
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium catalyzed reductive Heck coupling and its application in total synthesis of (−)-17-nor-excelsinidine†
Lisi Yuan,
Linrong Chen,
Xiaoxiao Yan,
Kun Gao* and
Xiaolei Wang *
*
College of Chemistry and Chemical Engineering, State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: wangxiaolei@lzu.edu.cn; npchem@lzu.edu.cn
First published on 17th February 2021
Abstract
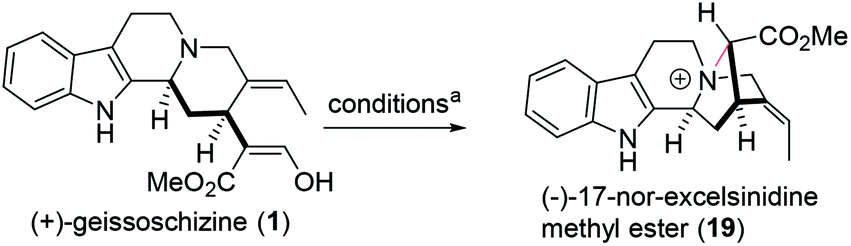
Monoterpene indole alkaloids, bearing a highly substituted piperidine ring, are a structurally diverse class of bioactive natural products, found in various parts of the world. Herein, we reported the construction of the key piperidine ring via palladium catalyzed reductive Heck coupling with a good syn selective manner, avoiding the usage of stoichiometric, highly toxic, air sensitive and moisture sensitive Ni(COD)2. To further showcase the value of this methodology, we realized the total synthesis of the structurally unique zwitterionic monoterpene indole alkaloid (−)-17-nor-excelsinidine in 9 steps, in which the key ammonium–acetate connection (N4–C16) of (−)-17-nor-excelsinidine was constructed via oxidative coupling in excellent yield and high regioselectivity under NBS/pyridine from the enolate of geissoschizine.
Monoterpene indole alkaloids are a structurally diverse class of bioactive natural products found in various parts of the world. Related biological investigations showed that these alkaloids have broad biological activities and medical properties, ranging from the treatment of headaches to that of cancer, pulmonary diseases, and various bacterial and fungal infections.1 Their characteristic biological activities and interesting architectures have stimulated synthetic efforts directed toward the total syntheses of which by many research groups.2
According to biosynthetic hypothesis, many of them can be accessed from geissoschizine (1) through oxidative cyclization with different interconnections.3 Inspired by the biosynthetic hypothesis, several monoterpene indole alkaloids were synthesized via a bioinspired strategy. Baran and coworkers invented an intermolecular oxidative coupling strategy between indole and carbonyl enolates to assemble these natural products.4 Ma and coworkers elaborate the core structure of communesin F via an intramolecular oxidative coupling reaction.5 Using a similar intramolecular oxidative coupling between indole and malonate moieties, Ma's group also achieved the total synthesis of another akuammiline alkaloid aspidophylline A.6 In all of these total synthesis of indole alkaloids, Ma,5,6 Zhu7 and co-workers constructed, at early stages, either the C7–C16 bond over the N1–C16 bond using LiHMDS/I2 oxidative conditions.
So far, most of this interconnection transformation are not completely confirmed via total synthesis study for many reasons, such as the difficulties to control the regioselective oxidation due to the density of functional groups, to adjust the spatial distance of two functional groups, or to maintain the stability of the related products and strong oxidants. For example, Lounasmaa's group did many synthetic study base on “biogenetic-type cyclization” but not getting desired ring system.8 So far, it's still quite difficult to explain these negative results.
Even today, synthetic approach towards these alkaloids via biosynthetic hypothesis is still quite challenging and demanding. In 2018, Vincent's group reported the first total synthesis of (−)-17-nor-excelsinidine via bioinspired oxidative cyclization strategy.9 Due to the tolerance of the indole ring, the desired cyclization products were only obtained in 25% yield. According to reported biosynthetic hypothesis, from the key precursor geissoschizine (1), nature products such as marvacurin (2), strychnos (3), rhazimal (4), meloyine B (5), ajmalicine (6) and (−)-17-nor-excelsinidine (7) might be synthesized via connections of different atoms (Fig. 1). Based on these hypotheses, herein we reported the total synthesis of (−)-17-nor-excelsinidine via palladium catalyzed reductive heck coupling and NBS promoted oxidative cyclization with high overall yield.
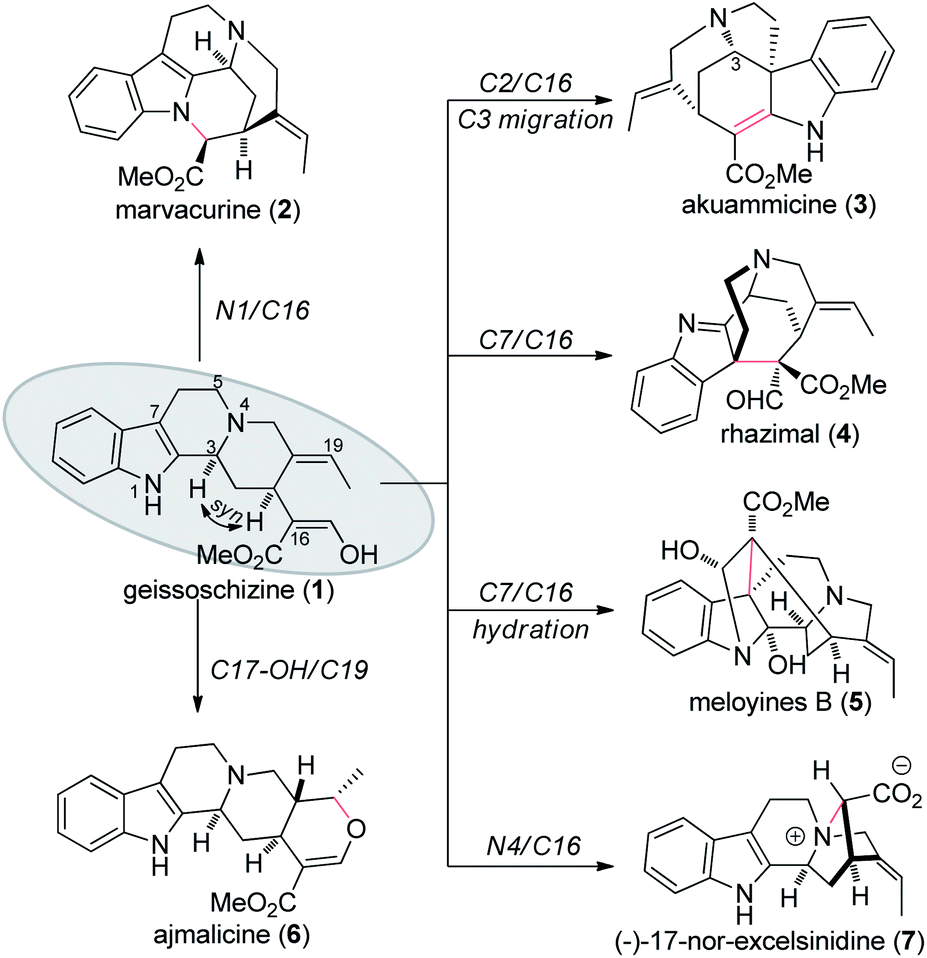 | ||
| Fig. 1 Postulated biosynthetic transformation of selected monoterpene indole alkaloids. | ||
From the skeleton of monoterpene indole alkaloids, most of them bear a highly substituted syn-piperidine ring. However, this key syn-piperidine ring was mainly synthesized through reductive heck coupling by using stoichiometric Ni(COD)2.6,9,10 Due to the highly toxic, air sensitive and moisty sensitive of Ni(COD)2, this type of reactions has to be handled in a glove box, which further limited its application. Yet, efficient preparation of this highly substituted 2,4-syn-piperidine ring through other less toxic, catalytic, air and moisty stable transition metal catalyst with a stereo-control manner remains quite challenging.
As is known to us, palladium-catalyzed reductive Heck coupling involves intercepting the alkylpalladium intermediate generated upon migratory insertion with a hydride source. This transformation has been investigated since the early 1980s, and pioneering work by Cacchi11 and others during that period led to effective strategies with several classes of C–C–π-bond-containing substrates that lack β-H atoms or that form stabilized π-allyl/π-benzyl/enolate intermediates.12 In contrast, application of this mode of reactivity to alkenes is comparatively underdeveloped, likely due to the rapid velocity of the aforementioned β-H elimination step with such substrates.
To circumvent this issue and test the validity of this proposal, the precursors for reductive Heck coupling was synthesized via Mannich addition and N-alkylation as shown in Scheme 1 as the known protocol. Iodo (±)-1210c was smoothly generated from imine 813a and enolate 9.
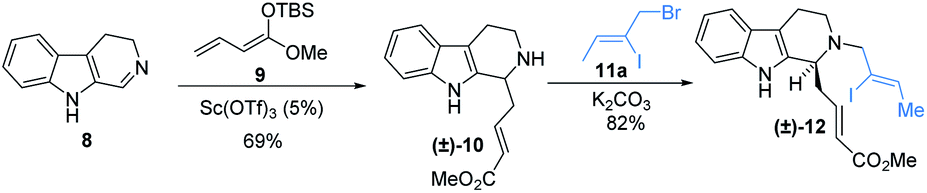 | ||
| Scheme 1 Synthesis of the starting materials for reductive Heck-coupling. | ||
With iodo (±)-12 in hand, we chose palladium as the transition metal source, which was frequently used for reductive heck coupling.12 After extension study of palladium systems, we can obtain the desired syn- (±)-13 in 10% yield with other inseparable anti and elimination mixture while using Pd(OAc)2 (0.01 equiv.) as catalyst, HCO2Na (5.0 equiv.) as the hydride source and n-Bu4NCl as the phase transfer reagent additive in DMF. The yield was considerably diminished under other reductants, additives or different palladium catalysts (entries 2–9). To our delight, the reaction yield was significant boosted while using halogen additive and increase the equivalent of HCO2Na (entries 10–13). After examination of different equivalents of the hydride sources and additives, we finally got the desired syn-(±)-1314 in 56% yield in 0.1 mmol scale of (±)-12. By comparing with previous work,15 the intermediate 13 was generated in 53% yield while using 3.0 equivalent of Ni(COD)2. To further test the application of this reductive Heck coupling, gram-scale synthesis of syn-(±)-13 was then achieved with an acceptable yield (Table 1).
|
||||
|---|---|---|---|---|
| Entry | Catalysts | Reductants | Additives | Yieldb |
| a Unless otherwise noted, the reaction of (±)-12 (0.1 mmol, 1.0 equiv.) was carried out using a catalytic of palladium (0.1 equiv.) under Ar atmosphere in the presence of reductant and an additive in DMF (2.0 mL) at 40 °C for 12 h.b Isolated yields.c HCO2Na (5.0 equiv.), n-Bu4NCl (7.5 equiv.).d HCO2Na (10.0 equiv.), n-Bu4NCl (15.0 equiv.) and LiCl (5.0 equiv.).e HCO2Na (10.0 equiv.), n-Bu4NCl (15.0 equiv.) and LiBr (5.0 equiv.).f HCO2Na (15.0 equiv.), n-Bu4NCl (22.5 equiv.) and LiBr (5.0 equiv.).g HCO2Na (15.0 equiv.), n-Bu4NCl (22.5 equiv.).h Gram-scale: (±)-12 (3.16 mmol, 1.42 g), 45% yield. DMF: N,N′-dimethylformamide; DIPEA: N,N-diisopropylethylamine; PMP: 1,2,2,6,6-pentamethylpiperidine. | ||||
| 1c | Pd(OAc)2 | HCO2Na | n-Bu4NCI | 10% |
| 2 | Pd2(dba)3CHCI3 | HCO2Na | — | 0 |
| 3 | Pd(OAc)2/PPh3 | HCO2H | Et3N | 0 |
| 4 | Pd(OAc)2/PPh3 | HCO2H | DIPEA | 0 |
| 5 | Pd(OAc)2/PPh3 | HCO2Na | — | 0 |
| 6 | Pd2(dba)3CHCI3 | HCO2H | DIPEA | 0 |
| 7 | Pd2(dba)3CHCI3/DPPE | HCO2H | DIPEA | 0 |
| 8 | Pd(OAc)2 | HCO2H | Et3N | 0 |
| 9 | Pd(OAc)2 | HCO2H | PMP | 0 |
| 10d | Pd(OAc)2 | HCO2Na | nBu4NCI/LiCI | 47% |
| 11e | Pd(OAc)2 | HCO2Na | n-Bu4NCI/LiBr | 55% |
| 12f | Pd(OAc)2 | HCO2Na | n-Bu4NCI/LiBr | 51% |
| 13g,h | Pd(OAc)2 | HCO2Na | n-Bu4NCI/LiBr | 56% |

Encouraged by this result, the scope with respect to the configuration and substitution of alkene was evaluated, as shown in Scheme 2, iodo 16a10c was smoothly generated from imine 1413b and enolate 9 via Mannich addition and N-alkylation. 16b and 16c bearing different configurations of alkene and substitutions were also obtained via a similar synthetic route. We found that both Z and E configuration are suitable in this reaction system to obtain 17a and 17b with acceptable yield. Other substitution of alkene is also well-tolerated in this reaction, giving desire syn-17c in 38% yield. Benzyloxycarbonyl substituent at C5 position increased the yield of reductive products in gram-scale. Therefore, starting from enantiopure tryptophan instead of tryptamine would allow us to perform an asymmetric synthesis of this series of natural products. Thus more than two-gram of ester 17a was prepared as the protocol in Scheme 2.
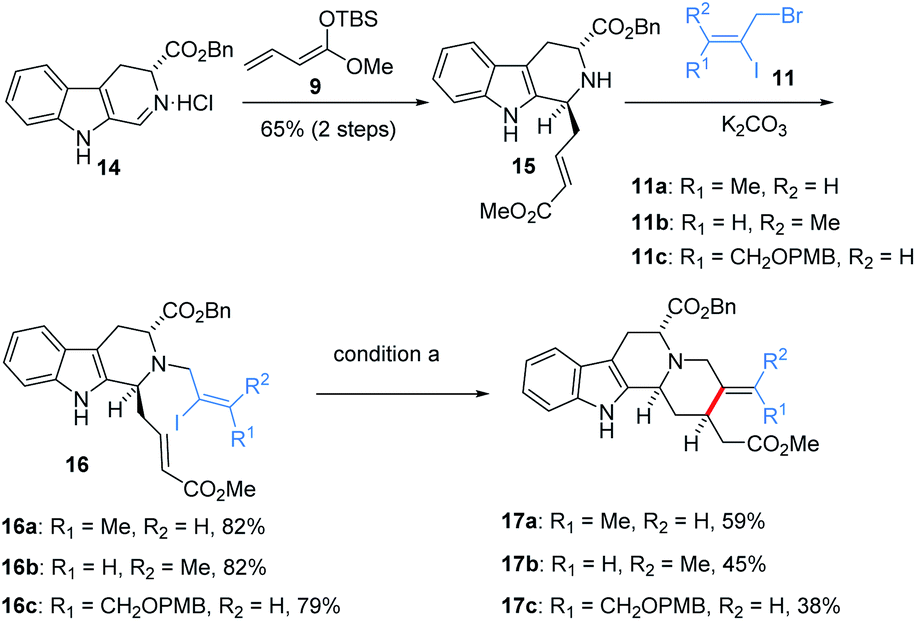 | ||
| Scheme 2 Substrate synthesis and the scope of reductive heck coupling. | ||
Subsequently, the benzyl ester was removed in a classical sequence,10c namely debenzylation into acid 18, then comes the formation of phenylselenoester, and decarboxylation under reductive radical conditions, to yield enantiopure 13. After the formylation of 13 under LDA/HCO2Me, geissoschizine (1) was synthesized as expected in seven steps in the longest linear sequence (Scheme 3).
 | ||
| Scheme 3 The synthesis of (−)-17-nor-excelsinidine via NBS oxidation. | ||
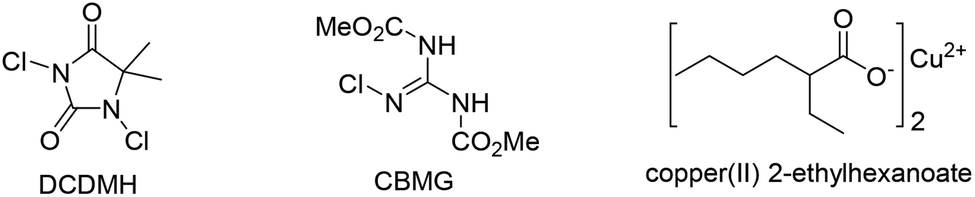
With geissoschizine (1) in hand and inspired by biosynthetic hypothesis as highlighted in Fig. 1, we investigated the potential transformation to yield natural products via different atom connections with C16. As shown in Fig. 1, ajmalicine (6) can be obtained through hydroxyl 1,4-addition to the imine intermediate. 17-nor-Excelsinidine can be accessed via carbon selective addition to the in situ generated ammonia salt and other types of monoterpene alkaloids via different connections. Initially, we use LiHMDS/I2 as oxidative conditions to forehead the synthesis of 17-nor-excelsinidine (entry 1). To our disappointment, less than 10% of 17-nor-excelsinidine was obtained with other inseparable decomposed mixture through this reaction system. Inspired by Baran's work, we screened other oxidants, such as Cu(II), Fe(III) and other organic oxidants as shown in Table 2 (entries 2–4). These oxidative condition only afforded a very complex mixture. The main reason might come from the intolerance of geissoschizine (1) under these oxidation conditions. The skeleton of natural product might be obtained by introducing acidic environment to reduce the activity of tertiary amine. With this propose in mind, we tested other acidic oxidation system (entries 5–10). We can only recovery the starting material while using Mn(OAc)3 system. While using t-BuOCl, DCDMH, CBMG16a and other similar oxidants,16b the starting material disappeared very quickly, yielding a complex reaction mixture. As photoredox oxidation catalysis17 system usually have a good functional group tolerance, we wondered whether the imine intermediate might be generated under this photoredox system. However, in our reaction system, the starting material was totally decomposed via previous conditions. Without getting positive results, we further screened basic oxidative system, and to our delight, 17-nor-excelsinidine precursor (19) was generated smoothly under NBS/pyridine system in 91% yield. The high yield is likely due to complete conversion and the region-selectivity of this coupling resulted from the inherent nucleophilicity of the lone pair electrons of the N-4 position. Methyl ester 19 was then saponified to afford (−)-17-nor-excelsinidine in 73% yield. The analytical and spectral data of synthetic (−)-17-nor-excelsinidine was in good agreement with previously reported.
|
||
|---|---|---|
| Entry | Conditions | Yield (19)b |
| a Unless otherwise noted, geissoschizine (1) (5.0 mg, 0.014 mmol, 1.0 equiv.) was used for optimization of the oxidative coupling.b Isolated yields.c RBr: dimethyl 2-bromomalonate.d Geissoschizine (1) (20.0 mg, 0.057 mmol, 1.0 equiv.) was used. | ||
| 1 | LiHMDS (2.2 equiv.), I2 (1.1 equiv.) THF, −78 °C | <10% (19) |
| 2 | LiHMDS (2.2 equiv.), Cu(II) (2.2 equiv.) THF, −78 °C | Trace (19) |
| 3 | LiHMDS (2.2 equiv.), Fe(acac)3 (2.2 equiv.) THF, −78 °C | Trace (19) |
| 4 | K2CO3 (20 equiv.), NBS (1.0 equiv.) THF/H2O, rt | <10% (19) |
| 5 | Mn(OAc)3 (2.0 equiv.) CH3CO2H, 60 °C | NR |
| 6 | TFA (1.1 equiv.), t-BuOCI (1.4 equiv.) CHCI3, 0 °C-rt | Decomp. |
| 7 | TfNH2 (1.0 equiv.), t-BuOCI (2.0 equiv.) CH2CI2, 0 °C-rt | Decomp. |
| 8 | TFA (1.1 equiv.), DCDMH (1.2 equiv.) CHCI3, 0 °C-rt | Decomp. |
| 9 | TFA (1.1 equiv.), CBMG (1.2 equiv.) CHCI3, 0 °C-rt | Decomp. |
| 10 | TFA (1.5 equiv.), DMDO (1.2 equiv.) CH2CI2, 0 °C | Decomp. |
| 11c | Ir(ppy)3 (0.05 equiv.), RBr (3 equiv.), hν, DMF, rt | Decomp. |
| 12 | Ru(bpy)3CI2 (0.05 equiv.), O2, hν, MeCN, rt | Decomp. |
| 13d | NBS (1.0 equiv.), pyridine/CH2CI2 (1/1), −40 °C | 91% |
 |
||
Conclusions
In conclusion, we successfully constructed the key piperidine ring via a palladium catalyzed reductive Heck coupling with a good syn selective manner, avoiding the usage of stoichiometric, highly toxic, air and moisty sensitive Ni(COD)2. From the key intermediate, we further built the key ammonium–acetate connection (N4–C16) of (−)-17-nor-excelsinidine via oxidative coupling in excellent yield and high regioselective under NBS/pyridine from the enolate of geissoschizine. Finally, racemic 17-nor-excelsinidine was synthesized in six steps with 11.8% overall yield, while the asymmetric synthesis was achieved in nine steps with 6.7% overall yield. Choosing a suitable oxidative system for realizing the selective oxidative coupling base on the biosynthetic route is still undergoing in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (051170001, 21772084), the Fundamental Research Funds for the Central Universities (lzujbky-2017-k06), Open Projects Funds of Shandong Key Laboratory of Carbohydrate Chemistry and Glycobiology, Shandong University (No. 2019CCG05). Xiaolei Wang thanks the Thousand Young Talents Program and Longyuan Talent Program for financial support.Notes and references
- (a) M. S. Baliga, G. C. Jagetia, J. N. Ulloor, M. P. Baliga, P. Venkatesh, R. Reddy, K. V. Rao, B. S. Baliga, S. Devi, S. K. Raju, V. Veeresh, T. K. Reddy and K. L. Bairy, Toxicol. Lett., 2004, 151, 317–326 CrossRef CAS; (b) Compiling group of Yunnan Traditional Chinese Medicine, Yunnan Traditional Chinese Medicinal Plant, Yunnan People's Press, Kunming, 1977 Search PubMed; (c) J. Leonard, Nat. Prod. Rep., 1999, 16, 319–338 RSC; (d) E. Saxton, J. Nat. Prod. Rep., 1997, 14, 559–590 RSC; (e) L. Zhang, C.-J. Zhang, D.-B. Zhang, J. Wen, X.-W. Zhao, Y. Li and K. Gao, Tetrahedron Lett., 2014, 55, 1815–1817 CrossRef CAS.
- (a) R. Vicente, Org. Biomol. Chem., 2011, 9, 6469–6480 RSC; (b) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef CAS; (c) T. Guo, F. Huang, L. Yu and Z. Yu, Tetrahedron Lett., 2015, 56, 296–302 CrossRef CAS; (d) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS; (e) N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159–184 CrossRef CAS.
- (a) D. Schmidt and J. Stöckigt, Planta Med., 1995, 61, 254–258 CrossRef CAS; (b) C. Kan-Fan and H. P. Husson, Tetrahedron Lett., 1980, 21, 1463–1466 CrossRef CAS; (c) J. Stöckigt, G. Hoefle and A. Pfitzner, Tetrahedron Lett., 1980, 21, 1925–1926 CrossRef; (d) A. Pfitzner and J. Stöckigt, Phytochemistry, 1982, 21, 1585–1588 CrossRef CAS; (e) M. Sottomayor, I. L. Cardoso, L. G. Pereira and A. R. Barcel, Phytochem. Rev., 2004, 3, 159–171 CrossRef CAS; (f) A. I. Scott and A. A. Qureshi, J. Am. Chem. Soc., 1969, 91, 5874–5876 CrossRef CAS; (g) E. Wenkert and B. Wickberg, J. Am. Chem. Soc., 1965, 87, 1580–1589 CrossRef CAS.
- (a) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2008, 130, 17938–17954 CrossRef; (b) P. S. Baran, T. J. Maimoneand and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS.
- Z. Zuo, W. Xie and D. Ma, J. Am. Chem. Soc., 2010, 132, 13226–13228 CrossRef CAS.
- M. Teng, W. Zi and D. Ma, Angew. Chem., Int. Ed., 2014, 53, 1814–1817 CrossRef CAS.
- W. Ren, N. Tappin, Q. Wang and J. Zhu, Synlett, 2013, 24, 1941–1944 CrossRef CAS.
- M. Lounasmaa and P. Hanhinen, Tetrahedron, 1996, 52, 14225–15242 CrossRef.
- M. Jarret, A. Tap, C. Kouklovsky, E. Poupon, L. Evanno and G. Vincent, Angew. Chem., Int. Ed., 2018, 57, 12294–12298 CrossRef CAS.
- (a) D. Sole, Y. Cancho, A. Llebaria, J. M. Moretd and A. Delgado, J. Am. Chem. Soc., 1994, 116, 12133–12134 CrossRef CAS; (b) J. Bonjoch, D. Sole and J. Bosch, J. Am. Chem. Soc., 1995, 117, 11017–11018 CrossRef CAS; (c) S. Yu, O. M. Berner and J. M. Cook, J. Am. Chem. Soc., 2000, 122, 7827–7828 CrossRef CAS; (d) J. Ma, W. Yin, H. Zhou and J. M. Cook, Org. Lett., 2007, 9, 3491–3494 CrossRef CAS; (e) J. Ma, W. Yin, H. Zhou, X. Liao and J. M. Cook, J. Org. Chem., 2009, 74, 264–273 CrossRef CAS; (f) T. Wang, X. Duan, H. Zhao, S. Zhai, C. Tao, H. Wang, Y. Li, B. Cheng and H. Zhai, Org. Lett., 2017, 19(7), 1650–1653 CrossRef CAS.
- For a review on early work, see S. Cacchi, The palladium-catalyzed hydroarylation and hydrovinylation of carbon-carbon multiple bonds: New perspectives in organic synthesis, Pure Appl. Chem., 1990, 62, 713–722 CAS.
- For selected reductive-Heck coupling, see: (a) M. Catellani, G. P. Chiusoli, W. Giroldini and G. Salerno, J. Organomet. Chem., 1980, 199, C21–C23 CrossRef CAS; (b) S. Cacchi and A. Arcadi, J. Org. Chem., 1983, 48, 4236–4240 CrossRef CAS; (c) A. Arcadi, S. Cacchi and F. Marinelli, Tetrahedron, 1985, 41, 5121–5131 CrossRef CAS; (d) A. Minatti, X. Zheng and S. L. Buchwald, J. Org. Chem., 2007, 72, 9253–9258 CrossRef CAS; (e) C. Shen, R.-R. Liu, R.-J. Fan, Y.-L. Li, T.-F. Xu, J.-R. Gao and Y.-X. Jia, J. Am. Chem. Soc., 2015, 137, 4936–4939 CrossRef CAS; (f) W. Kong, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 3987–3991 CrossRef CAS; (g) K. Semba, K. Ariyama, H. Zheng, R. Kameyama, S. Sakaki and Y. Nakao, Angew. Chem., Int. Ed., 2016, 55, 6275–6279 CrossRef CAS; (h) L.-J. Xiao, L. Cheng, W.-M. Feng, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461–464 CrossRef CAS; (i) J. A. Gurak and K. M. Engle, ACS Catal., 2018, 8, 8987–8992 CrossRef CAS.
- (a) S. F. Martin, C. W. Clark and J. W. Corbett, J. Org. Chem., 1995, 60, 3236–3242 CrossRef CAS; (b) S. F. Martin, K. X. Chen and C. Todd Eary, Org. Lett., 1999, 1, 79–82 CrossRef CAS.
- Due to the inseparable anti and elimination mixture, the trace anti isomer was detected via the 1H NMR of the inseparable mixture.
- (a) Y. Zheng, K. Wei and Y.-R. Yang, Org. Lett., 2017, 19, 6460–6462 CrossRef CAS; (b) K. Sato, N. Kogure, M. Kitajima and H. Takayama, Org. Lett., 2019, 21, 3342–3345 CrossRef CAS.
- (a) R. A. Rodriguez, C. M. Pan, Y. Yabe, Y. Kawamata, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 6908–6911 CrossRef CAS; (b) S. Song, X. Li, J. Wei, W. Wang, Y. Zhang, L. Ai, Y. Zhu, X. Shi, X. Zhang and N. Jiao, Nat. Catal., 2019, 3, 107–115 CrossRef.
- Selected reviews and literatures, see: (a) J. W. Beatty and C. R. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81(16), 6898–6926 CrossRef CAS; (c) X.-Y. Liu and Y. Qin, Acc. Chem. Res., 2019, 52, 1877–1891 CrossRef CAS; (d) J. W. Beatty and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 10270–10273 CrossRef CAS; (e) L. Furst, J. M. Narayanam and C. R. Stephenson, Angew. Chem., Int. Ed., 2011, 50, 9655–9659 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00015b |
| This journal is © The Royal Society of Chemistry 2021 |