
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled fibrinogen–fibronectin hybrid protein nanofibers with medium-sensitive stability†
Karl Scheuer‡
a,
Christian Helbing‡a,
Izabela Firkowska-Bodena and
Klaus D. Jandt *ab
*ab
aChair of Materials Science, Otto Schott Institute of Materials Research, Friedrich Schiller University Jena, Germany. E-mail: k.jandt@uni-jena.de
bJena Center for Soft Matter, Friedrich Schiller University Jena, Germany
First published on 16th April 2021
Abstract
Hybrid protein nanofibers (hPNFs) have been identified as promising nano building blocks for numerous applications in nanomedicine and tissue engineering. We have recently reported a nature-inspired, self-assembly route to create hPNFs from human plasma proteins, i.e., albumin and hemoglobin. However, it is still unclear whether the same route can be applied to other plasma proteins and whether it is possible to control the composition of the resulting fibers. In this context, to further understand the hPNFs self-assembly mechanism and to optimize their properties, we report herein on ethanol-induced self-assembly of two different plasma proteins, i.e., fibrinogen (FG) and fibronectin (FN). We show that by varying initial protein ratios, the composition and thus the properties of the resulting hPNFs can be fine-tuned. Specifically, atomic force microscopy, hydrodynamic diameter, and zeta potential data together revealed a strong correlation of the hPNFs dimensions and surface charge to their initial protein mixing ratio. The composition-independent prompt dissolution of hPNFs in ultrapure water, in contrast to their stability in PBS, indicates that the molecular arrangement of FN and FG in hPNFs is mainly based on electrostatic interactions. Supported by experimental data we introduce a feasible mechanism that explains the interactions between FN and FG and their self-assembly to hPNFs. These findings contribute to the understanding of dual protein interactions, which can be beneficial in designing innovative biomaterials with multifaceted biological and physical characteristics.
Introduction
Protein nanofibers (PNFs) are a subject of current research in the field of interdisciplinary nanoscience due to their unique properties, such as good biocompatibility, large surface area, as well as their ability to mimic individual structures of naturally occurring tissue.1–9 On account of the growing area of tissue engineering, several approaches to create PNFs have been introduced over the past years, including electrospinning,2,3,7,10 phase separation,11,12 and extrusion.4,9,12–14 These approaches allow the creation of nanofibers with uniform dimensions, i.e., diameter and length, yet with limited functionalities and consequently a narrow application range. A promising approach to modulate PNF properties is to combine different protein species and thus different functionalities into single nanofibers, so-called hybrid protein nanofibers (hPNFs).The incorporation of dual proteins into hybrid fibers has proven successful in creating fibers with synergistic properties, e.g., high biocompatibility and mechanical strength15,16 or high elongation and nerve regeneration capability.13 The majority of reported hybrid fibers, however, have diameters in the microscale, owing to the fabrication methods, such as wet spinning16 and extrusion.4,9 Given the nanoscale dimensions of protein fibers found in natural tissue, it is more desirable to create hybrid protein fibers with nanoscopic dimensions. In addition, stabilizers and cross-linking agents,10,17 as well as external stimuli,18,19 often involved in the fiber formation process, remain critical aspects for the preservation of the protein-specific properties.
Recently, we reported the creation of hPNFs composed of human plasma proteins (albumin and hemoglobin) via a nature-inspired self-assembly approach. In general, the self-assembly process is based on physicochemically or enzymatically induced protein unfolding.1,18,20–24 This allows previously blocked binding sites of the proteins to be exposed and thus to interact with each other.18,21,23 Computational bioinformatics analysis stated that at least 40% domain structure similarity is advantageous to facilitate co-aggregation of proteins.25 Our own research has shown that ethanol-induced unfolding of similar amino acid sequences in albumin and hemoglobin, and consequent hydrophobic interactions between them, was the driving force for the self-assembly mechanism of the hPNFs.26,27 Based on these findings, one may hypothesize that other plasma proteins can potentially self-assemble to hPNFs assuming similarity in the proteins' amino acid sequences and their exposition driven by controlled changes in the environmental conditions.
Considering human plasma proteins, fibronectin (FN) and fibrinogen (FG) in particular arise as potential dual protein system, adequate to form hPNFs. This is based on: (1) similarity in the primary structure; (2) ability to form individual PNFs via ethanol-induced denaturation;8,28 (3) the proteins' interaction in vivo, i.e., FN can bind to fibrin, the enzymatically activated form of FG.21,29–31 Given the role of both proteins, namely FG's key function in the blood clotting cascade, and FN's contribution to a variety of cellular processes (e.g., cell growth, migration, adhesion, and differentiation29,32), it is highly desirable to combine both proteins into hPNFs.7,32,33
In the present study, thus, we selected FN and FG to corroborate the hypothesis that an ethanol-induced self-assembly approach can be applied to create hPNFs from other plasma proteins. We characterized the structure and physical properties of resulting protein nano fibers by mixing FN and FG molecules under denaturing conditions. Using a combination of different techniques, including atomic force microscopy (AFM), dynamic light scattering (DLS) and immunogold labeling, we report a strong correlation of protein fiber composition to initial protein ratio, which suggests hPNF formation. Furthermore, we find that created hPNFs show medium-dependent stability. Based on the experimental results, we propose an interaction mechanism between FN and FG during their self-assembly into hPNFs.
Results and discussion
Nanofiber formation
The self-assembly process of proteins into nanofibers depends on partial protein unfolding and interactions of the unfolded proteins in solution. While the unfolding process itself can be induced by a denaturant like ethanol or temperature, other parameters like the assembly time and the protein concentration have a vital impact on the self-assembly kinetics. Thus, to investigate possible effects of the protein ratio on the fibers properties it is crucial to minimize the impact of these parameters. For this reason, an initial assembly test row was performed to find experimental conditions that reproducibly yield fibers (see ESI Table TS1†). According to these results, self-assembly of FN and FG was conducted by mixing different FN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FG molar ratios (Table 1) in 80 vol% ethanol and incubating the samples at 37 °C for 4 hours. Prior to AFM investigation, protein–ethanol mixtures were deposited on polystyrene substrates (PS) and characterized in the dry state. In the following, fibers that contained either 100% FN or FG are referred to as pure PNFs.
:FG molar ratios (Table 1) in 80 vol% ethanol and incubating the samples at 37 °C for 4 hours. Prior to AFM investigation, protein–ethanol mixtures were deposited on polystyrene substrates (PS) and characterized in the dry state. In the following, fibers that contained either 100% FN or FG are referred to as pure PNFs.
| Sample | FN 100 | FN 66 | FN 50 | FN 33 | FG 100 |
| FN:FG (mol mol−1) |
1:0 |
2:1 |
1:1 |
1:2 |
0:1 |
Fig. 1a shows AFM height images of PNF structures which were found for all FN–FG mixtures (Fig. S1†). The PNFs show ribbon-like structures composed of individual nanofibers aligned side-by-side (Fig. 1b). According to literature, fiber assembly to ribbons is driven by hydrophobic, van-der-Waals and also electrostatic interactions between individual protein fibers.34,35 The presence of ribbons indicates similar interactions in our system, likely due to ethanol-induced exposition of side chains and polar groups. To our knowledge, there are no reports about ribbon structures consisting of FG, FN and of self-assembled structures of FG–FN mixtures. Only single fibers and networks were previously reported for pure FN as well as FG.8,28 As shown in Fig. 1c, the single PNFs incorporated into the ribbons are composed of small protofibrils which measure 3 to 5 nm in height. The AFM images indicate that the protein fiber formation involves protofibril formation, followed by single fibers formation and their preferential alignment to ribbon structure. The self-assembled structures from FG–FN mixtures resemble our previously observed self-assembled hPNF-structures consisting of albumin and hemoglobin.27 Thus, the similarity in PNF structures is a first indication that hybrid fiber formation occurred.
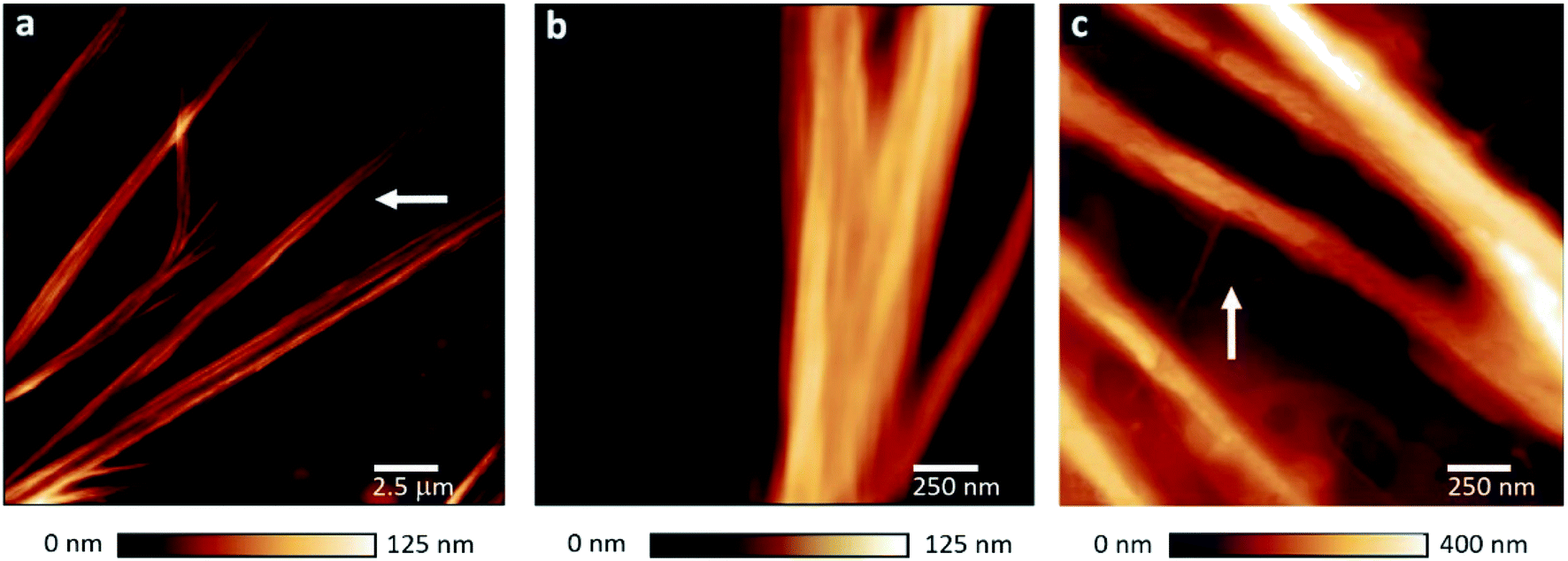 | ||
| Fig. 1 AFM images of FN–FG PNFs. (a) AFM height images of a typical ribbon-like fiber structure found for all FN–FG compositions. As marked with an arrow, the ribbons are composed of individual nanofibers. (b) Side-by-side aligned protein fibers in a ribbon. (c) Protofibrils (marked with an arrow) incorporated into a single nanofiber. | ||
Immunogold labeling
To further confirm the formation of hPNFs we used immunolabeling with secondary antibodies conjugated to gold nanoparticles (AuNPs) of different diameters. The same method was successfully used to investigate the composition of albumin-hemoglobin hPNFs.27 Accordingly, secondary antibodies conjugated with AuNPs with a diameter of 15 ± 2.5 nm and 10 ± 2.5 nm were used to label FN and FG, respectively. Fig. 2 shows a representative scanning transmission electron microscopy (STEM) image of labelled hPNFs as well as the normalized AuNP distribution. The low coverage of hPNFs with AuNPs is related to the general low labeling efficiency, which is about 10–15%.36 Despite of this, immunogold labeling is usually considered suitable for quantification purposes.37 The distribution of AuNPs shows that the ratio of both particles correlates well with the expected fiber compositions based on the initial protein ratio. This, on the other hand, strongly indicates that both proteins were integrated in the hPNFs according to their initial mixing ratios in solution. A small amount (less than 25%) of unspecific labeling occurred for pure PNFs which may be considered as error range for this method. This error is induced by the modified washing procedure during immunogold labeling. Ultrapure water, normally used to remove unattached antibodies from the nanofibers, was replaced by PBS due to the observed fiber instability in ultrapure water which will be discussed later. A cross reaction between the antibodies as well as an unexpected interaction of primary and secondary antibodies with denaturated regions of the proteins is ruled out as this would lead to a similar Au-NP distribution independent from the protein's ratio.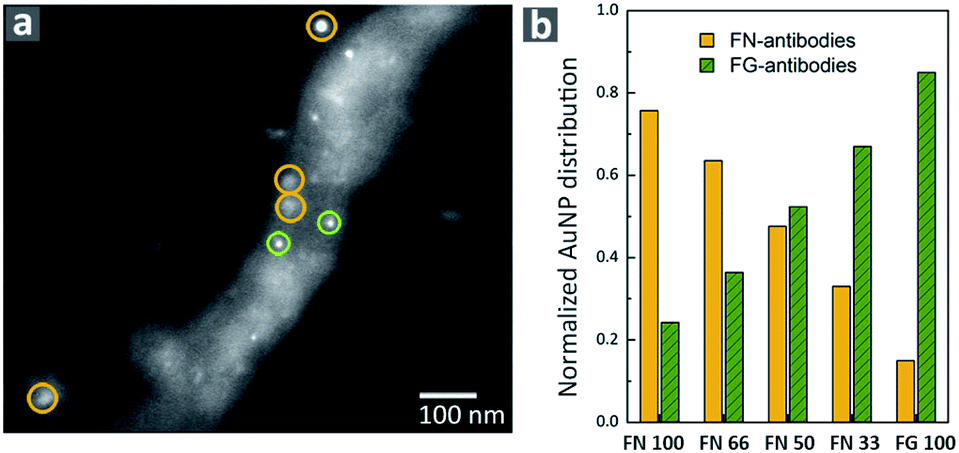 | ||
| Fig. 2 Immunogold labeling of hPNF. (a) Representative STEM image of immunogold-labeled hPNFs (FN 66) with AuNPs indicating FN (yellow) and AuNPs indicating FG (green). (b) Normalized AuNPs distribution as a function of the protein ratio. | ||
FN–FG fiber characteristics
To assess the influence of the proteins ratio on hPNF dimensions, the height as well as the hydrodynamic radii (RH) of the pure PNFs and hPNFs were determined via AFM and DLS. Fig. 3a shows the average fiber heights of the pure PNFs and hPNFs as a function of the FN to FG ratio. The pure PNFs show the lowest heights with 33 nm ± 17 nm (FN 100) and 38 nm ± 18 nm (FG 100). In comparison to this, the heights of the hPNFs increased with increasing content of the second protein species. The maximum average fiber height of 84 nm ± 35 nm was obtained for hPNFs with FN and FG ratio of 1:1.
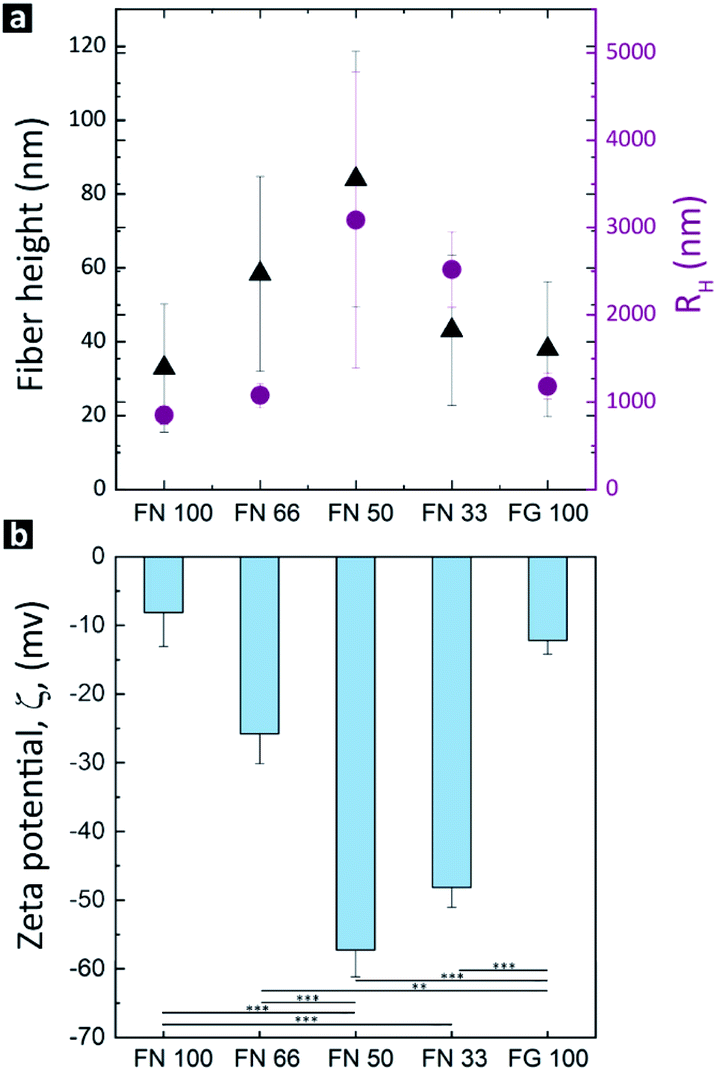 | ||
| Fig. 3 Structural and physical characteristics of hPNFs. (a) The average fiber heights and RH after 4 h of incubation in 80 vol% ethanol at 37 °C. (b) Composition dependent ζPot of the protein fibers measured at pH = 7.4. The lines beneath the columns mark statistically significant differences among the groups (**p = 0.002; ***p < 0.001). | ||
The data for the lateral dimensions of the dispersed pure PNFs and hPNFs was extracted from RH. Due to the fibers length of several μm, it was not possible to distinguish in the AFM images where individual fibers ended and started. The RH describes the radius of a theoretical sphere which has the same diffusion coefficient as the measured particle.38 Since fibers may entangle in dispersion, similar to polymer chains in solution, the RH corresponds to the size of the entangled fibers. Fig. 3a shows the resulting average values of the RH as a function of the fiber composition. Interestingly, the RH distribution correlates with the observed fiber heights. The highest RH was found for hPNFs with FN to FG ratio of 1:1 whereas the minimum was for pure PNFs. The observed differences in the RH can be explained by the fiber thickness and length. Thicker fibers are less flexible than thinner ones and therefore less prone to entangle. Also, longer fibers are able to form a larger coil leading to a higher RH. Based on the height measurements, it is safe to assume that the increase in RH results from a combination of both, increase in fiber thickness and length.
The observed correlation between the fiber dimensions (height and RH) and protein's ratio can be explained by favorable interactions between ethanol-induced partially unfolded proteins. Among others, Dubey et al. showed that the presence and amount of a partially unfolded protein species can influence the tendency of a second protein species to undergo unfolding.24,39 Moreover, it has been shown that the presence of another partially unfolded protein increases the total amount of possible binding sites and interactions, and thus helps to stabilize the other protein in the partially unfolded state.39–41 This, on the other hand, results in a more pronounced unfolding and fiber formation.24,39–41 Based on the above, it is safe to assume that adding FG to FN or vice versa accelerates the formation of protofibrils resulting in a rapid growth of hybrid fibers. Therefore, one would expect an increased height after the same assembly time compared to pure PNF, which is what was indeed observed in the current study. Since a higher amount of a partially unfolded protein species increases the assembly speed, it is logical to observe the highest values of the hPNF parameters, i.e., fiber height and RH, for hPNFs with a protein ratio of 1:1 (highest amount of the second protein species).
The self-assembly of FN–FG to the hPNFs according to their initial mixing ratio is further supported by the results of the ζPot-measurements. As depicted in Fig. 3b, the lowest ζPot was obtained for pure PNFs in contrast to the significantly increased ζPot of the hPNFs. The ζPot dependency on fiber composition is consistent with the fiber's height and RH. It is known, however, that ζPot does not depend on the particle size.42,43 This is confirmed by ζPot values of pure PNFs which correspond to the reported ζPot values of FN44,45 and FG46 molecules. An important factor influencing the ζPot is the surface charge of a material. Changes in surface charge of a protein and thus ζPot may imply protein unfolding and/or aggregation.18,46,47 Therefore, the variation in the ζPot as a function of fiber composition can be corroborated to changes in hPNFs surface charge due to various degrees of protein unfolding and thus the amount of amino acid sequences exposed by the proteins.
Fiber stability
A key factor for future biomedical and biotechnological applications of hPNFs is their behavior in different environments. As mentioned in the immunogold labeling section, we observed an unexpected and distinct instability of PNFs in ultrapure water. To investigate this issue closer, fibers were drop-cast on PS-substrates and exposed to ultrapure water for 5 min. As evident from AFM measurements (Fig. 4b), the pure PNFs and hPNFs disassembled within 5 min, leaving small residues on the substrate. The latter had a strongly decreased height, up to one order of magnitude lower than initial fibers. To ensure that the fibers were not washed away, the experiment was repeated by placing a drop of ultrapure water on a substrate. After evaporation the same result was observed. The fact that the fibers dissolved almost instantaneously in ultrapure water suggests that the observed fiber formation is reversible. A reversible fiber formation points towards non-covalent interactions between the proteins.48,49 Since fibrillogenesis of proteins is often associated with hydrophobic interactions between β-sheet structures of unfolded proteins, one might expect that these interactions prevail and stabilize the fibers in aqueous medium.18,39 For this reason we assume that the molecular arrangement of FN and FG within the hPNFs is partially based on electrostatic interactions.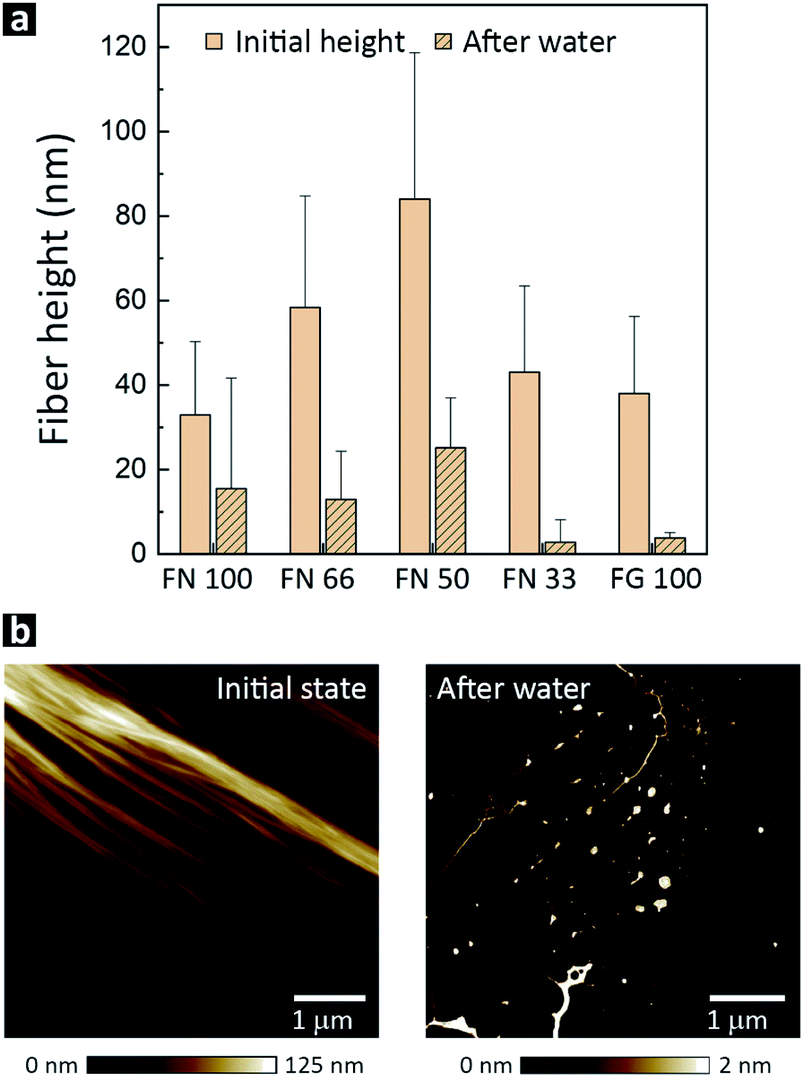 | ||
| Fig. 4 (a) Fiber height before adding ultrapure water and after storage in water for 100 min. (b) Representative AFM height image of hPNFs (FN 33) before and after 5 min exposure to ultrapure water. | ||
The role of electrostatic interactions in hPNFs stability can be explained by the isoelectric point of used proteins,50 which ranges between 5.8 (ref. 51) and 5.6–6.1 (ref. 52) for FG and FN, respectively. Ultrapure water has a pH of approximately 7. However, it's pH decreases to 6 and below after contact with air and adsorption of carbon dioxide.53 Therefore, the pH of ultrapure water is similar to the isoelectric points of both proteins. Consequently, the electrostatic interactions between the proteins are weakened by the change in the protonation of amino acids, resulting in the dissolution of the pure PNFs and hPNFs. This hypothesis is strongly supported by recent findings of Buttafoco et al. The authors reported the dissolution of electrospun protein fibers consisting of collagen and elastin without a cross linking agent in water.10 They argued that the inhibited natural fibrillogenesis of collagen during electrospinning is the reason for the weak protein–protein interactions.10
To test the effect of the medium's pH on the stability of hPNFs, the protein fibers were exposed to PBS at a pH of 7.4. To this end, PS-substrates coated with pure PNFs or hPNFs were immersed into PBS for 24 h and characterized by AFM. As shown in Fig. S2,† all fibers were stable in PBS. The slight variation in fibers heights, observed after PBS exposure, can be explained by the systematic error of the measurement. The stability of hPNFs in PBS at pH of 7.4 is analogues to findings of Stapelfeldt et al.,54 who observed FG fiber formation in PBS at a pH range between 7 and 9. Furthermore, they showed the lack of fiber formation at lower pH (5 and 6), which allow us to expect that hPNFs will disassemble at similar pH values. Overall, the stability test in PBS further supports our assumption that electrostatic interactions are responsible for the molecular assembly and that the dissolution can be triggered by shifts of the medium's pH.
Another factor that needs to be considered is the low ionic strength of ultrapure water compared to the used PBS solution. It is known that an increased ionic strength can lead to enhanced fibrillation of proteins.54,55 In case of reversible fiber formation, one would expect that decreasing the ionic strength would result in fiber disassembly. This assumption is confirmed by our results, i.e., the observed spontaneous fiber disassembly in ultrapure water. Interestingly, Stapelfeldt et al.,54 showed that in PBS of varying pH the FG fiber formation was independent of ionic strength.
Mechanism of FN–FG interaction and hPNF formation
In light of the above described results and the fact that both proteins self-assemble to pure PNFs by enzymatic activation in vivo as well as by denaturant-induced partial re- and unfolding,8,28,56 we propose that the FN–FG interaction mechanism is mediated by specific and non-specific protein interactions.Considering the specific FN–FG interactions, it is known that the fibrin-binding region Fib-1 of the FN molecule can bind covalently and non-covalently to two different sites in the Aα221-391 region of the FG's αC-domain (Fig. 5a).21,29–31 The Fib-2 region, on the other hand, can bind to Aα221-610 of FG's αC-domain with partial overlap of Fib-1 binding sites.31 Since these binding sites are only available in fibrin and not in the native state of FG,21,29–31 we suggest that the ethanol-induced unfolding of the FG molecule results in the detachment of the αC domains from the central E domain and exposes the binding sites for Fib-1 and Fib-2 (Fig. 5a). The detachment of the αC domains was reported for ethanol-induced FG-fiber formation as well as for fiber and network assembly of FG on hydrophobic surfaces.8,57
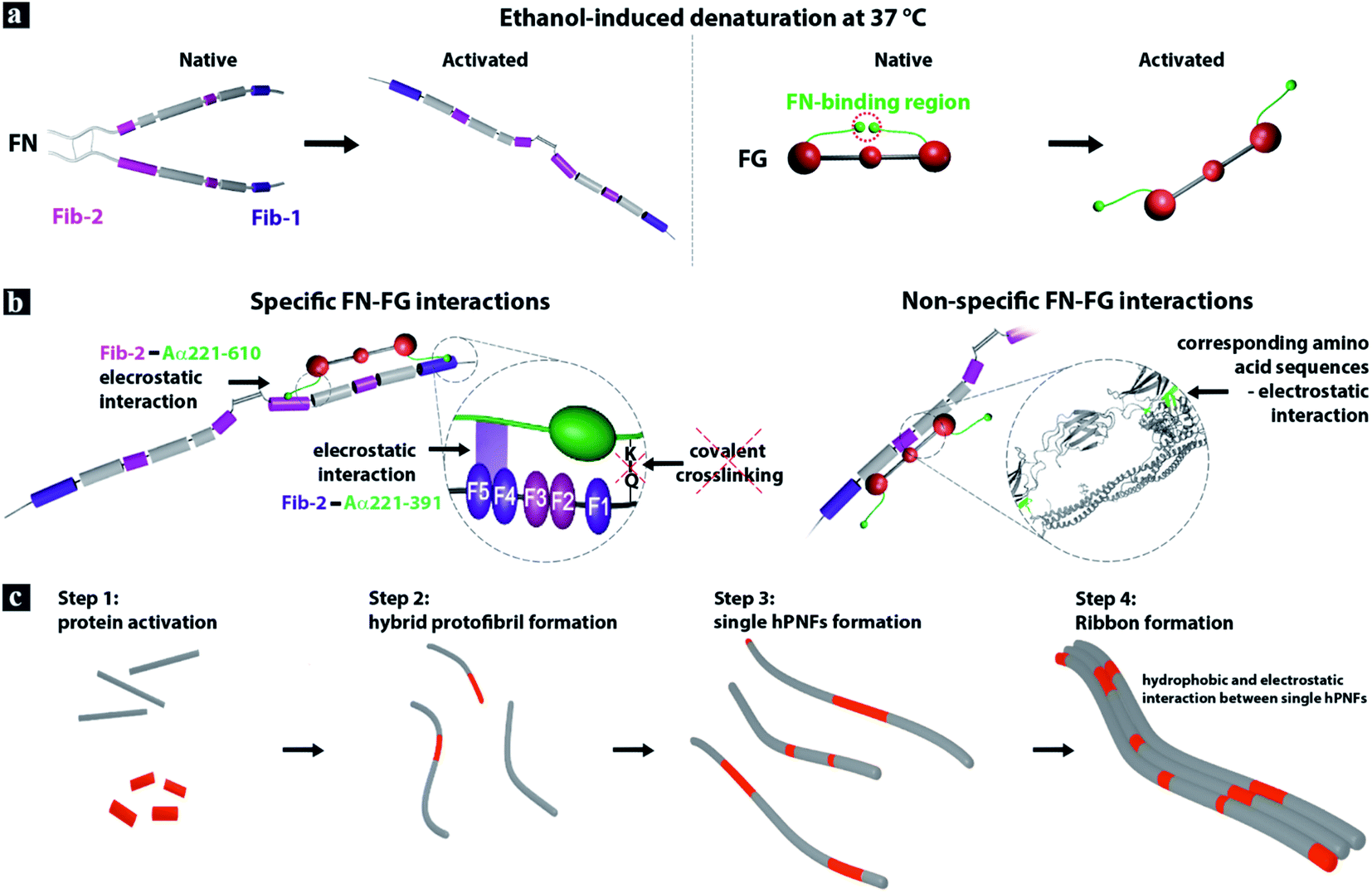 | ||
| Fig. 5 Model of hPNF formation. (a) FN consists of two almost identical polypeptide strands which are connected at their C-termini via disulfide bonds.32,33 The strands have a linear arrangement of different domains that contain several binding sites, e.g., for other FN-molecules and for fibrin (Fib-1 and Fib-2). The soluble plasma protein FG consists of six polypeptide chains, forming a central E-domain, two outer D-domains and two αC-domains attached to E-domain. Ethanol-induced denaturation results in the proteins unfolding and in case of FG in the detachment of the αC-domains from the central E-domain.21,28,31,32 (b) The interaction between the activated proteins is driven by specific and non-specific electrostatic interactions. (c) FN–FG self-assembly to hPNF consist of 4 steps. | ||
In vivo the enzymatic cleavage of the fibrinopeptides A and B in the FG molecule is accomplished by thrombin29,58 and the interaction with the blood coagulation factor XIIIa (plasma transglutaminase) is necessary to form the covalent bond between FN and fibrin.30,31 Due to the absence of both enzymes during FN–FG hPNF formation and observed fiber dissolution in ultrapure water, we suggest non-covalent electrostatic interactions between the proteins (Fig. 5b, left).
The non-specific electrostatic interactions between FN and FG can likely be facilitated by the presence of distinguished regions in the proteins primary structure with similar amino acid sequences (Fig. 5b, right). A comparison of FGs' and FNs' primary structure with the alignment tool from UniProt59 revealed that 34% of the primary structure has corresponding amino acids with similar properties (Fig. S3–S5†). The chemical formulas of the amino acids residual groups are given in Fig. S6.† While most of the similar amino acid sequences are of hydrophobic character, both proteins consist of considerable amounts of oppositely charged amino acids that can act as additional interaction sides. Note, FN contains roughly 20% while FG contains approximately 25% charged amino acids. This supports the assumption that not only hydrophobic interactions are involved in the self-assembly process. Although the verification of the exact amino acid sequences exposed by the proteins upon ethanol-triggered unfolding is not possible, we assume that the majority of the similar sequences are available on the activated proteins. A previous study of protein coaggregation indicated that a sequence similarity below 19% is sufficient to promote aggregation of mixed protein systems.39 Interestingly, the same study reported that the aggregation behavior between mixed protein samples shows faster kinetics than that of single protein systems. Built on this finding and FN–FG hPNFs characteristics, i.e., correlation between the fiber dimensions (height and RH) and protein's ratio, we propose the following hPNF formation steps (Fig. 5c). Step 1: ethanol-induced activation of proteins. Step 2: protofibril formation mediated by specific and non-specific electrostatic interactions between proteins. Protofibrils are likely composed of both proteins rather than individual ones, based on the faster assembly kinetics for dual proteins.39 Step 3: rapid growth of protofibrils to single hPNFs. Step 4: preferential side-by-side single hPNF alignment to ribbons. The latter is driven by hydrophobic and electrostatic interactions between individual protein fibers.34
Conclusions
This study introduced hPNFs from FN and FG that were created by self-assembly, supported by ethanol-induced protein activation. The heterogenic nature of protein fibers was deduced from a strong correlation of fibers dimensions (height and RH) and ζPot to the initial mixing ratio of proteins. The observed unexpected rapid degradation of hPNFs in ultrapure water, in contrast to their high stability in PBS, suggests the key role of electrostatic interactions in the hPNF formation. Based on the existence of binding sites for fibrin in FN and similarity in the proteins primary structure, an FN–FG interaction mechanism combining specific and non-specific electrostatic interactions was proposed.Furthermore, the results discussed herein not only support our previously proposed mechanism for self-assembly of dual plasma proteins but also highlight the ability to fine-tune the properties of the hPNFs, depending on the ratio of the two proteins. The latter is of great interest for future applications of hPNFs as building blocks in the rational design of smart biomaterials for tissue engineering and regenerative medicine.
Experimental
Fiber formation
Protein stock solutions of FN (Chemicon, Limburg an der Lahn, Germany) and FG (Calbiochem, Merck, Darmstadt, Germany) were prepared by dissolving the proteins in ultrapure water at 37 °C. The initial concentration was determined by UV-Vis spectroscopy (LAMBDA 35 UV/Vis, PerkinElmer, Waltham, USA) and further adjusted to concentrations of 20 ng μl−1 for each solution. In the next step, the stock solution of FN and FG were mixed to obtain protein solutions with the molar protein ratios shown in Table 1. After that, ethanol was added until an ethanol concentration of 80 vol% was reached. The ethanol/protein mixtures were incubated in a water bath at 37 °C for 4 hours. We chose these parameters after we tried to find self-assembly conditions to obtain reproducible fibers of FN and FG, based on our previous experience.27 Finally, 20 μl of the fiber dispersion was drop-cast onto cleaned PS. The coated substrates were dried in a vacuum. After drying, the protein nanofibers' morphology was investigated by AFM.Characterization of hPNFs
The morphology of self-assembled hPNFs was investigated by AFM in tapping mode. AFM measurements in air were performed by using NanoWizard 4 (JPK BioAFM, Bruker, Berlin, Germany), a Dimension 3100 and a MultiMode (both from Bruker, Santa Barbara, CA) equipped with a Nanoscope IV controller. Measurements were performed at room temperature by using standard tapping mode silicon cantilevers from Bruker (model RTESP, Bruker, Santa Barbara, CA) with a resonance frequency in the range of 315–364 kHz in air, a spring constant in the range of 20–80 N m−1, and a typical tip radius of less than 10 nm (typical 7 nm).Immunogold labeling
Tris-buffered solution (TBS) was prepared as block- and labeling buffer, by dissolving Tris (1.5 g) and NaCl (2.19 g) in ultrapure water. The solution was adjusted to a pH-value of 7.4 by adding 1 M HCl. Finally, the buffer solution was filled up to 250 ml with ultrapure water. Furthermore, 0.1 wt% gelatine of cold water fish skin was added (Sigma-Aldrich, Schnelldorf, Germany). Indirect labeling was performed with primary antibodies from rabbit against FN (sc-9068 Santa Cruz, Dallas, TX, USA) and mouse against FG (sc-69775 Santa Cruz, Dallas, TX, USA). To label FN and FG, secondary anti-rabbit antibodies conjugated with AuNPs with a diameter of 15 ± 2.5 nm (EM.GAR15 from BBI Solutions, Cardiff, United Kingdom) and secondary anti-mouse antibodies conjugated with AuNPs with a diameter of 10 ± 2.5 nm (EM.GMHL10 from BBI Solutions, Cardiff, United Kingdom) were used.To conduct the indirect immunolabeling, the PNFs were deposited by drop-casting 2 μl of protein dispersion on a TEM-grid which was then fixed with TBS–gelatine for 5 min. Afterwards, the TEM-grids were incubated for 1 h in a TBS–gelatine–primary antibody solution mixture (ratio 50:2) at room temperature. This was followed by washing of unbound primary antibodies in TBS–gelatine three times for 5 min. Analogous the secondary antibodies were added by incubating the TEM-grids for 30 min in a TBS–gelatine:secondary antibody solution (50:2). Finally, the TEM-grids were washed 3 times in TBS–gelatine and twice in phosphate 1× buffered solution (PBS) (Dulbecco) for 5 min each.
The immunolabeled nanofibers were investigated via STEM with an AURIGA 60 CrossBeam workstation (Carl Zeiss AG, Oberkochen, Germany). The analysis of the attached Au-nanoparticles was done in “ImageJ”.
Stability tests
Stability tests were performed in two different media, ultrapure water and PBS. The behavior of the fibers was examined by comparative AFM-measurements of the fiber-covered substrates before and after exposition to ultrapure water or PBS, respectively. The fiber coated samples where immersed for 5 min and 24 h in ultrapure water and PBS, respectively. Subsequently, they were investigated with AFM. Additionally, 2 μl ultrapure water was dropped on the fiber coated substrate, without immersing the complete sample. After evaporation of the ultrapure water at room temperature (t < 5 min), AFM measurements were performed as well. This was done to ensure that the fibers were not washed off from the surface by the water.RH and ζPot
RH and ζPot were measured as a function of the fiber composition. The measurements were carried out in the incubation media. RH was determined via DLS, whereas electrophoretic light scattering (ELS) was applied to measure ζPot. The measurements were performed on a Zetasizer Nano ZS (Malvern Instruments, Herrenberg, Germany). The instrument operated in the 173° backscatter mode at 25 °C and a wavelength of λ = 633 nm.Statistics
All measured values from the experiments are given as mean values ± standard deviation. The specific FG–FN PNF characteristics and the results of the stability tests were examined with One-Way ANOVA for their statistical differences. All statistical tests were performed with the software package Sigmaplot 13.0 (Stystat Software Inc.).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of the Deutsche Forschungsgemeinschaft (DFG) projects: “Novel functional materials based on self-assembled protein nanofibers (PNNF): creating and understanding nanofibers” and “Novel functional materials based on self-assembled protein nanofibers (PNNF): degradation dynamics of PNNFs/hybrid PNNFs and creation of PNNF/hybrid PNNF micro scaffolds (step 2)”. Further, we gratefully acknowledge the partial financial support of the Deutsche Forschungsgemeinschaft (DFG), grant number INST 275/389-1 FUGG.Notes and references
- C. Gong, S. Sun, Y. Zhang, L. Sun, Z. Su, A. Wu and G. Wei, Nanoscale, 2019, 11, 4147–4182 RSC.
- T. D. Stocco, N. J. Bassous, S. Zhao, A. E. C. Granato, T. J. Webster and A. O. Lobo, Nanoscale, 2018, 10, 12228–12255 RSC.
- M. Li, M. J. Mondrinos, M. R. Gandhi, F. K. Ko, A. S. Weiss and P. I. Lelkes, Biomaterials, 2005, 26, 5999–6008 CrossRef CAS PubMed.
- M. Raoufi, T. Das, I. Schoen, V. Vogel, D. Brueggemann and J. P. Spatz, Nano Lett., 2015, 15, 6357–6364 CrossRef CAS PubMed.
- K. Sanford and M. Kumar, Curr. Opin. Biotechnol., 2005, 16, 416–421 CrossRef CAS PubMed.
- T. Scheibel, Curr. Opin. Biotechnol., 2005, 16, 427–433 CrossRef CAS PubMed.
- R. J. Wade and J. A. Burdick, Nano Today, 2014, 9, 722–742 CrossRef CAS.
- G. Wei, J. Reichert and K. D. Jandt, Chem. Commun., 2008, 3903–3905, 10.1039/B806316H.
- M. Raoufi, Integr. Biol., 2016, 8, 1059–1066 CrossRef CAS PubMed.
- L. Buttafoco, N. G. Kolkman, P. Engbers-Buijtenhuijs, A. A. Poot, P. J. Dijkstra, I. Vermes and J. Feijen, Biomaterials, 2006, 27, 724–734 CrossRef CAS PubMed.
- M. Cui, X. Wang, B. An, C. Zhang, X. Gui, K. Li, Y. Li, P. Ge, J. Zhang, C. Liu and C. Zhong, Sci. Adv., 2019, 5, eaax3155 CrossRef CAS PubMed.
- K. G. DeFrates, R. Moore, J. Borgesi, G. Lin, T. Mulderig, V. Beachley and X. Hu, Nanomaterials, 2018, 8, 457 CrossRef PubMed.
- S. Underwood, A. Afoke, R. A. Brown, A. J. MacLeod, P. A. Shamlou and P. Dunnill, Biotechnol. Bioeng., 2001, 73, 295–305 CrossRef CAS PubMed.
- H. Bak, A. Afoke, A. J. McLeod, R. Brown, P. A. Shamlou and P. Dunnill, Chem. Eng. Sci., 2002, 57, 913–920 CrossRef CAS.
- J. Fu, P. A. Guerette, A. Pavesi, N. Horbelt, C. T. Lim, M. J. Harrington and A. Miserez, Nanoscale, 2017, 9, 12908–12915 RSC.
- M. M. Jacobsen, D. Li, N. Gyune Rim, D. Backman, M. L. Smith and J. Y. Wong, Sci. Rep., 2017, 7, 45653 CrossRef CAS PubMed.
- Y. Z. Zhang, J. Venugopal, Z. M. Huang, C. T. Lim and S. Ramakrishna, Polymer, 2006, 47, 2911–2917 CrossRef CAS.
- S. Bouhallab and T. Croguennec, Adv. Polym. Sci., 2014, 256, 67–101 CrossRef CAS.
- E. Freire, A. Schön, B. M. Hutchins and R. K. Brown, Drug Discovery Today, 2013, 18, 1007–1013 CrossRef CAS PubMed.
- R. Jurado, F. Castello, P. Bondia, S. Casado, C. Flors, R. Cuesta, J. M. Domínguez-Vera, A. Orte and N. Gálvez, Nanoscale, 2016, 8, 9648–9656 RSC.
- H. A. Scheraga, Biophys. Chem., 2004, 112, 117–130 CrossRef CAS PubMed.
- A. Aggell, M. Bell, N. Bdoen, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS PubMed.
- J. E. Gillam and C. E. MacPhee, J. Phys.: Condens. Matter, 2013, 25, 373101 CrossRef CAS PubMed.
- M. Sleutel, I. Van den Broeck, N. Van Gerven, C. Feuillie, W. Jonckheere, C. Valotteau, Y. F. Dufrêne and H. Remaut, Nat. Chem. Biol., 2017, 13, 902–908 CrossRef CAS PubMed.
- C. F. Wright, S. A. Teichmann, J. Clarke and C. M. Dobson, Nature, 2005, 438, 878–881 CrossRef CAS PubMed.
- C. Helbing and K. D. Jandt, in Artificial Protein and Peptide Nanofibers, ed. G. Wei and S. G. Kumbar, Woodhead Publishing, 2020, pp. 69–97, DOI:10.1016/B978-0-08-102850-6.00004-8.
- C. Helbing, T. Deckert-Gaudig, I. Firkowska-Boden, G. Wei, V. Deckert and K. D. Jandt, ACS Nano, 2018, 12, 1211–1219 CrossRef CAS PubMed.
- G. Wei, T. F. Keller, J. Zhang and K. D. Jandt, Soft Matter, 2011, 7, 2011–2018 RSC.
- M. W. Mosesson, J. Thromb. Haemostasis, 2005, 3, 1894–1904 CrossRef CAS PubMed.
- E. Makogonenko, G. Tsurupa, K. Ingham and L. Medved, Biochemistry, 2002, 41, 7907–7913 CrossRef CAS PubMed.
- E. Makogonenko, K. C. Ingham and L. Medved, Biochemistry, 2007, 46, 5418–5426 CrossRef CAS PubMed.
- R. Pankov and K. M. Yamada, J. Cell Sci., 2002, 115, 3861–3863 CrossRef CAS PubMed.
- M. J. Bradshaw and M. L. Smith, Acta Biomater., 2014, 10, 1524–1531 CrossRef CAS PubMed.
- J. Juarez, P. Taboada and V. Mosquera, Biophys. J., 2009, 96, 2353–2370 CrossRef CAS PubMed.
- K. M. M. Carneiro, H. Zhai, L. Zhu, J. A. Horst, M. Sitlin, M. Nguyen, M. Wagner, C. Simpliciano, M. Milder, C.-L. Chen, P. Ashby, J. Bonde, W. Li and S. Habelitz, Sci. Rep., 2016, 6, 23105 CrossRef CAS PubMed.
- G. Griffiths and H. Hoppeler, J. Histochem. Cytochem., 1986, 34, 1389–1398 CrossRef CAS PubMed.
- F. D'Amico and E. Skarmoutsou, J. Microsc., 2008, 230, 9–15 CrossRef PubMed.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- K. Dubey, B. G. Anand, M. K. Temgire and K. Kar, Biochemistry, 2014, 53, 8001–8004 CrossRef CAS PubMed.
- I. Horvath and P. Wittung-Stafshede, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12473 CrossRef CAS PubMed.
- S. J. Wood, J. Wypych, S. Steavenson, J. C. Louis, M. Citron and A. L. Biere, J. Biol. Chem., 1999, 274, 19509–19512 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- S. Usui, H. Sasaki and H. Matsukawa, J. Colloid Interface Sci., 1981, 81, 80–84 CrossRef CAS.
- D. E. MacDonald, B. Markovic, A. L. Boskey and P. Somasundaran, Colloids Surf., B, 1998, 11, 131–139 CrossRef CAS.
- I. Verma, I. Pani, D. Sharma, S. Maity and S. K. Pal, J. Phys. Chem. C, 2019, 123, 13642–13650 CrossRef CAS.
- M. Nattich-Rak, Z. Adamczyk, M. Wasilewska and M. Sadowska, J. Colloid Interface Sci., 2015, 449, 62–71 CrossRef CAS PubMed.
- R. Li, Z. Wu, Y. Wangb, L. Ding and Y. Wang, Biotechnology Reports, 2016, 9, 46–52 CrossRef PubMed.
- E. L. Guenther, Q. Cao, H. Trinh, J. Lu, M. R. Sawaya, D. Cascio, D. R. Boyer, J. A. Rodriguez, M. P. Hughes and D. S. Eisenberg, Nat. Struct. Mol. Biol., 2018, 25, 463–471 CrossRef CAS PubMed.
- M. P. Hughes, M. R. Sawaya, D. R. Boyer, L. Goldschmidt, J. A. Rodriguez, D. Cascio, L. Chong, T. Gonen and D. S. Eisenberg, Science, 2018, 359, 698–701 CrossRef CAS PubMed.
- G. Wei, J. Reichert, J. Bossert and K. D. Jandt, Biomacromolecules, 2008, 9, 3258–3267 CrossRef CAS PubMed.
- M. Wasilewska, Z. Adamczyk and B. Jachimska, Langmuir, 2009, 25, 3698–3704 CrossRef CAS PubMed.
- R. O. Hynes, Fibronectins, Springer, New York, NY, 2012 Search PubMed.
- H. T. Byck, Science, 1932, 75, 224 CrossRef CAS PubMed.
- K. Stapelfeldt, S. Stamboroski, P. Mednikova and D. Bruggemann, Biofabrication, 2019, 11, 025010 CrossRef CAS PubMed.
- L. N. Arnaudov and R. de Vries, Biomacromolecules, 2006, 7, 3490–3498 CrossRef CAS PubMed.
- B. Hämisch, A. Büngeler, C. Kielar, A. Keller, O. Strube and K. Huber, Langmuir, 2019, 35, 12113–12122 CrossRef.
- J. Koo, D. Galanakis, Y. Liu, A. Ramek, A. Fields, X. Ba, M. Simon and M. H. Rafailovich, Biomacromolecules, 2012, 13, 1259–1268 CrossRef CAS PubMed.
- J. W. Weisel and R. I. Litvinov, Fibrous Proteins: Structures and Mechanisms, Springer International Publishing, 2017 Search PubMed.
- T. Doğan, A. MacDougall, R. Saidi, D. Poggioli, A. Bateman, C. O'Donovan and M. J. Martin, Bioinformatics, 2016, 32, 2264–2271 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10749b |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |