
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of fully protected trinucleotide building blocks on a disulphide-linked soluble support†
Ruth Suchslanda,
Bettina Appel a,
Pasi Virtab and
Sabine Müller*a
a,
Pasi Virtab and
Sabine Müller*a
aUniversity Greifswald, Institute for Biochemistry, 17487 Greifswald, Germany
bUniversity of Turku, Department of Chemistry, 20014 Turku, Finland. E-mail: smueller@uni-greifswald.de
First published on 20th January 2021
Abstract
In recent years, preparation of fully protected trinucleotide phosphoramidites as synthons for the codon-based synthesis of gene libraries as well as for the assembly of oligonucleotides from blockmers has gained much attention. We here describe the preparation of such trinucleotide synthons on a soluble support using a disulphide linker.
The synthesis of fully protected trinucleotide synthons for codon-based assembly of oligonucleotides has a long history and originally was motivated by the need for methods of combinatorial and evolutionary protein engineering, which combine combinatorial gene synthesis with functional screening or genetic selection applied at the phenotype level to an ensemble of many structural variants generated in parallel.1–3 Among a host of related methods, the use of mixtures of pre-formed trinucleotide blocks representing codons for the 20 canonical amino acids stands out as allowing fully controlled randomization individually at any number of arbitrarily chosen codon positions of a given gene.4–6 The chance of functional proteins in such libraries is increased, as randomization independent from the degenerated genetic code is possible and thus bias to amino acids represented by more than one codon as well as stop codons can be avoided.6–9 The power of this method has been successfully demonstrated for randomization of immunoglobulins or at the example of a gene library of tHisF from the hyperthermophile Thermotoga maritima.7,10 Apart from the preparation of gene libraries with controlled randomization, fully protected trinucleotides have potential as building blocks for oligonucleotide synthesis from blockmers (n = 1, 2, 3, 4, …), in particular then, when the oligomer is composed of repetitive sequence patches. In an ideal case, one previously synthesized blockmer can be coupled several times to obtain the desired oligomer. Moreover, oligonucleotide assembly from blockmers is advantageous in terms of easier purification, since n − 1, −2, … side products cannot be formed. A number of routes to fully protected trinucleotide building blocks have been developed, based on strategies in solution, on solid phase or on soluble supports (Fig. 1).4,11,12 Traditionally, trinucleotide synthons have been prepared in solution, paying special attention to the pair of orthogonal protecting groups for the 5′- and 3′-OH functions, when synthesizing a dinucleotide that subsequently can be extended in 5′- or 3′-direction.1,13–19
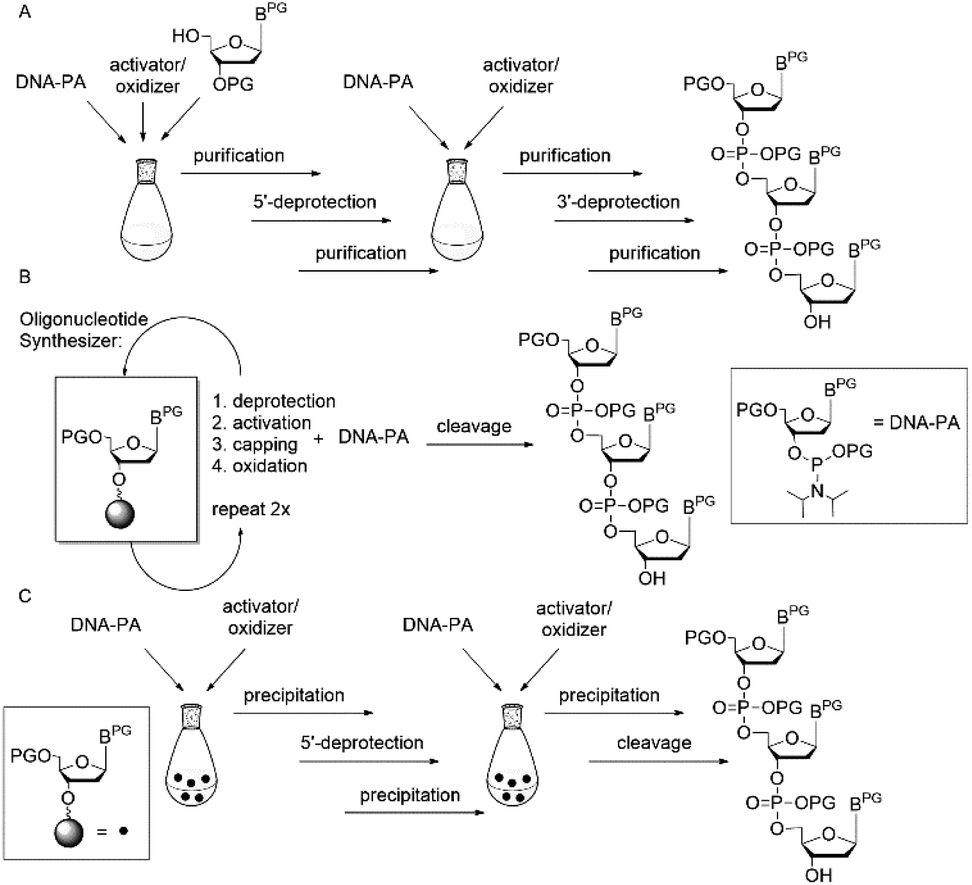 | ||
| Fig. 1 Strategies for preparation of fully protected trinucleotides in solution (A), on solid support (B) and on soluble phase (C); PG = protecting group. | ||
Synthesis in solution requires isolation of products after each synthesis step, which can become rather tedious. Therefore, we11,20 and others19 have developed strategies for trinucleotide synthesis on solid support as an attractive alternative to protocols in solution.
The key issue is the attachment of the 3′-start nucleoside to the solid support via a suitable linker, allowing to cleave off the trinucleotide after synthesis without loss of the protecting groups. With regard to trinucleotide synthesis, an oxalyl anchor19 or a disulphide linker have been described.11,21
In recent years, protocols for the synthesis of oligonucleotides on soluble supports have emerged,12,21–24 and those have also been used with particular attention to the preparation of fully protected trinucleotides.12,21 The general strategy involves iterative cycles of reaction steps in solution and precipitation for isolation/purification of reaction products. Several soluble supports have been used for oligomer synthesis (reviewed in ref. 25), among those, pentaerythritol-derived cores, which are easily precipitated from methanol.24 Fully protected trinucleotides have been prepared on the pentaerythritol-derived core with the start nucleoside being tethered to the polymer via a disulphide bridge21 or hydroquinone-O,O′-diacetic acid (Q-linker).12 In both strategies, phosphotriester chemistry has been used for trimer assembly, which in the disulphide strategy resulted in a trinucleotide with 3′-terminal ortho-chlorophenylphosphate.21 This 3′-remnant may be activated as a phosphotriester, but in standard automated DNA synthesis, where phosphoramidite coupling is strongly preferred, it would require to be selectively removed in order to convert the trinucleotide to the 3′-O-phosphoramidite building block. Moreover, the reductive cleavage of the disulphide bridge was performed in conditions that caused premature loss of the 5′-dimethoxytrityl (DMT) protecting group.21
Based on our previous experience,11,14,20 we here report on the preparation of fully protected trinucleotides on a pentraerythritol-derived soluble support using phosphoramidite chemistry for nucleotide coupling. As we have reported previously, tethering the start nucleoside to a solid support (polystyrene) via a dithiomethyl linkage is a superior strategy for assembly of blockmers that upon release from the support by reductive cleavage carry protecting groups at all functionalities, but offer a free 3′-OH group for conversion to the phosphoramidite building block.11,20 Application of this immobilization strategy to the pentaerythritol-derived core, first required appropriate functionalization of the core. This was achieved as described previously by conjugation of commercially available S-propargyl thioacetate to the tetrakis-O-[4-(azidomethylphenyl)pentaerythritol] support21 by Cu(I) catalyzed 1,3-dipolar cycloaddition,26,27 yielding tetrakis-O-{4-[4-[(acetylthiomethyl)]-1H-1,2,3-triazol-1-ylmethyl]-phenyl} pentaerythritol (1) (Scheme 1). The 3′-O-methylthiomethyl (MTM) modified start nucleosides (T and Bz-dA) were synthesized as described20 following the strategy originally developed for synthesis of 2′-O-DTM functionalized ribonucleotide building blocks.28 Aminolysis of the thioacetate on the support with butylamine in methanol delivered the free thiol function (2) required for immediate reaction with the nucleoside derivative to form the disulphide linkage. To this end, the 3′-O-MTM functionalized nucleoside (3a–b) was activated by treatment with sulfuryl chloride to give the 3′-O-chloromethyl ether (4a–b) in a Pummerer rearrangement, which immediately was converted in the presence of potassium thiotosylate to the reactive species (5a–b). This subsequently reacted with the support bound thiol (2) to form the desired loaded tetrapodal soluble support (6a–b, Scheme 1).
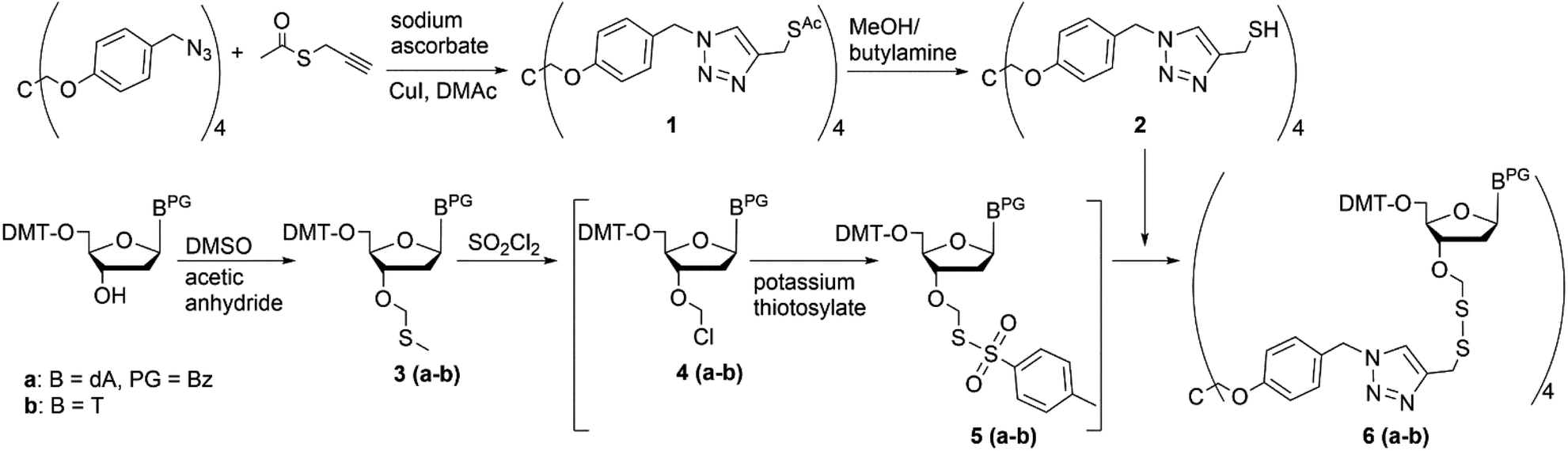 | ||
| Scheme 1 Reaction scheme for coupling of start nucleoside to soluble support. | ||
To start trinucleotide assembly, the support carrying the start nucleoside (6a–b) was treated with 4% dichloroacetic acid in ethylene dichloride to cleave off the 5′-O-DMT group (Fig. 2). The acid treatment was quenched by addition of pyridine, the solvents were evaporated, and the support linked with the deprotected start nucleoside (7a–b) was precipitated from methanol. The coupling reaction was carried out by taking up the precipitate in acetonitrile containing six equivalents of the N-acyl-5′-O-DMT protected nucleoside phosphoramidite in 0.1 M concentration, and addition of benzylmercapto-tetrazole29 as activator. The resulting dimer 8a–b was oxidized by addition of a 0.2 M solution of iodine in trimethylpyridine/ACN/H2O (1/11/5) to give the dinucleotide product 9a–b (Fig. 2). The support-linked dinucleotide 9a–b was directly precipitated from the reaction mixture by addition of methanol. The solid was filtered off and used for the next coupling cycle. All steps including deprotection, coupling and oxidation were repeated to obtain the fully protected trinucleotide on the soluble support (10a–b–12a–b, Fig. 2).
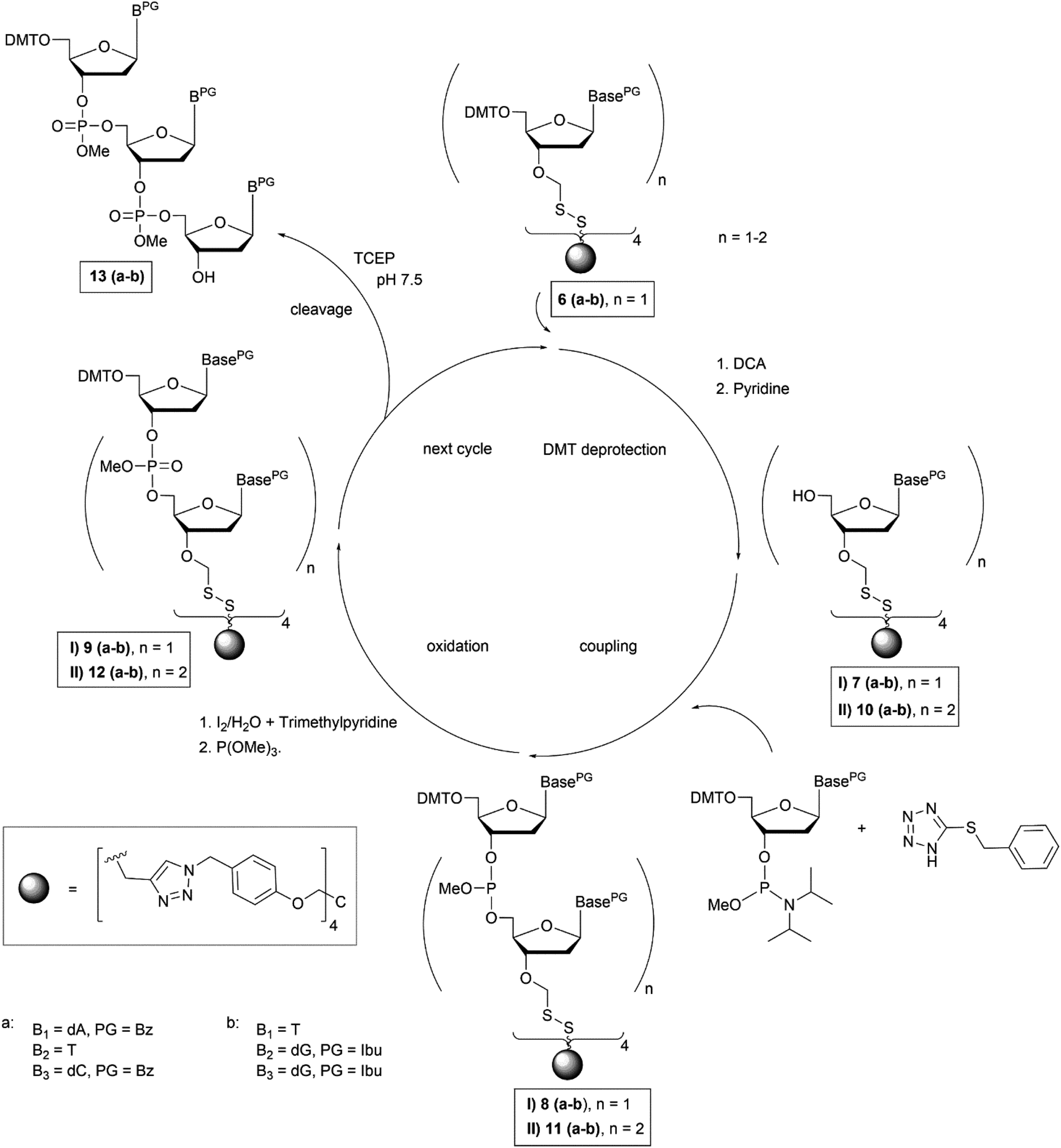 | ||
| Fig. 2 Reaction scheme of trinucleotide assembly on soluble support. | ||
NMR spectroscopy was used to analyse success of the synthesis. However, as long as the start nucleoside as well as the intermediate dinucleotide and the final trinucleotide were bound to the soluble support, complexity of the products and their modest solubility in common deuterated solvents retarded the analysis. Consequently, detailed NMR characterization was applicable only for the products released from the support. After assembly of the trinucleotide was finished, it was released in fully protected form from the support by reductive cleavage of the disulphide linkage with TCEP at pH 7.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 (13a–b, Fig. 2). After treatment with TCEP, the support was thoroughly washed with acetonitrile, acetone, ethanol and ethyl acetate, in order to separate the cleaved off trinucleotide. Traces of TCEP and of the tetrapodal support, which remained after this work up, were finally removed by chromatographic purification, which yielded sufficiently pure trinucleotide blockmers CTA and GGT in 43% and 35% overall isolated yield, respectively (both synthesized in tens of μmol scale). Extensive analysis by various NMR techniques (1H, 13C, DEPT, HSQC, DQF-COSY, for detailed information see ESI†) unambiguously confirmed the identity and purity of the desired products. For further application as synthons for DNA synthesis, the trinucleotides were converted to 3′-O-methylphosphoramidites following the standard procedure for phosphitylation (Fig. 3).31 The methyl group was chosen for protection of the phosphorous, because of its higher stability as compared with the β-cyanoethyl group and thus easier handling. Nevertheless, it should be noted, that also β-cyanoethyl protection can be used, both for the phosphate moieties in the trinucleotide and in the final trinucleotide 3′-O-phosphoramidite, if all steps of synthesis, purification and storage are carried out with particular care, partially requiring specific conditions.14 After aqueous work up and extensive drying, 0.1 M solutions of both trinucleotide phosphoramidites in appropriate solvents (see below) were prepared and used for coupling on the DNA synthesizer. Both trinucleotide synthons CTA (14a) and GGT (14b) were coupled in individual syntheses onto a short oligomer (CTT) on CPG. The 5′-terminal DMT group was left on, and the resulting 6mers 5′-CTACTT-3′ and 5′-GGTCTT-3′ were cleaved from the support by concentrated ammonia. RP-HPLC analysis was used to evaluate the coupling efficiency of the trinucleotide blockmers (Fig. 4).
30 (13a–b, Fig. 2). After treatment with TCEP, the support was thoroughly washed with acetonitrile, acetone, ethanol and ethyl acetate, in order to separate the cleaved off trinucleotide. Traces of TCEP and of the tetrapodal support, which remained after this work up, were finally removed by chromatographic purification, which yielded sufficiently pure trinucleotide blockmers CTA and GGT in 43% and 35% overall isolated yield, respectively (both synthesized in tens of μmol scale). Extensive analysis by various NMR techniques (1H, 13C, DEPT, HSQC, DQF-COSY, for detailed information see ESI†) unambiguously confirmed the identity and purity of the desired products. For further application as synthons for DNA synthesis, the trinucleotides were converted to 3′-O-methylphosphoramidites following the standard procedure for phosphitylation (Fig. 3).31 The methyl group was chosen for protection of the phosphorous, because of its higher stability as compared with the β-cyanoethyl group and thus easier handling. Nevertheless, it should be noted, that also β-cyanoethyl protection can be used, both for the phosphate moieties in the trinucleotide and in the final trinucleotide 3′-O-phosphoramidite, if all steps of synthesis, purification and storage are carried out with particular care, partially requiring specific conditions.14 After aqueous work up and extensive drying, 0.1 M solutions of both trinucleotide phosphoramidites in appropriate solvents (see below) were prepared and used for coupling on the DNA synthesizer. Both trinucleotide synthons CTA (14a) and GGT (14b) were coupled in individual syntheses onto a short oligomer (CTT) on CPG. The 5′-terminal DMT group was left on, and the resulting 6mers 5′-CTACTT-3′ and 5′-GGTCTT-3′ were cleaved from the support by concentrated ammonia. RP-HPLC analysis was used to evaluate the coupling efficiency of the trinucleotide blockmers (Fig. 4).
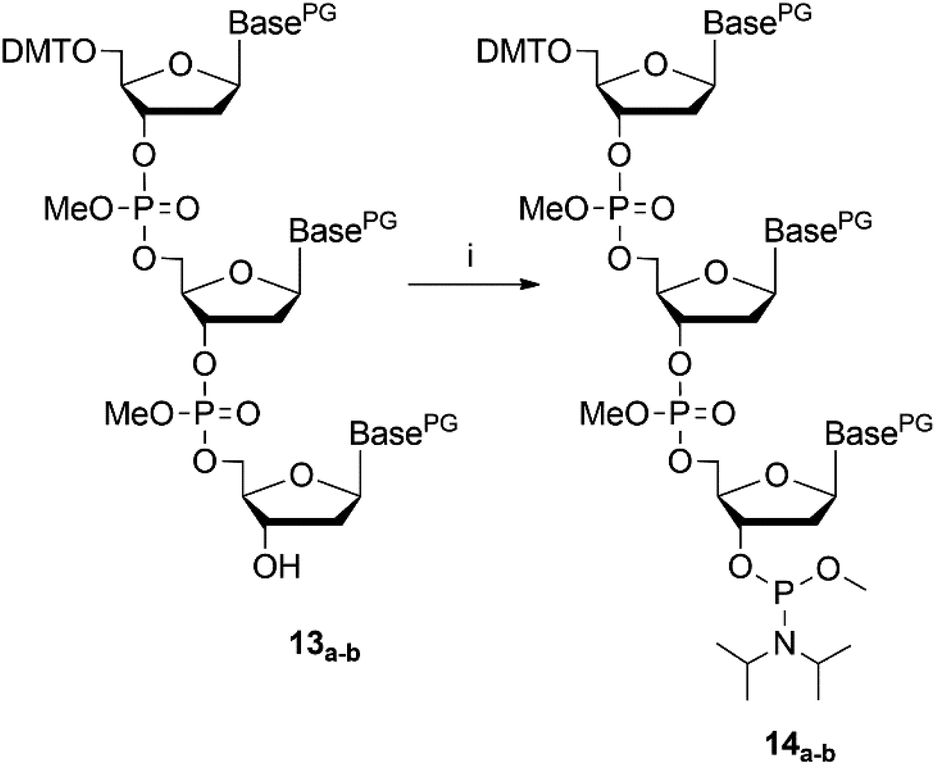 | ||
| Fig. 3 Reaction scheme of trinucleotide phosphitylation, (i) N,N-diisopropyl-methyl-phosphonamidic-chloride, TEA, DCM, 3.5 h, rt; PG = protecting group. | ||
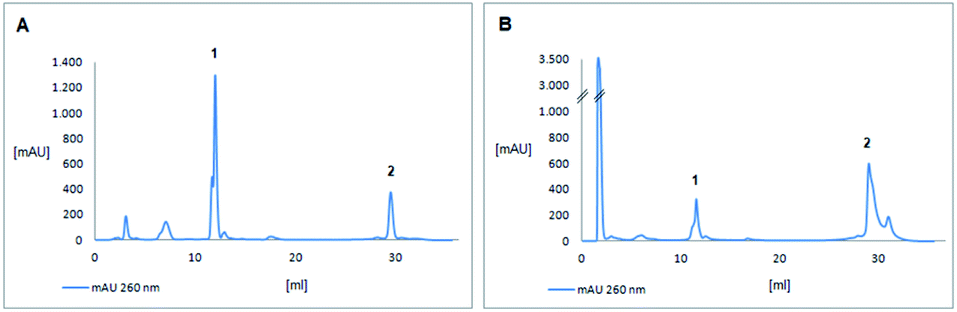 | ||
| Fig. 4 HPLC analysis of CTACTT (A) and GGTCTT (B) (DMT-on). (A) peak 1: abortive fragments, peak 2: CTACTT; (B) peak 1: abortive fragments, peak 2: GGTCTT; AU = absorption unit. Conditions for (A and B) nucleodur 125/4, CV = 1.571 ml, 1 ml min−1; buffer (A) 5% ACN, 0.1 M TEAAc; buffer (B) 30% ACN, 0.1 M TEAAc; gradient: starting with 0% buffer (B) for 4 CV, to 40% buffer (B) over 3 CV, to 60% buffer (B) over 7 CV, to 100% buffer (B) over 2 CV, then 100% buffer (B) for another 2 CV, to 0% buffer (B) over 3 CV. | ||
It has been discussed in the past that some but not all trinucleotide synthons are soluble in acetonitrile1,32 or various compositions of acetonitrile–dichloromethane mixtures12,16,19 with or without addition of DMF.18 The fully protected trinucleotide CTA showed poor solubility in acetonitrile and therefore was dissolved in dichloromethane. Solubility was clearly better than in acetonitrile, but not fully satisfying. Accordingly, HPLC analysis of the 6mer after coupling and deprotection showed a rather low coupling yield (Fig. 4A). This result may be interpreted as a consequence of a low effective concentration of the CTA blockmer and of the missing acetonitrile being the ideal solvent for phosphoramidite coupling. For dissolving the second trinucleotide GGT, we applied a 3:1 mixture of dichloromethane and acetonitrile, which allowed to prepare a 0.1 M solution of the trinucleotide phosphoramidite ready for coupling. A double coupling cycle was used to increase the coupling efficiency. The results are shown in Fig. 4B. A clearly higher coupling yield was achieved for the trinucleotide synthon GGT as compared to CTA (Fig. 4A). HPLC peak areas designated as final 6mers and abortive fragments would give rough estimation for the coupling yields, being about 17% for the CTA synthon and 72% for the GGT synthon under the described conditions (see ESI† for details). The identity of the assembled 6mers was confirmed by MS analysis (see ESI†). For GGT, the estimated coupling yield is well in the range of that for trinucleotide synthons reported in the literature,15,16,32 although the coupling yields given there are usually just concluded from detritylation values on the synthesizer,1,12,18,32 or it is not at all specified how those were determined.15,19 Therefore, it is difficult if not impossible to compare coupling yields among the different protocols. Trinucleotide synthons that we had prepared in the past, showed coupling yields in DNA synthesis comparable to that achieved here in the range of 70 to 90%.14 This certainly can be further optimized in a systematic study including various solvents and solvent compositions for trinucleotide phosphoramidites, which however would require larger amounts of trinucleotide synthons than produced here. The focus of this work is on preparation of the trinucleotides on a soluble support, which has been successfully achieved, and the general functionality of the prepared trinucleotide synthons in automated DNA synthesis was demonstrated, although suitable conditions for efficient coupling still need to be determined.
Conclusions
Based on our results, we conclude that the disulphide chemistry we had developed previously for the synthesis of fully protected trinucleotides on polystyrene,11,20 is a superior strategy also for synthesis on soluble support. S-propargyl thioacetate was efficiently clicked onto an azido-functionalized tetrapodal support, and upon deprotection of the thiol, the 3′-O-dithiomethyl functionalized start nucleoside was activated and immobilized onto the support via a disulphide bridge. Assembly of the trinucleotide and release from the support by reductive treatment delivered fully protected trinucleotides, which were further converted to phosphoramidite building blocks and used in standard DNA synthesis. Moderate, however at least for GGT yet satisfying coupling yields were obtained. Most likely, the lower coupling yields result from the insufficient solubility of the two trinucleotide synthons described herein in acetonitrile. However, the encountered solubility problems are independent of the strategy for trinucleotide preparation, and need to be addressed separately for each individual sequence prior to usage in standard DNA synthesis. This may also include empirical studies of nucleobase and phosphate protection schemes to gain end products that allow better solubility and efficient automated assembly.Trinucleotide synthons have potential for the preparation of gene and protein libraries as well as for the assembly of functional oligonucleotides from blockmers. Their synthesis on soluble support offers a way for economic synthesis at larger scale and thus would be advantageous over synthesis on solid support, which is more expensive, or completely in solution, which includes numerous working up and product isolation steps. The disulphide chemistry presented here allows for facile immobilisation of the starting nucleoside onto the support as well as for easy release of the assembled trinucleotide with preservation of all protecting groups and with generation of a free 3′-OH group for further conversion to a phosphoramidite.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Fonds der Chemischen Industrie (fellowship to R. S.) is gratefully acknowledged.Notes and references
- B. Virnekäs, L. Ge, A. Plückthun, K. C. Schneider, G. Wellnhofer and S. E. Moroney, Nucleic Acids Res., 1994, 22, 5600 CrossRef.
- S. Sen, V. Venkata Dasu and B. Mandal, Appl. Biochem. Biotechnol., 2007, 143, 212 CrossRef CAS.
- N. E. Labrou, Curr. Protein Pept. Sci., 2010, 11, 91 CrossRef CAS.
- R. Suchsland, B. Appel and S. Müller, Beilstein J. Org. Chem., 2018, 14, 397 CrossRef CAS.
- R. Raetz, B. Appel and S. Müller, Chim. Oggi, 2016, 34, 14 Search PubMed.
- T. S. Arunachalam, C. Wichert, B. Appel and S. Müller, Org. Biomol. Chem., 2012, 10, 4641 RSC.
- B. Popova, S. Schubert, I. Bulla, D. Buchwald, W. Kramer and T. Tuller, PLoS ONE, 2015, 10, e0136778 CrossRef.
- C. Neylon, Nucleic Acids Res., 2004, 32, 1448 CrossRef CAS.
- A. V. Shivange, J. Marienhagen, H. Mundhada, A. Schenk and U. Schwaneberg, Curr. Opin. Chem. Biol., 2009, 13, 19 CrossRef CAS.
- A. Knappik, L. Ge, A. Honegger, P. Pack, M. Fischer, G. Wellnhofer, A. Hoess, J. Wölle, A. Plückthun and B. Virnekäs, J. Mol. Biol., 2000, 296, 57 CrossRef CAS.
- R. Suchsland, B. Appel, M. Janczyk and S. Müller, Appl. Sci., 2019, 9, 2199 CrossRef CAS.
- V. Kungurtsev, H. Lönnberg and P. Virta, RSC Adv., 2016, 6, 105428 RSC.
- J. Yáñez, M. Argüello, J. Osuna, X. Soberón and P. Gaytán, Nucleic Acids Res., 2004, 32, e158 CrossRef.
- M. Janczyk, B. Appel, D. Springstubbe, H.-J. Fritz and S. Müller, Org. Biomol. Chem., 2012, 10, 1510 RSC.
- A. Zehl, A. Starke, D. Cech, T. Hartsch, R. Merkl and H.-J. Fritz, Chem. Commun., 1996, 23, 2677 RSC.
- A. Yagodkin, A. Azhayev, J. Roivainen, M. Antopolsky, A. Kayushin, M. Korosteleva, A. Miroshnikov, J. Randolph and H. Mackie, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 473 CrossRef CAS.
- M. H. Lyttle, E. W. Napolitano, B. L. Calio and L. M. Kauvar, BioTechniques, 1995, 19, 274 CAS.
- A. Ono, A. Matsuda, J. Zhao and D. V. Santi, Nucleic Acids Res., 1995, 23, 4677 CrossRef CAS.
- A. Kayushin, Nucleic Acids Res., 1996, 24, 3748 CrossRef CAS.
- R. Suchsland, B. Appel and S. Müller, Curr. Protoc. Nucleic Acid Chem., 2018, 75, e60 CrossRef.
- A. M. Jabgunde, A. G. Molina, P. Virta and H. Lönnberg, Beilstein J. Org. Chem., 2015, 11, 1553 CrossRef.
- Y. Matsuno, T. Shoji, S. Kim and K. Chiba, Org. Lett., 2016, 18, 800 CrossRef CAS.
- P. R. J. Gaffney, J. F. Kim, I. B. Valtcheva, G. D. Williams, M. S. Anson, A. M. Buswell and A. G. Livingston, Chem.–Eur. J., 2015, 21, 9535 CrossRef CAS.
- V. Kungurtsev, J. Laakkonen, A. G. Molina and P. Virta, Eur. J. Org. Chem., 2013, 2013, 6687 CrossRef CAS.
- H. Lönnberg, Beilstein J. Org. Chem., 2017, 13, 1368 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41 Search PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- A. Semenyuk, A. Földesi, T. Johansson, C. Estmer-Nilsson, P. Blomgren, M. Brännvall, L. A. Kirsebom and M. Kwiatkowski, J. Am. Chem. Soc., 2006, 128, 12356 CrossRef CAS.
- R. Welz and S. Müller, Tetrahedron Lett., 2002, 43, 795 CrossRef CAS.
- J. C. Han and G. Y. Han, Anal. Biochem., 1994, 220, 5 CrossRef CAS.
- L. J. McBride and M. H. Caruthers, Tetrahedron Lett., 1983, 24, 245 CrossRef CAS.
- J. Sondek and D. Shortle, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3581 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10941j |
| This journal is © The Royal Society of Chemistry 2021 |