
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon-coated SnO2 riveted on a reduced graphene oxide composite (C@SnO2/RGO) as an anode material for lithium-ion batteries†
Yao Daiae,
Fu Lide,
Yuan-Xiang Fube,
Dong-Chuan Mo ce and
Shu-Shen Lyu*ce
ce and
Shu-Shen Lyu*ce
aSchool of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: lvshsh@mail.sysu.edu.cn
bSchool of Chemical Engineering & Guizhou Provincial Key Laboratory of Energy Chemistry, Guizhou Institute of Technology, Guiyang, 550003, PR China
cSchool of Materials, Sun Yat-sen University, Guangzhou 510275, P. R. China
dSchool of Chemical Engineering and Technology, Sun Yat-sen University, Guangzhou, 510275, China
eGuangdong Engineering Technology Research Centre for Advanced Thermal Control Material and System Integration (ATCMSI), Sun Yat-sen University, Guangzhou, 510275, P. R. China
First published on 24th February 2021
Abstract
The research on graphene-based anode materials for high-performance lithium-ion batteries (LIBs) has been prevalent in recent years. In the present work, carbon-coated SnO2 riveted on a reduced graphene oxide sheet composite (C@SnO2/RGO) was fabricated using GO solution, SnCl4, and glucose via a hydrothermal method after heat treatment. When the composite was exploited as an anode material for LIBs, the electrodes were found to exhibit a stable reversible discharge capacity of 843 mA h g−1 at 100 mA g−1 after 100 cycles with 99.5% coulombic efficiency (CE), and a specific capacity of 485 mA h g−1 at 1000 mA g−1 after 200 cycles; these values were higher than those for a sample without glucose (SnO2/RGO) and a pure SnO2 sample. The favourable electrochemical performances of the C@SnO2/RGO electrodes may be attributed to the special double-carbon structure of the composite, which can effectively suppress the volume expansion of SnO2 nanoparticles and facilitate the transfer rates of Li+ and electrons during the charge/discharge process.
Introduction
Rechargeable lithium-ion batteries (LIBs) have been extensively applied in mobile electronic devices, electric automobiles, and storage systems for renewable energy sources (such as wind, tidal, and solar energy) due to their high energy density and excellent recyclability, among other advantages.1 However, with the explosive development of highly-integrated electric equipment, pure electric vehicles, and hybrid electric vehicles in the last decade, the increasing market requirements of high-performance LIBs are hindered by the relatively low theoretical capacity of commercial graphite (∼372 mA h g−1) anode materials.2,3 At the same time, it is extremely significant to develop anode materials with high theoretical capacities and excellent cycle stability in the industrial application of LIBs.4–6 Stannic oxide (SnO2) has aroused significant interest as one of the most promising anode electrodes for LIBs due to its high theoretical capacity (782 mA h g−1), environmental friendliness, and low cost.7–9 Nevertheless, some inherent defects, including the ∼300% volume expansion during repeated Li+ extraction and insertion, as well as low electron and ion conductivities, directly impede the practical application of SnO2 materials in LIBs. Approaches including the preparation of SnO2 carbon-based composites (with amorphous carbon, porous carbon, carbon nanotubes, and graphene sheets),10–13 the hybridisation of other inorganic matters,14–16 and the adjustment of the structure of SnO2 have been explored for mitigating its defects with remarkable effects.9,17,18However, the methods also have some non-ignorable problems. For SnO2 carbon-based composites, on the one hand, they can't give play to the characteristics for high capacity because of the carbon material.19,20 On the other hand, SnO2 nanocrystalline is difficult to be uniformly distributed on carbon materials, and agglomeration will occur in some areas, which weaken the buffer effect of carbon materials on volume expansion of SnO2 in the process of charging and discharging.21 The hybridization of SnO2 with other inorganic substances also has some disadvantages. In order to improve the capacity, researchers are more inclined to hybridize with inorganic materials of high capacity. However, such materials often have some defects, such as poor electrical conductivity and volume expansion, which are even worse than tin oxide.22–25 Whereas hybridizing with inorganic materials of low capacity, the problems are similar to those of carbon composites.26,27 For structural adjustment of SnO2, hollow structure is the focus of research at present.28,29 However, the existence of hollow structure will inevitably waste the effective space of the material, and the performance will decline sharply once the structure collapses. Compared with the above methods, in terms of operability, SnO2 carbon-based composite material is easier to achieve. Additionally, due to the inherently superior properties of graphene, the assembly of SnO2–graphene-based composites are considered to be a more promising strategy for the facilitation of the practical application of SnO2 anodes in LIBs.30–32 Graphene is a honeycomb-structured carbon lattice of single-layer carbon material, and is considered to be an ideal base material for the anodes and cathodes of LIBs due to its high electrical and thermal conductivities, large specific surface area, and excellent mechanical capacities.33–35 Based on these qualities, graphene sheets can be effectively applied to improve the performance of low-conductivity and large-expansion electrode composites for energy storage. Wu et al.36 synthesised SnO2/graphene oxide (SnO2/GO) anode material via a hydrothermal method, and the resulting electrode displayed a reversible capacity of 612.2 mA h g−1 after 100 cycles with 98.8% coulombic efficiency (CE) due to the assistance of GO. Wang et al. designed and successfully prepared sandwich-like mesoporous SnO2/graphene/mesoporous SnO2 nanosheets (C@SnO2–rGO–SnO2) that acted as a template for the assembly of SnO2 nanoparticles, and the resulting electrode was found to have a high reversible capacity (1211 mA h g−1 after 300 cycles at 200 mA g−1) and good stability (525 mA h g−1 after 1200 cycles at 2000 mA g−1).32 Zhou et al. fabricated three-dimensional hollow SnO2@TiO2 spheres with GO aerogels (HSTGAs) via a self-assembly technique, and the resulting electrode was found to maintain a reversible capacity of ∼668.2 mA h g−1 after 200 cycles at 100 mA g−1 for LIBs, which was higher than those of the pure SnO2@TiO2 spheres (HSTSs; 138 mA h g−1) and hollow SnO2 spheres (HSSs; 109.1 mA h g−1) under the same test conditions.37 It is therefore evident that previous studies have revealed that graphene sheets can improve the energy storage performance of pure SnO2.
The key of preparing SnO2 carbon-based composite lies in how to solve the dispersibility problem of SnO2 nanocrystalline. In the present study, carbon-coated SnO2 riveted on reduced graphene oxide composite (denoted as C@SnO2/RGO) was designed and successfully prepared as the anode material for LIBs using a sample hydrothermal method. Briefly, GO contains a large number of uniformly dispersed oxygen-containing functional groups. We used electrostatic adsorption to make the high valence Sn4+ ions fixed on these functional groups, which ensure the SnO2 riveted on GO and dispersed uniformly. At the same time, in the process of hydrothermal reaction, the hydroxyl and aldehyde groups on the glucose can also be adsorbed on the Sn4+, then the formation of carbon film to completely wrap it. The synergistic effect of flexible GO and hard glucose makes the tin dioxide firmly fixed after carbonization. It was also demonstrated that the carbon-coated SnO2 and RGO sheets exhibited an obvious synergistic effect, and the C@SnO2/RGO presented superior electrochemical performance as compared to both SnO2/RGO and pure SnO2 nanoparticles. The C@SnO2/RGO electrode was found to display a stable reversible discharge capacity of 843 mA h g−1 at 100 mA g−1 after 100 cycles, and 485 mA h g−1 after 200 cycles at 1000 mA g−1 with ∼100% CE.
Experimental section
Materials synthesis
Graphene oxide (GO) powder was produced using an improved Hummer's method with some adjustments.38,39 Then, the C@SnO2/RGO composite was synthesised via a hydrothermal method accompanied by heat calcination. In a typical process, 900 mg SnCl4·5H2O, 90 mg GO powder, 512 mg glucose, and 60 ml deionised water underwent ultrasonication for 45 min to form a homogeneous solution. The solution was then transferred to a Teflon-lined stainless-steel autoclave in an oven at 180 °C for 10 h. Subsequently, a black product was collected after washing several times with deionised water and freeze-drying, and the C@SnO2/RGO composite was obtained after the black product was calcined in an N2 atmosphere at 600 °C (denoted as C@SnO2/RGO-90). The C@SnO2/RGO-45 and SnO2@C/RGO-135 specimens were synthesised via the same process by adjusting the content of GO. For comparative study, SnO2/RGO-90 without glucose and pure SnO2 nanoparticle samples without GO or glucose were prepared using the same method.Characterization and measurements
The crystalline structures of all samples were measured by a Rigaku D-MAX 2200 VPC X-ray diffractometer (XRD) with Cu Ka radiation. The degree of defect in samples could be measured by microscopic confocal Raman spectrometer with an excitation wavelength of 514.5 nm and 785 nm. Morphologies of the samples were observed using a FEI-Q400F scanning electron microscope (SEM) equipped with an X-ray energy dispersive spectrometer (EDS) and a JEOL-2010 HR transmission electron microscope (TEM). X-ray photoelectron spectroscopy (XPS) was conducted on an ESCALAB 250 scanning X-ray microprobe spectrometer with an Al Kα X-ray source. Thermogravimetric analysis (TGA) was performed on a 209 F3 Tarsus TG instrument between room temperature (RT) and 850 °C in air. The Brunauer–Emmett–Teller (BET) nitrogen adsorption–desorption isotherms and pore size distribution was analysed by an accelerated surface area and porosimetry system (Micromeritics, ASAP2460).Electrochemical characterization
Electrochemical tests were performed via CR2032 coin-type half cells at room temperature within 0.01–3 V (vs. Li+/Li). The working electrodes were prepared by mixing the active materials (75%), carbon black (15%), and polyvinylidene fluoride (PVDF, 10%) in an N-methyl-2-pyrrolidinone (NMP) solution to form a homogeneous slurry. The slurry was then painted on copper foil and dried at 120 °C under a vacuum overnight. The CR2032 coin-type cells were assembled in a glove box using lithium foil as the counter-electrode, 1 M LiPF6 in ethylene carbonate/ethyl methyl carbonate/dimethyl carbonate (vol = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) as the electrolyte, Celgard 2400 polypropylene film as the separator, and the as-prepared working electrodes (active material mass ∼1.2 mg cm−2). Discharge/charge tests were conducted on a Neware battery testing system with a voltage range of 0.01 to 3 V (vs. Li/Li+). Cyclic voltammetry (CV) was performed on an electrochemical workstation (Chenhua, CHI760E) at a scan rate of 0.1 mV s−1 from 0.01 to 3 V (vs. Li/Li+). The electrochemical impedance spectrum (EIS) measurements (from 1 to 0.1 Hz) were tested using an electrochemical workstation (Chenhua, CHI760E).
:1:1) as the electrolyte, Celgard 2400 polypropylene film as the separator, and the as-prepared working electrodes (active material mass ∼1.2 mg cm−2). Discharge/charge tests were conducted on a Neware battery testing system with a voltage range of 0.01 to 3 V (vs. Li/Li+). Cyclic voltammetry (CV) was performed on an electrochemical workstation (Chenhua, CHI760E) at a scan rate of 0.1 mV s−1 from 0.01 to 3 V (vs. Li/Li+). The electrochemical impedance spectrum (EIS) measurements (from 1 to 0.1 Hz) were tested using an electrochemical workstation (Chenhua, CHI760E).
Results and discussion
The preparation process schematic of the C@SnO2/RGO-90 is presented in Fig. 1. The C@SnO2/RGO-90 sample was obtained after the hydrothermal process and heat treatment. X-ray diffraction (XRD) was used to confirm the crystalline structure of the resultant specimen. Fig. 2a presents the XRD patterns of the C@SnO2/RGO-90 and SnO2/RGO-90 samples, and reveals four broad sharp diffraction peaks located at 2θ ≈ 26.6°, 33.8°, 37.9°, and 51.8°, as well as some weak peaks that can be attributed to the tetragonal SnO2 phase (JCPDS 72-1147);36,40 the broad peaks of the samples indicate the small SnO2 particles. It was also found that a broad peak at about 2θ ≈ 20–30° appeared on the XRD curves of the C@SnO2/RGO-90 and SnO2/RGO-90 composites as compared with the SnO2 sample, implying the presence of carbonaceous material in the composites.36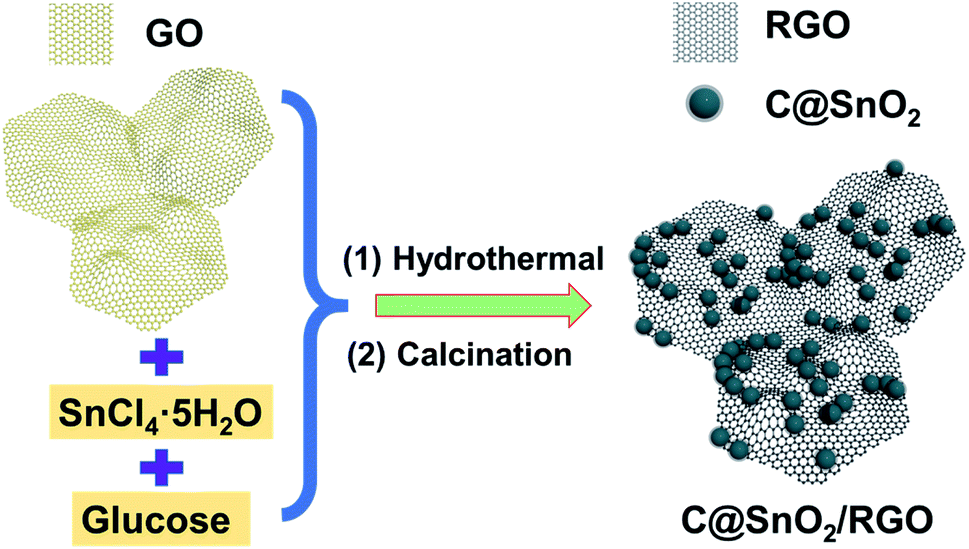 | ||
| Fig. 1 Schematic illustration of the preparation of the C@SnO2/RGO composite. | ||
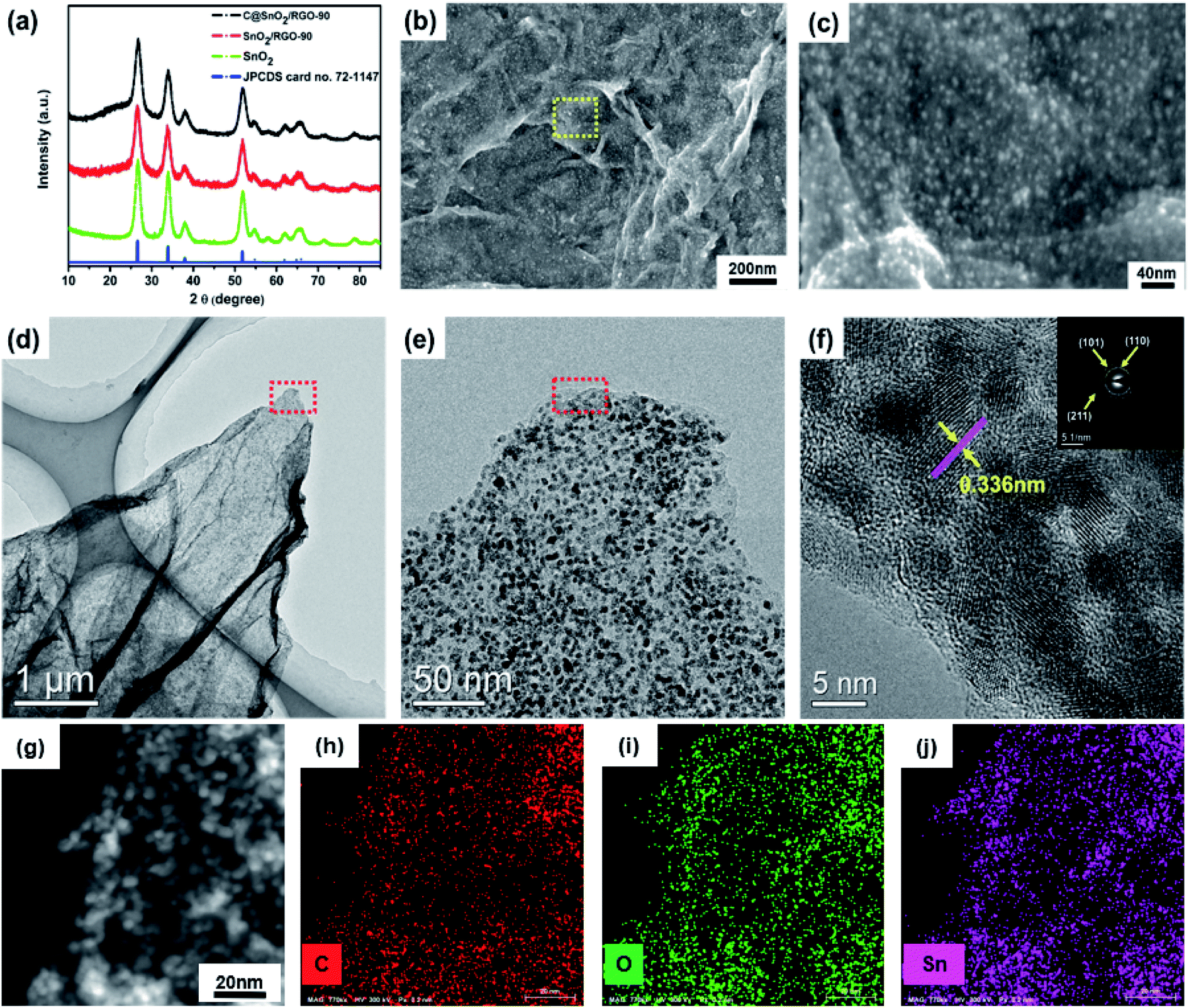 | ||
| Fig. 2 (a) XRD patterns of pure SnO2, SnO2/RGO-90, and C@SnO2/RGO-90. (b and c) SEM images and (d–f) TEM images of C@SnO2/RGO-90. (g) The corresponding EDX elemental mappings (h) C, (i) O and (j) Sn of the C@SnO2/RGO-90 composite. | ||
SEM images reveal the morphology of the C@SnO2/RGO-90 composite. As presented in Fig. 2b, numerous SnO2 nanoparticles were observed to be riveted on the surface of the RGO sheets, and the magnified image presented in Fig. 2c more clearly displays the SnO2 nanoparticles with sizes of 2–5 nm. The SnO2/RGO-90 sample displayed similar features to those of the C@SnO2/RGO-90 composite (Fig. S1†). The pure SnO2 particles were larger than those of the C@SnO2/RGO-90 and SnO2/RGO-90 composites (Fig. S2†), which may be attributed to the presence of GO. TEM and high-resolution TEM (HRTEM) images were observed to better explore the microstructure of the C@SnO2/RGO-90 composite. Fig. 2d displays RGO sheets with obvious wrinkles and a coating of many SnO2 nanoparticles; moreover, the magnified TEM image in Fig. 2e and S3† further confirms that the SnO2 nanoparticles were uniformly dispersed on the RGO sheet, which is in agreement with the magnified SEM image. The HRTEM image further clearly reveals that the lattice spacing of the tetragonal SnO2 nanoparticles was 0.336 nm, corresponding to the tetragonal SnO2 (110) planes.30,36 The carbon layer (red arrow) and RGO sheet (purple arrow) are also depicted in the image. The selected-area electron diffraction (SAED) pattern of the C@SnO2/RGO-90 composite presented in Fig. 2f reveals obvious diffraction rings in the top right corner, which verifies the XRD result of the tetragonal structure of SnO2 in the C@SnO2/RGO-90 composite. As shown in Fig. 2g–j, the X-ray EDX spectrum reveals the presence of C, O, and Sn in the C@SnO2/RGO-90 sample from the test specimen stage. As determined from the three elemental mapping figures, the Sn was dispersed homogeneously in the C@SnO2/RGO-90 composite.
The reduction degree of GO for the sample was analysed by Raman spectra. As shown in Fig. 3a, the GO structural disorders degree was confirmed by the ratio of D band at 1340 cm−1 and G band at 1580 cm−1. The ID/IG ratio for C@SnO2/RGO-90 composite (1.13) is close to SnO2/RGO-90 composite (1.05), but higher than that of GO (0.85), which specifies the increased degree of structural defects in the C@SnO2/RGO-90 and SnO2/RGO-90 composites in comparison with the GO. Fig. 3b presents the full survey XPS spectrum of the C@SnO2/RGO-90 specimen, which again authenticates the presence of C, O, and Sn. Fig. 3c presents the high-resolution spectrum of the C1s region, which can be divided into three peaks located at 284.7 eV, 285.7 eV, and 287.3 eV, which are respectively attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–OH, and CO bonds.31 The high-resolution spectrum of the O1s peak presented in Fig. 3d exhibits three peaks located at 531.0 eV, 531.4 eV, and 532.8 eV, which are respectively assigned to Sn–O–Sn, CO, and C–O–H groups.41 As shown in Fig. 3e, the Sn 3d peak is composed of the Sn 3d3/2 and Sn 3d5/2 peaks, which respectively correspond to 495.8 and 487.4 eV. This provides confirmation of the presence of SnO2 because the binding energy values are related to that of Sn4+.37 TGA was subsequently carried out to quantify the component contents in the products. Fig. 3f reveals a weight loss of 2.5 wt% of the C@SnO2/RGO-90 composite in air before the temperature reached 200 °C, which can be ascribed to the evaporation of gas or water from the specimen. With the increase of the temperature, the weight declined quickly in the range from 300 to 420 °C, and the final weight loss of the sample was found to be 17.7 wt% after the temperature reached 800 °C, which can be primarily attributed to the combustion of carbon (amorphous carbon and RGO). The SnO2/RGO-90 sample experienced a weight loss of about 10.6 wt% over a similar process as that of the C@SnO2/RGO-90 sample. Thus, the mass of amorphous carbon approximately 7.1 wt% in the SnO2/RGO-90 composite base on the mass loss of the pure SnO2 is 3.8 wt% in the same process.42 The specific surface area (BET) of the SnO2/RGO-90 sample was about 153.2 m2 g−1, as indicated by the adsorption/desorption isotherm in Fig. S4† and the corresponding average pore diameter of ∼3 nm was characterised via nitrogen sorption analysis.
C, C–OH, and CO bonds.31 The high-resolution spectrum of the O1s peak presented in Fig. 3d exhibits three peaks located at 531.0 eV, 531.4 eV, and 532.8 eV, which are respectively assigned to Sn–O–Sn, CO, and C–O–H groups.41 As shown in Fig. 3e, the Sn 3d peak is composed of the Sn 3d3/2 and Sn 3d5/2 peaks, which respectively correspond to 495.8 and 487.4 eV. This provides confirmation of the presence of SnO2 because the binding energy values are related to that of Sn4+.37 TGA was subsequently carried out to quantify the component contents in the products. Fig. 3f reveals a weight loss of 2.5 wt% of the C@SnO2/RGO-90 composite in air before the temperature reached 200 °C, which can be ascribed to the evaporation of gas or water from the specimen. With the increase of the temperature, the weight declined quickly in the range from 300 to 420 °C, and the final weight loss of the sample was found to be 17.7 wt% after the temperature reached 800 °C, which can be primarily attributed to the combustion of carbon (amorphous carbon and RGO). The SnO2/RGO-90 sample experienced a weight loss of about 10.6 wt% over a similar process as that of the C@SnO2/RGO-90 sample. Thus, the mass of amorphous carbon approximately 7.1 wt% in the SnO2/RGO-90 composite base on the mass loss of the pure SnO2 is 3.8 wt% in the same process.42 The specific surface area (BET) of the SnO2/RGO-90 sample was about 153.2 m2 g−1, as indicated by the adsorption/desorption isotherm in Fig. S4† and the corresponding average pore diameter of ∼3 nm was characterised via nitrogen sorption analysis.
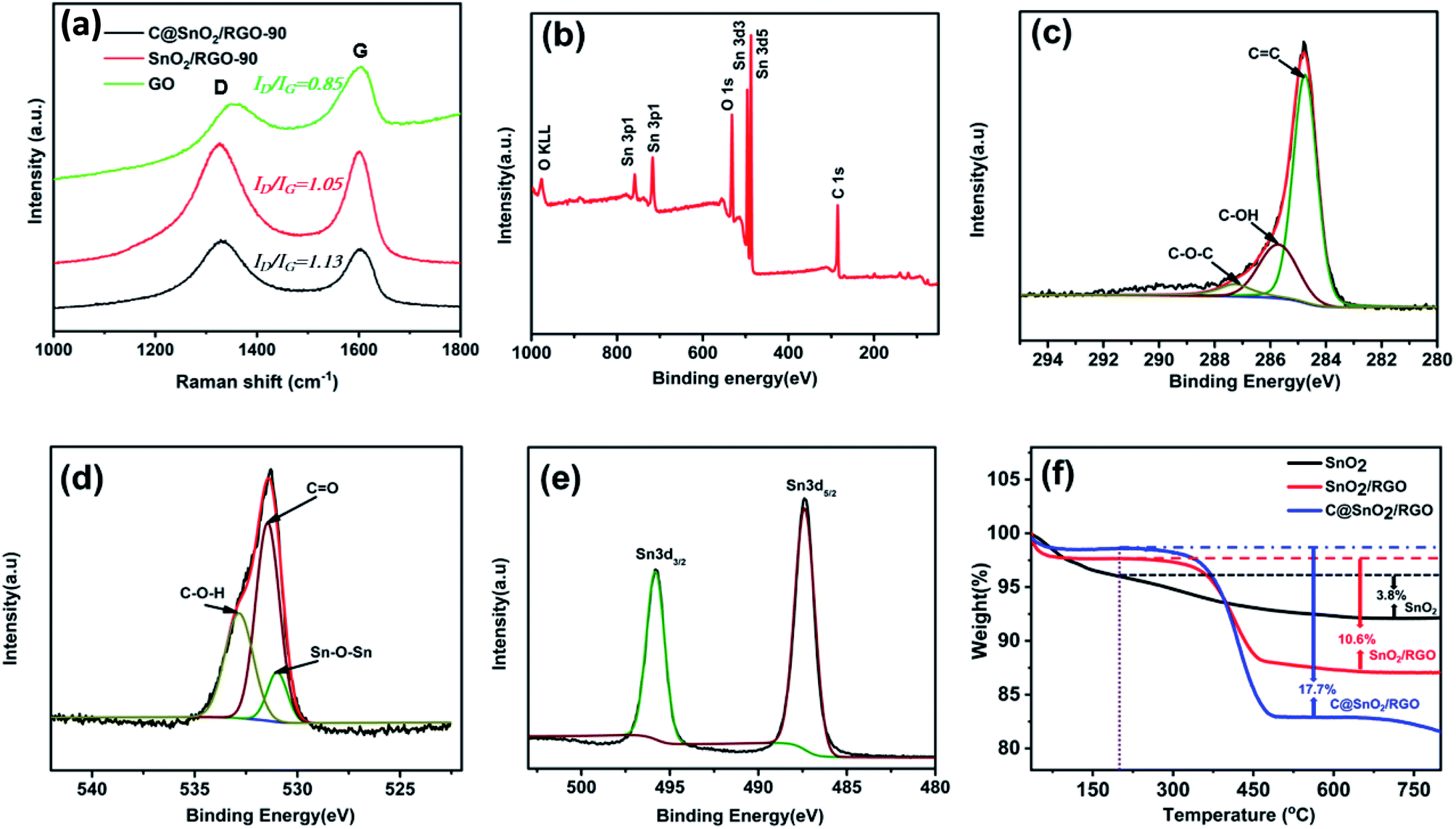 | ||
| Fig. 3 (a) Raman spectrum of the C@SnO2/RGO-90, SnO2/RGO-90, GO composite and (b) full survey XPS spectrum of the C@SnO2/RGO-90 composite, (c–e) high-resolution XPS spectra of C1s, O1s, and Sn 3d of the C@SnO2/RGO-90, respectively, and (f) TGA curves of SnO2, SnO2/RGO-90, and C@SnO2/RGO-90. | ||
The electrochemical performance of the C@SnO2/RGO-90 composite was investigated by various experimental tests. As presented in Fig. 4a, the CV curves of the composite were obtained in a range from 0.1 to 3.0 V at a scanning rate of 0.1 mV s−1. A broad peak at ∼1.25 V and a sharp peak at ∼0.52 V appeared in the first anodic scan. The ∼1.25 V peak can be ascribed to the formation of the SEI film and the SnO2 conversion reaction (SnO2 + 4Li+ + 4e− ⇌ 2Li2O + Sn), and the sharp peak at ∼0.52 V can be attributed to the Sn metal lithiation reaction.43 Moreover, broad peaks at 0.6 V and ∼1.25 V were observed in the first cathodic sweep derived from the Sn metal de-lithiation reaction and SnO2 conversion reaction, respectively.44 It was also found that the following CV curves almost overlapped, indicating that the C@SnO2/RGO-90 electrode was characterized by good reversibility. Fig. 4b exhibits the charge and discharge curves of the C@SnO2/RGO-90 electrode at 100 mA g−1. The first discharge/charge capacities of the C@SnO2/RGO-90 electrode was found to be 1528.9 mA h g−1 and 972.8 mA h g−1, respectively, and the initial coulombic efficiency (ICE) was 63.6%. The irreversible capacity may be ascribed to the consumption of SEI formation between the electrode interfaces. The second discharge/charge capacities were found to be 1000.4 mA h g−1 and 971.5 mA h g−1, respectively, and the CE was increased to 97.1%. Moreover, the discharge capacity of the electrode could be maintained at 842.7 mA h g−1 after 100 cycles. Fig. 4c presents the cycle performance of the C@SnO2/RGO-90 electrode after 100 cycles at 100 mA g−1; the electrode exhibited a high reversible capacity of 842.7 mA h g−1 after 100 cycles with good cycle stability (CE ∼99.5%), higher than some SnO2 -based anodes in early reports, as shown in Table 1. As a comparison, the reversible capacities of the SnO2/RGO-90 electrode gradually degraded to 568.2 mA h g−1 after 100 cycles at 100 mA g−1. It was also found that the pure SnO2 electrode only maintained a reversible capacity of 184 mA h g−1 under the same test conditions.
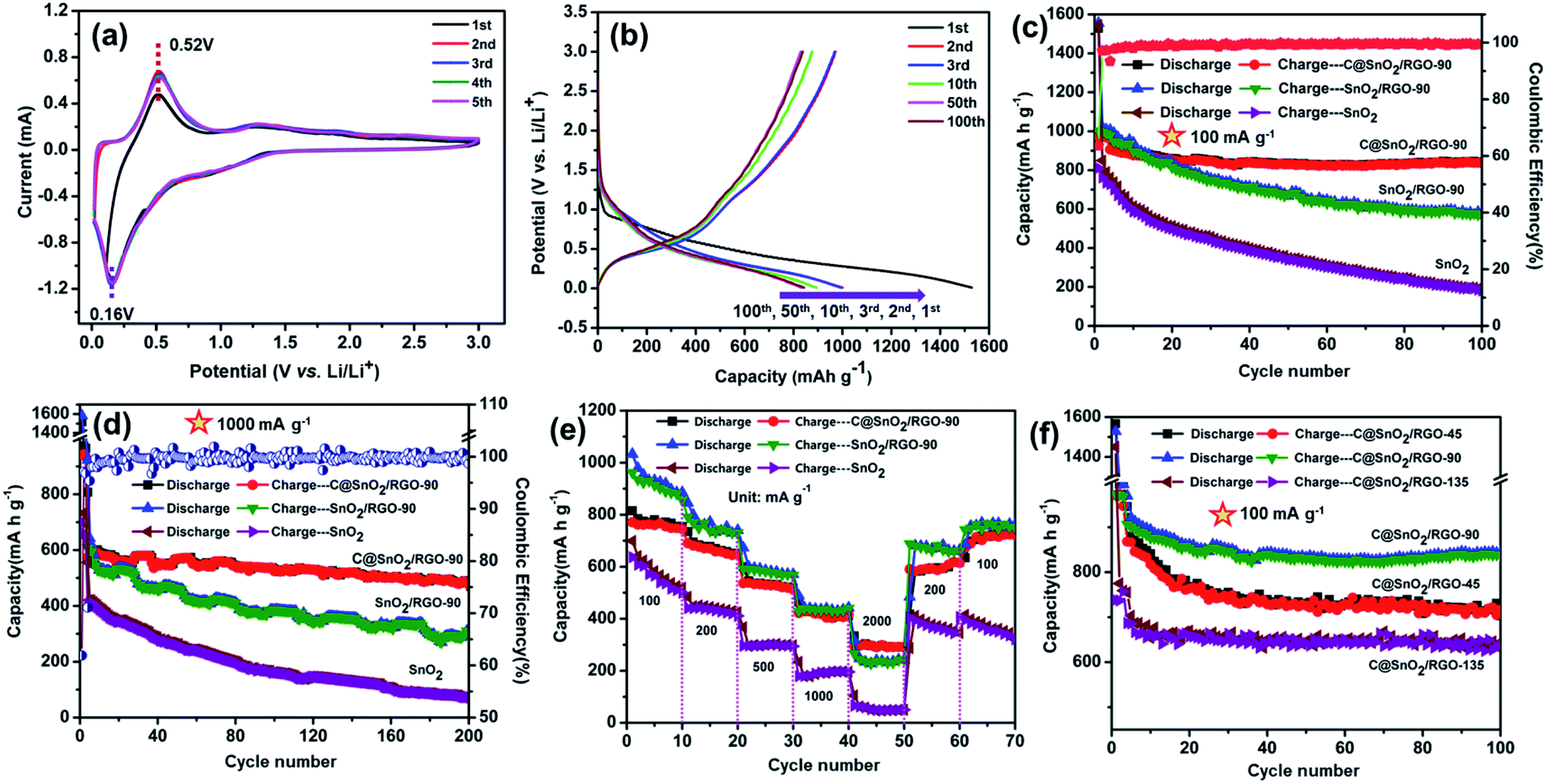 | ||
| Fig. 4 (a) CV curves of C@SnO2/RGO-90 at a scan rate of 0.1 mV s−1. (b) Discharge/charge curves of C@SnO2/RGO-90 at 100 mA g−1. (c) Cycling performances and CE patterns of the C@SnO2/RGO-90 sample at 100 and (d) 1000 mA g−1. (e) Rate capacities of the three samples. (f) Cycling performances of C@SnO2/RGO samples with different RGO contents. | ||
| Samples | Current density (mA g−1) | Cycle number | Capacity (mA h g−1) | Reference |
|---|---|---|---|---|
| SnO2/GO | 100 | 100 | 612.2 | 36 |
| 3D porous SnO2/C | 250 | 250 | 513 | 45 |
| Carbon-coated SnO2 nanotubes | 100 | 30 | 492.5, (0.01–2 V) | 46 |
| SnO2/C | 80 | 100 | 441.0 | 13 |
| CNT@SnO2@C | 720 | 40 | 698 | 47 |
| Carbon-riveted graphene/SnO2 | 100 | 150 | 815 | 48 |
| rGO/SnO2–C | 100 | 200 | 496.3 | 49 |
| SnO2 QDs@ C | 100 | 100 | 724.9 | 50 |
| CNC–SnO2 | 100 | 100 | 380 | 51 |
| SnO2−x nanoparticles | 500 | 100 | 380 | 52 |
| Hollow SnO2@C | 0.1C | 100 | 741 | 53 |
| ZnO/graphene | 0.5C | 100 | 404 | 54 |
| CuO–graphene | 1000 | 60 | 349 | 55 |
| C@SnO2/RGO | 100 | 100 | 843 | This work |
| 1000 | 200 | 485 |
It can be observed that the reversible capacities of the SnO2/RGO-90 sample were notably higher than those of the pure SnO2 specimen, which may be ascribed to the ultrathin RGO sheet, which could suppress the expansion of the SnO2 nanoparticles during the charge and discharge processes. The RGO sheet can also enhance the internal electron and Li-ion transport performance in the battery due to its inherent high electrical conductivity. Additionally, the C@SnO2/RGO-90 electrode exhibited favourable cycling stability and a higher reversible capacity than the SnO2/RGO-90 electrode. It is considered that the glucose transformed into amorphous carbon after the hydrothermal reaction and annealing treatment, and the amorphous carbon formed a protective film on the SnO2 nanoparticles; this protective film could more effectively rivet SnO2 nanoparticles on the surfaces of the RGO sheets to ultimately endure greater volume change during the charge and discharge processes. Therefore, the C@SnO2/RGO-90 sample exhibited superior cycling stability to that of the SnO2/RGO-90 specimen.
It is worth noting that the C@SnO2/RGO-90 electrode could deliver a reversible capacity of 485 mA h g−1 at 1000 mA g−1 after 200 cycles (Fig. 4d), which is around 1.6 times higher than that of the SnO2/RGO-90 electrode (310 mA h g−1), and 6.5 times higher than that of the pure SnO2 electrode (72 mA h g−1) under the same test conditions. Additionally, the results of the C@SnO2/RGO-90 electrode were also relatively higher than the results of SnO2 and others types of transition metal oxides (TMOs) graphene based anode materials, as shown in Table 1. The rate capabilities of the samples are presented in Fig. 4e; the discharge capacity of the C@SnO2/RGO-90 electrode was found to be 754, 648, 518, 411, and 292 mA h g−1 at increasing current rates of 100, 200, 500, 1000, and 2000 mA g−1, respectively. Furthermore, the reversible capacity of the electrode can be restored to 733 when the current is reverted to 100 mA g−1. In contrast, the SnO2/RGO-90 electrode exhibited low reversible capacity attained 242 mA h g−1 at 2000 mA g−1, and the pure SnO2 electrode delivered a poor capacity of only 50 mA h g−1 at 2000 mA g−1, which was lower than that of the C@SnO2/RGO-90 electrode. To explore the capacity effect of RGO sheets content in the C@SnO2/RGO sample, two C@SnO2/RGO-45 and C@SnO2/RGO-135 samples were synthesised by adjusting the GO content via the same preparation method. Fig. 4f presents the cycle performances of the three samples at 100 mA g−1, from which it is evident that the C@SnO2/RGO-90 electrode exhibited higher reversible capacities and more excellent cycle stability than both the C@SnO2/RGO-45 and C@SnO2/RGO-135 electrodes after 100 cycles at 100 mA g−1. In addition, the C@SnO2/RGO-90 electrode also displayed higher reversible capacities than the two other electrodes at 1000 mA g−1 after 200 cycles (Fig. S5†), and the rate performances of the C@SnO2/RGO-90 electrode were superior to those of the other two specimens (Fig. S6†). The reason for this could be ascribed to the low content of RGO sheets in the sample, which could not effectively inhibit the volume effect of the SnO2 nanoparticles during the charge/discharge processes.56 Meanwhile, the high content of RGO sheets may have decreased the content of SnO2, and the lithium storage capability of pure RGO sheets is not high. Therefore, the excellent lithium storage capability of the C@SnO2/RGO-90 specimen may be attributed to the rational collocation of SnO2 nanoparticles and the RGO content.57
The EIS results reveal the electronic resistance and Li+ diffusion properties of the C@SnO2/RGO-90 electrode. The Nyquist plots of the pure SnO2, SnO2/RGO-90, and C@SnO2/RGO-90 samples after 200 cycles at 1000 mA g−1 are presented in Fig. 5a. The electrolyte resistance (RSEI) and charge-transfer resistance (Rct) information was respectively indicated by the high and medium frequencies of the Nyquist plot. Additionally, the RSEI and the Rct values of the three electrodes as determined after calculation using the equivalent circuit are exhibited in Table 2. It can be observed that the RSEI value of the C@SnO2/RGO-90 electrode was less than those of the other two electrodes, which could be attributed to the synergistic effect of the RGO and amorphous carbon layer. Additionally, the RSEI value of the C@SnO2/RGO-90 electrode after testing was also less than that before testing, as shown in Fig. S7 and Table S1.†
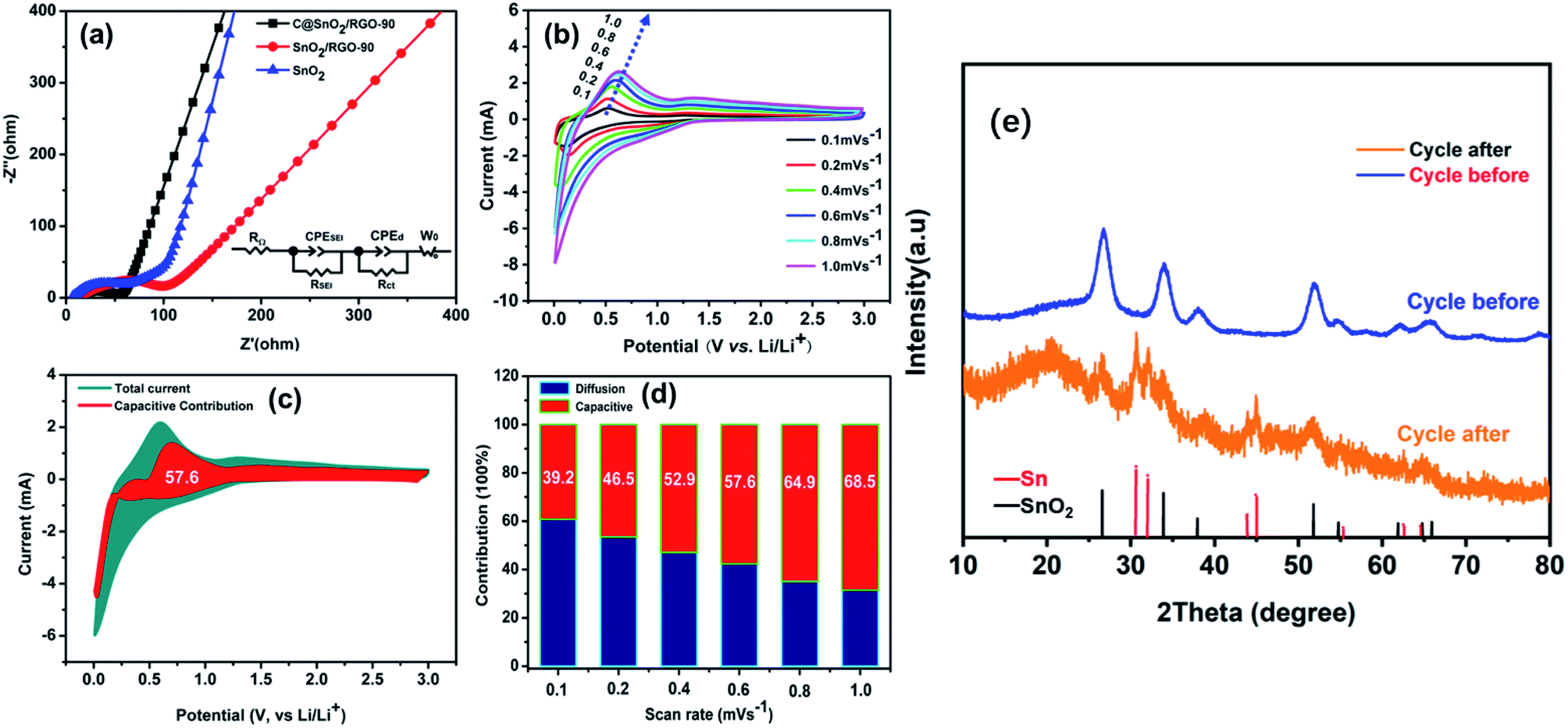 | ||
| Fig. 5 (a) The EIS curves of SnO2, SnO2/RGO-90, and C@SnO2/RGO-90 after 200 cycles at 1000 mA g−1. (b) The CV curves of C@SnO2/RGO-90 at scan rates of 0.1, 0.2, 0.4, 0.6, 0.8, and 1.0 mV s−1. (c) Capacitive capacity contribution ratio (red region) in comparison with the total current at 0.6 mV s−1. (d) Histograms of the capacitive capacity contribution ratios of C@SnO2/RGO-90 at various scan rates. (e) XRD patterns of C@SnO2/RGO-90 electrode at 1000 mA g−1 after 200 cycles. | ||
| Samples | RSEI (Ω) | Rct (Ω) | DLi (cm−2 s−1) |
|---|---|---|---|
| C@SnO2/RGO-90 | 29.12 | 5.092 | 5.28 × 10−12 |
| SnO2/RGO-90 | 81.15 | 10.92 | 2.87 × 10−12 |
| SnO2 | 42.11 | 2.52 | 1.67 × 10−12 |
The Li+ diffusion performances of the three electrodes were determined using eqn (1) according to the low frequency of the Nyquist plots in the linear Warburg regions (between 0.2 and 0.01 Hz).58 In the formula, F is the Faraday constant, A is the contact surface area of the electrode and electrolyte, σw is the Warburg pre-factor from the Warburg region of impedance response, and Vm is the molar volume of the specimen. The calculated DLi values of the three samples are presented in Table 2. The DLi value of the C@SnO2/RGO-90 sample was found to be about 1.8 times greater than that of the SnO2/RGO-90 sample, and about 3.16 times greater than that of the pure SnO2 specimen. It is clear that the improvement in the internal conductivity of the C@SnO2/RGO-90 sample was derived from the synergistic effect of the RGO sheets and the amorphous carbon layer.42,59,60
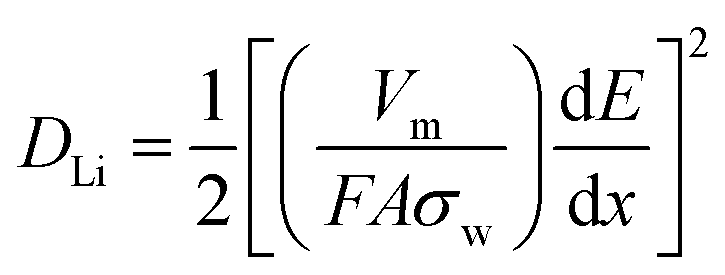 | (1) |
The diffusion and capacitive capacity contributions in the C@SnO2/RGO-90 were investigated at different CV scan rates to explain the favourable Li+ storage performance. Fig. 5b presents the CV curves obtained at five different scan rates. Additionally, the quantified capacitive contributions of the C@SnO2/RGO-90 electrode were obtained by eqn (2),61 where ν is the sweep rate in the formula, and k1 and k2 are constants that can be obtained by plotting iv−1/2 versus v1/2. It can be observed in Fig. 5c that the capacitive capacity contribution ratio (red region) of the C@SnO2/RGO-90 electrode was around 57.6% in comparison with the total current at a scan rate of 0.6 mV s−1. Moreover, with the increase of the scan rate, the capacitive capacity contribution ratio of the C@SnO2/RGO-90 electrode gradually increased (Fig. 5d), and attained 68.5% at a scan rate of 1 mV s−1. Thus, the C@SnO2/RGO-90 electrode delivered favourable lithium storage performance, which may be partially ascribed to the high capacitive capacity contribution.62 At the same time, ex situ XRD have been performed to investigate the C@SnO2/RGO-90 electrode at 1000 mA g−1 after 200 cycles (Fig. 5e). In addition to SnO2, elemental Sn was found after the C@SnO2/RGO-90 electrode cycling, which was consistent with the electrochemical reaction mechanism of SnO2-based LIBs.63 It showed that the C@SnO2/RGO-90 electrode was reactive after 200 cycles with high current density, the structure of the electrode material was not destroyed by the volume expansion of SnO2 during lithiation. It was also proved that the double-carbon effect significantly enhances the stability of electrode materials.
| i = k1ν + k2v1/2 | (2) |
The excellent lithium storage performance of the C@SnO2/RGO-90 electrode as an anode material for LIBs can be assigned to the following reasons. (a) The special sheet structure of the C@SnO2/RGO-90 sample was due to the ultrathin RGO sheets, which improved the electrical conductivity of the sample.36,64,65 In addition, the flexible structure was able to release the volume change of the SnO2 nanoparticles during the charge and discharge cycles. (b) The protective effect of SnO2 from the amorphous carbon layer was derived from glucose. First, the amorphous carbon layer could effectively fix SnO2 nanoparticles on the surface of RGO sheets, similar to rivets, which could provide additional force to restrain the volume change of the SnO2 nanoparticles during the lithiation and de-lithiation processes. Secondly, the electron transport and Li-ion transport performances of the sample were enhanced due to the existence of the amorphous carbon layer on the surfaces of the SnO2 nanoparticles.30,32 (c) The high capacitive capacity contribution in the specimen was due to the synergistic effect of amorphous carbon and RGO sheets, which allowed the C@SnO2/RGO-90 electrode to maintain excellent cycling stability under a large current density.44,60 For these reasons, the C@SnO2/RGO-90 sample delivered attractive lithium storage performance, and may be a promising anode material for the next generation of high-performance LIBs.
Conclusion
In summary, carbon-coated SnO2 riveted on reduced graphene oxide composite (C@SnO2/RGO-90) was prepared by a hydrothermal method after heat treatment. As an anode material for LIBs, the prepared composite exhibited a stable reversible capacity of 843 mA h g−1 at 100 mA g−1 after 100 cycles, and a specific capacity of 485 mA h g−1 at 1000 mA g−1 after 200 cycles. The fascinating lithium storage capability of the specimen can be ascribed to the synergistic effect of the unique double-carbon structure, which can more effectively improve the electrical conductivity of the SnO2 nanoparticles than can RGO sheets alone. Additionally, the electrode exhibited better cyclic stability due to the extra assistance from the amorphous carbon layer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 51676212, 51876226), and Guangdong Natural Science Foundation (Grant No. 2018A030313482) for financial support.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- A. Funabiki, M. Inaba, T. Abe and Z. Ogumi, J. Electrochem. Soc., 1999, 146, 2443–2448 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- Y. M. Sun, N. A. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- J. Liu, Y. L. Cai, C. Q. Xiao, H. Zhang, F. L. Lv, C. Z. Luo, Z. H. Hu, Y. T. Cao, B. Cao and L. Yu, Ind. Eng. Chem. Res., 2019, 58, 20491–20494 CrossRef CAS.
- L. Yuan, Z. P. Guo, K. Konstantinov, H. K. Liu and S. X. Dou, J. Power Sources, 2006, 159, 345–348 CrossRef CAS.
- H. E. Wang, L. J. Xi, R. G. Ma, Z. G. Lu, C. Y. Chung, I. Bello and J. A. Zapien, J. Solid State Chem., 2012, 190, 104–110 CrossRef CAS.
- M. X. Huang, Y. H. Sun, D. C. Guan, J. M. Nan and Y. P. Cai, Ionics, 2019, 25, 5745–5757 CrossRef CAS.
- J. Read, D. Foster, J. Wolfenstine and W. Behl, J. Power Sources, 2001, 96, 277–281 CrossRef CAS.
- Z. H. Wen, Q. Wang, Q. Zhang and J. H. Li, Adv. Funct. Mater., 2007, 17, 2772–2778 CrossRef CAS.
- Q. Y. Han, J. T. Zai, Y. L. Xiao, B. Li, M. Xu and X. F. Qian, RSC Adv., 2013, 3, 20573–20578 RSC.
- Y. F. Kong, Z. Ma, Y. J. Ye, G. P. He, Y. H. Sun, X. X. Zuo, X. Xiao and J. M. Nan, ACS Appl. Energy Mater., 2018, 1, 7065–7075 CrossRef CAS.
- Y. L. Wang, J. J. Xu, H. Wu, M. Xu, Z. Peng and G. F. Zheng, J. Mater. Chem., 2012, 22, 21923–21927 RSC.
- J. W. Deng, C. L. Yan, L. C. Yang, S. Baunack, S. Oswald, H. Wendrock, Y. F. Mei and O. G. Schmidt, ACS Nano, 2013, 7, 6948–6954 CrossRef CAS.
- J. H. Jeun, K. Y. Park, D. H. Kim, W. S. Kim, H. C. Kim, B. S. Lee, H. Kim, W. R. Yu, K. Kang and S. H. Hong, Nanoscale, 2013, 5, 8480–8483 RSC.
- L. Zhang, H. B. Wu, B. Liu and X. W. Lou, Energy Environ. Sci., 2014, 7, 1013–1017 RSC.
- H. D. Liu, J. M. Huang, X. L. Li, J. Liu, Y. X. Zhang and K. Du, Appl. Surf. Sci., 2012, 258, 4917–4921 CrossRef CAS.
- K. M. Liu, S. M. Zhu, X. F. Dong, H. Huang and M. Qi, Adv. Mater. Interfaces, 2020, 7 DOI:10.1002/admi.201901916.
- F. L. Zhang, X. L. Teng, W. K. Shi, Y. F. Song, J. Zhang, X. Wang, H. S. Li, Q. Li, S. D. Li and H. Hu, Appl. Surf. Sci., 2020, 527 Search PubMed.
- Y. S. Wang, X. Q. Zhang and G. W. Wen, Appl. Surf. Sci., 2020, 531 Search PubMed.
- B. J. Ma, B. Lu, J. Luo, X. L. Deng, Z. Y. Wu and X. Y. Wang, Electrochim. Acta, 2018, 288, 61–70 CrossRef CAS.
- F. Li, G. E. Luo, J. F. Yu, W. J. Huang, D. H. Xu, W. Y. Chen, X. Y. Huang, S. Y. Yang, Y. P. Fang and X. Y. Yu, J. Alloys Compd., 2019, 773, 778–787 CrossRef CAS.
- T. Li, T. Xin, Y. H. Ding, J. J. Zou, H. Q. Liu, B. Liu and Y. Q. Wang, J. Solid State Electrochem., 2019, 23, 379–387 CrossRef CAS.
- W. Li, F. Yang, B. Huang, Z. Liu, H. C. Meng, J. X. Zheng, B. Y. Zeng, J. W. Yang, Y. W. Li, S. H. Xiao, Q. Q. Chen and X. Zhao, Ionics, 2020, 26, 6125–6132 CrossRef CAS.
- Q. H. Tian, Y. B. Chen, F. Zhang, W. Zhang, Z. Y. Sui and L. Yang, Appl. Surf. Sci., 2020, 511 Search PubMed.
- Q. Liu, L. L. Wang, K. N. Zhao, W. Yan, M. M. Liu, D. H. Wei, L. L. Xi and J. J. Zhang, Electrochim. Acta, 2020, 354 CAS.
- S. Y. Kong, J. J. Xu, G. X. Lin, S. N. Zhang, W. J. Dong, J. C. Wang and F. Q. Huang, Chem. Commun., 2020, 56, 10289–10292 RSC.
- Z. W. Zhou, L. Pan, Y. T. Liu, X. D. Zhu and X. M. Xie, Chem. Commun., 2018, 54, 4790–4793 RSC.
- L. Yue, C. Xue, B. B. Huang, N. Xu, R. F. Guan, Q. F. Zhang and W. H. Zhang, Electrochim. Acta, 2016, 220, 222–230 CrossRef CAS.
- H. C. Tao, S. C. Zhu, L. Y. Xiong, X. L. Yang and L. L. Zhang, Chemelectrochem, 2016, 3, 1063–1071 CrossRef CAS.
- W. Q. Yao, S. B. Wu, L. Zhan and Y. L. Wang, Chem. Eng. J., 2019, 361, 329–341 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- T. J. Booth, P. Blake, R. R. Nair, D. Jiang, E. W. Hill, U. Bangert, A. Bleloch, M. Gass, K. S. Novoselov, M. I. Katsnelson and A. K. Geim, Nano Lett., 2008, 8, 2442–2446 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- S. Y. Zuo, D. R. Li, Z. G. Wu, Y. Q. Sun, Q. H. Lu, F. Y. Wang, R. F. Zhuo, D. Yan, J. Wang and P. X. Yan, Electrochim. Acta, 2018, 264, 61–68 CrossRef CAS.
- W. P. Xin, T. T. Gao, W. Q. Zhang, T. T. Hu, X. F. Sun and G. W. Zhou, J. Alloys Compd., 2019, 784, 157–164 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS.
- L. Ma, X. Y. Pei, D. C. Mo, S. S. Lyu and Y. X. Fu, Ceram. Int., 2018, 44, 22664–22670 CrossRef CAS.
- Y. X. Wan, X. Q. Xu, J. W. Liu, Y. Sha, Y. J. Chen, L. L. Li, G. Xue, X. L. Wang and D. S. Zhou, Controlling the Sn-C bonds content in SnO2@CNTs composite to form in situ pulverized structure for enhanced electrochemical kinetics, Adv. Mater. Technol., 2017, 2, 1600156 CrossRef.
- Y. Y. Cheng, J. F. Huang, H. Qi, L. Y. Cao, X. M. Luo, J. Y. Li, Z. W. Xu and J. Yang, Nanoscale, 2017, 9, 18681–18689 RSC.
- C. F. Zhang, X. Peng, Z. P. Guo, C. B. Cai, Z. X. Chen, D. Wexler, S. Li and H. K. Liu, Carbon, 2012, 50, 1897–1903 CrossRef CAS.
- Q. K. Tan, Z. Kong, X. J. Chen, L. Zhang, X. Q. Hu, M. X. Mu, H. C. Sun, X. C. Shao, X. G. Guan, M. Gao and B. H. Xu, Appl. Surf. Sci., 2019, 485, 314–322 CrossRef CAS.
- W. H. Chen, K. M. Song, L. W. Mi, X. M. Feng, J. M. Zhang, S. Z. Cui and C. T. Liu, J. Mater. Chem. A, 2017, 5, 10027–10038 RSC.
- Z. Y. Tian, J. W. Zhao, B. Li, Y. Y. Feng, J. X. Song, C. M. Niu, L. Shao and W. X. Zhang, Ionics, 2020, 26, 2773–2779 CrossRef CAS.
- P. Wu, N. Du, H. Zhang, J. X. Yu, Y. Qi and D. R. Yang, Nanoscale, 2011, 3, 746–750 RSC.
- S. Chen, Y. L. Xin, Y. Y. Zhou, F. Zhang, Y. R. Ma, H. H. Zhou and L. M. Qi, J. Mater. Chem. A, 2014, 2, 15582–15589 RSC.
- X. H. Liu, T. T. Ma, L. Sun, Y. S. Xu, J. Zhang and N. Pinna, Chemsuschem, 2018, 11, 1321–1327 CrossRef CAS.
- X. Q. Zhang, X. X. Huang, X. D. Zhang, L. Xia, B. Zhong, T. Zhang and G. W. Wen, Mater. Des., 2017, 114, 234–242 CrossRef CAS.
- C. P. Wu, K. X. Xie, J. P. He, Q. P. Wang, J. M. Ma, S. Yang and Q. H. Wang, Rare Metals, 2021, 40, 48–56 CrossRef CAS.
- Q. N. Tran, I. Kim, J. Hur, J. H. Kim, H. W. Choi and S. J. Park, Korean J. Chem. Eng., 2020, 37, 898–904 CrossRef.
- S. L. Zhu, J. X. Liu, S. Wang, Y. N. Zhang, Y. J. Zhang, P. Dong, Z. R. Zhou and S. B. Xia, Ionics, 2021, 27, 533–540 CrossRef CAS.
- Y. H. Zhou, X. G. Ming, L. Ren, H. M. Liu and X. Z. Yi, Ionics, 2020, 26, 5319–5324 CrossRef CAS.
- L. L. Xu, S. W. Bian and K. L. Song, J. Mater. Sci., 2014, 49, 6217–6224 CrossRef CAS.
- W. Zhou, F. F. Zhang, S. M. Liu, J. W. Wang, X. C. Du, D. M. Yin and L. M. Wang, RSC Adv., 2014, 4, 51362–51365 RSC.
- X. Xu, C. S. Rout, J. Yang, R. Cao, P. Oh, H. S. Shin and J. Cho, J. Mater. Chem. A, 2013, 1, 14548–14554 RSC.
- H. B. Liu, C. Miao, Y. Meng, Q. Xu, X. H. Zhang and Z. Y. Tang, Electrochim. Acta, 2014, 135, 311–318 CrossRef CAS.
- J. X. Qiu, S. Li, E. Gray, H. W. Liu, Q. F. Gu, C. H. Sun, C. Lai, H. J. Zhao and S. Q. Zhang, J. Phys. Chem. C, 2014, 118, 8824–8830 CrossRef CAS.
- S. Q. Liang, K. F. Yu, Y. Li and C. Liang, Mater Res Express, 2020, 7 DOI:10.1088/2053-1591/ab61a0.
- Y. J. Wang, H. Wu, Z. F. Liu, H. Zhao, H. Liu and Y. Zhang, Inorg. Chem., 2018, 57, 13693–13701 CrossRef CAS.
- D. L. Chao, C. R. Zhu, P. H. Yang, X. H. Xia, J. L. Liu, J. Wang, X. F. Fan, S. V. Savilov, J. Y. Lin, H. J. Fan and Z. X. Shen, Nat. Commun., 2016, 7, 12122 CrossRef CAS.
- Y.-X. Fu, Y. Dai, X.-Y. Pei, S.-S. Lyu, Y. Heng and D.-C. Mo, Appl. Surf. Sci., 2019, 497, 143553 CrossRef CAS.
- S. M. Jung, D. W. Kim and Y. Y. Jung, J. Mater. Chem. A, 2020, 8, 8244–8254 RSC.
- D. Zhou, W. L. Song, X. G. Li and L. Z. Fan, ACS Appl. Mater. Interfaces, 2016, 8, 13410–13417 CrossRef CAS.
- J. Liu, W. Li and A. Manthiram, Chem. Commun., 2010, 46, 1437–1439 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10912f |
| This journal is © The Royal Society of Chemistry 2021 |