme for the development of spirooxindoles 70 catalyzed by dinuclear nickel Schiff base was proposed by Matsunaga et al.57 The reaction between aldehydes 69 and 3-isothiocyanato oxindoles 40 was effected by Ni2/Schiff base in 1,4-dioxane at room temperature (Scheme 28). The enantioselectivity was a bit lower for α-branched aldehydes compared to the linear aliphatic ones. The diastereoselectivity, yield and enantioselectivity were 81:19, 99% and 96% respectively for aldehyde having silyl ether component. The outcomes were poor in the case of aromatic aldehydes. Isothiocyanato oxindoles bearing electron-releasing and electron-withdrawing substituents on aromatic ring underwent the reaction and those containing allyl as the N-protecting group also gave good results.
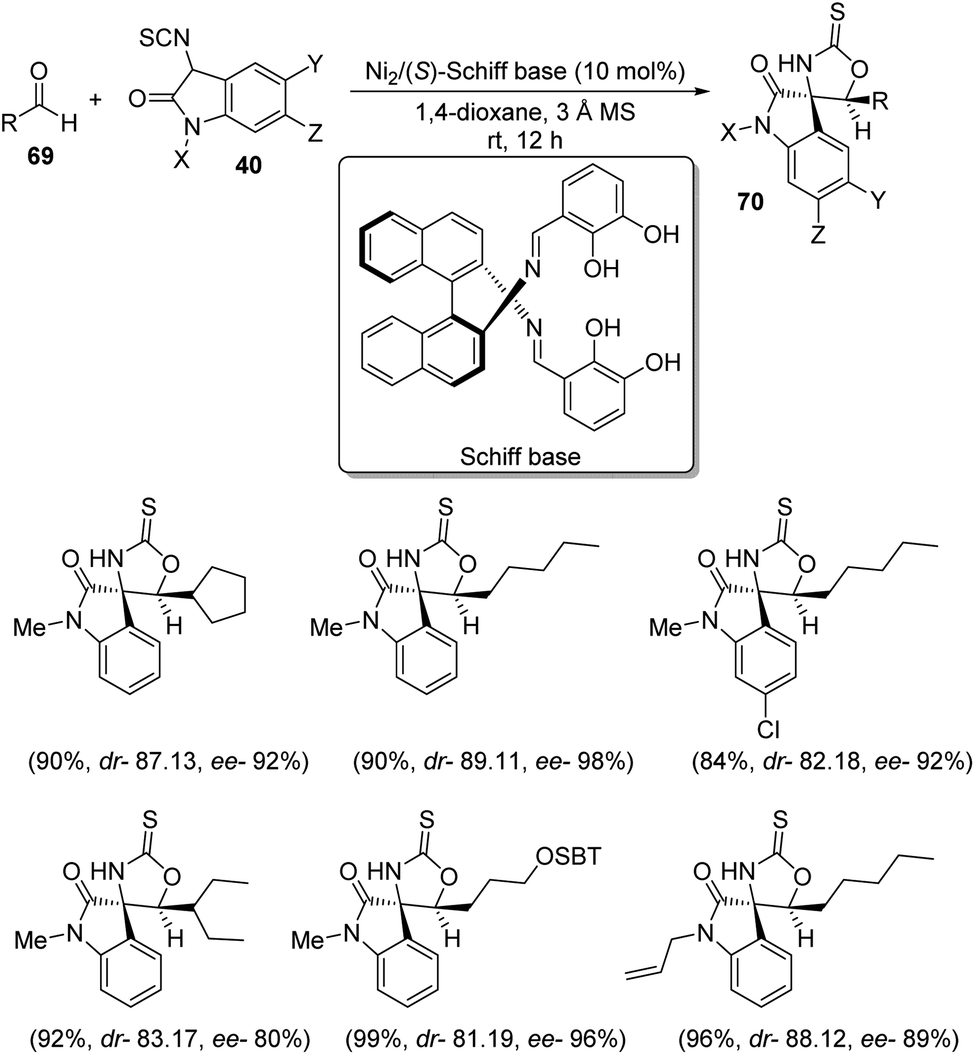 | ||
| Scheme 28 Synthesis of spirooxindoles using Ni2/(S)-Schiff base as the catalyst. | ||
Spirooxindoles with three stereocenters were achieved from isothiocyanatooxindole derivatives via catalysis by zinc and nickel. The other substrate for the reaction was: 3-nitro-2H-chromenes and aldehydes in the case of zinc- and nickel-catalyzed approaches respectively. Enantioselectivity was slightly higher for the zinc-catalyzed reaction in comparison to the nickel-catalyzed one.
6.3 Reactions involving methyleneindolinone derivatives
Arai et al. presented a plan for the formation of chiral thiochromanyl-spirooxindoles 73 by using bis-(imidazolidine)pyridine-Ni(OAc)2 catalyst.58 Here, spirooxindoles were evolved from thiosalicylaldehydes 71 and methyleneindolinones 72 via Michael/Aldol pathway. Various substrates reacted in the optimized conditions of 11 mol% PyBidine and 10 mol% Ni(OAc)2.4H2O in toluene at −40 °C (Scheme 29). Moderate to efficient yields were provided by thiosalicylaldehydes with different substituents. High diastereoselectivities and yields were endowed by methyleneindolinones bearing different substituents on the aromatic ring. Good yield and enantioselectivity were afforded by methyleneindolinone substituted with alkyl group and those with substituents on indolinone ring.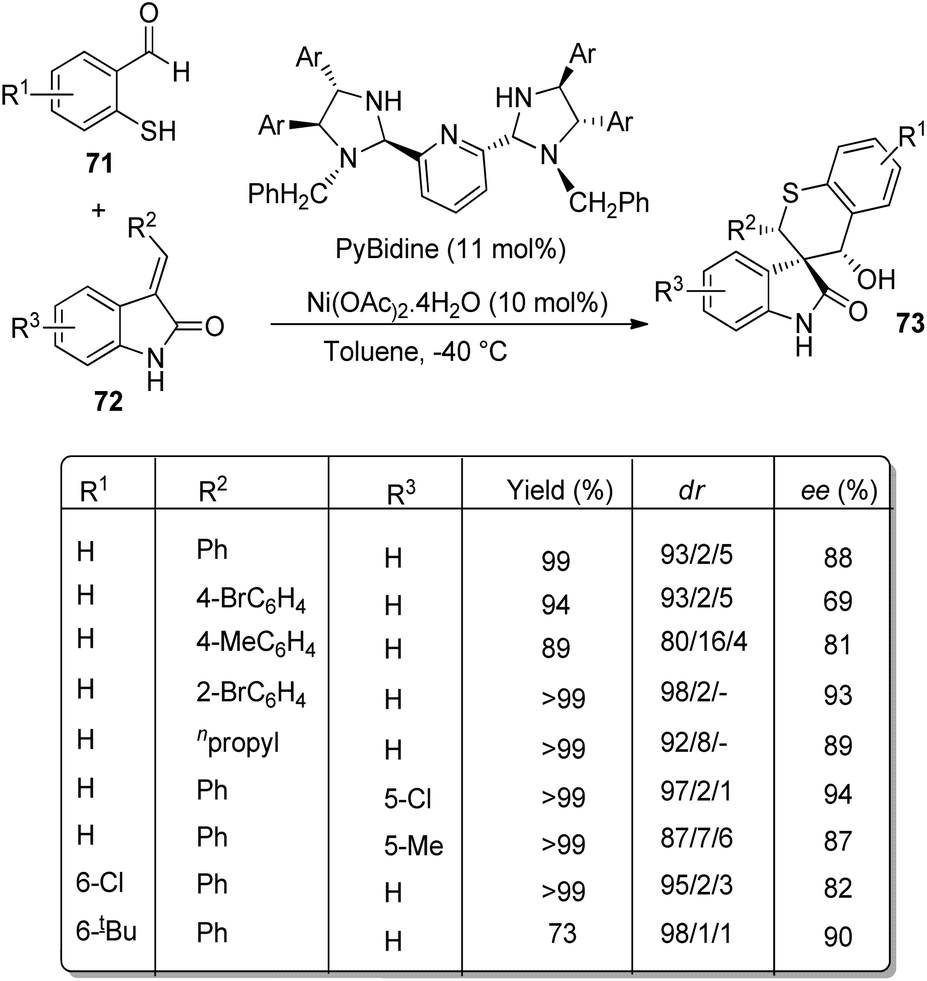 | ||
| Scheme 29 PyBidine-Ni(OAc)2 catalyzed thiochromanyl spirooxindole synthesis. | ||
Feng and co-workers introduced a procedure for the fabrication of spirooxindole cyclohexaneamides 75 catalyzed by N,N′-dioxide/nickel(II) complex.59 1,3-Dienyl carbamates 74 underwent a Diels–Alder reaction with methyleneindolinones 72. The reaction proceeded in DCM at 0 °C applying 10 mol% of L6-PiPr2/Ni(BF4)2·6H2O as the catalyst (Scheme 30). 87–99% yield, 93–99% ee and 93:7 to >95:5 dr were acquired from diverse substituents on the aromatic ring of methyleneindolinones. The result was also excellent with different substituents R2 on methyleneindolinones. When R4 = methyl, only one diastereoisomer with four chiral centers was obtained.
 | ||
| Scheme 30 Substrate scope for the Diels–Alder reaction between 1,3-dienylcarbamates and methyleneindolinones. | ||
Further, the reaction between 1,2-dihydropyridine 76 and methyleneindolinone 72 was conducted to access the corresponding spirooxindole 77 (Scheme 31). Efficient ee and dr values and high to excellent yield were brought by different protecting groups on N of 1,2-dihydropyridine. Excellent yield, ee and dr values were obtained when R8 = Ph, Me and Bn. Due to the steric effect between methyleneindolinone and methyl group (R9), the product was contributed only in tinge. The Diels–Alder reaction was also done on gram scale. The group could also confirm that the reaction followed a concerted pathway.
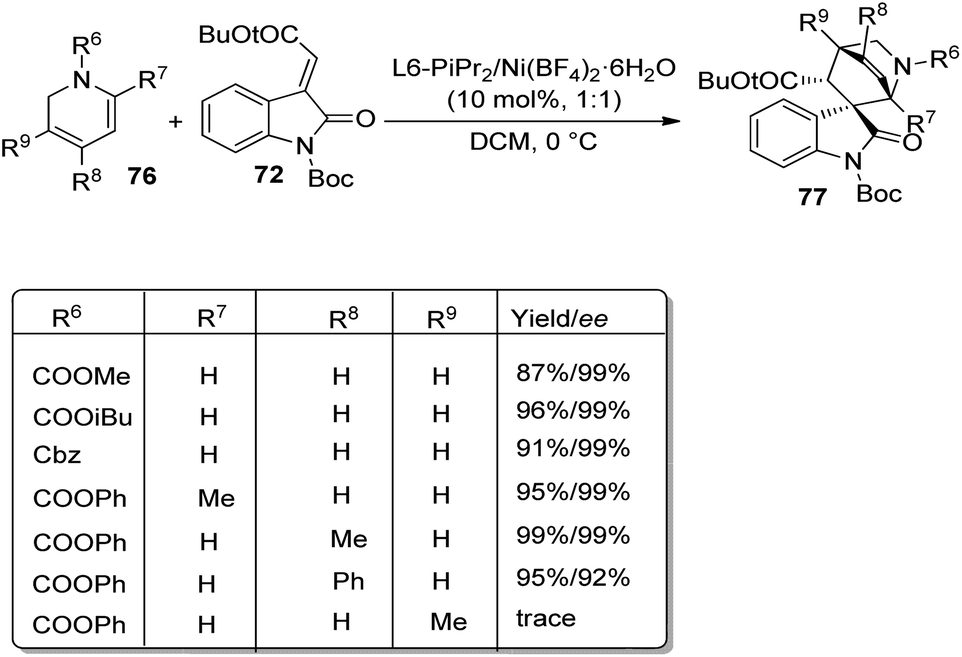 | ||
| Scheme 31 Substrate scope for the Diels–Alder reaction between 1,2-dihydropyridines and methyleneindolinone. | ||
7 Fe-catalyzed spirooxindole synthesis
Iron is superior to majority of the transition metals in terms of non-toxicity, earth abundance, cost-effectiveness and environmental benignity. Its redox potential range is broad and can exhibit variable electronic states. These properties led it to become a promising candidate as a catalyst in organic reactions. Fe catalysts keep the oxidation state varying from −2 to +6. Consequently, they are functional in both oxidation and reduction reactions.60 By changing the ligands, the Lewis acidity can be tuned, it is also dependent on the oxidation state.Different iron catalysts: FeCl3, Fe(OTf)2, CoFe2O4- and Fe3O4-nanoparticles with various modifications were used for the synthesis of spirooxindoles. The modifications in terms of morphology, size and shape is a way of tuning the activities of nano sized catalysts. Among the diverse synthetic approaches adopted, those dependent on isatin derivatives prevailed over others.
7.1 Reaction involving methyleneindolinone derivatives
Yuan and co-workers formulated a procedure for the generation of novel spirooxindole tetrahydroquinolines 78 by applying FeCl3 as catalyst.61 The reaction occurs through a 1,5-hydride transfer followed by ring closure in 1,2-dichloroethane with FeCl3 (10 mol%) (Scheme 32). The reaction was carried out using different methyleneindolinone substrates 72 including tetrahydroisoquinoline, morpholine, piperidine and pyrrolidine derivatives. High to efficient yields and diastereoselectivities were ensued from unprotected and protected methyleneindolinones bearing different substituents on the aromatic ring of oxindole. Piperidine derivatives manifested 87–94% yield and up to 91:9 dr values. The N-protected morpholines reacted rapidly than the unprotected ones. The dr values and yields were adequate with substrates derived from tetrahydroisoquinoline also.
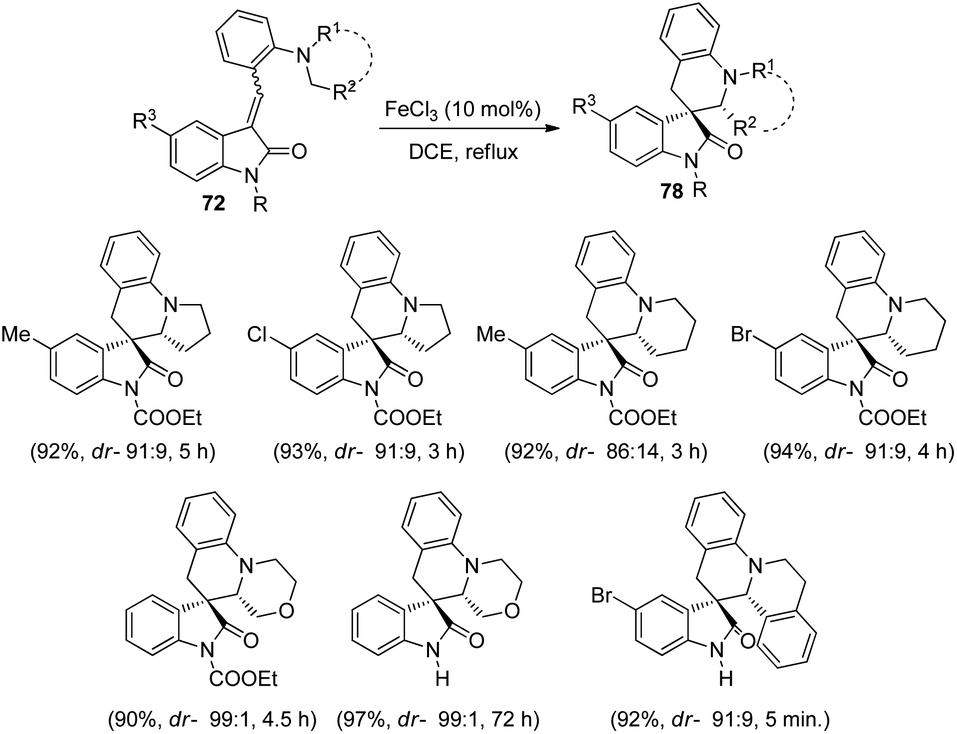 | ||
| Scheme 32 Synthesis of spirooxindole derivatives catalyzed by FeCl3. | ||
7.2 Reactions involving isatin derivatives
A green strategy for the production of pyrazolophthalazinyl spirooxindoles 61 catalyzed by surface functionalized Fe3O4 was worked out.62 The catalyst was prepared by the treatment of Fe3O4 NPs with tetraethyl orthosilicate which was further treated with 3-chloropropyltriethoxy silane and finally dipyridine-2-ylmethanol was employed for the base group loading on the surface. The reaction between hydrazine 80, malonic dinitrile 28, isatin 6 and phthalic anhydride 79 was effected by 0.0008 g of Fe3O4/dipyridine magnetic particles (MPs) in solvent free-condition at rt (Scheme 33). Isatins with various substituents resulted in high to excellent yields without notable electronic effects. The group could reuse the catalyst with almost similar efficiency.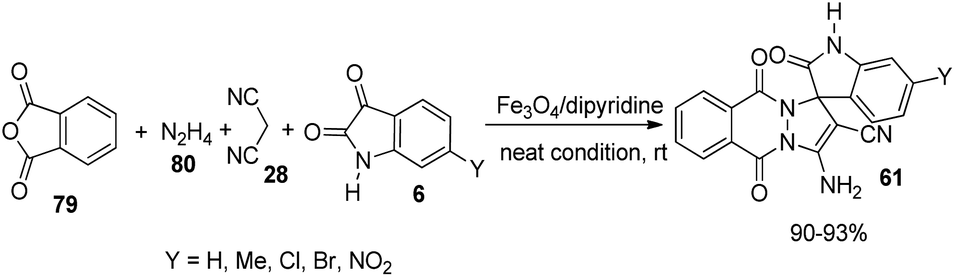 | ||
| Scheme 33 Fe3O4/dipyridine MPs catalyzed synthesis of pyrazolophthalazinyl spirooxindoles. | ||
Safaei-Ghomi et al. described a proposal for the emergence of spirooxindoles 81, 82 catalyzed by guanidine-functionalized Fe3O4 magnetic NPs.63 It is an eco-friendly procedure which involves a multi-component reaction between isatin, ethyl acetoacetate, hydrazine hydrate and active methylene compound or dimedone in ethanol. First the reaction of acetoacetic ester-derivative of 67, hydrazine 80 and dimedone 24 was carried out with variously substituted isatins 6. Then the reaction was done using active methylene compound 35 instead of dimedone (Scheme 34). High to efficient yields were provided by isatins with both electron-releasing and -withdrawing substituents.
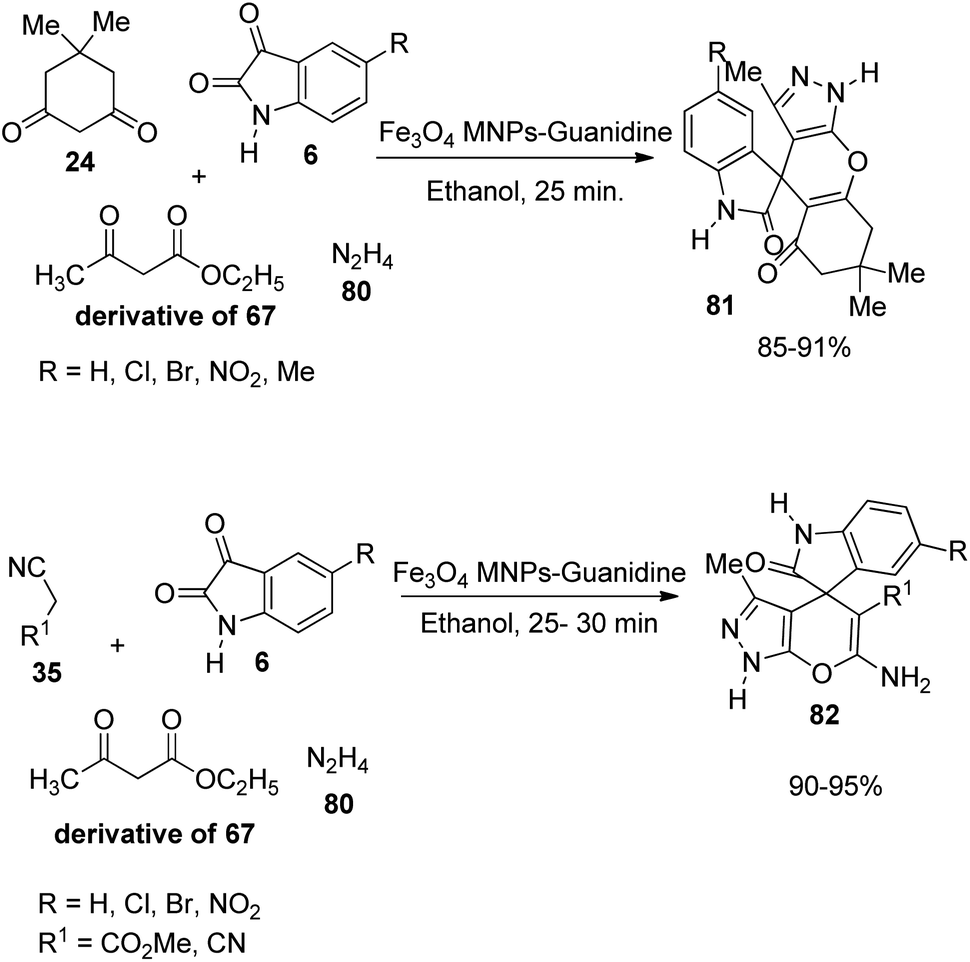 | ||
| Scheme 34 Multi-component reaction between acetoacetic ester, hydrazine hydrate, dimedone/active methylene compound and isatins catalyzed by Fe3O4 MNPs-guanidine. | ||
A design for the formation of derivatives of spirooxindoles 63, 64 catalyzed by CoFe2O4@SiO2 nanoparticles was confirmed by Hemmat et al.64 The catalyst was produced by coating silica on the surface of cobalt ferrite nanoparticles by the application of tetraethylorthosilicate, which helped to prevent the aggregation of CoFe2O4 NPs. Optimization of the reaction between cyanoacetonitrile 28, isatins 6 and cyclic 1,3-diketone 29 or 4-hydroxycoumarin 50 was carried out in water/ethanol mixture with 0.01 g of catalyst at 80 °C (Scheme 35). Good to excellent yields were given by different isatins and cyclic 1,3-diketones. The yield was slightly low for electron-releasing groups than electron-withdrawing ones on the aromatic ring of isatin. It was also observed that, 1,3-dicarbonyl compounds reacted rapidly than 4-hydroxycoumarin.
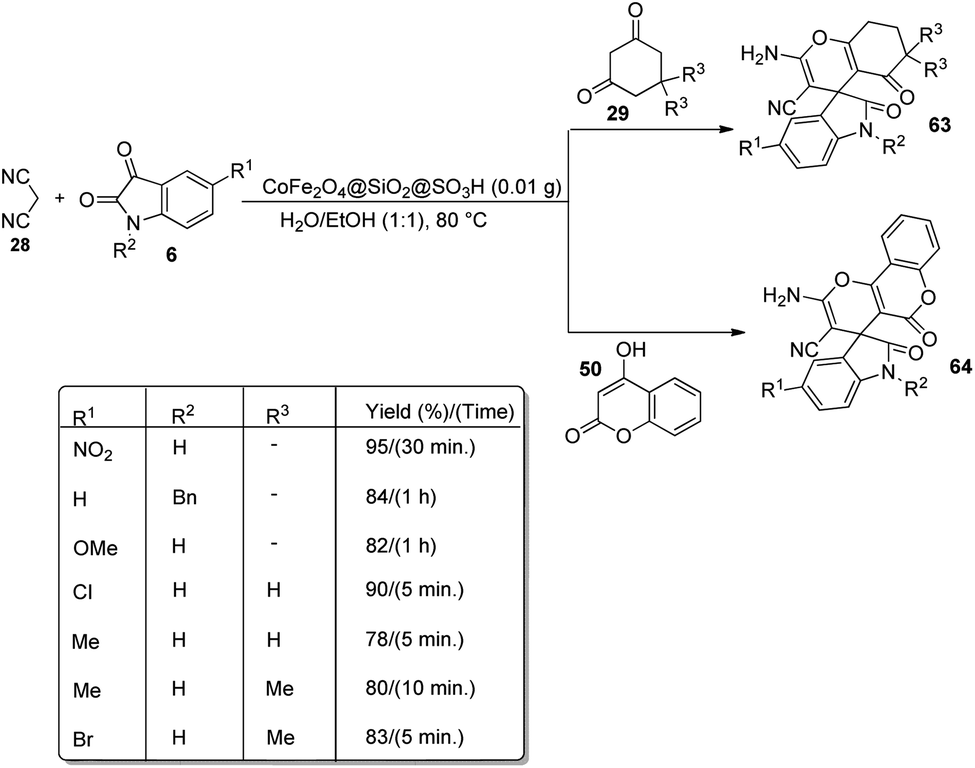 | ||
| Scheme 35 CoFe2O4@SiO2-catalyzed spirooxindole synthesis. | ||
The mechanistic studies suggest that cyanoacetonitrile 28 underwent a nucleophilic addition with isatin 6 whose carbonyl group was triggered by silica nanoparticles. Further, a Knoevenagel condensation occurred and the intermediate 83 thus formed was reacted with dimedone 24 to give the desired product 63. The mechanism is depicted in Scheme 36.
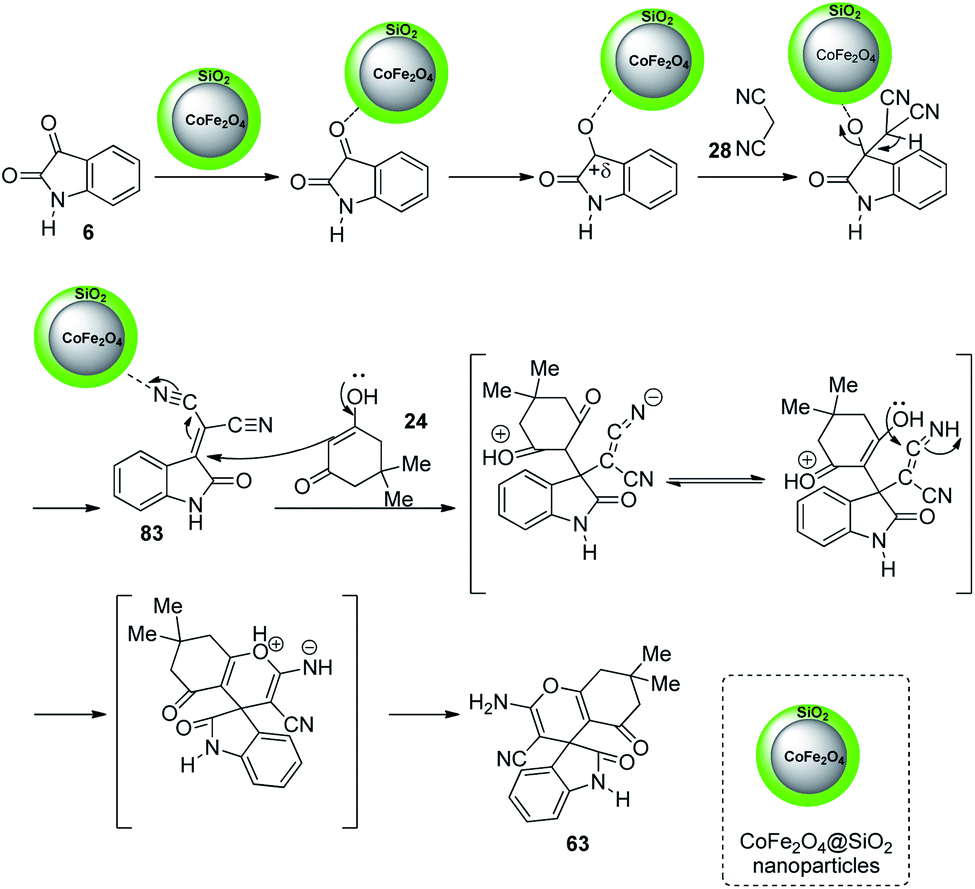 | ||
| Scheme 36 Suggested mechanism for the synthesis of spirooxindoles with CoFe2O4@SiO2 as catalyst [reproduced with permission from ref. 64]. | ||
In the same year Zamani-Ranjbar-Garmroodi and co-workers used the same approach for the development of spirooxindoles by making use of slightly modified CoFe2O4@SiO2@SO3H as the catalyst,65 which was prepared by treatment of CoFe2O4 with tetraethylorthosilicate which was further treated with chlorosulphonic acid. The one-pot reaction with the same substrates proceeded under the same conditions and provided almost similar results with respect to reactivity and electronic effects (Scheme 37).
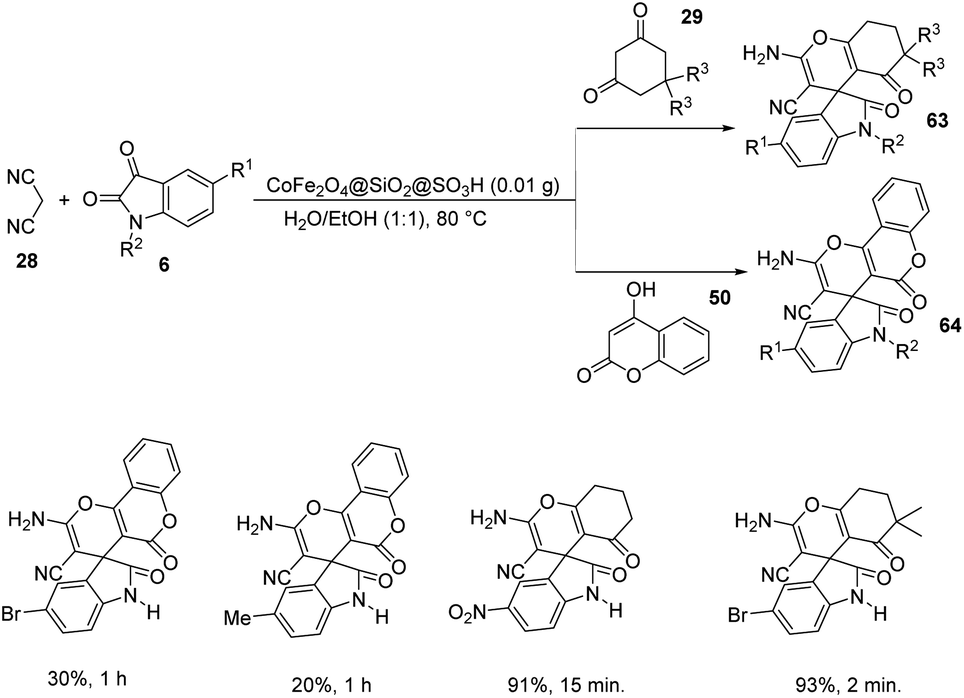 | ||
| Scheme 37 CoFe2O4@SiO2@SO3H-catalyzed spirooxindole synthesis. | ||
An environmentally benign approach for the development of derivatives of spirooxindoles 63, 64 by working with Fe(III)@graphitic carbon nitride was established by Allahresani et al.66 Substituted isatins 6, dicyanomethane 28 and 4-hydroxycoumarin 50 or cyclic 1,3-diketone 29 reacted in the optimized conditions of 0.07 g of Fe(III)@g-C3N4 in a mixture of water and ethanol in 1:1 ratio (Scheme 38). Various isatins afforded 70–98% of the products without significant effect on the electronic nature of the substituents. In the case of 4-hydroxycoumarin, the reaction time was longer and the yield was lower relative to 1,3-dicarbonyls. This approach shows benefits including short reaction time, eco-friendly reaction media and the catalyst can be reused without significant loss of efficiency.
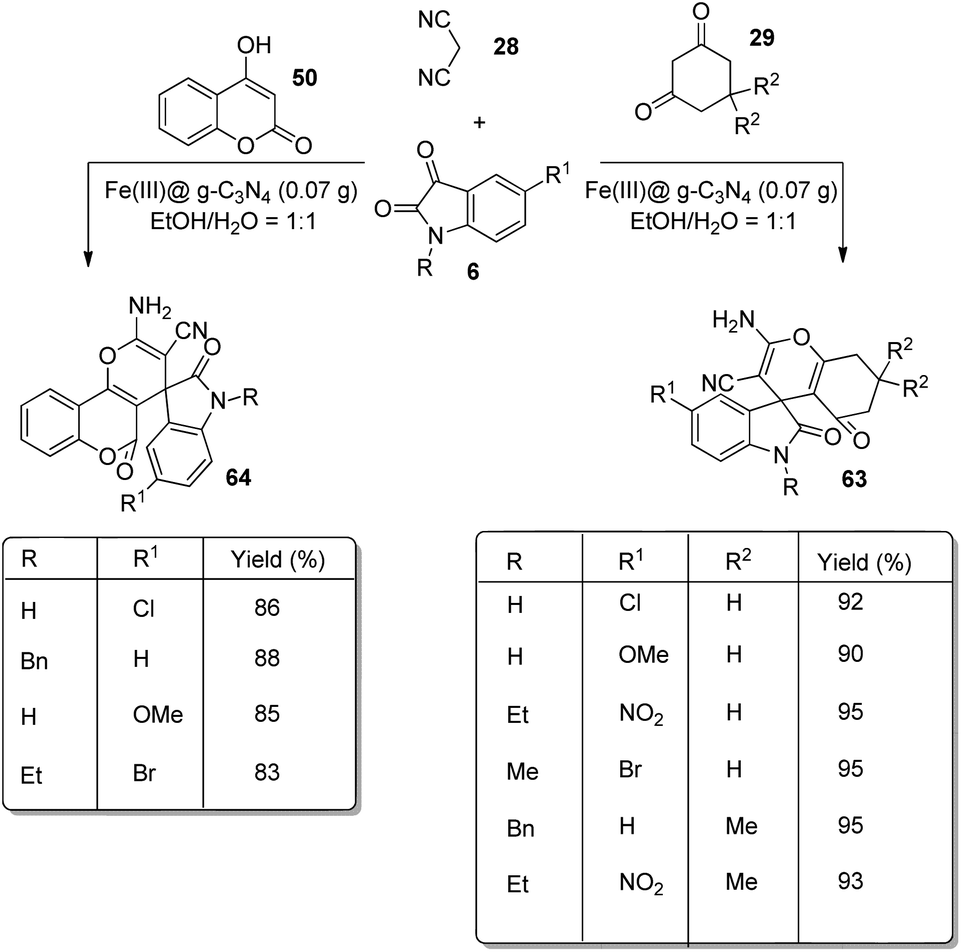 | ||
| Scheme 38 Substrate scope for spirooxindole synthesis with Fe(III)@g-C3N4. | ||
An idea for the growth of polyheterocyclic spirooxindoles 85 by utilizing environment friendly Fe(ClO4)3·6H2O as the catalyst was established by Pan et al.67 The product formation occurs through a hetero-Pictet–Spengler reaction. The reaction between variously substituted tryptophols 84 and isatins 6 was optimized under the conditions of Fe(ClO4)3·6H2O (10 mol%) with tetrahydrofuran as the solvent at 80 °C. First, N-methyl tryptophol-derivative of 84 was reacted with different isatins 6 (Scheme 39). Electronic nature of the substituents was not very noticeable with respect to product formation. The reaction was also promoted by disubstituents on the phenyl ring.
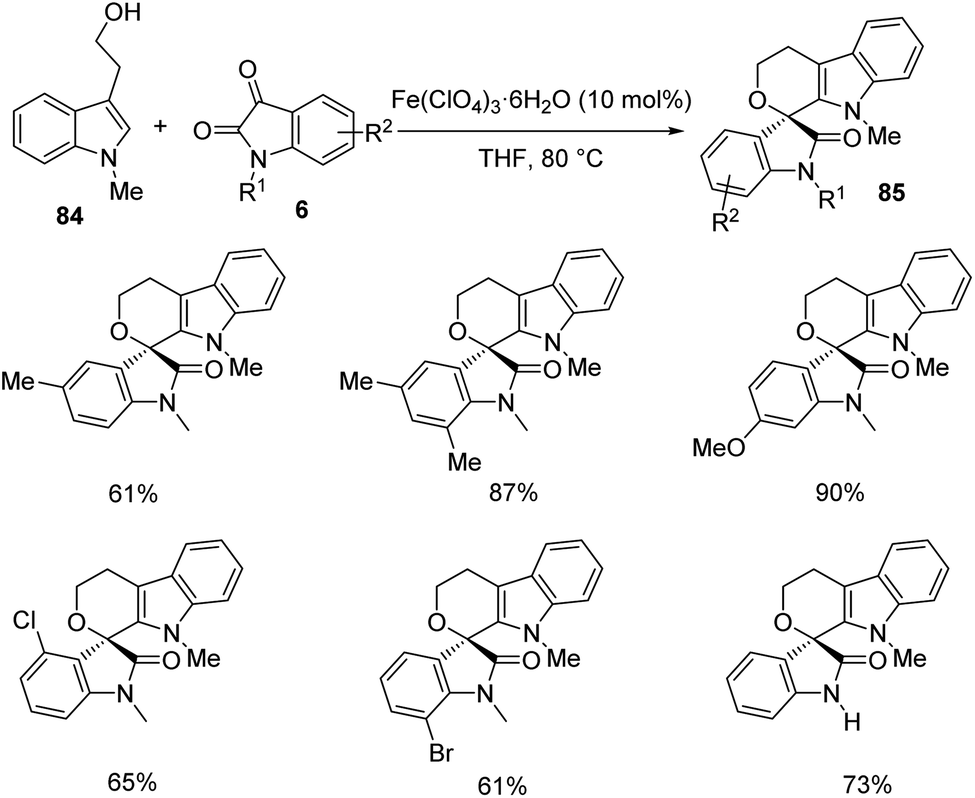 | ||
| Scheme 39 Synthesis of polyheterocyclic spirooxindoles using N-methyl tryptophols and substituted isatins. | ||
Then various tryptophols 84 were reacted with N-methyl isatin-derivative of 6 and rendered moderate to excellent yields (Scheme 40). The concept was further extended to produce a series of polyheterocyclic spirooxindoles 87, 88 by applying different substrates 86: 2-(benzofuran-3-yl)ethanol, 2-(thiophene-2-yl)ethanol, 2-(indole-3-yl)acetamide etc. with substituted isatins 6 (Scheme 41).
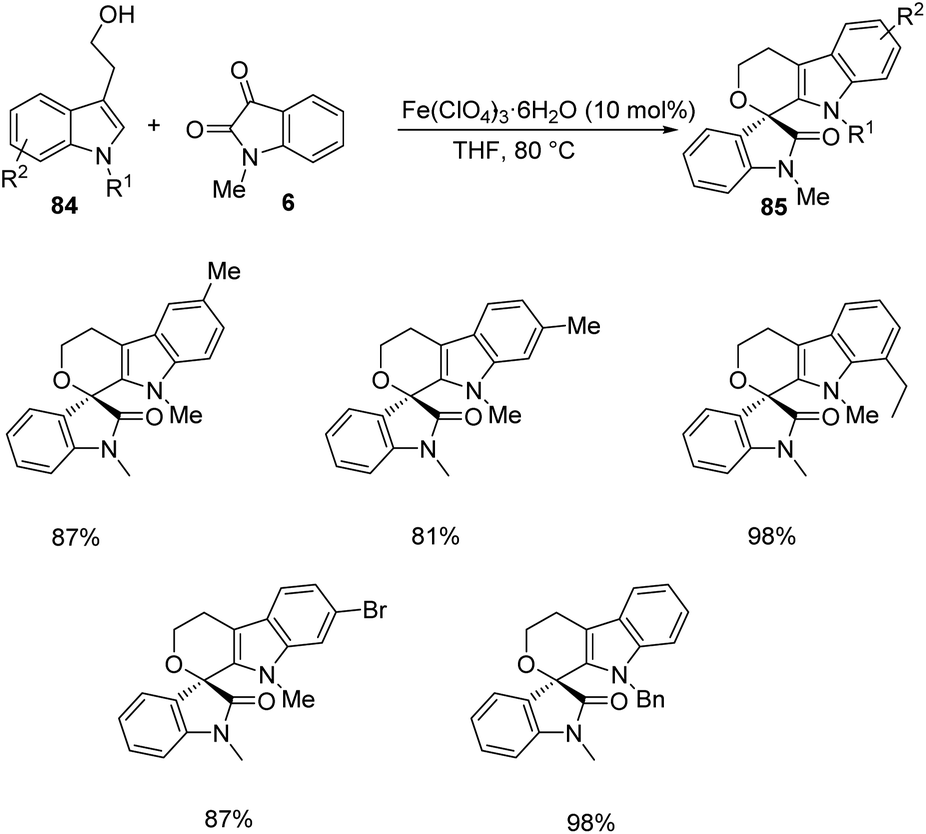 | ||
| Scheme 40 Synthesis of polyheterocyclic spirooxindoles using N-methyl isatin and substituted tryptophols. | ||
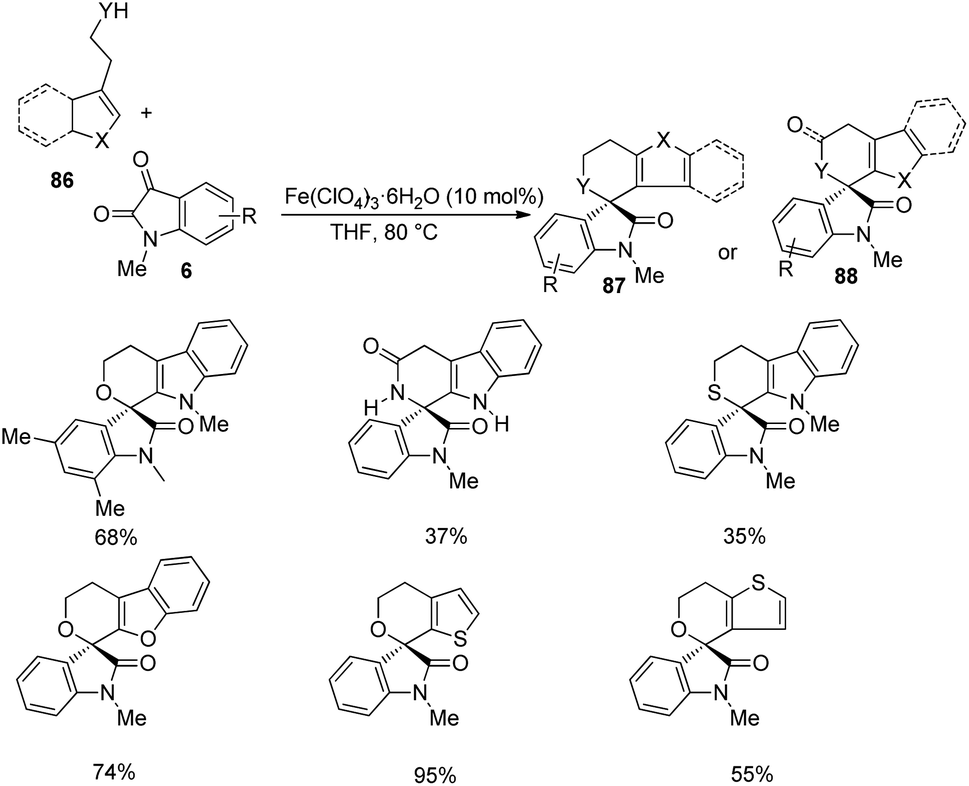 | ||
| Scheme 41 Substrate scope investigation for the synthesis of polyheterocyclic spirooxindoles catalyzed by Fe(ClO4)3·6H2O. | ||
Kavyani and Baharfar developed a concept for the generation of spirooxindole-dihydropyridines 90 catalyzed by Fe3O4/GO/Au–Ag.68 It was a novel catalyst made by the assembly of Au–Ag alloy NPs on the surface of Fe3O4/graphene oxide spheres. The optimized reaction conditions include water as solvent and Fe3O4/GO/Au–Ag as catalyst at room temperature, under which derivatives of barbituric acids 30, derivatives of isatin 6 and 6-amino uracil 89 underwent a one-pot reaction to afford spirooxindole products 90 in 81–93% yields (Scheme 42). The catalyst remained effective without losing its activity and chemical composition up to five cycles of reaction.
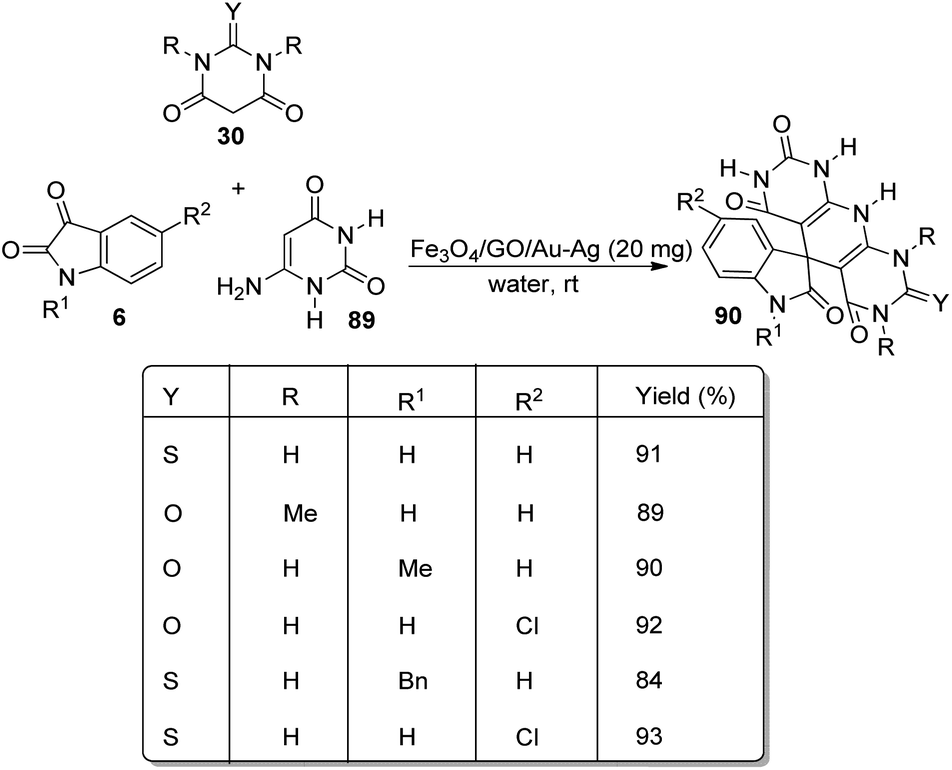 | ||
| Scheme 42 Fe3O4/GO/Au–Ag catalyzed spirooxindole-dihydropyridine synthesis. | ||
7.3 Reactions involving oxindole derivatives
An approach for the formation of derivatives of imidazolidinyl spirooxindoles 93 was reported by Wang and co-workers.69 Here, Fe(OTf)2, which is a Lewis acid, was used as the catalyst in the three-component reaction between diazo-oxindoles 91 and 1,4-oxazepins 92. The reaction was optimized with 10 mol% Fe(OTf)2 in tetrahydrofuran as the solvent at rt. This approach was evaluated by employing differently substituted diazo-oxindoles and 1,4-oxazepins. First, dibenzo[b,f][1,4]oxazepin-derivative of 92 was reacted with substituted diazo-oxindoles 91 (Scheme 43). Electron-releasing and electron-withdrawing groups at different positions on the aromatic ring of diazo-oxindoles underwent the reaction and the products were acquired in 80–91% yield. Different N-protecting groups were well tolerated in this reaction. Then, 3-diazoindoline-2-one, derivative of 91 was reacted with substituted dibenzo[b,f][1,4]oxazepins 92 (Scheme 44). Electron-rich and electron-deficient substituents and disubstituted dibenzo[b,f][1,4]oxazepins also underwent the reaction and afforded 80–89% of the corresponding product.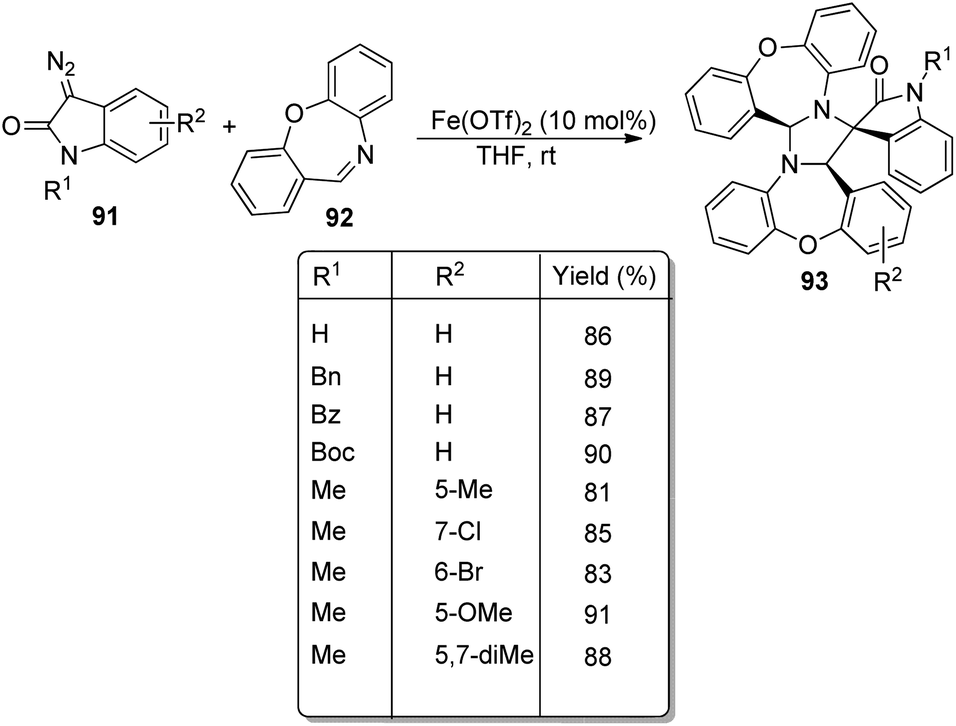 | ||
| Scheme 43 Synthesis of spirooxindoles using dibenzo[b,f][1,4]oxazepins and substituted diazo-oxindoles. | ||
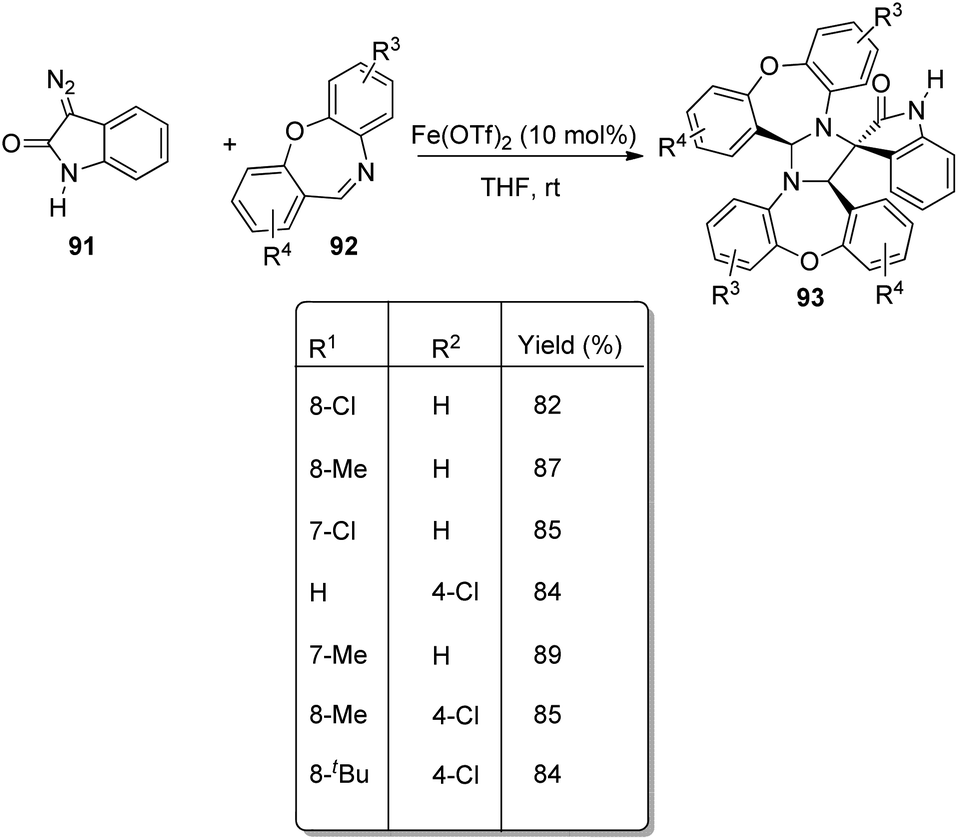 | ||
| Scheme 44 Synthesis of spirooxindoles using diazo-oxindole and substituted dibenzo[b,f][1,4]oxazepins. | ||
A novel procedure for the synthesis of spirooxindolo-2-iminothiazolines 96 catalyzed by FeCl3 was established.70 It involves a [3 + 2] cycloaddition reaction between aryl/alkyl isothiocyanates 94 and 1,3-dipoles produced from spirooxindole aziridines 95. The one-pot reaction proceeded with FeCl3 (10 mol%) in dichloromethane at −20 °C (Scheme 45). The substrate scope investigation was carried out utilizing different aryl/alkyl isothiocyanates and spirooxindole aziridines. The recovered yield of product was less with electron-deficient groups on the aromatic ring of oxindole relative to electron-rich groups. Compared to ethyl and benzyl protection on oxindole N, the yield was better for methyl substitution. Both aryl and alkyl isothiocyanates underwent the reaction without electronic and steric effect of the substituents on the aryl ring. Isothiocyanates which are non-aromatic and aliphatic were also compliant to the reaction. They could also obtain 72% of the products by employing the reaction on gram scale.
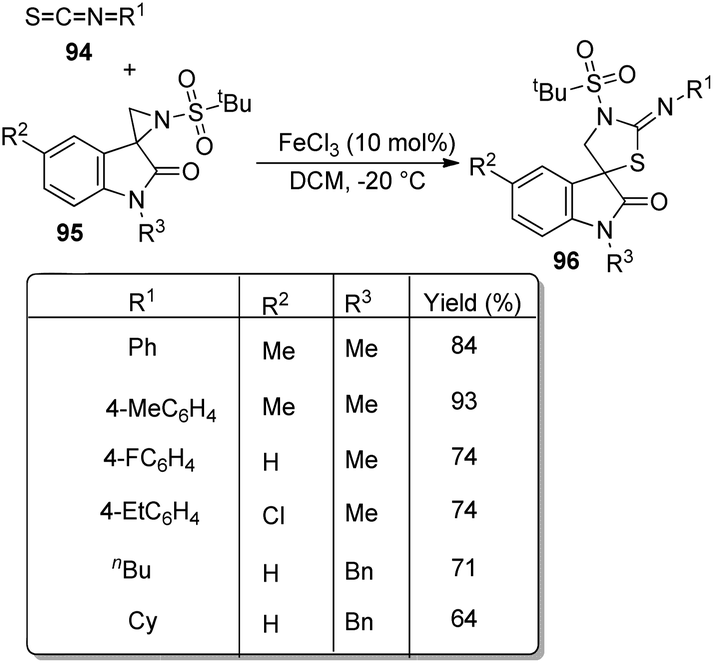 | ||
| Scheme 45 Synthesis of spirooxindolo-2-iminothiazolidines with FeCl3 as catalyst. | ||
8 Pd-catalyzed spirooxindole synthesis
Pd catalysts are used in reactions like Wacker process,71 carbon–carbon bond forming reactions72,73 etc. and are still obtaining popularity in novel reactions. In organic transformations, palladium plays ubiquitous role among other transition metals. In most of the Pd-catalyzed reactions, the usual steps involved are two-electron reductive elimination and oxidative addition.Spiropyrrolidinyl oxindoles, spirocyclooxindoles, cyclopropane-fused spirooxindoles etc. were produced by employing diverse Pd catalysts like Pd2(dba)3, Pd(OAc)2, PdCp(η3-C3H5), Pd(PPh3)4 and so on. In most of the reports palladium(II)acetate was used as the catalytic species owing to its higher reactivity.
8.1 Reactions involving aryl iodide derivatives
Overman et al. disclosed a pathway for the synthesis of spirooxindoles 98 facilitated by palladium catalyst with a diphosphine ligand.74 They used two different reaction conditions and they could obtain the two enantiomers of the product 98 from one enantiomer of the chiral ligand (R)-(+)-BINAP. Aryl iodides 97 were used as the substrates which underwent Heck reaction to give either enantiomer of the products. The (S)-enantiomer 98a was produced by providing the conditions of 5 mol% of tris(dibenzylidene acetone)dipalladium, 10 mol% of the ligand, N,N-dimethylacetamide and Ag3PO4 (2 equiv.) at 60–80 °C (Scheme 46). In order to obtain the (R)-enantiomer 98b, the following reaction conditions were used: 10 mol% of tris(dibenzylidene acetone)dipalladium, 20 mol% of the ligand, N,N-dimethylacetamide and 1,2,2,6,6-pentamethylpiperidine (5 equiv.) at 80–100 °C (Scheme 47). In both the cases, N-methyl-2-pyrrolidinone was used as the solvent and high to good yields were observed.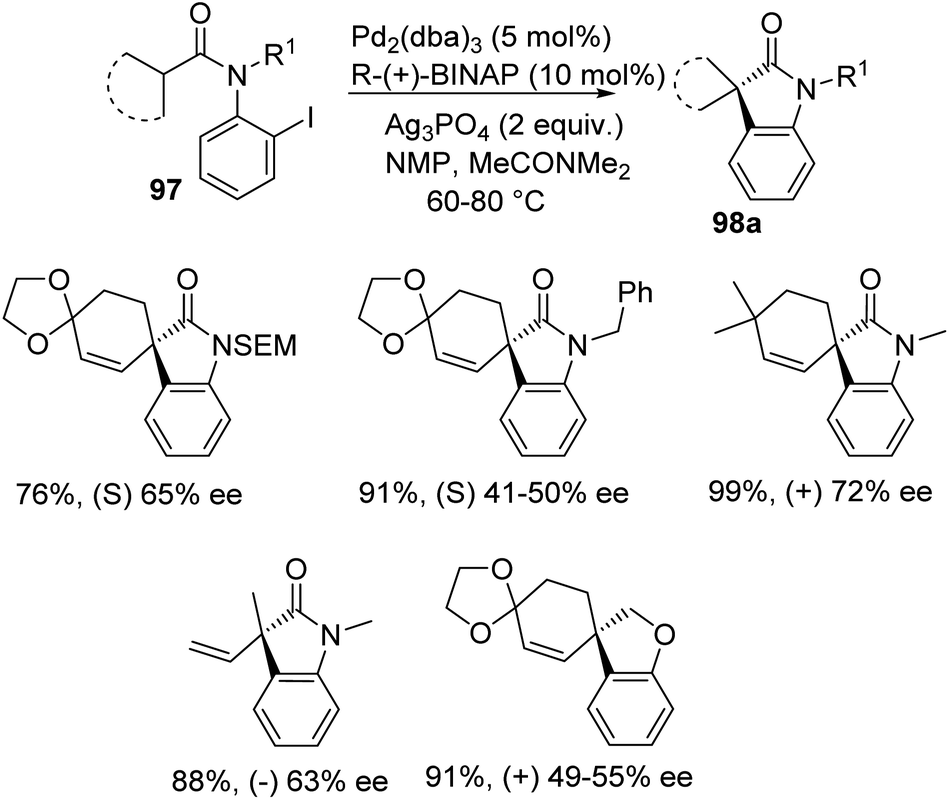 | ||
| Scheme 46 Asymmetric synthesis of (S)-enantiomer of spirooxindole derivative utilizing palladium and R-(+)-BINAP. | ||
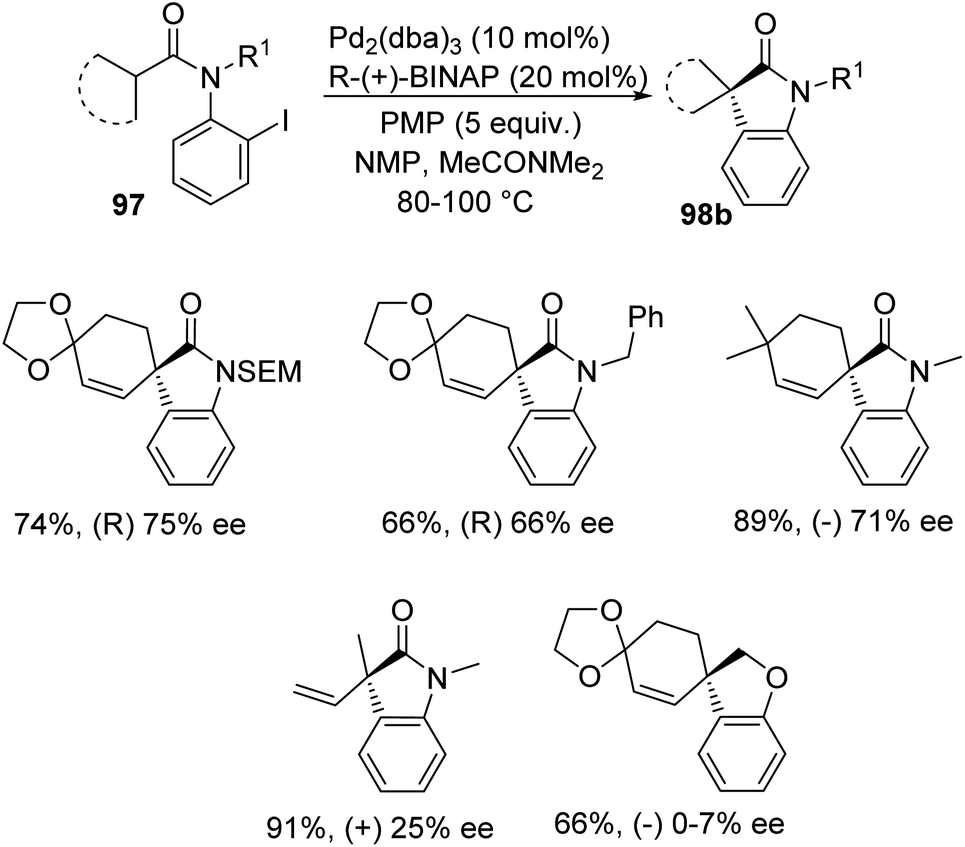 | ||
| Scheme 47 Asymmetric synthesis of (R)-enantiomer of spirooxindole derivatives utilizing Pd and R-(+)BINAP. | ||
Yang et al. designed a process which involves palladium-catalyzed triple C–H bond activation for the formation of spirooxindoles75 101, using diverse iodobenzene 100. The reaction between iodobenzene-derivative of 100 and substituted acrylamides 99 was optimized using 10 mol% of palladium(II)acetate, 20 mol% triphenylphosphine, 5 equiv. of caesium carbonate and 0.5 equiv. of TBAI in dimethyl sulfoxide as the solvent under N2 atmosphere at 120 °C (Scheme 48). Acrylamides with 4-methylbenzyl, 4-fluorobenzyl, benzyl and ethyl substituents on nitrogen underwent the reaction, but those with tosyl group and H atom on nitrogen were unable to perform the reaction. Electron-deficient and -rich groups on the 2-iodophenyl moiety was tolerated but the yield was lower with cyano substitution. Moderate to good yields were also afforded by various substituents on the benzene ring associated with double bond.
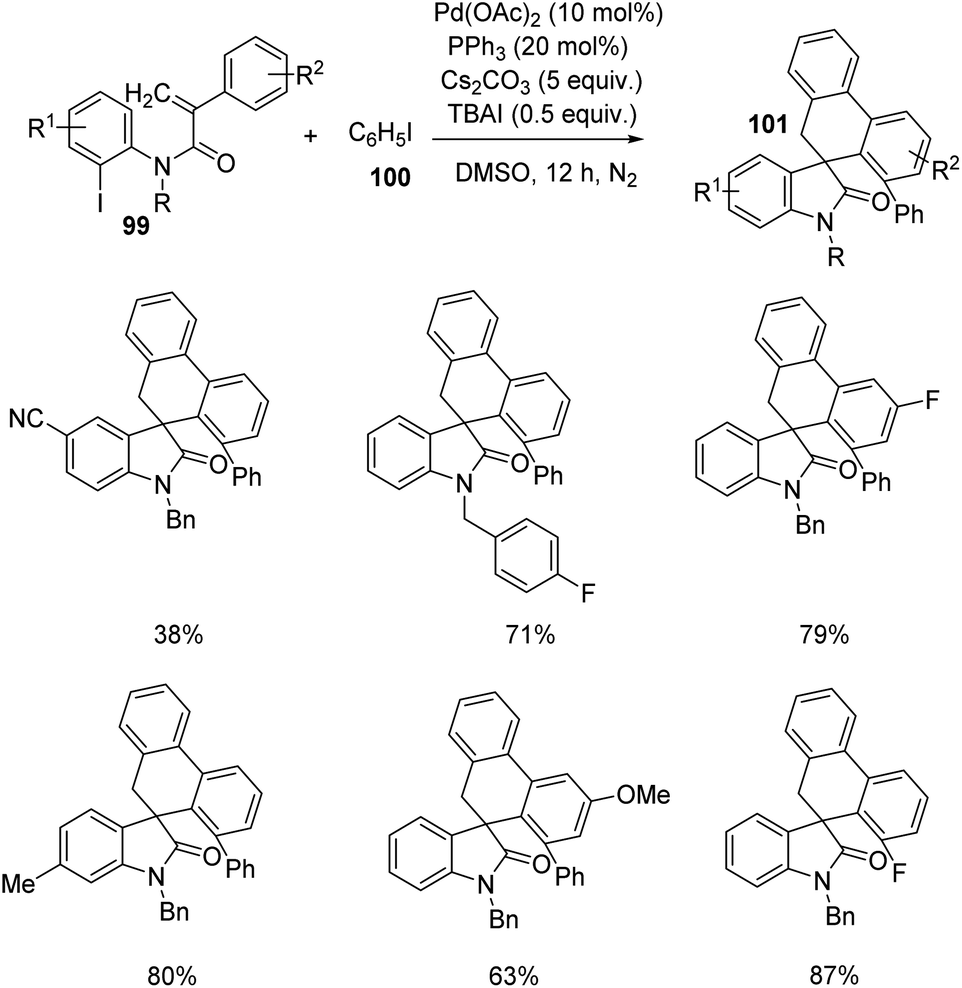 | ||
| Scheme 48 Synthesis of spirooxindoles from substituted N-(2-iodophenyl)-2-phenyl acrylamides and iodobenzene. | ||
Then the reaction was carried out with diverse iodobenzenes 100 under the same conditions (Scheme 49). The electronic nature of the substituents was insignificant in case of iodobenzenes with p-substitution. With an increased reaction time, 65% spirooxindole was obtained from substrate bearing two methyl substituents on the meta-position.
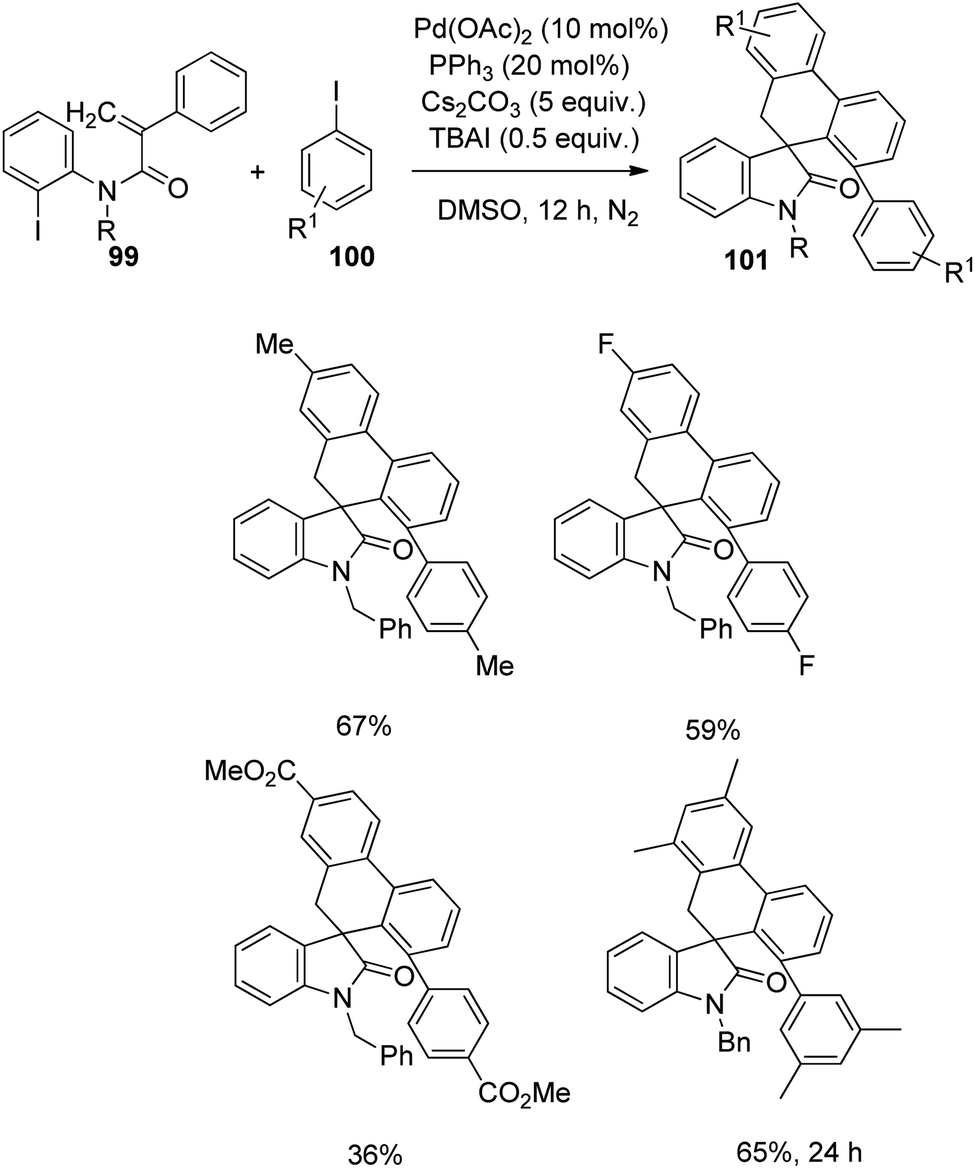 | ||
| Scheme 49 Synthesis of spirooxindole from substituted iodobenzenes and N-(2-iodophenyl)-2-phenyl acrylamides. | ||
Aryl iodide derivatives were used along with palladium catalysts Pd2(dba)3 and Pd(OAc)2 which furnished the spirooxindole product with up to 99% and 87% yields respectively.
8.2 Reactions involving isatin derivatives
A strategy for the formation of derivatives of spirooxindole 103 from γ-methylidene-δ-valerolactones 102 and isatins 6 catalyzed by a palladium complex was devised by Hayashi and co-workers.76 Here γ-methylidene-δ-valerolactones 102 underwent decarboxylative [4 + 2] cycloaddition with isatin 6 to afford spirooxindoles 103 which are highly stereoselective. The reaction between various isatins and γ-methylidene-δ-valerolactones was catalyzed with PdCp(η3-C3H5) (5 mol%) and phosphoramidite ligand (L8) (10 mol%) in tetrahydrofuran at 40 °C (Scheme 50).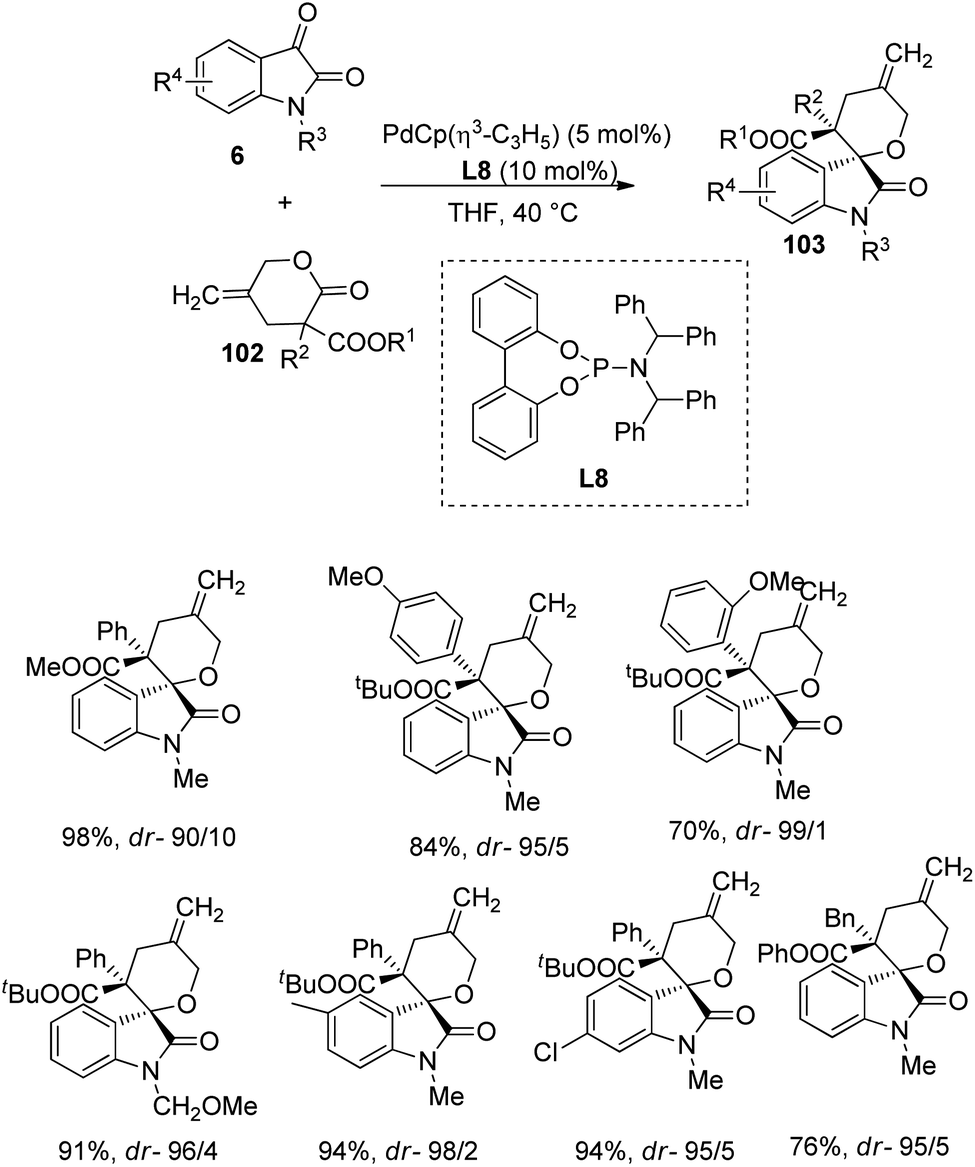 | ||
| Scheme 50 Substrate scope for the synthesis of spirooxindoles via decarboxylative cyclization catalyzed by Pd complex. | ||
Isatins with different groups on N atom and on aromatic ring were tolerated and rendered high to efficient yields and dr values. In the case of γ-methylidene-δ-valerolactone, better dr values were achieved from tBu compared to methyl as the ester groups. α-(Hetero)aryl-γ-methylidene-δ-valerolactones and α-alkyl lactones also underwent the reaction but in the case of α-alkyl lactones satisfactory yield was obtained only when phenyl was used as the ester group. They expanded the strategy to other ketones including diethylketomalonate. Scheme 51 provides the catalytic cycle for the reaction.
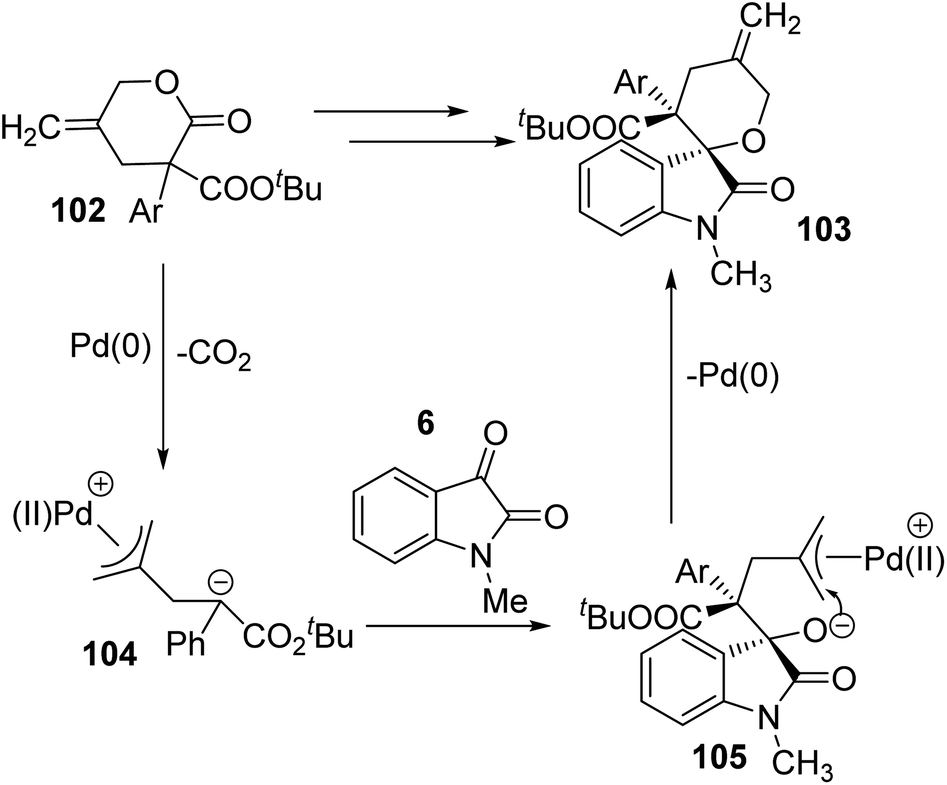 | ||
| Scheme 51 Possible catalytic cycle for the decarboxylative cyclization catalyzed by Pd [reproduced with permission from ref. 76]. | ||
The allyl ester group of γ-methylidene-δ-valerolactone 102 underwent oxidative addition to Pd(0). A 1,4-zwitterionic species 104 was generated via subsequent elimination of a CO2 molecule. An intermediate 105 was developed by the attack of anionic C of zwitterion 104 to electrophilic C of N-methyl isatin, derivative of 6. In the intermediate 105, the π-allyl palladium was attacked by nucleophilic O to give the spirooxindole product 103. Here, the catalyst was restored.
Synthesis of spirooxindoles 107 by the formal [5 + 3] cycloaddition reaction between aryl substituted vinylethylene carbonates 106 and α-(trifluoromethyl)imines derived from isatin 6 was established by Shi and co-workers.77 Making use of the conditions given in the Scheme 52, they synthesised spirooxindole fused with an 8-membered ring as individual diastereoisomer. 66–82% of the products were afforded by isatin-derived α-(trifluoromethyl)imines with various substituents on the aryl ring without significant electronic and positional effects. Diverse groups such as Me, Bn and iPr on the nitrogen atom of isatin underwent the reaction but the yield was only 27% without protection. The substrate scope for vinylethylene carbonates was also probed and moderate to good yields were contributed by those with electron-deficient and electron-donating groups on aromatic ring. The yield was 0 and 85% for vinylethylene carbonates without any substitution and with naphthyl substituent respectively. A chiral phosphine ligand was used to perform the asymmetric version of the [5 + 3] cycloaddition reaction.
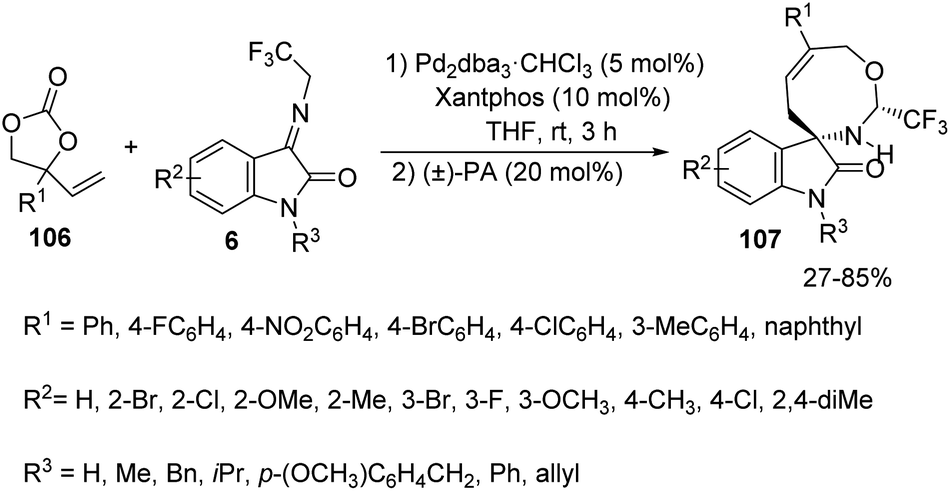 | ||
| Scheme 52 Synthesis of spirooxindoles through formal [5 + 3] cycloaddition using Pd catalyst. | ||
8.3 Reactions involving oxindole derivatives
A procedure for the formation of spirocyclooxindoles through the synthesis of 3-cyanomethyloxindoles was executed by Jaegli et al.78 Here spirocyclopropyl-, spiropiperidinyl- and spiropyrrolidinyl oxindoles were produced from 3′-alkyl-3-cyanomethyl-2-oxindoles which were made via Heck/cyanation catalyzed by palladium. Different heterocycles were further prepared by the functionalization of the products thus achieved, through Buchwald–Hartwig N-arylation, N-sulfonylation, N-acylation and so on.A procedure for the generation of cyclopentadiindolyl spirooxindoles 109 catalyzed by palladium through oxidative coupling reaction was reported by Kim et al.79 3,3-Diindolyl-2-oxindoles 108 upon treatment with 10 mol% Pd(OAc)2 as the catalyst, 3 equiv. AgOAc as the oxidant and 1 equiv. caesium carbonate as the base in pivalic acid at 120 °C afforded cyclopentadiindolyl spirooxindoles (Scheme 53). Good yields were furnished by N-methylisatins with electron-poor and electron-rich groups with mono- and disubstitution. Indoles with Me and OMe group on fifth position offered 58–87% of spirooxindoles. Low reaction temperature (80 °C) was used with dimethoxy derivative in order to avoid decomposition.
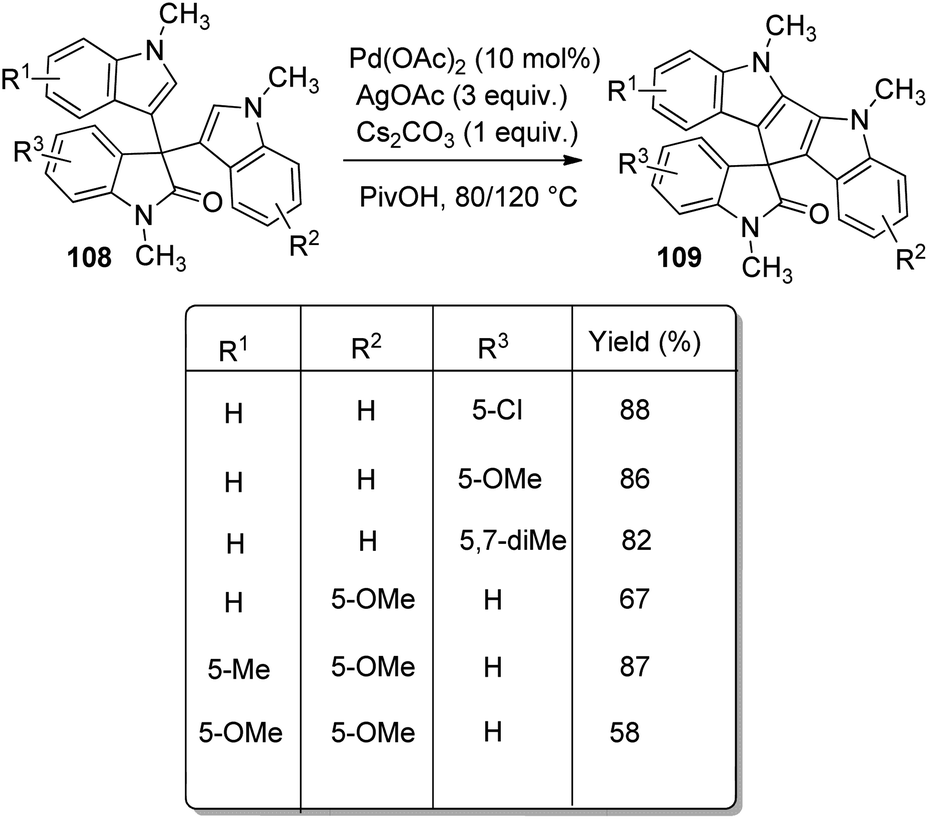 | ||
| Scheme 53 Substrate scope for cyclopentadiindolyl spirooxindoles syntheses catalyzed by palladium. | ||
According to the mechanism an intermediate 110 was generated by the electrophilic palladation of 3,3-diindolyl-2-oxindole 108 with Pd(OPiv)2 or Pd(OAc)2. Next electrophilic palladation took place in the intermediate 110 intramolecularly and subsequent reductive elimination provided the required cyclopentadiindolyl spirooxindole 109. The palladium catalyst was regenerated by silver acetate. The proposed reaction path is depicted in Scheme 54.
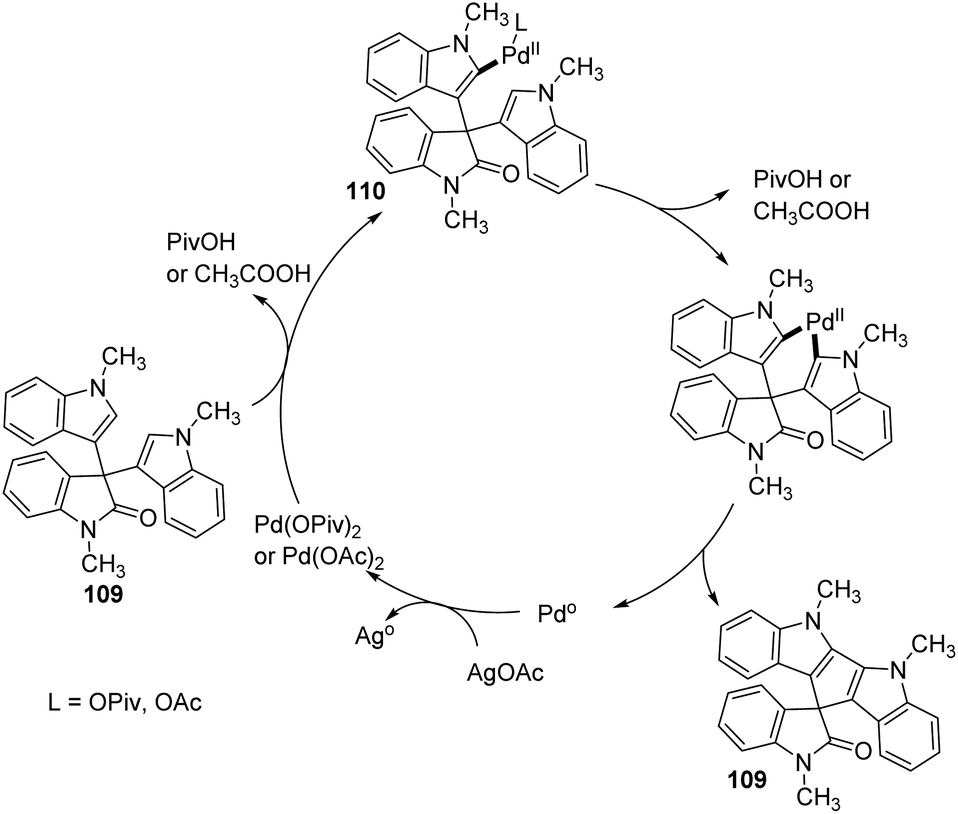 | ||
| Scheme 54 Plausible mechanism for the oxidative coupling reaction with Pd catalyst [reproduced with permission from ref. 79]. | ||
Pd-catalyzed carbenylative amination of o-vinylanilines 111 with diazooxindole-derivative of 91 was described by Anbarasan and co-workers.80 A set of indoline fused spirooxindoles 112 was achieved in good yields with proficient diastereoselectivity, utilizing differently substituted substrates. The reaction conditions involved 5 mol% [Pd(cinnamyl)Cl]2 in toluene at 120 °C for 5 h and the scope of o-vinylaniline was investigated (Scheme 55). The reaction proceeded with simple aniline, p-methoxy, p-phenyl, halo-, acetal-, acyloxy- and tosyloxy-substituted o-vinylanilines. Due to steric reasons, naphthyl-substituted o-vinylaniline was unable to perform the reaction. o-Vinylaniline with varied substituents on nitrogen also underwent the reaction. Different groups such as Ph, p-tbutylphenyl, p-tolyl, p-fluorophenyl and p-ethyl on the alkene fragment were tolerated (67–82%). Substrates with thiophenyl and cyclohexenyl substituents were also suitable for this reaction. The desired product was not obtained with simple styrene, but Me substituted one afforded the product.
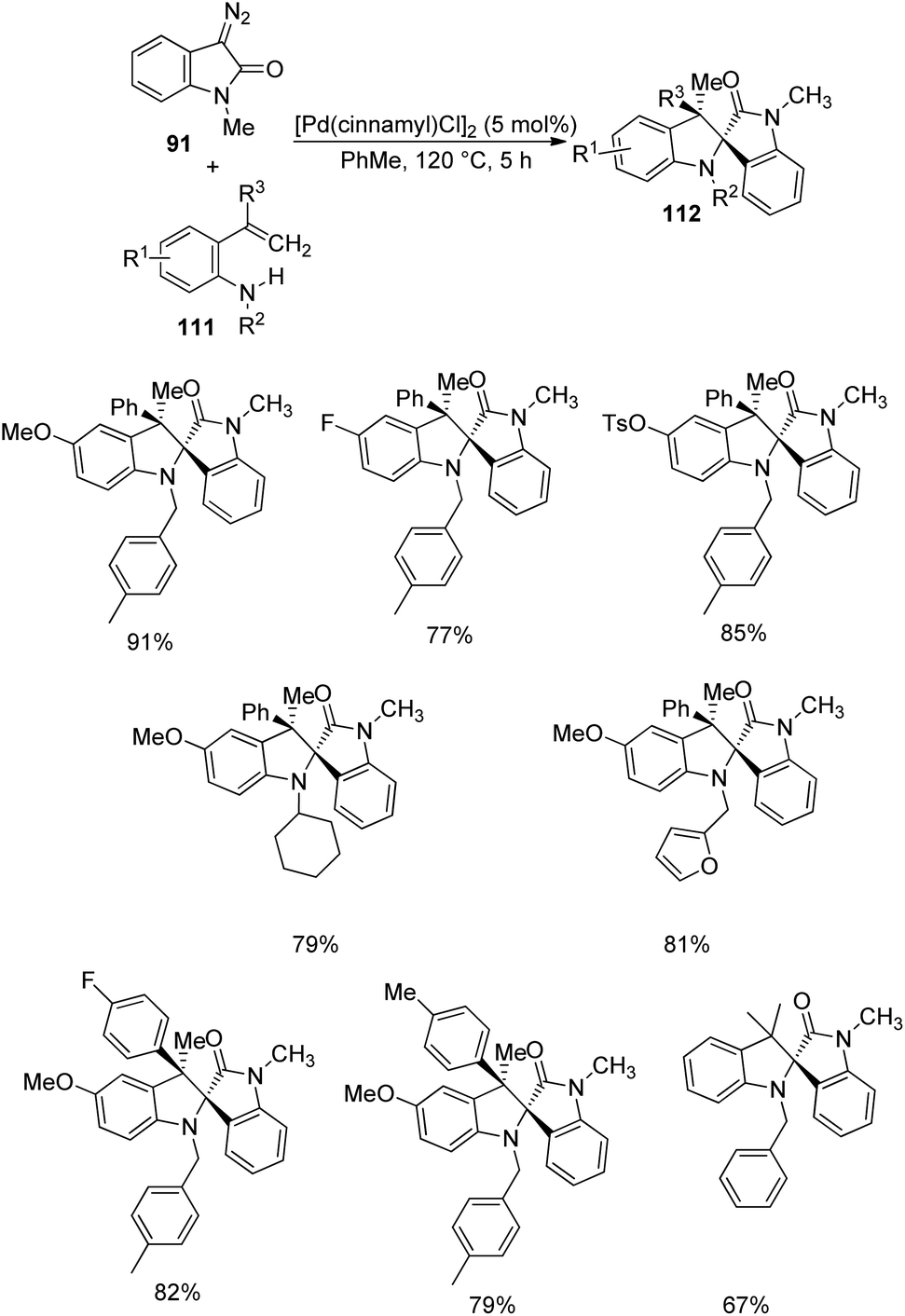 | ||
| Scheme 55 Diversification of o-vinylanilines in Pd-catalyzed carbenylative amination. | ||
Next, the reaction was performed using differently substituted diazo compounds 91 (Scheme 56). N-Alkyl, N-propargyl, N-allyl and N-benzyl substituted diazo compounds underwent the reaction. Good yields were given by those with electron-withdrawing substituents on the fifth position. Further, spirooxindoles 112 were produced by one-pot reaction between tosyl hydrazones 113 and o-vinylanilines- derivative of 111 under the conditions given in Scheme 57.
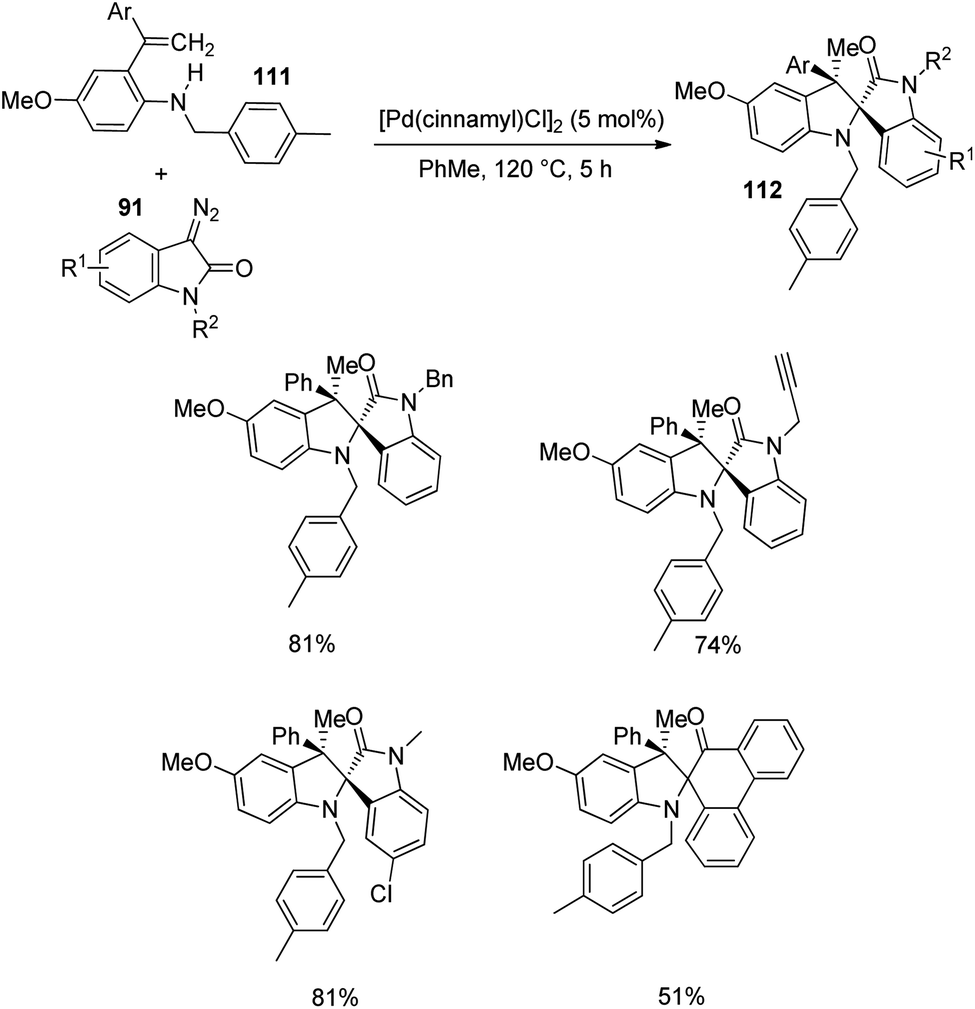 | ||
| Scheme 56 Diversification of diazo compounds in Pd-catalyzed carbenylative amination. | ||
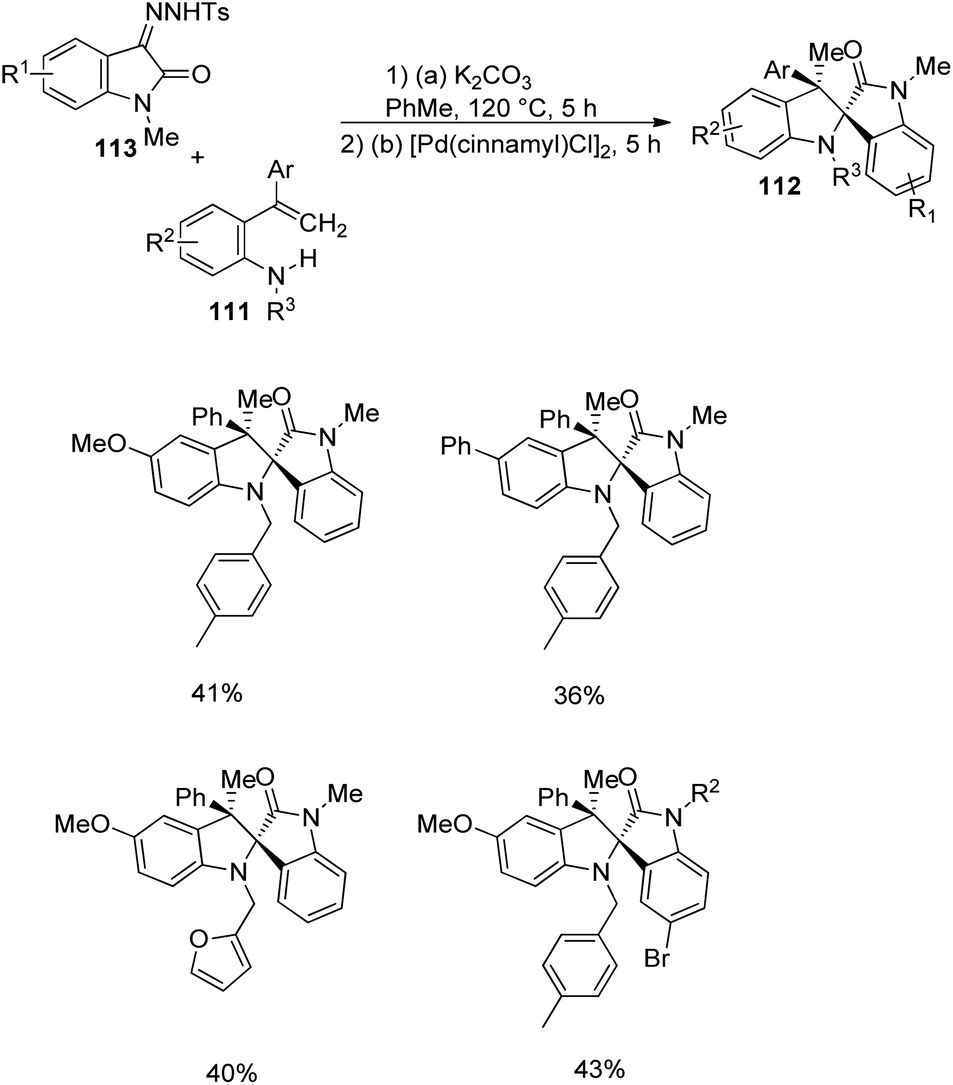 | ||
| Scheme 57 Synthesis of spirooxindoles via one-pot conversion of p-tosylhydrazone. | ||
Most of the reactions with oxindole derivatives employing Fe and Pd catalysts were atom economical and contributed products with excellent diastereoselectivities.
8.4 Reactions involving carbamoyl chloride derivatives
Takemoto and co-workers established a design for the preparation of spirooxindoles 117 catalyzed by palladium, in 2011.81 It involves a carbosilylation accompanied by a Sakurai-type cyclization, in which 1,3-dienes having carbamoyl chloride 114 were used as substrates. In the first step, carbamoyl chlorides 114 were reacted with hexamethyldisilane 115 and [Pd(η3-allyl)Cl]2 (5 mol%) in xylene at 100 °C to deliver the corresponding oxindoles 116. Then Sakurai-type cyclization was performed to produce 5-membered carbocycle-, tetrahydropyran- and piperidine-fused spirooxindoles 117 (Scheme 58).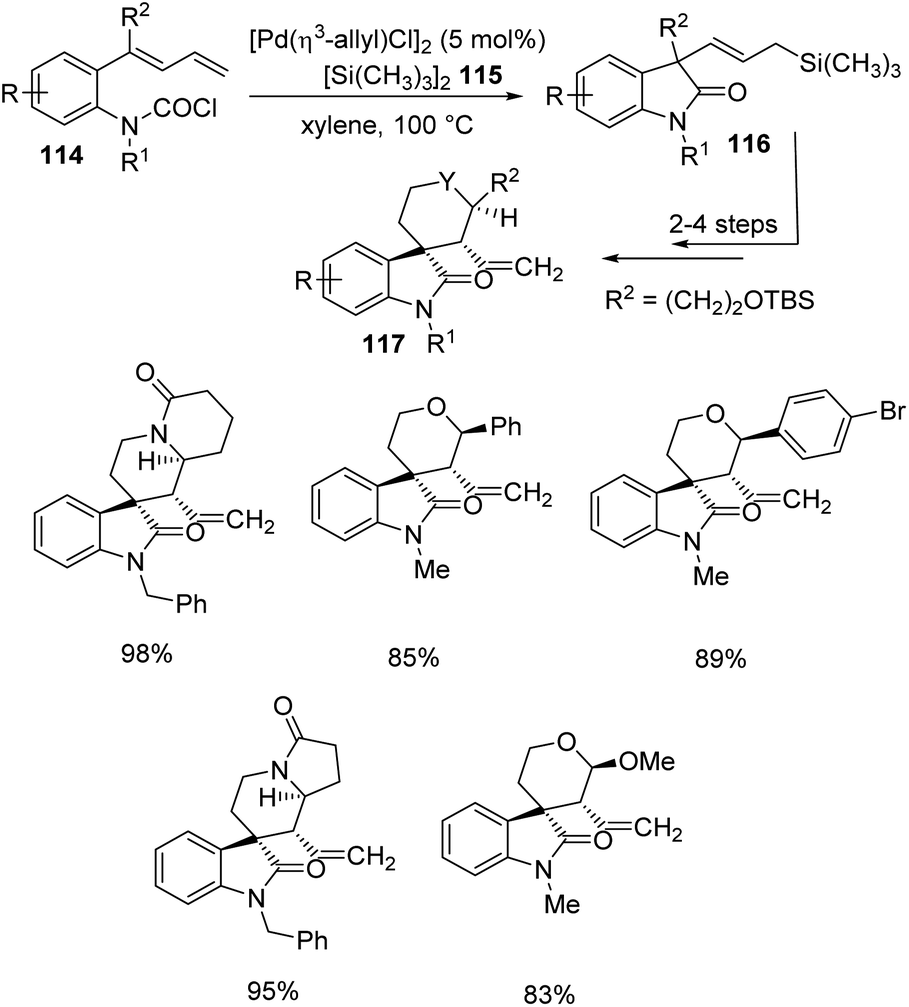 | ||
| Scheme 58 Pd-catalyzed carbosilylation and Sakurai-type cyclization to synthesise spirooxindoles. | ||
In 2013, Takemoto et al. proposed a methodology which involves a C(sp3)–H activation for the synthesis of cyclopropane-fused spirooxindoles 119 catalyzed by palladium.82 It entails the C(sp3)–H activation of carbamoyl chlorides having cyclopropyl group, 118. The reaction was optimized using 6–10 mol% di(1-adamantyl)-n-butylphosphine, 3–5 mol% palladium(II)acetate, 30 mol% N-hydroxypivalamide and 1.1 equiv. caesium carbonate under CO atmosphere in mesitylene at 120–135 °C (Scheme 59). Good yields were brought by ortho-ethyl and ortho-isopropyl substituted carbamoyl chlorides. In both the cases, activation happened only to the cyclopropyl methine C(sp3)–H bond. In the case of carbamoyl chloride having a naphthyl group, the activation was better for cyclopropyl methine C(sp3)–H bond. The yield granted by substrates with ortho-substituents was superior to that without o-substitution. Products were also afforded by carbamoyl chlorides holding siloxymethyl on the cyclopropane ring.
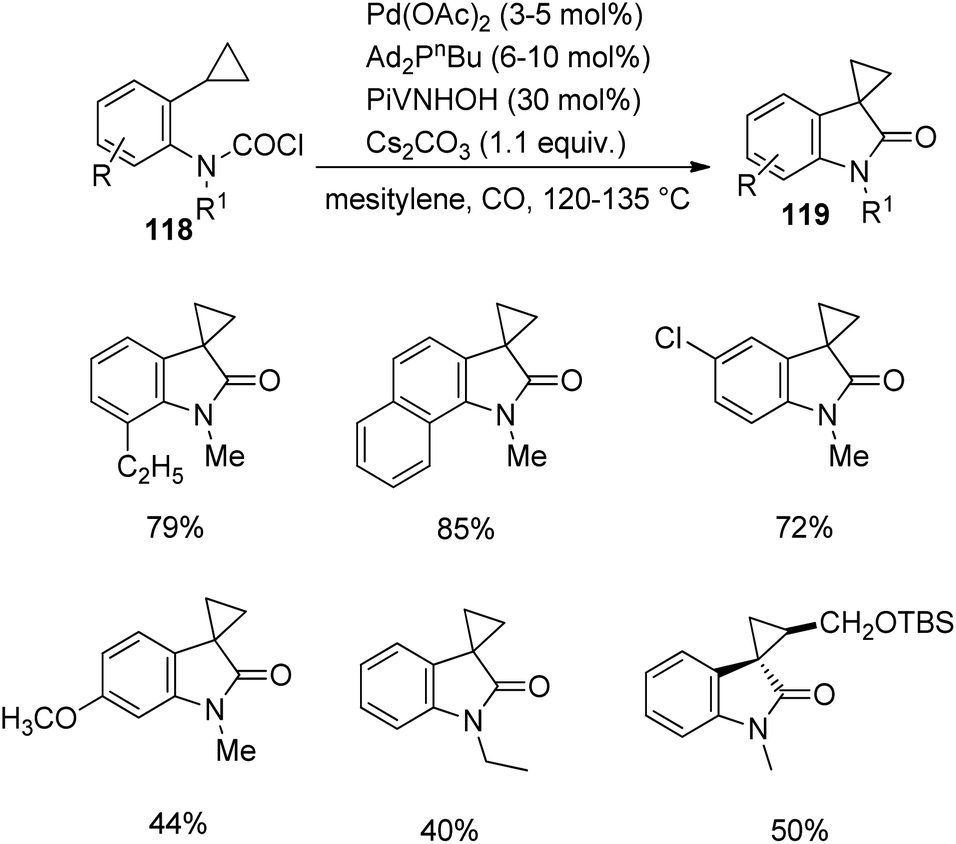 | ||
| Scheme 59 Cyclopropane-fused spirooxindole synthesis catalyzed by Pd. | ||
Studies related to regioselectivity was also done by the group. Heck reaction took place when N atom of carbamoyl chloride carried an alkyl group. Heck reaction > cyclopropyl methine C(sp3)–H activation > methyl C(sp3)–H activation > C(sp2)–H activation was the order of selectivity of the reaction.
[Pd(η3-allyl)Cl]2 and Pd(OAc)2 were used as catalysts for the syntheses of spirooxindoles starting with carbamoyl chloride derivatives. The temperature applied was 100 °C for the former and 120–135 °C for the latter.
8.5 Reactions involving methyeleneindolinones derivatives
A proposal for the generation of enantioenriched 3,3′-pyrrolinyl spirooxindoles 121 catalyzed by a palladium complex was presented by Lu and co-workers.83 Here, methyleneindolinones 72 underwent [3 + 2] cycloaddition with vinyl aziridines 120. The optimized reaction conditions include 12 mol% of ligand L9, 5 mol% of [Pd2(dba)3]·CHCl3 in toluene at 10 °C (Scheme 60). They first investigated the effects of alkene substituents on methyleneindolinones. Good yields were afforded by substrates with arylacyl and ester groups. Diastereo- and enantioselectivities were not affected by electronic nature of the substituents. Methyleneindolinones carrying cyclohexylacyl, 3-thienylacyl and 2-furylacyl groups also underwent the reaction. The influence of substituents on oxindole moiety was surveyed. Methyleneindolinones with mono- and disubstitutions on aromatic ring rendered 69–90% yield, 4:1–19:1 dr values and 82–99% ee values. High yield and stereoselectivities were also obtained from various substituents on nitrogen of vinyl aziridine and oxindole. Gram scale reaction and diversity-directed spirooxindole synthesis were also performed by the group.
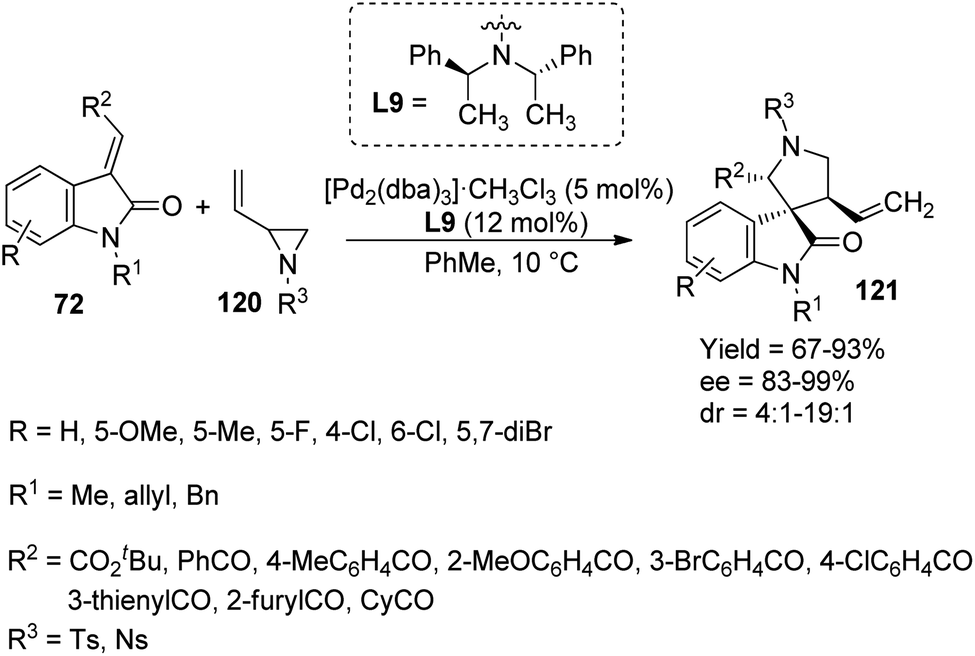 | ||
| Scheme 60 Synthesis of spirooxindoles via [3 + 2] cycloaddition catalyzed by palladium-complex. | ||
8.6 Reactions involving furan derivatives
Furans are conjugated dienes having low aromatic nature and they can function as analogues of 1,3-dienes in organic reactions.Yin and co-workers described 2,5-alkoxyarylation of furan rings for the synthesis of dispirooxindoles 123 by employing a Pd catalyst.84 Variously substituted furans 122 underwent intramolecular cyclization under the optimized conditions of 5 mol% of [Pd2(dba)3], 10 mol% of 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (L10) and 2 equivalent of LiOtBu in an atmosphere of nitrogen in tetrahydrofuran at 100–110 °C (Scheme 61). The yields were good with electron-withdrawing groups on the aromatic ring of furan substrate compared to electron-releasing ones. The products were obtained only in traces when hydrogen or iPr was present on N atom of the substrate instead of methyl and ethyl substituents. The yield was 65–80% and 50% with substrates bearing side chain of four-carbon and five-carbon atoms respectively. It was zero in case of six-carbon and two-carbon chains.
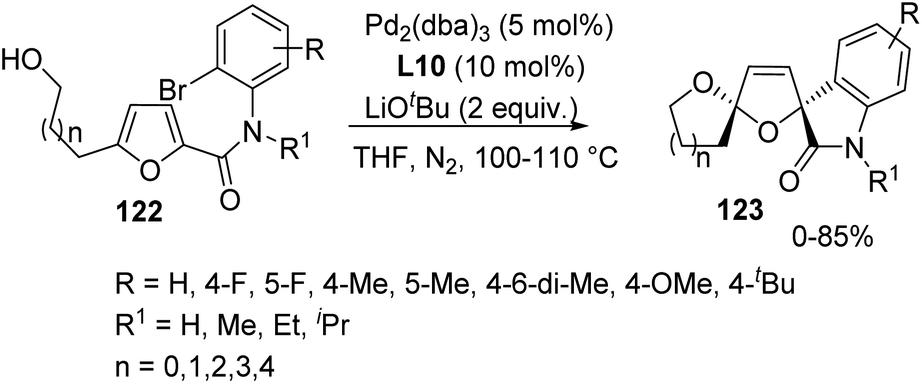 | ||
| Scheme 61 Dispirooxindole synthesis via intramolecular cyclization of furan. | ||
Further the intermolecular cyclization was examined by using variously substituted N-2-bromophenyl-2-furamides 124 with 5 mol% of Pd(OAc)2, 6 mol% of BINAP and 2 equivalent of TEA in excess alcohol 125 under N2 atmosphere at 100 °C and the corresponding spirooxindole 126 was obtained (Scheme 62). When tbutanol was employed as the solvent, spirooxindoles were not yielded. There was no significant effect for the electronic nature of the substituents on the aromatic ring of the substrate.
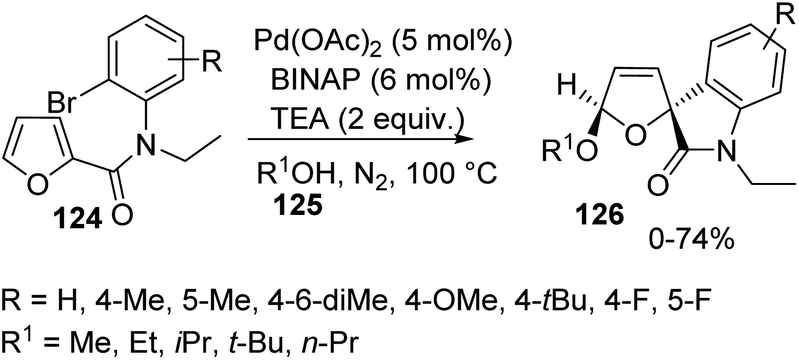 | ||
| Scheme 62 Spirooxindole synthesis via intermolecular cyclization of furan rings. | ||
In the same year, a method for the synthesis of spirooxindoles 127 catalyzed by Pd was put forward by the same group.85 Here also derivatives of furan-derivative of 122 were utilized which underwent intramolecular arylation and transformed into spirooxindoles 127 with high stereo- and regioselectivity. The Z-isomer was formed in higher amount when the reaction of N-(2-bromophenyl)-2-furancarboxamides proceeded with 5 mol% Pd(PPh3)4, 10 mol% PPh3 and 200 mol% K2CO3 in DCE at 80–90 °C (Scheme 63). A combination of 10 mol% of PPh3, 5 mol% of Pd(PPh3)4 and 200 mol% of tBuOLi in 1,4-dioxane as the solvent at 80 °C afforded the E-isomer in higher amounts (Scheme 64).
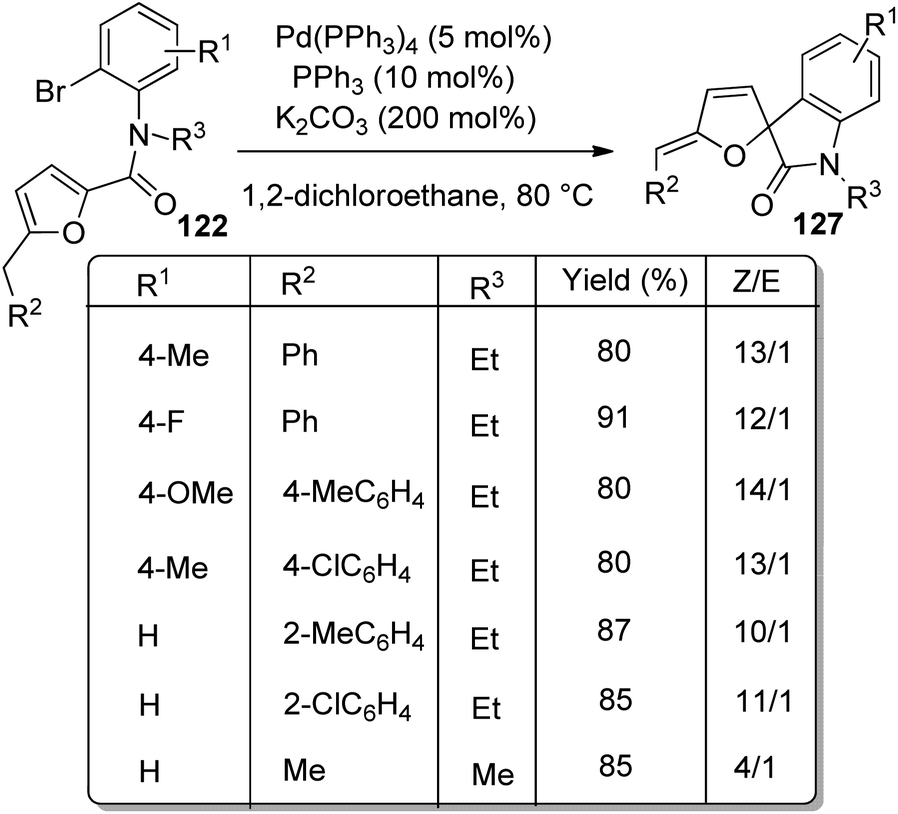 | ||
| Scheme 63 Preferential synthesis of Z-isomer of spirooxindole from N-(2-bromophenyl)-2-furancarboxamides. | ||
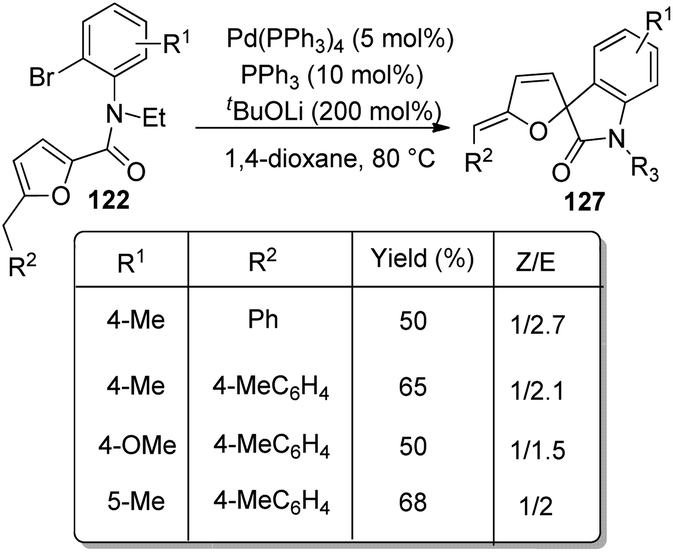 | ||
| Scheme 64 Preferential synthesis of E-isomer of spirooxindoles from N-(2-bromophenyl)-2-furancarboxamides. | ||
First the reaction scope for the preparation of Z-isomer was studied by employing substrates with different substituents on aryl and furan rings and also on nitrogen. Both electron-deficient and electron-rich groups on Ph ring was tolerated. When naphthyl, thiophenyl, phenyl or alkyl group other than Me was present as R2 the selectivity was higher. The desired product was not acquired when H was present on N of the substrate. Then the effect of substituents on formation of E-isomer was examined. Substrates with p-Tolyl and Ph as R2 gave the E-isomer preferentially. But in case of substrate with Me as R2, the selectivity towards Z-isomer was higher even with tBuOLi as the base.
Pd2(dba)3, Pd(OAc)2 and Pd(PPh3)4 were used as catalysts for spirooxindole synthesis starting from furan derivatives and they afforded the yield of up to 85%, 74% and 91% respectively.
8.7 Reactions involving acrylamide derivatives
A strategy for the formation of spirooxindoles 129 through C–H activation and subsequent benzyne insertion catalyzed by palladium was designed by Lautens and co-workers in 2016.86 The reaction between acrylamides 99 and benzyne precursors 128 was optimized with 10 mol% Pd(PPh3)4, 2 equiv. of CsF, 1.5 equiv. of Cs2CO3 in a mixture of toluene and acetonitrile (1:1) at 80 °C (Scheme 65). Small amount of triphenylene was also formed along with the desired spirooxindole product. Acrylamides with electron-releasing and -deficient substituents underwent the cyclization. High yields were obtained from acrylamide bearing a fluorine atom on the attached aryl group. The presence of CH2CO2C2H5 and MOM on the nitrogen atom was also tolerated, but the yield was lower with electron-deficient groups. Acrylamide with attached aryl group having ortho-substitution afforded 78% of the product. The yield was lower with electron-withdrawing benzyne precursors compared to the electron-releasing ones. The group could produce 85% of the spirooxindoles by performing the reaction on gram scale. Further ring expansion and aromatization of the spirooxindoles were also carried out.
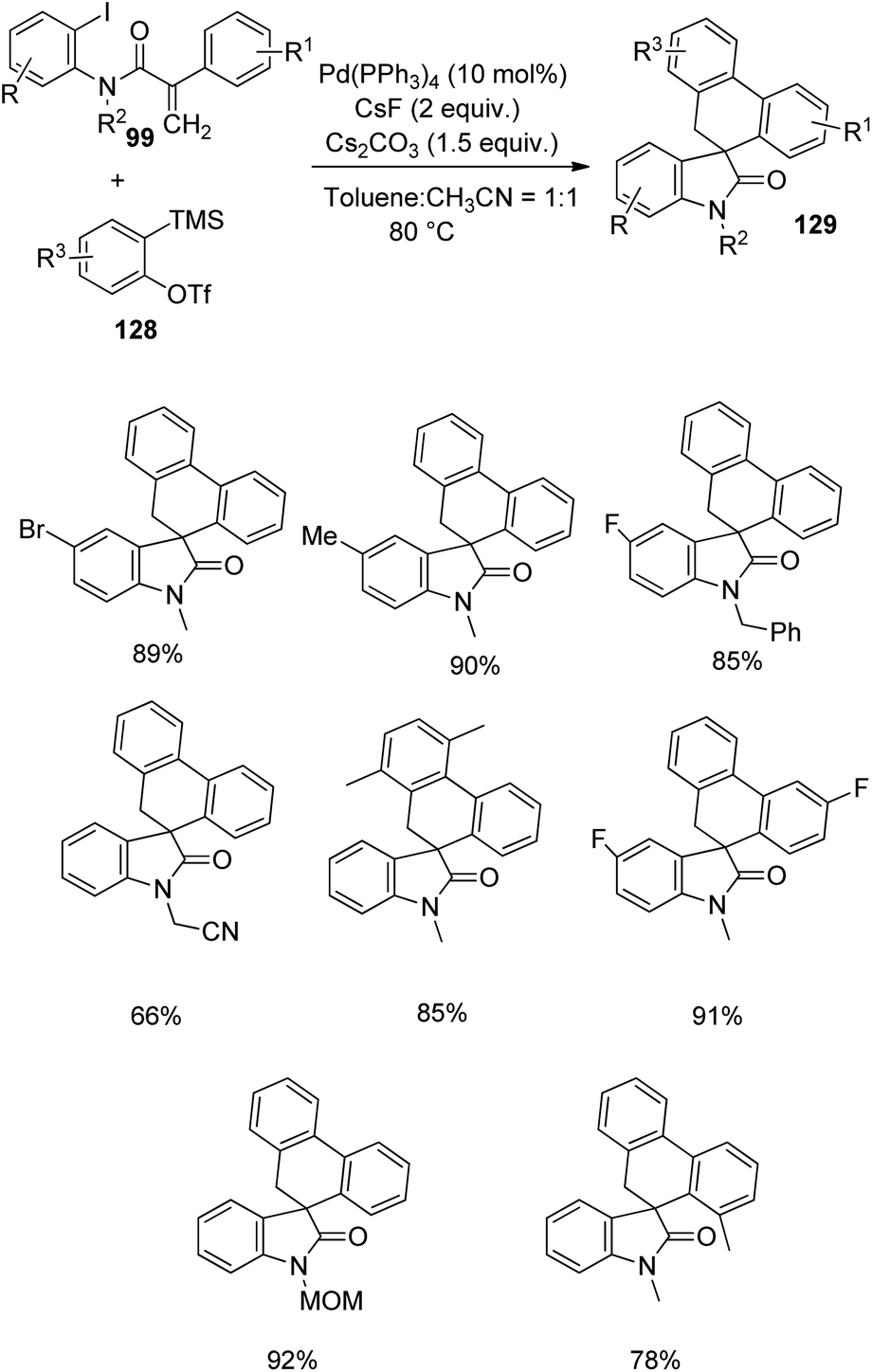 | ||
| Scheme 65 Spirooxindole synthesis via Heck spirocyclization catalyzed by palladium. | ||
In the next year, the same author reported the synthesis of spirooxindoles 131 through C–H activation and alkyne insertion, with palladium catalyst.87 The starting materials employed were acrylamides-bromo derivative of 99 and internal alkynes 130 and the optimized reaction conditions were 10 mol% Pd(PPh3)4 and 1.5 equiv. caesium carbonate in toluene as solvent at 100 or 120 °C which afforded regioselectivity >20:1. First ethyl phenylpropiolate-derivative of 130, was reacted with differently substituted acrylamides 99 (Scheme 66). Electron-releasing substituents on aromatic ring next to nitrogen atom and on the other aromatic ring gave 61–88% of the desired spirooxindole products. Fluorinated acrylamides and those with N-Bn and N-MOM protection also underwent the reaction. Acrylamide bearing a tethered thiophene and that with a pyridinylbromide substituent were also tolerated. The corresponding product was not provided by the substrate with electron-poor groups on the hanging aromatic ring.
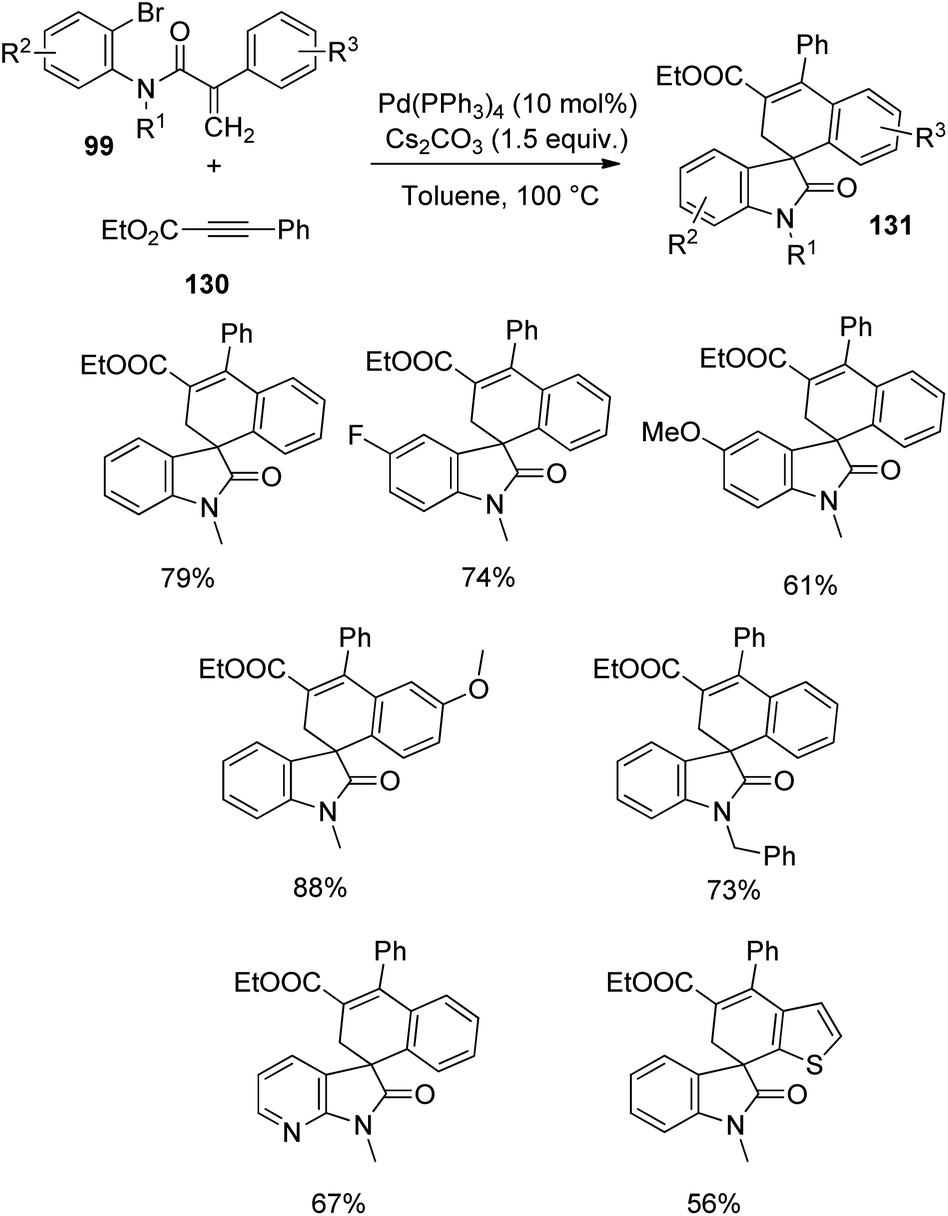 | ||
| Scheme 66 Spirocyclization with ethyl phenylpropiolate and different acrylamides catalyzed by palladium. | ||
Next the reaction was performed with different internal alkynes 130 under the same conditions (Scheme 67). Indole derived and ethyl 2-butynoate inserted alkynes underwent the reaction. Alkyne with a non enolizable ketone in place of the ester group afforded 79% spirooxindole. Weinreb amide, diaryl alkyne and alkyne containing 4-trifluromethyl-2-pyridine required higher temperature (120 °C). Regioselectivity was inferior (9:1) with phenylpropionitrile.
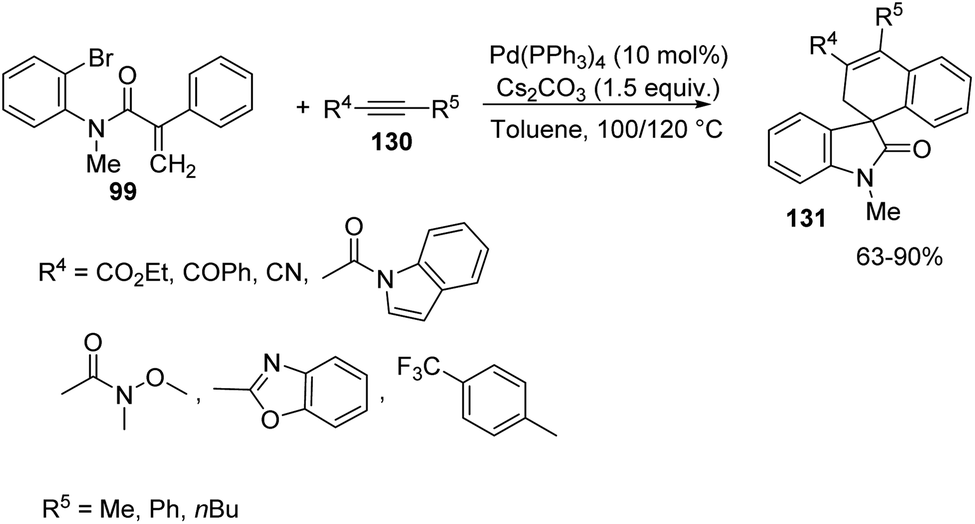 | ||
| Scheme 67 Spirocyclization with acrylamide and different internal alkynes catalyzed by Pd. | ||
Zhang and co-workers described the growth of spirooxindoles 134, 135 via functionalization of remote C–H, utilizing a Pd catalyst.88 They put forward a path in which dibromomethane 133 reacted with the palladacycle achieved through C–H activation of the acrylamide substrate. The substrate scope was then investigated by reacting acrylamides at optimized conditions using 20 mol% P(o-tol)3, 10 mol% Pd(OAc)2, 4 equiv. potassium carbonate, 2 equiv. dibromomethane, 2 equiv. triethylamine and 0.5 equiv. tetrabutylammonium iodide in DMA at 90 °C under N2 atmosphere. Ph, OMe, Me or halo substitutions on the aryl ring attached to double bond were tolerated. The yield with Br was less compared to Cl and F substitutions.
Acrylamides 132 having aryl iodides were reacted under the optimized conditions (Scheme 68). Phenyl ring containing halo, other electron-deficient as well as electron-rich groups underwent the reaction. Excellent yields were provided by 2-ethoxy-2-oxoethyl, benzyl, n-butyl or ethyl groups on nitrogen atom. N-Substituted acrylamides and the one bearing an ester linkage gave no yield but 35% product was obtained from the ether analogue. Acrylamide bearing aryl bromides-bromo derivative of 99, were examined under same conditions (Scheme 69). Due to steric hindrance, the yield was only 60% with ortho-methyl substituent. Trifluoromethyl and di-fluoro substituted acrylamides were tolerated in the reaction. Moderate yields were offered by substrates with N-cyanomethyl and N-2-methylallyl substituents.
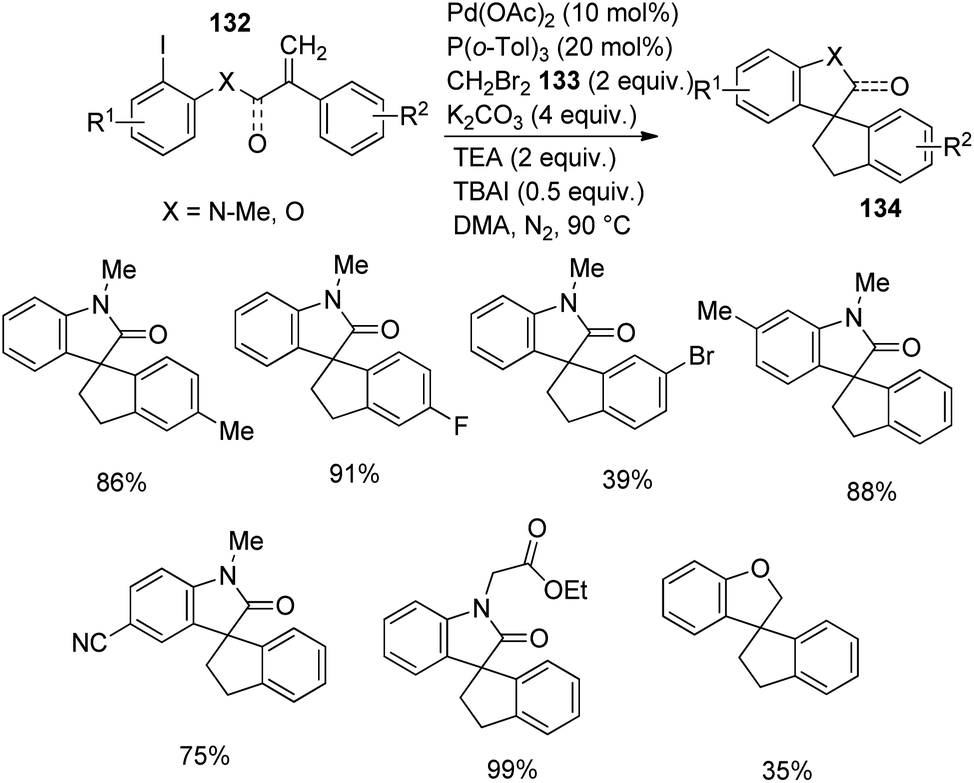 | ||
| Scheme 68 Spirooxindole synthesis through remote C–H activation of acrylamide containing aryl iodides. | ||
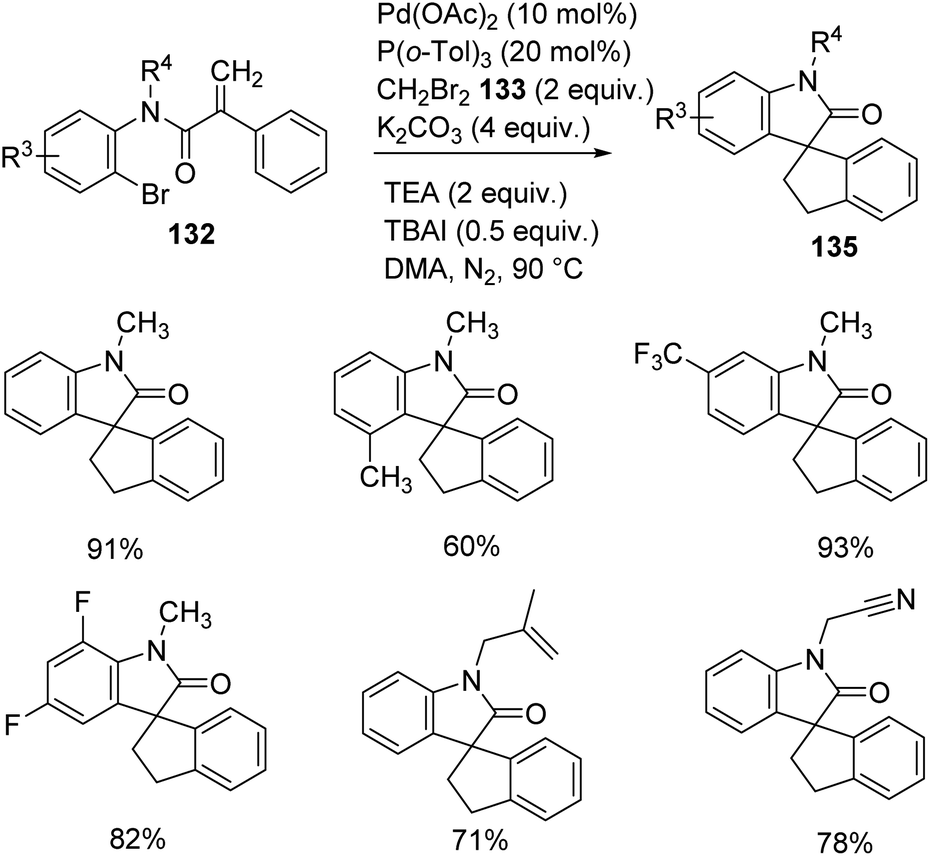 | ||
| Scheme 69 Spirooxindole synthesis through remote C–H activation of acrylamides containing aryl bromides. | ||
The reactions involving acrylamide derivatives were catalyzed by Pd(PPh3)4 and Pd(OAc)2, in which the reactions involving Pd(PPh3)4 were easily scalable.
8.8 Reactions involving other substrates
A comparison of the efficiency of palladium complexes with phosphinamine and (R)-BINAP as ligands in the intramolecular Heck cyclization of aryl triflates 136 to form spirooxindole derivatives 137 was put forth by Kiely and Guiry.89 The group screened different ligands including (R)-BINAP, tBu-substituted and iPr-substituted ferroceneoxazoline, tBu-derived and iPr-derived diphenylphosphinoaryloxazoline. The product yield, regioselectivity and enantioselectivity were investigated by employing solvents including benzene, toluene, dimethylacetamide and bases including proton sponge (PS) and 1,2,2,6,6-pentamethylpiperidine (PMP). The best enantioselectivity was gained from complex of palladium with tBu-derived ferrocenyloxazoline ligand (L7) in DMA as the solvent and proton sponge as base (Scheme 70). The regioselectivity was also better with phosphinamine ligands. The reactivity was higher and the reaction time was lower when BINAP was used as the ligand with pentamethylpiperidine as base. Solvents DMA and toluene gave isomeric ratios 25:75 and 75:25 respectively (Scheme 71).
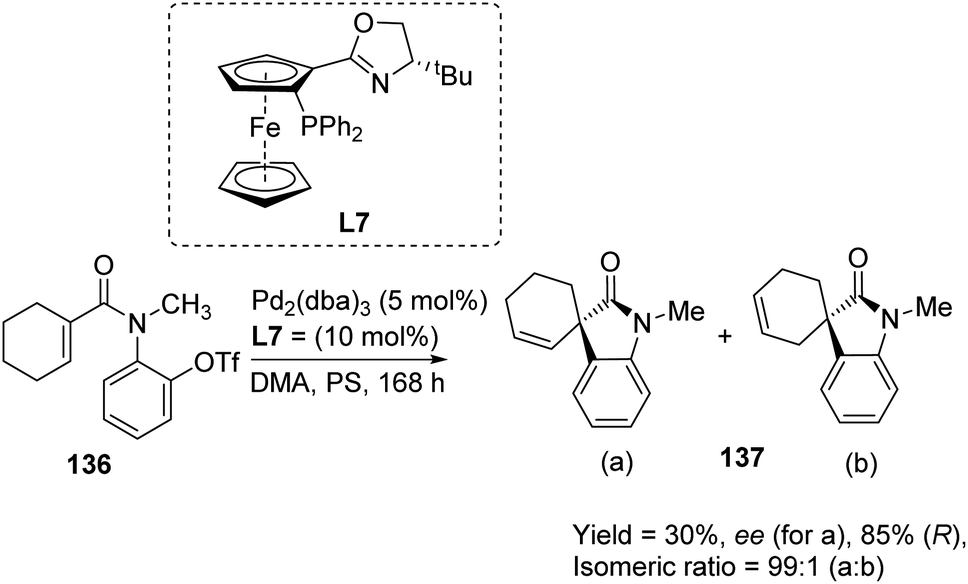 | ||
| Scheme 70 Heck cyclization of aryl triflates using palladium complex with tBu-derived ferrocenyloxazoline as the ligand. | ||
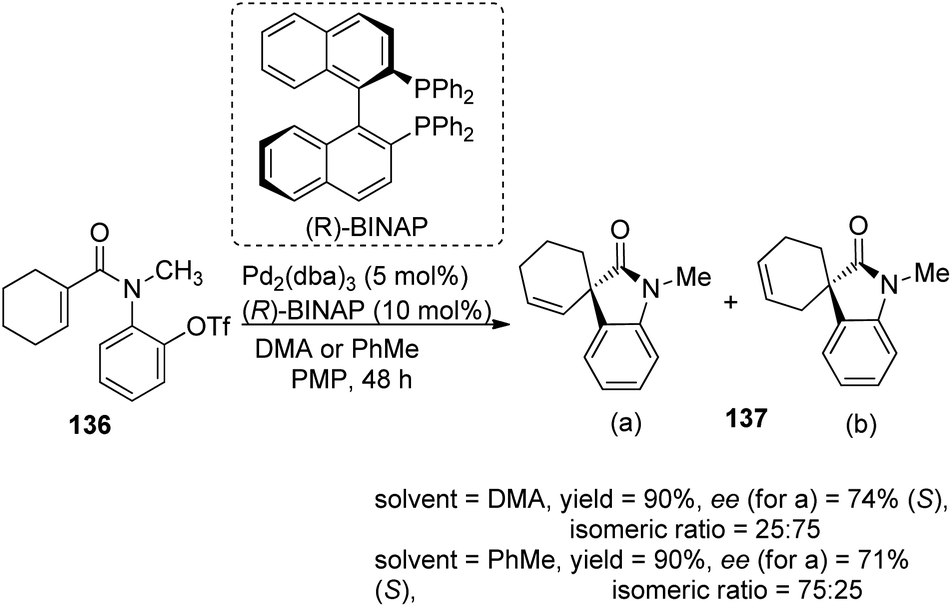 | ||
| Scheme 71 Heck cyclization of aryl triflates using Pd complex with (R)-BINAP as the ligand. | ||
Zhu et al. implemented a protocol for the development of spiropyrrolidinyl oxindoles 139 catalyzed by palladium from derivatives of anilides 138 via carbo-heterofunctionalization.90 The reaction was optimized with 0.1 equiv. of PdCl2 and 2 equiv. of PhI(OAc)2 as the oxidant in CH3CN at 80 °C (Scheme 72). Differently substituted anilide derivatives 138 underwent the reaction and afforded spirooxindoles 139 with 37–58% yield.
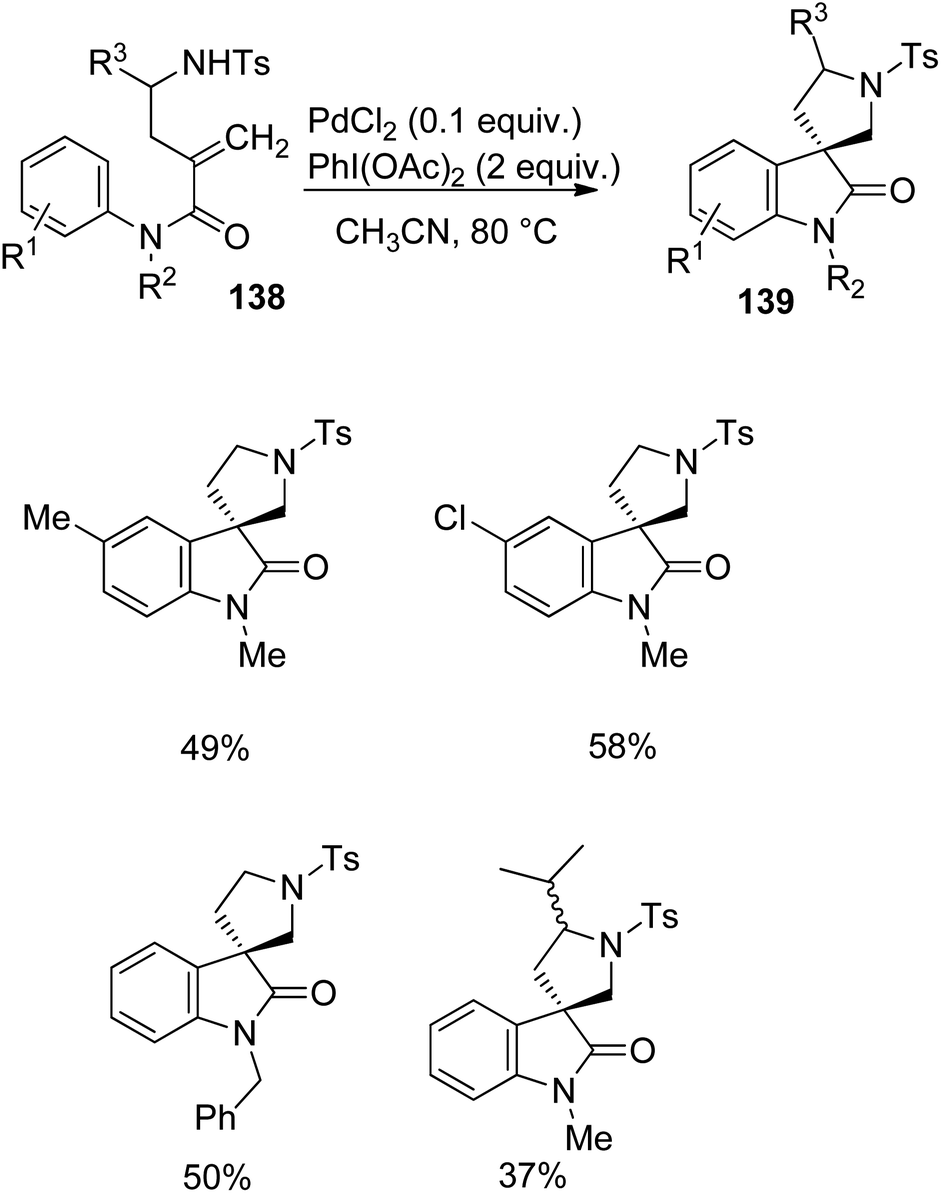 | ||
| Scheme 72 Spirooxindole synthesis through alkene carbo-heterofunctionalization. | ||
A plan for the synthesis of spirooxindoles 141 by making use of a sigma-alkyl palladium(II) complex, starting from an acrylamide derivative 140 was provided by Zhu et al.91 The preparation involves a carbopalladation followed by a C(sp3)–C(sp3) bond formation, via C(sp3)–H bond activation. The reaction was optimized with 0.1 equiv. of Pd(OAc)2, 0.2 equiv. of 1,3-bis(diphenylphosphino)propane as the ligand and 1.3 equiv. of K2CO3 in mesitylene at 140 °C (Scheme 73). In some cases, notable quantity of quinolinone 142 was also formed.
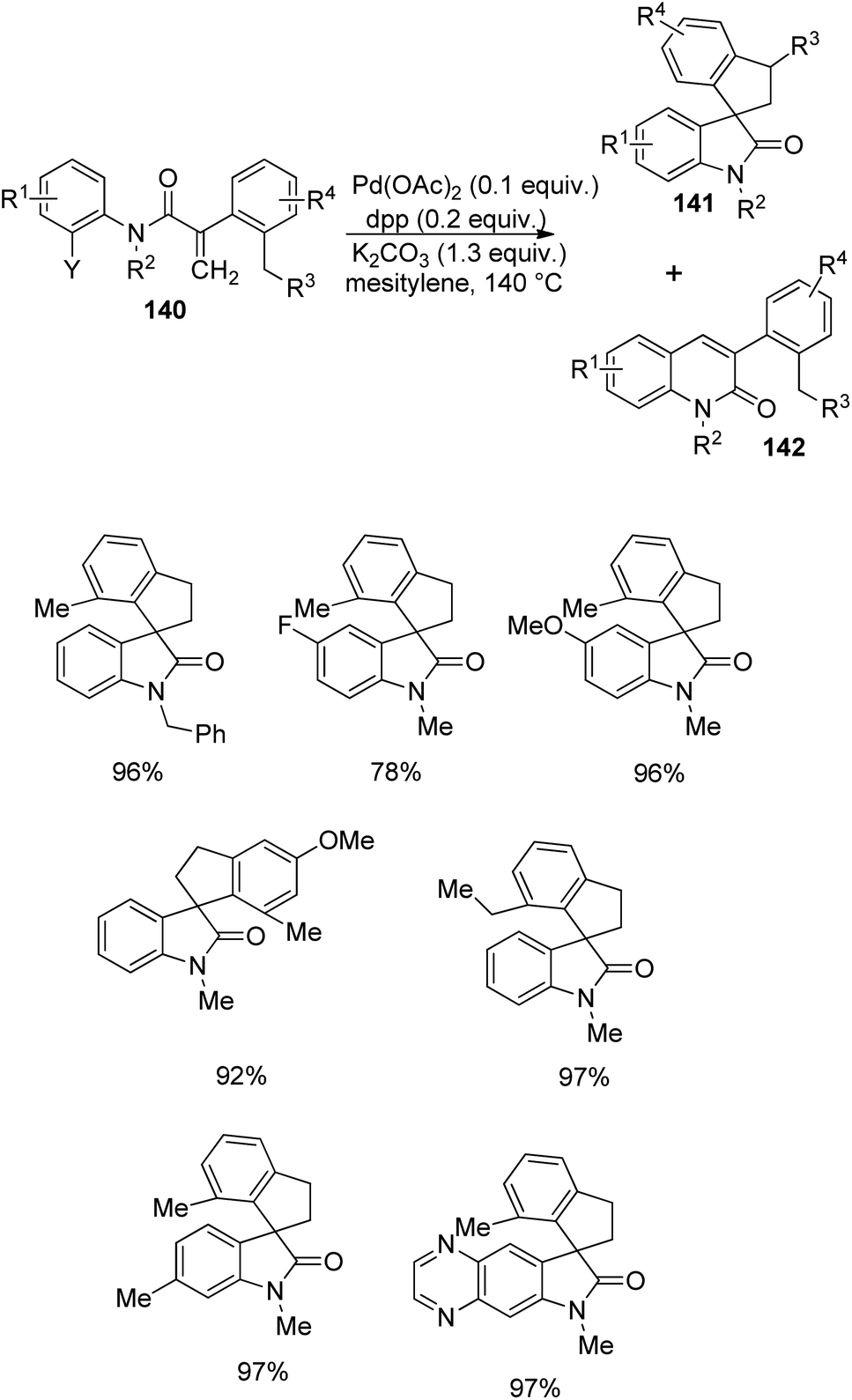 | ||
| Scheme 73 Substrate scope for the carbopalladation/C(sp3)–C(sp3) bond formation. | ||
Substrate bearing chloranilide was unable to perform the reaction, but that with iodoanilide and bromoanilide provided excellent yields. Secondary amide was also unsuccessful in the reaction. Substrate with various substituents on N underwent the reaction. Substrates having substituents o- to the halide gave quinolinone as the major product but 78–97% yields were obtained from m- and p-substituents. Substrates containing heterocycles were also tolerated in this reaction. In the case of acrylate α-aryl substituents excellent yields were contributed by OMe, Me and tBu groups whereas 2-(o-tolyl)acrylamide was fruitless in this reaction. Two diastereoisomers in 2:1 ratio was given by anilide with 2,5-diethyl substituted phenyl ring. The activation of methyl C(sp3)–H bond was lower compared to that of naphthyl C(sp2)–H bond when naphthalene was placed instead of benzene.
An approach for the synthesis of spirooxindoles 144 by using a Pd catalyst was developed by Kim et al.92 The substrates used were 3-(γ,δ-disubstituted)allylidene-2-oxindoles(a) 143 which undergoes an oxidative Heck (Fujiwara–Moritani) arylation and functionalisation of the C–H bond of aryl/arylation reaction. The products 144 were formed by utilizing 4 equiv. of AgOAc, 5 mol% of Pd(OAc)2 and 6 mol% of PivOH with benzene under reflux (Scheme 74). Both the isomers of the substrate 143, 143-ZE and 143-EE, gave similar results due to isomerisation of Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cβ double bond.
Cβ double bond.
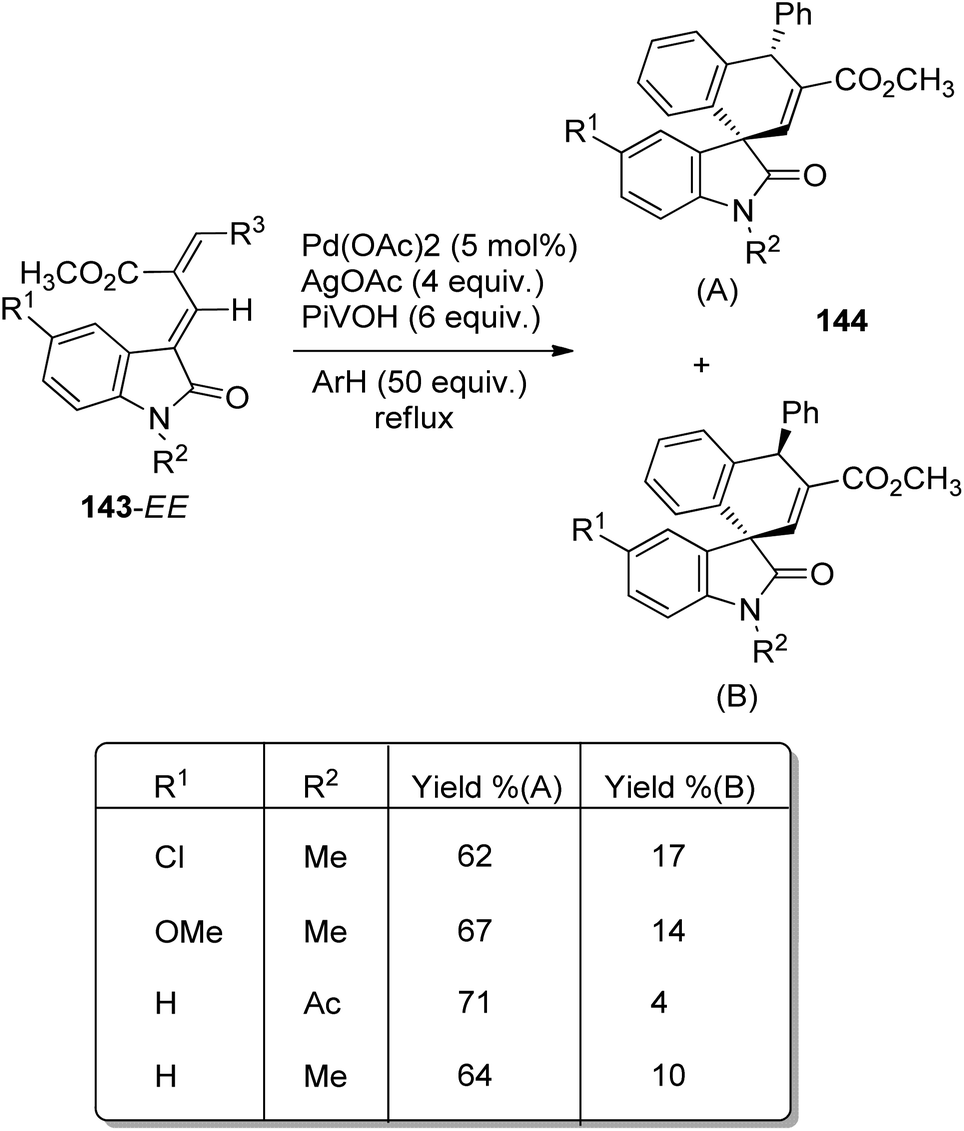 | ||
| Scheme 74 Substrate scope for spirooxindole synthesis from 3-(γ,δ-disubstituted)alkylidene-2-oxindoles. | ||
The substrate scope investigations were performed by employing 143-EE isomer. The results were alike in the case of substrates with electron-withdrawing or electron-releasing group on aromatic ring. Substrates with Ac and Me groups on nitrogen were tolerated but that without any protection was unable to perform the reaction. The major product was the one formed via naphthalene C–H bond activation in the case of 143-EE isomer with 1-naphthyl substituent. When the δ-position of the substrate carried 2,4-dichlorophenyl or 3,4-dichlorophenyl moiety, phenyl C–H activation occurred preferentially.
Jia et al. established a protocol for the formation of spirooxindoles 146 from indoles with C2-substitution 145, via reductive-Heck reaction with Pd catalyst.93 Optimization was carried out using 5 mol% Pd(OAc)2, 3 equiv. HCO2Na and 10 mol% PCy3·HBF4 in methanol at 100 °C (Scheme 75). Indoles with various substituents reacted easily without significant steric and electronic effects. 71–99% yield was accomplished with substituents at ortho-, meta- and para- to amino group of the 2-bromoaniline component. 90% of 7-azaspirooxindole was synthesised from indole having pyridine moiety. Product corresponding to β-H elimination was major for indole substrate with methyl at the third position. The spirooxindole product was not obtained with C2-substituted benzothiophene as the substrate but C2-substituted benzofuran 147 gave 66% yield for the corresponding product 148 (Scheme 76).
 | ||
| Scheme 75 Substrate scope for synthesis of spirooxindoles from indoles with C2-substitution. | ||
 | ||
| Scheme 76 Extension of reductive Heck reaction to benzofuran derived substrate. | ||
A procedure for the development of spirooxindoles 151 by employing a palladium catalyst was introduced by Yang et al. which involves a ring opening [3 + 2]-annualation.94 The reaction was implemented using α,β-unsaturated nitroalkenes 149 and spirovinylcyclopropyl oxindole 150 in presence of 10 mmol% Xantphos and 5 mmol% Pd(OAc)2 in toluene at rt for 12 h (Scheme 77). Both N-protected and N-unprotected spirovinylcyclopropyl oxindoles underwent the reaction but the dr was lower with the unprotected ones. C5-, C6- or C7-substituted spirocyclopropane were tolerated. Efficient dr and good yield were obtained when C7-position of the substrate was having a Cl group. The yield dropped with nitroalkenes having ortho-substitution and the diastereoselectivity dropped with strong electron-releasing groups at meta- or para-position. (1E)-2-Phenylethenyl nitroalkene gave dr value of only 70:30.
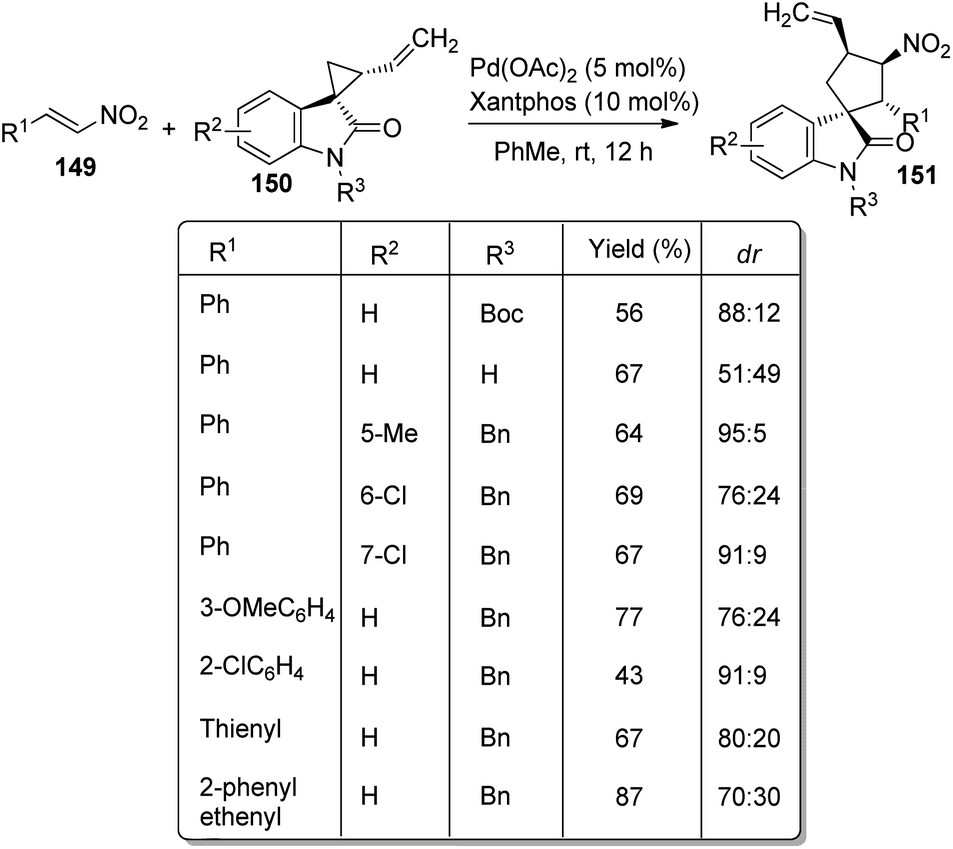 | ||
| Scheme 77 Spirooxindole synthesis via ring-opening [3 + 2]-annulation catalyzed by Pd. | ||
Liao and co-workers studied arylboronic acid 153 addition to nitriles 152, catalyzed by palladium, for the construction of spirooxindolyl oxazole-2(5H)-ones95 154. The optimized reaction conditions were 5 mol% palladium(II)acetate, 6 mol% 2,2′-bipyridine and 5 equiv. acetic acid in NMP as the solvent at 80 °C (Scheme 78). The substrate scope exploration was carried out by taking different nitriles and boronic acids. O-Ethoxycarbonyl cyanohydrins derived from isatins were used as the nitriles and high yields were obtained from those with different groups on the nitrogen and 80–95% yields of the products were contributed by substrates with different groups on Ph ring. In the case of arylboronic acids those having electron-releasing and electron-deficient groups, excluding nitro, underwent the reaction. 9-Phenanthrene, β-naphthyl and α-naphthyl boronic acids were tolerated, but alkyl and hetero-aromatic ones were not tolerated. The group extended the reaction for the creation of other spirocycles also.
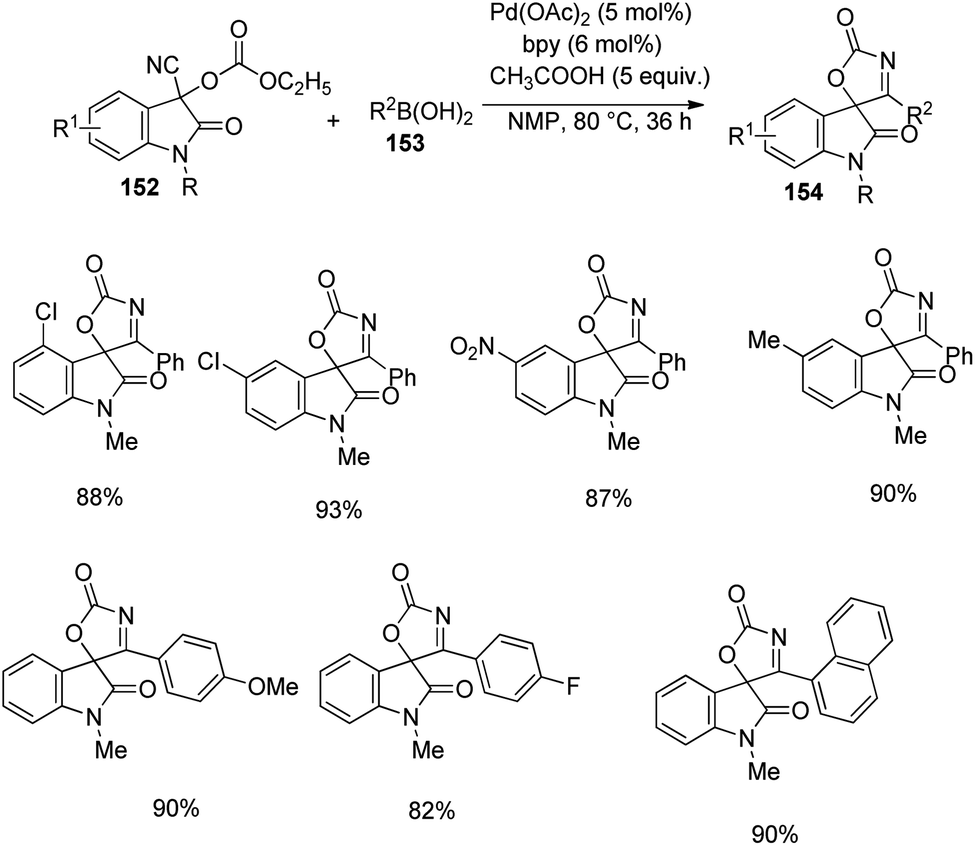 | ||
| Scheme 78 Synthesis spirooxindolyl oxazol-2(5H)-one catalyzed by Pd. | ||
Other substrates employed for Pd-catalyzed spirooxindole synthesis include derivatives of aryl triflates, anilides, indoles, nitroalkenes etc. and catalysts such as Pd2(dba)3, PdCl2, Pd(OAc)2 and so on. The respective yields of up to 58%, 98% and 87% were furnished by anilides, indoles and nitroalkenes.
9 Cu-catalyzed spirooxindole synthesis
Copper is highly sustainable, cheap and innocuous. Copper has high significance in the biological world. It is widely accepted by the synthetic community also. Through a two- and one-electron mechanism, copper can catalyze reactions and the coordination of Cu to π bonds and heteroatoms can be achieved easily.96Various investigations on the Cu-catalyzed spirooxindole synthesis was reported from 2014 to 2020. These were majorly carried out using copper triflate, copper sulphate and copper ferrite nanoparticles compared to other copper catalysts. The nuleophilicity of alkenes can be greatly increased by copper triflate which is a soft Lewis acid. The advantage of nano copper ferrite catalyst is its easy recoverability and recyclability.
9.1 Reactions involving isatin derivatives
A 1,3-dipolar cycloaddition catalyzed by copper for the synthesis of spirooxindolopyrrolizidines 157 was reported in 2014.97 The reaction between 3-(2-alkenoyl)-1,3-oxazolidin-2-ones 155, (S)-proline 156 and isatin 6 was effected by Cu(OTf)2 as the source of copper, cyclohexane-1,2-bis(arylmethyleneamine) carrying chloro-substituent as the ligand (L11) [Cu(OTf)2:L11 = 1:1.1] in a mixture of EtOH and DCM as the solvent at room temperature (Scheme 79). The desired spirooxindole product was obtained by the [3 + 2] cycloaddition between the dipolarophile and azomethine ylide formed in situ from (S)-proline and isatin. They could attain the yield of 88–99% and ee of 87–95%.
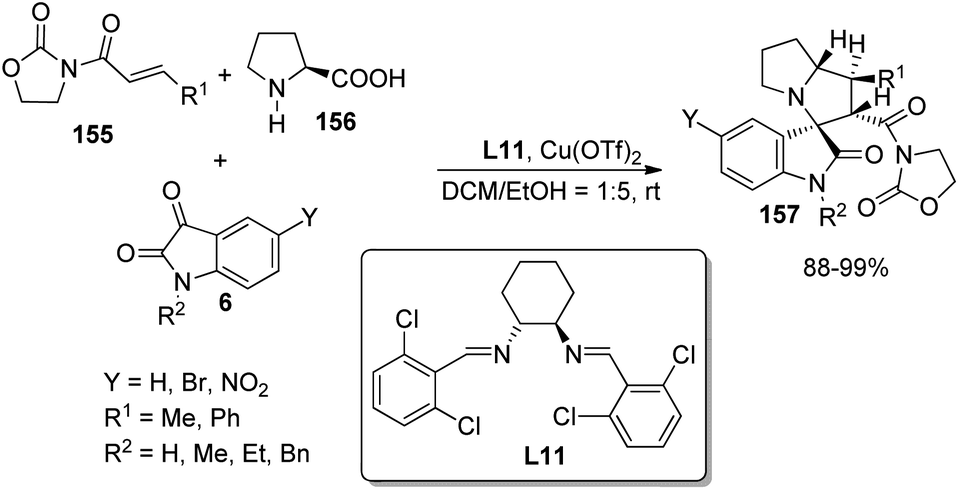 | ||
| Scheme 79 Synthesis of spirooxindolopyrrolizidine through 1,3-dipolar cycloaddition catalyzed by Cu(OTf)2 and cyclohexane-1,2-bis(arylmethyleneamine) ligand carrying chloro substitution. | ||
In the next year, the synthesis of spirooxindolopyrrolizidines using the same method by changing the ligand and solvent was reported.98 Here they used cyclohexane-1,2-bis(arylmethyleneamine) having tBu substituent as the ligand (L12) and ethanol as the solvent which afforded 85–98% yield and 82–92% ee (Scheme 80).
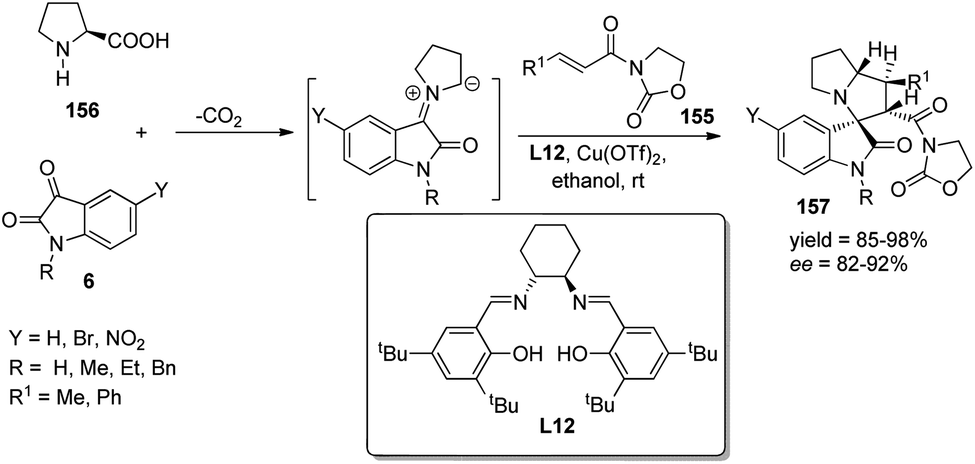 | ||
| Scheme 80 Spirooxindolopyrrolizidine synthesis through 1,3-dipolar cycloaddition catalyzed by Cu(OTf)2 salen complex. | ||
A green method for the production of indan-1,3-dione grafted spirooxindolopyrrolizidines linked 1,2,3-triazoles 161 with Cu(II) catalyst was narrated by Khurana et al. in 2015.99 Here, azides 158, N-propargylated isatins-derivative of 6, sarcosine 159, aldehydes 69 and indan-1,3-diones 160 undergo a one-pot reaction which was proceeded with 10 mol% of aq. CuSO4·5H2O and 20 mol% of aq. sodium ascorbate in PEG-400 at 80 °C (Scheme 81). Here sodium ascorbate will convert Cu(II) into Cu(I) and the reaction proceeds through Knoevenagel condensation/two consecutive 1,3-dipolar cycloaddition pathway. The scope of the reaction was investigated by using different azides and aldehydes.
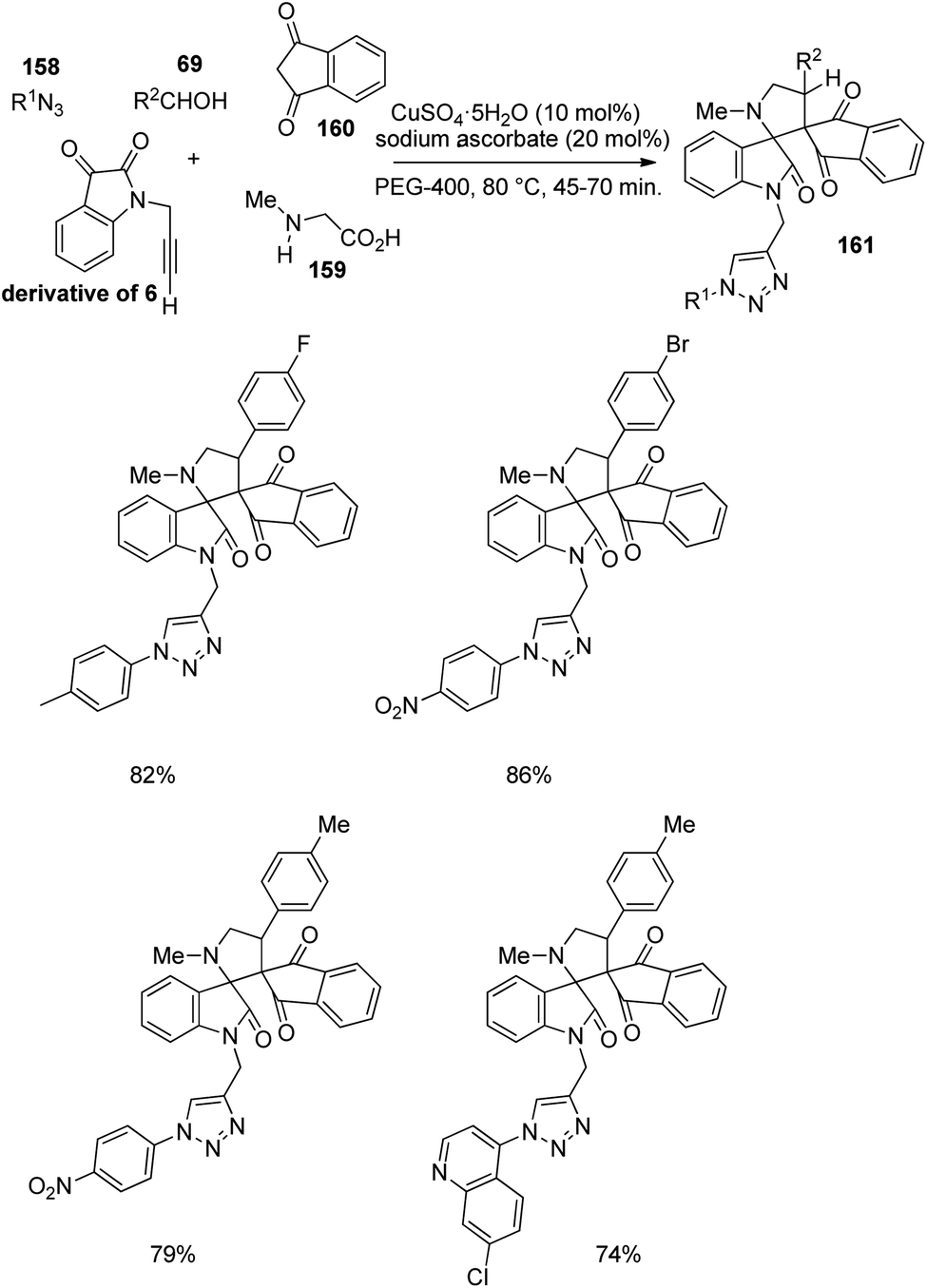 | ||
| Scheme 81 Substrate scope for Cu(I)-catalyzed indan-1,3-dione grafted spirooxindolopyrrolizidine linked 1,2,3-triazoles synthesis. | ||
In 2016, they described a four-component reaction of aryl azides-derivative of 158, sarcosine 159 or L-proline 156, N-propargylated isatin-derivative of 6 and coumarin-3-carboxylic acid derivatives 162 through copper-catalyzed [3 + 2] cycloaddition for the building up of spirooxindole pyrrolizidine linked 1,2,3-triazoles 163.100 The reaction is found to have increased regio- and stereoselectivity when catalyzed by CuSO4·5H2O and sodium ascorbate in presence of acetic acid (glacial) at 60 °C (Scheme 82). Differently substituted aryl azides were utilized to investigate the generality of the procedure. Electron-withdrawing as well as electron-releasing groups afforded the spirooxindole products in 71–90% yield.
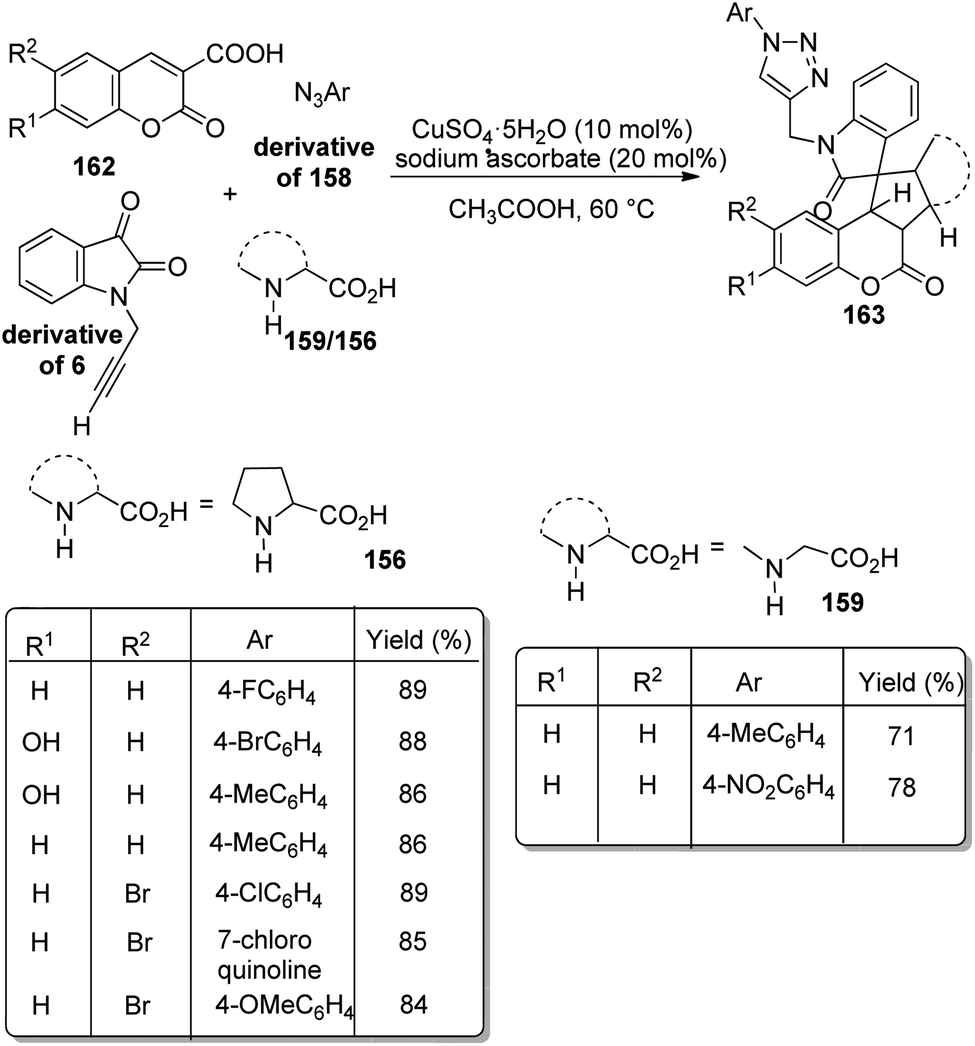 | ||
| Scheme 82 Spirooxindole pyrrolizine linked 1,2,3-triazole synthesis through copper-catalyzed four-component reaction. | ||
An eco-friendly magnetic copper ferrite NPs catalyzed one-pot reaction for the synthesis of spirooxindoles derivative of 63, 64, 164, 165 was reported.101 The three-component reaction of dicyanomethane 28, Michael-donors and isatin 6 involved 7 mol% of CuFe2O4 NPs as the catalyst and water–ethanol mixture as solvent under reflux temperature (Scheme 83). 80–92% yields were obtained by using 4-hydroxycoumarin 50, 1,3-cyclohexanedione-derivative of 29, barbituric acid-derivative of 30 and thiobarbituric acid-derivative of 30 as Michael-donors. Mechanistically the reaction follows a Knoevenagel condensation/Michael addition/enolization sequence. Simple separation and reusability of the catalyst are the advantages of this green protocol.
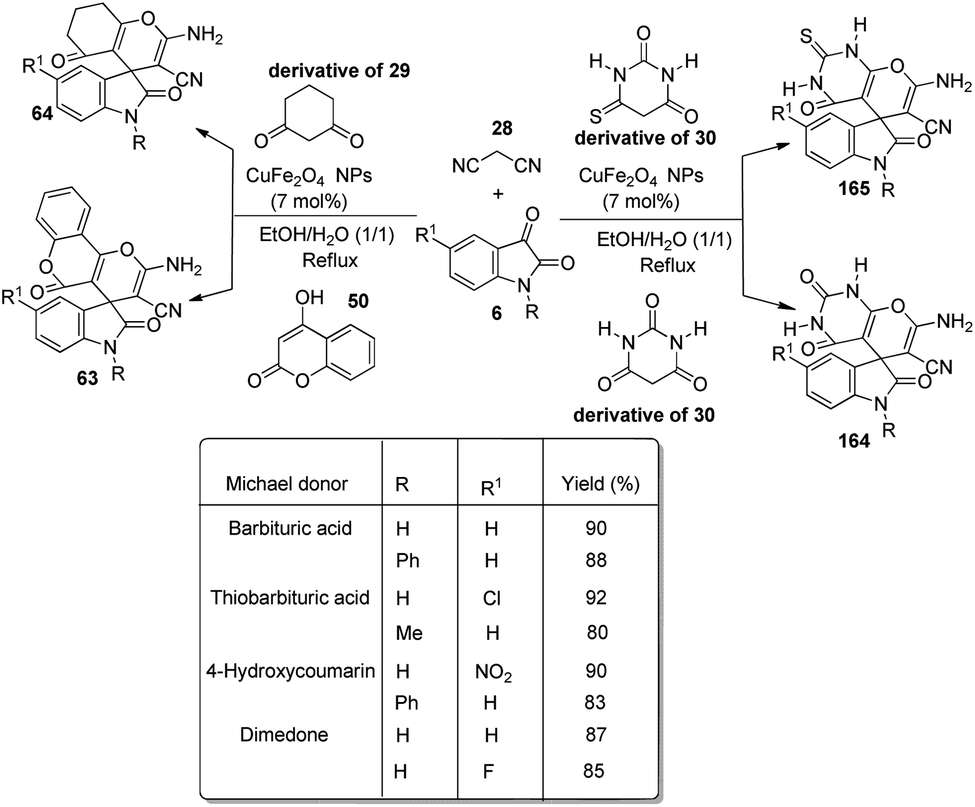 | ||
| Scheme 83 Synthesis of spirooxindoles using magnetic copper ferrite NPs as the catalyst. | ||
Mo and co-workers developed a method for the synthesis of spirooxindoles via Cu-catalyzed reaction between alkenyl boronic acids 166 and 3-(hydroxyimino)indolin-2-ones 167.102 In this method isatin oximes were N-vinylated selectively and the N-vinyl nitrones 168, 169 thus obtained were rearranged to the corresponding spirooxindoles 170, 171 by heating (Scheme 84). The reaction conditions of 10 mmol% Cu(OAc)2, 3 equiv. pyridine and 6 equiv. sodium sulphate in methanol at room temperature offered mono N-vinylated product 168. When the catalyst amount and the solvent were changed to 2 equiv. and 1,2-dichloroethane respectively, the corresponding double N-vinylated products 169 were obtained. These N-vinyl nitrones offered 32–80% of the spirooxindole products by heating in toluene at 120–140 °C. Decomposition happened to mono N-vinylated products when R1 = n-Pr, n-Bu, (CH2)4Cl, (CH2)3CO2Me, p-FC6H4, p-MeOC6H4 and Ph.
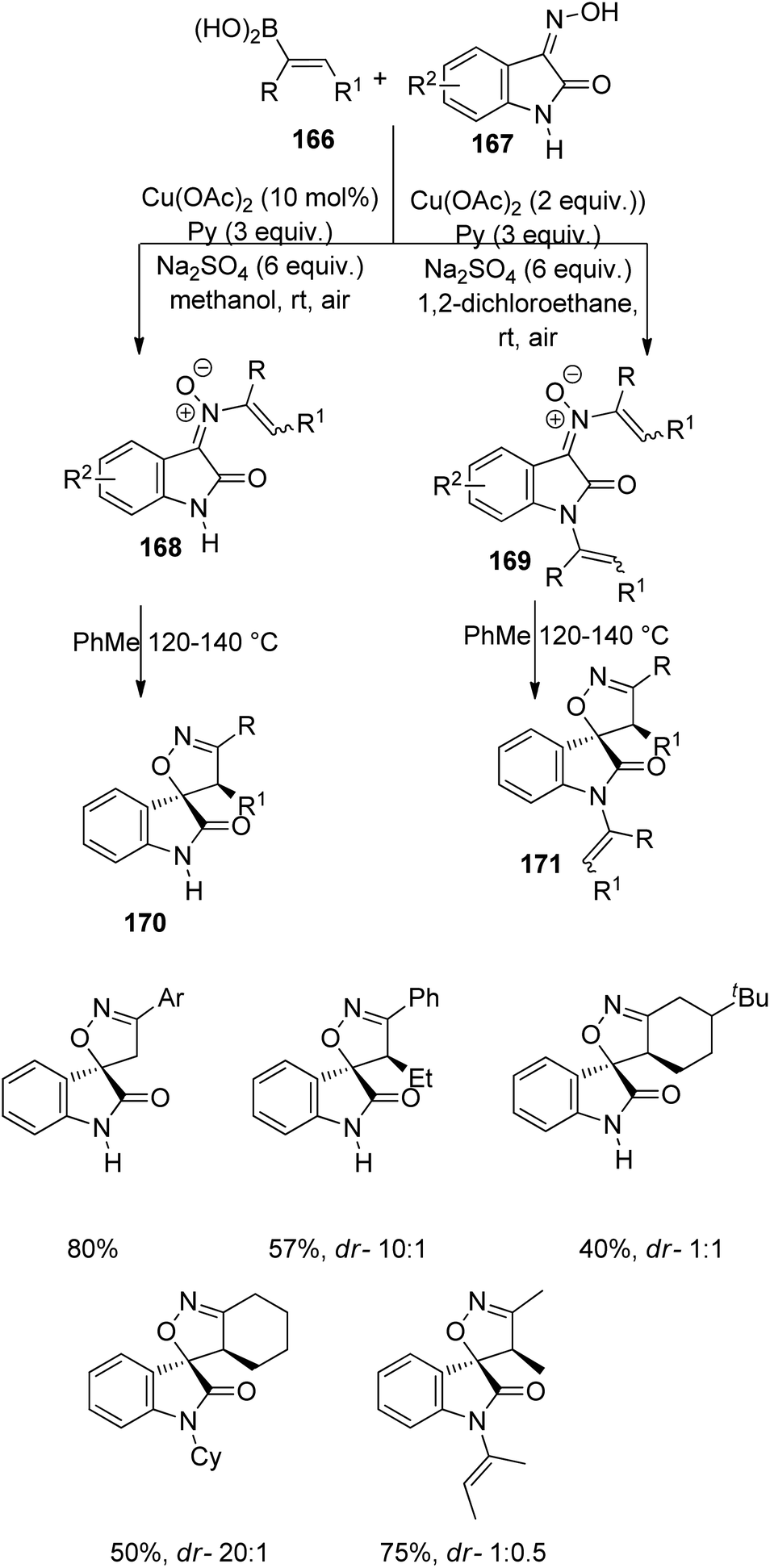 | ||
| Scheme 84 Copper-catalyzed N-vinyl nitrone synthesis and subsequent conversion to spirooxindoles. | ||
A powerful protocol for the construction of pyrrolo[1,2-a]indole spirooxindoles 174 using a Cu catalyst via Friedel–Crafts alkylation/cyclization path was demonstrated by Bu et al.103 The reaction between oxodienes derived from isatin 172 and 3-substituted indoles 173 was optimized in presence of catalyst Cu(OTf)2 (20 mol%) at 35 °C with acetonitrile solvent (Scheme 85). The scope of the reaction was examined by using isatin-derived oxodienes and different 3-alkyl/aryl indoles. The yield was lower with electron-deficient groups on the oxodiene aromatic ring. The substituent position also affected the yield but the nature of the substituents on N did not. Indoles with various aryl and alkyl substituents on the C3-position underwent the reaction and those with amino, hydroxyl, ester and carboxylic acid groups were well tolerated. Upto 99% spirooxindole product was obtained by executing the reaction in gram scale.
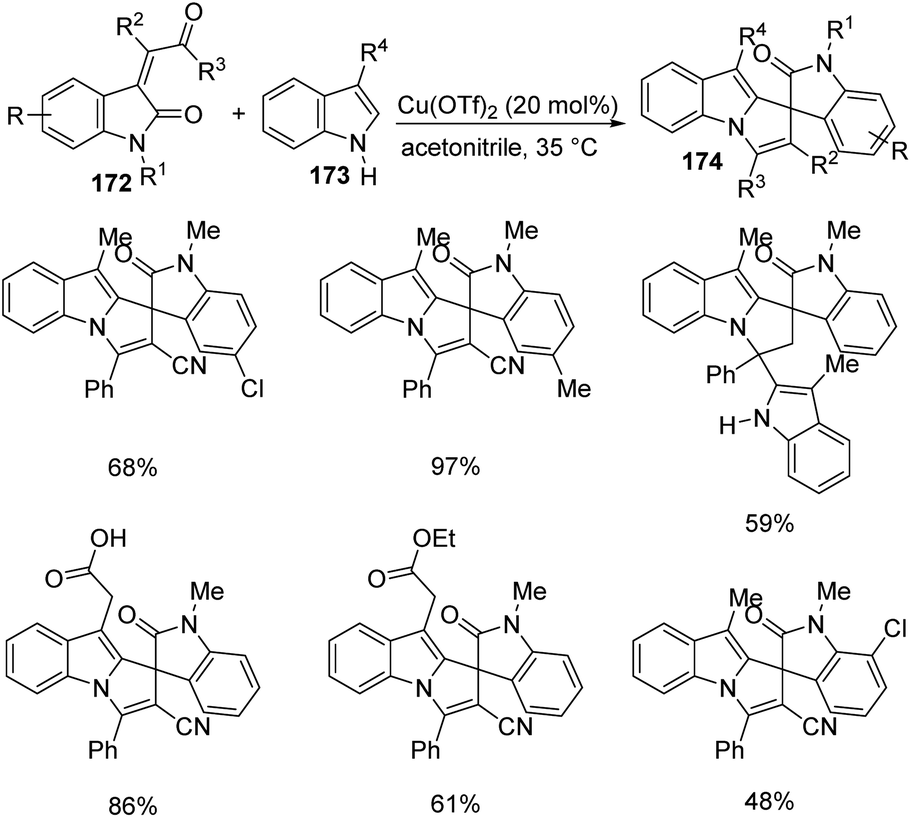 | ||
| Scheme 85 Synthesis of pyrrolo[1,2-a]indole spirooxindole employing Cu(OTf)2 as the catalyst. | ||
A possible mechanism for the reaction is interpreted in Scheme 86. Here, Cu(OTf)2 which is a Lewis acid, caused activation of oxodiene derived from N-methyl isatin 172. An intermediate 175 was formed through Friedel–Crafts alkylation in which 3-methyl indole-derivative of 173, attacked the activated oxodiene. Another intermediate 176 was obtained by the attack of electrophilic N of indole on the carbonyl adjacent to the aryl ring. The pyrrolo[1,2-a]indole spirooxindole 174 was acquired from this intermediate by the elimination of a water molecule. The catalyst was regenerated in the last step.
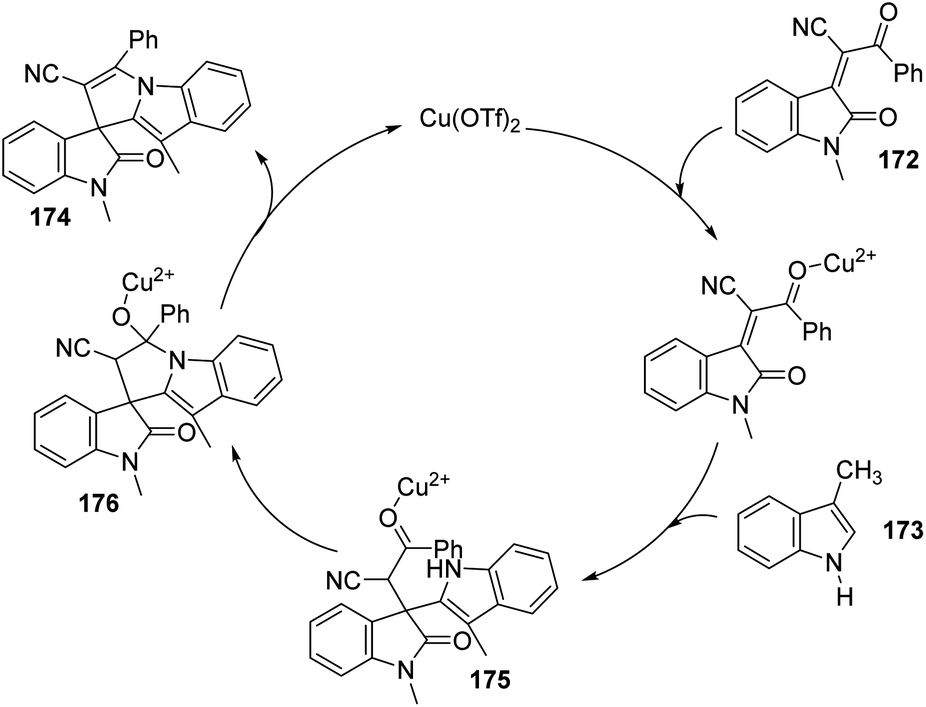 | ||
| Scheme 86 The possible mechanism for the reaction [reproduced with permission from ref. 103]. | ||
An efficient method for spirooxindole 178, 179 synthesis from derivatives of isatin 6, cyclic 1,3-diketone 177, 29 and activated methylene compound 35, using CuO NPs as catalyst was reported by Moradi and Ataei.104 The reaction was effected by CuO NPs (4 mol%) in ethanol solvent at rt (Scheme 87). Dicyanomethane gave higher yields than ethylcyanoacetate. 5-Bromoisatin gave the lowest yield compared to other isatin derivatives. In the case of various 1,3-diketones used, the yield was lowest with N,N-dimethyl barbituric acid.
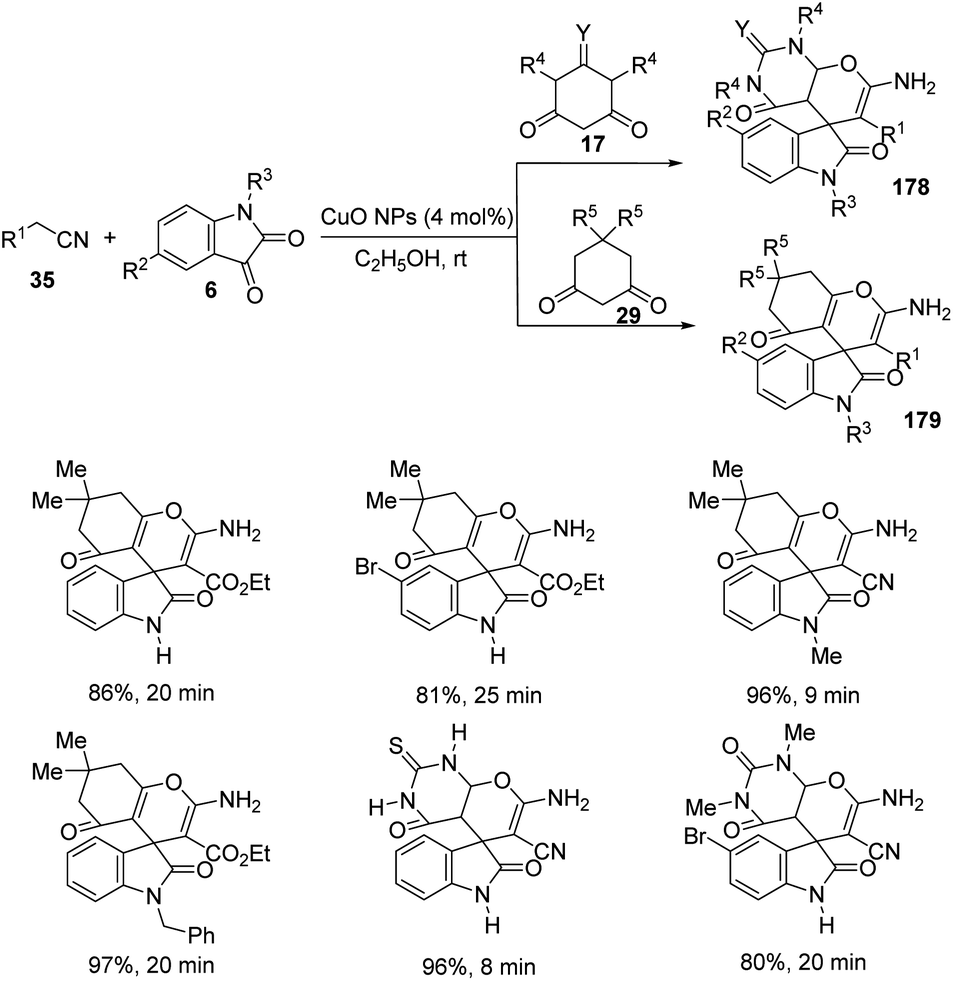 | ||
| Scheme 87 Substrate scope for CuO nanoparticle-catalyzed spirooxindole synthesis. | ||
An efficient and novel approach for the formation of derivatives of spirooxindole 183, 184 through environmentally benign copper triflate catalyzed multi-component reaction of 1,3-dicarbonyl compounds 182 or β-oxo-benzenepropanenitrile 181, 5-aminopyrazole 180 and isatin 6 has been put forward.105 The reaction in the presence of Cu(OTf)2 as the catalyst and ethanol as solvent ensued in different spirooxindole derivatives in 75–90% yield (Scheme 88).
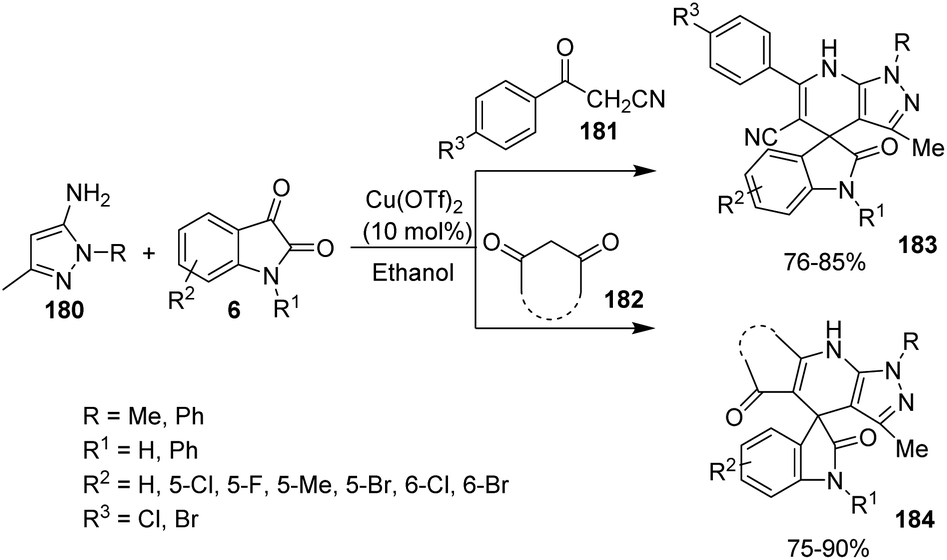 | ||
| Scheme 88 Synthesis of spirooxindoles through Cu(OTf)2-catalyzed three-component reaction. | ||
Khan and co-workers introduced a Cu/TEMPO catalyzed generation of spirooxindoles 187, 188 through a dehydrogenative cycloaddition.106 First alkylated ketones 185 were used as the substrates which undergo dehydrogenation in presence of 10 mol% of Cu(OAc)2 as the catalyst, 0.1 mmol of TEMPO and 0.1 mmol of 2,2′-bipyridyl as the additives in TBAA (tetrabutylammonium acetate) as the solvent at 80 °C, to give an alkene 186. After dehydrogenation isatin-derivative of 6 and sarcosine 159 or L-proline 156 were added which resulted in desired products 187, 188 through 1,3-dipolar cycloaddition reaction (Scheme 89). Both electron-deficient and electron-releasing groups in the substrate afforded 80–87% of the spirooxindole products. Then quinolinyl-alkylated ketones of type 1, 189 and 2, 191 were used as substrates under the same reaction conditions and the desired products 190, 192 were obtained in 65–84% yields (Schemes 90 and 91). Anti-diabetic and anti-oxidant properties were exhibited by almost all of the compounds produced.
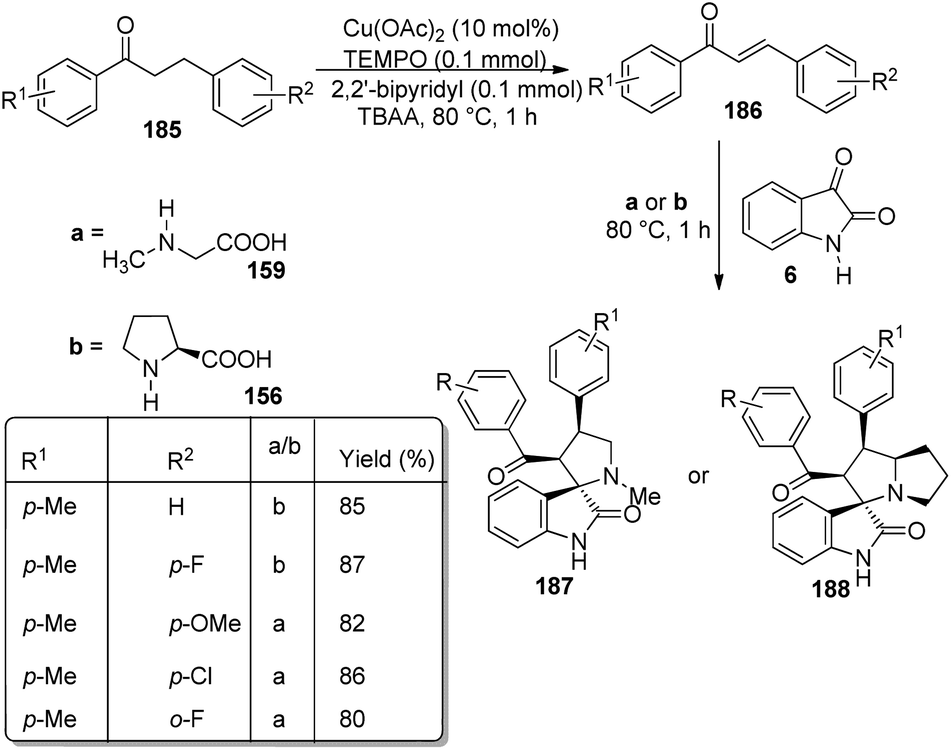 | ||
| Scheme 89 Spirooxindole synthesis through dehydrogenative cycloaddition of alkylated ketones using Cu/TEMPO catalyst. | ||
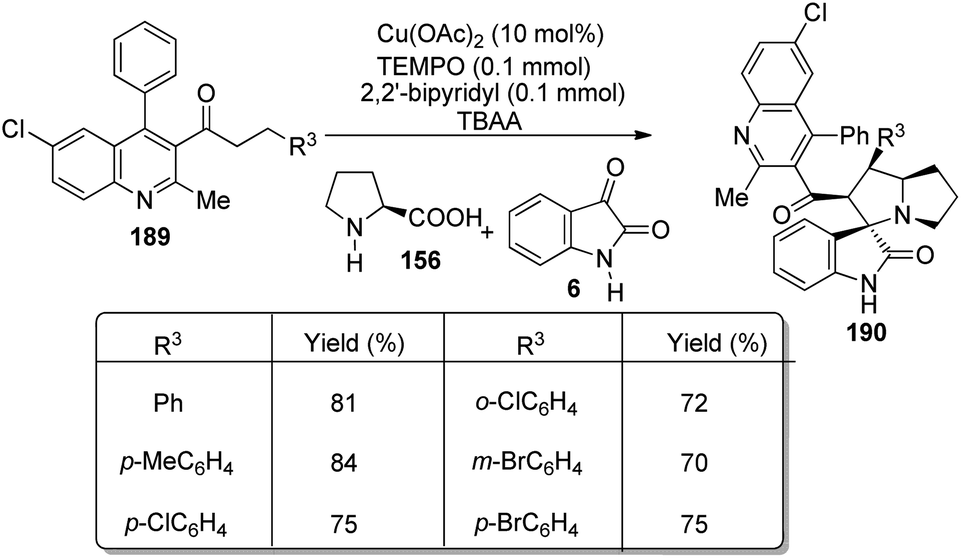 | ||
| Scheme 90 Spirooxindole synthesis through dehydrogenative cycloaddition of quinolinyl alkylated ketones of type 1 using Cu/TEMPO catalyst. | ||
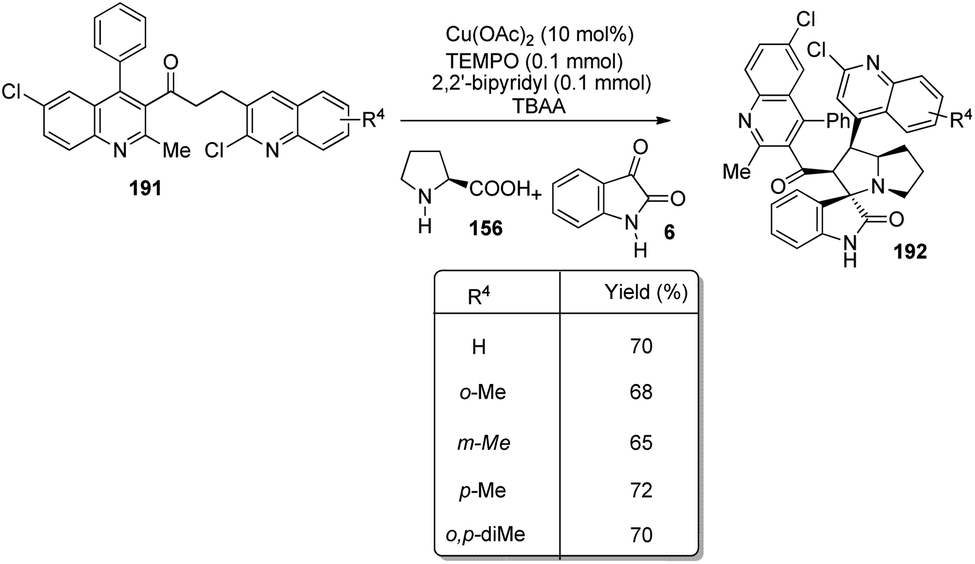 | ||
| Scheme 91 Spirooxindole synthesis through dehydrogenative cycloaddition of quinolinyl alkylated ketones of type 2 using Cu/TEMPO catalyst. | ||
Isatins were availed as the starting materials in all of the metal classes mentioned above. Majority of the approaches were eco-friendly and provided excellent enantio- and diastereoselectivities and yields.
9.2 Reaction involving benzoic acid derivatives
Zhang et al. established an atom-economical procedure which involves Cu-catalyzed oxidative annulation for the fabrication of 3-spirooxindole benzofuranones107 194. The reaction of derivatives of benzoic acid 193 under the optimized conditions of 20 mol% CuBr2, 2 equiv. sodium methoxide in DMF at 150 °C under O2 atmosphere afforded the required product 194 in 55–86% yield (Scheme 92). The yield was lower for substrates with strong electron-withdrawing substituents on aniline ring compared to strong electron-releasing ones. Various N-substituents like Me, Bn and iPr used to give the anticipated product in good yields. Substituted benzoic acid ring with electron-deficient and -rich groups was explored and was found fruitful under the reaction.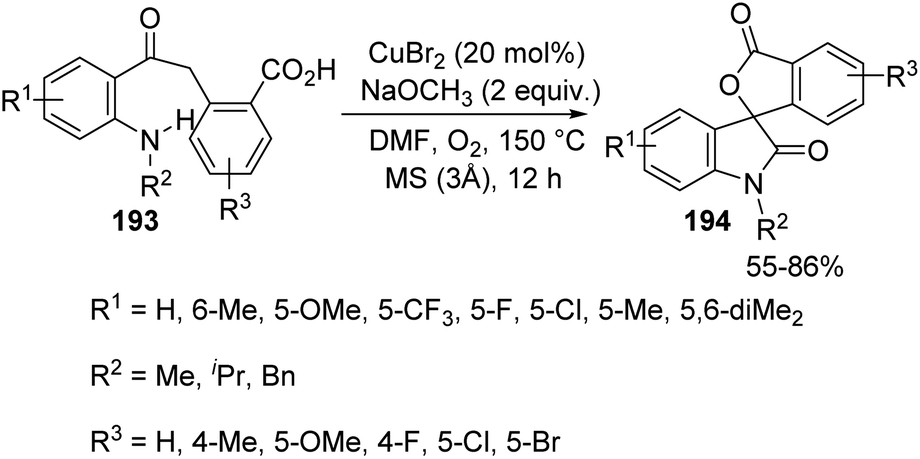 | ||
| Scheme 92 Synthesis of 3-spirooxindole benzofuranone via copper-catalyzed oxidative annulation. | ||
While considering copper-catalyzed spirooxindole synthesis, the yields given by benzoic acid derivatives were slightly less compared to other derivatives-isatins and methyleneindolinones.
9.3 Reaction involving methyleneindolinone derivatives
A method for the production of spirooxindoles 196 catalyzed by Cu(I)/Ganphos was established by Duan et al.108 Diverse spirooxindoles 196 were produced by the reaction between 3-methylene-2-oxindoles- derivatives of 72 which are alkyl substituted and glycine imino esters 195 under the conditions of 3 mol% Cu(MeCN)4BF4, 6 mol% Ganphos which is a chiral phosphine ligand, 20 mol% triethylamine and 4 equiv. ethanol in THF at −20 °C (Scheme 93). In the case of 3-methylene-2-oxindoles, the enantioselectivities were affected by the alkyl substituents, and the methyl substituents gave only 70% ee. The yield, diastereo- and enantioselectivities were good with substrates emanated from aliphatic ketones. In the case of imino esters, decreased enantioselectivities and yields were obtained from those bearing Ph ring with o-substitution, but the electronic nature of the substituents had no significant effect. 91% ee was given by methyleneindolinone with thienyl group as substituent.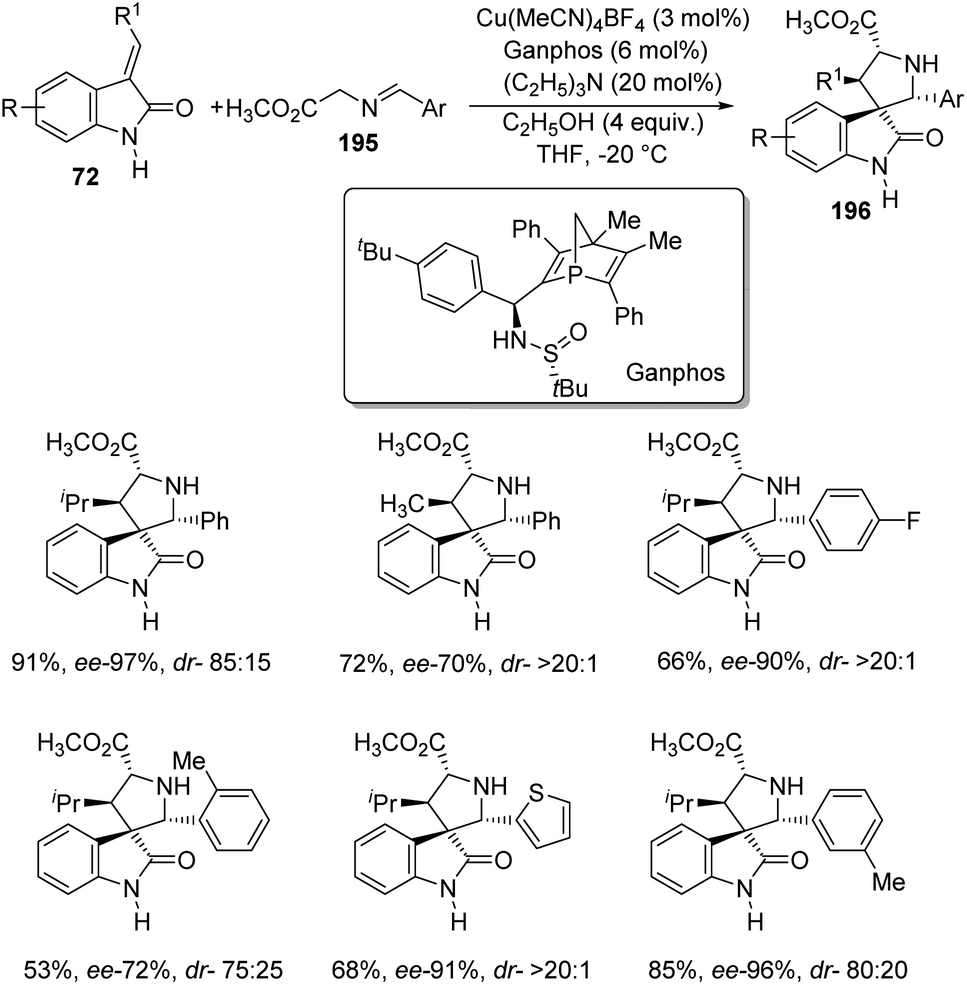 | ||
| Scheme 93 Synthesis of spirooxindoles using [Cu(I)/Ganphos] catalytic system. | ||
Next the reaction of iminoesters was performed with 3-methylene-2-oxindoles- derivative of 72 which are substituted with aryl groups. Here they changed the ligand from Ganphos to Kephos, and kept the other conditions as such (Scheme 94). Diversely substituted iminoesters 197 and benzylidene indolinones underwent the reaction even those including aliphatic iminoesters. The group further examined the amount of catalyst and found that the reaction proceeded even with 0.1 mol% of the catalyst. Excellent yield and good enantioselectivities were acquired by doing the reaction in gram scale.
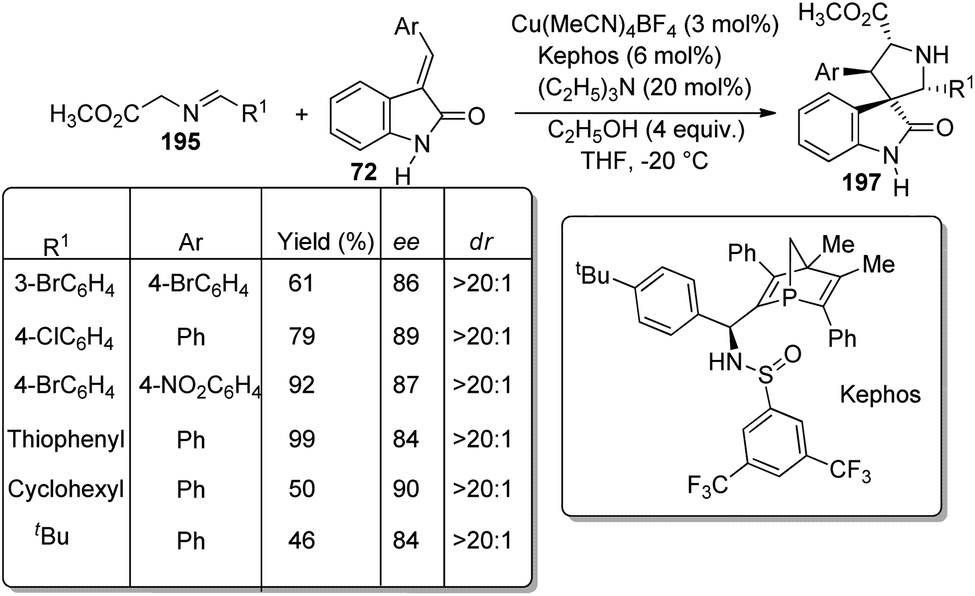 | ||
| Scheme 94 Synthesis of spirooxindoles using [Cu(I)/Kephos] catalytic system. | ||
In the case of methyleneindolinone derivatives, Ni, Fe, Pd and Cu were used as the catalysts and could achieve the yield up to 99% with Ni- and Cu based catalysts. A diastereoselectivity of up to 99:1 and enantioselectivity of >99% were provided by FeCl3 and Pd2(dba)3·CH3Cl3 respectively. The catalyst loading of 3 mol% was enough for the copper(I)/Ganphos catalyst system and it could be reduced to even 0.1 mol%.
10 Conclusion
Spirooxindoles form an extensive class of alkaloids and other artificial compounds. They have appreciable biological and pharmaceutical applications. In organic chemistry, the evolution of synthetic procedures for spirooxindole derivatives is having soaring importance. Transition metal-catalyzed synthesis is the highest probed one among numerous described synthetic approaches. Here we have given an account of transition metal-catalyzed synthesis of spirooxindoles. There is copper, palladium, iron, nickel etc. catalyzed approaches through which nitrile-, pyrrolidine-, benzofuranone-, etc. containing spirooxindoles were synthesized efficiently. Different tactics such as oxidative annulation, 1,3-dipolar cycloaddition, Heck/cyanation, hetero-Pictet–Spengler approach and so on were applied, most of them involving wide functional group tolerability and one-pot multi-component pathways. Several green and nano catalysts were employed and diverse spirooxindole derivatives, with up to eight-membered rings, were produced with high diastereo- and enantioselectivities. The advantage of catalyst recoverability with the aid of magnet was utilized in case of catalysts like MnFe2O4, CoFe2O4@SiO2 etc. In the case of catalysts like Cu(OTf)2, MnFe2O4 and Fe(OTf)2, the Lewis acidic sites in them triggered the reaction. In greater number of the reactions, the catalyst employed was nanoparticles which possess individual characteristics compared to the bulk, because of their large surface area to volume ratio and nano size. Derivatives of isatins, isothiocyanatooxindoles, methyleneindolinones etc. were generally used as the starting materials in contrast to derivatives of benzoic acids, aryl triflates, anilides and so on. It is anticipated that in the close future, transition metal-catalyzed spirooxindole synthesis would undergo huge investigations to fine tune the reaction for better regio-, diastereo- and enantioselectivities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PVS and TA thank the Council of Scientific and Industrial Research (CSIR, New Delhi) for junior research fellowships. MN thanks the University Grants Commission (UGC, New Delhi) for a research fellowship. GA thanks the Kerala State Council for Science, Technology and Environment (KSCSTE, Trivandrum) for a research grant (Order No. 341/2013/KSCSTE dated 15.03.2013).References
- N. Deppermann, H. Thomaneck, A. H. G. P. Prenzel and W. Maison, J. Org. Chem., 2010, 75, 5994 CrossRef CAS.
- T. Watanabe, M. Arisawa, K. Narusuye, M. S. Alam, K. Yamamoto, M. Mitomi, Y. Ozoe and A. Nishida, Bioorg. Med. Chem., 2009, 17, 94 CrossRef CAS.
- F. Y. Miyake, K. Yakushijin and D. A. Horne, Org. Lett., 2004, 6, 711 CrossRef CAS.
- Y. Arun, K. Saranraj, C. Balachandran and P. T. Perumal, Eur. J. Med. Chem., 2014, 74, 50 CrossRef CAS.
- B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673 CrossRef CAS.
- R. S. Kumar, P. Antonisamy, A. I. Almansour, N. Arumugam, G. Periyasami, M. Atlaf, H. R. Kim and K. B. Kwon, Eur. J. Med. Chem., 2018, 152, 417 CrossRef CAS.
- K. Parthasarathy, C. Praveen, K. Saranraj, C. Balachandran and P. S. Kumar, Med. Chem. Res., 2016, 25, 2155 CrossRef CAS.
- G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79 CrossRef CAS.
- S. T. Al-Rashood, A. R. Hamed, G. S. Hassan, H. M. Alkahtani, A. A. Almehizia, A. Alharbi, M. M. Al-Sanea and W. M. Eldehna, J. Enzyme Inhib. Med. Chem., 2020, 35, 831 CrossRef CAS.
- T. E. Ali and R. M. Abdel-Rahman, J. Sulfur Chem., 2014, 35, 399 CrossRef CAS.
- L. Chen, J. Xie, H. Song, Y. Liu, Y. Gu, L. Wang and Q. Wang, J. Agric. Food Chem., 2016, 64, 6508 CrossRef CAS.
- S. Haddad, S. Boudriga, T. N. Akhaja, J. P. Raval, F. Porzio, A. Soldera, M. Askri, M. Knorr, Y. Rousselin, M. M. Kubicki and D. Rajani, New J. Chem., 2015, 39, 520 RSC.
- N. R. Ball-Jones, J. J. Badillo and A. K. Franz, Org. Biomol. Chem., 2012, 10, 5165 RSC.
- Y. Tan, E.-L. Feng, Q.-S. Sun, H. Lin, X. Sun, G.-Q. Lin and X.-W. Sun, Org. Biomol. Chem., 2017, 15, 778 RSC.
- Z.-Y. Song, K.-Q. Chen, X.-Y. Chen and S. Ye, J. Org. Chem., 2018, 83, 2970 Search PubMed.
- L.-L. Wang, J. Bai, L. Peng and L.-W. Qi, Chem. Commun., 2012, 48, 5175 RSC.
- R. Mishra, A. Jana, A. K. Panday and L. H. Choudhury, New J. Chem., 2019, 43, 2920 RSC.
- K. Ramakumar, T. Maji, J. J. Partridge and J. A. Tunge, Org. Lett., 2017, 19, 4017 CrossRef.
- G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79 CrossRef CAS.
- Y.-C. Wang, J.-L. Wang, K. S. Burgess, J.-W. Zhang, Q.-M. Zheng, Y.-D. Pu, L.-J. Yan and X.-B. Chen, RSC Adv., 2018, 8, 5702 RSC.
- Y. T. Gao, X. Y. Jin, Q. Liu, A. D. Liu, L. Cheng, D. Wang and L. Liu, Molecules, 2018, 23, 2265 CrossRef.
- B. Wang, X. H. Wang, W. Huang, J. Zhou, H. P. Zhu, C. Peng and B. Han, J. Org. Chem., 2019, 84, 10349 CrossRef.
- R. Sridhar, B. Srinivas, B. Madhav, B. P. Reddy, Y. V. D. Nageswar and K. P. Rao, Can. J. Chem., 2019, 87, 1704 CrossRef.
- Y. Li, H. Chen, C. Shi, D. Shi and H. Ji, J. Comb. Chem., 2010, 12, 231 CrossRef CAS.
- G. S. Hari and Y. R. Lee, Synthesis, 2010, 3, 453 Search PubMed.
- M. S. Khan, D. K. Parmar and H. B. Bhatt, Asian J. Green Chem., 2019, 3, 470 CAS.
- Z. Karimi-Jaberi and A. Fereydoonnezhad, Iran. Chem. Commun., 2017, 5, 407 Search PubMed.
- A. Deepthi, N. V. Thomas and V. Sathi, Curr. Green Chem., 2019, 6, 210 CrossRef CAS.
- X. Fang and C.-J. Wang, Org. Biomol. Chem., 2018, 16, 2591 RSC.
- D. Cheng, Y. Ishihara, B. Tan and C. F. Barbas, ACS Catal., 2014, 4, 743 CrossRef CAS.
- G.-J. Mei and F. Shi, Chem. Commun., 2018, 54, 6607 RSC.
- H. Wang, S. Jiao, K. Chen, X. Zhang, L. Zhao, D. Liu, Y. Zhou and H. Liu, J. Org. Chem., 2015, 11, 416 CAS.
- M. Font, F. Acuña-Parés and T. Parella, Nat. Commun., 2014, 5, 4373 CrossRef CAS.
- T.-L. Liu, Z.-Y. Xue, H.-Y. Tao and C.-J. Wang, Org. Biomol. Chem., 2011, 9, 1980 RSC.
- L. Wu, P. Chen and G. Liu, Chin. J. Chem., 2014, 32, 681 CrossRef CAS.
- Z. Rashid, T. Moadi and R. Ghahremanzadeh, New J. Chem., 2016, 40, 3343 RSC.
- E. L. Millington, H. A. Dondas, C. W. G. Fishwick, C. Kilner and R. Grigg, Tetrahedron, 2018, 74, 3564 CrossRef CAS.
- R. Ghahremanzadeh, Z. Rashid, A.-H. Zarnani and H. Naeimi, Appl. Catal., A, 2013, 467, 270 CrossRef CAS.
- R. Ghahremanzadeh, Z. Rashid, A.-H. Zarnani and H. Naeimi, RSC Adv., 2014, 4, 43661 RSC.
- H. Naeimi, Z. Rashid, A.-H. Zarnani and R. Ghahremanzadeh, New J. Chem., 2014, 38, 5527 RSC.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770 CrossRef CAS.
- A. Guijarro, D. M. Rosenberg and R. D. Rieke, J. Am. Chem. Soc., 1999, 121, 4155 CrossRef CAS.
- A. Dandia, V. Parewa, A. K. Jain and K. S. Rathore, Green Chem., 2011, 13, 2135 RSC.
- Y.-H. Miao, Y.-Z. Hua and M.-C. Wang, Org. Biomol. Chem., 2019, 17, 7172 RSC.
- F. Tan, L.-Q. Lu, Q.-Q. Yang, W. Guo, Q. Bian, J.-R. Chen and W.-J. Xiao, Chem.–Eur. J., 2014, 20, 3415 CrossRef CAS.
- J.-Q. Zhao, Z.-J. Wu, M.-Q. Zhou, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2015, 17, 5020 CrossRef CAS.
- Y.-J. Guo, X. Guo, D.-Z. Kong, H. Lu, L. Liu, Y.-Z. Hua and M.-C. Wang, J. Org. Chem., 2020, 85, 4195 CrossRef CAS.
- V. K.-Y. Lo, A. O.-Y. Chan and C.-M. Che, Org. Biomol. Chem., 2015, 13, 6667 RSC.
- H. Huang, Y. Zhou and H. Liu, J. Org. Chem., 2011, 7, 897 CAS.
- K. Parthasarathy, C. Praveen, J. C. Jayaveeran and A. A. M. Prince, Bioorg. Med. Chem. Lett., 2016, 26, 4310 CrossRef CAS.
- J. Cai, B. Wu, G. Rong, C. Zhang, L. Qui and X. Xu, Org. Lett., 2018, 20, 2733 CrossRef CAS.
- V. P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef CAS.
- X.-N. Zhang, Y.-X. Li and Z.-H. Zhang, Tetrahedron, 2011, 67, 7426 CrossRef CAS.
- M. A. Nasseri, F. Kamali and B. Zakerinasab, RSC Adv., 2015, 5, 26517 RSC.
- S. Yagnum, A. M. Akondi, R. Trivedi, B. Rathod, R. S. Prakasham and B. Sridhar, Synth. Commun., 2018, 48, 255 CrossRef.
- Z. A. Moqadam, A. Allahresani and H. Hassani, Res. Chem. Intermed., 2020, 46, 299 CrossRef.
- S. Kato, M. Kanai and S. Matsunaga, Chem.–Asian J., 2013, 8, 1768 CrossRef CAS.
- T. Arai, T. Miyazaki, H. Ogawa and H. Masu, Org. Lett., 2016, 18, 5824 CrossRef CAS.
- Y. Zhou, Y. Lu, X. Hu, H. Mei, L. Lin, X. Liu and X. Feng, Chem. Commun., 2017, 53, 2060 RSC.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778 CrossRef.
- Y.-Y. Han, W.-Y. Han, X. Hou, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2012, 14, 4054 CrossRef CAS.
- S. M. Zadeghzadeh and M. A. Nasseri, Catal. Today, 2013, 217, 80 CrossRef.
- F. A. Tameh, J. Safaei-Ghomi, M. M. Hashemi and H. S. Alavi, RSC Adv., 2016, 6, 74802 RSC.
- K. Hemmat, M. A. Nasseri, A. Allahresani and S. Ghiami, J. Org. Chem., 2019, 903, 120996 CrossRef.
- B. Zamani-Ranjbar-Garmroodi, M. A. Nasseri, A. Allahresani and K. Hemmat, Res. Chem. Intermed., 2019, 45, 5665 CrossRef CAS.
- A. Allahresani, B. Taheri and M. A. Nasseri, Iran. J. Catal., 2019, 9, 163 CAS.
- J. R. Song, Z. Y. Li, G. D. Wang, N. Zhang, C. Chen, J. Hen, H. Ren and W. Pan, Adv. Synth. Catal., 2020, 362, 500 CrossRef CAS.
- S. Kavyani and R. Baharfar, Appl. Organomet. Chem., 2020, 34, e5560 CrossRef CAS.
- J.-Q. Zhang, Z.-H. Qi, S.-J. Yin, H.-Y. Li, Y. Wang and X.-W. Wamg, ChemCatChem, 2016, 8, 1 CrossRef.
- S. Bhandari, A. P. Sakla and N. Shankaraiah, ChemistrySelect, 2020, 5, 2886 CrossRef CAS.
- J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038 CrossRef CAS.
- W. A. Herrmann, V. P. W. Böhm and C.-P. Reisinger, J. Chem. Educ., 2000, 77, 92 CrossRef CAS.
- M. Nambo and K. Itami, Chem.–Eur. J., 2009, 15, 4760 CrossRef CAS.
- A. Ashimori and L. E. Overman, J. Org. Chem., 1992, 57, 4571 CrossRef CAS.
- X. Luo, Y. Xu, G. Xiao, W. Liu, C. Qian, G. Deng, J. Song, Y. Liang and C. Yang, Org. Lett., 2018, 20, 2997 CrossRef CAS.
- R. Shintani, S. Hayashi, M. Murakami, M. Takeda and T. Hayashi, Org. Lett., 2009, 11, 3754 CrossRef CAS.
- B. Niu, X. Y. Wu, Y. Wei and M. Shi, Org. Lett., 2019, 21, 4859 CrossRef CAS.
- S. Jaegli, J. P. Vors, L. Neuville and J. Zhu, Tetrahedron, 2010, 66, 8911 CrossRef CAS.
- D. Y. Seo, G. Kim, H. Y. Jo and J. N. Kim, Bull. Korean Chem. Soc., 2018, 39, 587 CrossRef CAS.
- A. C. S. Reddy, P. M. Reddy and P. Anbarasan, Adv. Synth. Catal., 2020, 362, 801 CrossRef CAS.
- S. M. Hande, M. Nakajima, H. Kamisaki, C. Tsukano and Y. Takemoto, Org. Lett., 2011, 13, 1828 CrossRef CAS.
- C. Tsukano, M. Okuno and Y. Takemoto, Chem. Lett., 2013, 42, 753 CrossRef CAS.
- T. R. Li, B. Y. Cheng, S. Q. Fan, Y. N. Wang, L. Q. Lu and W. J. Xiao, Chem.–Eur. J., 2016, 22, 6243 CrossRef CAS.
- J. Liu, X. Xu, J. Li, B. Liu, H. Jiang and B. Yin, Chem. Commun., 2016, 52, 9550 RSC.
- J. Liu, H. Peng, Y. Yang, H. Jiang and B. Yin, J. Org. Chem., 2016, 81, 9695 CrossRef CAS.
- H. Yoon, A. Lossouarn, F. Landau and M. Lautens, Org. Lett., 2016, 18, 6324 CrossRef CAS.
- H. Yoon, M. Rolz, F. Landau and M. Lautens, Angew. Chem., Int. Ed., 2017, 56, 10920 CrossRef CAS.
- C. Shao, Z. Wu, X. Ji, B. Zhou and Y. Zhang, Chem. Commun., 2017, 53, 10429 RSC.
- D. Kiely and P. J. Guiry, Tetrahedron Lett., 2002, 43, 9545 CrossRef CAS.
- S. Jaegli, J. Dufour, H. Wei, T. Piou, X. H. Duan, J. P. Vors, L. Neuville and J. Zhu, Org. Lett., 2010, 12, 4498 CrossRef CAS.
- T. Piou, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2012, 51, 11561 CrossRef CAS.
- K. H. Kim, H. R. Mooon, J. Lee and J. N. Kim, Adv. Synth. Catal., 2015, 357, 701 CrossRef CAS.
- R. R. Liu, Y. Xu, R. X. Liang, B. Xiang, H. J. Xie, J. R. Gao and Y. X. Jia, Org. Biomol. Chem., 2017, 15, 2711 RSC.
- J. A. Xiao, X. L. Cheng, Y. C. Li, Y. M. He, J. L. Li, Z. P. Liu, P. J. Xia, W. Su and H. Yang, Org. Biomol. Chem., 2019, 17, 103 RSC.
- H. Song, N. Cheng, L. Q. She, Y. Wu and W. W. Liao, RSC Adv., 2019, 9, 29424 RSC.
- X. Zhu and S. Chiba, Chem. Soc. Rev., 2016, 45, 4504 RSC.
- F. Salahi, M. J. Taghizadeh, H. Arvinnezhad, M. Moemeni, K. Jadidi and B. Notash, Tetrahedron Lett., 2014, 55, 1515 CrossRef CAS.
- M. J. Taghizadeh, A. Javidan and K. Jadidi, J. Korean Chem. Soc., 2015, 59, 205 CrossRef CAS.
- M. Rajeswari, J. Sindhu, H. Singh and J. M. Khurana, RSC Adv., 2015, 5, 39686 RSC.
- M. Rajeswari, S. Kumari and J. M. Khurana, RSC Adv., 2016, 16, 9297 Search PubMed.
- M. Baghernejad, S. Khodabakhshi and S. Tajik, New J. Chem., 2016, 40, 2704 RSC.
- C. H. Chen, Q. Q. Liu, X. P. Ma, Y. Feng, C. Liang, C. X. Pan, G. F. Su and D. L. Mo, J. Org. Chem., 2017, 82, 6417 CrossRef CAS.
- Y. S. Zhu, B. B. Yuan, J. M. Guo, S. J. Jin, H. H. Dong, Q. L. Wang and S. W. Bu, J. Org. Chem., 2017, 82, 5669 CrossRef CAS.
- L. Moradi and Z. Ataei, Green Chem. Lett. Rev., 2017, 10, 380 CrossRef CAS.
- C. Wu, J. Liu, D. Kui, Y. Lemao, X. Yingjie, X. Luo, X. Meiyang and R. Shen, Polycyclic Aromat. Compd., 2020 DOI:10.1080/10406638.2020.1726976.
- C. Teja, S. N. Babu, A. Noor, J. A. Daniel, S. A. Devi and F. R. N. Khan, RSC Adv., 2020, 10, 12262 RSC.
- C. Zhang, M. Liu, M. Ding, H. Xie and F. Zhang, Org. Lett., 2017, 19, 3418 CrossRef CAS.
- H. Kui, K. Li, Y. Wang, M. Song, C. Wang, D. Wei, E. Q. Li, Z. Duan and F. Mathey, Org. Biomol. Chem., 2020, 18, 3740 RSC.
| This journal is © The Royal Society of Chemistry 2021 |