
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA reduction-responsive drug delivery with improved stability: disulfide crosslinked micelles of small amiphiphilic molecules†
Man Li,
Longbing Ling,
Qing Xia and
Xinsong Li *
*
School of Chemistry and Chemical Engineering, Southeast University, Nanjing, 214122, China. E-mail: lixs@seu.edu.cn
First published on 1st April 2021
Abstract
Micelles self-assembled from small amphiphilic molecules are unstable in biological fluids, and thus are poor drug carriers. In contrast, amphiphilic polymer micelles can encapsulate hydrophobic drugs in their core to greatly enhance their aqueous solubility and extend their retention time in blood circulation owing to their hydrophilic shell. However, the major disadvantages of conventional polymer micelles are the heterogeneity of the amphiphilic polymer structure and premature drug leakage. Thus, herein, to address these shortcomings, disulfide crosslinked micelles composed of a small amphiphilic molecule, di-lipoyl-glycerophosphorylcholine (di-LA-PC), were developed as redox-responsive drug carriers. Specifically, di-LA-PC was synthesized and self-assembled to form crosslinked micelles under catalysis by dithiothreitol. The disulfide crosslinked micelles maintained high stability in a simulated physiological environment, but rapidly disassembled under reductive conditions. Furthermore, paclitaxel (PTX), as a model drug, was encapsulated in the core of the crosslinked micelles with a high loading content of 8.13%. The in vitro release studies indicated that over 80% of PTX was released from the micelles in the reductive environment, whereas less than 20% PTX was released without reduction in the 68 h test. Benefiting from their nanoscale characteristics, the PTX-loaded micelles showed efficient cellular internalization and effectively induced the death of cancer cells, as revealed in the MTT, apoptosis and cell cycle tests. Moreover, pharmacokinetic studies demonstrated that the crosslinked micelles prolonged the circulation of the incorporated PTX in the bloodstream and increased its accumulation in the tumor tissue via the EPR effect. Finally, the PTX-loaded micelles displayed prominent in vivo anti-tumor activity in a 4T1 xenograft tumor model. In summary, the di-LA-PC crosslinked micelle platform possesses excellent stability, high loading capacity and reduction-responsive release profile, which may have applications in the delivery of PTX and other anti-cancer drugs.
1. Introduction
In last few decades, amphiphilic polymer micelles have been extensively investigated due to their promising potential for delivering therapeutic agents in cancer therapy.1–3 These micelle formulations usually possess greatly enhanced aqueous solubility for hydrophobic drugs, extended retention time in the blood circulation, precise accumulation at tumor sites due to the enhanced permeability and retention (EPR) effect and altered pharmacological profiles with reduced side effects.4–7 However, conventional polymer-based micelles often suffer from premature drug leakage because of their poor physical stability in vivo and insufficient intracellular drug release at tumor sites, thereby limiting their applications in clinic.8,9 Another disadvantage of polymer micelles is the heterogeneity of the chemical structure of polymers including their molecular weight and distribution.A successful strategy to stabilize polymeric micelles is covalent crosslinking.10–12 Disulfide (–SS-) cross-linking is continuously being explored for the reversible stabilization of micelles.13,14 It has been reported that –SS- cross-linkages sustain prominent stability in blood circulation and in extracellular tissues with lower glutathione (GSH) contents but are susceptible to disassembly under reductive tumor milieu to release their payload.15,16 Accordingly, numerous cysteine-containing structures have been introduced into the polymer backbone to construct disulfide crosslinked micelles.17,18 Additionally, external disulfide-bearing linkers can be also applied to prepare crosslinked micelles for drug delivery.19 Disulfide crosslinked polymeric micelles possess superior serum stability, exhibiting better therapeutic efficacy compared to uncrosslinked micelles. Recently, lipoic acid (LA) was conjugated to polymer chains20 to prepare disulfide crosslinked micelles. These lipoic acid–based micelles exhibited noteworthy depolymerization character upon exposure to reductive conditions,21–23 which showed a superior doxorubicin (Dox) loading capacity1 and controlled release in a reductive environment.
Phosphorylcholine (PC) is a zwitterionic molecular segment obtained from biological phospholipids, which is a key constituent of cell membranes.24 Its zwitterionic lipid-like structure can maintain effective uptake by cells via fusion with the cell membrane. Accordingly, several PC-based functional molecules and polymers with favorable biocompatibility and biodegradability have been reported for use in drug delivery for cancer chemotherapy.25–27 Covalently attaching a PC moiety to a reactive hydrophobic drug or by polymerizing vinyl monomers containing PC such as 2-(methacryloyloxy)-ethyl phosphorylcholine (MPC) can significantly improve the water solubility and enhance the stability of drugs, thereby leading to better therapeutic potential.28 Moreover, some studies displayed the superior in vivo performance of PC-containing polymer micelles compared to PEG-based nanocarriers, including longer circulation in the blood, more efficient internalization and better inhibition versus tumor tissues.28–30
Thus, herein, to address the shortcomings of conventional polymer micelles including the heterogeneity of amphiphilic polymer structures and premature drug leakage, disulfide crosslinked micelles of a small amphiphilic molecule, di-lipoyl-glycerophosphorylcholine (di-LA-PC), were developed. The amphiphilic conjugate di-LA-PC, a phosphorylcholine derivative, was self-assembled to form micelles in aqueous medium and subsequently stabilized by disulfide crosslinking. PTX as a model drug was encapsulated in the crosslinked micelles using a conventional method (Scheme 1). The size, loading capacity and reduction release behavior of the PTX-loaded disulfide crosslinked micelles were characterized in detail. Their cellular uptake, cell cycle, in vitro cytotoxicity and apoptotic activities were further investigated. Moreover, the in vivo pharmacokinetics and anticancer activity of the PTX-loaded crosslinked micelles were analyzed.
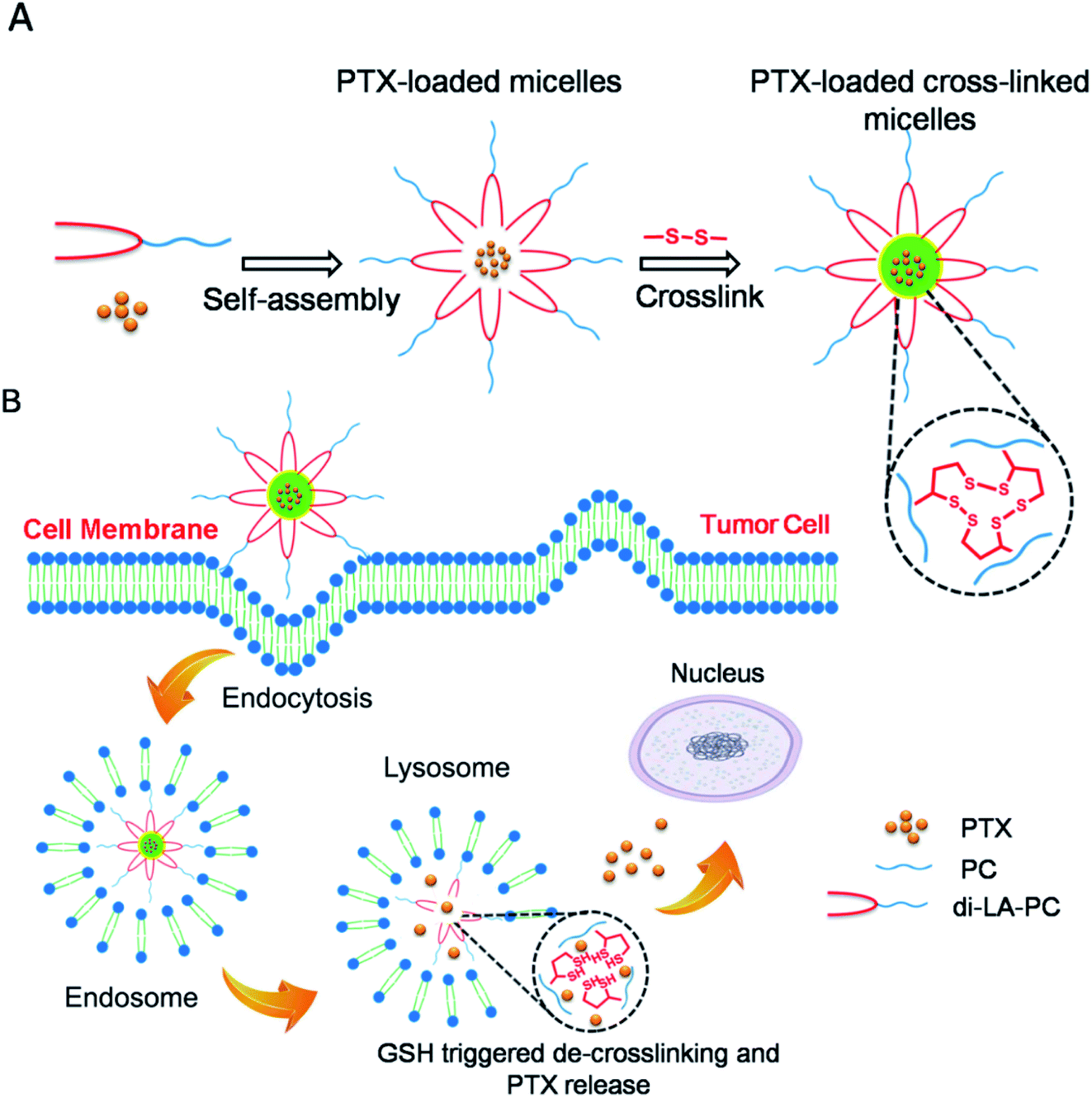 | ||
| Scheme 1 Schematic illustration of the reduction-responsive crosslinked di-LA-PC micelles for drug loading and GSH-triggered release of PTX. (A) Self-assembly of amphiphilic di-LA-PC conjugate and PTX loading, followed by disulfide crosslinking. (B) Cellular uptake via endocytosis and GSH-triggered de-crosslinking, micelle disruption and PTX release. | ||
2. Materials and methods
2.1. Reagents
Lipoic acid (LA, purity ≥ 98%) and glycerophosphorylcholine (PC, purity ≥ 98%) were obtained from Fushilai Pharmaceutical Co., Ltd (Suzhou, China). ε-Caprolactone, 4-(dimethylamino)pyridine (DMAP), N,N′-carbonyl-diimidazole (CDI), dicyclohexylcarbodiimide (DCC) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were provided by Aladdin Biochemical Technology Co., Ltd (Shanghai, PR China). Imidazole (purity ≥ 99%), tert-butyl diphenyl chlorosilane (TBDPSCl, purity ≥ 98%), and tetra-butylammonium fluoride (TBAF) were obtained from Meryer Chemical Technology Co., Ltd (Shanghai, China). Methyl tetrazolium (MTT), fetal-bovine serum (FBS), Roswell Park Memorial Park medium (RPMI, 1640 medium), cyanine (Cy5.5), 0.1% trypsin–EDTA and annexin V-FITC/PI apoptosis kit were purchased from KeyGEN Ltd (Nanjing, China). Unless otherwise noted, all chemicals/reagents were of analytical grade and used as received in the following experiments without further purification.2.2. Instruments
Reversed phase high performance liquid chromatography (RP-HPLC) characterization was performed on an Agilent 1100 Infinity Quaternary HPLC System using an analytical column (ZORBAX SB-C18, 4.6 × 150 mm, 5 μm). An Agilent 7200B Quadruple Mass Spectrometer (MS) was used to obtain the mass spectrum of the final products in the range of 100 to 4200 m/z (Agilent, CA). 1H-NMR and 13C-NMR analysis were performed on a Bruker Advance DPX spectrometer at 500 MHz (Bruker, WI). The chemical shifts are based on the peaks of the deuterated solvent of CDCl3 or CH3OH-d4 using TMS as an internal standard.2.3. Synthesis of di-LA-PC conjugate
Scheme 2 summarizes the synthetic route employed for the preparation of the di-LA-PC conjugate. The detailed synthetic procedure and structural characterizations are described as follows: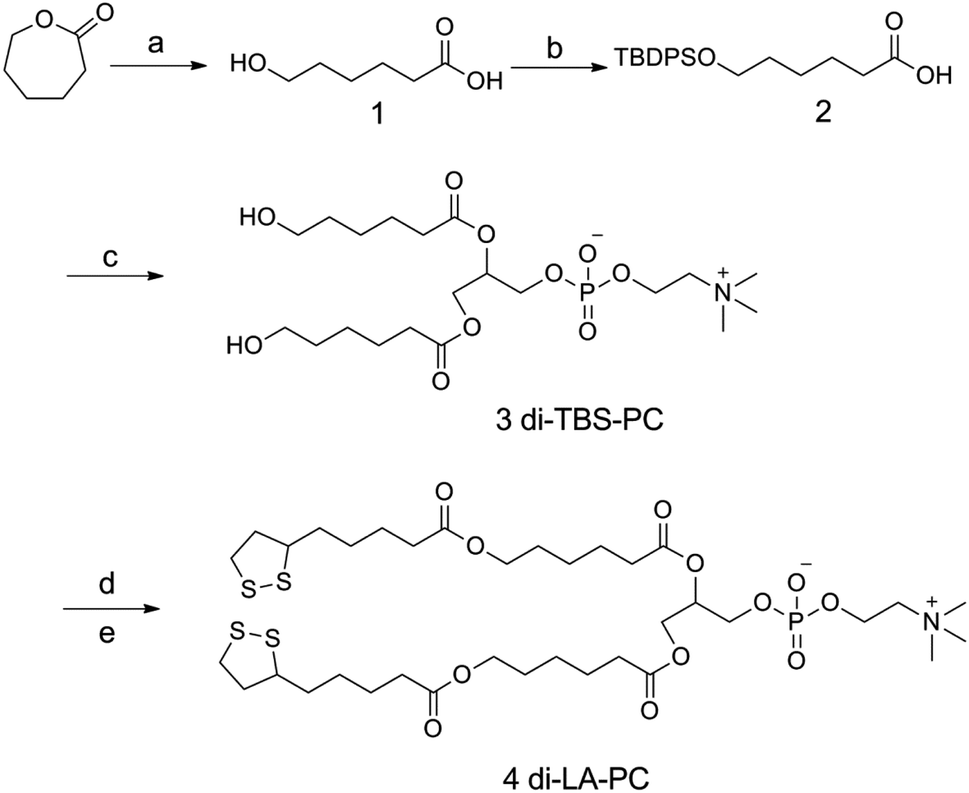 | ||
| Scheme 2 Synthetic route for di-LA-PC conjugate. Reagents and conditions: (a) NaOH/H2O, rt, 12 h, 98%; (b) TBDPSCl, imidazole, DMF, 0 °C to rt, 8 h, 67%; (c) L-α-glycerophosphorylcholine, CDI, DBU, CH2Cl2, DMSO, rt to 45 °C, 12 h, 71%; (d) TBAF, THF, rt, 4 h, 96%; and (e) lipoic acid, DCC, DMAP, CH2Cl2, 35 °C, 23%. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (1:3, v/v) as the eluent, finally providing compound 2 as a colorless viscous liquid (4.2 g, yield: 77%). MS (m/z): calcd for C22H30O3Si, 370.20; found, 371.20 [M + H]+, 393.18 [M + Na]+. 1H NMR (500 MHz, CDCl3): δ 7.76 (d, 4H, H-1, 1′, 5, 5′, J = 6.5 Hz), 7.44 (t, 6H, H-2, 2′, 3, 3′, 4, 4′, J = 7.4 Hz), 3.75 (t, 2H, H-9, J = 6.5 Hz), 2.41 (t, 2H, H-13, J = 7.5 Hz), 1.71–1.65 (m, 2H, H-10), 1.54–1.48 (m, 2H, H-12), 1.34–1.29 (m, 2H, H-11), 1.14 (s, 9H, H-6, 7, 8) ppm.:CH3OH, 10:1; solvent B: CH2Cl2:CH3OH:H2O 65:25:4) and gradient elution to obtain a white solid (1.68 g, yield: 71%). MS (m/z): calcd for C52H76NO10PSi2, 961.4; found, 962.4 [M + H]+, 984.4 [M + Na]+. 1H NMR (500 MHz, CDCl3): δ 7.69 (d, 8H, H-3, 3′, 4, 4′, 6, 6′, 10, 10′, J = 6.5 Hz), 7.41 (t, 12H, H-1, 1′, 2, 2′, 5, 5′, 7, 7′, 8, 8′, 9, 9′, J = 7 Hz), 5.24 (d, 2H, H-20, J = 5.5 Hz), 4.44 (t, 2H, H-22, J = 7.4 Hz), 4.16 (m, 1H, H-19), 3.99 (t, 2H, H-23, J = 7.5 Hz), 3.93 (t, 4H, H-14, 14′, J = 7.2 Hz), 3.72 (d, 1H, H-21, J = 7.4 Hz), 3.29 (s, 9H, H-24, 25, 26), 2.33–2.26 (m, 4H, H-17, 17′), 1.61–1.54 (m, 4H, H-16, 16′), 1.42–1.35 (m, 4H, H-15, 15′), 1.26 (t, 4H, H-18, 18′, J = 7.0 Hz), 1.06 (s, 18H, H-11, 11′, 12, 12′, 13, 13′) ppm.:CH3OH, 10:1) and then solvent B (CH2Cl2:CH3OH:H2O 65:25:4), providing compound 4 as a yellowish solid with the yield of 23% (0.20 g). MS (m/z): calcd for C36H64NO12PS4, 861.3; found, 862.3 [M + H]+, 884.3 [M + Na]+. 1H NMR (500 MHz, CH3OH-d4): δ 5.26 (d, 2H, H-16, J = 5.5 Hz), 4.47 (t, 2H, H-18, J = 7.4 Hz), 4.23–4.16 (m, 1H, H-15), 4.09 (t, 4H, H-9, 9′, J = 6.2 Hz), 4.02 (t, 2H, H-19, J = 7.5 Hz), 3.65 (d, 2H, H-17, J = 7.4 Hz), 3.36 (s, 9H, H-20, 21, 22), 2.54–2.41 (m, 12H, H-1, 1′, 7, 7′, 13, 13′), 2.39–2.33 (m, 2H, H-3, 3′), 1.97–1.89 (m, 4H, H-2, 2′), 1.77–1.61 (m, 16H, H-4, 4′, 6, 6′, 10, 10′, 12, 12′), 1.50–1.42 (m, 8H, H-5, 5′, 11, 11′) ppm. 13C NMR (500 MHz, CH3OH-d4): δ 173.41, 172.50, 70.06, 64.97, 63.42, 63.04, 61.89, 58.61, 55.70, 52.83, 39.46, 37.51, 34.91, 33.83, 33.06, 27.91, 27.56, 24.70, 23.96, 23.69 ppm.
:hexane (1:3, v/v) as the eluent, finally providing compound 2 as a colorless viscous liquid (4.2 g, yield: 77%). MS (m/z): calcd for C22H30O3Si, 370.20; found, 371.20 [M + H]+, 393.18 [M + Na]+. 1H NMR (500 MHz, CDCl3): δ 7.76 (d, 4H, H-1, 1′, 5, 5′, J = 6.5 Hz), 7.44 (t, 6H, H-2, 2′, 3, 3′, 4, 4′, J = 7.4 Hz), 3.75 (t, 2H, H-9, J = 6.5 Hz), 2.41 (t, 2H, H-13, J = 7.5 Hz), 1.71–1.65 (m, 2H, H-10), 1.54–1.48 (m, 2H, H-12), 1.34–1.29 (m, 2H, H-11), 1.14 (s, 9H, H-6, 7, 8) ppm.:CH3OH, 10:1; solvent B: CH2Cl2:CH3OH:H2O 65:25:4) and gradient elution to obtain a white solid (1.68 g, yield: 71%). MS (m/z): calcd for C52H76NO10PSi2, 961.4; found, 962.4 [M + H]+, 984.4 [M + Na]+. 1H NMR (500 MHz, CDCl3): δ 7.69 (d, 8H, H-3, 3′, 4, 4′, 6, 6′, 10, 10′, J = 6.5 Hz), 7.41 (t, 12H, H-1, 1′, 2, 2′, 5, 5′, 7, 7′, 8, 8′, 9, 9′, J = 7 Hz), 5.24 (d, 2H, H-20, J = 5.5 Hz), 4.44 (t, 2H, H-22, J = 7.4 Hz), 4.16 (m, 1H, H-19), 3.99 (t, 2H, H-23, J = 7.5 Hz), 3.93 (t, 4H, H-14, 14′, J = 7.2 Hz), 3.72 (d, 1H, H-21, J = 7.4 Hz), 3.29 (s, 9H, H-24, 25, 26), 2.33–2.26 (m, 4H, H-17, 17′), 1.61–1.54 (m, 4H, H-16, 16′), 1.42–1.35 (m, 4H, H-15, 15′), 1.26 (t, 4H, H-18, 18′, J = 7.0 Hz), 1.06 (s, 18H, H-11, 11′, 12, 12′, 13, 13′) ppm.:CH3OH, 10:1) and then solvent B (CH2Cl2:CH3OH:H2O 65:25:4), providing compound 4 as a yellowish solid with the yield of 23% (0.20 g). MS (m/z): calcd for C36H64NO12PS4, 861.3; found, 862.3 [M + H]+, 884.3 [M + Na]+. 1H NMR (500 MHz, CH3OH-d4): δ 5.26 (d, 2H, H-16, J = 5.5 Hz), 4.47 (t, 2H, H-18, J = 7.4 Hz), 4.23–4.16 (m, 1H, H-15), 4.09 (t, 4H, H-9, 9′, J = 6.2 Hz), 4.02 (t, 2H, H-19, J = 7.5 Hz), 3.65 (d, 2H, H-17, J = 7.4 Hz), 3.36 (s, 9H, H-20, 21, 22), 2.54–2.41 (m, 12H, H-1, 1′, 7, 7′, 13, 13′), 2.39–2.33 (m, 2H, H-3, 3′), 1.97–1.89 (m, 4H, H-2, 2′), 1.77–1.61 (m, 16H, H-4, 4′, 6, 6′, 10, 10′, 12, 12′), 1.50–1.42 (m, 8H, H-5, 5′, 11, 11′) ppm. 13C NMR (500 MHz, CH3OH-d4): δ 173.41, 172.50, 70.06, 64.97, 63.42, 63.04, 61.89, 58.61, 55.70, 52.83, 39.46, 37.51, 34.91, 33.83, 33.06, 27.91, 27.56, 24.70, 23.96, 23.69 ppm.2.4. Micelle formation and critical micelle concentration (CMC)
Uncrosslinked di-LA-PC micelles were formulated via the dropwise addition of a DMF solution of di-LA-PC conjugate to deionized (DI) water while stirring at rt and dialyzed against freshly DI water for 24 h with a membrane (MWCO 1000 Da). During dialysis, the DI water was replaced every 3 h.CMC of di-LA-PC conjugate was examined by a fluorescent pyrene method.32 In brief, 10 μL of pyrene dissolved in acetone (5 × 10−4 M) was injected into a di-LA-PC conjugate dispersion with a series of concentrations ranging from 1 mg mL−1 to 1.25 μg mL−1. Subsequently, the acetone was evaporated under ambient environment, while the final concentration of pyrene was kept at 6 × 10−7 M in each tube. The fluorescence emission spectra were measured on a FluoroMax®-4 system (HORIBA Jobin Yvon Inc., France). The fluorescence emissions at 372 nm (I1) and 383 nm (I3) were checked under the excitation wavelength of 330 nm. The CAC value was evaluated by plotting log(concentration) vs. the intensity ratio I3/I1.
2.5. Preparation and characterization of crosslinked micelles
The crosslinked micelles based on di-LA-PC were formed by using a catalytic quantity of DTT.3 Briefly, 10 mol% DTT relative to the molar ratio of lipoic acid was added to the above-prepared dispersion and subjected to stirring for 2 h at 37 °C. Then, the resulting solution was dialyzed for 48 h (MWCO 1000 Da) to give crosslinked di-LA-PC micelles as a pale blue emulation.The five-membered ring of lipoic acid possesses a specific absorption peak at 330 nm, which was detected using a Thermo UV-vis spectrophotometer (Thermo Fisher Scientific, MA). The hydrodynamic diameter, polydispersity and zeta-potential of the di-LA-PC micelles were determined via dynamic light scattering (DLS) on a Zetasizer Nano ZS90 detector (Malvern, UK). The micellular nanostructure was imaged through transmission electron microscopy (TEM, JEM 2100, JEOL, Japan) performed at an accelerating voltage of 200 kV. The crosslinked micelle samples were transferred to a carbon film copper grid (300 mesh), air dried and negatively stained using 2% (w/v) phosphomolybdic acid before testing.
The in vitro colloidal stabilities of the crosslinked di-LA-PC micelles and non-crosslinked micelles against × 100 dilutions and high salt concentration (2 M NaCl) were monitored by DLS. Moreover, the reduction-triggered destabilization of the di-LA-PC micelles was similarly tested using DLS equipment. Briefly, the crosslinked di-LA-PC micelles in water were treated with 10 mM of reductive DTT, and then incubated at 37 °C with a rotation of 200 rpm. At 2 h intervals, the size changes were checked by DLS.
2.6. Encapsulation of PTX
PT-loaded di-LA-PC micelles were prepared via the slow addition of DMF solution containing PTX and di-LA-PC conjugate to deionized water at rt under constant stirring. Subsequently, the prepared samples were dialyzed for 48 h (MWCO 1000 Da) to remove the unincorporated PTX cross-linked via the addition of a catalytic amount of DTT. Accordingly, crosslinked micelles loaded with PTX drug and a concentration of 0.5 mg PTX per mL was obtained.The PTX loading content was established via RP-HPLC (Agilent, CA). The detection was performed using a ZORBAX SB-C18 column (150 mm × 4.6 mm, 5 μm) at 25 ± 2 °C and a wavelength of 254 nm. The flow rate was set at 1.0 mL min−1, applying a gradient mobile phase comprised of methanol in water (0–12 min: 60–80%; and 12–20 min: 80–80%) containing 0.1% TFA. The drug loading capacity (DLC) and drug loading efficiency (DLE) were determined based on the following formula:
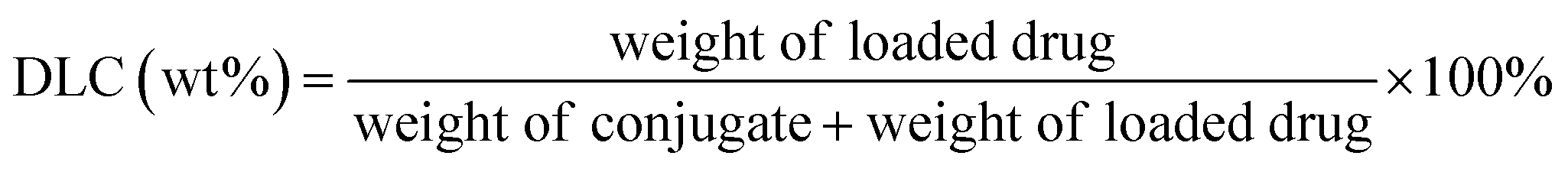
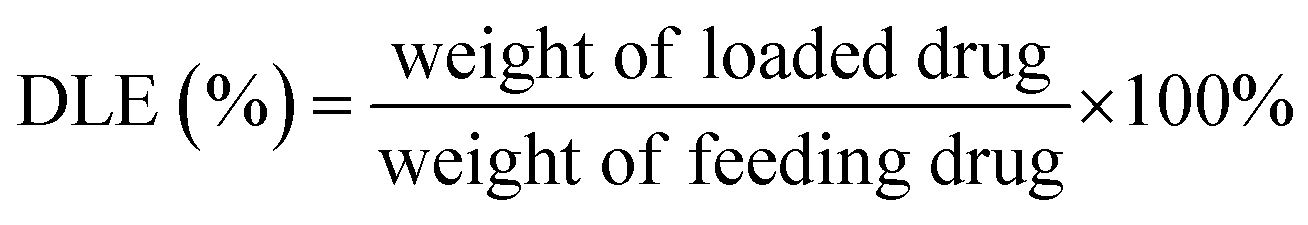
2.7. Reduction-triggered release of PTX
PTX release from the crosslinked di-LA-PC micelles was investigated via a dialysis method (MWCO 1000) upon incubation in different media for 68 h at 37 °C.33 The release study was conducted at a lower micellar concentration of 20 μg mL−1. Typically, 5 mL PTX-loaded crosslinked micelles was shifted to a dialysis tube and kept in 100 mL PBS (pH = 7.4) solution with 2 mM or 10 mM DTT together with Tween-80 (0.5%, w/v). At various timepoints, the release medium (1 mL) was collected, and the equivalent amount of external buffer was added to maintain the sink condition. The quantity of released PTX was determined via RP-HPLC, as mentioned earlier. Accordingly, the cumulative release of PTX was calculated and the percentage of total PTX was plotted against incubation time.2.8. Cell culture
MCF-7 cells, A549 cells and HepG-2 cells were obtained from the Public Heath Department of Southeast University. These three carcinomas were cultured in complete medium (RPMI-1640 comprising FBS (10%)) at 37 °C under a humidified atmosphere with 5% CO2. When the cells grew to appropriately 80% confluency, they were digested by trypsin/EDTA solution and passage before the tests.2.9. Cellular uptake of Cy5.5-loaded micelles
To aid the visualization under a fluorescent microscope, Cy5.5 dye as a model near-infrared fluorescent probe was applied to label the crosslinked di-LA-PC micelles. Cy5.5-loaded di-LA-PC micelles with a concentration of Cy5.5 at 10 μg mL−1 were prepared via similar process to that for the PTX loaded in micelles described above. For qualitative assessment of the cellular uptake by MCF-7 cells, a logarithmic growth of cells was cultured in a 6-well plate containing 1.0 × 104 cells in each well and attached overnight at 37 °C, followed by the addition of the Cy5.5-loaded crosslinked micelles. Afterward, the incubated cells were rinsed with cold PBS solution thrice, treated with paraformaldehyde (4%) for 30 min, and finally stained using 4′,6-diamidino-2-phenylindole (DAPI) for 15 min at rt. Then, the obtained cells were imaged via scanning confocal laser microscopy (CLSM, Leica, Germany).2.10. Subcellular location
A density of 1.0 × 104 MCF-7 cells per well was plated in a 6-well plate. After culturing for 12 h, the Cy5.5-loaded micelles were applied to each well and further incubated for 1 h. Then, the cells were treated with 20 nM of LysoTracker Green (Gibco Life Technology, Switzerland) for 30 min before the end of the incubation period. The cells were cleaned with PBS twice and imaged using the fluorescence emitted by LysoTracker Green (505 nm) and Cy5.5 (707 nm) at 400× magnification.2.11. MTT assay
The in vitro tumor cell inhibition by the blank crosslinked di-LA-PC micelles and PTX-loaded di-LA-PC micelles was evaluated against HepG-2, MCF-7 and A549 cells using the MTT assay. Briefly, the cells were seeded in a 96-well plate (5 × 103 cells per well) in 100 μL RPMI-1640 medium supplemented with 10% FBS. After attachment overnight, the medium was removed and substituted with 150 μL of freshly prepared medium comprised of various concentrations of blank di-LA-PC micelles, Taxol-like (6 mg paclitaxel dissolved in 527 mg polyoxyethyl castor oil and 527 mg absolute ethanol) and PTX-loaded di-LA-PC micelles. The concentration of the blank di-LA-PC crosslinked micelles varied in the range of 0.5–100 μg mL−1, while the PTX-loaded di-LA-PC micelles had a PTX concentration in the range of 0.156–10 μg mL−1. After 24 h incubation, 20 μL of 5 mg mL−1 MTT solution in PBS was supplemented in all the wells and incubated for a further 4 h at 37 °C. After aspiration of the residual medium, DMSO (150 μL) was added and the absorbance at a wavelength of 490 nm of the generated MTT-formazan was checked using a microplate reader (Bio-Rad, CA). The percentage cellular viability was determined as follows: (OD of test group)/(OD of control group) × 100%, where OD is the optical density.2.12. Cell apoptosis and cell cycle assay
The cell apoptosis and cycle distribution analyses were performed as previously described.34,35 About 5 × 105 MCF-7 cells per well were placed in a 6-well plate and cultured for 24 h. Then, Taxol-like and PTX-loaded crosslinked micelles at an equivalent concentration of 5 μg PTX per mL in RPMI-1640 medium were added to the wells separately and incubation was continued for another 24 h. Untreated MCF-7 cells were applied as a negative control. For the determination of apoptosis quantitatively, the cells after treatment were collected and rinsed with cold PBS twice, followed by re-suspension in binding buffer (200 μL). Thereafter, annexinV-FITC (5 μL) and PI (10 μL) were added and stained in the dark for 15 min based on the standard protocol. To analyze the cell cycle, the collected carcinomas were fixed with 70% pre-cooled alcohol at 4 °C for 12 h, incubated with 100 μg mL−1 of RNase A for 20 min and stained by PI solution (50 μg mL−1) for 30 min in the dark. Finally, both cell apoptosis and cell cycle were measured using a flow cytometer (BD FACSCalibur, NJ).2.13. Animals
Female, 5 week-old BALB/c mice (16–18 g) were obtained from Lingchang Biotechnology Co., Ltd (Shanghai, China). According to AAALAC guidelines, the mice were housed under a pathogen-free environment and the study protocols on animal-related tests were approved by the Institutional Animal Care and Use Committee of Southeast University.2.14. Pharmacokinetic analysis
Female BALB/c mice weighting 16–18 g were assigned to the Taxol-like and PTX-loaded crosslinked micelles groups (n = 3). All the mice received an intravenous injection of PTX-loaded crosslinked micelles or Taxol-like formulation with an equivalent dose of 5 mg PTX per kg through their tail vein. At specified intervals, the plasma was acquired from the blood samples after centrifugation at 3000 rpm for 15 min and 4 °C. Subsequently, 100 μL of plasma sample was added into 1 mL of methanol. Methanol was used for both protein precipitation and extraction of PTX. After centrifugation, the supernatant was obtained and aliquots of 20 μL of the supernatant were subjected to analysis using an RP-HPLC system. The amount of PTX was acquired from the standard curves of previously analyzed of blood samples containing known amounts of free PTX. The pharmacokinetic parameters were calculated using the PKSolver program available in Microsoft Excel (China Pharmaceutical University, Nanjing).2.15. In vivo anticancer activity
Female BALB/c mice with a weight of 16–18 g were tumorized in the right armpit region subcutaneously with 4T1 mammary carcinoma cells at 1 × 107 viable cells (0.1 mL). After the tumor sizes grew to the appropriate size of 80–100 mm3, the mice (n = 3) were randomly grouped into Taxol-like and PTX-loaded crosslinked micelles. The tested mice were administered i.v. injection of Taxol-like formulation and PTX-loaded micelles through their tail vein at a PTX dosage of 10 mg kg−1 every two days, while a normal saline-treated group was employed as a negative control. The tumor volume and weight of each mouse was recorded for a period of 21 days. After treatment, the 4T1 tumor-bearing mice were sacrificed, and their tumors extirpated and weighed, followed by fixing in 4% paraformaldehyde. The tumor inhibition ratio (IR%) was determined according to the following equation:where Wsaline represents the mean tumor weight of the control (saline group) and Wtested is that of the drug-administered group.
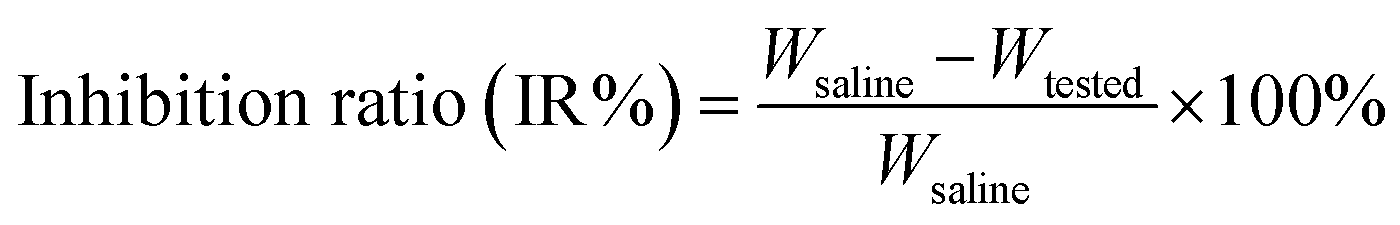
For histological analyses, the tumors were embedded in paraffin. The sliced tumor with a thickness of 4 mm was affixed on a glass slide and double stained using hematoxylin and eosin (H&E), followed by observation using light microscopy.
2.16. Statistical analysis
Numerical results are presented as mean ± SD. The comparison between different groups was analyzed via the Student's t test using the SPSS software (version 17.0), and p less than 0.05 was regarded as statistically significant.3. Results and discussion
3.1. Synthesis of di-LA-PC conjugate
The preparation of the di-LA-PC conjugate was carried out via a facile approach through covalently attaching lipoic acid (LA) to glycerophosphorylcholine (PC), as illustrated in Scheme 2. Initially, compound 2 was prepared via the base-catalyzed hydrolysis of ε-caprolactone, followed by the step-wise protection of the C6 hydroxy group by tert-butyl diphenyl chlorosilane (TBDPS). Esterification of the glycerophosphorylcholine with compound 2 using CDI/DBU as the catalyst generated the corresponding intermediate of di-TBS-PC (71% overall yield). The chemical structure of the di-TBS-PC conjugate was determined via MS and 1H NMR spectroscopy (see ESI, Fig. S5 and S6,† respectively). In the next step, the intermediate di-TBS-PC was deprotected from the TBDPS group in TBAF solution and lipoic acid was added in the presence of the DCC and DMAP catalytic system, affording the di-LA-PC conjugate. RP-HPLC and MS were used to determine the purity and exact mass of the product di-LA-PC conjugate, respectively.As revealed in Fig. 1A (inset), the purity of the conjugate was 98.6% with a retention time of 5.85 min. The MS data showed the molecular iron peaks of di-LA-PC at 862.3 (m/z, [M + H]+) and 884.3 (m/z, [M + Na]+), which are consistent with the calculated value of 861.3 for C60H68NO26P (Fig. 1A). Analysis of the di-LA-PC conjugate via 500 MHz 1H NMR spectroscopy (Fig. 1B) confirmed that the methylene protons in the dithiolane appeared at 1.98 ppm and 2.52 ppm, and the prominent typical singlet at δ 3.31 ppm is attributed to the methyl protons in the –N+(CH3)3 group from the PC moiety, thereby powerfully indicating the coupling of LA and PC. In addition, the new carbon signal at 53.50 ppm is the typical peak of the –N+(CH3)3 group in the 13C NMR spectrum of the di-LA-PC conjugate (Fig. 1C). Thus, all the experimental results successfully confirmed the synthesis of the di-LA-PC conjugate.
 | ||
| Fig. 1 Characterization of di-LA-PC conjugate: (A) mass spectrum, inset: HPLC result of di-LA-PC eluted with 60% CH3OH in H2O comprising 0.1% TFA in 11 min at 5.85 min. (B) 1H NMR and (C) 13C NMR spectra recorded at 500 MHz in CH3OH-d4. | ||
3.2. Formulation and reduction-triggered disruption of crosslinked micelles
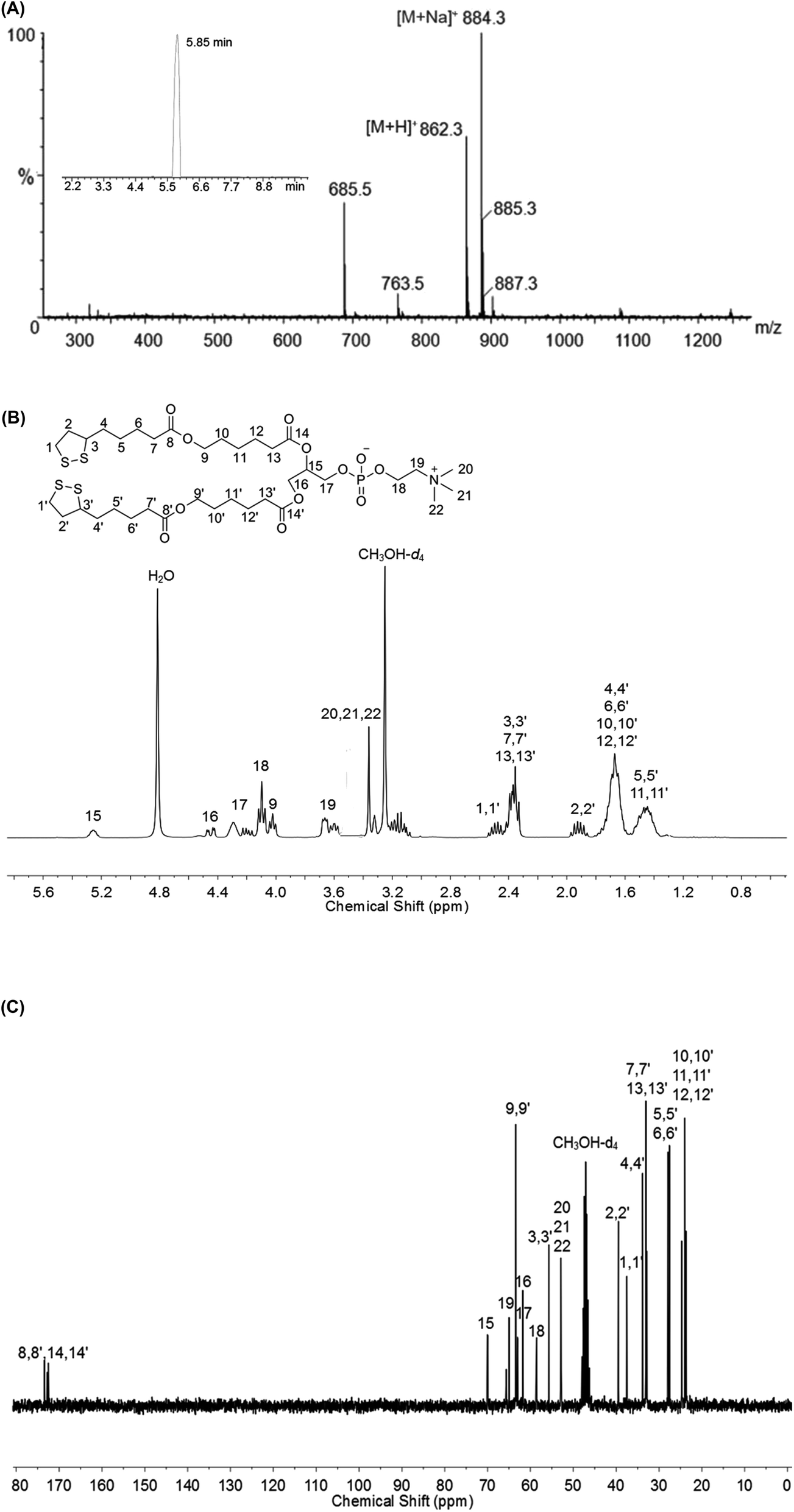
The ability of the di-LA-PC conjugate to assemble into small-molecule micelles was examined by determining its CMC using the pyrene probe method. The I3/I1 value remained unchangeable at a low di-LA-PC concentration and increased dramatically at a certain concentration. According to the junction of the curve, the CMC value was determined to be about 63.09 μg mL−1 (Fig. 2A). This small CMC value for the di-LA-PC micelles indicate their good ability to assemble in aqueous medium.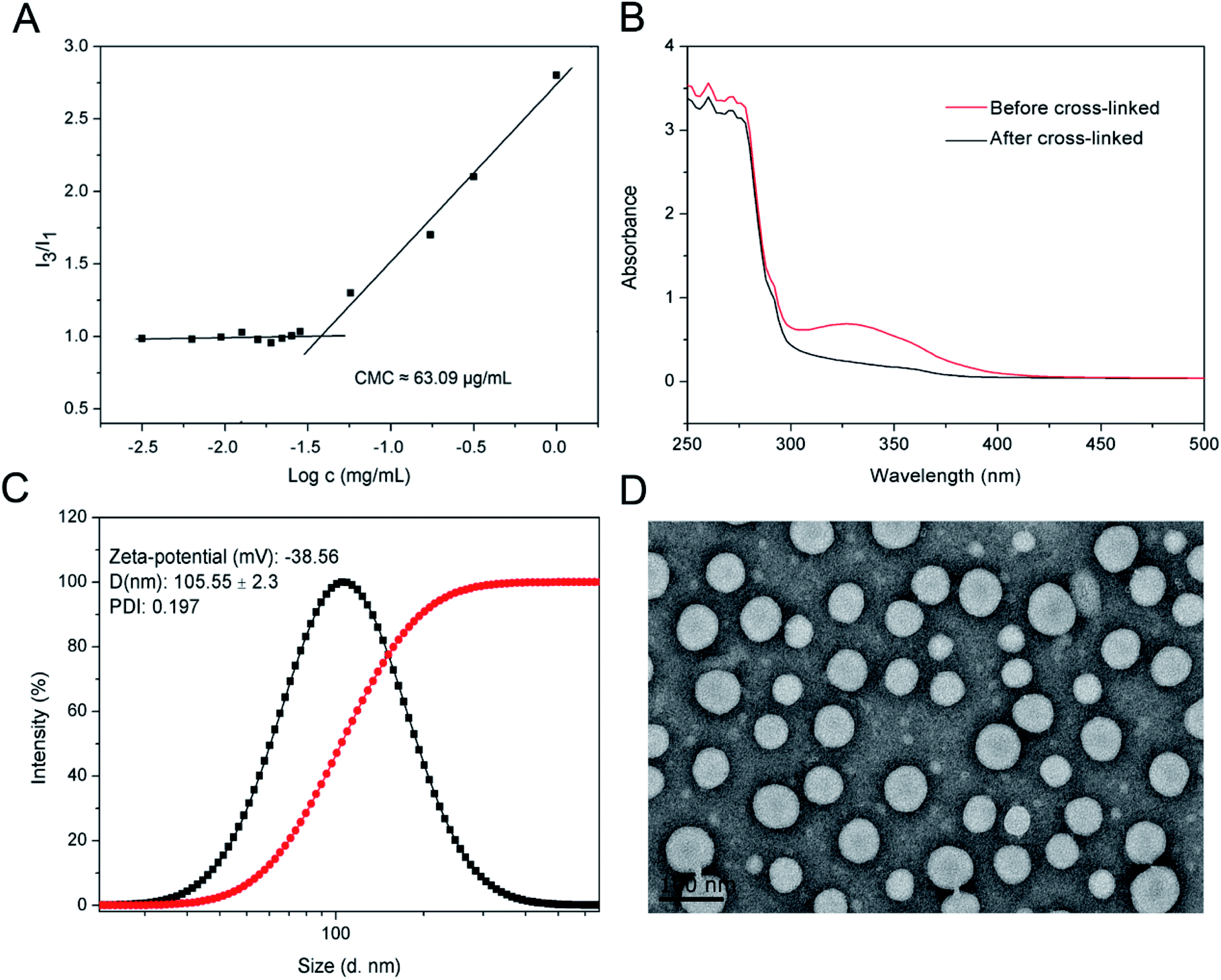 | ||
| Fig. 2 Characterization of crosslinked di-LA-PC micelles: (A) relationship between the fluorescent intensity ratio (I3/I1) and di-LA-PC concentration dispersed in water. The CMC value is about 63.09 μg mL−1. (B) UV spectral change in the micelles after the introduction of 10 mol% DTT and incubation at 37 °C. (C) Size distribution and zeta-potential investigated by DLS and (D) TEM image. | ||
The di-LA-PC conjugates and crosslinked di-LA-PC micelles were further characterized via DLS and TEM. The DLS data exhibited that the di-LA-PC conjugates assembled into nano-sized micelles with a mean hydrodynamic diameter of 122.42 ± 3.6 nm and small polydispersity of 0.148 (Fig. S7†). Further morphological observation by TEM demonstrated the development of spherical vesicles with an identical morphology and size (Fig. S8†). Subsequently, the di-LA-PC micelles were exposed to DTT (10 mol%) relative to the amount of lipoyl units in water to carry out their cross-linking upon incubation for 2 h at 37 °C. Complete polymerization was estimated by the loss of the UV absorbance peak at 330 nm, which is attributed to the characteristic five-membered cyclic disulfide ring (Fig. 2B). The di-LA-PC micelles following crosslinking process had a slightly reduced size (105.55 ± 2.3 nm) and a narrow distribution (PDI: 0.197). The obtained crosslinked micelles were negatively charged with a zeta-potential value of −38.56 mV in water, indicating their promising stability (Fig. 2C). In addition, the TEM image revealed the spherical structure of the di-LA-PC conjugate assembled micelles having a mean diameter of 110 nm close to that monitored by DLS (Fig. 2D).
The crosslinked di-LA-PC micelles treated with 100-fold dilution were observed to exhibit no significant change in hydrodynamic diameter, while the non-crosslinked micelles were monitored with nearly no particular particle size under identical conditions, suggesting the enhanced stability of the crosslinked micelles (Fig. 3A). Moreover, the addition of 2 M NaCl solution induced severe aggregation in the non-crosslinked micelles with a size increase of up to ∼1000 nm, while it had no significant influence on the size of the crosslinked micelles (Fig. 3B). Apparently, covalent crosslinking effectively inhibited the dissociation of the di-LA-PC micelles. After incubation under simulated physiological conditions (PBS, pH 7.4 with 10% FBS) at 37 °C for 48 h, the micelles displayed no significant variation in hydrodynamic diameter. Thus, the combined results demonstrate that the di-LA-PC crosslinked micelles possess excellent colloidal stability in vitro. After treatment with 10 mM DTT at 37 °C for 2 h, the diameter of the crosslinked di-LA-PC micelles quickly decreased (less than 1 nm), which showed that the crosslinked di-LA-PC micelles completely disintegrated into unimers (Fig. 3C).
 | ||
| Fig. 3 Stability and release profiles of small-molecule micelles: (A) in vitro colloidal stability of di-LA-PC micelles after crosslinking versus 100× dilution and (B) 2 M salt condition, where the non-crosslinked micelles were applied as the control. (C) Size changes in the crosslinked di-LA-PC micelles under 10 mM DTT in water at 37 °C. Data is presented as mean ± standard deviation of three independent experiments (n = 3). (D) Characterization of PTX-loaded crosslinked di-LA-PC micelles. (E) In vitro PTX release of crosslinked di-LA-PC micelles in PBS with or without DTT. | ||
3.3. Loading and in vitro release of PTX
Loading of PTX into the non-crosslinked di-LA-PC micelles was performed via a solvent exchange method, and subsequently crosslinking with 10 mol% DTT. The average size and drug loading efficiency (DLE%) of the crosslinked micelles were enhanced with an increase in the initial feed molar ratio of PTX (2:100, 5:100, 10:100, and 20:100). The mean size of the PTX-loaded crosslinked micelles ranged from 137.02 ± 6.67 nm to 168.97 ± 3.12 nm, which is larger than the blank crosslinked micelles (105.55 ± 2.3 nm) (Fig. 3D). This slight increase in size is probably because of PTX encapsulation in the core of the micelles, where the inert space of these micelles expanded. Moreover, the crosslinked micelles with the initial drug:conjugate ratio of 10:100 exhibited a higher DLE% (88.6%) and favorable DLC% (8.13%).
The in vitro release profiles of the PTX-loaded crosslinked micelles showed that rapid release behavior occurred in the first 20 h both at the concentration of 2 μM and 10 mM DTT, followed by slow release steadily over the next 48 h. Moreover, PTX released at a higher concentration of DTT (10 mM) exhibited a faster release rate than that at the low DTT concentration of 2 mM, while in the absence of DTT, the release of PTX was very slow and only about 20% was released after 68 h incubation in PBS at pH 7.4 (Fig. 3E). This demonstrates that the reduction-sensitive character of the micelles for the release of the drug resulted from the cleavage of the disulphide bond.
3.4. Cellular uptake of di-LA-PC micelles
Near-infrared fluorescence (NIRF) Cy5.5 was utilized to label the crosslinked di-LA-PC micelles and the nucleus was stained by DAPI, which emitted blue fluorescence. As shown in Fig. 4, the cells exhibited definite uptake of the crosslinked micelles within 1 h of incubation at 37 °C and their uptake increased over a period of 4 h. Thus, these results significantly confirm that the time-dependent internalization of these crosslinked micelles will allow the intracellular accumulation of antitumor drugs to enhance the chemotherapeutic efficiency.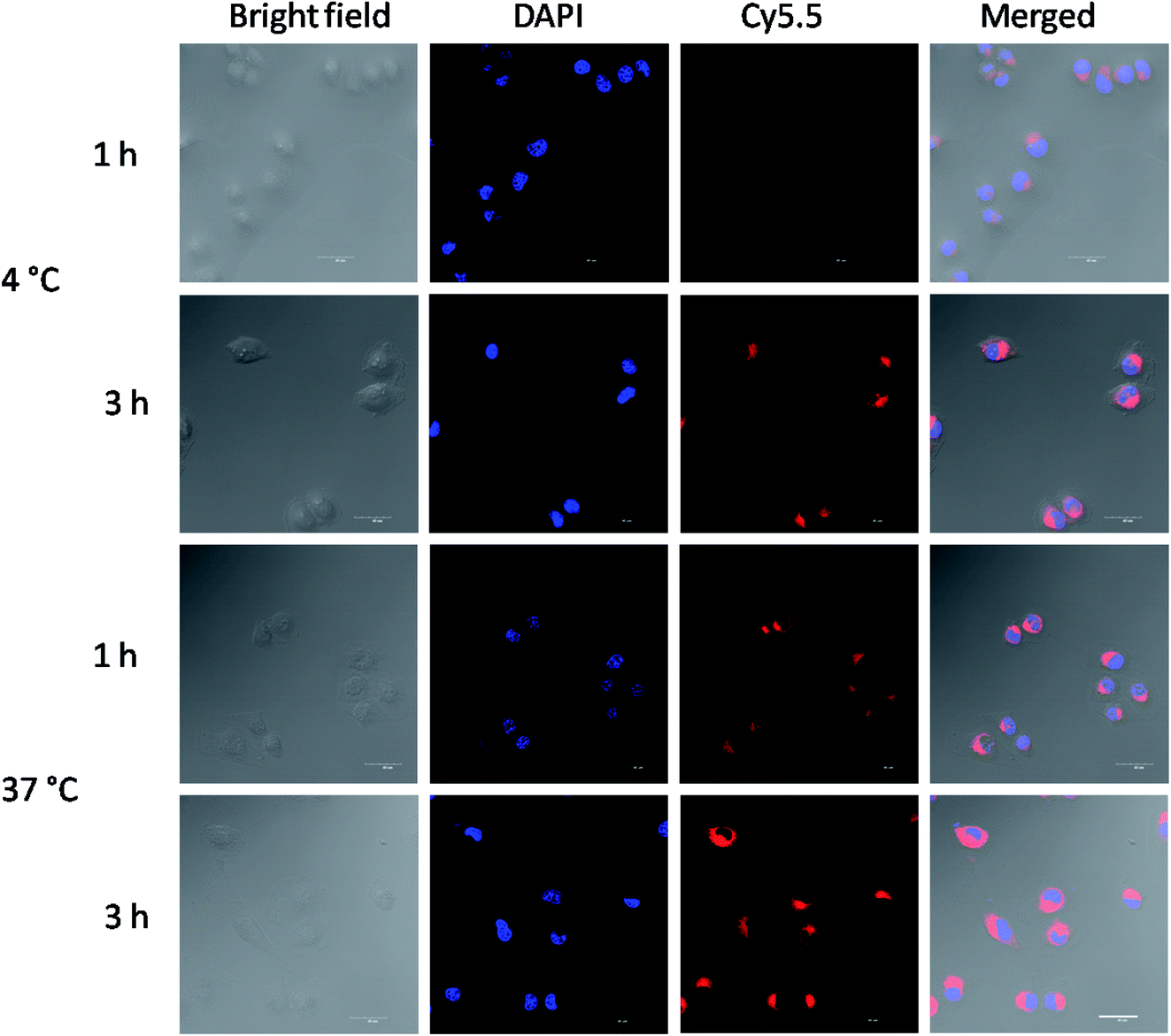 | ||
| Fig. 4 CLSM images of MCF-7 cells cultured with Cy5.5-loaded crosslinked micelles at 4 °C or 37 °C for 1 h and 3 h. Cell nuclei were stained by DAPI (blue). Scale bar: 25 μm. | ||
The cellular uptake mechanism of the Cy5.5-loaded crosslinked micelles against MCF-7 cells was further investigated by counterstaining with LysoTracker Green. The Cy5.5-loaded crosslinked micelles were internalized into the MCF-7 cells (red), while the LysoTracker Green was observed as green fluorescence (Fig. 5). In the merged images, the resulting yellowish color from the co-localization of Cy5.5 red and LysoTracker Green clearly demonstrated the retention of di-LA-PC micelles in the lysosomes. Furthermore, the low fluorescence intensity after 3 h incubation at low temperature (4 °C) compared with the behavior at 37 °C confirmed the internalization of the crosslinked micelles via energy-dependent endocytosis from endosomes towards lysosomes in the MCF-7 cells.36–38
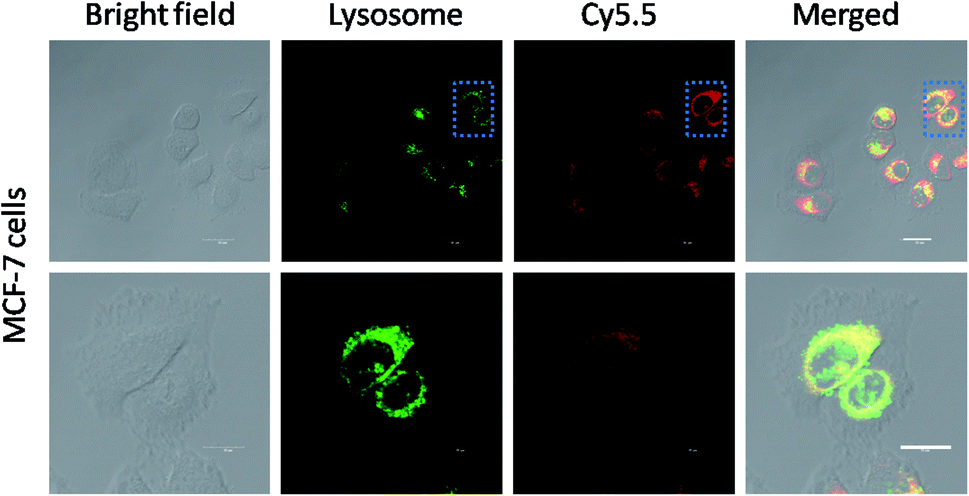 | ||
| Fig. 5 Colocalization of Cy5.5-loaded crosslinked micelles with lysosomes. MCF-7 cells were treated with Cy5.5-loaded micelles for 1 h and then incubated with LysoTracker Green for specific staining of the lysosomes. The white bars represent 25 μm. | ||
3.5. In vitro cytotoxic activity of PTX-loaded crosslinked micelles
The in vitro cytotoxicity of the blank crosslinked and PTX-loaded crosslinked micelles was evaluated versus MCF-7, HepG-2 and A549 cells using the MTT assay. The excellent biocompatibility of the blank crosslinked di-LA-PC micelles as a nanocarrier was proven by their nontoxicity to the MCF-7, HepG-2 and A549 cells (cell viabilities: 92.7–98.1%) at a concentration in the range of 31.25 μg mL−1 to 2 mg mL−1 (Fig. 6A).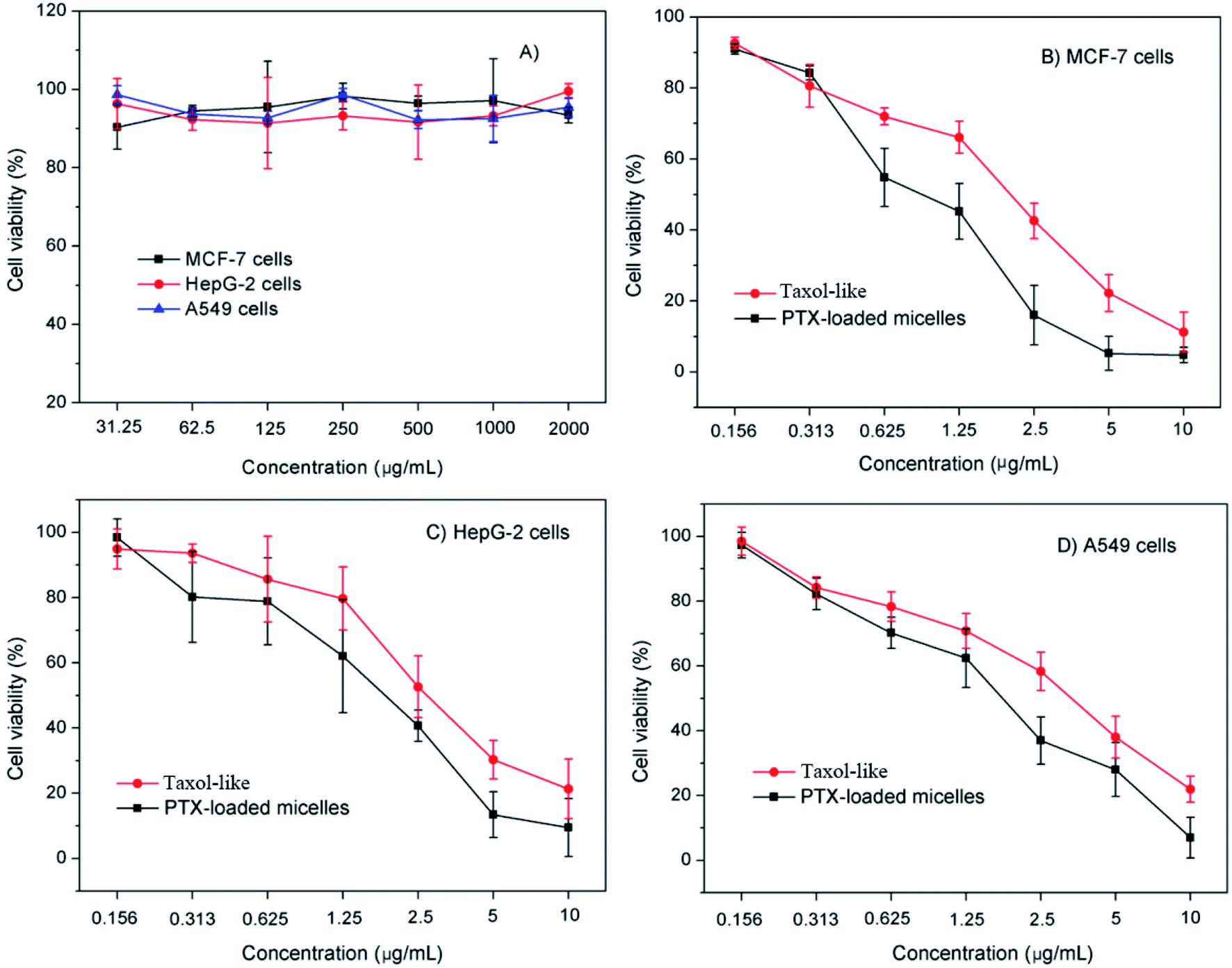 | ||
| Fig. 6 In vitro cytotoxic activities of (A) blank crosslinked micelles and crosslinked micelles loaded with PTX in cell culture of (B) MCF-7, (C) HepG-2 and (D) A549 cells. Error bar represents mean ± standard deviation (n = 4). | ||
The cytotoxicity of the PTX-loaded crosslinked micelles increased in a dose-dependent manner. It is important that the PTX-loaded micelles induced pronounced antitumor effects higher than that of their Taxol-like counterparts against the tested tumor cell lines (Fig. 6B–D). The half maximal inhibitory concentration (IC50) of the MCF-7, HepG-2 and A549 cells subjected to the PTX-loaded micelles was determined to be 1.1 ± 0.08 μg mL−1, 2.1 ± 0.13 μg mL−1, and 1.9 ± 0.42 μg mL−1, which was significantly lower than that of the Taxol-like group (1.8 ± 0.26 μg mL−1, 3.3 ± 0.04 μg mL−1 and 3.5 ± 0.07 μg mL−1 for MCF-7, HepG-2 and A549 cells), respectively.
3.6. Cell apoptosis and cell cycle assay
The annexin V-FITC/PI apoptosis detection assay was applied to measure the ratio of apoptotic cells. MCF-7 carcinomas were treated with the Taxol-like and PTX-loaded crosslinked micelles utilizing an equivalent concentration of 5 μg mL−1 for 24 h incubation and exposed to annexin V-FITC/PI staining, and the cell lines without any treatments were used as the negative control. As displayed in Fig. 7A, early and late apoptotic cells appeared in Q3 and Q2, respectively, and the normal cells were located in Q4. After 24 h incubation, the ratio of apoptosis (early and late apoptosis) from PTX-loaded micelles treatment was 18.11%, which is higher than that with the Taxol-like micelles (13.26%). In both the cytotoxicity and apoptosis assays, the PTX-loaded crosslinked di-LA-PC micelles displayed the best antitumor activity. The data significantly verified that the PTX-loaded crosslinked di-LA-PC micelles can enhance PTX-induced apoptosis compared with Taxol-like at the same concentration.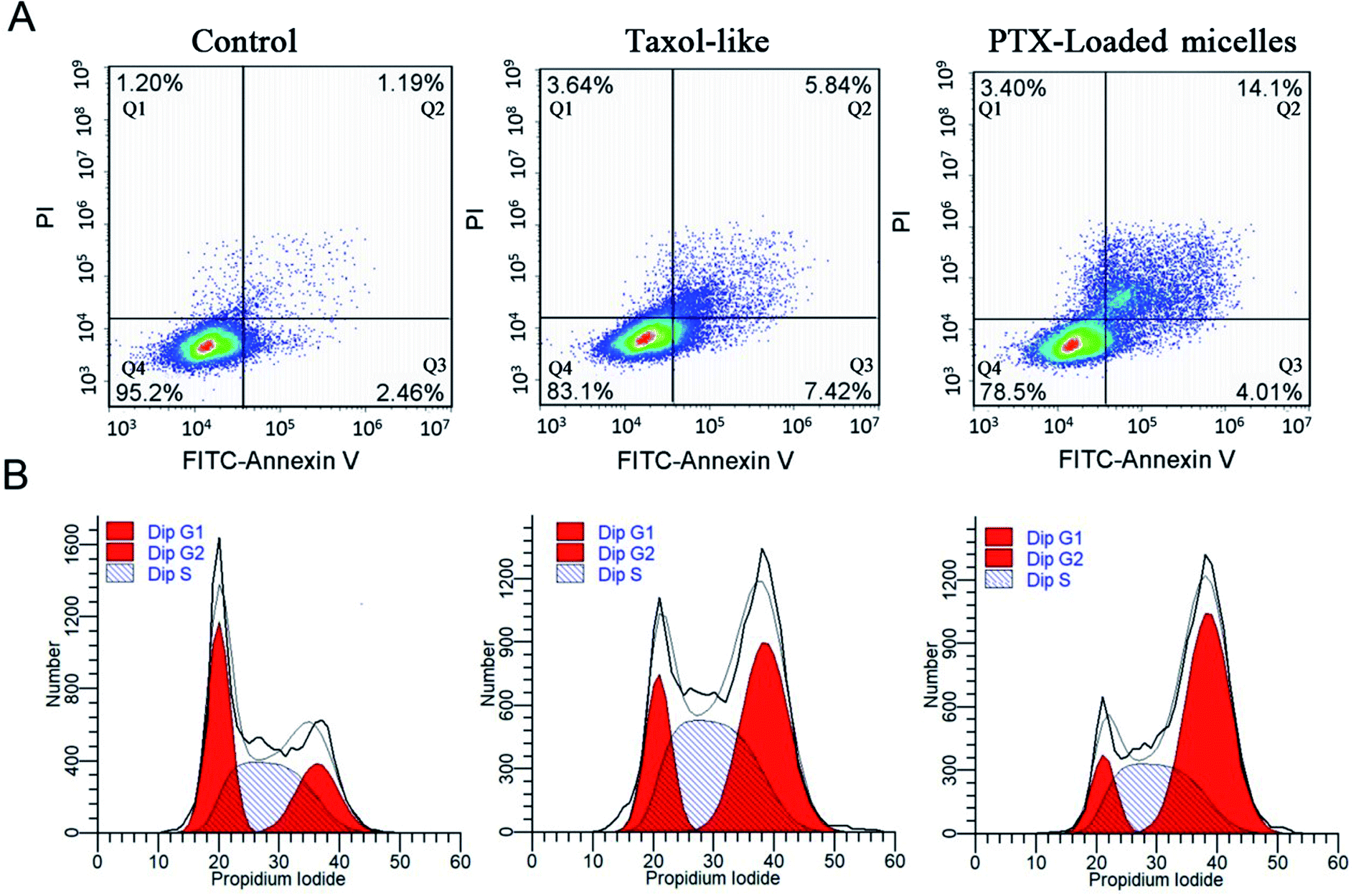 | ||
| Fig. 7 Flow cytometry analysis for apoptosis (A) and cell cycle (B) against MCF-7 cells induced by Taxol-like and PTX-loaded crosslinked micelles at the same concentration of 5 μg mL−1 for 24 h. The untreated cells were used as a control. | ||
Cell cycle arrest was examined using the PI/RNase staining buffer detection kit. MCF-7 cells were treated with PTX-loaded crosslinked micelles and Taxol-like micelles at the equivalent dose of 10 μg mL−1 for 24 h, and subsequently stained with PI to evaluate their DNA content by flow cytometry. The percentage of MCF-7 cells in the G2/M phase when treated with the Taxol-like and PTX-loaded crosslinked micelles underwent a significant increase, while most of the non-treated cells were distributed in the G1 and S phases (Fig. 7B). The G2/M phase treated with PTX-loaded crosslinked micelles for 24 h significantly increased to 56.4% ± 2.35% compared with that of the Taxol-like micelles (45.7% ± 1.78%), demonstrating their strong antitumor efficacy. PTX can induce cell cycle block in the G2/M phase, and thereby increase cell apoptosis because PTX can promote intracellular tubulin polymerization. The Taxol-like micelles retained the characteristic of PTX and blocked the cell cycle in the G2/M phase. It is worth noting that the PTX-loaded crosslinked micelles further blocked the cell cycle in the G2/M phase, which means that they further inhibited mitosis and promoted apoptosis by enhancing the polymerization of tubulin. Therefore, the apoptosis rate of MCF-7 treated with the PTX-loaded crosslinked micelles was higher than that treated with the Taxol-like micelles. These results are in accordance with the apoptosis analysis.
3.7. Pharmacokinetics analysis
The in vivo pharmacokinetics of the PTX-loaded crosslinked micelles vs. Taxol-like micelles was studied in female BALB/c mice after i.v. administration at a dosage of 5 mg PTX per kg. Significantly, the developed PTX-loaded crosslinked micelles underwent a longer circulation time in the blood compared to that of the Taxol-like micelles (Fig. 8). The corresponding pharmacokinetic parameters were calculated using the PKSolver software (China Pharmaceutical University, Nanjing) following non-compartmental models and listed in Table 1. The overall of AUC0–∞ of PTX loaded in the crosslinked micelles was 1.27-fold higher than that of the Taxol-like group, while the CL of the PTX-loaded micelles (0.010 L h−1 kg−1) was significantly lower compared with the Taxol-like micelles (0.032 L h−1 kg−1). Accordingly, the elimination half-life (t1/2) and mean residence time (MRT0–inf) demonstrated the prolonged blood circulation with values of 11.296 h and 1.634 h for the PTX-loaded micelles, which are 1.228-fold and 1.623-fold higher than that of the Taxol-like micelles, respectively. This result is likely ascribed to the nanoscale characteristics of PTX loaded in the crosslinked micelles, which are beneficial to avoid rapid uptake by the reticular endothelial system (RES), ultimately facilitating the accumulation of the drug in tumor tissues.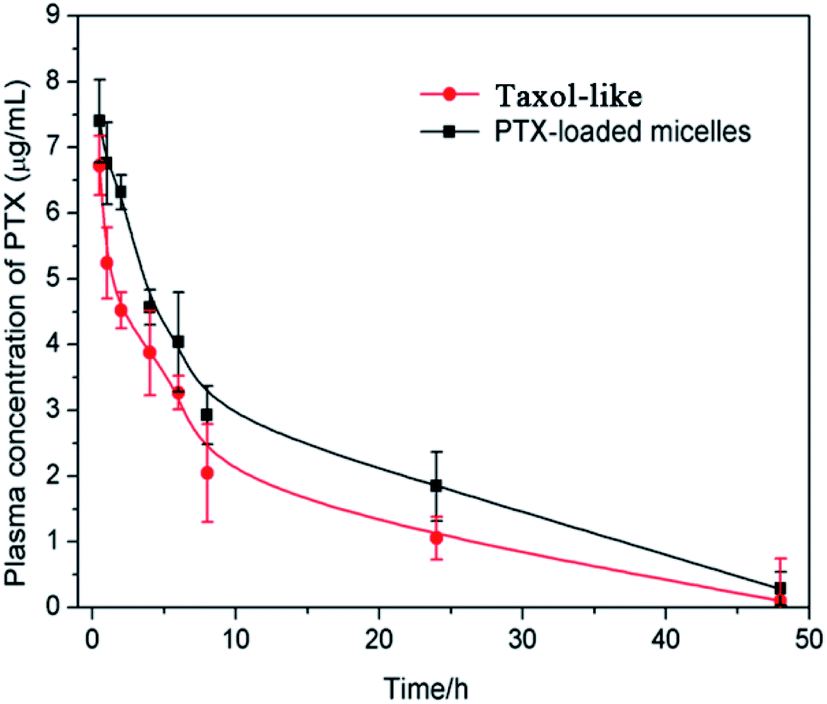 | ||
| Fig. 8 Plasma concentration-time profiles of Taxol-like and PTX-loaded crosslinked micelles after i.v. administration in BALB/c mice at the dosage of 5 mg PTX per kg. Each group contains three mice, and the data is shown as average ± standard error (n = 3). | ||
| Parametersa | Unit | Taxol-like micelles | PTX-loaded micelles |
|---|---|---|---|
| a Parameters: AUC, area under the curve; MRT, mean residence time; t1/2, elimination half-life; Vd, apparent volume of distribution; CL, plasma clearance; and Cmax, peak plasma concentration. | |||
| AUC0–t | mg L−1 h−1 | 77.469 | 96.073 |
| AUC0–∞ | mg L−1 h−1 | 79.048 | 100.685 |
| MRT0–inf | h | 10.323 | 16.634 |
| t1/2 | h | 9.202 | 11.296 |
| CL | L h−1 kg−1 | 0.032 | 0.010 |
| Vd | L kg−1 | 0.017 | 0.016 |
| Tmax | h | 0.5 | 0.5 |
| Cmax | mg L−1 | 6.724 | 7.402 |
| C0 | mg L−1 | 8.109 | 8.628 |
3.8. In vivo antitumor activity evaluation
In vivo antitumor activity was evaluated in xenograft models of 4T1 breast tumor, which were intravenously injected with normal saline as a control, Taxol-like micelles and PTX-loaded micelles via the tail vein. According to Fig. 9A, the relative tumor volume of the formulated PTX-loaded micelles was significantly smaller than that in the mice treated with 0.9% saline (**p < 0.01) and Taxol-like micelles (**p < 0.01), suggesting that the PTX-loaded micelles after disulfide crosslinking showed the strongest tumor growth inhibition among the therapeutic groups. After 21 days treatment, the tumors were harvested, weighed and photographed (Fig. 9C and D, respectively). The tumor inhibitory rate (TIR) for the micellar formulation was calculated from the excised tumor weight and found to be approximately 78.9% ± 3.65%, considerably higher compared with that treated with the Taxol-like micelles (59.0 ± 4.32%, **p < 0.01).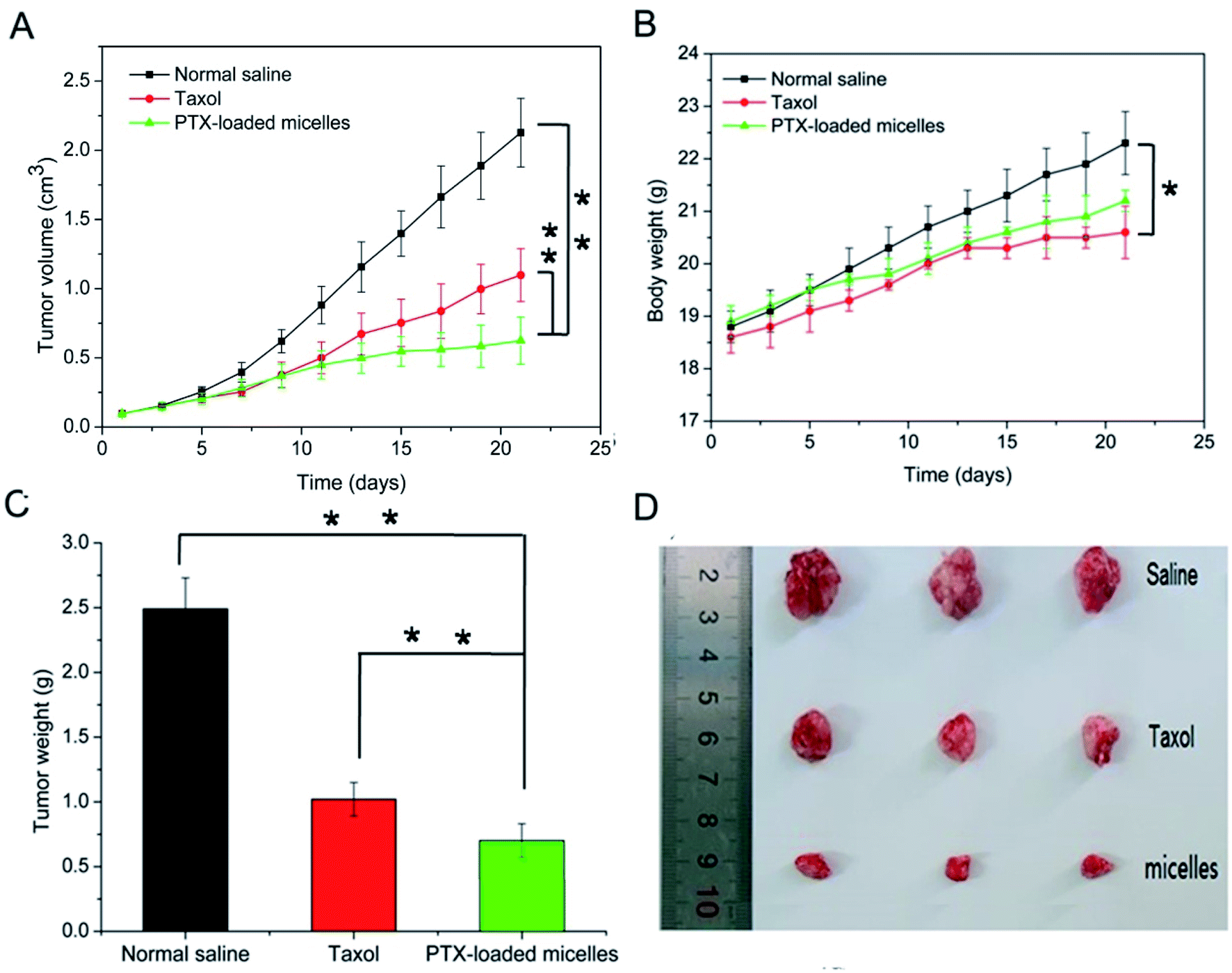 | ||
| Fig. 9 In vivo therapeutic efficacy of Taxol-like and PTX-loaded crosslinked micelles after intravenous administration of the equivalent PTX dose (10 mg kg−1) in 4T1 tumor-bearing BALB/c mice. (A) Tumor volume and (B) body weight changes in the tested mice. (C) Excised tumor weight and (D) photos of the tumor tissues. The data is presented as (mean ± SD, n = 3), where * represents p < 0.05 and **p < 0.01. | ||
Fig. 9B reveals that no significant decrease in body weight in the mice occurred due to the prepared PTX-loaded micelles in the experimental period, confirming the low systemic toxicity or side effects at the dosage used (10 mg kg−1). Finally, histopathological hematoxylin and eosin (H&E) analysis of the tumor tissues was performed to assess the antitumor efficacy between the Taxol-like and PTX-loaded micelles. As shown in Fig. 10, the cancer cells treated with normal saline apparently exhibited the histological characteristics of malignant tumors, such as giant, hyperchromatic nuclei and spindle-shaped cells, proving their violent growth. The arrangement of the tumor cells was still tight, but the cells were edema like in shape and the capillaries between the cells were reduced after treatment with the Taxol-like and PTX-loaded micelles, indicating that the tumor structure was destroyed. Due to the degenerative changes in the cells resulting in cytoplasmic vacuolation, together with lymphocytes, a few cells with acidophilic degeneration of the cytoplasm and karyopyknosis were found. These changes demonstrated cell apoptosis. Compared with the Taxol-like group, more cytoplasmic vacuolation, acidophilic degeneration and karyopyknosis were observed in the PTX-loaded crosslinked micelle group. Thus, these results further demonstrate the enhanced antitumor efficacy of the drug PTX delivered by the developed reduction-responsive crosslinked di-LA-PC micelles in the xenograft 4T1 breast tumor model.
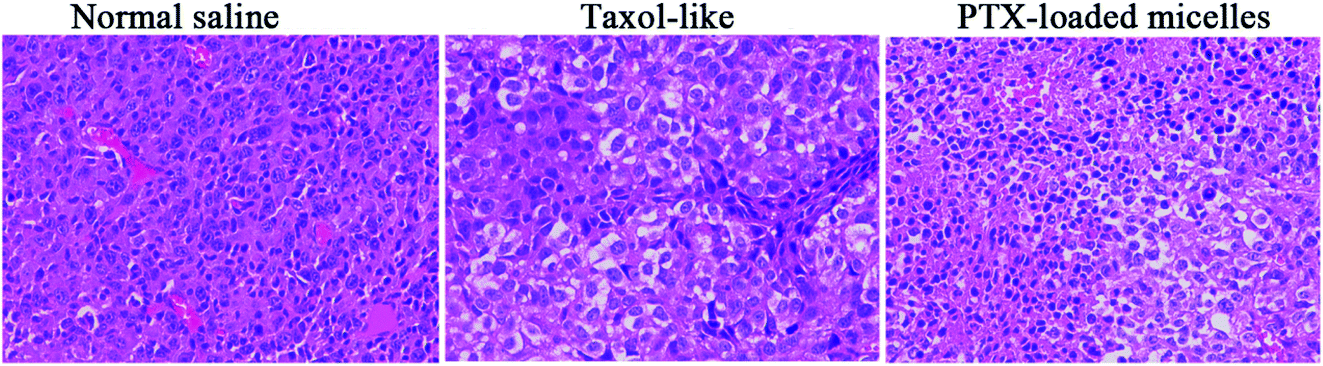 | ||
| Fig. 10 H&E staining images of the excised tumor after intravenous administration of normal saline, Taxol-like and PTX-loaded crosslinked micelles (magnification ×400). | ||
4. Conclusion
In summary, small-molecule di-LA-PC phospholipid-based disulfide crosslinked micelles were developed as a novel PTX delivery system. Due to their features of excellent colloidal stability and rapid micelle disassembly and drug release in response to reductive conditions, the developed crosslinked di-LA-PC micelles could enter tumor cells effectively to induce high in vitro cytotoxicity. Moreover, the nanoscale characteristics of the small-molecule micelles facilitated a longer retention time in blood circulation, which led to the effective accumulation of the incorporated PTX in the tumor sites, thereby exhibiting better in vivo antitumor efficacy than the Taxol-like formulation. Therefore, crosslinked di-LA-PC micelles may provide a versatile platform for the targeted and triggered intracellular release of clinical chemotherapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Major National Science and Technology Program of China for Innovative Drug (2017ZX09101002-001-004); National Natural Science Foundation of China (Project 51373034); and Department of Science & Technology of Jiangsu Province, China (Projects BA2013037 and BY2015070-11).References
- R. Wei, L. Cheng, M. Zheng, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2012, 13, 2429–2438 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- L. Wu, Y. Zou, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomaterials, 2013, 34, 5262–5272 CrossRef CAS PubMed.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2009, 61, 364 CrossRef CAS.
- F. Li, W. L. Chen, B. G. You, Y. Liu, S. Di Yang, Z. Q. Yuan, W. J. Zhu, J. Z. Li, C. X. Qu, Y. J. Zhou, X. F. Zhou, C. Liu and X. N. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 32146–32158 CrossRef CAS PubMed.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- J. Y. Yhee, S. Lee and K. Kim, Nanoscale, 2014, 6, 13383–13390 RSC.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed.
- B. G. Amsden and D. Marecak, Mol. Pharm., 2016, 13, 3004–3012 CrossRef CAS PubMed.
- J. T. Peters, S. S. Hutchinson, N. Lizana, I. Verma and N. A. Peppas, Chem. Eng. J., 2018, 340, 58–65 CrossRef CAS.
- Y. Zhu, J. Zhang, F. Meng, L. Cheng, J. Feijen and Z. Zhong, J. Mater. Chem. B, 2018, 6, 3040–3047 RSC.
- S. Y. Wu, H. Y. Chou, C. H. Yuh, S. L. Mekuria, Y. C. Kao and H. C. Tsai, Adv. Sci., 2018, 5, 170339 Search PubMed.
- L. Zhou, F. Lv, L. Liu, G. Shen, X. Yan, G. C. Bazan and S. Wang, Adv. Mater., 2018, 30, 1–8 Search PubMed.
- A. E. Ekkelenkamp, M. R. Elzes, J. F. J. Engbersen and J. M. J. Paulusse, J. Mater. Chem. B, 2018, 6, 210–235 RSC.
- H. Qiao, Z. Zhu, D. Fang, Y. Sun, C. Kang, L. Di, L. Zhang and Y. Gao, J. Drug Targeting, 2018, 26, 75–85 CrossRef CAS PubMed.
- T. Fuoco, A. Finne-Wistrand and D. Pappalardo, Biomacromolecules, 2016, 17, 1383–1394 CrossRef CAS PubMed.
- C. Iacobucci, C. Piotrowski, A. Rehkamp, C. H. Ihling and A. Sinz, J. Am. Soc. Mass Spectrom., 2018, 139–148 Search PubMed.
- F. F. Sahle, M. Gulfam and T. L. Lowe, Drug Discovery Today, 2018, 23, 992–1006 CrossRef CAS PubMed.
- Y. L. Li, L. Zhu, Z. Liu, R. Cheng, F. Meng, J. H. Cui, S. J. Ji and Z. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914–9918 CrossRef CAS PubMed.
- E. K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed.
- G. Gasparini, G. Sargsyan, E. K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328–7331 CrossRef CAS PubMed.
- A. Tschiche, B. N. S. Thota, F. Neumann, A. Schäfer, N. Ma and R. Haag, Macromol. Biosci., 2016, 811–823 CrossRef CAS PubMed.
- C. Giacomelli, L. Le Men, R. Borsali, J. Lai-Kee-Him, A. Brisson, S. P. Armes and A. L. Lewis, Biomacromolecules, 2006, 7, 817–828 CrossRef CAS PubMed.
- K. Ishihara, Y. Goto, M. Takai, R. Matsuno, Y. Inoue and T. Konno, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 268–275 CrossRef CAS PubMed.
- Q. Jin, Y. Chen, Y. Wang and J. Ji, Colloids Surf., B, 2014, 124, 80–86 CrossRef CAS PubMed.
- Y. Yang, D. Guan, L. Lei, J. Lu, J. Q. Liu, G. Yang, C. Yan, R. Zhai, J. Tian, Y. Bi, F. Fu and H. Wang, Toxicol. Appl. Pharmacol., 2018, 341, 98–105 CrossRef CAS PubMed.
- L. Ling, Y. Du, M. Ismail, R. He, Y. Hou, Z. Fu, Y. Zhang, C. Yao and X. Li, Int. J. Pharm., 2017, 526, 11–22 CrossRef CAS PubMed.
- L. Jia, J. P. Xu, H. Wang and J. Ji, Colloids Surf., B, 2011, 84, 49–54 CrossRef CAS PubMed.
- W. Zong, B. Thingholm, F. Itel, P. S. Schattling, E. Brodszkij, D. Mayer, S. Stenger, K. N. Goldie, X. Han and B. Städler, Langmuir, 2018, 34, 6874–6886 CrossRef CAS PubMed.
- M. Rajabi, M. Lanfranchi, F. Campo and L. Panza, Synth. Commun., 2014, 44, 1149–1154 CrossRef CAS.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed.
- J. C. Cuggino, M. L. Picchio, A. Gugliotta, M. Bürgi, L. I. Ronco, M. Calderón, M. Etcheverrigaray, C. I. Alvarez Igarzabal, R. J. Minari and L. M. Gugliotta, Eur. Polym. J., 2021, 145, 110237 CrossRef CAS.
- F. Gu, C. Hu, Z. Tai, C. Yao, J. Tian, L. Zhang, Q. Xia, C. Gong, Y. Gao and S. Gao, Sci. Rep., 2016, 6, 1–15 CrossRef PubMed.
- Y. Wu, Q. Chu, S. Tan, X. Zhuang, Y. Bao, T. Wu and Z. Zhang, Int. J. Nanomed., 2015, 10, 5219–5235 CAS.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- Y. Li, J. Lin, J. Ma, L. Song, H. Lin, B. Tang, D. Chen, G. Su, S. Ye, X. Zhu, F. Luo and Z. Hou, ACS Appl. Mater. Interfaces, 2017, 9, 34650–34665 CrossRef CAS PubMed.
- A. A. R. Mota, P. H. P. R. Carvalho, B. C. Guido, H. C. B. De Oliveira, T. A. Soares, J. R. Corrêa and B. A. D. Neto, Chem. Sci., 2014, 5, 3995–4003 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00079a |
| This journal is © The Royal Society of Chemistry 2021 |