DOI:
10.1039/D0RA08905B
(Paper)
RSC Adv., 2021,
11, 3897-3916
Design, synthesis, characterization and in vitro, in vivo and in silico antimicrobial and antiinflammatory activities of a new series of sulphonamide and carbamate derivatives of a nebivolol intermediate†
Received
19th October 2020
, Accepted 27th December 2020
First published on 20th January 2021
Abstract
A series of new sulphonamide and carbamate derivatives of Nebivolol drug intermediate (5) were designed and synthesized by reacting various biopotent sulphonylchlorides and chloroformates. The synthesized compounds are structurally characterized by spectral (IR, 1H & 13C NMR and mass) and screened for their in vitro antimicrobial activity against four bacterial and three fungal strains, in vitro and in vivo antiinflammatory activity against LPS-induced inflammation in RAW 264.7, in vitro COX-1 and COX-2 inhibition potentiality, antagonistic profiles of carrageenan induced paw edema and cotton pellet induced granuloma in rat. Further, the compounds were screened for their antimicrobial and antiinflammatory activity against DNA gyrase A, COX-1 and COX-2 by using molecular docking approach. The bioactivity and toxicity risks were analysed through Molecular Operating Environment. The results revealed that the compounds 8b, 8c, 8d, 8e, 8f, 8g and 9a exhibited the most promising antimicrobial activity against all the bacterial and fungal strains tested when compared with the standard drugs streptomycin and fluconazole. In view of in antiinflammatory activity, the compounds, 8b, 8c, 8d, 8e, 8f, 8g and 9a have shown potent antiinflammatory activity by inhibiting the LPS-induced inflammation in RAW 264.7 cell line, concentration dependent inhibition of COX-1 and COX-2, dose response dependent antagonism of carrageenan induced paw edema and granuloma tissue in rat. Molecular docking, ADMET and QSAR studies predicted that the recorded in silico profiles are in strong correlation with in vitro and in vivo antimicrobial and antiinflammatory results. In addition, the elevated toxicology risks of the title compounds are identified with in the potential limits of drug candidates. Hence, it is suggested that the synthesized derivatives will stand as the promising antimicrobial and anti-inflammatory drug candidates in future.
1. Introduction
Fast and effective relief of pain and inflammation in human beings is a major challenge for medical researchers. Non-steroidal antiinflammatory drugs (NSAIDs) are important therapeutic agents for the alleviation of pain and inflammation associated with a number of pathological conditions.1 A major mechanism of action of NSAIDs is lowering prostaglandin (PG) production through the inhibition of cyclooxygenase (COX); a key enzyme in PGs biosynthesis that catalyzes the conversion of arachidonic acid into PGs and thromboxanes.2 It was discovered that COX exists in two isoforms COX-1 and COX-2 which are regulated differently. COX-1 is a constitutive isoform; responsible for the production of cytoprotective prostaglandins in the gastrointestinal tract (GI) and proaggregatory thromboxane in blood platelets. However, COX-2 is an inducible isoform which is induced in response to the release of several proinflammatory mediators.3–5 Most of the classical NSAIDs such as aspirin act as nonselective inhibitors of COX, inhibiting both the COX-1 and COX-2. The chronic use of NSAIDs to treat pain and inflammation is often accompanied by side effects such as gastric ulceration, bleeding and renal impairment. It is believed that the COX-2 selective inhibitors will greatly reduce these side effects.6–8 Thus, the development of COX-2 selective inhibitors has led attention to improve the therapeutic potency and to reduce the classical side effects associated with the use of conventional NSAIDs.
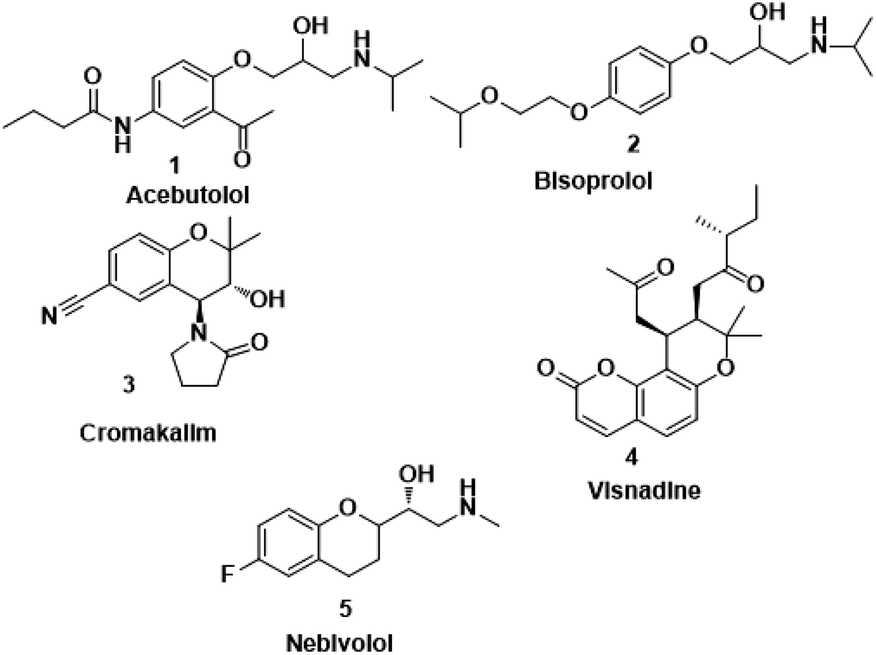
Nebivolol is a highly cardioselective 3rd generation long acting-1 blocker which is usually prescribed once a day as monotherapy or in combination with other antihypertensive drugs. It is only beta-blocker that has vasodilating properties and is safely prescribed in diabetics patients too as it does not have any adverse effect on insulin sensitivity and serum lipid profile. It also does not cause sexual dysfunction like other beta-blockers even though it improves sexual dysfunction in diabetic patients. It also inhibits platelets aggregation, improves endothelial and left ventricular systolic dysfunction, and improves arterial stiffness, coronary and peripheral blood flow in patients of angina, myocardial infarction, chronic heart failure, and various arrhythmias.9,10 Apart from their beneficial effects on blood pressure, nebivolol has a very strong antiinflammatory and antioxidant potential. Nebivolol reduces the level of various oxidative and inflammatory markers like oxidized LDL, 8-isoprostanes, CRP, interleukin 6 (IL-6), adiponectin, leptin, intercellular adhesions molecule (ICAM-1), vascular cell adhesions molecule (VCAM-1), and tumor necrosis factor-α in various clinical studies.11,12 Some of the important beta blockers such as Acebutolol (Sectral) (1), Bisoprolol (Zebetal) (2), Cromakalim (3), Visnadine (4), Nebivolol (Bystolic) (5) are shown in Fig. 1. In addition, numerous sulphonamide derivatives found to retain various types of pharmacological activities such as antibacterial,13 antitumor,14 antihypersensitive,15 anti-inflammatory,16 antiprotozoal,17 antimitotic,18 hypoglycemic,19 carbonic anhydrase enzyme inhibitors,20 etc. besides, organic carbamates are also valuable synthetic intermediates which are widely used for a number of applications, including drug synthesis. They constitute a versatile class of compounds used as insecticides,21 fungicides, antibacterial,22 acaricides,23 molluscicides,24 sprout inhibitors or herbicides.25 Among the highly marketed COX-2 inhibitors, celecoxib is the one which is treated as a safe anti-inflammatory and analgesic agent. It is considered as a typical model of the diaryl heterocyclic template that is known to selectively inhibit the COX-2 enzyme. On the other hand, the compounds with the backbone of nebivolol have been reported to possess various biological activities, including anti-inflammatory, antimicrobial, antifungal, antioxidant, and anticancer activities.26,27 The combination of both the active scaffolds, nebivolol and analogues of nebivolol, may provide synergistic effect to improve the antiinflammatory activity. These predictions encouraged us to synthesize hybrid compounds containing nebivolol scaffold. Hence, we have designed the synthesis of a series of nebivolol derivatives, synthesized, and studied the antimicrobial, antiinflammatory activity profiles using in vitro, in vivo and in silico approaches.
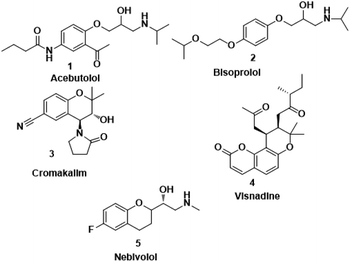 |
| | Fig. 1 A few of the selective β1-blockers. | |
2. Materials and methods
2.1. Chemistry
All the chemicals were purchased from Sigma-Aldrich and the solvents from Merck. Thin layer chromatography was performed on Merck 60F254 plates. Infrared spectra were recorded on Bruker ALPHA Interferometer. Melting points of the compounds were determined on Guna digital melting point apparatus using open capillary tubes and are uncorrected. NMR spectra were recorded on a Bruker instrument operating at 400 MHz for 1H and 100 MHz for 13C in DMSO-d6, TMS was used as an internal standard. Chemical shift (δ) and coupling constant (J) were expressed in ppm and hertz respectively. LC mass spectra were recorded on Agilent LCMS-model 2010A Shimadzu instrument in positive mode. HRMS spectra were recorded on Agilent HRMS instrument in positive mode.
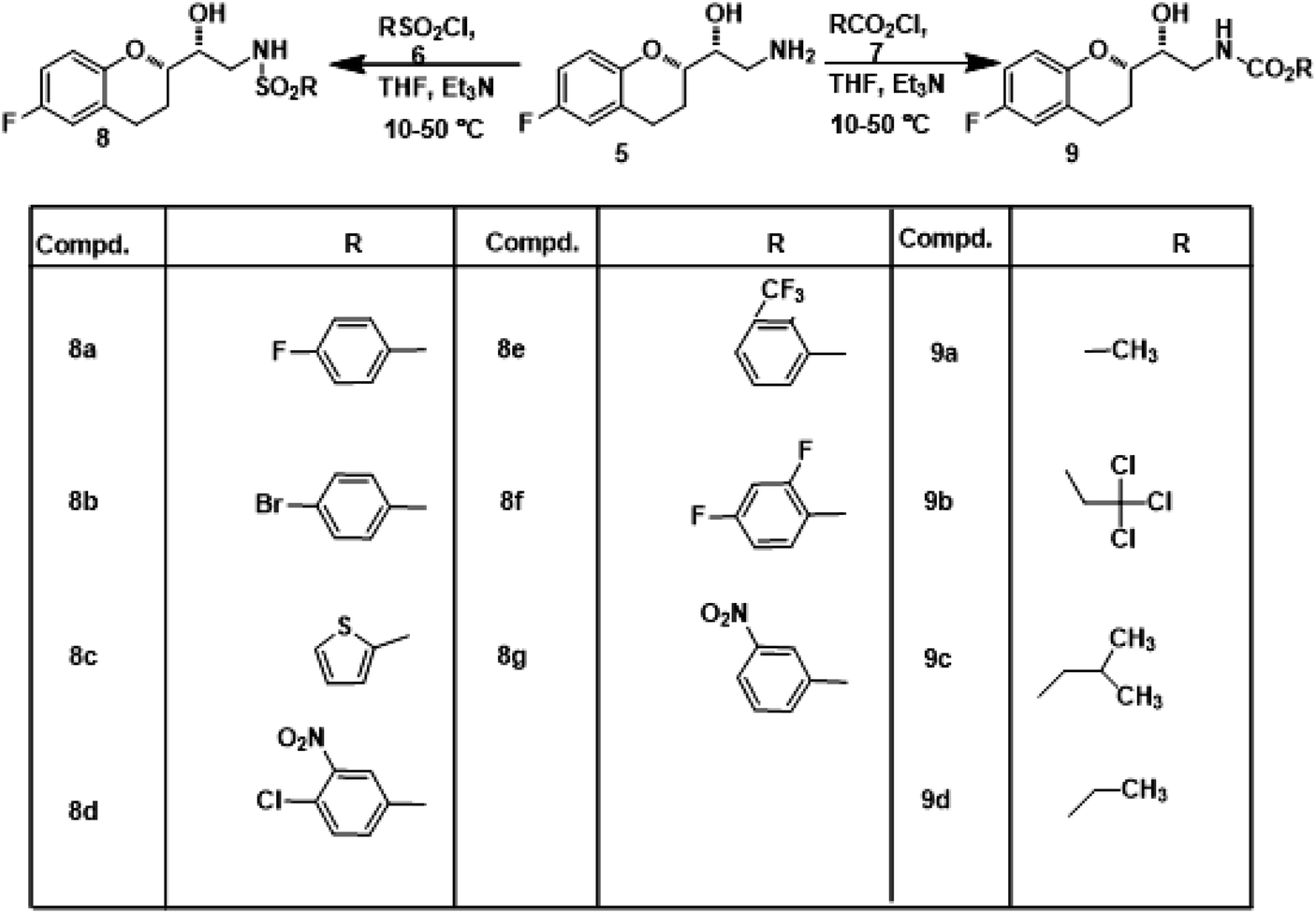
2.1.1. General procedure for the synthesis of sulphonamide and carbamate derivatives of Nebivolol (8a–g & 9a–d). Nebivolol intermediate (5) (211 mg, 0.001 mol) was dissolved in dry THF (10 mL) and cooled to 10 °C. Triethylamine (0.0012 mol) in 5 mL of THF was added. 4-Fluorobenzenesulfonyl chloride (6a) (302 mg, 0.001 mol) in 10 mL of THF was added and then the reaction mixture was stirred at 40 °C for 4 h. Reaction progress was monitored by TLC using n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (2:1). After completion of the reaction, it was filtered to remove Et3N·HCl and the filtrate was concentrated in a rota-evaporator. The crude product was partitioned between ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The crude product thus obtained was purified by column chromatography using n-hexane:ethyl acetate (4:1). Same procedure was adopted for the synthesis of the remaining compounds (8b–g & 9a–d).
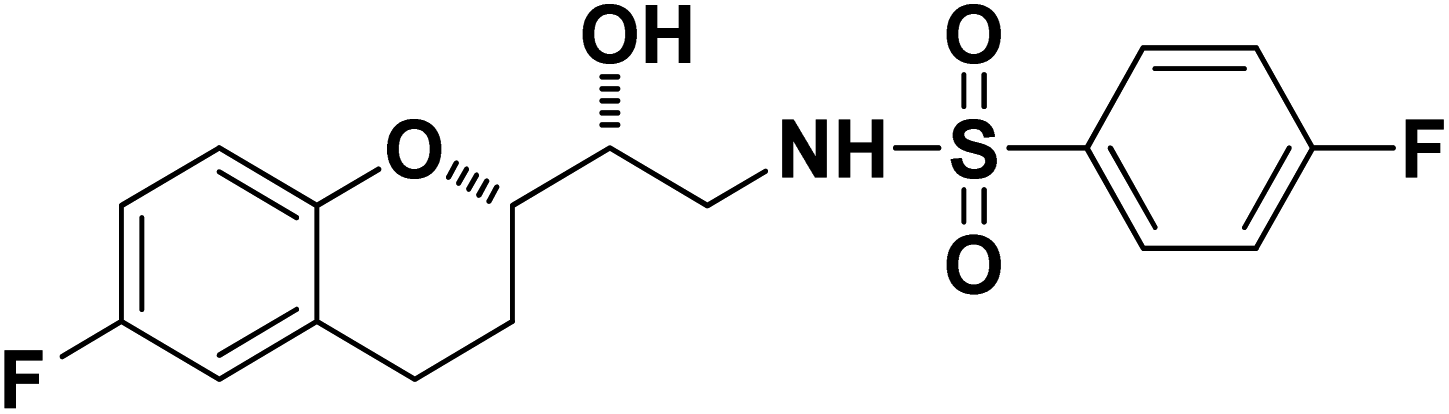
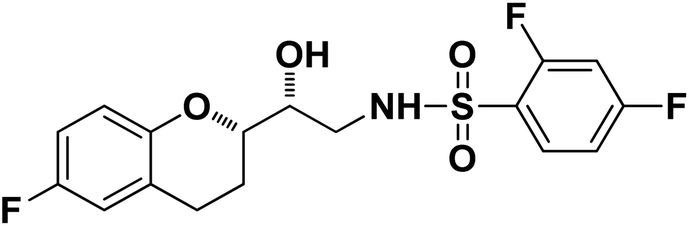
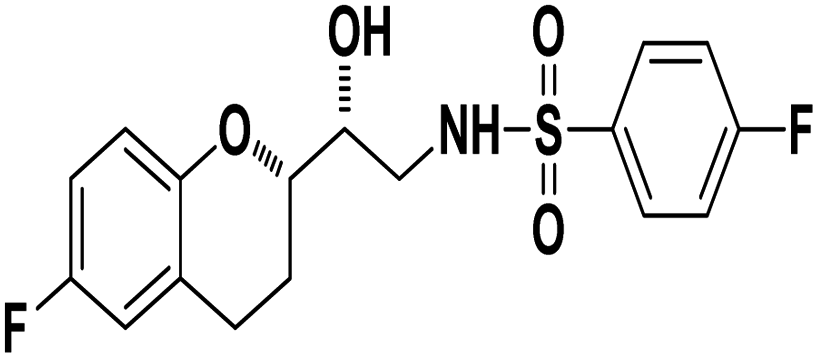
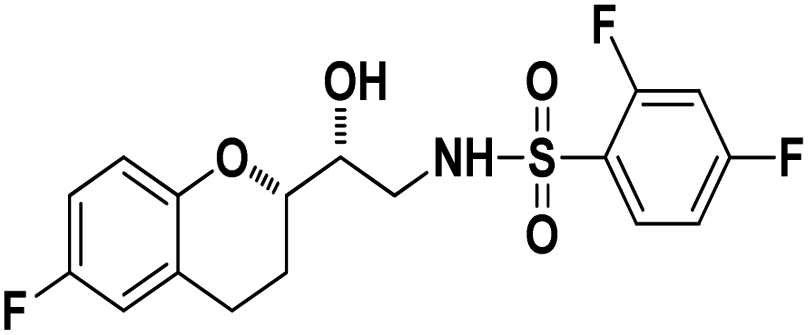
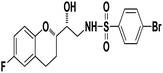
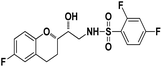
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-4-fluorobenzenesulfonamide (8a). White solid; yield: 85%; mp 167–170 °C; IR (vmax/cm−1): 3503(OH), 3318(N–H), 3072(Ar–C–H), 1396, 1158(S
:ethyl acetate (2:1). After completion of the reaction, it was filtered to remove Et3N·HCl and the filtrate was concentrated in a rota-evaporator. The crude product was partitioned between ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The crude product thus obtained was purified by column chromatography using n-hexane:ethyl acetate (4:1). Same procedure was adopted for the synthesis of the remaining compounds (8b–g & 9a–d).
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-4-fluorobenzenesulfonamide (8a). White solid; yield: 85%; mp 167–170 °C; IR (vmax/cm−1): 3503(OH), 3318(N–H), 3072(Ar–C–H), 1396, 1158(S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1221, 976 cm−1; 1H-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.24 (1H, s, N–H),7.15–7.25 (4H, m, Ar–H), 6.71–6.86 (2H, m, Ar–H), 6.63–6.34 (1H, t, J = 5.0 Hz), 5.33 (1H, s, OH), 3.77–3.84 (2H, m, –CH), 3.09–3.11 (2H, m, –CH2), 2.73–2.78 (2H, m, –CH2), 2.09–2.18 (2H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 167.4, 154.6, 151.0, 141.3, 130.6, 125.7, 119.8, 115.9, 115.2, 113.1, 77.6, 71.5, 46.4, 23.8, 22.2.; LC-MS (m/z): 370 [M + H]+ .; anal. calcd for C17H17F2NO4S: C 55.28, H 4.64, N 3.76; found: C 54.62, H 4.25, N 3.29.
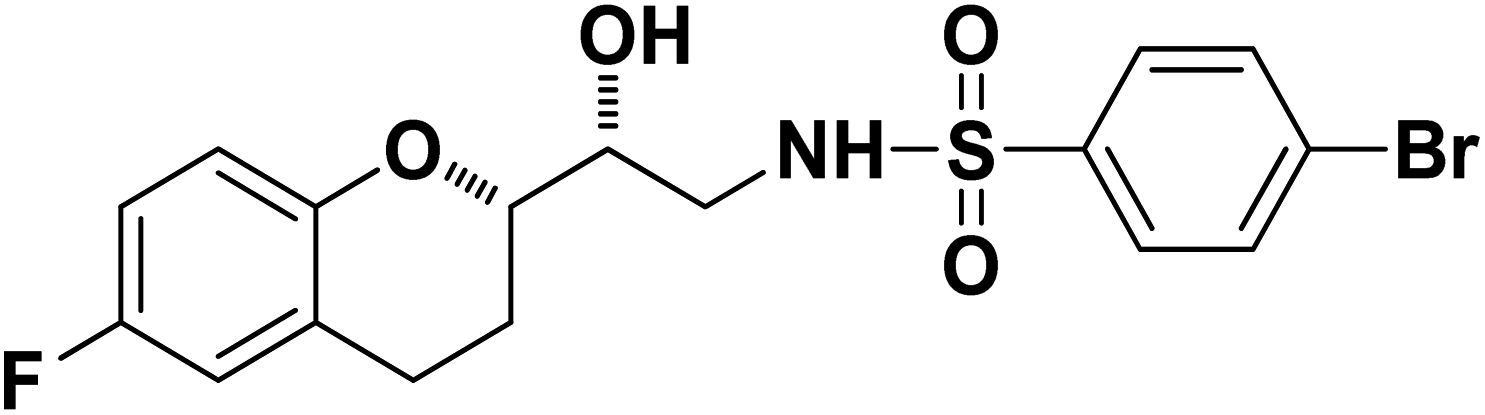
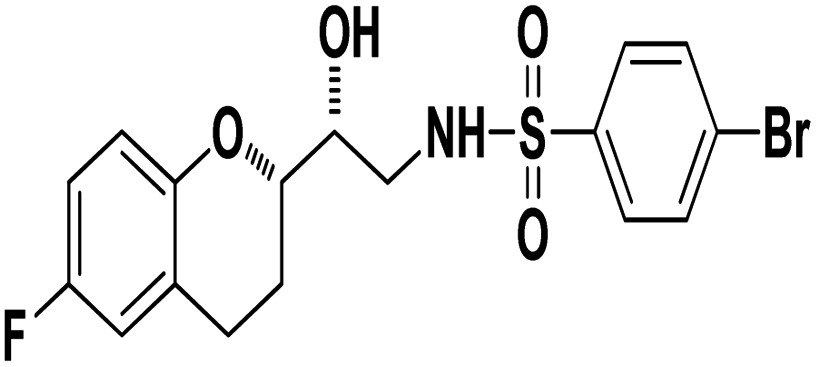
4-Bromo-N-((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)benzenesulfonamide (8b). White solid; yield: 79%; mp 195–198 °C; IR (vmax/cm−1): 3583(OH), 3388, 3203(N–H), 2930(Ar–C–H), 1393, 1143(SO), 1213, 964(S–N str) cm−1; H1-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.23 (1H, s, N–H),7.87–7.72 (4H, m, Ar–H), 6.89–6.83 (2H, m, Ar–H), 6.69–6.34 (1H, t, J = 5.0 Hz), 5.19 (1H, s, OH), 3.89–3.83(1H, d, J = 2.9 Hz), 3.83–3.79 (1H, m, –CH), 3.69–3.53 (2H, m, –CH2), 2.77–2.71 (2H, m, –CH2), 2.10–2.18 (1H, m, –CH2), 1.74–1.69 (1H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 155.0, 149.8, 141.6, 131.5, 128–5, 126.3, 119.8, 115.1, 113.3, 78.0, 70.8, 41.9, 23.8, 22.4.
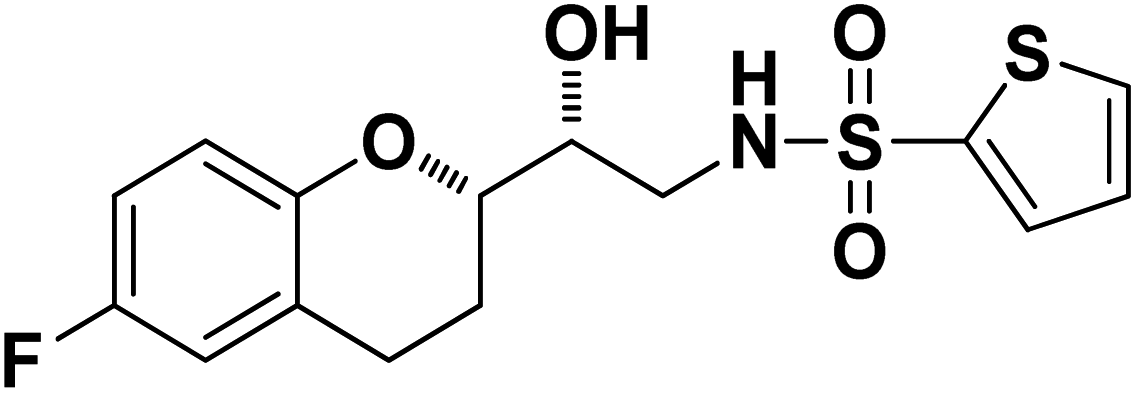
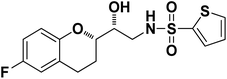
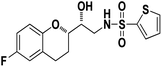
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)thiophene-2-sulfonamide (8c). White solid; yield: 80%; mp 132–135 °C; IR (KBr) (vmax/cm−1) 3574, 3412, 3205, 2945, 1336, 1163, 1236, 987; H1-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.44 (1H, s, N–H),7.59–7.43 (1H, d, J = 5.9 Hz, Ar–H),7.35–7.12 (2H, m, Ar–H), 6.94–6.89 (2H, m, Ar–H), 6.53–6.34 (1H, t, J = 4.9 Hz), 5.30 (1H, s, OH), 3.78–3.74 (1H, m, –CH), 3.34–3.58 (1H, m,–CH), 3.34–3.12 (2H, m, –CH2), 2.73–2.68 (2H, m, –CH2), 1.96–1.78 (2H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 155.4, 150.5, 127.7, 125.9, 125.6, 124.6, 119.8, 115.4, 113.0, 82.5, 71.5, 41.0, 23.5, 22.7.
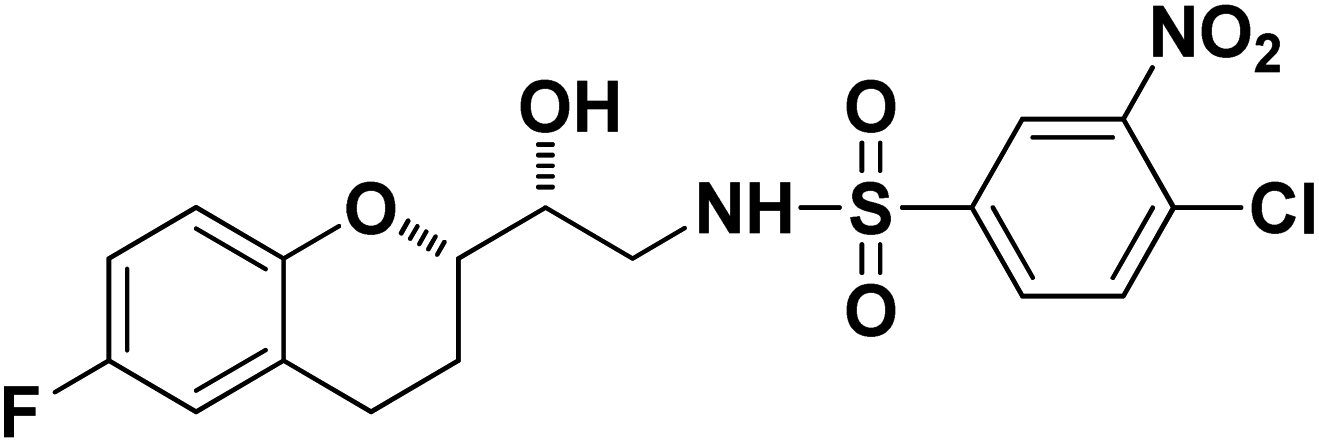
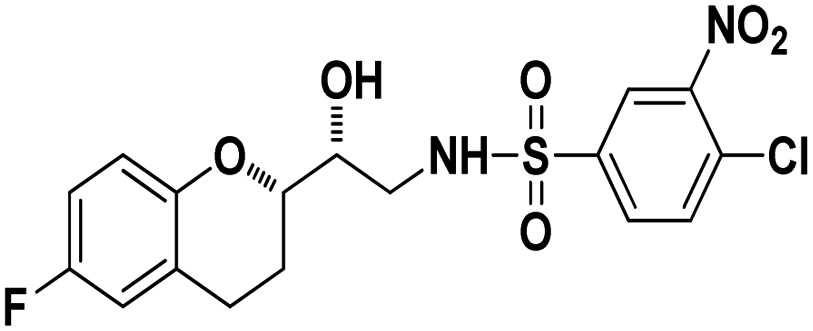
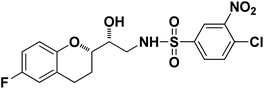
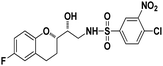
4-Chloro-N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-3-nitrobenzenesulfonamide (8d). White solid; yield: 76%; mp 126–129 °C; IR (KBr) (vmax/cm−1): 3523, 3454, 3298,3198, 1539, 1331, 1147, 1228, 983. H1-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.46 (1H, s, N–H),8.08–8.06 (2H, d, J = 6.5 Hz, Ar–H), 7.97–7.99 (1H, t, J = 6.7 Hz, Ar–H), 6.90–6.84 (2H, m, Ar–H), 6.68–6.64 (1H, m, Ar–H), 5.25 (1H, s, OH), 3.78–3.74 (1H, t, J = 2.8 Hz, –CH), 3.39 (1H, s, –CH), 3.10–2.94 (2H, m, –CH2), 2.70–2.50 (2H, m, –CH2), 2.03–1.67 (1H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 155.3, 147.3, 138.0, 134.2, 131.7, 129.8, 125.8, 124.8, 111.8, 110.3, 81.5, 72.9, 41.9, 25.9, 21.9. LC-MS (m/z): 431 [M + H]+ .; anal. calcd for C17H16FN2O6S: C 47.39, H 3.74, N 6.50; found: C 46.82, H 3.23, N 6.22.
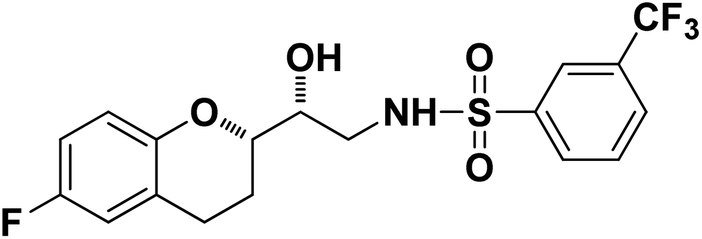
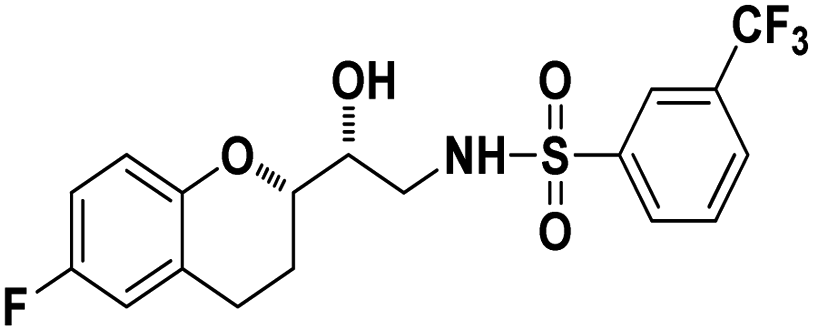
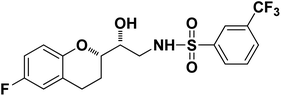
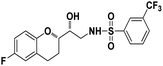
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-3-(trifluoromethyl)benzenesulfonamide (8e). White solid; yield: 79%; mp 155–158 °C; IR (KBr) (vmax/cm−1): 3533, 3267, 3065, 1321, 1121, 987; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.10 (1H, s, N–H),7.96(1H, s, Ar–H), 7.75–7.64 (3H, m, Ar–H), 6.94–6.86 (2H, m, Ar–H), 6.72–6.69 (1H, t, J = 5.0 Hz), 5.36 (1H, s, OH), 3.91 (1H, brs), 3.83–3.71 (1H, m, –CH), 3.41–3.29 (2H, m, –CH2), 2.78–2.69 (2H, m, –CH2), 2.09–2.18 (1H, m, –CH2),; 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz):154.7, 149.5, 141.2, 130.3, 129.6, 129.2, 128.1, 125.9, 123.8, 122.2, 119.9, 115.4, 113.5, 77.9, 69.9,44.8, 23.8, 22.8.
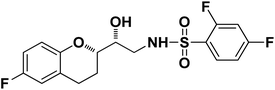
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-2,4-difluorobenzenesulfonamide (8f). White solid; yield: 81%; mp 135–138 °C; IR (KBr) (vmax/cm−1): 3537, 3249, 3106, 1324, 1144, 1210, 969; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.58 (1H, s, N–H), 7.94–7.85 (1H, m, Ar–H) 7.74–7.29 (1H, m, Ar–H), 6.85–6.98 (1H, m, Ar–H), 6.79–6.65 (3H, m, Ar–H), 5.26 (1H, s, OH), 3.87–3.84 (1H, m, –CH), 3.57–3.53 (1H, m, –CH), 3.12–3.02 (2H, m, –CH2), 2.81–2.69 (2H, m, –CH2), 2.14–1.91 (1H, m, –CH2),; 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 157.3, 154.8, 152.6, 150.5, 129.1, 124–9, 121.2, 118.4, 115.4, 113.5, 110.9, 103.3, 76.9, 70.1, 41.1, 23.8, 22.5.
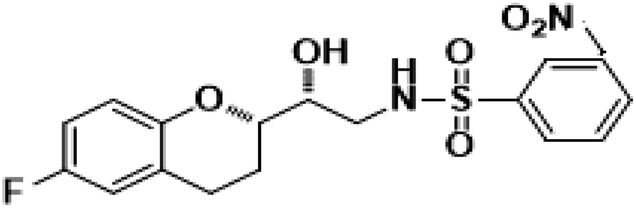
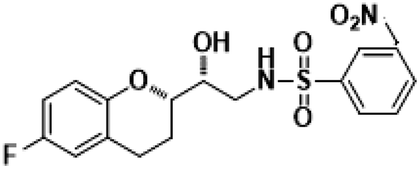
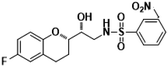
N-((R)-2-((S)-6-Fluorochroman-2-yl)2-hydoxyethyl)-2-nitrobenzenesulfonamide (8g). White solid; yield: 80%; mp 134–137 °C; IR (KBr) (vmax/cm−1): 3543, 3294, 3160, 1506, 1347, 1154, 1238, 984; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.56 (1H, s, N–H),7.85–7.34 (4H, m, Ar–H), 6.89–6.75 (2H, m, Ar–H), 6.72–6.69 (1H, m, Ar–H), 5.38 (1H, s, OH), 3.86–3.77(1H, t, J = 2.9 Hz, –CH), 3.76–3.62 (1H, m, –CH), 3.42–3.39 (2H, m, –CH2), 2.84–2.76 (2H, brs, –CH2), 2.10–1.77 (2H, m, –CH2),; 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 156.4, 150.97, 148.6, 136.4, 132.5, 130.0, 126.7, 123.8, 121.2, 119.4, 115.3, 112.6, 77.0, 70.8, 42.1, 23.2, 21.9.
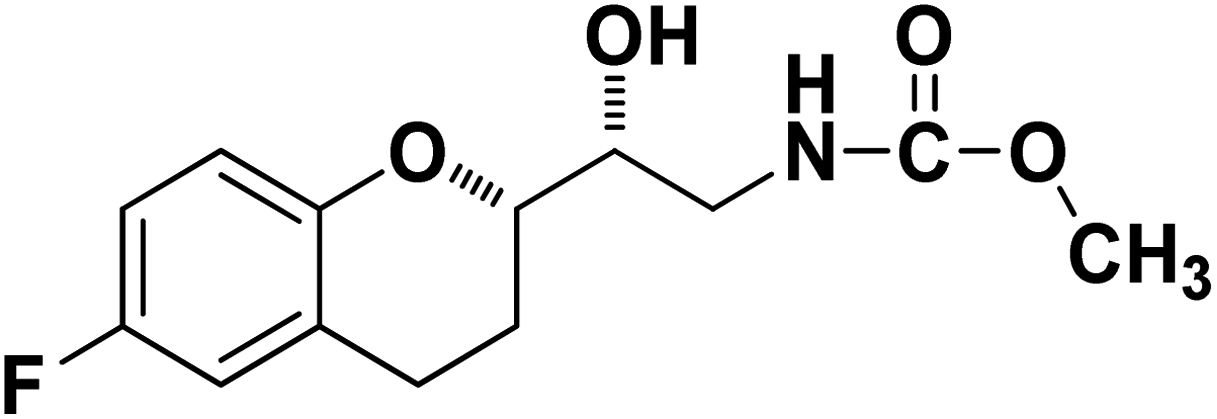
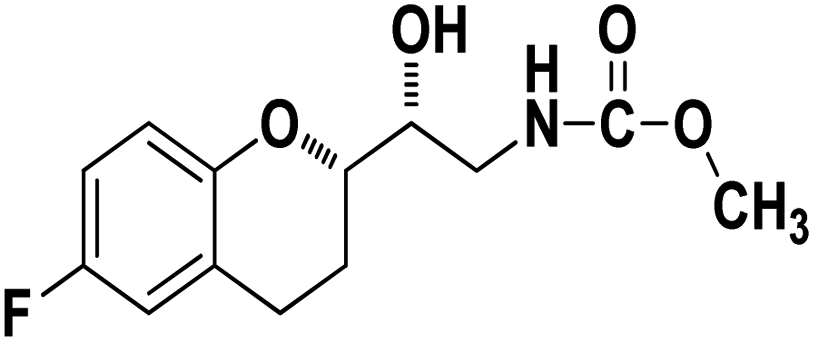
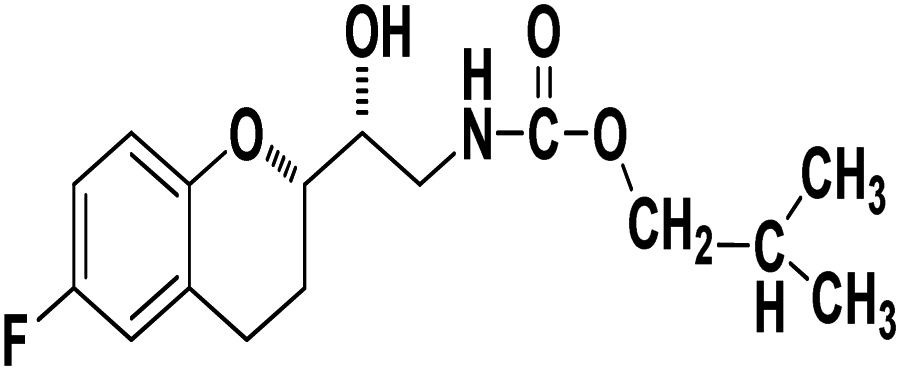
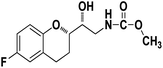
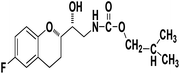
Methyl ((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9a). White solid; yield:79%; mp 132–135 °C; IR (KBr) (vmax/cm−1): 3524,3246, 3148, 1717, 1232, 1080; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.61 (1H, s, N–H),7.08–7.05 (1H, m, Ar–H), 6.92–6.85 (1H, m, Ar–H), 6.74–6.71 (1H, m, Ar–H), 5.16 (1H, s, OH), 4.01–3.81 (1H, m, –CH), 3.79–3.61 (1H, m, –CH), 3.59–3.32 (3H, m, –CH3), 3.05–3.00 (2H, m, –CH), 2.76–2.51 (2H, m, –CH2), 2.18–2.07 (1H, m, –CH2), 1.73–1.66 (1H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 156.8, 154.8, 149.3, 123.8, 117.4, 114.9, 113.8, 77.3, 69.9, 46.8, 24.0, 21.5; LCMS calculated for C13H16FNO4: 269.11; found m/z 270 [M + H]+.
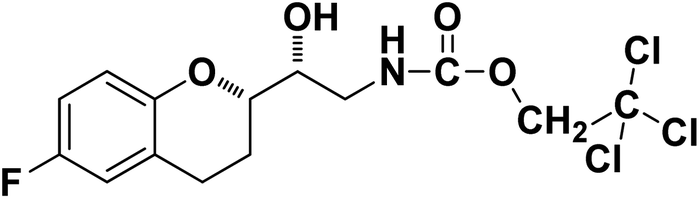
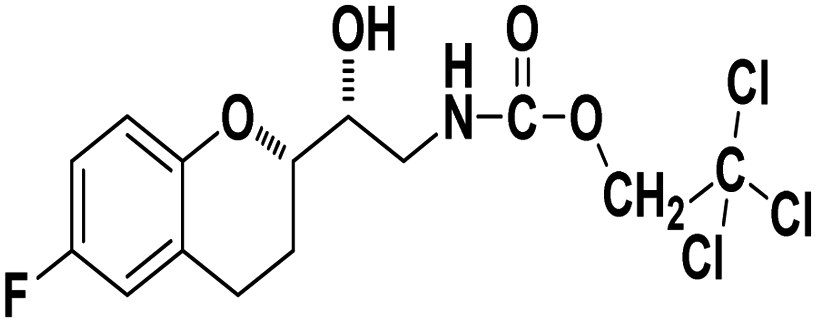
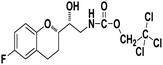
2,2,2-Trichloroethyl ((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9b). White solid; yield: 82%; mp 167–170 °C; IR (KBr) (vmax/cm−1): 3592, 3364, 3284, 2947, 1678, 1284, 1216; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.38 (1H, s, N–H), 6.93–6.71 (3H, m, Ar–H), 5.14 (1H, s, OH), 4.85(2H, s,–CH2), 4.09 (1H, brs, –CH), 3.83–3.81 (1H, m, –CH), 3.00–3.07 (2H, m, –CH2), 2.74–2.50 (2H, m, –CH2), 1.76–1.71 (2H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 154.6, 152.4, 149.9, 123.7, 117.4, 114.3, 110.5, 82.6, 78.6, 73.6, 69.0, 41.9, 23.5, 21.7.
Isobutyl ((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9c). White solid; yield: 78%; mp 126–129 °C; IR (KBr) (vmax/cm−1): 3531, 3335, 3277, 3029, 2929, 1694, 1296, 1226; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.58 (1H, s, N–H), 6.76–6.68 (3H, m, Ar–H), 5.19 (1H, s, OH), 4.34–4.26 (1H, t, J = 3.1 Hz, –CH), 3.85–3.83 (2H, m, –CH2), 3.76–3.35 (2H, m, –CH), 3.24–3.05 (2H, m, –CH2), 2.77–2.75 (2H, m, –CH2), 1.84–1.76 (2H, m, –CH2), 0.91(6H, s, –CH3); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 157.9, 155.7, 150.3, 123.4, 117.5, 117.4, 115.6, 113.9, 76.6, 73.3, 71.6, 43.7, 28.0, 24.5, 22.9, 19.1.; LC-MS (m/z): 312 [M + H]+; anal. calcd for C16H22FNO4: C 61.72, H 7.12, N 4.50; found: C 61.20, H 6.62, N 4.05.
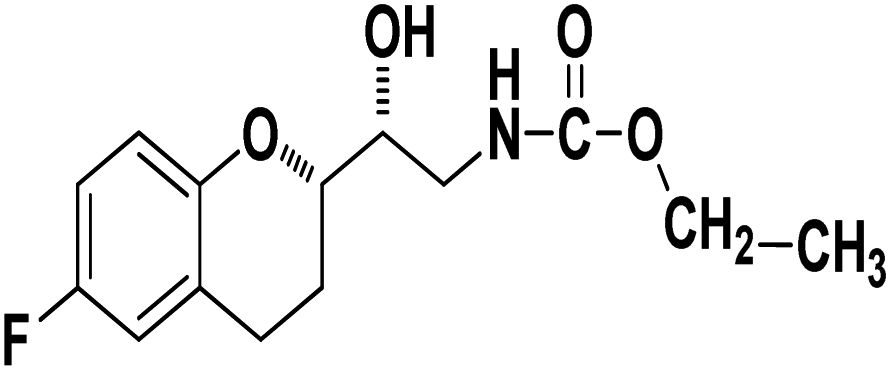
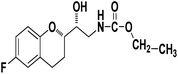
Ethyl((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9d). White solid; yield: 74%; mp 132–135 °C; IR (KBr) (vmax/cm−1): 3624, 3294, 3142, 2914, 1678, 1296, 1234; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.53 (1H, s, N–H), 6.92–6.85 (2H, m, Ar–H), 6.74–6.71 (1H, d, J = 5.0 Hz, Ar–H), 5.14 (1H, s, OH), 4.29–4.17(1H, t, J = 3.2 Hz, –CH), 4.08–3.98 (2H, m, –CH2), 3.79–3.62 (1H, m, –CH), 3.29–3.23 (2H, m, –CH2), 2.98–2.74 (2H, m, –CH2), 1.73–1.70 (2H, m, –CH2), 1.23–1.18 (3H, m, –CH3); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 156.7, 153.9, 149.9, 124.4, 118.5, 114.7, 111.4, 78.6, 73.3, 59.9, 42.2, 23.86, 22.1, 11.9.; LCMS calculated for C14H18FNO4: 283.12; found m/z 284 [M + H]+.
O), 1221, 976 cm−1; 1H-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.24 (1H, s, N–H),7.15–7.25 (4H, m, Ar–H), 6.71–6.86 (2H, m, Ar–H), 6.63–6.34 (1H, t, J = 5.0 Hz), 5.33 (1H, s, OH), 3.77–3.84 (2H, m, –CH), 3.09–3.11 (2H, m, –CH2), 2.73–2.78 (2H, m, –CH2), 2.09–2.18 (2H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 167.4, 154.6, 151.0, 141.3, 130.6, 125.7, 119.8, 115.9, 115.2, 113.1, 77.6, 71.5, 46.4, 23.8, 22.2.; LC-MS (m/z): 370 [M + H]+ .; anal. calcd for C17H17F2NO4S: C 55.28, H 4.64, N 3.76; found: C 54.62, H 4.25, N 3.29.
4-Bromo-N-((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)benzenesulfonamide (8b). White solid; yield: 79%; mp 195–198 °C; IR (vmax/cm−1): 3583(OH), 3388, 3203(N–H), 2930(Ar–C–H), 1393, 1143(SO), 1213, 964(S–N str) cm−1; H1-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.23 (1H, s, N–H),7.87–7.72 (4H, m, Ar–H), 6.89–6.83 (2H, m, Ar–H), 6.69–6.34 (1H, t, J = 5.0 Hz), 5.19 (1H, s, OH), 3.89–3.83(1H, d, J = 2.9 Hz), 3.83–3.79 (1H, m, –CH), 3.69–3.53 (2H, m, –CH2), 2.77–2.71 (2H, m, –CH2), 2.10–2.18 (1H, m, –CH2), 1.74–1.69 (1H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 155.0, 149.8, 141.6, 131.5, 128–5, 126.3, 119.8, 115.1, 113.3, 78.0, 70.8, 41.9, 23.8, 22.4.
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)thiophene-2-sulfonamide (8c). White solid; yield: 80%; mp 132–135 °C; IR (KBr) (vmax/cm−1) 3574, 3412, 3205, 2945, 1336, 1163, 1236, 987; H1-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.44 (1H, s, N–H),7.59–7.43 (1H, d, J = 5.9 Hz, Ar–H),7.35–7.12 (2H, m, Ar–H), 6.94–6.89 (2H, m, Ar–H), 6.53–6.34 (1H, t, J = 4.9 Hz), 5.30 (1H, s, OH), 3.78–3.74 (1H, m, –CH), 3.34–3.58 (1H, m,–CH), 3.34–3.12 (2H, m, –CH2), 2.73–2.68 (2H, m, –CH2), 1.96–1.78 (2H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 155.4, 150.5, 127.7, 125.9, 125.6, 124.6, 119.8, 115.4, 113.0, 82.5, 71.5, 41.0, 23.5, 22.7.
4-Chloro-N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-3-nitrobenzenesulfonamide (8d). White solid; yield: 76%; mp 126–129 °C; IR (KBr) (vmax/cm−1): 3523, 3454, 3298,3198, 1539, 1331, 1147, 1228, 983. H1-NMR (400 MHz, DMSO-d6), δ, ppm (J, Hz): 8.46 (1H, s, N–H),8.08–8.06 (2H, d, J = 6.5 Hz, Ar–H), 7.97–7.99 (1H, t, J = 6.7 Hz, Ar–H), 6.90–6.84 (2H, m, Ar–H), 6.68–6.64 (1H, m, Ar–H), 5.25 (1H, s, OH), 3.78–3.74 (1H, t, J = 2.8 Hz, –CH), 3.39 (1H, s, –CH), 3.10–2.94 (2H, m, –CH2), 2.70–2.50 (2H, m, –CH2), 2.03–1.67 (1H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 155.3, 147.3, 138.0, 134.2, 131.7, 129.8, 125.8, 124.8, 111.8, 110.3, 81.5, 72.9, 41.9, 25.9, 21.9. LC-MS (m/z): 431 [M + H]+ .; anal. calcd for C17H16FN2O6S: C 47.39, H 3.74, N 6.50; found: C 46.82, H 3.23, N 6.22.
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-3-(trifluoromethyl)benzenesulfonamide (8e). White solid; yield: 79%; mp 155–158 °C; IR (KBr) (vmax/cm−1): 3533, 3267, 3065, 1321, 1121, 987; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.10 (1H, s, N–H),7.96(1H, s, Ar–H), 7.75–7.64 (3H, m, Ar–H), 6.94–6.86 (2H, m, Ar–H), 6.72–6.69 (1H, t, J = 5.0 Hz), 5.36 (1H, s, OH), 3.91 (1H, brs), 3.83–3.71 (1H, m, –CH), 3.41–3.29 (2H, m, –CH2), 2.78–2.69 (2H, m, –CH2), 2.09–2.18 (1H, m, –CH2),; 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz):154.7, 149.5, 141.2, 130.3, 129.6, 129.2, 128.1, 125.9, 123.8, 122.2, 119.9, 115.4, 113.5, 77.9, 69.9,44.8, 23.8, 22.8.
N-((R)-2-((S)-6-Fluorochroman-2-yl)-2-hydroxyethyl)-2,4-difluorobenzenesulfonamide (8f). White solid; yield: 81%; mp 135–138 °C; IR (KBr) (vmax/cm−1): 3537, 3249, 3106, 1324, 1144, 1210, 969; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.58 (1H, s, N–H), 7.94–7.85 (1H, m, Ar–H) 7.74–7.29 (1H, m, Ar–H), 6.85–6.98 (1H, m, Ar–H), 6.79–6.65 (3H, m, Ar–H), 5.26 (1H, s, OH), 3.87–3.84 (1H, m, –CH), 3.57–3.53 (1H, m, –CH), 3.12–3.02 (2H, m, –CH2), 2.81–2.69 (2H, m, –CH2), 2.14–1.91 (1H, m, –CH2),; 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 157.3, 154.8, 152.6, 150.5, 129.1, 124–9, 121.2, 118.4, 115.4, 113.5, 110.9, 103.3, 76.9, 70.1, 41.1, 23.8, 22.5.
N-((R)-2-((S)-6-Fluorochroman-2-yl)2-hydoxyethyl)-2-nitrobenzenesulfonamide (8g). White solid; yield: 80%; mp 134–137 °C; IR (KBr) (vmax/cm−1): 3543, 3294, 3160, 1506, 1347, 1154, 1238, 984; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.56 (1H, s, N–H),7.85–7.34 (4H, m, Ar–H), 6.89–6.75 (2H, m, Ar–H), 6.72–6.69 (1H, m, Ar–H), 5.38 (1H, s, OH), 3.86–3.77(1H, t, J = 2.9 Hz, –CH), 3.76–3.62 (1H, m, –CH), 3.42–3.39 (2H, m, –CH2), 2.84–2.76 (2H, brs, –CH2), 2.10–1.77 (2H, m, –CH2),; 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 156.4, 150.97, 148.6, 136.4, 132.5, 130.0, 126.7, 123.8, 121.2, 119.4, 115.3, 112.6, 77.0, 70.8, 42.1, 23.2, 21.9.
Methyl ((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9a). White solid; yield:79%; mp 132–135 °C; IR (KBr) (vmax/cm−1): 3524,3246, 3148, 1717, 1232, 1080; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.61 (1H, s, N–H),7.08–7.05 (1H, m, Ar–H), 6.92–6.85 (1H, m, Ar–H), 6.74–6.71 (1H, m, Ar–H), 5.16 (1H, s, OH), 4.01–3.81 (1H, m, –CH), 3.79–3.61 (1H, m, –CH), 3.59–3.32 (3H, m, –CH3), 3.05–3.00 (2H, m, –CH), 2.76–2.51 (2H, m, –CH2), 2.18–2.07 (1H, m, –CH2), 1.73–1.66 (1H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 156.8, 154.8, 149.3, 123.8, 117.4, 114.9, 113.8, 77.3, 69.9, 46.8, 24.0, 21.5; LCMS calculated for C13H16FNO4: 269.11; found m/z 270 [M + H]+.
2,2,2-Trichloroethyl ((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9b). White solid; yield: 82%; mp 167–170 °C; IR (KBr) (vmax/cm−1): 3592, 3364, 3284, 2947, 1678, 1284, 1216; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.38 (1H, s, N–H), 6.93–6.71 (3H, m, Ar–H), 5.14 (1H, s, OH), 4.85(2H, s,–CH2), 4.09 (1H, brs, –CH), 3.83–3.81 (1H, m, –CH), 3.00–3.07 (2H, m, –CH2), 2.74–2.50 (2H, m, –CH2), 1.76–1.71 (2H, m, –CH2); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 154.6, 152.4, 149.9, 123.7, 117.4, 114.3, 110.5, 82.6, 78.6, 73.6, 69.0, 41.9, 23.5, 21.7.
Isobutyl ((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9c). White solid; yield: 78%; mp 126–129 °C; IR (KBr) (vmax/cm−1): 3531, 3335, 3277, 3029, 2929, 1694, 1296, 1226; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.58 (1H, s, N–H), 6.76–6.68 (3H, m, Ar–H), 5.19 (1H, s, OH), 4.34–4.26 (1H, t, J = 3.1 Hz, –CH), 3.85–3.83 (2H, m, –CH2), 3.76–3.35 (2H, m, –CH), 3.24–3.05 (2H, m, –CH2), 2.77–2.75 (2H, m, –CH2), 1.84–1.76 (2H, m, –CH2), 0.91(6H, s, –CH3); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 157.9, 155.7, 150.3, 123.4, 117.5, 117.4, 115.6, 113.9, 76.6, 73.3, 71.6, 43.7, 28.0, 24.5, 22.9, 19.1.; LC-MS (m/z): 312 [M + H]+; anal. calcd for C16H22FNO4: C 61.72, H 7.12, N 4.50; found: C 61.20, H 6.62, N 4.05.
Ethyl((R)-2-((S)-6-fluorochroman-2-yl)-2-hydroxyethyl)carbamate (9d). White solid; yield: 74%; mp 132–135 °C; IR (KBr) (vmax/cm−1): 3624, 3294, 3142, 2914, 1678, 1296, 1234; H1-NMR (400 MHz, DMSO-d6) δ, ppm (J, Hz): 8.53 (1H, s, N–H), 6.92–6.85 (2H, m, Ar–H), 6.74–6.71 (1H, d, J = 5.0 Hz, Ar–H), 5.14 (1H, s, OH), 4.29–4.17(1H, t, J = 3.2 Hz, –CH), 4.08–3.98 (2H, m, –CH2), 3.79–3.62 (1H, m, –CH), 3.29–3.23 (2H, m, –CH2), 2.98–2.74 (2H, m, –CH2), 1.73–1.70 (2H, m, –CH2), 1.23–1.18 (3H, m, –CH3); 13C NMR (100 MHz, DMSO-d6) δ, ppm (J, Hz): 156.7, 153.9, 149.9, 124.4, 118.5, 114.7, 111.4, 78.6, 73.3, 59.9, 42.2, 23.86, 22.1, 11.9.; LCMS calculated for C14H18FNO4: 283.12; found m/z 284 [M + H]+.
2.2. Anti-microbial activity
Disk diffusion method was used for evaluation of antibacterial activity.28 The cultures were grown in nutrient agar media and subcultured for log phasic cultures in a liquid nutrient broth medium for MIC studies and further subcultured onto the media in Petri plates for the experimental purposes. The broth cultures were diluted with sterilized saline to bring the final size of inoculum approximately to 105–106 CFU mL−1. The compounds were diluted in acetone, DMSO and diethyl ether for biological assays. Among the three solvents, diethyl ether is preferred over the other two solvents due to its non-turbidity and high dissolving capacity. The bacterial cultures were placed on the media and incubated at 37 °C for 24 h to 48 h which is a suitable period for their rapid growth along with the diluted compounds introduced through disks dipped and placed over the nutrient media is effective for the determination of antibacterial efficacy of the compounds at a constant time interval. For disk diffusion method, the diluted test compounds were introduced onto the disk, and once the disk was found completely saturated, it was immediately transferred onto the surface of the medium with bacterial inoculums spread by spread plate method evenly. The Petri dishes were incubated at 37 °C for 24 h with title compounds, and MIC was determined by maintaining at different concentrations of the compounds against microbial cultures. The MIC of the synthesized compounds against three pathogenic fungi, Aspergillus niger, Aspergillus flavus and Rhizopus arrhizus was determined using the poison plate technique.29 Test compounds were dissolved in DMSO (10 mL) before mixing with potato dextrose agar medium (PDA, 90 mL). The 12 wells of each row were filled with 0.5 mL sterilized potato dextrose agar medium. Sequentially, wells 2–11 received an additional 0.5 mL of a mixture of culture medium, and compounds were serially diluted to create a concentration sequence from 5 to 50 μg mL−1. Well 1 served as growth control and well 12 as antibiotic control. The deep wells were incubated for 24 h at 37 °C. The resulting turbidity was observed, and after 24 h MIC was determined to be where growth was no longer visible by assessment of turbidity by optical density readings at 600 nm with a Beckman DU-70 UV-Vis spectrophotometer. The results were compared with the activity of the standard antibiotics, Streptomycin and Fluconazole.
2.3. In vitro anti-inflammatory activity
2.3.1. Maintenance of cell lines and toxicity studies. RAW 264.7 murine macrophage cell lines were procured from NCCS Pune, India. RAW macrophages were plated into T75 flasks and cultured in Dulbecco's Modified Eagle Medium (DMEM) with high glucose, stable glutamine and sodium pyruvate, supplemented with 10% fetal bovine serum heat inactivated (65 °C for 20 min) and 1% penicillin–streptomycin antibiotics (100 IU/mL penicillin and 100 μg mL−1 streptomycin) at 37 °C in 5% CO2 atmosphere. The toxicity study was conducted in accordance with OECD guidelines (Testing of Chemical Number 423). Healthy RAW 264.7 murine macrophage cell lines were used in this investigation. Cell lines were treated with different concentrations (i.e., 200 μM and 400 μM) of title compounds and observed for 48 h from the time of administration. The median non-toxic concentration (MNTC) and cytotoxic concentration (CC50) were determined. The minimum concentration of the test compounds which has not shown any toxic effect on healthy RAW 264.7 cell lines were selected for further anti-inflammatory analysis.
2.3.2. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cell viability was measured based on the formation of purple formazan, which is metabolised from colourless MTT (Sigma-Aldrich) by mitochondrial dehydrogenases, enzymes that are only active in live cells. RAW 264.7 cells (5 × 105 cells per mL) were seeded in a 96-well plate. The cells were pre-treated with various concentrations (100 μM and 200 μM) of synthesized compounds for 1 h and then stimulated by 100 μM LPS for 24 h. Following the incubation with title compounds and LPS, the cultured media was replaced with fresh media and the cells were incubated with 0.5 mg mL−1 MTT solution for 3 h. The supernatant was then discarded and the formazan blue, which was formed in the cells, was dissolved in dimethyl sulfoxide (Sigma-Aldrich). The optical density was measured at 540 nm with an ELISA plate reader. Percentage of inflammation was calculated by subtracting the percentage of cell viability over control cells.30
2.3.3. Determination of nitrite production. Nitrite accumulation (NO2) in cell culture media was determined by the Pekarova method.31 Briefly, 1 × 106 cells were seeded in a T-75 flask, allowed to adhere overnight, and then treated as previously mentioned in MTT assay. At the end of the different incubations in the CO2 incubator at 37 °C with 5% CO2, the different cell supernatants were collected, the samples (1 mL) were mixed with an equal volume of Griess reagent (1 mL of 1:1 0.1% naphthyl-ethylenediamine and 1% sulfanilamide in 5% phosphoric acid) in a tube, and incubated in the dark for 10 min at room temperature. Then the absorbance of the reaction mixture was measured at 540 nm on a microplate reader (Thermo Scientific Multiskan EX). The NO2 concentration was determined using a sodium nitrite (NaNO2) standard curve (working range: 0.1–6.25 μM). Each treatment was carried out in triplicates and the final results were expressed as μmol NO2-/mg protein.
2.4. COX inhibition assay
The assay was performed by using colorimetric COX (ovine) inhibitor Screening assay kit (Catalogue No. 760111, Cayman Chemicals Co., Ann Arbor, MI).32 Briefly, the reaction mixture contains, 150 mL of assay buffer, 10 mL of heme, 10 mL of enzyme (either COX-1 or COX-2), and 50 mL of sample (0.1 mM). The assay utilizes the peroxidase component of the COX catalytic domain. The peroxidase activity can be assayed calorimetrically by monitoring the appearance of oxidized N,N,N,N-tetramethyl-p-phenylenediamine (TMPD) at 590 nm. Celecoxib (0.1 mM) was used as a standard drug. The percent COX inhibition was calculated using following equation.
| COX inhibition activity (%) = 1 − T/C × 100 |
where T is the absorbance of the inhibitor well at 590 nm, C is the absorbance of the 100% initial activity without inhibitor well at 590 nm.
2.5. In vivo anti-inflammatory activity
2.5.1. Maintenance of animals. Animals were assigned into several groups randomly and each group consists of six animals. The animals were kept in appropriate cages at temperature controlled (25 ± 2 °C) rooms, under a 12 h light and dark cycle and they were fed standard rodent pellet. All the animals were acclimatized for a week before the experiment. Experimental protocols were reviewed and approved by the Institutional Animal Ethics Committee (No.10/(i)/a/CPCSEA/IAEC//SVU/Zool/WR/dt.08.07.2012.) and conform to the Indian National Science Academy Guidelines for the use and care of experimental animals in research. The animal house registration number with Government of India is 45/c/06/CPCSEA.
2.5.2. Carrageenan induced hind paw edema assay. Rats were divided into various groups (n1/46) and allowed for free access to water ad libitum. Different groups of rats administered with celecoxib (100 mg kg−1, b.w.) and various test compounds (100 mg kg−1, v) orally. One group of rats served as a control and administered with saline (1% (w/v); 10 mL kg−1, b.w., p.o.). One hour after the drug administration, to all rats, hind paw edema was induced by the method of Winter et al.33 by injecting 0.1 mL of 1% (w/v) solution of carrageenan subcutaneously into the sub planter region of hind paw. The hind paw edema volume was measured by volume displacement method using plethysmometer by immersing the paw till the level lateral malleolus at various time intervals (1, 3 and 6 h) after carrageenan injection.
2.5.3. Cotton pellets induced granuloma assay. Two sterilized cotton pellets (10 mg) were implanted on ventral regions of rats, procedure described by winter and porter34 and divided into various four groups (six rats/group). Different groups of rats administered with celecoxib (100 mg kg−1), and test compounds (100 mg kg−1) orally for the duration of 8 d. Control group received saline (1% (w/v); 10 mL kg−1, b.w., p.o.). On the day 9, rats were sacrificed with excess ether anesthesia. The cotton pellets were removed and freed from extraneous tissue and used for granular tissue formation by recording wet (immediately) and dry weight of pellets. The granular tissue formation was studied by drying cotton pellets at 55 °C for 6 h or till the weight of pellets remains constant. The dry weight was calculated after deducting cotton pellet weight and taken as a granular tissue formation.
2.6. Molecular docking studies
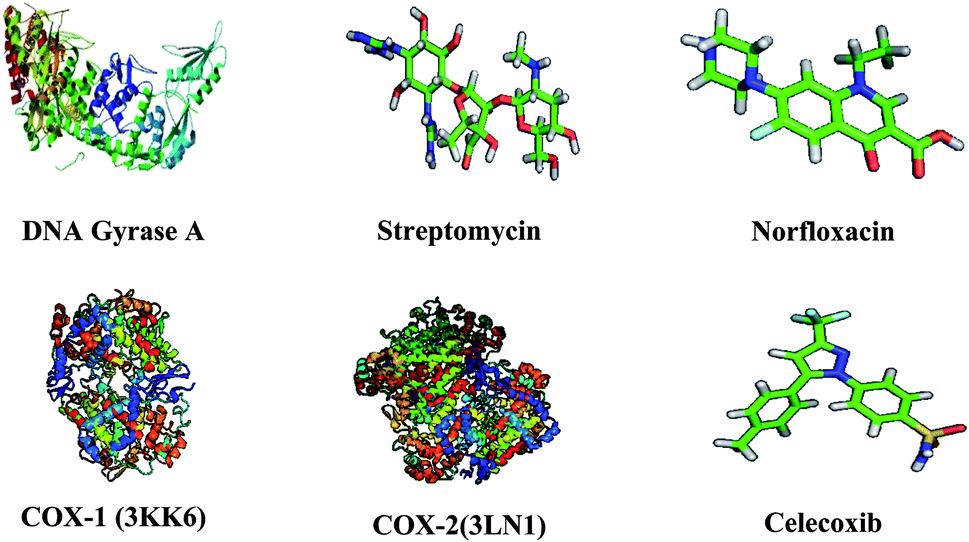
The three-dimensional structure of DNA gyrase A (3LPX), COX-1 (3KK6), COX-2 (PDB: 3LN1), and the reference drugs, such as streptomycin, norfloxacin and celecoxib were downloaded from the RCSB protein Data Bank and Pub Chem Data bank.35 The atomic coordinates of the protein was estranged and geometry optimization was done using Argus Lab 4.0.1. The chemical structures of compounds were prepared using ChemDraw Ultra and converted all the ligands into Pdbqt file format and atomic coordinates were generated using Pyrx 2010.12. The active sites are the coordinates of the ligand in the original target protein grids, and these active binding sites of target protein were analysed using the Drug Discovery Studio version 3.0 and 3D Ligand Site virtual tools.36 The structures of target protein and reference compound were shown in Fig. 2. Molecular docking studies were carried against DNA gyrase A, COX-1 and COX-2 protein with compounds 8a–g and 9a–d and the reference drugs using the docking module implemented in Pyrx 2010.12. Initially the protein structures were protonated with the addition of polar hydrogens, followed by energy minimization with the MMFF94x force field, in order to get the stable conformer of the proteins. Flexible docking was employed, the inhibitor binding site residues were softened and highlighted through the “Site Finder” module implemented in the Pymol software.37 The grid dimensions were predicted as X: 28.27, Y: 27.13, Z: 28.51 for DNA gyrase A, COX-1 and COX-2 respectively. The docking was carried out with the default parameters i.e., placement: triangle matcher, recording 1: London dG, refinement: force field, and a maximum of 10 conformations of each compound were allowed to be saved in a separate database file in a.mdb format. After the docking process, the binding energy and binding affinity of the protein-ligand complexes were calculated using Pymol viewer tool.38
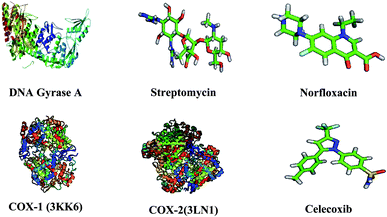 |
| | Fig. 2 Three dimensional structures of target proteins and reference compounds. | |
2.7. Bioactivity and toxicity risk studies
The bioactivity & toxicity risks like GPCR (G protein-coupled receptor) ligand property, ICM (ion channel modulator), KI (kinase inhibitor) and NRL (nuclear receptor ligand) interactions, PI (protease inhibitor) and EI (enzyme inhibitor) inhibitions, drug-likeness and drug scores of title compounds were determined using Molecular Operating Environment (MOE) software.39
2.8. ADMET properties
ADMET properties of title compounds have envisioned on pre ADMET online server,40 which helped to realize their potentialities with regard to in vivo BBB (blood brain barrier) penetration, in vitro PPB (plasma protein binding), in vitro Caco-2 cell permeability, %HIA (human intestinal absorption) and in vitro MDCK (Maden Darby Canine Kidney) cell permeability properties. In continuation, the toxicity properties viz., mutagenic, tumarogenic, irritant and reproductive effects also helped to ensure the drug-likeness. The BBB is fulfilled with intensely bound endothelial cells which restrict the ability of a compound that is to be carried into the bloodstream by passing through the administered route. The study of BBB penetration rate (BBB = [Brain]/[Blood]) assists us to examine the capability of a compound to percolate through blood–brain barrier, which is a vital in ascribing central nervous system (CNS) activity to the archives of pharmaceutical properties of a compound. The compounds which are capable to pass through the BBB are designated as CNS active (BBB penetration rate > 0.40) and which are inept are called CNS-inactive (BBB penetration rate < 0.40) compounds. On the other hand Caco-2 (human colon adenocarcinoma based cells) cells inter relates with intestinal epithelium system in multiple drug transport pathways like transcellular, paracellular and active efflux transports. The compounds conquering in vitro Caco-2 cell permeability value <4 are poor permeable, compounds in midst of 4–70 are moderately permeable and compounds with value >70 are extremely permeable and are easily transported to cellular biochemical processes. Besides, the degree of PPB impacts the level of distribution of unbound compound in body tissues and the amount of unbound compound which is distributed over the cellular sites of action, will further metabolizes and excreted from the system. The percentage of PPB (in vitro) categorizes the compounds as strongly bound if % PPB > 90% and as weakly bound if % PPB <90% and this degree of PPB of a compound determines its action as well as efficiency. Furthermore the MDCK cell system is identified as a useful tool to screen out the rapid permeability of a compound and to decide its potentiality as its cellular life span is smaller than the life span of Caco-2 cells and therefore the correlation will be so high. Mainly, in vitro MDCK permeability value <25 designates the compounds as poor permeable, value in between the range of 25–500 designates the compounds are reasonably permeable and value >500 designates the compounds are greatly permeable. Similarly the % HIA is considered as the percentage of a compound that is getting orally administered in to the hepatic portal vein. Mostly, it is expressed as comprehensive bioavailability and absorption which are measured from the ratio of aggregate excretion in bile, urine and feces. The % HIA in the range of 0–20 designates poor absorbance, 20–70 designates moderate absorbance and 70–100 designates good absorbance of a compound. In toxicology perception, it is significant to concern about the toxicology aspects of a compound as in to exclude the toxicity risks in its design. The negative toxicology result sustain the molecules under study as safer drugs as they are prone to mutagenicity, carcinogenicity and human ether-a-go-go related gene (HERG) channel inhibition on its administration in vivo.
2.9. QSAR studies
The QSAR descriptors of synthesized compounds have been evaluated from Molinspiration online server41 specifically, Molinspiration engine v2018.10 is used for property exploration and Molinspiration engine v2018.03 is used for bioactivity score exploration. Similarly, drug properties were predicted from OSIRIS online property explorer42 and the consolidated descriptors were given under the heads of Lipinski & Veber Parameters respectively. These predictions are very much helpful in interpreting the physico–chemical interactions of title compounds with their targets and ultimately assisted in ascertaining their drug properties by correlating with the bioactivity & toxicity risks studies.
2.10. Statistical analysis
All experiments were repeated at least three times. Results are expressed as the means ± standard deviation (SD) of three experiments. Statistical calculations were performed using prism Graph Pad 6.0.
3. Results and discussion
3.1. Chemistry
Nebivolol intermediate (5) was treated with various biopotent sulphonyl chlorides and chloroformates 6 (a-g & 7a-d) to obtain corresponding title compounds (8a–g, 9a–d) in high yields, with simple workup procedure. The synthetic route for the target compounds is depicted in Scheme 1. Product yields, melting points, reaction times and structures of the title compounds are given in Table 1.
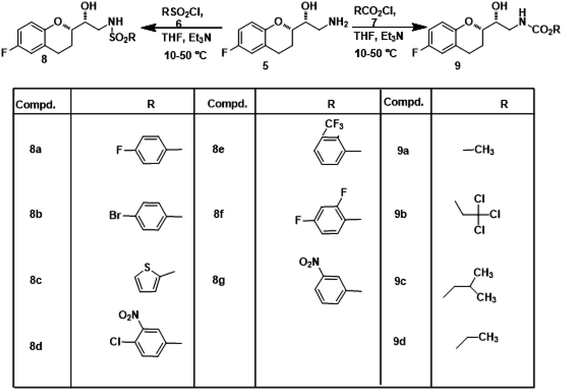 |
| | Scheme 1 Synthetic route for the synthesis of sulphonamide and carbamate derivatives of Nebivolol intermediate (8a–g & 9a–d). | |
Table 1 Physical data of the title compounds
| Comp. |
Structure |
Time (h) |
Yield (%) |
Melting point (°C) |
Purity profile |
| 8a |
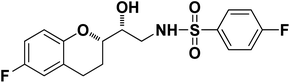 |
3.5 |
82 |
167–170 |
96% |
| 8b |
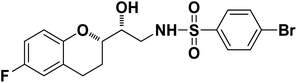 |
2.5 |
84 |
195–198 |
90% |
| 8c |
 |
4 |
82 |
132–135 |
91% |
| 8d |
 |
3.5 |
84 |
126–129 |
89% |
| 8e |
 |
4 |
79 |
155–158 |
89% |
| 8f |
 |
3 |
81 |
135–138 |
93% |
| 8g |
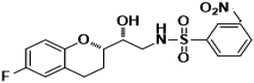 |
3.5 |
80 |
134–137 |
94% |
| 9a |
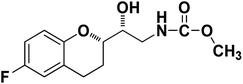 |
4 |
79 |
132–135 |
89% |
| 9b |
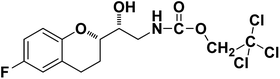 |
3.5 |
82 |
167–170 |
92% |
| 9c |
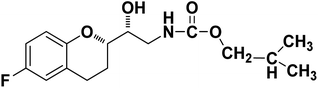 |
3 |
78 |
126–129 |
94% |
| 9d |
 |
4 |
74 |
132–135 |
86% |
The structures of the newly synthesized compounds were characterized by IR, 1H and 13C-NMR and LCMS. IR spectra of the title compounds displayed strong absorption bands at 1717–1678 cm−1 (CO sym str) and 1232–1216 cm−1 (C–O str) indicating the formation of carbamates. The absorption bands at 1396–1324 cm−1 and 1163–1121 cm−1, correspond to SO asymmetric stretch, SO symmetric stretching respectively. In 1H NMR, a singlet peak at δ 8.61–8.10 ppm is assigned for NH proton, a broad multiplet peak was observed in the range of 8.08–6.53 ppm, ascribed for aromatic protons, the hydroxyl proton (OH) resonated as a broad singlet at δ 5.38–5.14 ppm and multiplet peaks in the range of 4.85–0.91 ppm correspond to aliphatic protons. In 13C NMR spectra, a peak at δ 157.8–154.6 ppm corresponds to CO of carbamates function. Aromatic carbon signals appeared from δ 113.1 to 138.4. Besides NMR studies, mass spectra displayed the correct molecular ions for which measured LCMS and the data are in good agreement with calculated values of the newly synthesized compounds.
3.2. Anti-microbial activity
Antibacterial data revealed that the newly synthesized sulphonamide derivatives 8(a–g) showed better antibacterial activity than that of the carbamate derivatives 9(a–d). Out of all the title compounds, 8b, 8c, 8d, 8e, 8f, 8g and 9a have exhibited higher activity against both the Gram positive and Gram negative bacterial strains compared to the standard, streptomycin. In sulphonamide derivatives, the compounds having withdrawing groups on aromatic ring exhibited higher activity. For example the compound 8d exhibited high activity, it might be due to the presence of chlorine atom and nitro group, followed by the compounds 8e and 8b which may be due to the presence of m-tri fluoro methyl and p-bromo on the phenyl group. Whereas all the title compounds (8a–g & 9a–d) showed good to moderate activity against pathogenic fungi. The compounds which have shown significant activity against the bacterial strains were also exhibited the same extent of activity against the fungal strains also. Out of total, majority of the compounds displayed high activity with lowest MICs against A. flavus, A. flavus and R. arrhizus respectively when compared to the standard, fluconazole. These results suggest that small changes in the R group induced significant differences in antimicrobial activity. The MICs of title compounds against bacterial and fungal strains are shown in Tables 2 and 3.
Table 2 In vitro antimicrobial activity of the sulphonamide and carbamate derivatives of Nebivolol intermediate (8a–g & 9a–d)
| Compound |
MIC in μg mL−1 |
| Bacillus subtillis |
Staphylococcus aureus |
Escherichia coli |
Pseudomonas aeruginosa |
| Streptomycin |
25.40 ± 0.48 |
18.64 ± 1.07 |
26.40 ± 1.12 |
28.18 ± 0.80 |
| 8a |
31.60 ± 1.03 |
21.88 ± 0.33 |
30.14 ± 0.72 |
31.88 ± 1.04 |
| 8b |
15.08 ± 0.08 |
17.88 ± 0.61 |
12.68 ± 0.87 |
22.42 ± 0.40 |
| 8c |
21.60 ± 0.44 |
19.66 ± 0.62 |
17.12 ± 0.47 |
22.18 ± 0.63 |
| 8d |
18.30 ± 0.21 |
17.44 ± 0.40 |
24.17 ± 0.72 |
27.40 ± 0.65 |
| 8e |
19.66 ± 0.36 |
20.42 ± 0.62 |
26.26 ± 0.28 |
26.72 ± 0.88 |
| 8f |
16.22 ± 0.94 |
19.88 ± 0.66 |
24.06 ± 1.02 |
26.84 ± 0.55 |
| 8g |
14.32 ± 0.27 |
10.22 ± 0.52 |
12.06 ± 0.36 |
18.46 ± 0.12 |
| 9a |
20.49 ± 0.76 |
16.56 ± 0.35 |
21.70 ± 1.06 |
19.48 ± 1.25 |
| 9b |
25.98 ± 0.42 |
20.32 ± 0.06 |
21.14 ± 0.45 |
30.36 ± 0.22 |
| 9c |
26.08 ± 0.30 |
19.04 ± 1.08 |
22.72 ± 0.26 |
28.81 ± 0.60 |
| 9d |
27.66 ± 0.44 |
18.08 ± 0.80 |
27.22 ± 0.92 |
31.66 ± 0.88 |
Table 3 In vitro antifungal activity of the sulphonamide and carbamate derivatives of Nebivolol intermediate (8a–g & 9a–d)
| Compound |
MIC in μg mL−1 |
| Aspergillus niger |
Aspergillus flavus |
Rhizopus arrhizus |
| Fluconazole |
19.40 ± 0.17 |
18.38 ± 0.32 |
22.32 ± 0.45 |
| 8a |
22.32 ± 0.27 |
23.06 ± 0.25 |
22.80 ± 0.54 |
| 8b |
20.40 ± 0.87 |
17.81 ± 0.16 |
20.21 ± 0.65 |
| 8c |
18.10 ± 0.46 |
16.72 ± 0.70 |
22.22 ± 0.83 |
| 8d |
15.30 ± 0.15 |
17.04 ± 0.70 |
18.65 ± 0.65 |
| 8e |
11.71 ± 0.35 |
14.08 ± 0.55 |
16.64 ± 0.52 |
| 8f |
15.66 ± 0.69 |
16.18 ± 0.82 |
19.04 ± 0.20 |
| 8g |
14.06 ± 0.72 |
15.18 ± 0.80 |
14.36 ± 0.66 |
| 9a |
11.20 ± 1.08 |
12.66 ± 0.80 |
14.18 ± 0.69 |
| 9b |
24.72 ± 0.70 |
23.81 ± 0.67 |
19.24 ± 0.48 |
| 9c |
19.32 ± 0.47 |
19.81 ± 0.37 |
25.56 ± 0.48 |
| 9d |
28.46 ± 0.14 |
25.20 ± 0.66 |
31.28 ± 0.45 |
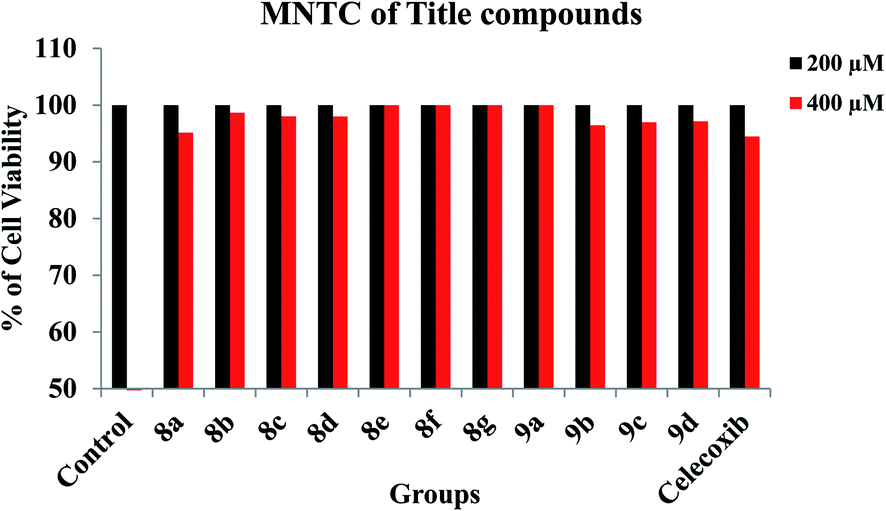
3.3. In vitro cell line studies
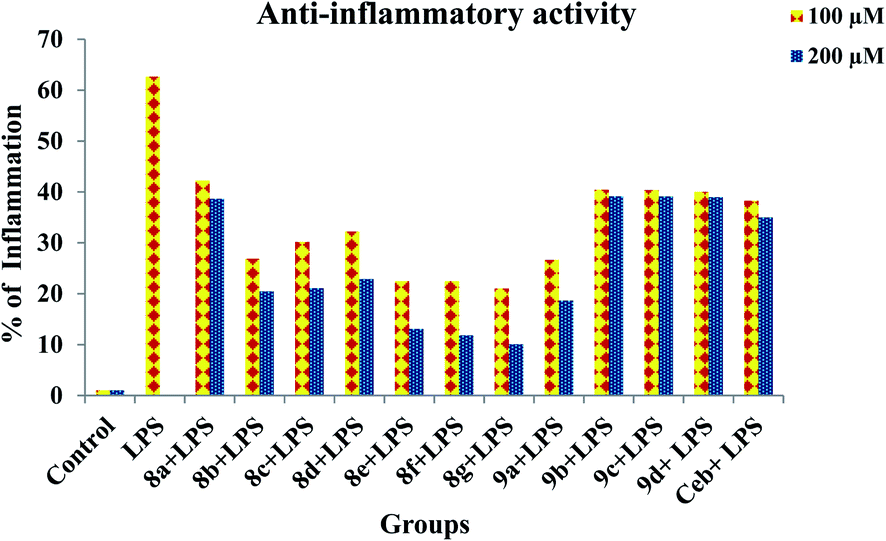
Prior to the evaluation of anti-inflammatory effect of title compounds, cytotoxicity was determined using RAW 264.7 cell lines. The cytotoxicity (cell death) was not observed for the test compounds up to 400 μM. The minimum lowest concentrations such as 100 μM and 200 μM were taken into consideration to test the anti-inflammatory effect against LPS-induced inflammatory cell death in RAW 264.7 cell lines respectively. The cytotoxicity of test compounds is shown in Fig. 3. In order to determine the anti-inflammatory effect of synthesized compounds, MTT assay was performed in RAW 264.7 cell lines. Cells were treated with test compounds with different concentrations (100 μM and 200 μM) followed by LPS. As demonstrated in Fig. 4, all the test compounds exhibited strong anti-inflammatory activity against LPS-stimulated inflammation in RAW 264.7 cell lines. Higher than 50% of inflammation was observed during the treatment with LPG at 100 μM. Whereas significant reduction in cell death was observed during the pre-treatment with tested compounds. The percentage of inflammation was reduced on dose dependent manner during the treatment with synthesized compounds. Among all the title compounds, 8g, 8e, 8f, 9a, 8b, 8d and 8c have shown strong anti-inflammatory activity against LPS induced cell death in RAW 264.7 cell lines.
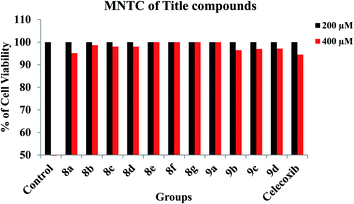 |
| | Fig. 3 Cytotoxic effect of title compounds during the treatment with different concentrations in RAW 264.7 macrophage cell lines. | |
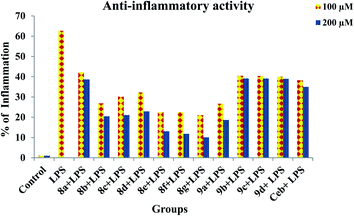 |
| | Fig. 4 Anti-inflammatory effects of title compounds with different concentrations during the pre-treatment in LPS-induced inflammation in RAW 264.7 macrophage cell lines. | |
3.4. NO production
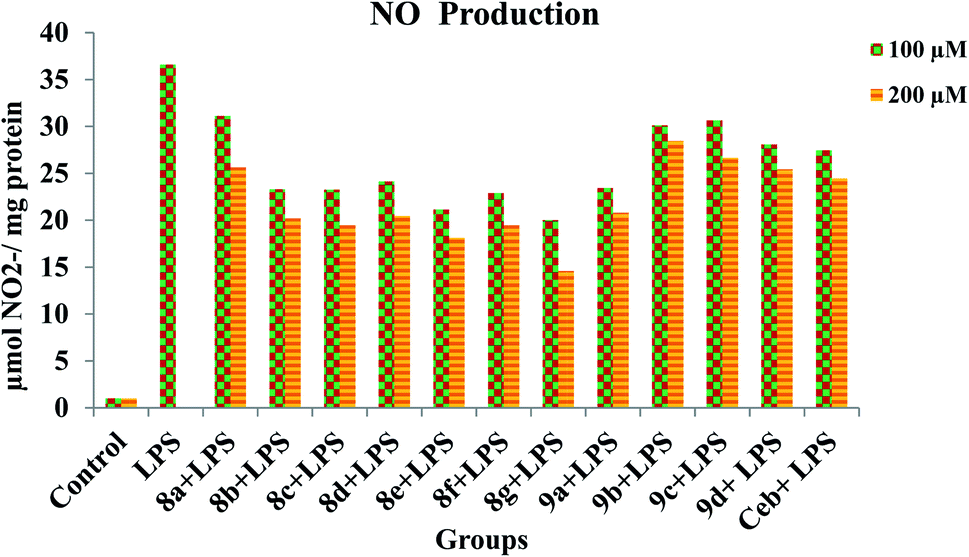
In order to provide strength to the synthesized compounds as to be anti-inflammatory agents the effect of synthesized compounds on NO production stimulated by LPS was also investigated using RAW 264.7 cell lines. The addition of LPS to RAW 264.7 cells resulted in an increase in NO production levels (Fig. 5). However, on pre-treatment with synthesized compounds significantly suppress LPS induced NO production in a concentration-dependent manner. Concomitant to the previous report, out of all title compounds 8g, 8e, 8f, 9a, 8b, 8c and 8d have shown strong inhibition against LPS-induced NO production in RAW 264.7 cell lines. Significantly reduced levels of NO production were observed in all compounds during pretreatment.
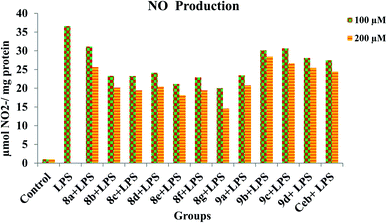 |
| | Fig. 5 Inhibition of NO production by title compounds and celecoxib with different concentrations in LPS-stimulated RAW 264.7 macrophages. | |
3.5. In vitro COX inhibition
All the synthesized compounds (8a–g and 9a–d) were screened for their inhibitory potential against COX-1 and COX-2 enzymes at 100 μM and 200 μM using colorimetric COX (ovine) inhibitor screening assay kit. Celecoxib was used as a reference compound. The results are given in Table 4. The results showed that most of the synthesized compounds had an inhibitory profile against both COX-1 and COX-2 enzymes, some were found to be moderate inhibitors when compared to the reference compound, celecoxib (8a, 9c and 9d). Among all, 8d, 8c and 9a at a concentration of 100 μM; 8f, 8g and 9a at a concentration of 200 μM against COX-1 and 8c, 8d and 8g at 100 μM; 8d, 8g and 9a at 200 μM against COX-2 exhibited higher content of inhibition akin to the rest of the compounds and the inhibition was highly significant when compared to the celecoxib respectively (Table 4). The structure activity relationship (SAR) study revealed that, compound by consisting with –F, –Cl –NO2, and –OCH3 substituents in their structure represents their higher inhibitory activity against COX-1 and COX-2 enzymes.
Table 4 In vitro COX-1 and COX-2 enzyme inhibitory activities of title compounds (8a–g & 9a–d)
| Compound |
% inhibition of COXa |
| COX-1 |
COX-2 |
| 100 μM |
200 μM |
100 μM |
200 μM |
| The determination was performed in triplicate for two independent experiments. |
| 8a |
37.30 ± 0.32 |
45.52 ± 0.77 |
23.53 ± 0.55 |
38.64 ± 0.33 |
| 8b |
46.10 ± 0.22 |
68.78 ± 0.43 |
34.43 ± 0.66 |
60.04 ± 0.24 |
| 8c |
51.08 ± 0.62 |
70.07 ± 0.70 |
40.40 ± 0.10 |
61.20 ± 0.72 |
| 8d |
53.88 ± 0.30 |
69.64 ± 0.18 |
39.32 ± 0.77 |
63.88 ± 0.46 |
| 8e |
48.42 ± 0.84 |
71.55 ± 0.15 |
35.55 ± 0.22 |
57.72 ± 0.36 |
| 8f |
33.87 ± 1.06 |
73.85 ± 0.54 |
28.70 ± 0.07 |
52.08 ± 0.78 |
| 8g |
47.69 ± 0.63 |
71.08 ± 0.16 |
39.92 ± 0.51 |
67.60 ± 0.73 |
| 9a |
50.66 ± 0.33 |
75.33 ± 0.14 |
18.06 ± 0.88 |
52.42 ± 0.21 |
| 9b |
43.08 ± 0.80 |
69.02 ± 0.50 |
37.70 ± 0.35 |
63.60 ± 0.32 |
| 9c |
39.60 ± 0.08 |
51.64 ± 0.32 |
24.30 ± 0.20 |
41.66 ± 0.23 |
| 9d |
41.40 ± 0.62 |
52.16 ± 0.30 |
29.73 ± 0.75 |
38.59 ± 0.29 |
| Celecoxib |
46.52 ± 0.40 |
68.07 ± 0.50 |
31.60 ± 0.44 |
51.32 ± 0.17 |
3.6. Carrageenan-induced rat paw edema bioassay
Compounds which have exhibited higher percentage of inhibition of COX-2/1 were selected for further anti-inflammatory evaluation by using carrageenan-induced hind paw edema in rats using celecoxib as a reference standard. The results of all the tested compounds are shown in Table 5. The results revealed that among the all tested compounds, compounds 8b, 8c, 8e, 8f and 8g inhibited the edema formation significantly at 3 h and 6 h than other compounds when compared to carrageenan control rats. Edema formation due to carrageenan is a biphasic event. The first phase begins immediately after injection and diminishes in 1 h and the second phase begins after 1 h. The initial phase of edema is attributed to the release of histamine and serotonin and second phase is due to the release of prostaglandins. All the tested compounds (8b, 8c, 8e, 8f and 8g) showed greater inhibition of paw edema formation at 6 h due to the inhibition of prostaglandins release, which are known to be released at 4–6 h after the carrageenan injection.
Table 5 Anti-inflammatory activity of the title compounds against the carrageenan-induced paw edema in rats with different time intervalsa
| Compound |
Dose (mg kg−1 p.o.) |
Paw volume (mL) |
| 1 h |
3 h |
6 h |
| *Each value is the mean ± SEM for six rats. #p < 0.05, ##p < 0.01 when compared with normal control group. **p < 0.01, *p < 0.05 when compared with carrageenan control group. |
| Control |
— |
0.42 ± 0.40 |
0.38 ± 0.14 |
0.40 ± 0.60 |
| Carrageenan |
— |
0.94 ± 0.15## |
1.17 ± 0.22## |
1.69 ± 0.84# |
| 8b |
100 |
0.52 ± 0.18* |
0.80 ± 0.38** |
1.06 ± 0.27* |
| 8c |
100 |
0.60 ± 0.64** |
0.88 ± 0.31** |
1.11 ± 0.13** |
| 8d |
100 |
0.78 ± 0.88* |
1.08 ± 0.40* |
1.50 ± 0.64* |
| 8e |
100 |
0.68 ± 0.96* |
0.74 ± 0.29* |
1.19 ± 0.65* |
| 8f |
100 |
0.62 ± 0.10** |
0.86 ± 0.32* |
1.10 ± 0.13* |
| 8g |
100 |
0.65 ± 0.20* |
0.77 ± 0.28** |
1.20 ± 0.38** |
| 9a |
100 |
0.72 ± 0.72* |
1.06 ± 0.52* |
1.56 ± 0.33* |
| 9b |
100 |
0.76 ± 0.48* |
0.88 ± 0.16** |
1.50 ± 0.25* |
| Celecoxib |
100 |
0.80 ± 0.50** |
0.98 ± 0.92* |
1.58 ± 0.22** |
3.7. Cotton pellet-induced granuloma bioassay
Cotton pellet implantation is the most suitable method for studying the efficacy of drugs against proliferative phase of inflammation. Compounds showing promising anti inflammatory activity in the carrageenan-induced rat paw edema bioassay were further evaluated for their in vivo cotton pellet-induced granuloma bioassay in rats using celecoxib as a reference standard. In this assay, selected test compounds 8b, 8c, 8e, 8f, and 8g inhibited both the exudatory and granulatory phases of inflammation (Table 6). During the repair process of inflammation, there is a proliferation of macrophages, neutrophils, fibroblasts and multiplication of small blood vessels, which are the basic sources of forming a highly vascularized radish mass called as granular tissue (granular tissue formation). Inhibition of this granular phase of inflammation by 8b, 8c, 8e, 8f, and 8g suggested significant antiinflammatory property was observed in the experimental animal models.
Table 6 Effect of test compounds on cotton pellet induced granuloma in ratsa
| Treatment and dose (100 mg kg−1, p.o.) |
Weight (mg) mean ± SEM |
| Wet |
Dry |
| **p < 0.01, *p < 0.05 when compared with normal control group. ##p < 0.01, #p < 0.05 when compared with standard group. |
| Control |
250.30 ± 1.48 |
123.30 ± 1.48 |
| 8b |
182.50 ± 1.77*# |
84.10 ± 1.67*# |
| 8c |
148.62 ± 1.85*## |
91.30 ± 1.72*# |
| 8e |
152.40 ± 1.57*## |
76.45 ± 1.95*## |
| 8f |
139.82 ± 2.42*## |
70.66 ± 2.33*## |
| 8g |
190.90 ± 1.84*# |
78.42 ± 2.04*# |
| Celecoxib |
150.14 ± 3.46** |
78.80 ± 3.24** |
3.8. Acute toxicity study
There were no significant behavioral changes observed with all the employed doses of various test compounds. To all the mice, neither toxic reaction nor mortality was observed. Therefore, 2000 mg kg−1, body weight (b.w.), dose was considered as maximum tolerated dose. Based on acute toxicity study, we have selected 100 mg kg−1 for entire anti-inflammatory activity in vivo.
3.9. Molecular docking
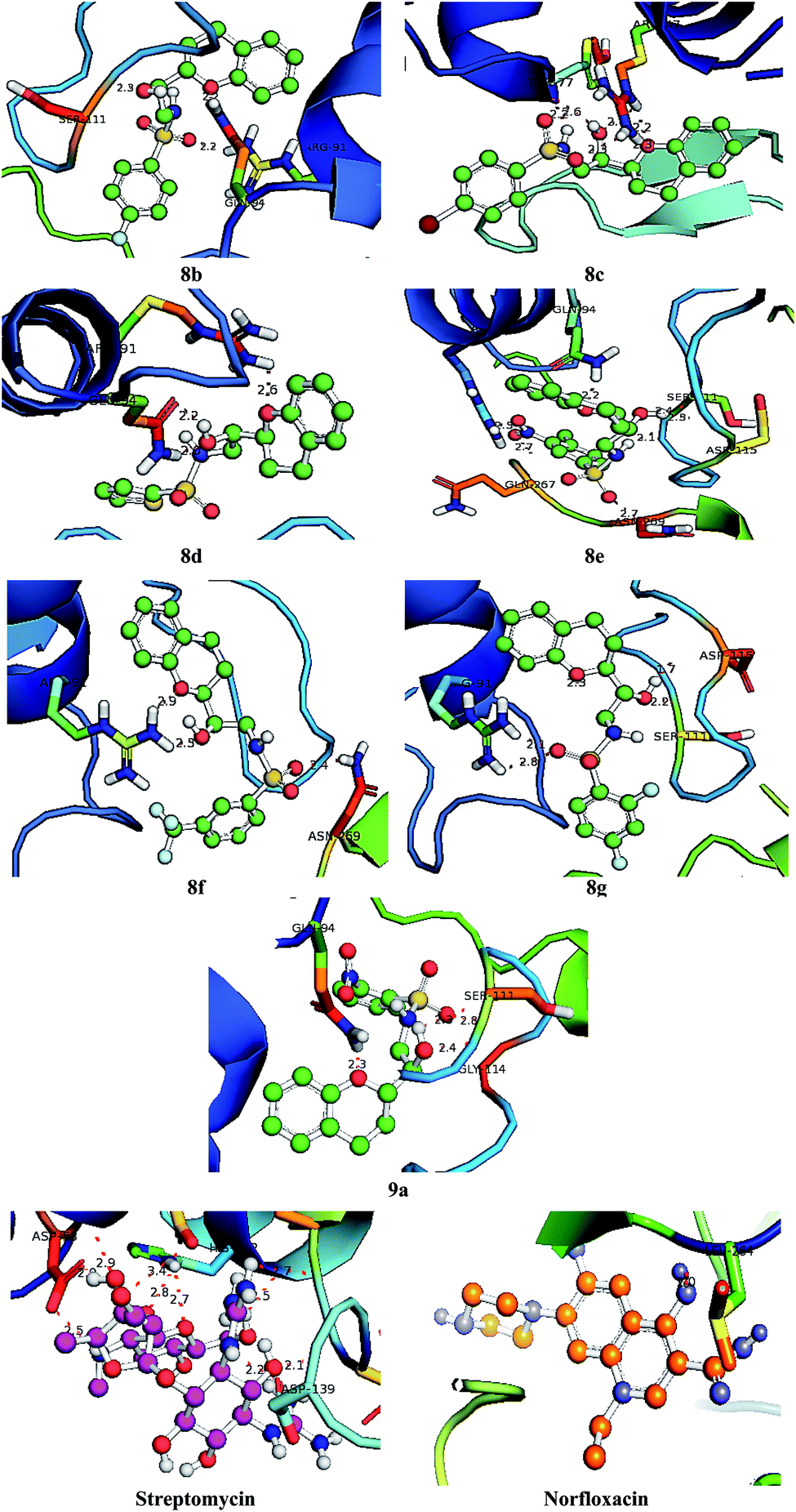
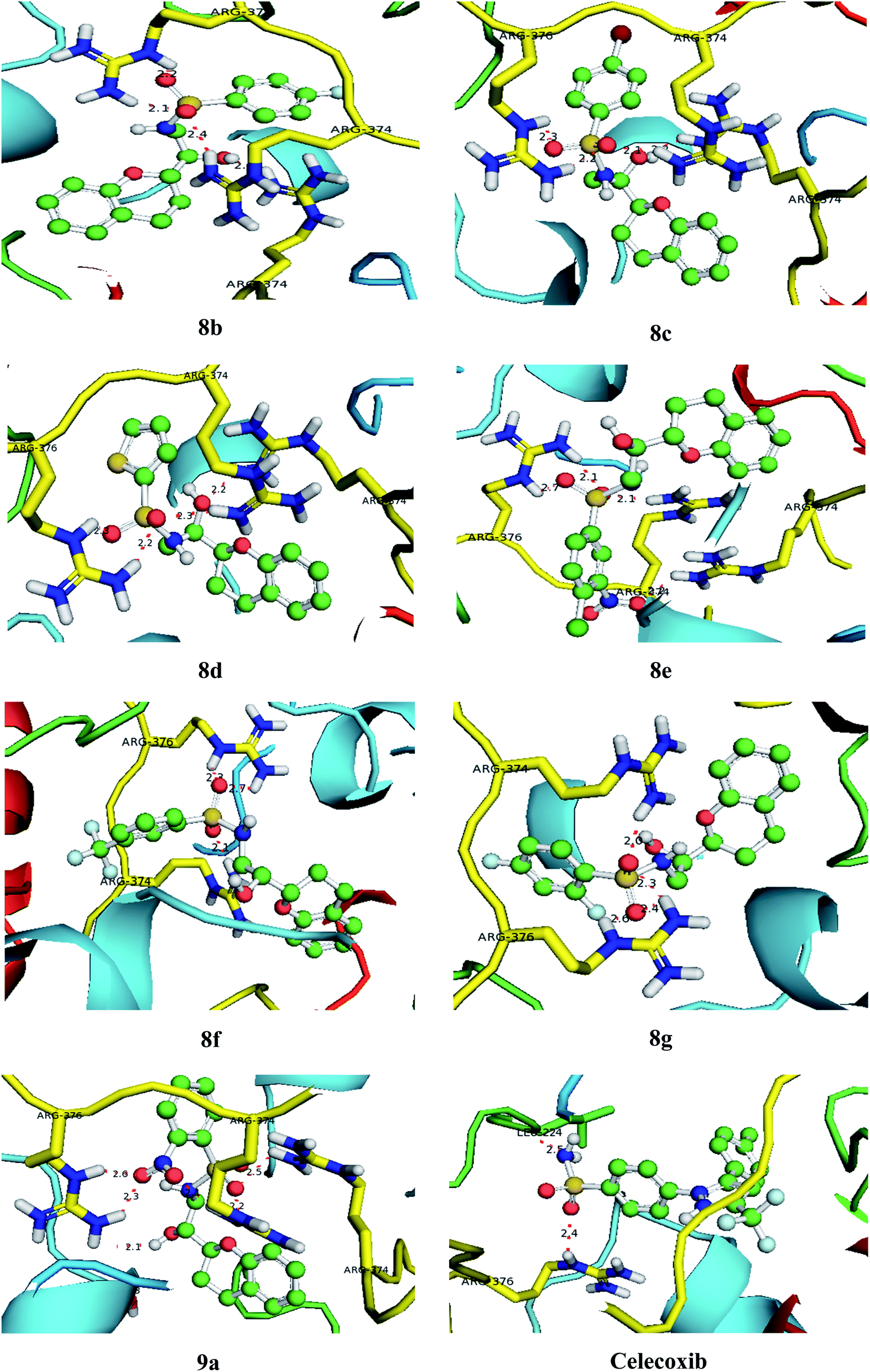
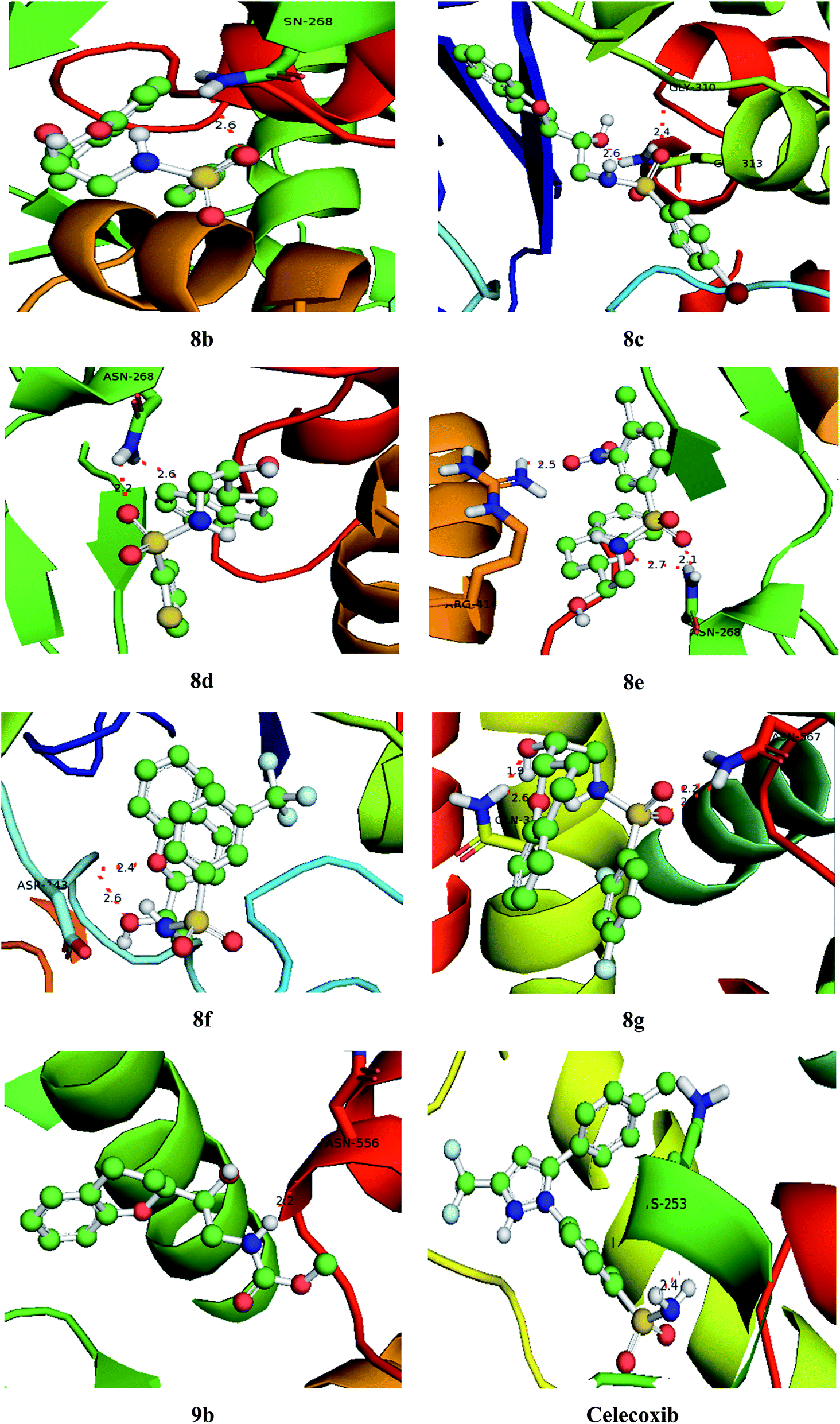
With an aim to investigate binding interactions and understand the potent activity of the active compounds, the compounds were selected for docking study by using Auto Dock 4.2 software. All the title compounds were docked into the active site of the enzyme DNA gyrase A, COX-1, and COX-2 which are suitable targets for antibacterial and antiinflammatory activity in silico. With reference to antibacterial activity, all the title compounds exhibited higher dock scores than the reference compound, streptomycin and norfloxacin except 8a, 9b, 9c and 9d. Out of all, 8e, 8f, 8g and 9a have formed the highest dock score akin to the rest of the title compounds. Whereas with reference to the inflammatory targets, except 8a, 9b and 9d against COX-1 and 8a and 9c against COX-2 have formed higher dock scores ranging from −6.3 to −9.4 (COX-1) and −5.6 to −9.4 (COX-2) when compared to the reference, celecoxib (−7.2 to −7.4) respectively. The H-bonds, binding affinities and energy profiles of compounds 8a–8g and 9a–9d along with reference drugs, towards the active site amino acids such as DNA gyrase A, COX-1, COX-2 and 3D interactions of the best lead compounds are summarized in Tables 7–9 and Fig. 6–8. In overall conclusion drawn from the in silico studies stated that the compounds 8b, 8c, 8d, 8e, 8f, 8g and 9a and 9b in common have shown the highest binding energies against all the selected target proteins and were actively fitted in the active sites of the target genes. Majority of the H-accepted type of bonds were formed by the ligands than the H-donor type. This reveals the active interaction of the compounds with the active site of target protein. Among the title compounds, 9b and 9d with COX-1 protein and 9c with COX-2 protein have formed hydrophobic interactions.
Table 7 Binding characteristics of synthesized compounds (8a–8g and 9a–9d) and references against DNA gyrase A protein
| Com. |
BE |
Binding interaction |
Bond length(Å) |
Bond angle (°) |
Bond type |
| Streptomycin |
−6.9 |
Arg 139 CG⋯HN |
2.2 |
124.4 |
H–don |
| Leu 135 CD⋯HN |
2.7 |
125.7 |
H–don |
| His 132 CB⋯OH |
2.5 |
125.0 |
H–acc |
| Asp 53 CG⋯OC |
3.4 |
116.7 |
H–acc |
| Asp 53 OC⋯OC |
2.9 |
118.9 |
H–acc |
| Asp 58 OD⋯OH |
2.0 |
118.6 |
H–acc |
| Asp 58 OD⋯HN |
2.5 |
116.4 |
H–don |
| His 132 ND⋯OC |
2.8 |
126.2 |
H–acc |
| His 132 ND⋯OC |
2.7 |
120.0 |
H–acc |
| His 132 OC⋯OH |
2.5 |
119.8 |
H–acc |
| Norfloxacin |
–7.3. |
Leu 264 CA⋯OC |
2.0 |
114.7 |
H–acc |
| 8a |
–6.0 |
Tyr 266 CZ⋯HN |
2.2 |
119.9 |
H–don |
| Thr 171 CA⋯HO |
2.2 |
114.6 |
H–don |
| Asn 169 OC⋯HN |
2.5 |
120.7 |
H–don |
| 8b |
−8.2 |
Ser 111 CA⋯ OC |
2.3 |
122.5 |
H–acc |
| Arg 91 CZ⋯ OS |
2.2 |
119.0 |
H–acc |
| Asn 169 OC⋯HN |
2.2 |
118.4 |
H–acc |
| 8c |
−8.1 |
Arg 47 CZ⋯OS |
2.3 |
119.4 |
H–acc |
| Arg 47 NE⋯OC |
2.2 |
116.2 |
H–acc |
| Arg 47 NE⋯OC |
2.6 |
118.9 |
H–acc |
| Arg 47 CZ⋯OC |
2.3 |
120.4 |
H–acc |
| Tyr 77 CA⋯HO |
2.6 |
118.7 |
H–don |
| Tyr 77 CA⋯HN |
2.2 |
122.0 |
H–don |
| 8d |
−8.0 |
Gln 94 CD⋯OH |
2.2 |
124.1 |
H–acc |
| Gln 94 CD⋯OC |
2.0 |
118.4 |
H–acc |
| Arg 91 CZ⋯OC |
2.6 |
119.1 |
H–acc |
| 8e |
−8.5 |
Arg 91 CZ⋯ON |
2.3 |
121.8 |
H–acc |
| Gln 267 CA⋯ON |
2.7 |
114.3 |
H–acc |
| Gin 94 CD⋯OC |
2.2 |
118.4 |
H–acc |
| Asp115 CA⋯HO |
2.3 |
124.2 |
H–don |
| Ser 111 CG⋯HO |
2.4 |
122.5 |
H–don |
| Ser 111 OC⋯HN |
2.1 |
123.1 |
H–don |
| Asg 269 HD⋯OS |
2.7 |
120.3 |
H–acc |
| 8f |
−8.4 |
Arg 91 CZ⋯OC |
2.9 |
120.8 |
H–acc |
| Arg 91 CZ⋯OC |
2.3 |
119.0 |
H–acc |
| Asn 269 CG⋯OS |
2.4 |
122.3 |
H–acc |
| 8g |
−8.8 |
Arg 91 CZ⋯OS |
2.1 |
119.0 |
H–acc |
| Arg 91 CZ⋯OS |
2.8 |
121.8 |
H–acc |
| Gln 94 CD⋯OC |
2.3 |
118.4 |
H–acc |
| Ser 111 HN⋯OC |
2.2 |
116.0 |
H–acc |
| Asp 115 CA⋯HO |
1.7 |
120.6 |
H–don |
| 9a |
−8.3 |
Gly 114 CA⋯OS |
2.8 |
112.3 |
H–acc |
| Ser 111 OC⋯HO |
2.3 |
120.4 |
H–don |
| Ser 111 HN⋯OC |
2.4 |
122.5 |
H–acc |
| Gln 94 CD⋯OC |
2.3 |
118.4 |
H–acc |
| 9b |
−6.5 |
Met 301 CA⋯OC |
2.1 |
115.2 |
H–acc |
| His 262 CA⋯HN |
2.1 |
121.2 |
H–don |
| Leu 264 CA⋯OC |
2.1 |
114.7 |
H–acc |
| Thr 219 CA⋯HO |
2.0 |
119.9 |
H–don |
| 9c |
−6.5 |
Met 301 HN⋯OC |
2.3 |
115.2 |
H–acc |
| Leu 264 HN⋯OC |
2.1 |
114.7 |
H–acc |
| 9d |
−6.6 |
Arg 91 CZ⋯OC |
2.5 |
119.0 |
H–acc |
| Arg 91 CZ⋯OC |
2.7 |
119.0 |
H–acc |
| Gln 94 NE⋯OC |
2.1 |
118.4 |
H–acc |
| Ser 111 CA⋯OC |
2.3 |
116.2 |
H–acc |
| Ser 111 CA⋯HO |
2.2 |
120.4 |
H–don |
Table 8 Binding characteristics of synthesized compounds (8a–8g and 9a–9d) and references against COX-1 protein
| Compound |
BE |
Bonding interaction |
Bond length(Å) |
Bond angle (°) |
Bond type |
| Celecoxib |
−7.4. |
Leu 324 CB⋯HN |
2.5 |
91.3 |
H–don |
| Arg 37 CA⋯OS |
2.4 |
103.1 |
H–acc |
| 8a |
−6.3 |
Gly 536 CZ⋯HO |
2.8 |
130.7 |
H–don |
| 8b |
−8.5 |
Arg 376 CZ⋯OS |
2.2 |
89.7 |
H–acc |
| Arg 376 CZ⋯OS |
2.1 |
90.3 |
H–acc |
| Arg 374 CB⋯OS |
2.4 |
93.8 |
H–acc |
| Arg 374 CB⋯OC |
2.1 |
99.3 |
H–acc |
| 8c |
−8.7 |
Arg 376 CZ⋯OS |
2.2 |
110.3 |
H–acc |
| Arg 376 CB⋯OS |
2.3 |
91.8 |
H–acc |
| Arg 374 CB⋯OS |
2.1 |
107.0 |
H–acc |
| Arg 374 CB⋯OH |
2.2 |
121.8 |
H–acc |
| 8d |
−8.0 |
Arg 376 CA⋯OS |
2.3 |
113.2 |
H–acc |
| Arg 376 CB⋯OS |
2.2 |
100.7 |
H–acc |
| Arg 374 CB⋯OS |
2.3 |
109.3 |
H–acc |
| Arg 374 CZ⋯OH |
2.2 |
121.0 |
H–acc |
| 8e |
−9.0 |
Arg 376 CZ⋯OS |
2.7 |
88.7 |
H–acc |
| Arg 376 CB⋯OS |
2.1 |
71.0 |
H–acc |
| Arg 374 CB ⋯.OS |
2.1 |
137.7 |
H–acc |
| Arg 374 CZ⋯ON |
2.2 |
118.7 |
H–acc |
| 8f |
−9.4 |
Arg 376 CZ⋯OS |
2.3 |
120.3 |
H–acc |
| Arg 376 CB⋯OS |
2.7 |
118.7 |
H–acc |
| Arg 374 CB⋯OS |
2.14 |
127.0 |
H–acc |
| 8g |
−8.7 |
Arg 376 CZ⋯OS |
2.6 |
94.6 |
H–acc |
| Arg 376 CB⋯OS |
2.4 |
80.7 |
H–acc |
| Arg 374 CB⋯OS |
2.3 |
98.1 |
H–acc |
| Arg 374 CZ⋯OC |
2.0 |
97.8 |
H–acc |
| 9a |
−8.7 |
Arg 374 CB⋯OS |
2.5 |
120.7 |
H–acc |
| Arg 374 CZ⋯OS |
2.2 |
96.3 |
H–acc |
| Arg 374 CB⋯OS |
2.1 |
81.8 |
H–acc |
| Arg 376 CZ⋯ON |
2.6 |
120.1 |
H–acc |
| Arg 376 CB⋯ON |
2.3 |
118.5 |
H–acc |
| Ser 143 CA⋯HO |
2.1 |
91.1 |
H–don |
| 9b |
−6.9 |
Arg 376 CB⋯OC |
2.5 |
133.1 |
H–acc |
| 9c |
−7.4 |
Arg 374 CB⋯OC |
2.3 |
91.6 |
H–acc |
| 9d |
−7.0 |
Arg 376 CZ⋯OC |
2.6 |
93.5 |
H–acc |
Table 9 Binding characteristics of synthesized compounds (8a–8g and 9a–9d) and references against COX-2 protein
| Compound |
BE |
Bonding interaction |
Bond length(Å) |
Bond angle (°) |
Bond type |
| Celecoxib |
−7.2. |
Lys 235 CZ⋯HN |
2.4 |
118.3 |
H–don |
| 8a |
−5.6 |
Arg 293 CB⋯OH |
2.5 |
108.8 |
H–acc |
| Asn 556 CZ⋯HN |
2.7 |
112.7 |
H–don |
| 8b |
−8.7 |
Asn 268 CB⋯OS |
2.6 |
100.3 |
H–acc |
| 8c |
−9.4 |
Gly 310 CB⋯ON |
2.6 |
119.3 |
H–acc |
| Gln 313 CB⋯OS |
2.4 |
101.0 |
H–acc |
| 8d |
−8.6 |
Asn 268 CB⋯OC |
2.6 |
86.1 |
H–acc |
| Asn 268 CA⋯OS |
2.2 |
106.3 |
H–acc |
| 8e |
−9.0 |
Asn 268 CB⋯OS |
2.1 |
91.1 |
H–acc |
| Asn 268 CA⋯OC |
2.7 |
104.7 |
H–acc |
| Arg 414 CZ⋯ON |
2.5 |
116.0 |
H–acc |
| 8f |
−8.5 |
Asp 143 CA⋯OC |
2.4 |
117.9 |
H–acc |
| Asp 143 CZ⋯OC |
2.6 |
104.9 |
H–acc |
| 8g |
−8.6 |
Asn 567 CA⋯OS |
2.5 |
66.7 |
H–acc |
| Asn 567 CZ⋯OS |
2.2 |
78.4 |
H–acc |
| Gln 336 CZ⋯OC |
2.6 |
90.3 |
H–acc |
| Gln 336 CZ⋯HO |
1.9 |
98.7 |
H–don |
| 9a |
−8.0 |
Asp 143 CB⋯OH |
2.3 |
89.9 |
H–acc |
| 9b |
−8.7 |
Asn 556 CZ⋯HN |
2.2 |
99.7 |
H–don |
| 9c |
−7.1 |
Ser 457 CB⋯HN |
2.6 |
120.0 |
H–don |
| 9d |
−7.3 |
Asn 556 CZ⋯OC |
2.2 |
117.3 |
H–acc |
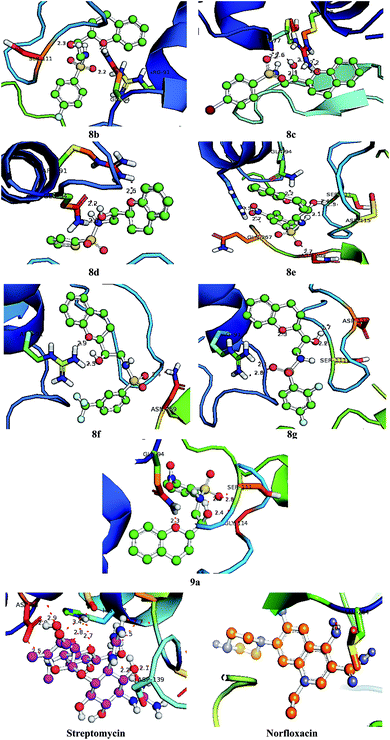 |
| | Fig. 6 Bonding interactions of the lead compounds and standards with DNA gyrase A. | |
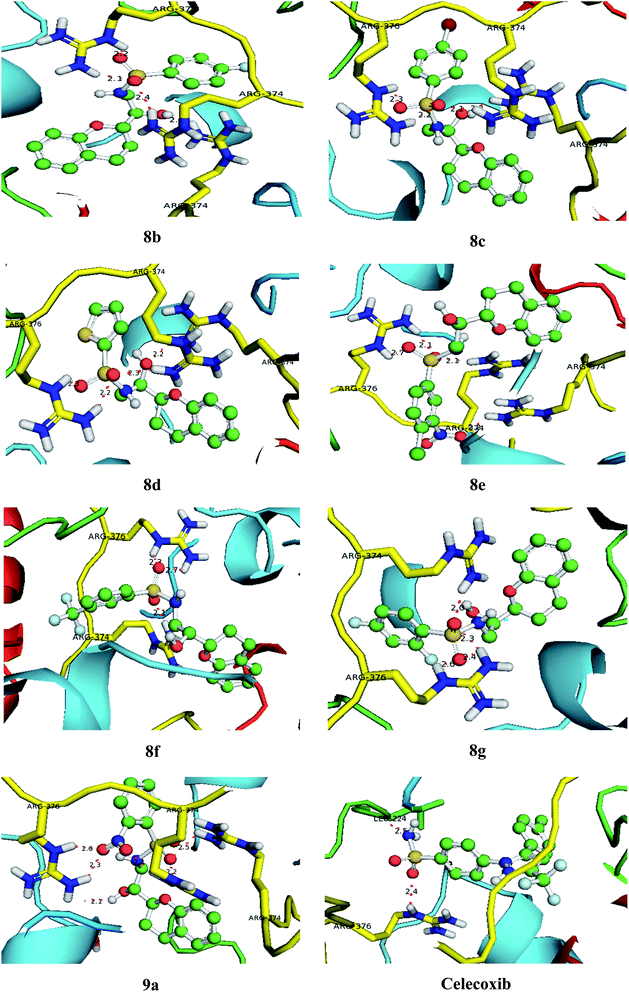 |
| | Fig. 7 Bonding interactions of the lead compounds and standard with COX-1. | |
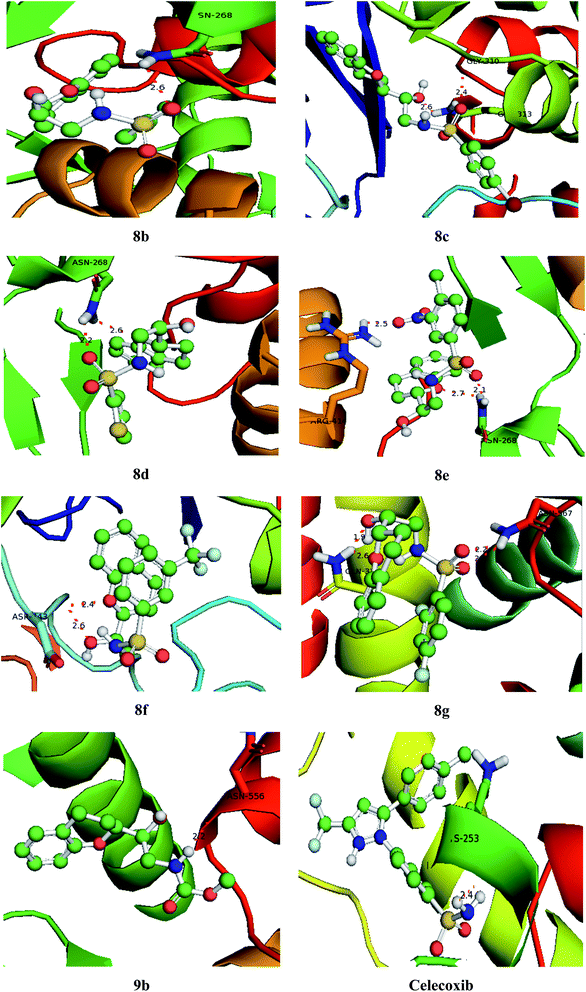 |
| | Fig. 8 Bonding interactions of the lead compounds and standard with COX-2. | |
3.10. Bioactivity & toxicity risk studies
The bioactivity properties like G protein-coupled receptor ligand (GPCRL) property, ion channel modulator (ICM) property, kinase inhibitor (KI) property, protease inhibitor (PI) property, nuclear receptor ligand (NRL) interaction property & enzyme inhibitor (EI) property and toxicity risk properties like drug-likeness and drug score were predicted for the title compounds (Table 10). The GPCRL property is identified in the limits of 0.64–0.18, ICM property is identified ranging from −0.18 to 0.27, KI property is identified limiting from −0.13 to 0.23, NRL interaction property is enriched with values ranging from −0.03 to 0.23, PI property is reputed in the range of 0.33–0.04 and EI property is explored in the limits of 0.14–0.04. All these properties are superior when compared standard, celecoxib. Similarly, it is found that all the compounds are in the range of −9.3 to −5.8 of drug-likeness and is comparable with celecoxib having −6.3 of drug-likeness. Likewise, the drug score is identified in the range of 0.92 to 0.29, which are superior to the celecoxib standard with 0.42 of drug score value. From the bioactivity assessment, it is also renowned that all the synthesized compounds are nonmutagenic, non-tumorigenic, non-irritant and they doesn't adhere the negative impacts on reproductive system and hence ascertained them as safer drugs with extensive binding affinities to pair up potentially with reactive species in the domains of the specific targets considered and established a robust host–receptor relation of compounds with the all the above ligands.
Table 10 Bioactivity & toxicity risk of the title compounds (8a–8g and 9a–9d)a
| Structure |
Bioactivity |
Toxicity risks |
| GPCRL |
ICM |
KI |
NRL |
PI |
EI |
MUT |
TUMO |
IRRI |
REP |
DL |
DS |
| GPCRL: G protein-coupled receptor ligand; ICM: ion channel modulator; KI: kinase inhibitor; NRL: nuclear receptor ligand; PI: protease inhibitor; EI: enzyme inhibitor; MUT: mutagenic; TUMO: tumorigenic; IRRI: irritant; REP: reproductive effect [a dash (−) indicates no effect (+) indicates the effect]; DL: drug likeness; DS: drug score. |
| 8a |
 |
0.22 |
−0.18 |
−0.36 |
−0.03 |
0.28 |
0.04 |
− |
− |
− |
+ |
−6.0 |
0.38 |
| 8b |
 |
0.18 |
0.10 |
−0.43 |
0.06 |
0.31 |
0.08 |
− |
− |
− |
− |
−9.4 |
0.56 |
| 8c |
 |
0.47 |
0.17 |
0.21 |
0.09 |
0.30 |
0.08 |
− |
− |
− |
− |
−8.9 |
0.62 |
| 8d |
 |
0.41 |
0.11 |
0.23 |
0.10 |
0.27 |
0.05 |
− |
− |
− |
− |
−9.2 |
0.70 |
| 8e |
 |
0.20 |
0.24 |
0.12 |
0.13 |
0.33 |
0.12 |
− |
− |
− |
− |
−9.3 |
0.82 |
| 8f |
 |
0.29 |
0.27 |
0.10 |
0.08 |
0.31 |
0.11 |
− |
− |
− |
− |
−9.2 |
0.84 |
| 8g |
 |
0.31 |
−0.20 |
−0.13 |
0.08 |
0.27 |
0.14 |
− |
− |
− |
− |
−9.0 |
0.92 |
| 9a |
 |
0.27 |
0.17 |
−0.08 |
0.21 |
0.23 |
0.06 |
− |
− |
− |
− |
−8.8 |
0.91 |
| 9b |
 |
0.38 |
0.19 |
−1.02 |
0.23 |
0.21 |
0.07 |
− |
− |
− |
+ |
−8.8 |
0.88 |
| 9c |
 |
0.64 |
−0.21 |
−1.21 |
−0.03 |
0.15 |
0.07 |
− |
− |
− |
− |
−6.1 |
0.37 |
| 9d |
 |
0.26 |
−0.38 |
−1.30 |
−0.04 |
0.12 |
0.05 |
− |
− |
− |
− |
−5.8 |
0.29 |
| Cel. |
 |
−0.63 |
−0.49 |
−1.42 |
−1.48 |
−0.79 |
−0.09 |
− |
− |
− |
+ |
−6.3 |
0.42 |
3.11. ADMET properties
This significant study helps to distinguish the pharmacokinetic properties of title compounds and to comprehend their drug-like interactions with in the cell. The human intestinal absorption discursively helps in carrying the active compounds to target cell tissues via blood stream to interact mutually. In oral administration, degree of absorption of a potential compound depends on the properties of its inherent bioavailability. Then the absorbed portion will itself be distributed into muscles from there to other organs by circulation via extracellular sites. As such the distribution of a compound lowers its plasma concentration and metabolizes it and then distributes its metabolites by enzymatic redox reactions. Some of the distributed active metabolites work efficiently in pharmacologically on cellular systems, on contrary the inactive metabolites deactivate the administered compound and reduce its effect in vivo and inert metabolites will be excreted from kidneys. The scrutiny of predicted ADMET properties (Table 11) of 8a–g and 9a–d revealed that the in vivo BBB penetration efficiency identified in the range of 0.394438–0.974248 ascertains their high significance of CNS activity approved their permeability to distribute themselves in vivo compared to celecoxib with 0.271436. It is reinforced on the grounds of their in vitro Caco-2 cell permeability observed in the range of 14.64320–82.36421 nm s−1, which are benign compared to celecoxib with 13.56965. This efficiency establishes their sustained permeability to bind with the plasma proteins to enable the penetration of these compounds in to BBB system. The in vitro PPB affinity observed in the range of 68.361450–97.664402% confirms their strong binding ability to the plasma proteins, which are worthy, compared to celecoxib with 38.972004. The in vitro MDCK cell permeability acknowledged in the range of 14.02520–66.45872 nm s−1 and hence discloses them as moderately permeable to interact with the concerned active species, compared to celecoxib with 22.82750. The %HIA recognized is proficient as they are in the range of 46.184572–88.624180 which are comparatively better than celecoxib with 45.302456% and promises their interactions with the reactive species in the expecting target of domains. The negative toxicity results indicate that the title compounds are safer and non-toxic drugs as omitted from the positive toxicity results. This study concludes the good physico–chemical interactions of 8a–g & 9a–d and their drug-likeness properties.
Table 11 Predicted ADMET profiles of the title compounds (8a–g and 9a–d)
| Com. |
BBB |
Caco2 |
PPB |
MDCK |
HIA% |
Toxicity |
| Rule |
>0.40 |
4–70 |
>90 |
25–500 |
20–100 |
Negative |
| 8a |
0.394438 |
15.49632 |
70.650233 |
25.10452 |
46.184572 |
Negative |
| 8b |
0.846620 |
60.36412 |
95.854201 |
31.65632 |
79.564202 |
Negative |
| 8c |
0.798246 |
71.55320 |
97.664402 |
38.48920 |
88.624180 |
Negative |
| 8d |
0.557214 |
65.43210 |
95.366320 |
43.56974 |
83.554630 |
Negative |
| 8e |
0.974248 |
80.42152 |
92.187240 |
51.69202 |
80.968742 |
Negative |
| 8f |
0.881146 |
82.36421 |
97.320045 |
60.45325 |
73.568940 |
Negative |
| 8g |
0.955226 |
78.99124 |
91.664025 |
66.45872 |
73.202145 |
Negative |
| 9a |
0.687236 |
69.10145 |
85.366240 |
59.88724 |
81.664524 |
Negative |
| 9b |
0.421078 |
14.64320 |
68.442052 |
14.65234 |
66.387552 |
Negative |
| 9c |
0.604482 |
22.74322 |
88.924210 |
55.46320 |
70.338564 |
Negative |
| 9d |
0.388460 |
15.32104 |
68.361450 |
14.02520 |
50.345892 |
Negative |
| Celecoxib |
0.271436 |
13.56965 |
38.972004 |
22.82750 |
45.302456 |
Negative |
3.12. QSAR studies
The study of biochemical interactions of compound under analysis with it pharmacological target is a vital step in realizing its drug-like properties. In such predicted QSAR properties of compounds revealed their effective scorings of molecular weights are in 283.1–431.0 range, which are less than 500 Da and supports their safer drug activity (Table 12). The number of hydrogen bond donating atoms or atom containing groups is 5, which is less than or equal to 5. Similarly, they are deserved with partition coefficient (logP) of 1.05–3.72, which is less than 5 and the molecular refractivity values in the range of 53.62–97.24 cm3 mol−1 are identified in the limits of potential drugs only. These observed results confirm that compounds 8a–g & 9a–d are with virtuous pharmaceutical parameters with zero Lipinski property violations. On compatible with Lipinski parameters we have predicted the total polar surface area (TPSA) values which are less than 140, number of rotatable bonds in the range of 5–6 which are less than 10 empowers them as potential molecules with zero Veber property violations to interact well with the target cells. The consistent correlation of Lipinski and Veber properties esteems title compounds as noble administrable drugs to effectively bind with host to assure their pharmacological activity. On the other hand, the number of hydrophobic atoms presents on the ligand molecules in the range of 16–19 assures their capacity to form complexes with target cell receptors to institute their ligand-receptor binding relations. The additional properties like van der Waals volume identified in the range of 122.6–288.6, density in the range of 1.12–1.62 g cm3 and solubility ranging from −1.02 to −4.66 supports the of ligand interactions with molecules with the hosting receptors and their proficient affinity with target cells within binding domain. This entire study of QSAR study provided an established support the evaluated biological activities respectively.
Table 12 QSAR profiles of the title compounds (8a–g and 9a–d)a
| Com. |
Lipinski Assets |
Veber Assets |
Other Assets |
| MW |
HD |
HA |
logP |
MR |
L Vio. |
TPSA |
RB |
V Vio. |
HP |
VV |
D |
Solu. |
| MW: molecular weight; HD: hydrogen bond donors; HA: hydrogen bond acceptors; logP: octanol to water partition coefficient; MR: molecular refractivity (cm3 mol−1); L Vio: Lipinski violations; TPSA: total polar surface area; RB: number of rotatable bonds; V Vio: Veber violations; HP: number of hydrophobic atoms; VV: van der Waals volume; D: density (g cm3); Solu.: solubility. |
| Rule |
<500 |
<5 |
<10 |
<5 |
<100 |
<1 |
<140 |
<10 |
<1 |
5–20 |
100–300 |
1–2 |
−1 to −5 |
| 8a |
370.0 |
4 |
6 |
2.80 |
53.62 |
0 |
130.44 |
5 |
0 |
16 |
140.6 |
1.62 |
−1.23 |
| 8b |
326.4 |
5 |
6 |
2.10 |
94.10 |
0 |
120.36 |
5 |
0 |
19 |
288.6 |
1.44 |
−3.45 |
| 8c |
388.1 |
5 |
6 |
1.89 |
97.24 |
0 |
115.42 |
5 |
0 |
19 |
246.6 |
1.30 |
−4.18 |
| 8d |
431.0 |
5 |
6 |
1.62 |
91.32 |
0 |
110.08 |
6 |
0 |
17 |
244.8 |
1.28 |
−4.66 |
| 8e |
296.4 |
5 |
6 |
1.05 |
90.72 |
0 |
108.46 |
6 |
0 |
19 |
238.4 |
1.26 |
−4.10 |
| 8f |
338.3 |
5 |
6 |
1.44 |
93.14 |
0 |
101.88 |
6 |
0 |
18 |
224.5 |
1.36 |
−3.98 |
| 8g |
401.1 |
5 |
6 |
2.04 |
93.58 |
0 |
100.05 |
6 |
0 |
18 |
278.9 |
1.40 |
−3.76 |
| 9a |
269.1 |
5 |
6 |
1.30 |
92.77 |
0 |
103.84 |
5 |
0 |
18 |
268.1 |
1.14 |
−4.22 |
| 9b |
363.0 |
4 |
6 |
3.02 |
63.78 |
0 |
132.56 |
6 |
0 |
13 |
122.6 |
1.50 |
−1.02 |
| 9c |
312.0 |
5 |
6 |
1.88 |
95.62 |
0 |
120.64 |
6 |
0 |
17 |
280.4 |
1.12 |
−4.66 |
| 9d |
283.1 |
5 |
6 |
3.72 |
60.14 |
0 |
138.60 |
5 |
0 |
11 |
128.0 |
1.58 |
−1.56 |
| Celecoxib |
381.3 |
4 |
5 |
2.89 |
46.23 |
0 |
128.42 |
5 |
0 |
9 |
132.7 |
1.66 |
0.18 |
Blood–Brain Barrier (BBB) penetration = brain/blood; Caco-2 cells are derived from human colon adenocarcinoma; possess multiple drug transport pathways through intestinal epithelium; PPB: % of drug binds to plasma protein; MDCK cell system used as tool for rapid in vitro permeability screening; human intestinal absorption is the sum of bioavailability and absorption evaluated from ratio of excretion or cumulative excretion in urine, bile and feces; In vitro Ames test by metabolic and non-metabolic activated TA100 and TA1535 strains collected from rat liver homogenate.
4. Conclusion
In conclusion, we successfully designed and synthesized a series of novel sulphonamide and carbamate derivatives of nebivolol intermediate in simple and convenient way. All the title compounds were investigated for anti-microbial activity and anti-inflammatory activities by applying the suitable in vitro, in vivo and in silico methods. Out of all the title compounds, 8b, 8c, 8d, 8e, 8f, 8g and 9a exhibited significant and promising activity against all the strains of bacterial, fungal and inhibited the percentage of inflammation and NO production against LPS-induced inflammation in RAW 246.7 cell lines during the treatment. Besides, 8b, 8c, 8d, 8e, 8f, 8g and 9a have significantly attributed for their efficient COX-1 and COX-2 inhibition in vitro and antagonistic activity against carrageenan induced paw edema and granular tissue formation. Molecular docking studies also disclosed that the majority of the synthesized compounds exhibited noteworthy binding modes with high dock scores ranging from −8.8 to −6.0 against DNA gyrase A, −9.3 to −6.3 against COX-1 and −9.4 to −5.6 against COX-2 protein when compared with the standard drugs. In addition to the docking studies, ADMET, QSAR, Bioactivity and Toxicity analysis of compounds are also provided an insight regarding the therapeutic potentiality and confirmed the drug likeness of the title compounds. Hence, the idea of synthesis of sulphonamide and carbamate derivatives of nebivolol intermediate has proved as a commending assignment in designing these potential antiinflammatory agents and proved nebivolol group as a potential pharmacophore of the synthesized derivatives. Therefore, the current study will open new vistas and the lead compounds will stand as the drug candidates for the discovery of new and highly potent pharmacologically active antiinflammatory agents which will have multiple applications in the fields of medicinal and synthetic chemistry.
Conflicts of interest
The authors declared that there are no conflicts of interest in this work.
Acknowledgements
The authors are grateful to the Department of Chemistry, Sri Venkateswara University and Department of Zoology, Faculty of Humanities and Sciences, Sri Venkateswara Vedic University for providing necessary lab facilities to carry out this work.
References
- A. A. Bekhit, H. T. Fahmy, S. A. Rostom and A. M. Baraka, Eur. J. Med. Chem., 2003, 38, 27–36 CrossRef CAS.
- I. K. Khanna, Y. Yu, R. M. Huff, R. M. Weier, X. Xu, F. J. Koszyk, P. W. Collins, J. N. Cogburn, P. C. Isakson, C. M. Koboldt and J. L. Masferrer, J. Med. Chem., 2000, 43, 3168–3185 CrossRef CAS.
- M. M. Frank and L. F. Fries, Immunol. Today, 1991, 12, 322–326 CrossRef CAS.
- H. O. Collier, Nature, 1971, 232, 17–19 CrossRef CAS.
- B. Cryer and M. Feldman, Arch. Intern. Med., 1992, 152, 1145–1155 CrossRef CAS.
- D. M. Clive and J. S. Stoff, N. Engl. J. Med., 1984, 310, 563–572 CrossRef CAS.
- M. C. Allison, A. G. Howatson, C. J. Torrance, F. D. Lee and R. I. Russell, N. Engl. J. Med., 1992, 327, 749–754 CrossRef CAS.
- D. E. Griswold and J. L. Adams, Med. Res. Rev., 1996, 16(2), 181–206 CrossRef CAS.
- J. E. Toblli, F. DiGennaro, J. F. Giani and F. P. Dominici, Vasc. Health Risk Manage., 2012, 8, 151 CrossRef CAS.
- J. Fongemie and E. Felix-Getzik, Drugs, 2015, 75, 1349–1371 CrossRef CAS.
- N. Merchant, S. T. Rahman, K. C. Ferdinand, T. Haque, G. E. Umpierrez and B. V. Khan, J. Hum. Hypertens., 2011, 25, 196–202 CAS.
- M. Serg, P. Kampus, J. Kals, M. Zagura, M. Zilmer, K. Zilmer, T. Kullisaar and J. Eha, Scand. J. Clin. Lab. Invest., 2012, 72, 427–432 CrossRef CAS.
- I. Edes, Z. Gasior and K. Wita, Eur. J. Heart Failure, 2005, 7, 631–639 CrossRef CAS.
- C. L. Fox, S. Modak, J. W. Stanford and P. L. Fox, Scand. J. Plast. Reconstr. Surg., 1979, 13, 89–94 CrossRef CAS.
- C. T. Supuran, F. Briganti, S. Tilli, W. R. Chegwidden and A. Scozzafava, Bioorg. Med. Chem., 2001, 9, 703–714 CrossRef CAS.
- T. Takenaka, K. Shiono, K. Honda, M. Asano, I. Miyazaki and H. Maeno, Clin. Exp. Hypertens., Part A, 1982, 4, 125–137 CAS.
- D. E. Roberts and J. G. Curd, Arthritis Rheumatol., 1990, 33, 1590–1593 CrossRef CAS.
- A. Salahuddin, A. Inam, R. L. van Zyl, D. C. Heslop, C. T. Chen, F. Avecilla, S. M. Agarwal and A. Azam, Bioorg. Med. Chem., 2013, 21, 3080–3089 CrossRef CAS.
- L. Hu, Z. R. Li, Y. Li, J. Qu, Y. H. Ling, J. D. Jiang and D. W. Boykin, J. Med. Chem., 2006, 49, 6273–6282 CrossRef CAS.
- R. Bouasla, M. Berredjem, S. Hessainia, Z. Chereait, H. Berredjem and N. E. Aouf, J. Chem. Chem. Eng., 2011, 5, 512–518 Search PubMed.
- W. H. Miller, A. M. Dessert and R. O. Roblin Jr, J. Am. Chem. Soc., 1950, 72, 4893–4896 CrossRef CAS.
- I. Zadrazilova, S. Pospisilova, M. Masarikova, A. Imramovsky, J. M. Ferriz, J. Vinsova, A. Cizek and J. Jampilek, Eur. J. Pharm. Sci., 2015, 77, 197–207 CrossRef CAS.
- T. R. Fukuto, Environ. Health Perspect., 1990, 87, 245–254 CrossRef CAS.
- M. H. Weiden, H. H. Moorefield and L. K. Payne, J. Econ. Entomol., 1965, 58, 66–70 CrossRef.
- A. E. Essawy, N. E. Abdelmeguied, M. A. Radwan, S. S. Hamed and A. E. Hegazy, Cell Biol. Toxicol., 2009, 25, 275 CrossRef CAS.
- D. W. Elias, M. A. Beazely and N. M. Kandepu, Curr. Med. Chem., 1999, 6, 1125 Search PubMed.
- M. L. Go, X. Wu and X. L. Liu, Curr. Med. Chem., 2005, 12, 483–499 CrossRef CAS.
- B. Song, H. Zhang, H. Wang, S. Yang, L. Jin, D. Hu, L. Pang and W. Xue, J. Agric. Food Chem., 2005, 53, 7886–7891 CrossRef CAS.
- A. G. Al-Bakri and F. U. Afifi, J. Microbiol. Methods, 2007, 68, 19–25 CrossRef CAS.
- J. S. Park, E. M. Park, D. H. Kim, K. Jung, J. S. Jung, E. J. Lee, J. W. Hyun, J. L. Kang and H. S. Kim, J. Neuroimmunol., 2009, 209, 40–49 CrossRef CAS.
- M. Pekarova, J. Kralova, L. Kubala, M. Ciz, I. Papezikova, T. Macickova, J. Pecivova, R. Nosal and A. Lojek, Acta Phys. Pol., 2009, 60, 143 CAS.
- M. Murias, N. Handler, T. Erker, K. Pleban, G. Ecker, P. Saiko, T. Szekeres and W. Jäger, Bioorg. Med. Chem., 2004, 12, 5571–5578 CrossRef CAS.
- C. A. Winter, E. A. Risley and G. W. Nuss, Proc. Soc. Exp. Biol. Med., 1962, 111, 544–547 CrossRef CAS.
- C. A. Winter and C. C. Porter, J. Am. Pharm. Assoc., 1957, 46, 515–519 CrossRef CAS.
- A. D. White, M. W. Creswell, A. W. Chucholowski, C. J. Blankley, M. W. Wilson, R. F. Bousley, A. D. Essenburg, K. L. Hamelehle, B. R. Krause, R. L. Stanfield and M. A. Dominick, J. Med. Chem., 1996, 39, 4382–4395 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 9, 1639–1662 CrossRef.
- E. ter Haar, J. T. Coll, D. A. Austen, H. M. Hsiao, L. Swenson and J. Jain, Nat. Struct. Mol. Biol., 2001, 8, 593–596 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- S. Vilar, G. Cozza and S. Moro, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS.
- http://preadmet.bmdrc.org/, (accessed 20 December 2019).
- http://www.molinspiration.com/cgi-bin/properties, (accessed 10 December 2019).
- http://www.organic-chemistry.org/prog/peo/, (accessed 01 December 2019).
Footnotes |
| † Electronic supplementary information (ESI) available: Spectra of 1H, 13C and LCMS reports of 8a and 9c compounds are given. See DOI: 10.1039/d0ra08905b |
| ‡ The first and second authors are equally contributed to the work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc,
N. Y. Sreedhara and
C. Naga Raju*a
*bc,
N. Y. Sreedhara and
C. Naga Raju*a