
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResearch developments in the syntheses, anti-inflammatory activities and structure–activity relationships of pyrimidines†
Haroon ur Rashid ab,
Marco Antonio Utrera Martines*a,
Adriana Pereira Duartea,
Juliana Jorgea,
Shagufta Rasoolb,
Riaz Muhammadb,
Nasir Ahmadc and
Muhammad Naveed Umar*d
ab,
Marco Antonio Utrera Martines*a,
Adriana Pereira Duartea,
Juliana Jorgea,
Shagufta Rasoolb,
Riaz Muhammadb,
Nasir Ahmadc and
Muhammad Naveed Umar*d
aInstitute of Chemistry, Federal University of Mato Grosso do Sul, Campo Grande, MS, Brazil. E-mail: marcomartines@gmail.com
bDepartment of Chemistry, Sarhad University of Science and Information Technology, Peshawar, Khyber Pakhtunkhwa, Pakistan
cDepartment of Chemistry, Islamia College University, Peshawar, Khyber Pakhtunkhwa, Pakistan
dDepartment of Chemistry, University of Malakand, Chakdara, Dir (L), Khyber Pakhtunkhwa, Pakistan. E-mail: m.naveedumar@uom.edu.pk
First published on 3rd February 2021
Abstract
Pyrimidines are aromatic heterocyclic compounds that contain two nitrogen atoms at positions 1 and 3 of the six-membered ring. Numerous natural and synthetic pyrimidines are known to exist. They display a range of pharmacological effects including antioxidants, antibacterial, antiviral, antifungal, antituberculosis, and anti-inflammatory. This review sums up recent developments in the synthesis, anti-inflammatory effects, and structure–activity relationships (SARs) of pyrimidine derivatives. Numerous methods for the synthesis of pyrimidines are described. Anti-inflammatory effects of pyrimidines are attributed to their inhibitory response versus the expression and activities of certain vital inflammatory mediators namely prostaglandin E2, inducible nitric oxide synthase, tumor necrosis factor-α, nuclear factor κB, leukotrienes, and some interleukins. Literature studies reveal that a large number of pyrimidines exhibit potent anti-inflammatory effects. SARs of numerous pyrimidines have been discussed in detail. Several possible research guidelines and suggestions for the development of new pyrimidines as anti-inflammatory agents are also given. Detailed SAR analysis and prospects together provide clues for the synthesis of novel pyrimidine analogs possessing enhanced anti-inflammatory activities with minimum toxicity.
1. Introduction
The word inflammation is derived from the Latin “flamma” meaning flame. It is normal feedback of the body to safeguard tissues against disease or infection. The inflammatory reaction initiates with the generation and discharge of chemical agents from the cells in the diseased, infected, or wounded tissue. Inflamed tissues produce extra signals that use leukocytes (white blood cells) at the position of inflammation. Leukocytes damage any infective or harmful agent and eliminate cellular residues from damaged tissue. This inflammatory reaction generally stimulates the curing process. However, an uncontrolled inflammatory response may become detrimental.1,2 Inflammation represents a portion of the body's immune response. An inflammatory reaction is liable for curing wounds, infections, and any damage to the tissues. Numerous feedbacks produced by the defense mechanism as a response to a physical injury or an infection result in inflammation.3 Acute inflammation grows rapidly and turns out to be severe in a brief time. Its symptoms continue for a few days but may last for several weeks under certain circumstances. Common indications of acute inflammation include swelling, redness, pain, immovability, and heat. Acute inflammation can be caused by certain conditions and diseases including acute bronchitis, abrasion or cut on the skin, achy throat from flu or cold, infected ingrown toenail, acute appendicitis, sinusitis, tonsillitis, dermatitis, infective meningitis, high-intensity exercise, and physical trauma.4,5 Chronic inflammation persists for a prolonged duration (for several months and even years) in which active inflammation, tissue damage, and repair occur concurrently. Usually, the extent and effects of chronic inflammation differ with the source of the injury and the efficiency of the body to heal and control the damage. Chronic inflammation is characterized by typical symptoms such as body pain, fever, rash, weight gain or weight loss, fatigue, joint pain, and mouth sores. Chronic inflammation may lead to the progression of certain diseases namely diabetes, cancer, cardiovascular diseases, rheumatoid arthritis, allergies, chronic obstructive pulmonary disease (COPD), tuberculosis, asthma, hepatitis, periodontitis, and chronic peptic ulcer.1,6 Fig. 1 indicates the health complications associated with acute and chronic inflammations. Nonsteroidal anti-inflammatory drugs (NSAIDs e.g. ibuprofen, aspirin, or naproxen), herbal supplements (e.g. curcumin, capsaicin, and Boswellia serrata) and corticosteroids (e.g. prednisone) can be utilized to lower the pain and fever resulting from numerous inflammatory disorders.7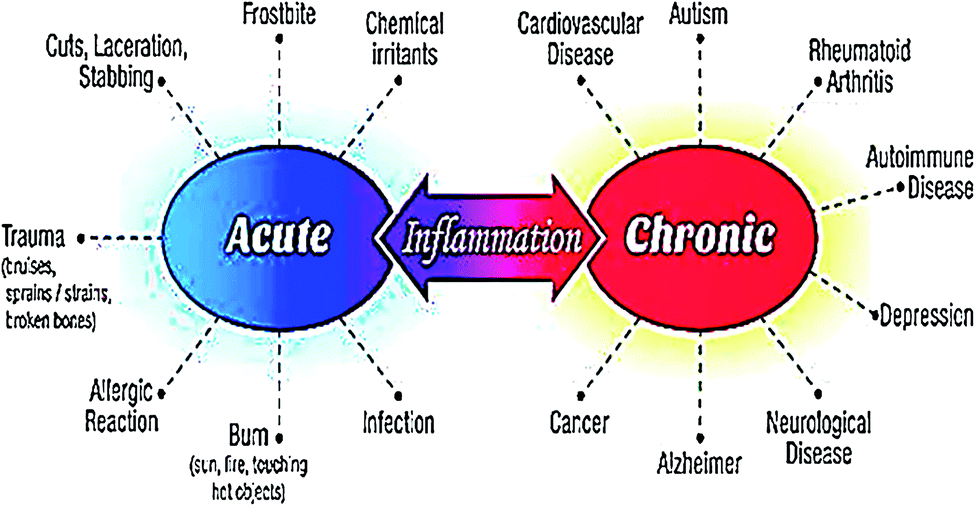 | ||
| Fig. 1 Serious health problems resulting from acute and chronic inflammations. | ||
Presently, NSAIDs are the most widely used to alleviate inflammatory fever and pain. The key mechanism of action of NSAIDs involves the suppression of the cyclooxygenase (COX) enzymes. COX enzymes are needed to convert arachidonic acid into thromboxanes, prostaglandins (PGE2), and prostacyclins.8 The beneficial effects of NSAIDs are credited to the deficiency of these eicosanoids. Particularly, thromboxanes cause platelet adhesion, PGE2 plays a role in vasodilation, enhances the temperature set-point in the hypothalamus, and lead to anti-nociception.9 Prostaglandins (PGE2) are lipid compounds that are generated by COX enzymes in nearly every human tissue and are responsible for inducing inflammation via their role in vasodilation. Two isoforms of COX enzyme namely COX-1 and COX-2 are responsible for the generation of PGE2 from arachidonic acid. NSAIDs inhibit the activity of COXs, and thus reduce the amount of PGE2 throughout the body. Consequently, existing inflammation, fever, and pain are alleviated.10,11 Traditional NSAIDs act non-selectively by inhibiting both COX-1 and COX-2 enzymes. On the other hand, coxibs (celecoxib, rofecoxib, and etoricoxib) represent a class of NSAIDs that selectively target the COX-2 without influencing COX-1. Coxibs possess anti-inflammatory effects similar to traditional nonselective NSAIDs, but there is some proof that they may be weaker analgesics.9,12,13 Important NSAIDs approved by Food and drug administration (FDA) include aspirin, diclofenac, etodolac, fenoprofen, flurbiprofen, indomethacin, ibuprofen, ketoprofen, ketorolac, mefenamic acid, meloxicam, nabumetone, naproxen, oxaprozin, piroxicam, sulindac, and tolmetin14 (Fig. SI-1†). However, the use of both traditional NSAIDs and coxibs has been linked with several adverse effects including cardiovascular and gastrointestinal (GI) disorders. They harm the upper and lower bowel by corroding COX-1 derived prostaglandins and producing local injury to the mucosa.15 Moreover, kidney toxicity has been reported in approximately 1–5% NSAIDs users. Studies show that the use of NSAIDs can cause both acute and chronic renal failures.16 Toxic nature of NSAIDs restricts their use to cure inflammatory disorders. Therefore, the discovery of novel and cost-effective anti-inflammatory agents carrying minimum adverse effects is imperative in the field of pharmacological study and presents a laborious task to scientists.1 Naturally-occurring bioactive compounds have long been used as traditional medicines to cure inflammatory complications such as pain, fever, arthritis, and migraine.17 Heterocyclic compounds occur abundantly in nature and are of immense importance to living things. Their basic subunits are found in numerous natural products for example hormones, pigments, antibiotics, and vitamins.18,19 Consequently, they have attracted significant attention from the researchers for the synthesis of new bioactive compounds.20,21
Among heterocycles, nitrogen-containing compounds represent an important class and have contributed substantially to the research field of medicinal chemistry.22
2. Pyrimidines and their medicinal applications
Pyrimidine is an aromatic heterocyclic compound analogous to pyridine (Fig. 2a). It is one of the three diazines (unsaturated six-membered ring containing two nitrogen atoms) that has two nitrogen atoms at positions-1 and -3 in the ring. Heterocyclic compounds carrying pyrimidine rings are of enormous importance because they represent a vital family of natural and synthetic products, several of which display valuable clinical applications and bioactivities.23,24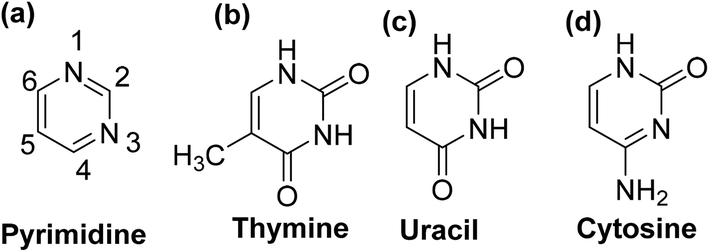 | ||
| Fig. 2 Chemical structures of (a) pyrimidine, (b) thymine, (c) uracil, and (d) cytosine. | ||
Substituted pyrimidines and purines are extensively found in living things and are among the leading compounds investigated by chemists.25 Pyrimidines represent the most abundant members of the diazine class with thymine (Fig. 2b), uracil (Fig. 2c), and cytosine (Fig. 2d) being key components of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). Moreover, pyrimidine moiety occurs in several natural products, for instance, vitamin B1 (thiamine) (Fig. 3a) and various synthetic products, such as barbituric acid (Fig. 3b) and veronal (Fig. 3c), which are used as soporific drugs (sleeping pills).26
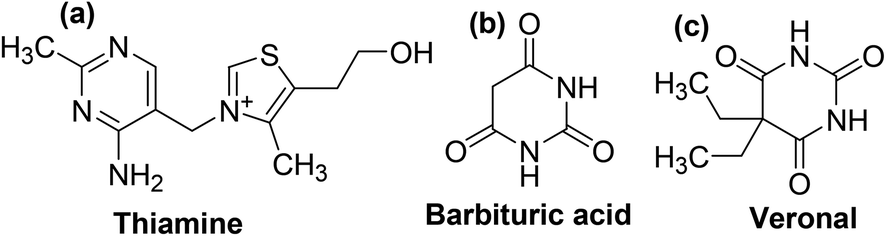 | ||
| Fig. 3 Chemical structures of (a) thiamine (b) barbituric acid, and (c) veronal. | ||
Literature survey indicates that pyrimidine derivatives demonstrate a variety of pharmacological activities (Fig. SI-2†) comprising antifungal,27–30 antibacterial,31–35 analgesic,36 antileishmanial,37 antihypertensive,38,39 antiviral,40–42 antipyretic43 antidiabetic,44 antioxidant,45 anticonvulsant,46 antihistaminic47 and anti-inflammatory.48 However, literature related to the synthesis, anti-inflammatory activities, and SAR studies of the pyrimidine derivatives is selected for this review.
3. Advances in the synthesis of pyrimidines
Pyrimidines have simple chemistry which facilitates several substitutions on their core ring through facile synthesis. Currently, numerous methodologies are used for the synthesis of pyrimidine analogs. A few of them are reported from the recent literature below.3.1. Synthesis via ZnCl2-catalyzed three-component coupling reaction
A three-component coupling reaction comprising substituted enamines, triethyl orthoformate, and ammonium acetate under ZnCl2 catalysis yielded numerous 4,5-disubstituted pyrimidine analogs in a single step (Scheme 1).49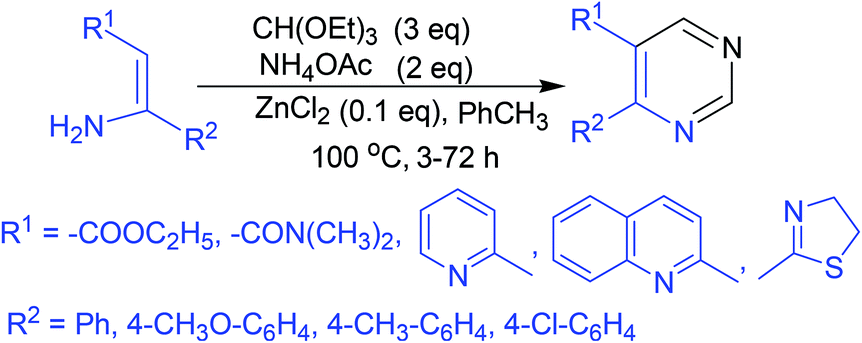 | ||
| Scheme 1 Synthesis of 4,5-disubstituted pyrimidine analogs via ZnCl2-catalyzed three-component coupling reaction. | ||
3.2. Synthesis via K2S2O8-facilitated oxidative annulation reaction
This approach consists of a K2S2O8-facilitated oxidative annulation reaction involving formamide as a route towards pyrimidines. Activation of acetophenone–formamide conjugates resulted in the formation of 4-arylpyrimidines (Scheme 2).50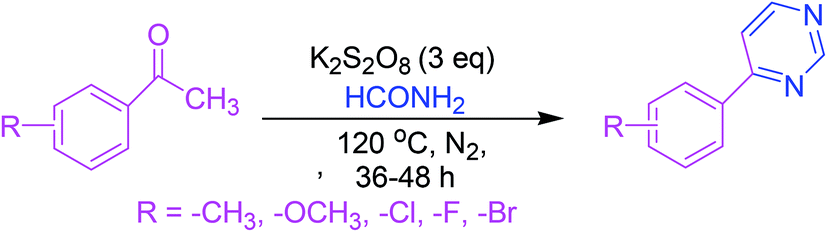 | ||
| Scheme 2 Synthesis of 4-arylpyrimidines via K2S2O8-facilitated oxidative annulation reaction. | ||
3.3. Synthesis via cyclization of ketones with nitriles
Distinctly substituted pyrimidines are synthesized via a simple and cost-efficient procedure that involves the cyclization of ketones with nitriles under Cu-catalysis in the presence of a base (Scheme 3).51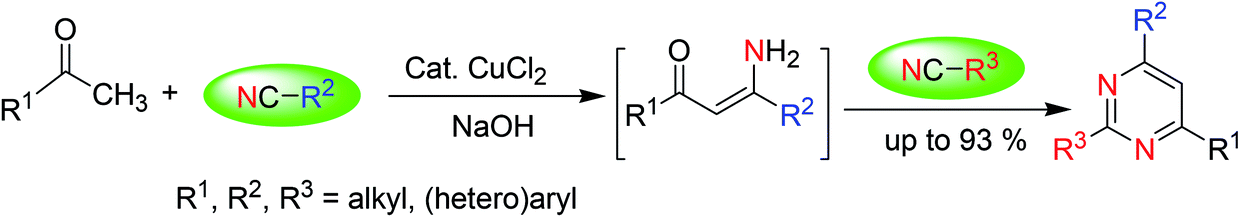 | ||
| Scheme 3 Pyrimidines synthesis via cyclization of ketones with nitriles under base catalysis. | ||
3.4. Synthesis via the reactions of carbonyl compounds with amidines
Synthesis of numerous pyrimidine analogs has been reported by the regioselective reaction of carbonyl compounds (esters, aldehydes, and ketones) with amidines in the presence (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and an in situ prepared recyclable iron(II)-complex. The mechanism indicated that the reactions progressed via a TEMPO complexation/enamine addition/transient α-occupation/β-TEMPO elimination/cyclization order (Scheme 4).52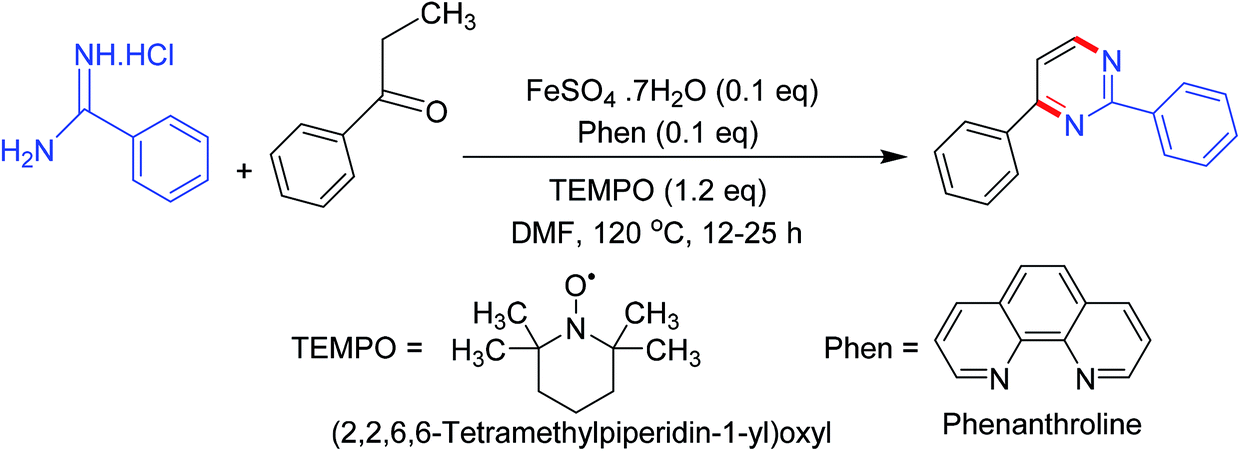 | ||
| Scheme 4 Pyrimidines synthesis via the reactions of carbonyl compounds with amidines. | ||
3.5. Synthesis via base-facilitated intermolecular oxidative C–N bond fabrication
A base-facilitated intermolecular oxidative C–N bond fabrication of allylic C(sp3)–H and vinylic C(sp2)–H of allylic compounds with amidines allows an easy synthesis of polysubstituted pyrimidines under oxygen as a solitary oxidant. This strategy ensures the supply of protective group free nitrogen, high efficiency, worthy functional group leniency, and environmental sustainability (Scheme 5).53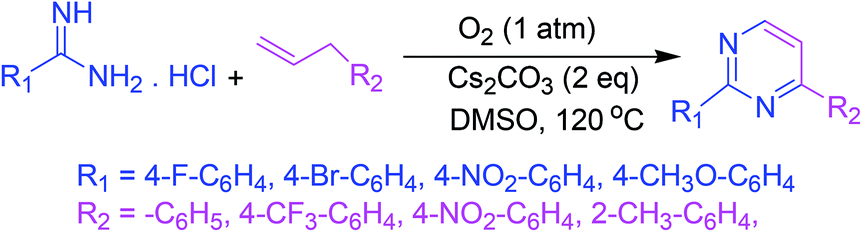 | ||
| Scheme 5 Synthesis of polysubstituted pyrimidines via base-facilitated intermolecular oxidative C–N bond fabrication. | ||
3.6. Synthesis via 4-HO-TEMPO-mediated [3 + 3] annulation of amidines with saturated ketones under Cu-catalysis
An effective and smooth synthesis of functionally vital pyrimidines has been reported by 4-HO-TEMPO-facilitated [3 + 3] annulation of commercial-grade amidines with saturated ketones under Cu-catalysis. This method gave a new protocol for the synthesis of pyrimidine derivatives via a cascade reaction of oxidative dehydrogenation/annulation/oxidative aromatization using direct β-C(sp3)–H functionalization of saturated ketones succeeded by annulation with amidines (Scheme 6).54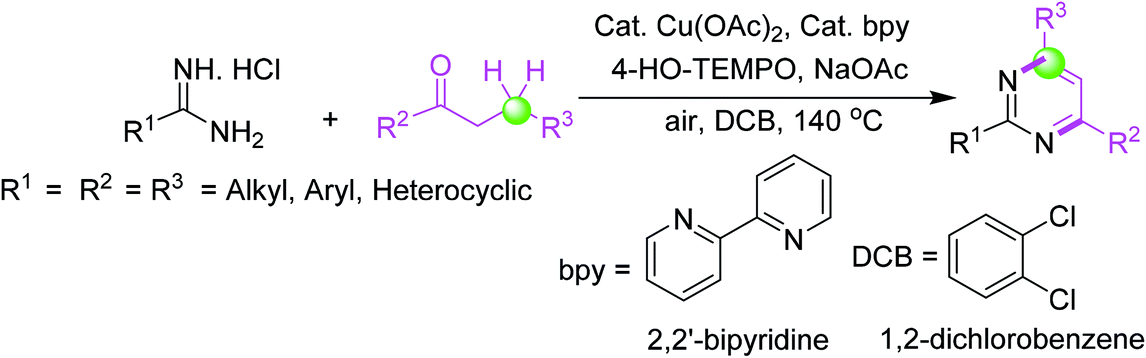 | ||
| Scheme 6 Pyrimidines synthesis via 4-HO-TEMPO-mediated [3 + 3] annulation of amidines with saturated ketones under Cu-catalysis. | ||
3.7. Viable multicomponent synthesis
Pyrimidines are also obtained from amidines and up to three (dissimilar) alcohols under iridium-catalysis through a regiose2lective, multicomponent synthetic approach. The reaction involves a series of condensation and dehydrogenation phases that produce a particular C–C and C–N bond configuration. Deoxygenation of the alcohols is accomplished through condensation, whereas aromatization is achieved via dehydrogenations. This sustainable multicomponent synthesis is catalyzed by PN5P–Ir–pincer complexes with high efficiency (Scheme 7).55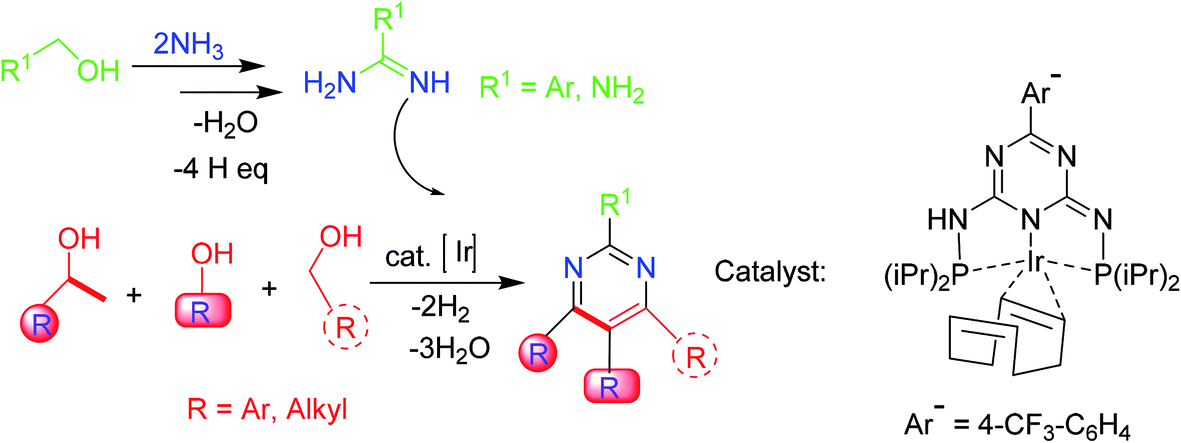 | ||
| Scheme 7 Pyrimidines synthesis from amidines and different alcohols via a multicomponent synthetic methodology. | ||
3.8. Synthesis of 2-substituted pyrimidine-5-carboxylic esters
A simple and convenient method for the synthesis of several 2-substituted pyrimidine-5-carboxylic esters has been reported. In this approach, the sodium salt of 3,3-dimethoxy-2-methoxycarbonylpropen-1-ol is treated with various amidinium salts to produce the target compounds (Scheme 8).56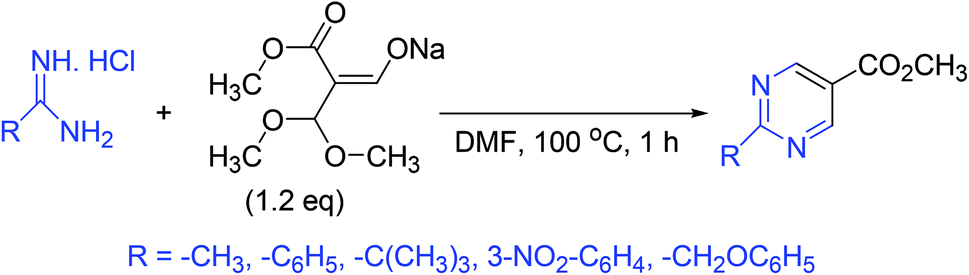 | ||
| Scheme 8 Synthesis of 2-substituted pyrimidine-5-carboxylic esters. | ||
3.9. Synthesis of densely substituted pyrimidines
Synthesis of C4-heteroatom derivatized pyrimidines is accomplished by condensation of cyanic acid analogs with N-vinyl/aryl amides. In this reaction, the utilization of cyanic bromide and thiocyanatomethane offers flexible azaheterocycles poised for additional substitution (Scheme 9).57,58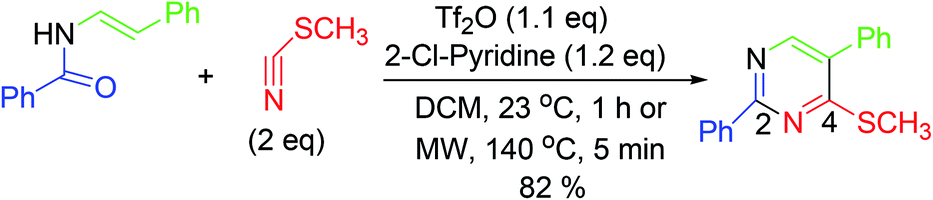 | ||
| Scheme 9 Synthesis of C4-heteroatom substituted pyrimidines. | ||
3.10. Synthesis via stereoselective access to β-enaminones under NaOH catalysis
This strategy consists of a rearrangement of propargylic hydroxylamines under NaOH catalysis, which permits efficient stereoselective access to Cbz-protected β-enaminones. Subsequent preparation of pyrimidines displays the synthetic application of these β-enaminones (Scheme 10).59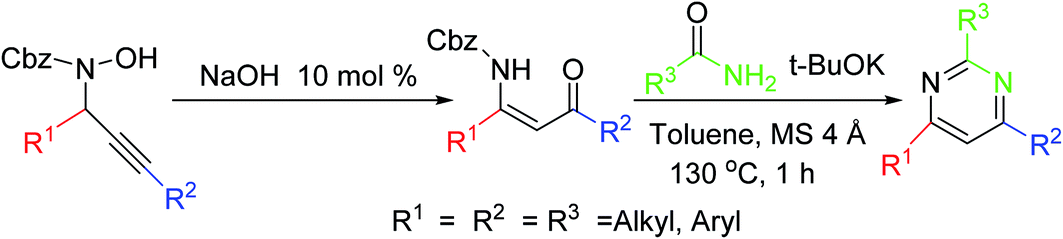 | ||
| Scheme 10 Pyrimidine synthesis via stereoselective access to β-enaminones. | ||
3.11. Synthesis via a flexible N-vinyl tertiary enamide formation
A straightforward and productive one-pot process can give a range of tetrasubstituted fused pyrimidine analogs. The tactical use of the N-p-methoxybenzyl (N-PMB) protective group ensured the production of an array of N-vinyl tertiary enamide as starting materials. It works as an efficient strategy for the synthesis of functionalized pyrimidine derivatives with the ability to control either acid chloride, nitrile, or ketone reaction participants (Scheme 11).60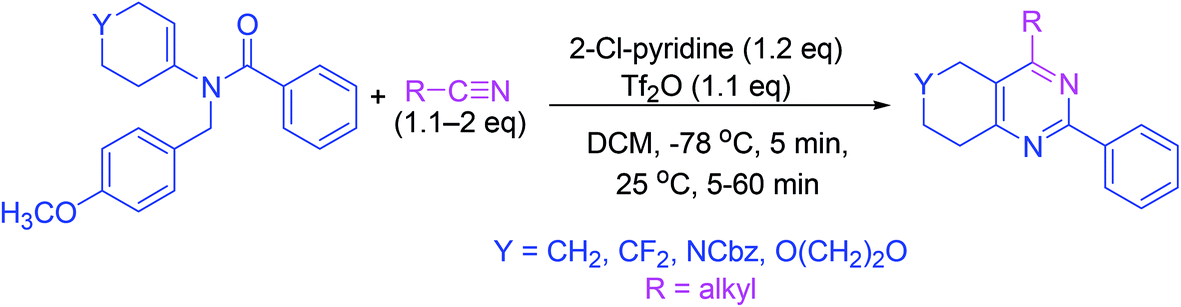 | ||
| Scheme 11 Synthesis of fused pyrimidine derivatives. | ||
3.12. Synthesis from β-formyl enamides under Lewis acid catalysis
Pyrimidine synthesis has been reported from β-formyl enamide which undergoes cyclization in the presence of samarium chloride (as a catalyst) and urea (as a source of ammonia) under microwave irradiation (Scheme 12).61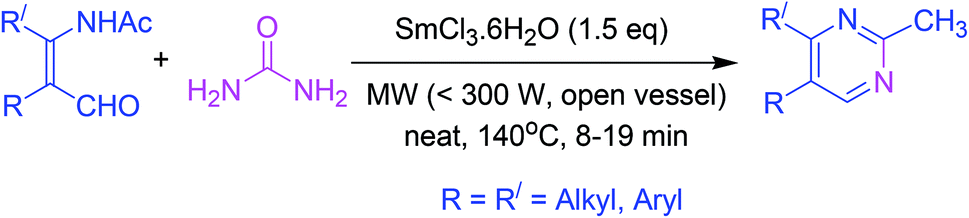 | ||
| Scheme 12 Lewis acid-catalyzed pyrimidines synthesis. | ||
3.13. One-pot synthesis by coupling-addition–cyclocondensation order
The strategy involves a coupling reaction between acid chlorides and terminal alkynes in the presence of one equivalent of triethylamine under Sonogashira conditions. Subsequently, amines or amidinium salts are added to the intermediate alkynones to accomplish the synthesis of pyrimidines in excellent yields under mild conditions (Scheme 13).62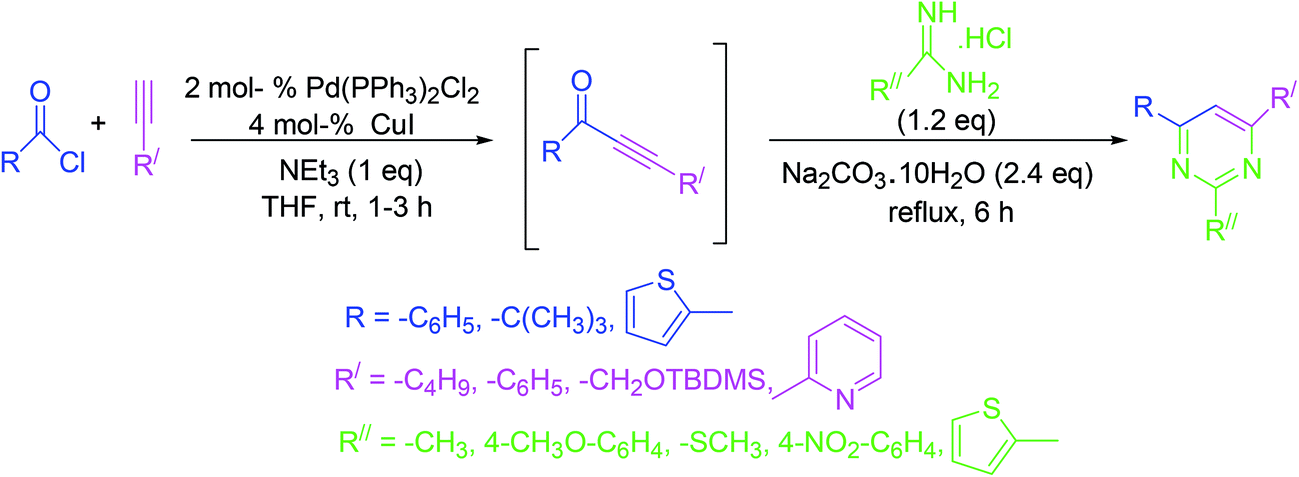 | ||
| Scheme 13 One-pot pyrimidine synthesis by the pairing of acid chlorides and terminal alkynes. | ||
3.14. Single-step synthesis from N-vinyl amides
Synthesis of pyrimidine analogs from N-vinyl amides is accomplished in a single step. In this procedure, 2-chloropyridine and trifluoromethanesulfonic anhydride are used for the activation of amides. Subsequently, nitrile is added to the reactive intermediate which ensures cycloisomerization (Scheme 14).63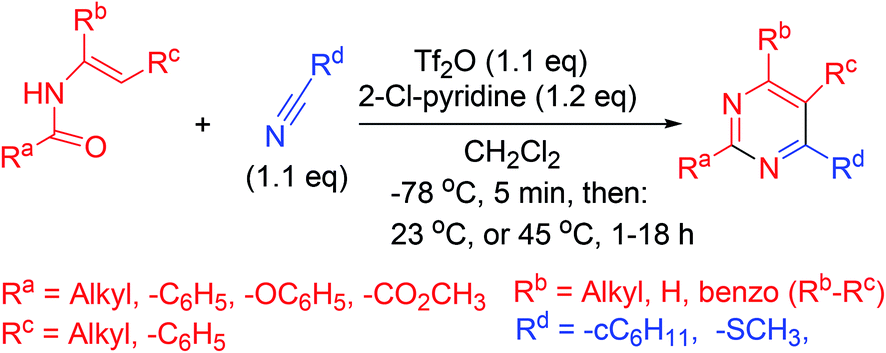 | ||
| Scheme 14 Single-step pyrimidines synthesis from N-vinyl amides. | ||
3.15. Synthesis via catalytic inverse electron demand Diels–Alder reaction
Highly functionalized pyrimidine derivatives are synthesized in good yields by inverse electron demand Diels–Alder (IEDDA) reaction of electron-deficient 1,3,5-triazines and electron-deficient ketones or aldehydes under trifluoroacetic acid (TFA)-catalysis. The mechanism indicates that the reaction involves step-by-step progress of inverse electron demand hetero-Diels–Alder (ihDA) reactions, succeeded by retro-Diels–Alder (rDA) reactions with the elimination of water (Scheme 15).64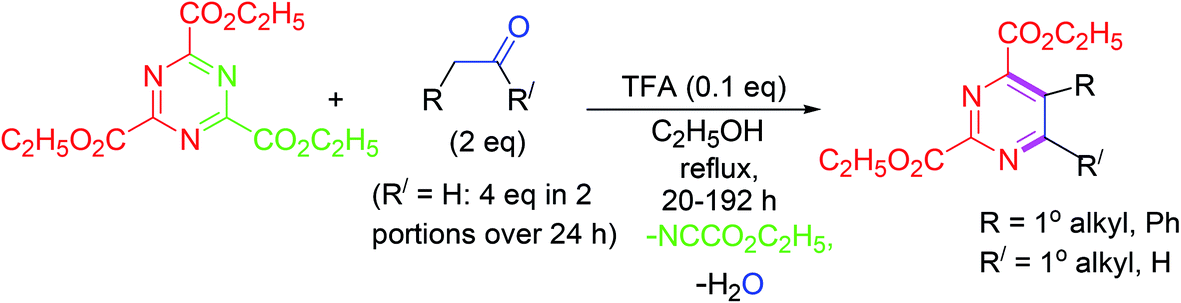 | ||
| Scheme 15 Synthesis of highly functionalized pyrimidines by IEDDA reaction under TFA catalysis. | ||
3.16. Ultrasound-driven synthesis
Highly substituted 4-pyrimidinols are synthesized in excellent yields via cyclocondensation of β-keto esters and amidines under ultrasound irradiations. Subsequently, the 4-pyrimidinols are tosylated to produce 4-pyrimidyl tosylates in another ultrasound-driven reaction. In the final step, 4-arylpyrimidines are obtained by Suzuki–Miyaura cross-coupling of 4-pyrimidyl tosylates with phenylboronic acid (Scheme 16).65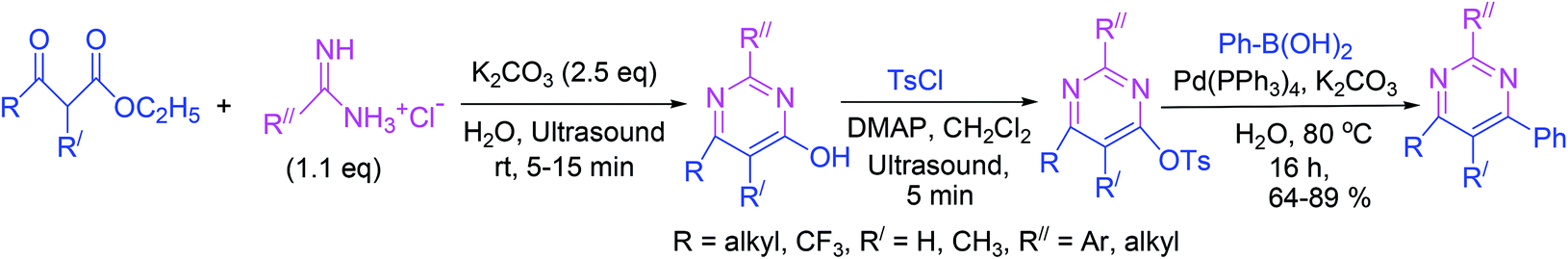 | ||
| Scheme 16 Synthesis of 4-arylpyrimidines under ultrasound irradiation. | ||
3.17. Synthesis of substituted pyrimidines via greener [3 + 3] tandem annulation–oxidation approach
Synthesis of partially and completely substituted pyrimidines has been reported via a cost-effective and eco-friendly approach. The procedure involves a reaction between easily available chalcones and benzamidine hydrochloride in the presence of eco-friendly choline hydroxide as both a catalyst and a reaction medium. The reaction proceeds in an [3 + 3] annulation–oxidation order, and the procedure is suitable for the synthesis of a wide array of biologically important pyrimidines. The noteworthy characteristics of this protocol include moderate reaction conditions, short reaction times, convenient workup method, recovery of the catalyst, and high yields of the products. This eco-friendly approach afforded high yields of the desired pyrimidines ranging from 82–93%(Scheme 17).66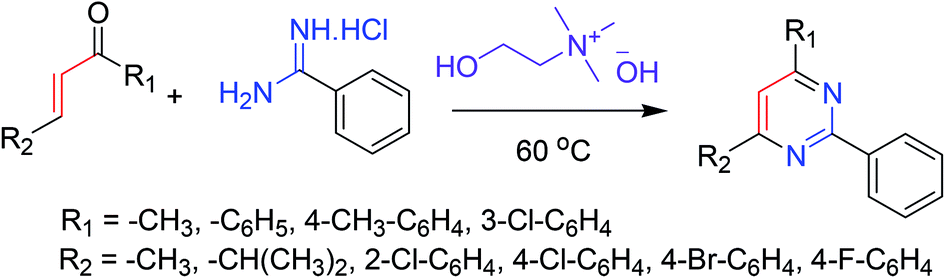 | ||
| Scheme 17 Synthesis of substituted pyrimidines via greener [3 + 3] tandem annulation–oxidation. | ||
3.18. Synthesis of fully substituted pyrimidines armed with an oxy-functionalized acetate chain
A novel strategy for the synthesis of totally substituted pyrimidine analogs equipped with an oxy-derivatized acetate moiety at the ring has been reported. Amidines provide nitrogen and the activated skipped diynes act as electrophilic reactive pardners in a coupled cascade approach. The first cascade process results in the assemblage of the six-membered heterocyclic ring via two successive aza-Michael addition reactions. In the second cascade reaction, a [H]-transfer and a [3,3]-sigmatropic rearrangement causes the aromatization of the product (Scheme 18).67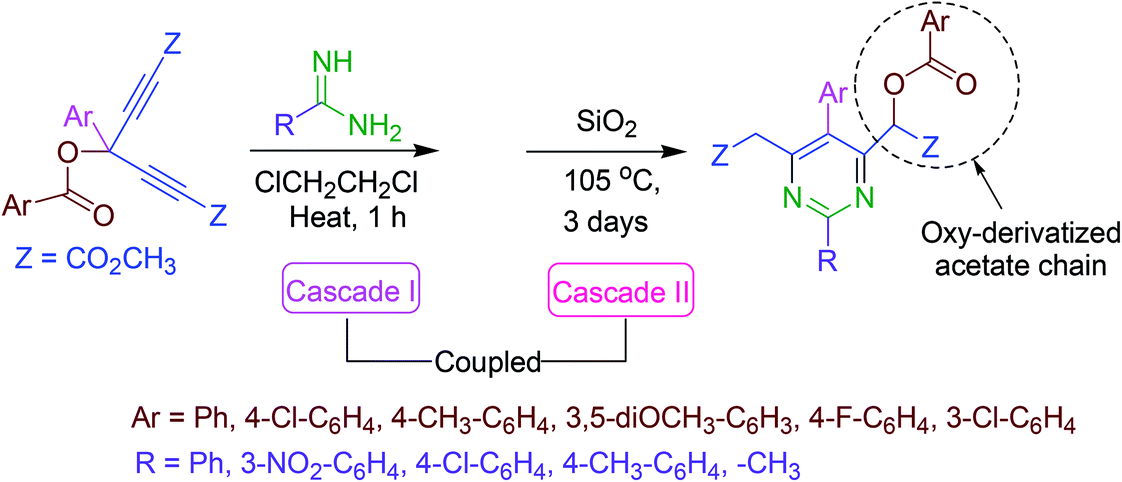 | ||
| Scheme 18 Synthesis of fully substituted pyrimidine derivatives. | ||
3.19. Synthesis of 5-substituted pyrimidine derivatives
The chemical characteristics and reactivities of 4-hydroxy- and 2,4-dihydroxy-5-(β-hydroxyethyl)pyrimidine derivatives, and the products of their modifications have been investigated. In the first step of the synthetic approach (Scheme 19), 4-chloro- and 2,4-dichloro-5-(β-chloroethyl)pyrimidine analogs were obtained. In the next step, the synthesis of several 4-alkyl(aryl)amino-5-(β-chloroethyl)pyrimidine derivatives was reported, which were subsequently transformed into 5,6-dihydropyrrolo[2,3-d]pyrimidine derivatives (Scheme 19).68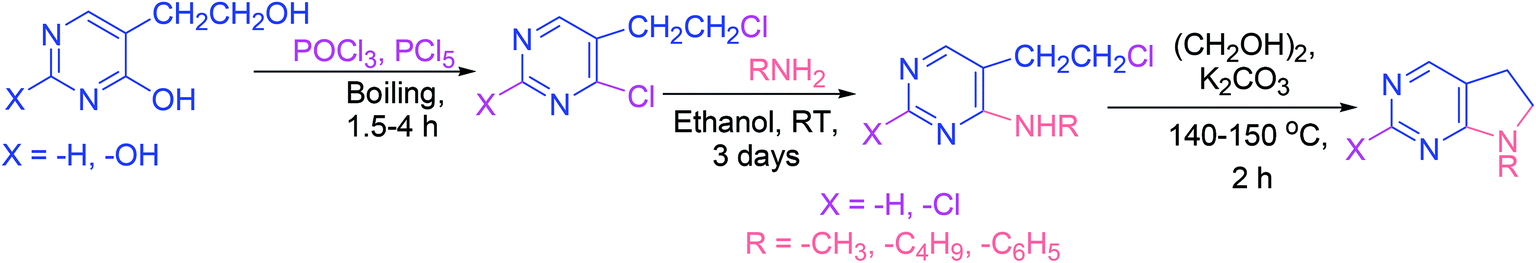 | ||
| Scheme 19 Synthesis of 5,6-dihydropyrrolo[2,3-d]pyrimidines. | ||
4. Advances in the anti-inflammatory activities of pyrimidines
Owing to the noteworthy pharmacological potential of pyrimidine derivatives, extensive research has been directed to their anti-inflammatory effects.69 Several pyrimidine analogs such as afloqualone, proquazone, epirizole, and tofacitinib (Fig. 4) have already been approved as anti-inflammatory drugs and are in clinical use.70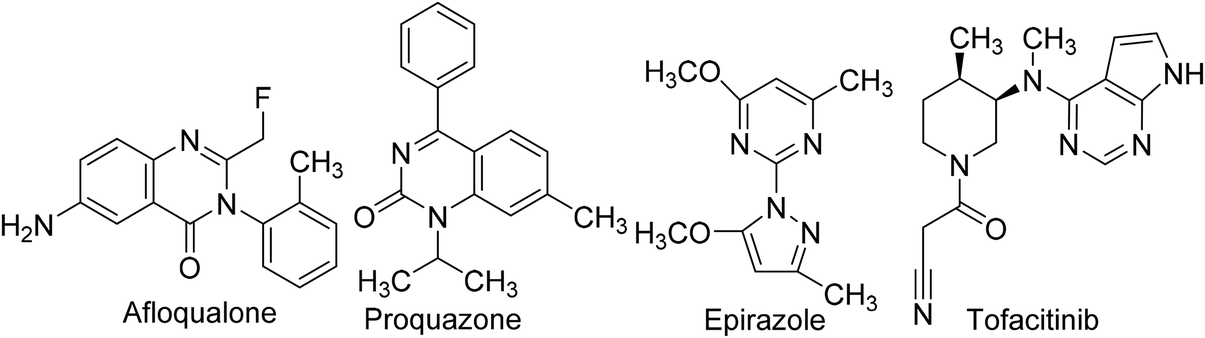 | ||
| Fig. 4 Clinically used pyrimidine-based anti-inflammatory drugs. | ||
Numerous research teams described the anti-inflammatory effects of various synthetic and natural pyrimidines. Their findings suggest that pyrimidines exhibit anti-inflammatory effects by inhibiting vital inflammatory mediators such as PGE2, nitric oxide (NO), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), chemokines, and cytokines. Consequently, this section of the article is focused on the inhibitory effects of pyrimidine derivatives against some key inflammatory mediators.
4.1. Cyclooxygenase inhibition
Generally, the mechanism of action of pyrimidine-based anti-inflammatory agents is associated with the inhibition of PGE2 generated by COX enzymes.71,72 Like other NSAIDs, pyrimidine-based anti-inflammatory agents function by suppressing the activity of COX-1 and COX-2 enzymes and thus reduce the generation of PGE2.73,74 Sufficient investigations have been performed to discover new and more efficient pyrimidine-based anti-inflammatory agents that can inhibit the activity of COX enzymes.Atatreh et al. synthesized several new pyrazolo[3,4-d]pyrimidine derivatives (3a–3d and 4a–4f) (Scheme 20). The inhibitory potential of the synthesized compounds versus COX enzymes was tested via COX inhibitor screening assay kit. Diclofenac and celecoxib were used as a positive control versus COX-1 and COX-2 enzymes respectively. Preliminary results (Table 1) revealed that four derivatives (3a, 3b, 4b, and 4d) suppressed the activity of COX enzymes, and thus inhibited the production of PGE2. The potency of the target analogs was reported in the form of half-maximal inhibitory concentration (IC50 values). The IC50 values of (3b, 4b, and 4d) against the activity of COX-1 were noted to be 19.45 ± 0.07, 26.04 ± 0.36, and 28.39 ± 0.03 μM, respectively. On the other hand, the IC50 values of (3a, 3b, 4b, and 4d) against the activity of COX-2 were reported to be 42.1 ± 0.30, 31.4 ± 0.12, 34.4 ± 0.10, and 23.8 ± 0.20 μM, respectively. The rest of the compounds (3c, 3d, 4a, 4c, 4e, and 4f) did not exhibit any inhibitory effects against COX enzymes. Furthermore, docking studies indicated an interaction of these derivatives with several vital residues of COX-2. Moreover, it was observed that compound (4d) had comparable binding styles to the typical COX-2 suppressor, celecoxib. In vivo analysis demonstrated that four compounds (3a, 3d, 4d, and 4f) possessed similar anti-inflammatory effects to that of widely used COX-2 suppressor, celecoxib. The potency for the majority of the target compounds was observed to be greater than diclofenac sodium.75
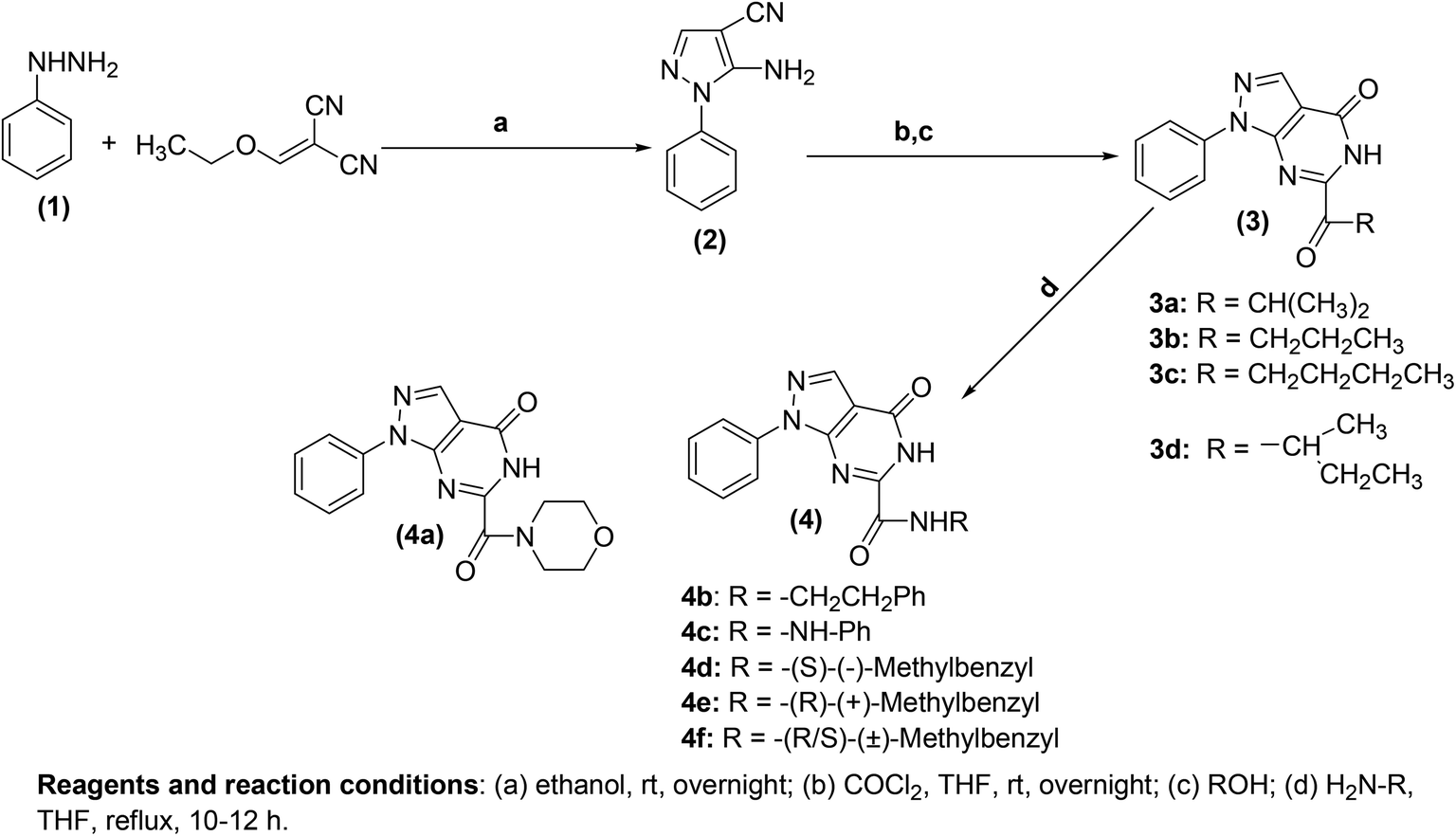 | ||
| Scheme 20 Synthesis of pyrazolo[3,4-d]pyrimidines (3a–3d and 4a–4f). | ||
| Comp. no. | COX-1, IC50 (μM) | COX-2, IC50 (μM) |
|---|---|---|
| 3a | — | 42.1 ± 0.30 |
| 3b | 19.45 ± 0.07 | 31.4 ± 0.12 |
| 3c | — | — |
| 3d | — | — |
| 4a | — | — |
| 4b | 26.04 ± 0.36 | 34.4 ± 0.10 |
| 4c | — | — |
| 4d | 28.39 ± 0.03 | 23.8 ± 0.20 |
| 4e | — | — |
| 4f | — | — |
| Celecoxib | 11.7 ± 0.23 | |
| Diclofenac | 8.72 ± 0.28 |
A convenient, cost-efficient, and green method was used for the synthesis of pyrano[2,3-d]pyrimidine derivatives (Fig. 5). The protocol involves one-pot three-component condensation performed by the treatment of p-chlorobenzaldehyde, malononitrile, and thiobarbituric acid in the presence of zinc oxide, ferrosoferric oxide or trimanganese tetraoxide nanoparticles as a catalyst. The anti-inflammatory activities of several target derivatives were examined. The results indicated that derivatives (5 and 6) showed a noteworthy in vitro anti-inflammatory activity as they potently suppressed the COX-2 activity. The IC50 values of the two compounds (5 and 6) against COX-2 inhibition were reported to be 0.04 ± 0.09 μmol and 0.04 ± 0.02 μmol, respectively as compared to the standard drug celecoxib (IC50 = 0.04 ± 0.01 μmol).76
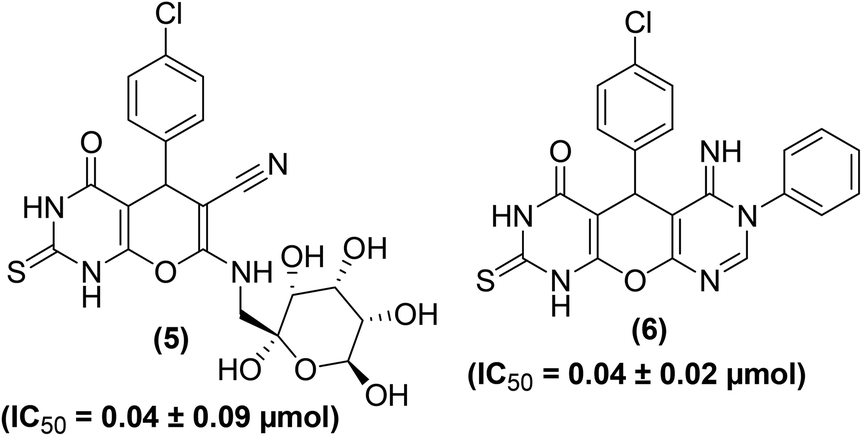 | ||
| Fig. 5 Chemical structures of selected pyrano[2,3-d]pyrimidines (5 and 6). | ||
Synthesis of several substituted 1H-pyrazolyl-thiazolo[4,5-d]pyrimidine derivatives has been reported. The results of two bioassays namely carrageenan-induced paw edema and cotton pellet-induced granuloma in rats indicated that pyrimidine derivatives (7–9) (Fig. 6) possessed anti-inflammatory effects similar to that of the widely practiced drug indomethacin. The ED50 (50% effective dose) values of compounds (7–9) were calculated to be 11.60, 8.23, and 9.47 μM, respectively as compared to indomethacin (ED50 = 9.17 μM). Moreover, the three pyrimidine derivatives (7–9) demonstrated stronger in vitro inhibitory effects versus COX-2 than COX-1 enzymes. Human COX enzymatic assessment showed that the investigated compounds (7–9) showed weak inhibitory effect against the activity of COX-1 enzyme (IC50 = between 95.0 and >100 μM) as compared to the reference drug, indomethacin (IC50 = 0.21 μM). However, compounds (7 and 9) exhibited better inhibitory effects against COX-2 enzyme with their IC50 values reported to be 0.36, and 0.29 μmol, respectively as compared to indomethacin (IC50 = 2.60 μmol). The IC50 values of compounds (7–9) and indomethacin for COX-1 and COX-2 enzymes are given in (Table 2).77
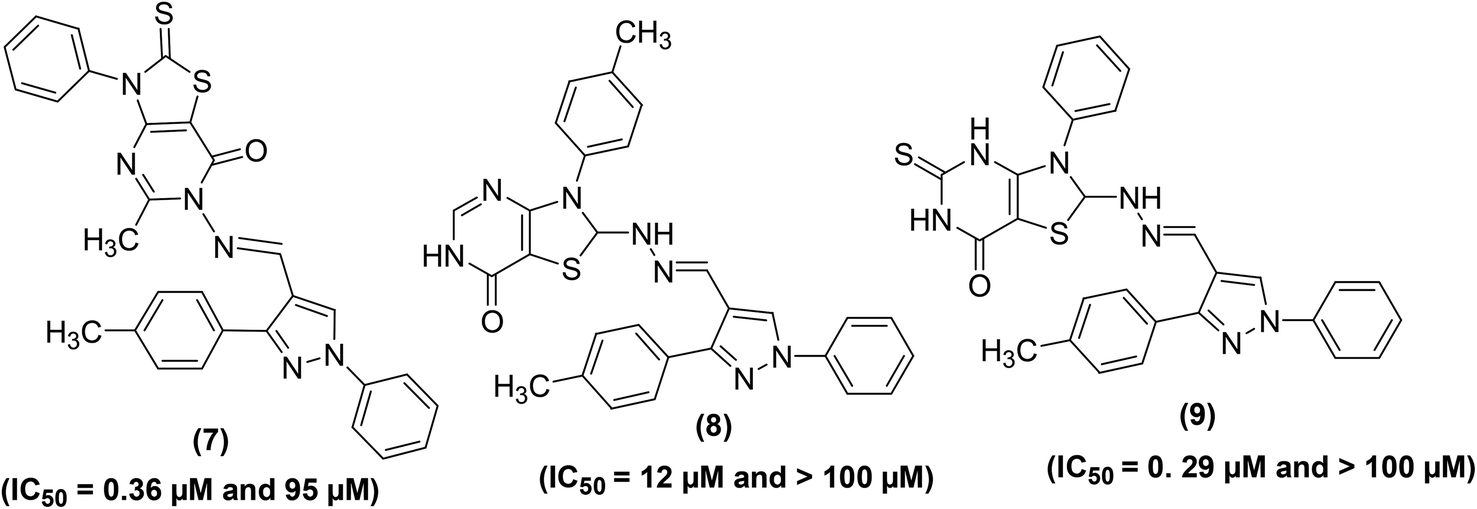 | ||
| Fig. 6 Chemical structures of substituted 1H-pyrazolyl-thiazolo[4,5-d]pyrimidines (7–9). | ||
Tageldin et al. reported the synthesis of novel pyrazolo[3,4-d]pyrimidine derivatives substituted with numerous functional groups or linked to a substituted pyrazole ring through various bonds (Fig. 7). The target pyrimidine derivatives were screened for their anti-inflammatory potential via in vitro COX-1/COX-2 inhibition analysis and in vivo formalin-induced paw edema and cotton pellet-induced granuloma tests. The preliminary assessment revealed that two derivatives (10 and 11) possessed larger COX-1/COX-2 selectivity indexes (SI values) than celecoxib and diclofenac sodium. Whereas, two other derivatives (12 and 13) showed larger SI values than diclofenac sodium and approximately equal to celecoxib. Compound (14) showed selectivity indexation similar to diclofenac sodium. In contrast, three pyrazolyl structural analogs (10, 13, and 14) exhibited anti-inflammatory potential about 2–2.5-times that of diclofenac sodium and approximately 8–10.5-times that of celecoxib in the cotton pellet-induced granuloma analysis. Moreover, compounds (10–12) displayed a nice gastrointestinal safety profile. The potency (in the form of IC50 and SI values) of five selected analogs (10–14), celecoxib, and diclofenac sodium is provided in Table 3.78
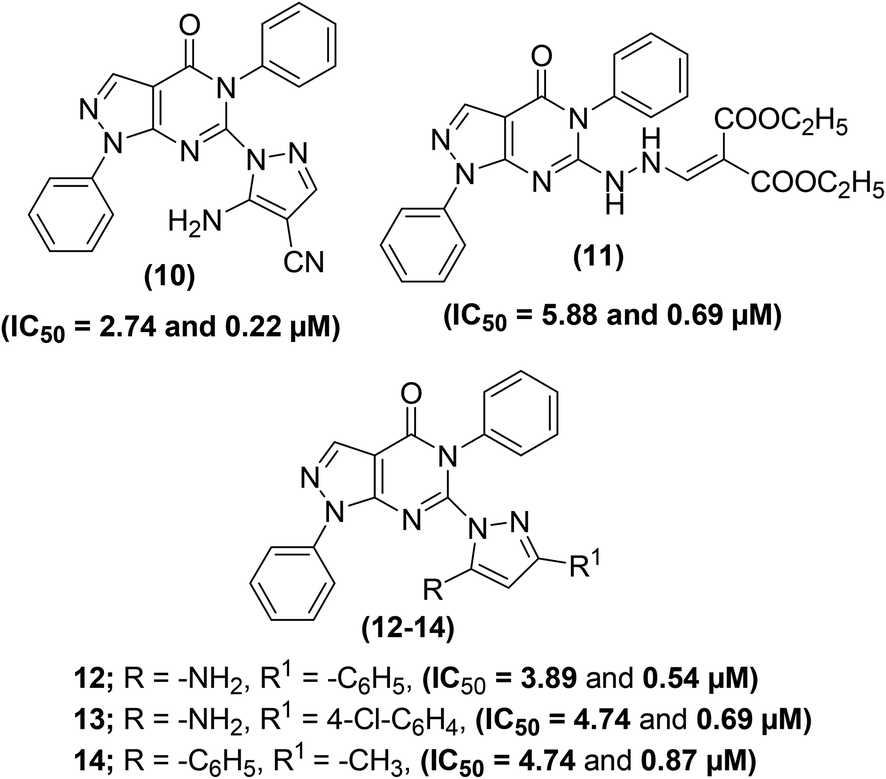 | ||
| Fig. 7 Chemical structures of selected pyrazolo[3,4-d]pyrimidines (10–14). | ||
| Tested compound | COX-1, IC50 (μM)a | COX-2, IC50 (μM) | SIb (COX-1/COX-2) |
|---|---|---|---|
| a Values are average of three readings obtained and the variation from the average is ±10% of the average value.b In vitro COX-2 selectivity indices (COX-1 IC50/COX-2 IC50). | |||
| 10 | 2.74 | 0.22 | 12.45 |
| 11 | 5.88 | 0.69 | 8.52 |
| 12 | 3.89 | 0.54 | 7.20 |
| 13 | 4.74 | 0.69 | 6.86 |
| 14 | 4.74 | 0.87 | 5.44 |
| Celecoxib | 5.46 | 0.78 | 7.23 |
| Diclofenac sodium | 6.74 | 1.10 | 6.12 |
Synthesis and anti-inflammatory activities of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine analogs have been reported. Western blotting and reverse transcription-polymerase chain reaction (RT-PCR) examinations were performed to study the influence of the synthesized compounds on messenger ribonucleic acid (mRNA) and protein levels of iNOS and COX-2 enzyme in RAW264.7 cells. Three compounds (15–17) (Fig. 8) were noticed to be the most potent. Experimental results indicated that exposure to derivatives (15–17) significantly decreased the iNOS and COX-2 mRNA expressions, with higher suppressive effects as compared to indomethacin. Additionally, the three derivatives also significantly reduced the protein levels of COX-2 and iNOS enzymes. Preliminary SAR investigation indicated that the existence of electron-releasing substituents such as pyridine (15) and chloromethyl group (16) on position-2 of pyrimidine skeleton facilitated the enhancement of anti-inflammatory activity. Moreover, the improved anti-inflammatory action of the compound (17) is credited to the existence of naphthyl moiety at position-3 of the pyrimidine skeleton. Naphthyl group can provide a multiple-π–π conjugated system, which leads to coupling with inflammatory cytokines and consequently causes better inhibition against the expression of COX-2 and iNOS enzymes.79
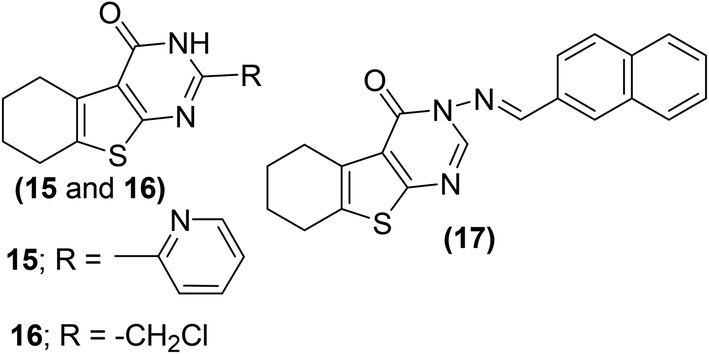 | ||
| Fig. 8 Chemical structures of selected tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines (15–17). | ||
Mohamed et al. reported the synthesis and anti-inflammatory effects of pyrrolo[2,3-d]pyrimidine analogs. Rat paw edema test was performed to measure the in vivo anti-inflammatory effects of the synthesized compounds. Preliminary data indicated that four compounds (18–21) (Fig. 9) showed significant inhibition against the activity of COX-2 enzyme. The activity profiles of the four derivatives showed that they had similar anti-inflammatory effects to ibuprofen. Analog (21) was observed to be the most potent derivative since it exhibited better anti-inflammatory activity compared to ibuprofen. The potency of compound (21) in terms of its (%) inhibition was reported to be 63.24 and 74.60 after 3 and 4 h intervals, respectively as compared to that of ibuprofen (60.66% and 69.52% after 3 and 4 h intervals, respectively). SAR study indicated that pyrrolo[2,3-d]pyrimidines (18–21) had better anti-inflammatory activity than pyrrolo[3,2-e][1,2,4]triazolo[4,3-c]pyrimidines reported in the same work. The introduction of 4-methoxyphenyl group at position-7 of pyrrolo[2,3-d]pyrimidines (18–21) contributed to activity enhancement. Furthermore, the coupling of 4-hydrazino-pyrrolopyrimidines and benzaldehyde produced two compounds (19 and 20) which also exhibited noticeable anti-inflammatory activity.80
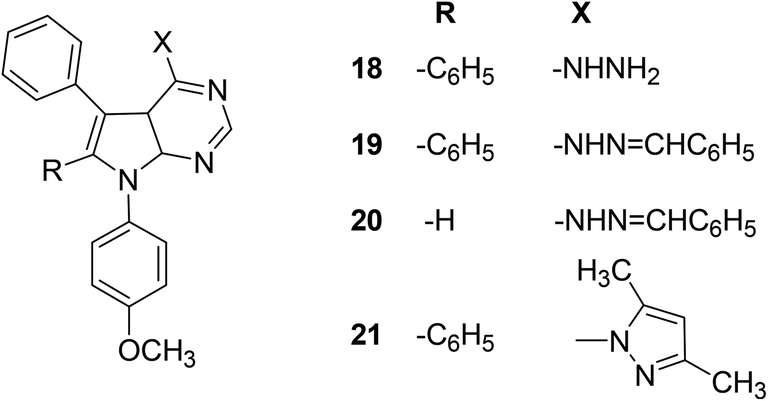 | ||
| Fig. 9 Chemical structures of selected pyrrolo[2,3-d]pyrimidines (18–21). | ||
Several novel pyrimidine-benzoxazole/benzimidazole hybrids (Scheme 21) have been synthesized and investigated for their COX inhibitory effects via an enzyme immunoassay (EIA) kit. Experimental data revealed that the two hybrids (24a and 24b) potently suppressed the activity of COX-1 enzyme with their IC50 values calculated to be 2.76 and 1.92 μM respectively. However, the two compounds exhibited moderate inhibitory effects versus COX-2 enzyme with their IC50 values to be 7.47 and 8.21 μM, respectively. Moreover, molecular docking studies of the two derivatives (24a and 24b) with COX-2 enzyme confirmed their mechanism of action. Docking of derivative (24a) in COX-2 enzyme showed that the derivative was well-tailored with the receptor via three hydrogen bonds having score energy (S) = −11.50 kcal mol−1. Furthermore, docking of derivative (24b) in COX-2 binding position revealed that the derivative had docking score = −11.50 kcal mol−1. Additionally, derivative (24b) displayed two hydrogen bonds; (1) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O with Gln192 (2.79 Å), and (2) CO with Arg513 (2.51 Å).81
O with Gln192 (2.79 Å), and (2) CO with Arg513 (2.51 Å).81
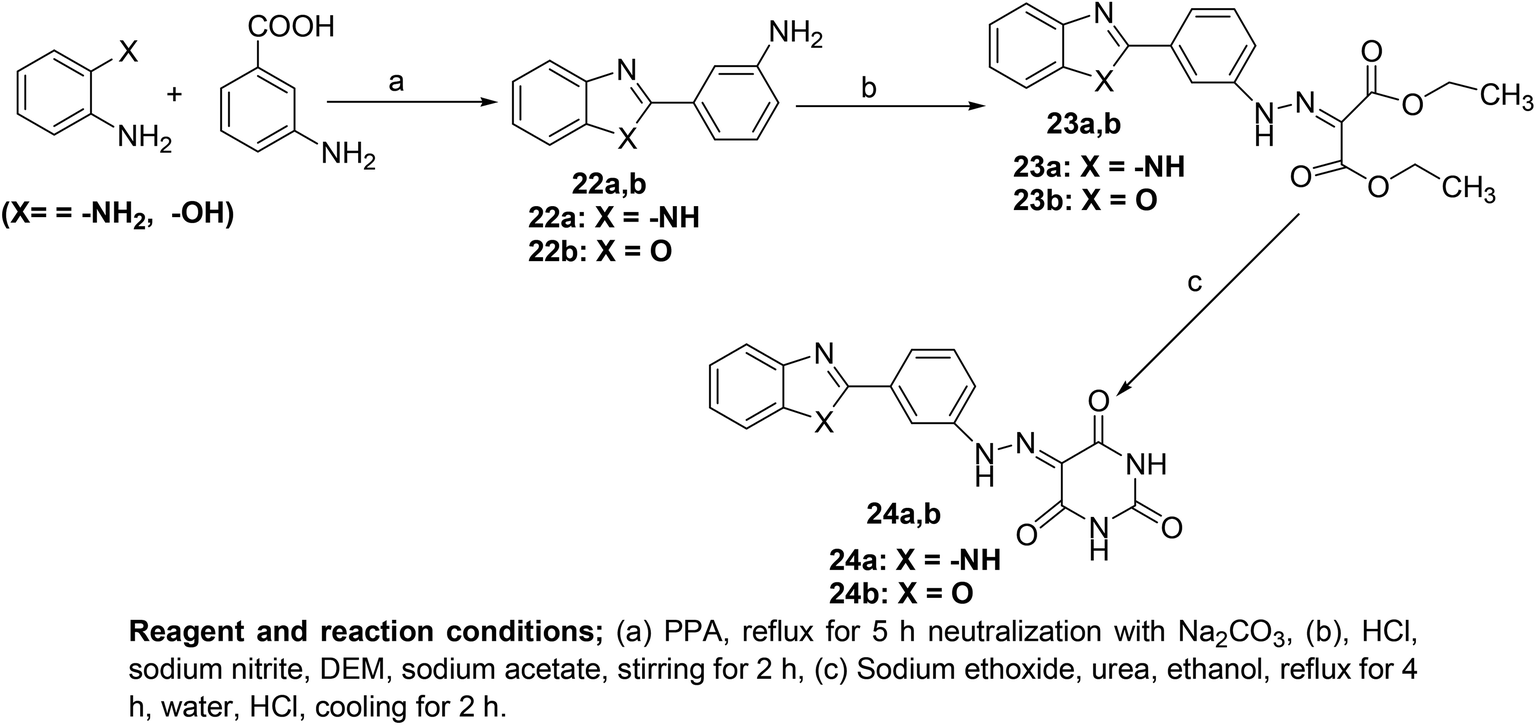 | ||
| Scheme 21 Synthetic methodology of new pyrimidine-benzoxazole/benzimidazole hybrids (24a and 24b). | ||
New pyrimidine-pyridine hybrids (Fig. 10) have also been reported for their potential use as anti-inflammatory candidates. The in vivo and in vitro anti-inflammatory activities were evaluated via the carrageenan-induced rat paw oedema model and EIA kit, respectively. Celecoxib was applied as a reference drug during activity analysis. Preliminary results indicated that the pyrido[2,3-d]pyrimidine derivative (25), bearing a trimethoxyphenyl group at site 4 of the pyridine ring, was the most efficient compound with its edema inhibiting potential of 74% over 1 hour. SAR analysis indicated that pyridodipyrimidine derivatives carrying electron-releasing groups (26–29) exhibited better oedema inhibitory effects than the pyrimidines of the same class carrying electron-withdrawing groups (30 and 31). Among all the synthesized derivatives, three compounds (25, 27, and 29) showed better inhibitory effect versus COX-2 enzyme with their IC50 values recorded to be 0.89, 0.62, and 0.25 μM, respectively in comparison to the standard drug celecoxib (IC50 = 1.11 μM).82
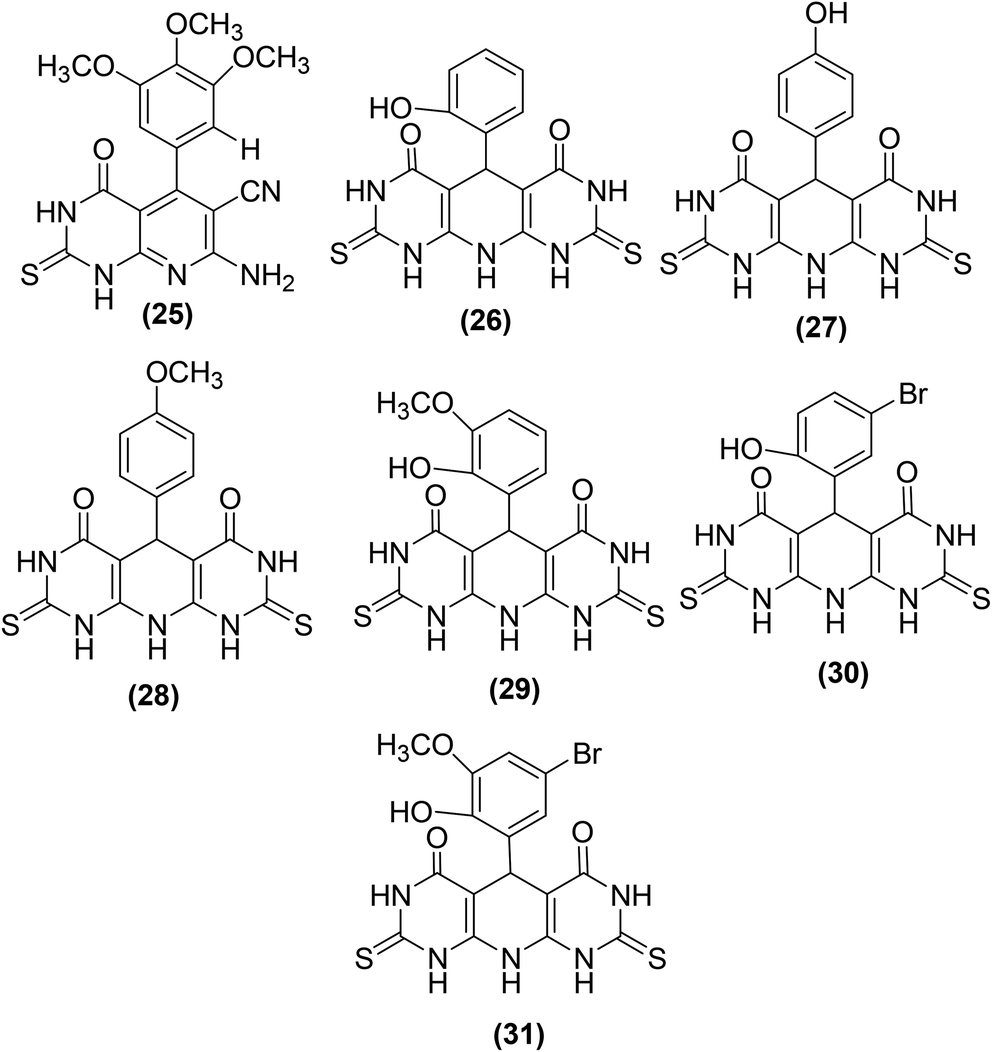 | ||
| Fig. 10 Chemical structures of selected pyrimidine-pyridine hybrids (25–31). | ||
The effect of C-5 substitution (in polysubstituted pyrimidine derivatives) on the suppression of PGE2 generation has been examined. The synthesis of polysubstituted pyrimidines was reported from the related 2-amino-4,6-dichloropyrimidines via the Suzuki cross-coupling reaction. The target pyrimidines were assessed for their ability to suppress the in vitro PGE2 generation from COX enzymes in C57BL6 mouse peritoneal cells. The results indicated that both monoaryl – as well as bisarylsubstituted 2-aminopyrimidines potently inhibited the PGE2 generation regardless of the length of C-5 substituents in polysubstituted pyrimidines. Moreover, greater potency against the suppression of PGE2 generation was observed for pyrimidines with shorter C-5 substituents. Compounds (32–35) (Fig. 11) displayed the most powerful inhibitory activities versus the production of PGE2 from COX enzymes with their IC50 values of 0.003–0.033 μM as compared to the two widely used drugs indomethacin (IC50 = 0.005 μM) and aspirin (IC50 = 4.08 μM). All of the four compounds are C-5 unsubstituted, i.e. analogs carry hydrogen atoms at the C-5 position. Among all the target pyrimidines, analog (32) was observed to be the strongest inhibitor of PGE2 generation with its IC50 value of 0.003 μM. The initial results support further preclinical investigations of the selected derivatives as promising anti-inflammatory agents. However, the precise mode of action of the compounds (32–35) remains to be illustrated.83
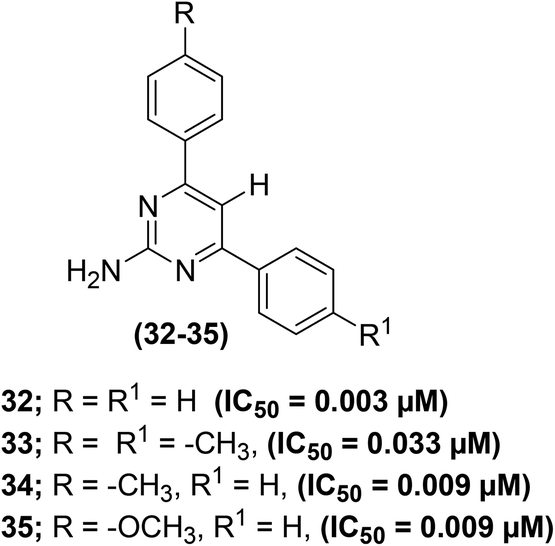 | ||
| Fig. 11 Chemical structures of selected polysubstituted pyrimidines (32–35). | ||
Raffa et al. synthesized pyrazolo[3,4-d]pyrimidine derivatives (Fig. 12) by the reaction of 5-aminopyrazole-4(N-benzoyl)carbohydrazide derivatives with appropriate triethylorthoesters. The synthesized analogs were examined for their potential to suppress COX-2 activity via a COX-1/COX-2 inhibitor screening assay kit. The (%) inhibition results (Table 4) indicated that the majority of the compounds exhibited greater inhibitory effects versus COX-2 enzyme in comparison to reference drugs indomethacin and NS398. The results indicated that the mode of action of the selected candidates involved the inhibition of PGE2 production from arachidonic acid.84
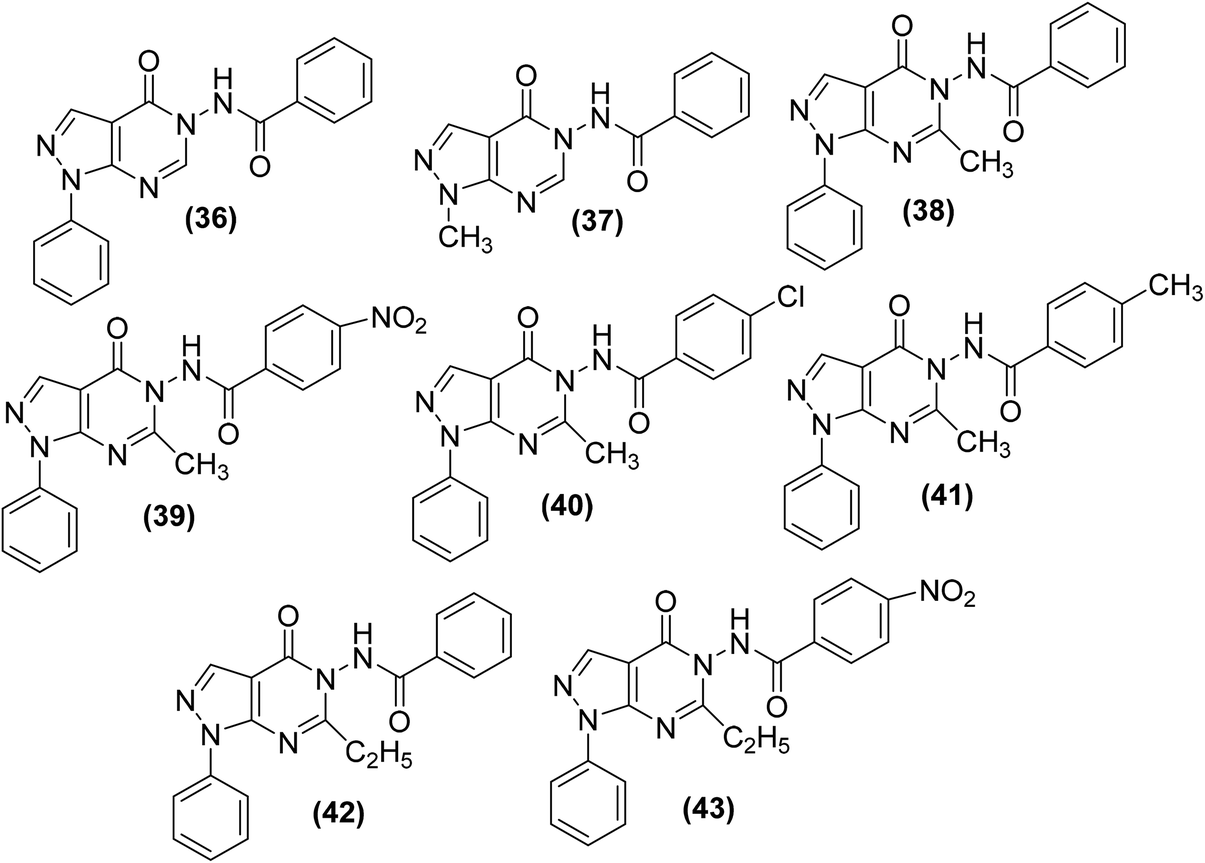 | ||
| Fig. 12 Chemical structures of selected pyrazolo[3,4-d]pyrimidines (36–43). | ||
The synthesis of new thiazolo[4,5-d]pyrimidine analogs for their potential use as COX-inhibitors (48a–l) (Scheme 22) has also been reported. The target pyrimidines were screened for their in vitro COX inhibitory potential, and in vivo anti-inflammatory effects via an EIA kit and formalin-induced rat paw oedema model, respectively. Experimental data indicated that most of the synthesized derivatives possessed substantial anti-inflammatory activities. However, compound (48g) was discovered to be the most efficient anti-inflammatory candidate with its (%) inhibitions of 57, 88, and 88 after 1, 3, and 5 h, respectively. The anti-inflammatory effect of the compound (48g) was recorded to be greater than that of reference drug celecoxib [(%) inhibition = 43, 43 & 54 after 1, 3, and 5 h, respectively]. Furthermore, twelve synthesized derivatives (48a–l) exhibited modest to potent suppressive effects against COX-2 enzyme (IC50 = 0.87–3.78 μM). The IC50 values of compounds (48e, 48g, and 48k) were recorded to be 0.92 μM, 0.87 μM, and 1.02 μM, respectively as compared to the selective COX-2 inhibitor, celecoxib (IC50 = 1.11 μM). Docking studies of the three compounds (48e, 48g, and 48k) with COX-2 enzyme were performed to elucidate their mode of action. The results indicated that the selected derivatives (48e, 48g, and 48k) displayed better score energy (S = −12.65 to −13.76) than the cocrystallized ligand (S = −11.93 kcal mol−1). Moreover, the selected candidates (48e, 48g, and 48k) also tailored well with the active site of COX-2 enzyme. Thus, docking studies confirmed the binding approach of the three analogs (48e, 48g, and 48k) with COX-2 enzyme.85
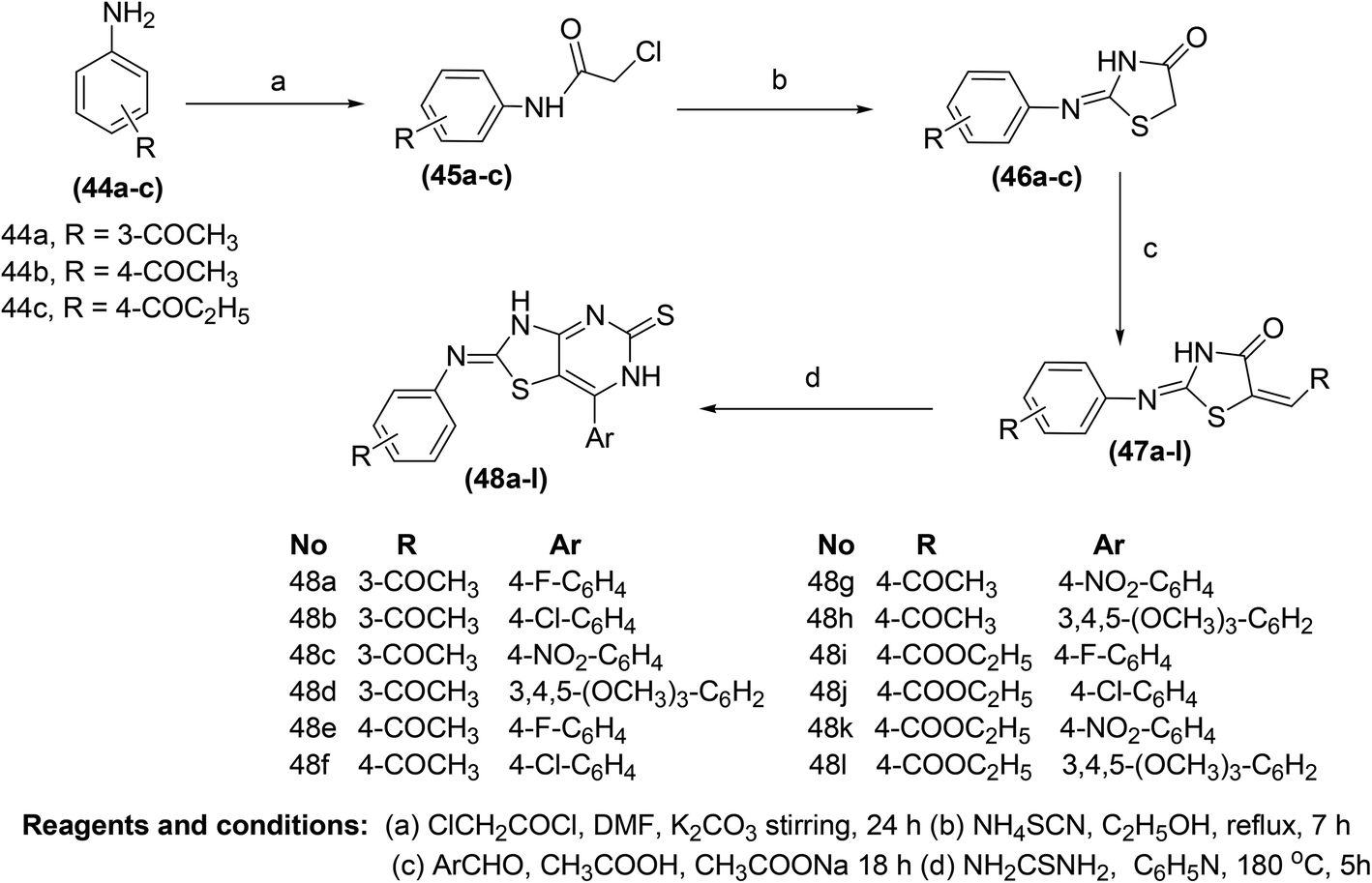 | ||
| Scheme 22 Synthesis of the thiazolo[4,5-d]pyrimidines (48a–l). | ||
The synthesis of several imidazo[1,2-a]pyrimidine analogs substituted with two aryls at positions-2 and -3 has been described. The target analogs were investigated for their anti-inflammatory effects through xylene-induced ear swelling in mice. Preliminary data revealed that most of the target pyrimidines demonstrated selective COX-2 inhibitory effects. Analog (49) (Fig. 13) was observed to be the most powerful (IC50 = 13 μM) COX-2 inhibitor. However, the inhibitory effect of compound (49) against COX-1 activity was observed to be much smaller (IC50 = 170 μM) than its inhibitory effect against COX-2 activity. Moreover, the anti-inflammatory potential of the compound (49) was recorded to be greater (63.8%) than that of ibuprofen (44.3%). SAR analysis verified that the introduction of two aryl moieties on the adjacent positions 2 and 3 in imidazo[1,2-a]pyrimidine analogs selectively improved the inhibitory effects of the target derivatives against COX-2 activity.86
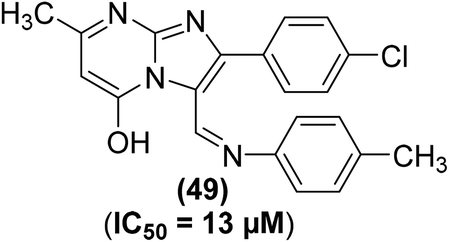 | ||
| Fig. 13 Chemical structure of selected imidazo[1,2-a]pyrimidine analog (49). | ||
Singour and coworkers reported the synthesis of five 2-chloropyrimidine derivatives (50–54) by the reaction of 2,4,6 trichloropyrimidine either with aromatic amines or amides (Scheme 23). The synthesized 2-chloropyrimidines were subsequently investigated for their COX-2 inhibitory activities. Moreover, the target compounds were also examined for their anti-inflammatory potential via the carrageenan-induced hind paw edema model in mice as compared to a standard drug (indomethacin). Amongst the tested compounds, derivative (50) exhibited the strongest activity with its (%) inhibition reported to be 68.85 after 3 hours in comparison to indomethacin (% inhibition = 69.83). SAR analysis indicated that the presence of five-member thiophene moiety at positions-4, 6, and electron releasing chlorine atom at position-2 contributed to the enhanced anti-inflammatory activity of the derivative (50).87
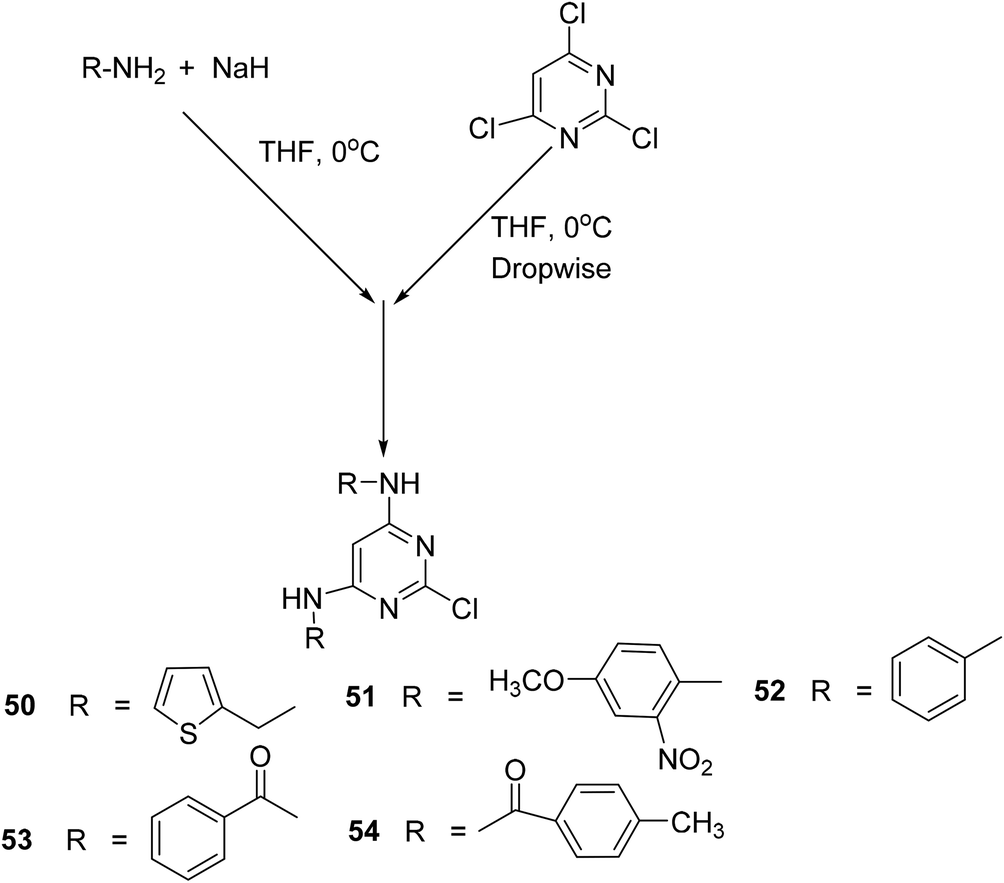 | ||
| Scheme 23 Synthesis of 2-chloropyrimidine derivatives (50–54). | ||
Bruni et al. synthesized several pyrazolo[l,5-a]pyrimidin-7-ones (57a–q) (Scheme 24) via chemical transformations on the 2-phenyl site of 4,7-dihydro-4-ethyl-2-phenylpyrazolo[l,5-a]pyrimidin-7-one (FPP028), known to be a weak PGE2 inhibitor. In vivo anti-inflammatory effects of the synthesized pyrimidine analogs were measured by carrageenan-induced rat paw edema test. Experimental data revealed that pyrimidine analogs inhibited the biosynthesis of PGE2 to different extents owing to the existence of various substituents at their 2-phenyl site. All the derivatives exhibited different anti-inflammatory activities. Among the investigated pyrimidines, compound (57g) was observed to be the most efficient derivative. At a concentration of 25 mg kg−1, compound (57g) caused 92.1 ± 15.6% inhibition of carrageenan-induced rat paw edema in comparison to its parent compound (FPP028) and standard drug indomethacin. Moreover, SAR analysis indicated that the replacement of the phenyl ring with a thien-2-yl moiety at position-2 caused a noteworthy enhancement in the anti-inflammatory effect for the isomer (57g). The mechanism of action revealed that compound (57g) was able to control cellular action in inflammation to various levels by suppressing both granulocyte superoxide generation and myeloperoxidase (MPO) discharge.88
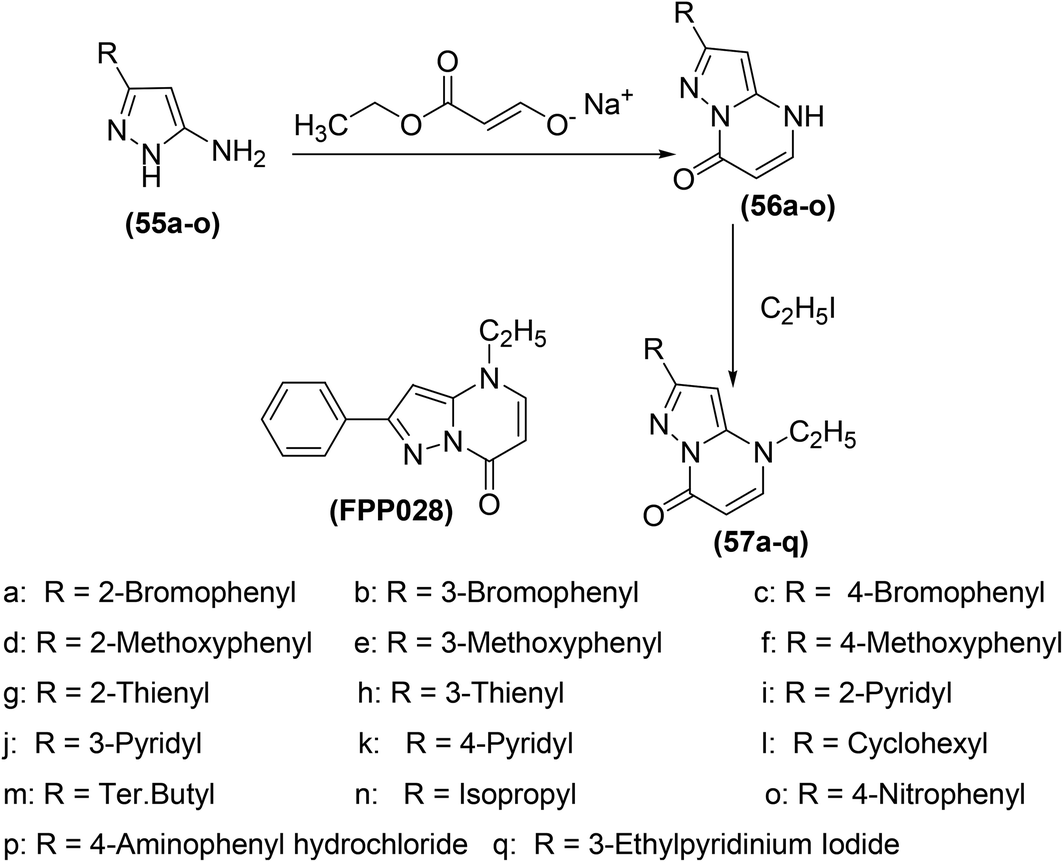 | ||
| Scheme 24 Synthesis of pyrazolo[l,5-a]pyrimidin-7-ones (57a–q). | ||
Many pyrazole derivatives are known to exhibit COX-2 inhibitory activity. Nofal et al. perceived that joining pyrazole and pyrimidine scaffolds together can result in the formation of pyrazolopyrimidine derivatives of enhanced COX-2 inhibitory activity. Based on the idea, several substituted derivatives were synthesized and subsequently evaluated via carrageenan-induced paw edema. Amongst all the investigated derivatives, pyrazolediazenylpyrimidine derivative (58) was noticed to be the most efficient anti-inflammatory candidate, exhibiting approximately 63% inhibitory effect in edema development after 1 hour. Moreover, compound (59) was observed to be the most viable anti-inflammatory agent over the entire experimental period with the usual change in paw volume of around 25% (67% inhibitory effect in edema development), which was similar to indomethacin (10 mg kg−1). SAR analysis indicated that the introduction of 3-chlorophenyl moiety on site-1 of pyrazolone ring in pyrazolediazenylpyrimidines (58) caused a substantial enhancement in anti-inflammatory activity. Similarly, the introduction of the carboxamide group at site-4 of pyrazolone (59) also contributed to the activity enhancement. Chemical structures of compounds (58 and 59) are shown in (Fig. 14).89
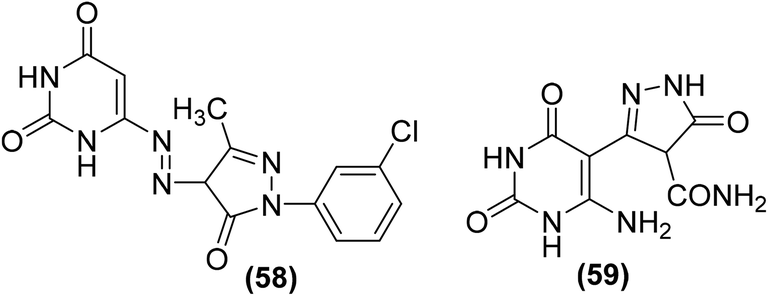 | ||
| Fig. 14 Chemical structures of pyrazolediazenylpyrimidine derivative (58) and pyrazolone derivative (59). | ||
Several pyrazolo[3,4-d] pyrimidine analogs were synthesized and investigated for their anti-inflammatory effects and selective COX-2 inhibitory potential via carrageenan-induced rat's paw edema and PGE2 EIA kit, respectively. Experimental data revealed that compounds (60–63) (Fig. 15) possessed better anti-inflammatory effects than the reference drug diclofenac. The four analogs were also observed to be the strongest COX-2 inhibitors. The underlying mechanism indicated that the selected candidates caused strong PGE2 suppression at the 2.5 and 5 mg kg−1 p.o. dose levels. The anti-inflammatory activities of the compounds (60–63) in terms of their (%) inhibitions of oedema and PGE2 (at the two dose levels) are given in Table 5.90
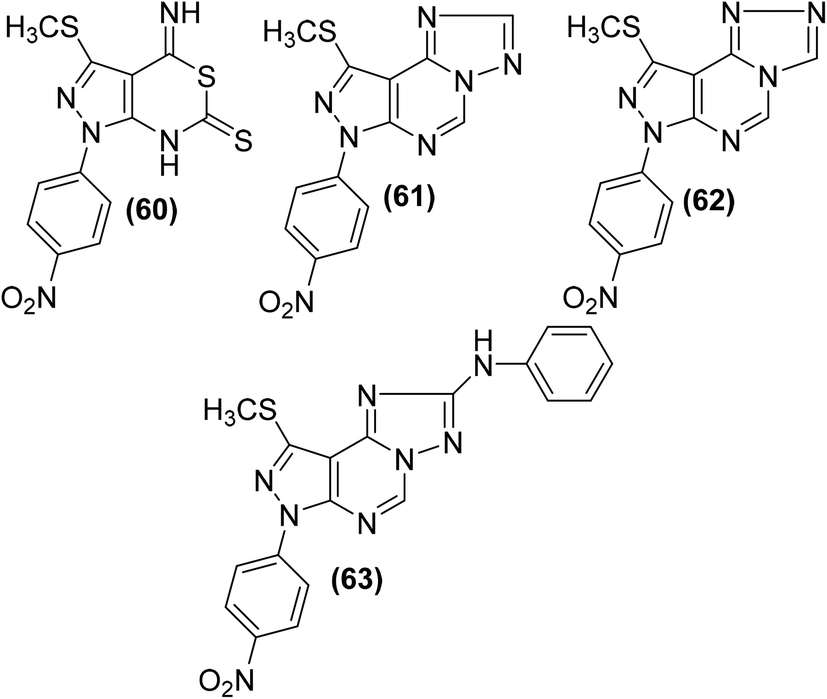 | ||
| Fig. 15 Chemical structures of selected pyrazolo [3,4-d] pyrimidines (60–63). | ||
| Compound no. | Anti-inflammatory activity | ||
|---|---|---|---|
| Dose, mg kg−1 p.o. | % inhibition of oedema | % inhibition of PGE2 | |
| 60 | 2.5 | 80.55 | 90.98 |
| 5 | 89.35 | 96.89 | |
| 61 | 2.5 | 85.32 | 83.98 |
| 5 | 91.06 | 90.00 | |
| 62 | 2.5 | 82.98 | 77.98 |
| 5 | 87.98 | 86.00 | |
| 63 | 2.5 | 94.20 | 85.31 |
| 5 | 95.88 | 92.09 | |
| Diclofenac potassium | 2.5 | 81.09 | 75.89 |
| 5 | 85.44 | 84.87 | |
Bakr et al. reported the synthesis of several new 1-phenylpyrazolo[3,4-d]pyrimidine analogs. The target compounds were screened for their COXs enzymes inhibitory effects and anti-inflammatory activities via an EIA kit and in vivo carrageenan-induced paw edema method in rats, respectively. Preliminary data indicated that all the tested analogs displayed stronger inhibitory effects versus COX-2 enzyme (IC50 = 0.56–5.89 μM) than COX-1 enzyme (IC50 = 3.97–10.11 μM). SAR study revealed that the derivatives carrying a pyrazolyl scaffold in a hybrid configuration with the pyrazolo[3,4-d]pyrimidine moiety (64–69) were typically stronger inhibitors of COX-2 enzyme than the compounds having other scaffolds. Moreover, the six hybrids were also noticed to be more COX-2 selective. The 4,5-dimethylpyrazole analog (69) was observed to be the strongest COX-2 inhibitor (IC50 = 0.56 μM), whereas the 5-aminopyrazole analog (65) was noticed to be the most COX-2 selective (SI = 11.99). Moreover, all the six analogs containing pyrazolyl moiety (64–69) and the acetohydrazide analog (70) exhibited noteworthy anti-inflammatory effects (ED50 = 87.9–10.1 μmol kg−1). The 5-aminopyrazole analog (66) was noticed to be stronger (ED50 = 87.9 μmol kg−1) anti-inflammatory agent than reference drug celecoxib (ED50 = 91.9 μmol kg−1). Chemical structures of compounds (64–70) are shown in (Fig. 16).91
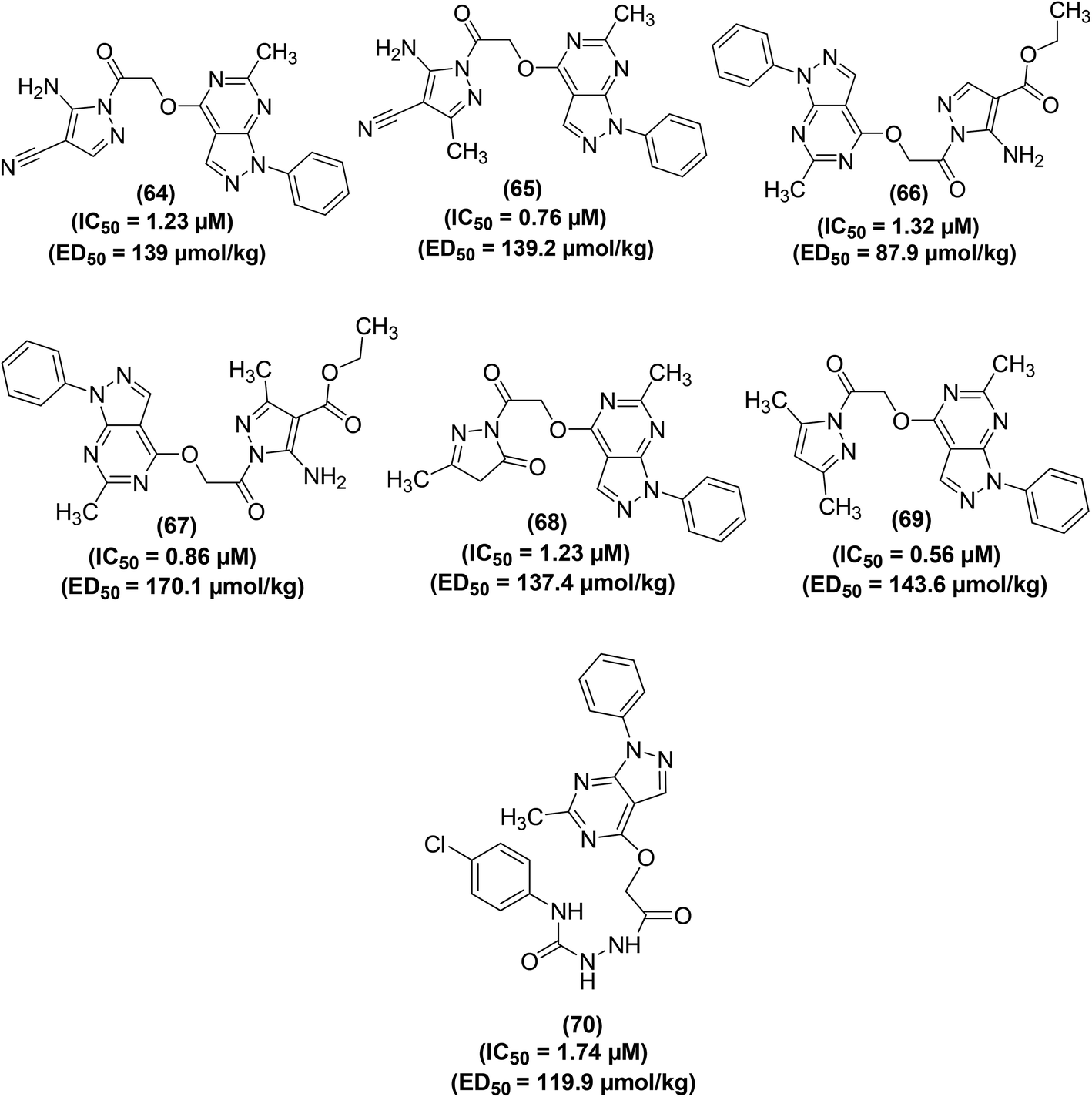 | ||
| Fig. 16 Chemical structures of selected 1-phenylpyrazolo[3,4-d]pyrimidine analogs (64–70). | ||
Beswick et al. applied a biotransformation methodology to identify and optimize a new set of pyrimidine-based COX-2 inhibitors. They used a biotransformation strategy together with conventional synthetic chemistry to get efficient access to a set of vital target compounds. In vivo anti-inflammatory experimental results indicated that the two isomeric benzyl pyrimidine analogs (71 and 72) and derivative (73) potently inhibited the activity of COX-2 enzyme. The IC50 values of compounds (71–73) were reported to be 0.104 μM, 0.114 μM, and 0.057 μM, respectively. SAR studies revealed that the replacement of the benzyl moiety with hydrophobic functional groups in benzyl pyrimidines was well-suited for anti-inflammatory enhancement. Elimination of the benzyl moiety and substitution with hydrogen or small-sized alkyl groups caused activity loss. Similarly, an increase in the length of the linker also contributed to decreased activity. Additionally, the substitution of the methylene linking group with a nitrogen linker caused activity enhancement, whereas sulfur and oxygen linkers led to activity loss. Chemical structures of compounds (71–73) are shown in (Fig. 17).92
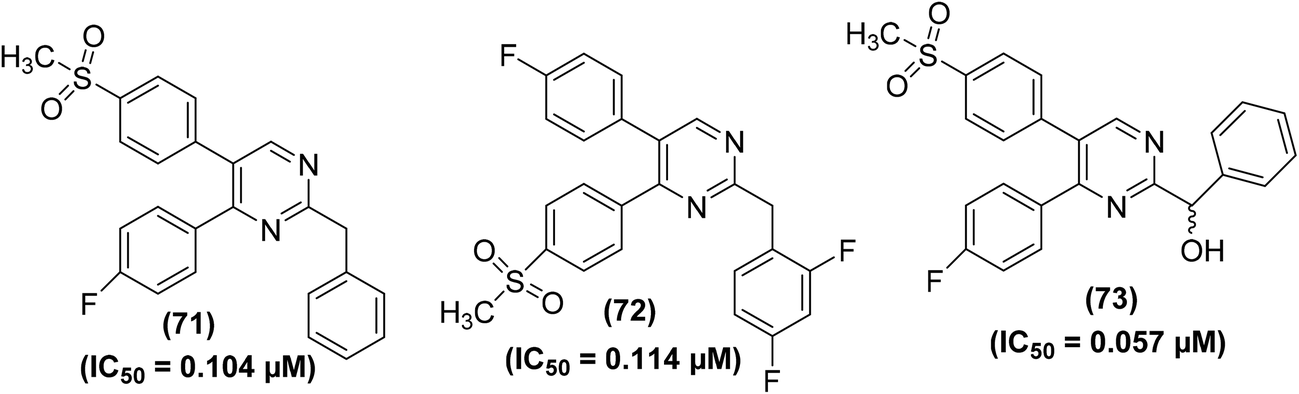 | ||
| Fig. 17 Chemical structures of compounds (71–73). | ||
Synthesis of several bicyclic pyrazolo[1,5-a]pyrimidines for their potential application as selective COX-2 inhibitors has been depicted. The target compounds were examined for their COXs inhibitory action both by in vivo (air-pouch model and carrageenan-induced paw edema) and in vitro (COXs suppression in human whole blood) analyses. SAR analysis of the target compounds revealed that 6,7-arrangement gave the best activity, and the existence of a nitrogen atom at site 4 of the core pyrazolo[1,5-a]pyrimidine was useful for selective inhibition of COX-2 activity. Among all the tested compounds, the 6,7-dimethylated analog (74) (Fig. 18) was noticed to be the strongest and most selective inhibitor of COX-2 enzyme. The in vitro (%) inhibitions of compound (74) versus COX-2 activity in human whole blood (HWB) were noted to be 96.4 and 72.8 at 10 μM and 1 μM doses, respectively. From in vivo analysis, the IC50 value of compound (74) was calculated to be 0.012 μM.93
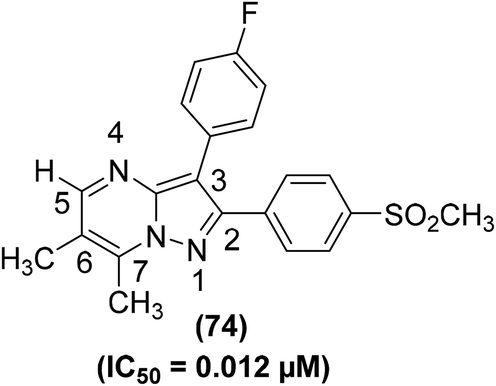 | ||
| Fig. 18 Chemical structure of 6,7-dimethylated analog of bicyclic pyrazolo[1,5-a]pyrimidine (74). | ||
The synthesis of a series of pyrimidine and thiopyrimidine heterocyclic analogs (75–79) (Fig. SI-3†) has been illustrated. The target pyrimidines were subsequently investigated for their COX-2 inhibitory effects and anti-inflammatory activities via in vitro enzymatic analysis and in vivo cotton pellet-stimulated granuloma bio-analysis, respectively. The results demonstrated that the tested analogs (75–79) potently suppressed the COX-2 activity with their IC50 values measured to be 0.25 ± 0.0005, 0.14 ± 0.0002, 0.12 ± 0.0001, 0.17 ± 0.0003, and 0.20 ± 0.0005 μM, respectively as compared to the potency of commonly used NSAIDs indomethacin (IC50 = 2.63 ± 0.0005 μM) and celecoxib (IC50 = 0.30 ± 0.0004 μM). Moreover, the five derivatives (75–79) also showed noteworthy anti-inflammatory activities with their ED50 values calculated to be 1.81 ± 0.0002 μM, 1.61 ± 0.0001 μM, 1.56 ± 0.0001 μM, 1.67 ± 0.0001 μM, and 1.71 ± 0.0002 μM, respectively as compared to indomethacin (ED50 = 9.568 ± 0.00078 μM). All the investigated derivatives demonstrated better GI safety outlines when measured against indomethacin. SAR analysis revealed that derivatives containing cycloheptenes fused to pyrimidine ring (76 and 77) showed better COX-2 inhibition and anti-inflammatory effects than the derivatives containing cyclohexenes fused to pyrimidine ring (78 and 79). The introduction of chlorine and several other small molecular fragments on the core thiopyrimidine caused a decrease in the anti-inflammatory activity.94
The synthesis and COX-2 inhibitory activities of several 2-(4-sulfamoylphenyl) and 2-(4-methylsulfonylphenyl) pyrimidines were reported by Orjales and coworkers. The purified enzyme (PE) and HWB analyses were performed to measure the COX-1 and COX-2 inhibitory effects of the target analogs, respectively. Experimental data indicated that numerous analogs potently inhibited (IC50 = 0.0024–0.003 μM) the COX-2 activity. Moreover, they also exhibited high selectivity for the inhibition of COX-2 activity (80- to 780-times greater selectivity than rofecoxib). Among all the tested pyrimidines, compound (80) (Fig. 19) was noticed to be one of the most efficient and selective COX-2 inhibitors with its IC50 value of 0.0012 μM and HWB SI value of 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 (780-times greater than rofecoxib). Detailed SAR investigations of the diarylheterocycle group indicated that the introduction of p-sulfonamide or p-methylsulfone on one of the aromatic rings is needed for maximum COX-2 inhibition and selectivity. The polar moieties were recognized to encourage COX-2 selectivity by plugging into its auxiliary sack connecting spot that is missing in COX-1 enzyme.95
300 (780-times greater than rofecoxib). Detailed SAR investigations of the diarylheterocycle group indicated that the introduction of p-sulfonamide or p-methylsulfone on one of the aromatic rings is needed for maximum COX-2 inhibition and selectivity. The polar moieties were recognized to encourage COX-2 selectivity by plugging into its auxiliary sack connecting spot that is missing in COX-1 enzyme.95
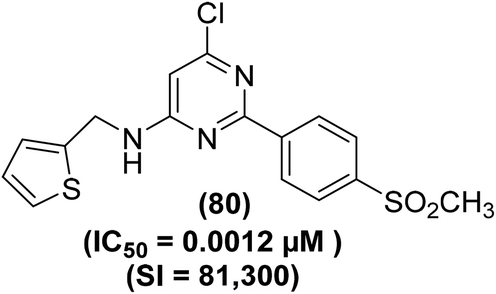 | ||
| Fig. 19 Chemical structure of a selected 2-(4-methylsulfonylphenyl) pyrimidine (80). | ||
Abbas and coworkers described the synthesis of quinazolinone–pyrimidine hybrids for their potential application as COX inhibitors and anti-inflammatory candidates. Carrageenan-induced rat paw oedema model and COX inhibitor screening analysis kit were applied to measure the anti-inflammatory effect and COX-2/COX-1 selectivity assay respectively. Among the target derivatives, five compounds (81–85) (Fig. 20) exhibited efficacy above 90% as compared to the reference drug diclofenac-sodium. Particularly, compounds (83 and 84) were noticed to be equipotent to diclofenac-sodium. The IC50 values of compounds (81–85) were recorded to be 143.26 μmol kg−1, 130.44 μmol kg−1, 114.03 μmol kg−1, 83.36 μmol kg−1, and 81.31 μmol kg−1, respectively as compared to diclofenac-sodium (IC50 = 141.03 μmol kg−1). Moreover, the COX-2/COX-1 proportion of the five hybrids revealed that three compounds (82–84) exhibit selectivity towards COX-2 inhibition having a ratio of 0.62, 0.40, and 0.44, respectively. SAR investigations indicated that the quinazolinone–pyrimidines carrying an anilino moiety at position-4 of the pyrimidine ring (83–85) were the most potent hybrids.96
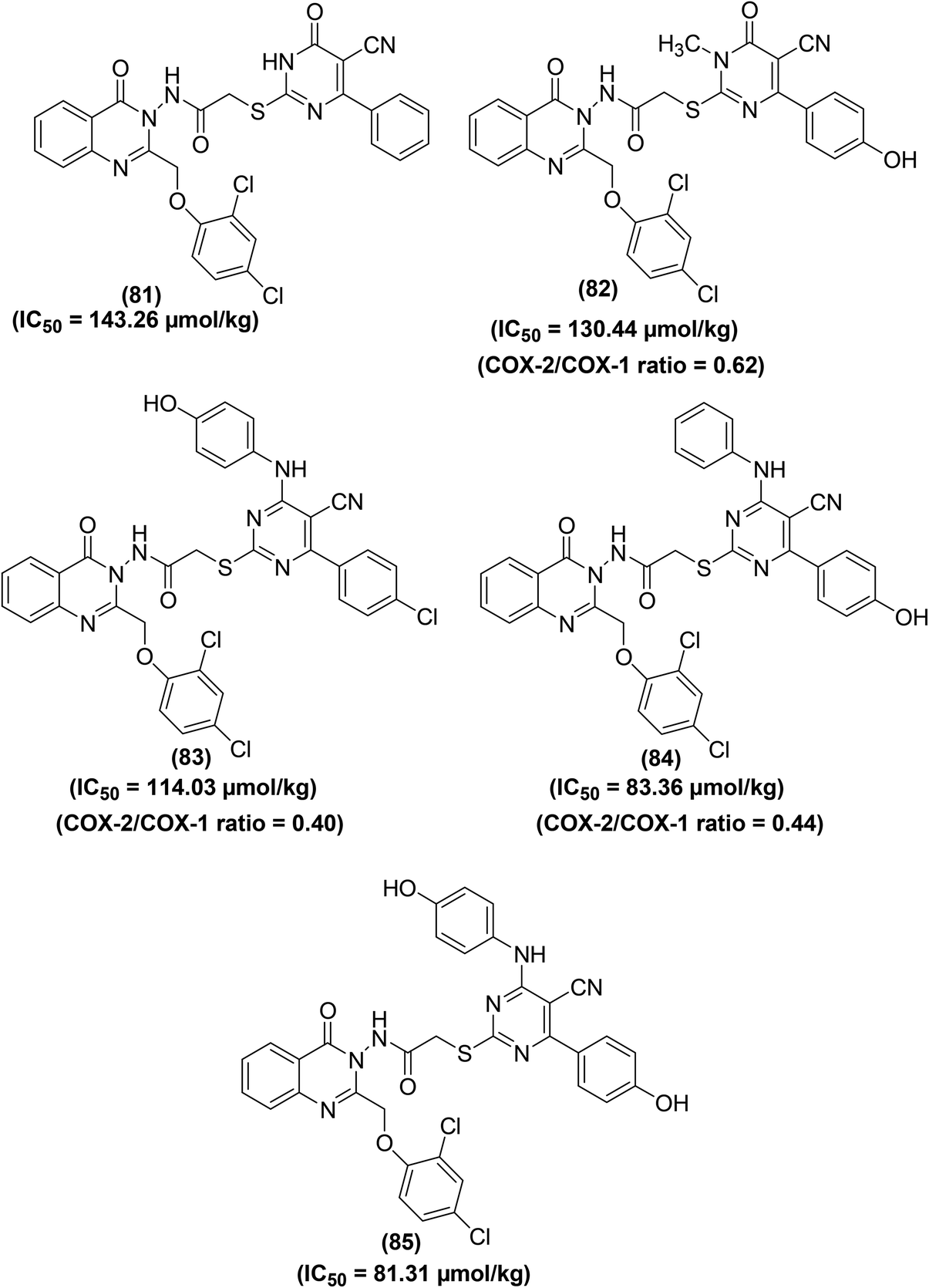 | ||
| Fig. 20 Chemical structures of selected quinazolinone–pyrimidine hybrids (81–85). | ||
Bukhari et al. synthesized several pyrimidine analogs and studied their inhibitory effects versus the activities of COX-1 and COX-2 enzymes via COX inhibition assay. Two compounds (86 and 87) (Fig. 21) were noticed to be the most potent pyrimidine derivatives against COX-2 enzyme with their (%) inhibition calculated to be 58.74 and 74.99, respectively as compared to the positive control aspirin [COX-2 (%) inhibition = 34.71%]. SAR investigations revealed that pyrimidine derivatives carrying methoxy and nitro moieties exhibit better COX-2 inhibitory potential.97
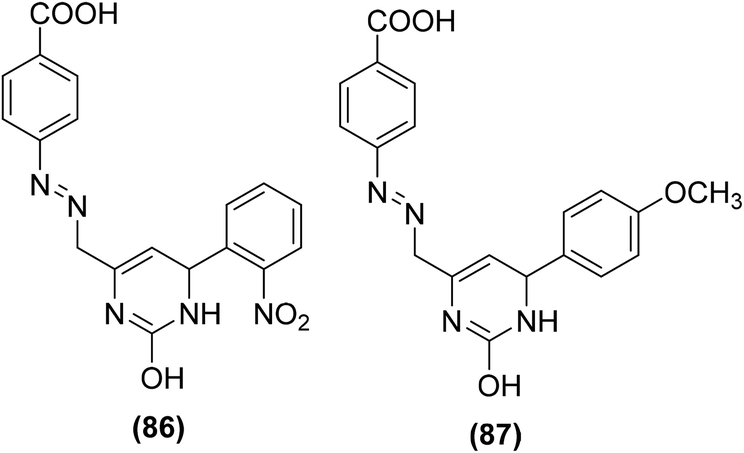 | ||
| Fig. 21 Chemical structures of selected pyrimidine analogs (86 and 87). | ||
Synthesis of numerous thieno[2,3-d]pyrimidines has also been reported in the literature. The target compounds were examined for their anti-inflammatory effects and PGE2 suppression through in vivo carrageenan-induced paw edema model and PGE2 rat specific enzyme-linked immunosorbent assay (ELISA) kit, respectively. Experimental data revealed that all the tested derivatives reduced the carrageenan-induced paw edema similar to diclofenac sodium. Furthermore, they also reduced the PGE2 concentration in blood serum. Thienopyrimidine (88) (Fig. 22) displayed the strongest in vivo anti-inflammatory effect with the protection of 35%, 36%, and 42% versus carrageenan-induced paw edema after 1 hour, 2 hours, and 3 hours, demonstrating 92%, 86%, and 88%, respectively of diclofenac efficacy. It also reduced the PGE2 concentration in blood serum to 19 pg mL−1 that is similar to diclofenac (PGE2 concentration = 12 pg mL−1).98
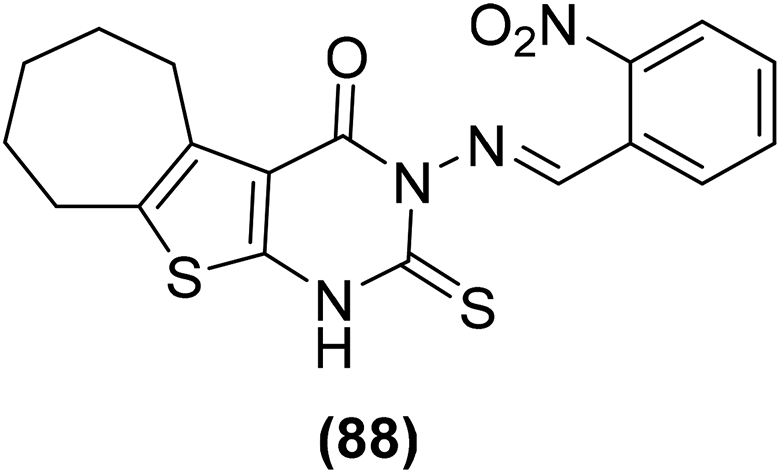 | ||
| Fig. 22 Chemical structure of a selected thienopyrimidine derivative (88). | ||
Synthesis of several pyrido[1,2-c]pyrimidines containing a sulfur, oxygen, or nitrogen functionality at C-1 (Fig. 23) was reported on solid-phase via the iminophosphorane procedure. The anti-inflammatory potential of the synthesized compounds was evaluated via the carrageenan paw edema analysis in mice. The results revealed that four derivatives (89–92) potently suppressed the production of PGE2 from COX-2 in RAW 264.7 macrophages activated with lipopolysaccharide (LPS). The mechanism of action of the target compounds (89–92) was attributed to the suppression of PGE2 generation and superoxide scavenging.99
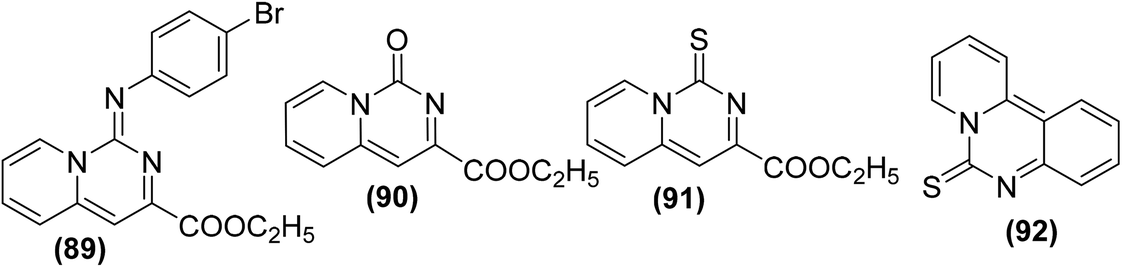 | ||
| Fig. 23 Chemical structures of selected pyrido[1,2-c]pyrimidine derivatives (89–92). | ||
Tietz and coworkers reported the synthesis of some new and selective pyrimidine-based fluorescent COX-2 inhibitors. In vitro COX-1/COX-2 inhibition investigations revealed that all derivatives were selective inhibitors of COX-2. However, compound (93) (Fig. 24) was identified as the most potent (IC50 = 1.8 μM) and a selective COX-2 inhibitor. Furthermore, compound (93) was tested for fluorescent COX-2 imaging in human colon cancer cell lines. The results indicated that compound (93) was able to label the COX-2 enzyme in human colon cancer cells.100
 | ||
| Fig. 24 Chemical structure of a selective pyrimidine-based fluorescent COX-2 inhibitor (93). | ||
Synthesis of several trifluoromethyl-substituted pyrimidine derivatives as selective COX-2 inhibitors has been reported. In vitro COX enzyme inhibition investigations indicated that most of the synthesized derivatives possessed high potency and selectivity as compared to the internal reference celecoxib. The lead compound (94) (Fig. 25) exhibited remarkable suppressive potency (IC50 = 7 nM) against the activity of COX-2 enzyme and did not display inhibitory potential against COX-1 in the dose range tested. Docking studies were carried out to discover the probable interaction of compound (94) with the active spot of COX enzymes. Promising inhibitory potency of compound (94) proposes a satisfactory orientation inside the COX-2 binding position. The SO2CH3 functionality in compound (94) entirely penetrates the secondary pocket zone of the COX-2 active position, where it is directed to R513, Q192, A516, and H90 residues. One of the oxygen atoms of the SO2CH3 functionality forms hydrogen bonding with the nitrogen atom of H90. The second oxygen atom shows hydrogen bonding with the nitrogen atom of Q192 residue. Furthermore, the –C6H5F functionality is positioned in the neighborhood of A527, V349, and R120 residues. On the contrary, docking investigations of compound (94) with COX-1 enzyme showed that compound (94) was unable to penetrate fully inside the COX-1 active position. SAR studies indicated that trifluoromethyl-substituted pyrimidines with large substituents (tert-butyl and phenyl groups) at the para position of the benzyl ring did not exhibit considerable COX-2 inhibitory potential. On the other hand, trifluoromethyl-substituted pyrimidines with electron-releasing (–CH3, –OCH3) or electron-withdrawing (–Cl, –F, –Br, –NO2, –CF3) groups at the para position of the benzyl ring exhibited considerable COX-2 inhibitory effects.101
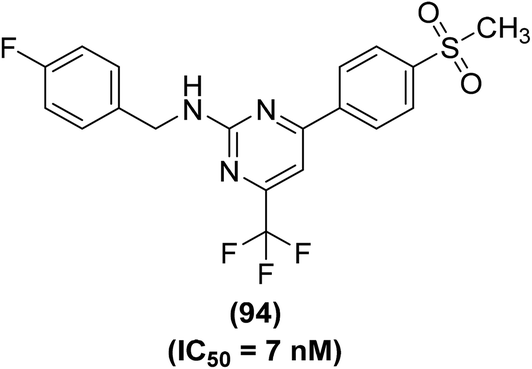 | ||
| Fig. 25 Chemical structure of a selected trifluoromethyl-substituted pyrimidine (94). | ||
Lokwani and coworkers designed and synthesized ten novel COX inhibitors by transforming the substitution model around 1,4-dihydropyrimidine skeleton. Docking studies revealed that the target compounds must have a ‘V’ shaped structure to bind impeccably in the active position of the COX-2 enzyme. Moreover, they must also possess an electronegative moiety to bind to the electropositive amino acid in the active position of the COX-1 enzyme. The synthesized derivatives were screened in vitro for their inhibitory effects versus COX-1 and COX-2 enzymes The results indicated that compound (95) (Fig. 26) potently suppressed both COX-1 and COX-2 enzymes with its inhibitory effects of 70.08% and 35.12%, respectively. The IC50 value of compound (95) against COX-1 enzyme was calculated to 254.82 μM.102
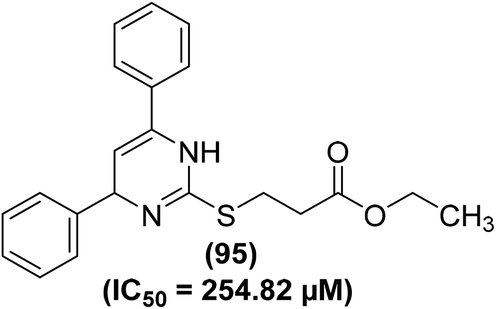 | ||
| Fig. 26 Chemical structure of compound (95). | ||
Abdelall et al. synthesized numerous pyrazolo [3,4-d]pyrimidines for their potential application as anti-inflammatory agents. Docking investigations were carried out to study the mode of action of the synthesized derivatives within the COX-2 active position. It was observed that the existence of an extra lateral pocket on COX-2 active position enhances its size to fit the large structures. The pocket permits further interaction with Arg513 that was substituted by His513 in COX-1. The ligand SC-558 (bromocelecoxib) attached to the COX-2 active position by making two hydrogen bonds between amino acids (Arg513 and His90) and –SO2CH3 moieties at a distance of 2.47 and 2.35 Å, respectively. Furthermore, the energy score was calculated to be −13.39 kcal mol−1. The anti-inflammatory effect of the target derivatives was evaluated by in vitro COX inhibition assay with reference to the standard drugs (indomethacin, diclofenac sodium, and celecoxib). The data confirmed that all the investigated derivatives had a more potent inhibitory effect against COX-2 enzyme (IC50 = 0.10–0.38 μM) than against COX-1 isoform (IC50 = 5.28–13.11 μM). Compounds (96–100) (Fig. 27) were noted to be the most potent (IC50 = 0.10–0.11 μM) against the activity of COX-2 enzymes as compared to celecoxib (IC50 = 0.049 μM). The COX-2 SI values (14.84–131.10) for all the investigated derivatives were noted to be greater than that of both indomethacin (SI = 0.080) and diclofenac sodium (SI = 4.52).103
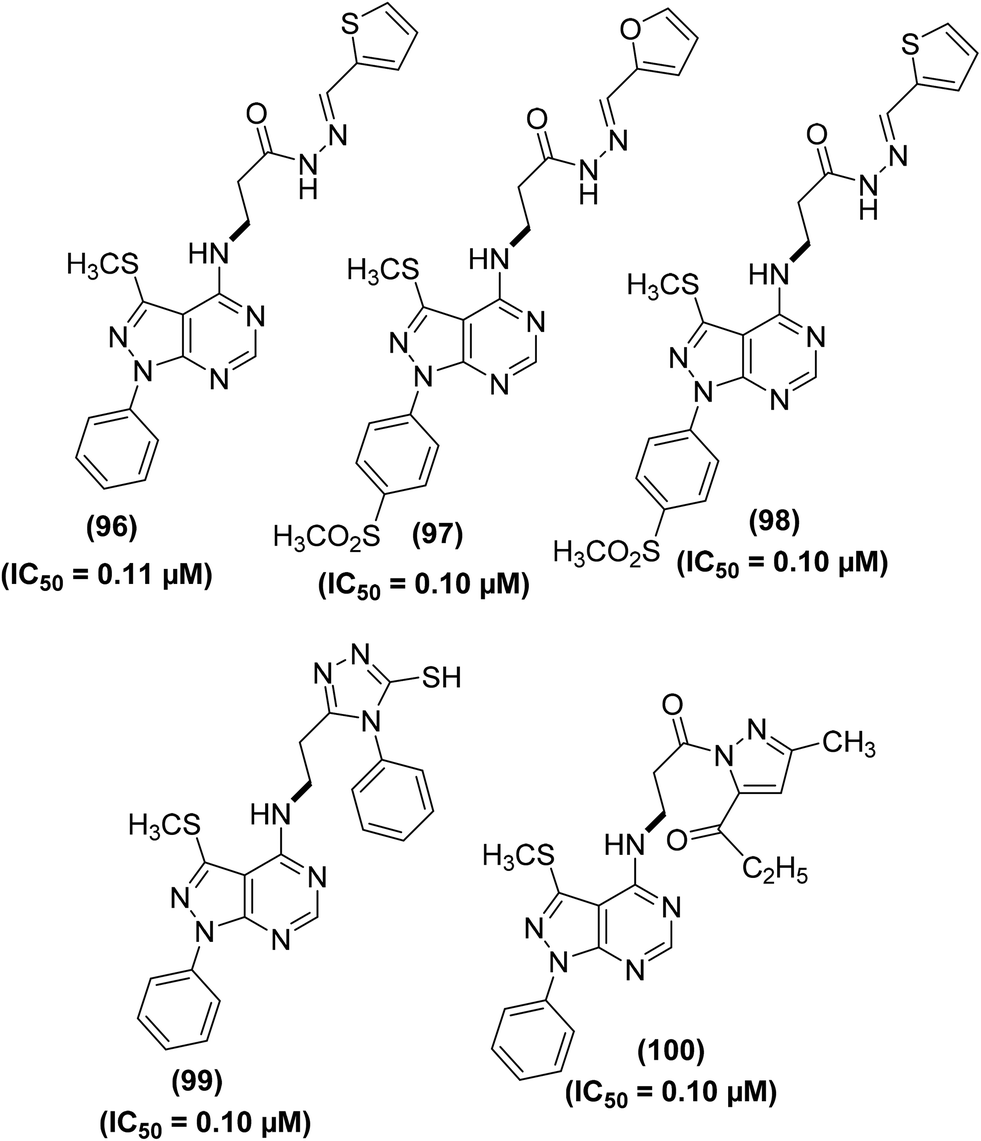 | ||
| Fig. 27 Chemical structures of selected pyrazolo [3,4-d]pyrimidines (96–100). | ||
Synthesis of novel 4-phenylpyrimidine-2(1H)-thiones has been reported for their potential use as COX-1 and COX-2 inhibitors. All the target derivatives were screened via in vitro COX inhibition assay for their inhibitory effects against the activities of both COX-1 and COX-2 enzymes. Preliminary results indicated that the 4-methoxy derivative (101) and the 4-nitro substituted analog (102) suppressed the activity of COX-1 enzyme only with their (%) inhibition calculated to be 33.23 and 29.81, respectively. However, the 2-nitro analog (103) exhibited noteworthy suppression against both cyclooxygenases. The activity of the compound (103) in terms of (%) inhibition against COX-1 and COX-2 enzymes was calculated to be 53.00 and 51.00, respectively. The binding mechanism of compounds (101–103) was studied by performing their docking into the binding position of COX enzymes. The results indicated that all derivatives formed H-bonds with Ser503 and Tyr385. Compounds (101 and 102) were also expected to interact with Arg120. However, only the derivative (103) made an extra H-bond with Tyr355. The rest of the derivatives did not show such an interaction, as they lacked H-bond acceptor substitution on the aromatic ring. Chemical structures of derivatives (101–103) are shown in (Fig. 28).104
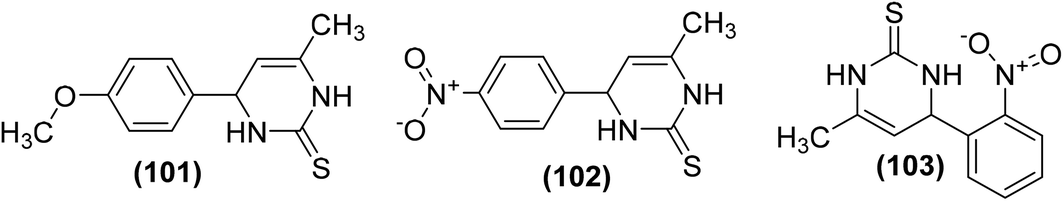 | ||
| Fig. 28 Chemical structures of 4-phenylpyrimidine-2(1H)-thiones (101–103). | ||
Kalčic et al. reported the synthesis of 28 new polysubstituted pyrimidines for their potential role as inhibitors against PGE2 generation and COX activity. An extensive SAR study was performed to discover the most potent inhibitors against COX activity and PGE2 production. The results revealed that compound (104) with the 4-(4-benzyloxy)phenyl moiety at C-4 position of pyrimidine exhibited strong inhibitory activity against the PGE2 production. Moreover, compound (105), the difluorinated derivative of compound (104), significantly inhibited the activity of COX-2. At a concentration of 20 μM, compound (105) did not suppress the COX-1 activity, however, it substantially (by 47%, P < 0.001) inhibited the COX-2 activity. Other pyrimidines investigated did not exhibit inhibitory effects against COX-1/2 enzymes. Additionally, the two most potent compounds (104 and 105) were observed to be noticeably efficient in vivo in a model of acute inflammation, inhibiting the carrageenan-induced rat paw edema by 36% and 46%, respectively. Chemical structures of compounds (104 and 105) are given in (Fig. 29).105
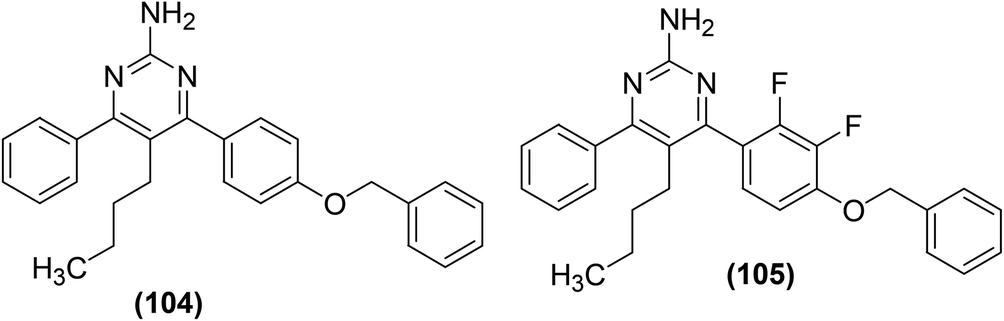 | ||
| Fig. 29 Chemical structures of selected polysubstituted pyrimidines (104 and 105). | ||
Synthesis and anti-inflammatory activities of several new pyrimidin-2-thione derivatives have been reported. COX-inhibitory effects of the synthesized compounds were measured by Cayman colorimetric COX (ovine) inhibitor screening assay kit. Whereas, the anti-inflammatory effect was evaluated employing the carrageenan-induced rat paw edema assay as compared to the reference drug (ibuprofen). The results showed that the majority of the target pyrimidine derivatives exhibited potent anti-inflammatory activity (61 to 86%) as compared to ibuprofen (69%). The results further revealed that compounds (106–108) (Fig. 30) possessed greater potency to COX-2 over COX-1. Derivative (106) showed greater potency towards COX-2 (IC50 = 0.046 μM) than derivatives (107) (IC50 = 0.21 μM), and (108) (IC50 = 0.11 μM) in addition to ibuprofen (IC50 = 43.628 μM). SAR analysis revealed that the existence of acetyl moiety at para-position in the side chain at C-5 of derivatives (106 and 108) contributed to greater anti-inflammatory activity. The acetyl moiety in derivatives (106 and 108) enhanced their binding interaction to COX-2 enzyme via hydrogen bonding. Docking investigations of derivatives (106–108) into the active position of COX-2 were performed by SYBLYL-X v.2.1 software. The results revealed that the tested derivatives (106–108) exhibited appropriate fitting to the active site of COX-2, with their scoring bond energies of 6.0459, 5.5331, and −3.6329 kcal mol−1, respectively in comparison to 9.0092 kcal mol−1 for ibuprofen. The binding mode of compound (107) into the active site of COX-2 did not show any hydrogen bonding. Therefore, its binding mode was explained via extensive van der Waals interaction with several hydrophobic residues. The binding modes of both compounds (106 and 108) into the active site of COX-2 showed 2 hydrogen bonds with the guanido side chain of Arg514. Overall, the docking investigations confirmed a good agreement with anti-inflammatory findings.106
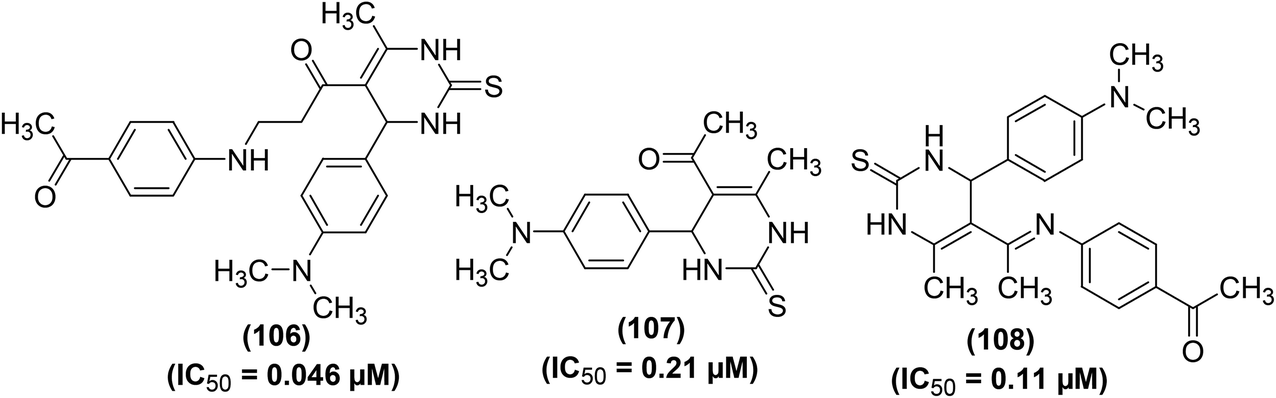 | ||
| Fig. 30 Chemical structures of selected pyrimidin-2-thione derivatives (106–108). | ||
Synthesis, COX-2 inhibitory potential, and anti-inflammatory effects of eighteen novel 6-(4-fluorophenyl)-pyrimidine-5-carbonitrile derivatives were reported. In vivo anti-inflammatory activity and COX-2 inhibitory potential of the target compounds were investigated via carrageenan-induced rat paw edema model and EIA kit, respectively. Two derivatives (109 and 110) (Fig. 31) showed significant anti-inflammatory activity and COX-2 inhibitory effect. Inhibition to the inflammation for the two compounds (109 and 110) was recorded to be 87% and 74%, respectively as compared to the standard drugs, celecoxib (52%) and ibuprofen (78%) after 3 h. Compound (109) was found to be the most potent with 79% and 87% suppression after 2 h and 3 h, respectively. COX-2 inhibitory results indicated that derivatives (109 and 110) had selective COX-2 inhibitory potential with their SI values of 105 and 85, respectively as compared to the standard drug, celecoxib (SI = 96). SAR analysis indicated that derivatives containing aromatic rings exhibited superior activity as compared to those bearing non-aromatic rings. Additionally, the anti-inflammatory activity decreased with the increase in the length of the side chain e.g., derivatives with isobutyl side chain were more potent than those with isopentyl side chain. Molecular docking studies revealed that most of the derivatives exhibited decent interactions with the COX-2 protein skeleton. The most potent compound (109) showed the best docking score of −8.34 kcal mol−1. Docking results further indicated that compound (109) interacts at the binding crack of COX-2 via polar interactions with Ser 353, Ser 530, Gln 192, and His 90 amino acid residuals of the enzyme.107
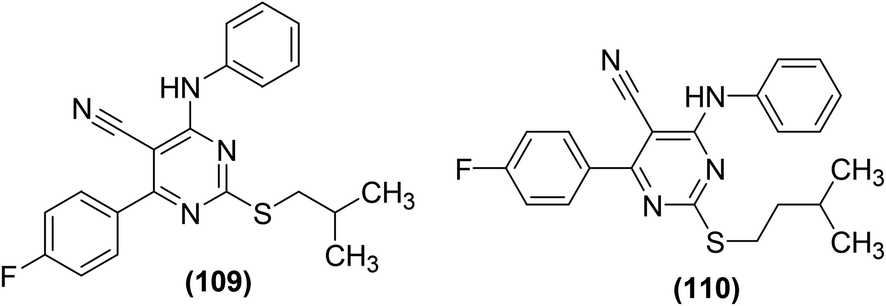 | ||
| Fig. 31 Chemical structures of selected 6-(4-fluorophenyl)-pyrimidine-5-carbonitrile derivatives (109 and 110). | ||
The potency of the selected pyrimidine derivatives versus the target enzyme (COX-1/COX-2) is provided in Table SI-1.†
4.2. Inhibition of nitric oxide (NO) generation
NO is a vital intercellular mediator that promotes inflammation. Three kinds of NOs have been recognized namely, endothelium nitric oxide synthase (eNOS), neural nitric oxide synthase (nNOS), and inducible nitric oxide synthase (iNOS) in different mammalian cells.108–110 Excessive NO secretion results in tissue damage and has been known to cause various types of acute and chronic inflammations. For example, excessive NO production by stimulated macrophages has been noticed in rheumatoid arthritis. Therefore, efforts were made in the recent past to develop novel compounds as possible inhibitors against NO production in the direction of treatment for inflammatory disorders.1,111 Several research groups also investigated pyrimidines as possible inhibitors against NO production.Bluhm and coworkers synthesized and evaluated several novel 3-aroylpyrido[1,2-a]pyrimidine derivatives (111–117) (Fig. SI-4†) as potential NOS inhibitors. Griess reagent kit was utilized to investigate the inhibitory effects of the synthesized derivatives versus the activity of NOS. Preliminary results indicated that some of the pyrido[1,2-a]pyrimidines possessed favorable inhibitory activities with moderate selectivity for one isoform of NOS. Few of them were observed to be as active as the known inhibitors of NOS namely, Nω-nitro-L-arginine (L-NNA), N-(3-(aminomethyl)benzyl)acetamidine, and S-ethyliso-thiourea (SEITU). However, no obvious specificity for any isoform was noted via this assay. Therefore, a more sensitive radioactive assay was executed to evaluate the inhibitory activities of a small number of derivatives. Experimental results indicated that all the investigated compounds more potently suppressed iNOS as compared to nNOS or eNOS. Moreover, nNOS was partially suppressed by 100 μM of four derivatives (111–114). The eNOS was most potently inhibited by three derivatives (113–115) (all at 100 μM). Compounds (112 and 114) were noticed to be more potent inhibitors than 7-nitro-1H-indazole (7-NI) versus nNOS and eNOS, respectively. Similarly, all the six pyrido[1,2-a]pyrimidines (112–117) potently suppressed the iNOS, with derivatives (114 and 115) being noticed to be stronger inhibitors than L-NNA under the same experimental conditions. The IC50 values of the tested derivatives were calculated to be 10–100 μM with reference to L-NNA, and 7-NI. Experiments were carried out to better understand the mode of action of the synthesized pyrimido[1,2-a]pyrimidines. The results revealed that the suppression of iNOS by few of the target derivatives was competitive with the L-arginine (substrate) and reversible. SAR analysis indicated that derivatives carrying a benzyloxybenzoyl, biphenyloyl or naphthoyl moiety exhibited the most potent inhibitory activities that were further enhanced by the incorporation of a methyl moiety on position-8 of the pyrido[1,2-a]pyrimidine scaffold.112
Synthesis of numerous 5-substituted 2-amino-4,6-dihydroxypyrimidine derivatives was reported for their possible inhibitory effects against immune-induced nitric oxide generation. The target compounds were evaluated in mouse peritoneal cells via the in vitro NO analysis. Regardless of the substitution pattern at position-5, 2-amino-4,6-dichloropyrimidine derivatives suppressed the immune-induced NO generation. Compound (118) (Fig. 32) carrying a fluorine atom at position-5 of the pyrimidine skeleton was noticed to be the most potent inhibitor against the production of immune-stimulated NO. The potency (IC50 = 2 μM) of compound (118) was observed to be greater than the most potent standard compound (N-[3-(aminomethyl)-benzyl]acetamide). The IC50 values for the rest of the derivatives in the series were calculated to be 9–36 μM. Experimental results further revealed that 2-amino-4,6-dihydroxypyrimidine analogs did not exhibit any NO-inhibitory effects. Additionally, they had no suppressive effects on the survival of the cells.113
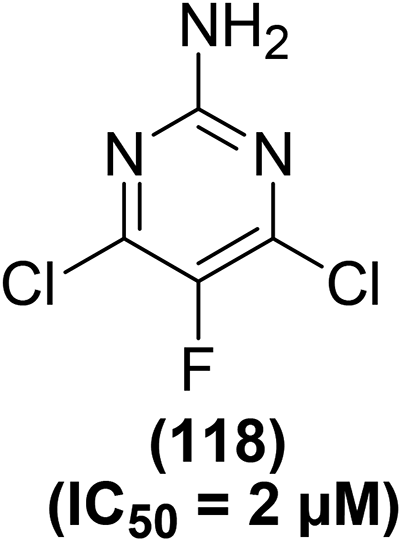 | ||
| Fig. 32 Chemical structure of a selected 5-substituted 2-amino-4,6-dihydroxypyrimidine (118). | ||
Numerous polysubstituted pyrimidines carrying several substituents (–H, –CH3, –C2H5, –C3H7, –CH(CH3)2, –CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, –CH2–HCCH2, –C4H9, –CH(CH3)–CH2–CH3, –C6H5, –CH2–C6H5, and –F) at position-5 of the ring were studied for their potential to suppress the immune-induced NO production. The inhibitory effects of such pyrimidines were evaluated against the in vitro generation of immune-stimulated NO in mouse peritoneal cells. The results revealed that all of the 5-substituted 4,6-dichloro-2-[(N,N-dimethylamino)methyleneamino]pyrimidine derivatives potently suppressed NO generation. The IC50 values for most of the derivatives were recorded to be less than 5 μM. Maximum suppression (IC50 = 2.57 μM) to NO production was detected for the derivative bearing 5-sec-butyl moiety (119), whereas minimum inhibition (IC50 = 11.49 μM) was caused by compound (120). Chemical structures of the two compounds (119 and 120) are given in (Fig. 33).114
CH, –CH2–HCCH2, –C4H9, –CH(CH3)–CH2–CH3, –C6H5, –CH2–C6H5, and –F) at position-5 of the ring were studied for their potential to suppress the immune-induced NO production. The inhibitory effects of such pyrimidines were evaluated against the in vitro generation of immune-stimulated NO in mouse peritoneal cells. The results revealed that all of the 5-substituted 4,6-dichloro-2-[(N,N-dimethylamino)methyleneamino]pyrimidine derivatives potently suppressed NO generation. The IC50 values for most of the derivatives were recorded to be less than 5 μM. Maximum suppression (IC50 = 2.57 μM) to NO production was detected for the derivative bearing 5-sec-butyl moiety (119), whereas minimum inhibition (IC50 = 11.49 μM) was caused by compound (120). Chemical structures of the two compounds (119 and 120) are given in (Fig. 33).114
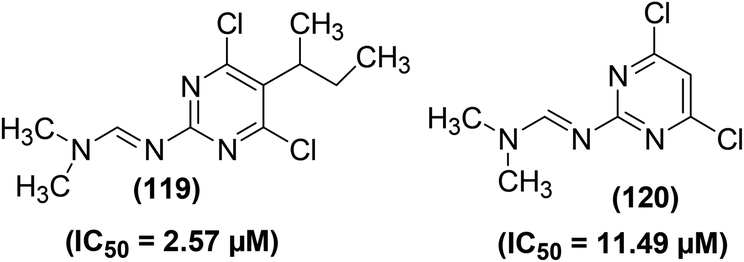 | ||
| Fig. 33 Chemical structures of selected polysubstituted pyrimidines (119 and 120). | ||
Zídek and coworkers reported the inhibitory actions of polysubstituted 2-aminopyrimidine derivatives (Fig. 34) against the production of both NO and PGE2, activated by interferon-γ and LPS in peritoneal macrophages of mouse and rat. A relationship between the inhibitory activities and chemical structures of pyrimidine derivatives was noticed. SAR analysis indicated that pyrimidines carrying –OH functional group at the C-4 and C-6 positions of their rings did not exhibit any inhibitory activity against NO and PGE2 production. Substitution of –OH moieties with chlorine resulted in significant inhibitory activity, the 4,6-dichloro analogs were more potent than the monochloro derivatives. Changing the amino functional group at the C-2 position to the (N,N-dimethylamino)methyleneamino or the formamido further enhanced the inhibitory effects. No significant variation was detected in the expression of NO-suppressive activities among analogs with characteristic kinds of substituents at the C-5 position (–H, –CH3, –C–2H5, –C3H7, –C4H9, –C6H5, and –CH2C6H5). The IC50 values of the most potent derivatives were noticed to be 2–10 μM. The NO-suppressive effects of these derivatives were observed to be more potent than those of indomethacin and aspirin. The NO and PGE2 inhibitory effect of the investigated derivatives was attributed to the reduced expression of iNOS mRNA and COX-2 mRNA, respectively. The results suggested that the mechanism of suppressive mode of polysubstituted 2-aminopyrimidine action is more complicated, involving post-translation interactions too. Moreover, particular NO-/PGE2-inhibitory pyrimidines reduced the intensity of intestinal inflammatory disorder in murine model of ulcerative colitis.115
 | ||
| Fig. 34 Chemical structures of selected polysubstituted 2-aminopyrimidines. | ||
Several 5-nitropyrimidine-2,4-dione analogs were synthesized and subsequently screened for their inhibitory effects against NO and iNOS activities. The synthesized compounds were screened against the generation of NO in LPS-stimulated RAW246.7 macrophages and iNOS activity as compared to indomethacin as a positive control. Experimental data revealed that derivative (121) bearing a meta-nitrophenyl moiety (Fig. 35) considerably suppressed (IC50 = 6.2 μM) the iNOS activity and iNOS-facilitated NO generation (IC50 = 8.6 μM) in LPS-stimulated RAW264.7 cells. Molecular docking of compound (121) into the active binding position of iNOS was performed. The results revealed that the nitrophenyl group of compound (121) was wrapped up in a pocket fabricated between ASN364, VAL346, PRO344, and a HEME. Two π–π interactions were noted between porphyrins of HEME with –C6H5 ring and –NO2 moiety. Additionally, four hydrogen-bonding interactions were established between 5-nitropyrimidine-2,4(1H,3H)-dione framework and amino acids GLU371, ASP367, and TYR341. Docking analysis confirmed that derivative (121) is firmly linked to the active sites of iNOS via several hydrogen bonds and π–π interactions. SAR analysis indicated that the introduction of electron-releasing p-substituents (such as –OH group) enhanced the inhibitory potency against NO production as compared to the unsubstituted styryl derivative. However, p-fluorinated as well as m-fluorinated derivatives exhibited enhanced inhibitory activities due to the characteristic electronegativity and small atomic radius of fluorine as compared to other halogens (Cl and Br).116
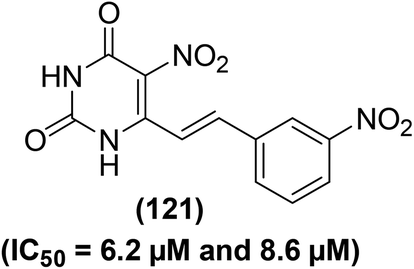 | ||
| Fig. 35 Chemical structure of a selected 5-nitropyrimidine-2,4-dione (121) with its potency (IC50 values) against the iNOS activity and NO generation. | ||
Zhang et al. synthesized and evaluated the anti-inflammatory effects of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines. Griess assay, western blot analysis, ELISA, and RT-PCR were carried out to assess the inhibitory activities of the synthesized derivatives against the secretion of NO, inflammation-associated proteins, cytokines, and mRNA, respectively in LPS-induced RAW264.7 cells. It was observed that compounds (122–124) inhibited the liberation of NO and inflammatory cytokines dose-dependently in RAW264.7 cells, without obvious toxicity, and prohibited nuclear translocation of NF-κB p65 by suppressing the degeneration of p50 and IκBα. Additionally, the three derivatives (122–124) considerably suppressed the phosphorylation of mitogen-activated protein kinase (MAPKs) in LPS-stimulated RAW264.7 cells. Compound (123) exhibited a NO-inhibitory effect similar to the reference drug indomethacin. Furthermore, the three derivatives also potently inhibited the iNOS expression and COX-2 activity. SAR investigations indicated that the incorporation of electron-releasing moieties (pyridine and chloromethyl) on position-2 of the pyrimidine ring enhanced the inhibitory activities of the resultant derivatives (122 and 123) against the NO secretion and iNOS expression. Moreover, derivative (124) with naphthyl moiety on position-3 of the pyrimidine ring exhibited improved inhibitory activity due to the presence of an extended π–π conjugation system. Chemical structures of compounds (122–124) are given in (Fig. 36).117
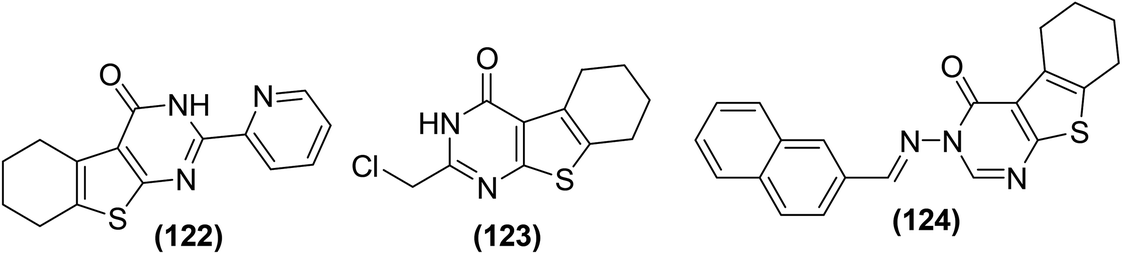 | ||
| Fig. 36 Chemical structures of selected tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine derivatives (122–124). | ||
Shi et al. reported the synthesis of 90 new pyrazolo[4,3-d]pyrimidine derivatives. Subsequently, the synthesized compounds were evaluated for their anti-inflammatory effects by suppression of the LPS-induced NO generation. Preliminary SAR analysis revealed that substituted 3,4,5-trimethoxystyryl at C-5 and alkylamine at C-7 pyrazolo[4,3-d]pyrimidine framework were useful for its anti-inflammatory activity. Among the target pyrimidines, compound (125) (Fig. 37) with an isopropylamine side chain was found to be the most potent. It exhibited a strong inhibitory effect (IC50 = 3.17 μM) against NO production in RAW 264.7 cells. Compound (125) also showed a strong inhibitory activity (IC50 = 1.12 μM) against iNOS. Initial studies on the mechanism of action revealed that the designated pyrimidine derivative could block iNOS dimerization. The anti-inflammatory activity of the compound (125) was evaluated through adjuvant-induced arthritis (AIA) in the rat model. The results revealed that the administration of the compound (125) caused a noteworthy suppression to hind paw swelling on day 30, which was comparable to the effect noticed in the aspirin-exposed group.118
 | ||
| Fig. 37 Chemical structure of most potent pyrazolo[4,3-d]pyrimidine (125). | ||
The potency of the selected pyrimidine derivatives versus inflammatory mediator (NO) is provided in Table SI-2.†
4.3. Inhibition of NF-κB and cytokines
NF-κB is a protein complex that regulates DNA replication, cytokine secretion, and cell viability. NF-κB passageway had long been regarded as a perfect proinflammatory signaling path, principally based on its use in the expression of proinflammatory cytokines, adhesion molecules, and chemokines. NF-κB is a favorable target for designing new anti-inflammatory agents and has been related to proinflammatory cytokines such as interleukin-1(IL-1) IL-12, IL-18, interferon-gamma (IFNγ) and Tumor Necrosis Factor-α (TNF-α) signaling pathways.119–121 Proinflammatory cytokines (small glycoproteins) are signaling molecules that are produced by immune cells and macrophages. They fulfill their vital role in the initiation of inflammation. Therefore, cytokine inhibitors are used to cure chronic inflammatory disorders.122Based on this concept, several research groups attempted to design novel pyrimidine-based NF-κB and cytokines inhibitors for their possible application as anti-inflammatory agents.
Kim and coworkers reported the solid-phase synthesis of several 1,6,8-trisubstituted tetrahydro-2H-pyrazino[1,2-a]pyrimidine-4,7-diones. The synthesized compounds were subsequently examined for their in vitro NF-κB inhibitory effects. Preliminary results indicated that most of the synthesized compounds possessed NF-κB-inhibitory potential. SAR analysis indicated that the inhibitory effect was dependent on the type of moieties introduced on positions-1, -6, and -8 of the basic bicyclic structure. Among all the tested compounds, two pyrimidine derivatives (126 and 127) (Fig. 38), bearing the fluorobenzyl and methoxybenzyl substituents at carbon 1 and carbon 8 exhibited the most potent NF-κB-inhibitory activities. The two derivatives (at 10 μM concentration) exhibited a 60% inhibitory activity versus the target NF-κB.123
 | ||
| Fig. 38 Chemical structures of selected 1,6,8-trisubstituted tetrahydro-2H-pyrazino[1,2-a]pyrimidine-4,7-diones (126 and 127). | ||
Comparative molecular field analysis (CoMFA) and docking techniques were jointly used to investigate the mechanism of action of several new pyrimidine analogs as binary suppressors of activator protein 1 (AP-1) and NF-κB transcriptional stimulation. Through CoMFA, a three-dimensional quantitative structure–activity relationship (3D-QSAR) model with decent numerical excellence and substantial anticipating capability was recognized, which facilitated the discovery of activity-influencing key elements. Docking results showed slightly lesser normal values for the elastic and stiff energy scores on the connecting sites. Moreover, positions-3 and -4 (Fig. 39) appeared to be the most anticipated connecting sites for AP-1 and NF-κB, respectively. The fundamental features of the most feasible connecting sites (from docking investigation) corresponded to the CoMFA field dispersions. Both CoMFA and docking analyses proposed that (a) the R2 group chosen should be a CH3–N–thienyl- or H–N–thienyl-kind moiety (b) the R5 group chosen should be a COOEt or COO-tBu moiety because the two choices are suitable for both steric and electrostatic fields and, (c) the R4 group chosen should be either an ethyl or 2-thienyl moiety, since their size matches the steric field, fulfilling the crucial part at this site. The docking analysis also revealed that the most feasible binding positions were exactly inside the zone where AP-1 (or NF-κB) and DNA coincide. Overall, the results suggested that this type of pyrimidine derivatives might behave as NF-κB-DNA and AP-1-DNA binding inhibitors that can prohibit free AP-1 and NF-κB from binding to DNA.124
 | ||
| Fig. 39 Chemical structures of 2,4,5-trisubstituted pyrimidine derivatives. | ||
Through solution-phase parallel synthesis and high throughput evaluation, several new 2-chloro-4-(trifluoromethyl)pyrimidine-5-N-(3′,5′-bis(trifluoromethyl)phenyl) carboxamides were recognized as NF-κB and AP-1 inhibitors. The tested derivatives were noticed to inhibit both IL-2 and IL-8 levels as well. Compound (128) was detected to be the most potent inhibitor (IC50 = 0.05 μM) against the stimulation of NF-κB and AP-1. To understand the mechanism of action of compound (128), experiments were performed on Jurkat T cell line and chronic model of inflammatory arthritis in mice. The results revealed that the compound (128) suppressed NF-κB guided expression of a luciferase reporter gene. This impact could not be described by general lethality as it did not influence β-actin transcription. Additionally, expression of endogenous NF-κB-controlled cytokines genes was correspondingly inhibited at the mRNA and protein extents by compound (128). Although compound (128) hindered NF-κB stimulation in T cell lines, it did not have any influence on cytokine generation by monocytoid cells, synoviocytes, fibroblasts, epithelial cells, endothelial cells, and osteoblasts. SAR analysis showed that the presence of chlorine atom (128) or methyl moiety (129) at position-2 of core pyrimidine was key for the inhibitory effects of the target analogs against the activation of both NF-κB and AP-1. The introduction of other substituents (such as –H, –N(CH3)2, –NH2, –nBuNH, –OH, –OCH3, –NHCH2C6H5 or –piperidyl) at this particular position caused complete loss of inhibitory action. Moreover, the conversion of amide nitrogen to N-benzyl or N-methyl derivatives caused a tenfold decrease in their activity. It was further observed that the presence of a substituted aromatic moiety at the amide nitrogen was crucial for enhanced inhibitory activity.125,126 Compound (128) showed good anti-inflammatory activity in several models. However, it exhibited low oral activity, compatible with reduced absorption in the gastrointestinal tract (Caco-2 permeability = 11 ± 4 × 10−7 cm s−1). Therefore, further SAR analysis of compound (128) was carried out to enhance its possible oral bioavailability. The synthesized derivatives of compound (129) were investigated for possible gastrointestinal absorption via Caco-2 (intestinal epithelial cell line). The solution-phase combinatorial technique was applied for the substitution of certain groups at positions-2, -4, and -5 of the pyrimidine skeleton. The replacement of 2-chlorine with 2-florine in the pyrimidine ring of the compound (128) did not affect the activity. Moreover, the replacement of trifluoromethyl moiety (at position-4) with chlorine, phenyl, methyl, or ethyl caused a minor decrease in the activity. The existence of the carboxamide group at position-5 was key for activity, and no activity was observed when the moiety was moved to position-6. Compound (129) exhibited similar in vitro NF-κB inhibitory activity (IC50 = 0.05 μM) but enhanced Caco-2 permeability as compared to its parent compound (128). Chemical structures of the two compounds (128 and 129) are shown in (Fig. 40).127
 | ||
| Fig. 40 Chemical structures of selected substituted pyrimidines (128 and 129). | ||
Another substituted pyrimidine analog (130) has been reported to exhibit inhibitory effect (IC50 = 2 μM) against AP-1 and NF-κB facilitated transcriptional triggering in the Jurkat T cell analyses. Attempts were made to further optimize the activity of derivative (130) via substitution of numerous moieties at the positions-2, -4, and -5 of the pyrimidine skeleton. SAR study indicated that N-methylation at 2-amino moiety of compound (130) resulted in the formation of compound (131) which exhibited six-times greater potency (IC50 = 0.3 μM). Furthermore, replacement of the trifluoromethyl moiety with 2-(5-methylthienyl) at position-4 of compound (130) produced derivative (132) which showed 44-times greater potency (IC50 = 0.045 μM). However, N-methylation at the 2-amino moiety of derivative (132) did not further enhance the efficacy. The replacement of the trifluoromethyl moiety with ethyl at position-4 of compound (130) gave derivative (133) which showed 5-times greater potency (IC50 = 0.4 μM). On this occasion, methylation at 2-amino moiety of derivative (133) caused an eleven-times increase in the potency (IC50 = 0.035 μM) of the resulting derivative (134). Overall, the two compounds (132 and 134) were identified as stronger binary inhibitors of AP-1 and NF-κB facilitated transcription. Chemical structures of five compounds (130–134) are given in (Fig. 41).128
 | ||
| Fig. 41 Chemical structures of substituted pyrimidine derivatives (130–134). | ||
Ha et al. synthesized several new heterocycle-substituted pyrimidines and subsequently evaluated them for their potential inhibitory effects versus the NF-κB transcription. NF-κB reporter gene analysis showed that several derivatives potently inhibited NF-κB transcription modulation linked to TNF-α cytokine discharge. Compound (135) (Fig. 42) was noticed to be the most potent (IC50 = 0.6 ± 0.05 μM) derivative for its suppression of NF-κB facilitated transcriptional stimulation in A-549 cells. Experimental results revealed that phenyl moiety in compound (135) did not contribute much towards its activity. However, aromatic nitrogen atoms contributed substantially to the activity enhancement of compound (135).129
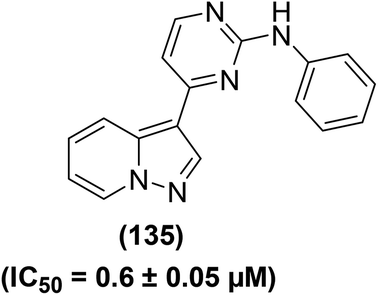 | ||
| Fig. 42 Chemical structure of a selected heterocycle-substituted pyrimidine derivative (135). | ||
Synthesis and TNF-α-inhibitory activity of several N-2,4-pyrimidine-N-phenyl-N′-phenyl ureas were reported. Most of the synthesized derivatives exhibited potent inhibitory effects versus the LPS-activated TNF-α generation. Among the pyrimidine-ureas, analogs bearing N-4-fluorophenyl and N′-substituted phenyl moieties were evaluated. SAR analysis indicated that derivatives carrying 2-chloro (136) and 2-methyl (137) substituents were the most efficient compounds against TNF-α with their IC50 values noticed to be 0.122 μM and 0.176 μM, respectively. Subsequently, derivatives of N′-phenyl-substituted ureas bearing different N-substituents were also investigated for TNF-α inhibitory effect. In this case, the results also confirmed the derivatives bearing 2-chloro (138) and 2-methyl (139) moieties to be the most potent inhibitors with their IC50 values of 0.092 μM and 0.135 μM respectively. X-ray crystallography investigations with mutated p38α indicated a binding mode for this group of urea suppressors that simulates conventional vicinal bis-aryl MAPK suppressors. Moreover, it was detected that the urea acquires a pseudo-bicyclic orientation due to the compound's capability to establish an inner hydrogen bond. Chemical structures of the four compounds (136–139) are shown in (Fig. 43).130
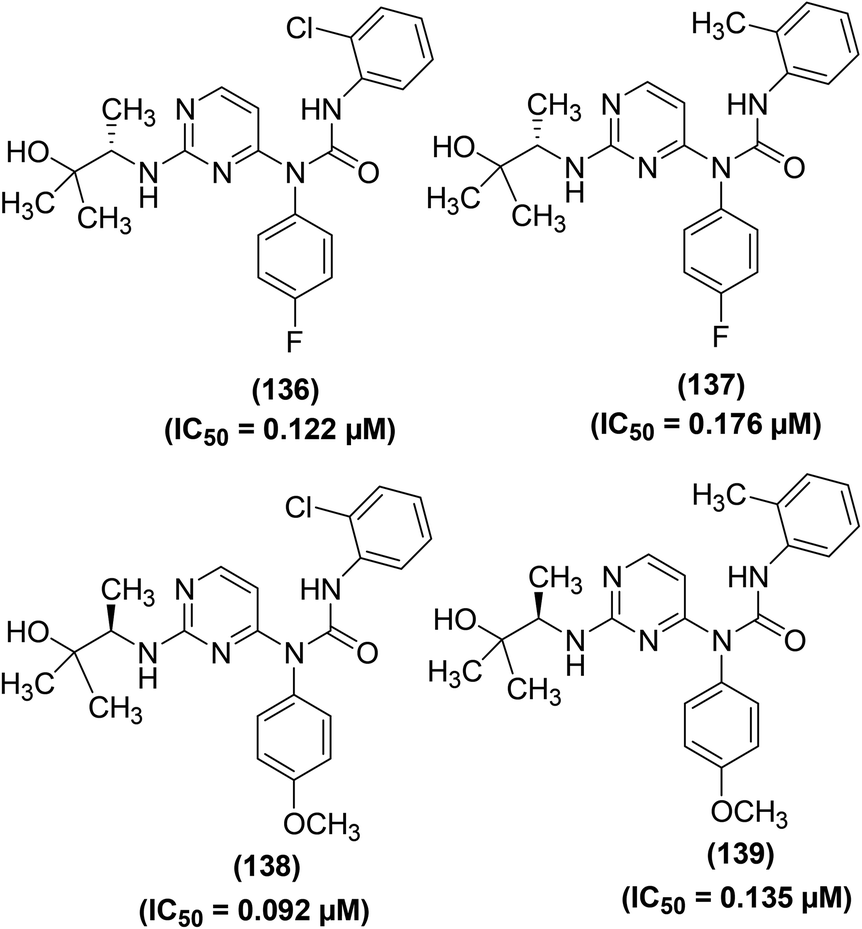 | ||
| Fig. 43 Chemical structures of selected pyrimidine-ureas (136–139). | ||
Laufer et al. synthesized several new pyridinyl pyrimidine analogs for their possible application to suppress the secretion of TNF-α and IL-1β from HWB and peripheral blood mononuclear cells (PBMC). It was observed that the oxidation state of the ultimate sulfur atom in pyridinyl pyrimidines greatly influenced the suppression of TNF-α secretion. The inhibitory effects of sulfoxides and sulfones were noted to be 4-times greater than sulfides, indicating that only polar groups at position-4 of the benzylsulfanyl scaffold enhance the inhibitory efficacy. SAR analysis further revealed that the ring NH of imidazole derivatives was stimulated by the introduction of an extra hydrogen-releasing substructure at position-4 of the parent pyrimidine skeleton. This modification caused a decrease in the inhibitory potential of the resultant derivative. The most potent derivative (140) (Fig. 44) was obtained by modifying the substituent at position-2 of the pyrimidine skeleton. The IC50 values of derivative (140) were recorded to be 3.2 μM and 2.3 μM versus the release of TNF-α and IL-1β, respectively. The results further revealed that the prolongation of the linker between the central pyrimidine and the methylsulfinylphenyl ring reduced the inhibitory efficacy compared to 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole (SB 203580). The presence of 4-fluoro-phenyl and pyridin-4-yl moieties was crucial for the bioactivity of this group of compounds. Furthermore, the benzyl group was expelled out of the suitable π–π stacking with Tyr35, which resulted in the overlapping of derivative (140) and SB 203580 in complex with p38 MAPK. The unfavorable configuration of such pyrimidine analogs can be controlled to some degree if the contribution of the methanesulfinyl group to biological activity is increased by its placement at the position-2 of the benzylsulfanyl moiety (140). The whole data propose a mode of action for pyridinyl pyrimidine inhibitors of cytokine liberation comparable to those for typical imidazole compounds.131
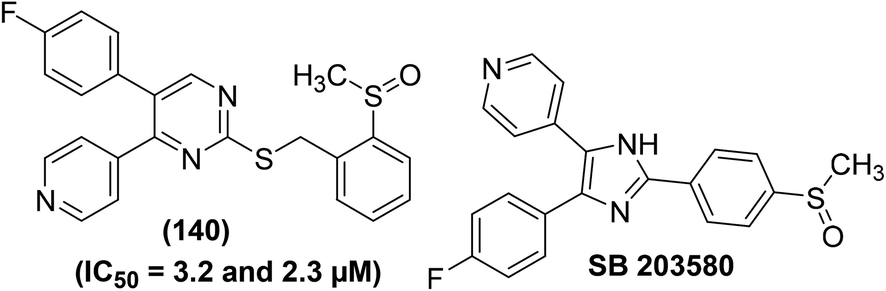 | ||
| Fig. 44 Chemical structures of pyridinyl pyrimidine derivative (140) (with its potency against TNF-α and IL-1β) and SB 203580. | ||
The enzymatic action of the MAPK p38 is necessary for the liberation of TNF-α and IL-1β from monocytes. Consequently, p38-inhibitors are useful for in vivo simulations of inflammatory disorders. Based on this concept, the synthesis of several novel imidazopyrimidines has been reported for their inhibitory actions versus the stimulation of MAPK and TNF-α production in vivo. SAR analysis indicated that the incorporation of benzylamine moiety on core imidazopyrimidine produced compound (141) which caused a significant decrease in p38α enzymatic action along with cellular suppression of TNF-α. Compound (141) was noticed to be more potent (IC50 = 0.006 μM) than compound (142) (IC50 = 0.570 μM) in p38α-inhibitory enzymatic assay. Likewise, compound (141) was also noticed to be more potent (IC50 = 0.006 μM) than compound (142) (IC50 = 0.040 μM) in cellular TNF-α inhibitory assay. It was assumed that the lipophilic interaction of the benzyl moiety with the enzyme and an extra hydrogen bonding interaction from the amino moiety might have improved the potency of the compound (141). However, further investigation would be needed to verify the enhanced potency of compound (141) due to these two types of interactions. On the contrary, the unsubstituted pyrimidine derivative (143) (IC50 = 0.990 μM) was noticed to be considerably less potent than compound (142) (IC50 = 0.570 μM) against p38α activity. Likewise, compound (143) was observed to be less potent (IC50 = 0.304 μM) than compound (142) (IC50 = 0.040 μM) in cellular TNF-α inhibitory assay too. Compound (144), containing (S)-a-methylbenzylamino moiety, was observed to be the most potent derivative. It suppressed p38a activity and TNF-α generation in cells with its IC50 values calculated to be 0.008 μM and 0.0006 μM, respectively. The extraordinary in vitro and in vivo efficacy of this class of compounds deserves additional studies in more sophisticated models of inflammatory disorders. Chemical structures of the selected four analogs (141–144) are provided in (Fig. 45).132
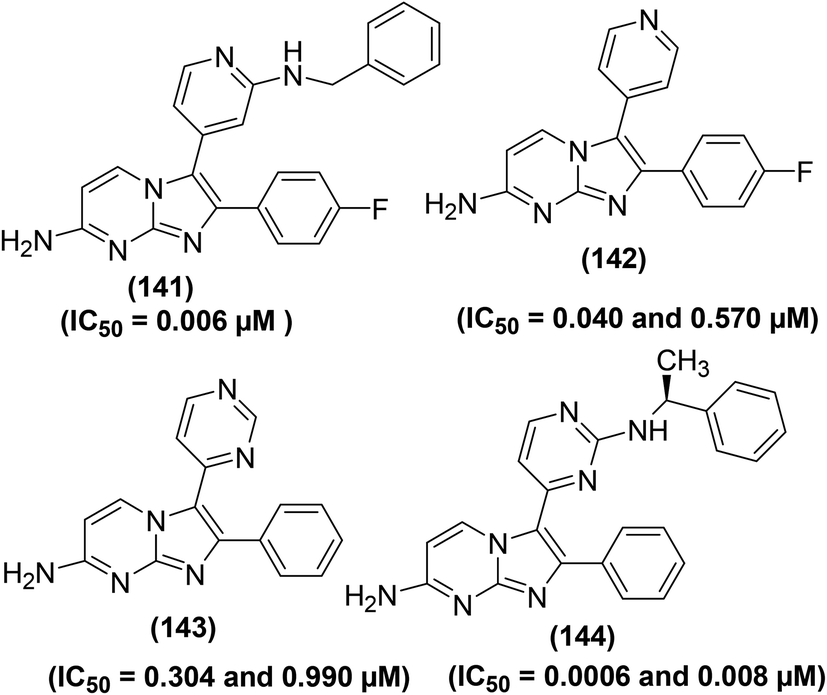 | ||
| Fig. 45 Chemical structures of selected imidazopyrimidines (141–144) and their potency (IC50 values) against p38a activity and TNF-α generation. | ||
Methotrexate (145) (Fig. 46) is a chemical analog of pyrimidine and is used to treat rheumatoid arthritis. Gerards et al. studied the inhibitory potential of methotrexate against the production of inflammatory cytokines utilizing mononuclear cells and whole blood of ten rheumatoid arthritis patients and twenty healthy volunteers. Initial results indicated that methotrexate efficiently suppressed the generation of proinflammatory cytokines namely IL-4, IL-13, IFNγ, TNF-α, and colony-stimulating factor 2 in whole blood cultures after T-cell activation. The suppression was noted at doses conveniently attained in plasma of rheumatoid arthritis patients taking the drug. Methotrexate slightly affected the IL-8 generation. Additionally, the suppression was reliant on the stimulant; IL-6, IL-8, IL-1β, and TNFα generation promoted by LOS or SAC was reduced to some extent by methotrexate. The mechanism of action revealed that the suppression was caused by the intervention with folate-reliant purine and pyrimidine synthesis. It was further observed that methotrexate had a negligible effect on cytokines generated via monocytes.133
 | ||
| Fig. 46 Chemical structure of methotrexate (145). | ||
Synthesis of several thienopyrimidine analogs for their potential inhibitory action against signal transducer and activator of transcription 3 (STAT3) stimulation triggered by IL-6 has been reported. Most of the target thienopyrimidine analogs potently (IC50 = 5.73–0.45 μM) inhibited the STAT3 expression stimulated by IL-6. Seven compounds (146–152) (Fig. SI-5†) were noticed to be the potent inhibitors against the phosphorylation of STAT3 and extracellular-signal-regulated kinases (ERK1/2) stimulated by IL-6 in Hep3B cells. SAR analysis indicated that substitution of a hydrogen atom at position-2 with chloride in the triazolopyridine moiety resulted in the formation of compound (147) which exhibited 9.9-times greater inhibitory activity (IC50 = 0.58 ± 0.01 μM) than compound (146) (IC50 = 5.73 ± 0.04 μM). Alternatively, derivative (148) with a methyl substituent at the p-position of the benzene ring exhibited only 1.8 times greater potency (IC50 = 3.24 ± 0.04 μM) than compound (146). Similarly, compound (149) bearing chlorine at the p-position of the benzene ring, exhibited greater inhibitory activity (IC50 = 2.94 ± 0.11 μM) than both compounds (146 and 148). Furthermore, compound (150) carrying chlorine at the m-position of the benzene ring, exhibited a 6.5-times greater inhibitory effect (IC50 = 0.45 ± 0.01 μM) than that of compound (149), proposing that substitution at m-position of the benzene ring can affect the activity on IL-6-stimulated STAT3 activation. Compound (151) exhibited 1.6-times greater potency (IC50 = 3.61 ± 0.02 μM) as compared to compound (146), signifying that insertion of a [3,2-d]thienopyrimidine functionality can enhance the IL-6-stimulated STAT3 suppression. Compound (152) with a methyl group at the m-position of the benzene ring exhibited an even more potent (IC50 = 0.88 ± 0.03 μM) suppressive effect than compound (151).134
Wang et al. synthesized several novel pyrazolo[4,3-d]pyrimidine analogs and subsequently assessed them for their anti-inflammatory effects in RAW264.7 cells. SAR investigations showed that the incorporation of 3-morpholinopropan-1-amine moiety into pyrazolo[4,3-d]pyrimidine might enhance the anti-inflammatory activity. Amongst the target pyrimidines, derivative (153) (Fig. 47) was noticed to be the strongest suppressor, which reduced the in vitro generation of cytokines namely TNF-α, IL-6, and NO with its IC50 values recorded to be 5.63 μM, 4.38 μM, and 2.64 μM, respectively. The underlying mechanism revealed that compound (153) dose-dependently blocked LPS-induced NO, IL-6, and TNF-α release in macrophages via inhibiting TLR4/p38 signaling pathway.135
 | ||
| Fig. 47 Chemical structure of a selected pyrazolo[4,3-d]pyrimidine (153) with its potency (IC50 values) against the generation of TNF-α, IL-6, and NO. | ||
Novel pyrazolo pyrimidines (154–156) (Fig. 48) have been synthesized and evaluated via LPS-activated TNF-α generation in mice. Analog (154) bearing a 4-chlorophenyl moiety was noticed to be the most potent (ID50 = 8.23 mg kg−1 and % inhibition = 62.27) inhibitor against LPS-induced TNF-α generation in mice as compared to the standard inhibitor (SB 203580) (ID50 = 26.38 mg kg−1 and % inhibition = 53.44%). The ID50 values of the derivatives (155 and 156) were recorded to be 13.64 mg kg−1 and 11.79 mg kg−1, respectively. SAR analysis indicated that substituted phenyl moieties (4-chlorophenyl, 4-methylphenyl, and 4-methoxyphenyl) at the amide bond of pyrazolo-pyrimidine scaffold increase the inhibitory effects. Compounds (155 and 156) exhibited weaker inhibitory activities against LPS-induced TNF-α generation as compared to compound (154), credited to the existence of electron releasing methyl and methoxy groups on the fourth position of the phenyl moiety. Docking studies of the target derivatives against p38α MAPK receptor were also carried out to recognize ligand–receptor interaction. The results revealed that compound (154) exhibited the maximum docking score of −9.824, and established π-cation and hydrogen bonding interactions with Lys53 and Asn115, respectively. Initial studies revealed that compound (154) can act as an encouraging framework in the exploration of novel and safer anti-inflammatory agents.136
 | ||
| Fig. 48 Chemical structures of selected pyrazolo pyrimidines (154–156). | ||
Chen et al. reported the synthesis, SAR analysis, and anti-inflammatory activity of new thiazolo[3,2-a]pyrimidins. SAR analysis facilitated the discovery of two potent inhibitors (Fig. 49) of IL-6 and TNF-α. Experimental results revealed that compounds (157 and 158) suppressed LPS-induced IL-6 and TNF-α discharge dose-dependently in mouse primary peritoneal macrophages (MPMs). The IC50 values of compound (157) against the inhibition of IL-6 and TNF-α were recorded to 0.60 μM and 1.16 μM respectively. While, the IC50 values of compound (158) against the inhibition of IL-6 and TNF-α were calculated to be 0.56 μM and 0.89 μM respectively. The results further indicated that the dosage of the two derivatives caused lung histopathological enhancements and reduced LPS-caused acute lung injury in vivo. The initial SAR analysis showed that the introduction of an electron-releasing moiety for instance allyloxy at the para-position of the phenyl group (157) or heterocycles such as thiophene at the core skeleton increases the anti-inflammatory effect. Further optimization showed that the oxygen atom of morpholinyl moiety is key to sustain the activity. Overall, the results revealed that compounds (157 and 158) are potent anti-inflammatory pyrimidines to cure acute inflammatory diseases for example acute lung injury and sepsis.137
 | ||
| Fig. 49 Chemical structures of selected thiazolo[3,2-a]pyrimidins (157 and 158). | ||
Zhang and coworkers reported the synthesis of several ursolic acid-based 1,2,4-triazolo[1,5-a]pyrimidines for their potential use as anti-inflammatory agents. LPS-stimulated inflammation model was used to assess the anti-inflammatory activity of the target compounds at three different doses (3 μM, 10 μM, and 30 μM) compared to dexamethasone (as a positive control). The results indicated that compounds (159–162) (Fig. SI-6†) significantly inhibited the production of IL-6 at 30 μM in LPS-induced RAW 264.7 macrophages. Moreover, compounds (159 and 160) also showed noteworthy inhibitory effects against the expression of TNF-α at 30 μM. These findings suggested that the introduction of a 1,2,4-triazolo[1,5-a]pyrimidine scaffold to ursolic acid could afford unexpected enhancement in the anti-inflammatory effect of ursolic acid derivatives. Strong IL-6 suppressive effects of compounds (159–162) were attributed to the presence of –COOH functional group at the 17 position. The incorporation of various substituents (a naphthalene ring, distinctively substituted benzene rings, and a heterocyclic moiety) to the C-2 of ursolic acid enhanced the anti-inflammatory effect. Additionally, the substitution of the benzene ring derivatives at the p-position caused greater efficacy, as illustrated by the derivatives (159, 160, and 162).138 Patel et al. have reported structure-based design and synthesis of 2,4-diaminopyrimidines (Fig. 50). The target derivatives were subsequently examined for their caspase-1 inhibitory effects via luminescence-based assay. The results revealed that six compounds (163–168) displayed noteworthy inhibitory activities with their IC50 values of 0.035, 0.027, 0.022, 0.078, 0.052, and 0.045 μM respectively. Furthermore, the six compounds were also investigated for their cell-based activity. The results showed that compounds (163–168) had moderate cell-based efficacy with their IC50 values of 6.25, 3.55, 3.20, 8.0, 6.84, and 6.37 μM, respectively. The initial SAR studies revealed that the electron-withdrawing moieties at distant phenyl ring (A) showed the best enzymatic suppression (163–165). The introduction of alkyl substituents at different positions of phenyl ring (B) also showed analogous potency (166–168). Briefly, the sizes and electronic features of the substituents may describe the structural criteria for strong caspase-1 suppression. Furthermore, a relative docking analysis of the six compounds was executed employing caspase-1, 3, 7, and 8. All the compounds were enclosed in the same zone as that of the co-crystal ligand. Their binding energy ranged from −7.508 to −11.260 kcal M−1 for caspase-1. However, lower binding energy from −1.518 to −4.969 kcal M−1 was calculated for caspase-3, 7, and 8. In short, molecular docking investigations described the selectivity and provided significant understanding for synthesizing novel series of derivatives.139
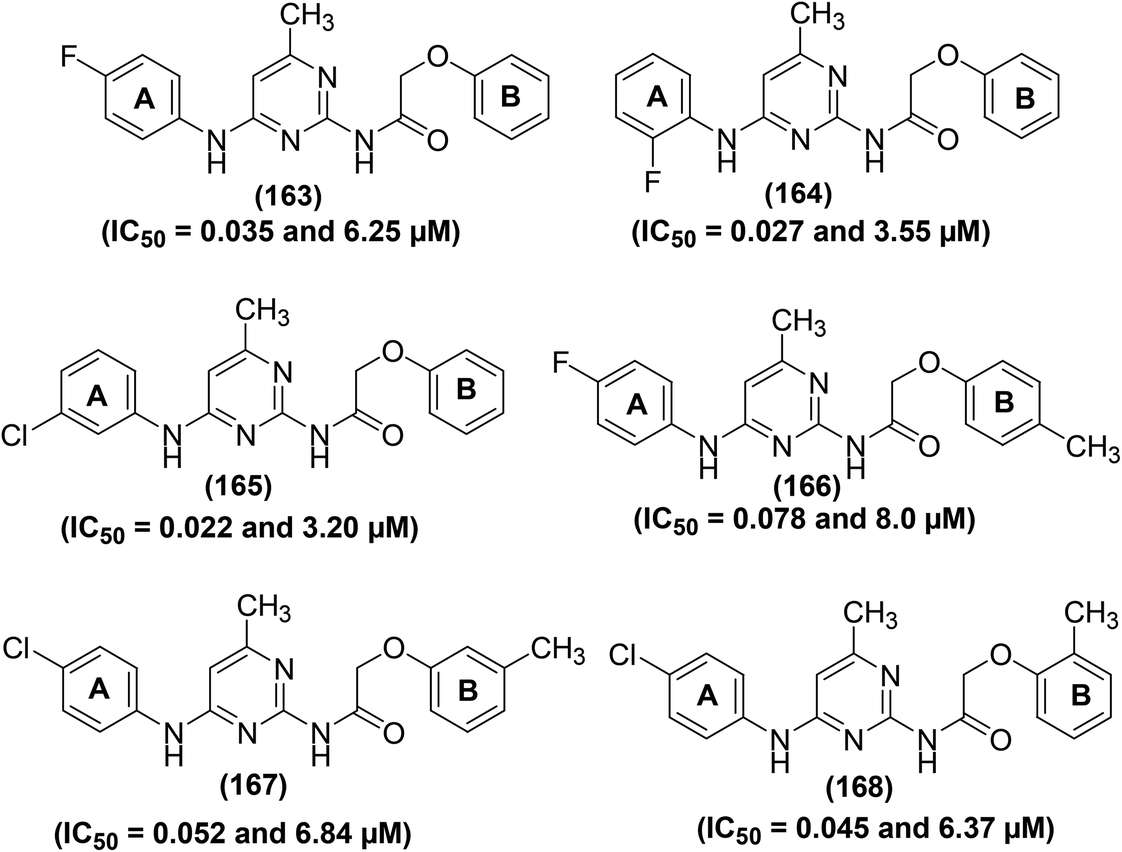 | ||
| Fig. 50 Chemical structures of selected 2,4-diaminopyrimidines (163–168). | ||
The potency of the selected pyrimidine derivatives versus NF-κB and cytokines is provided in Table SI-3.†
4.4. Inhibition of phosphodiesterase-4 and lipoxygenases
Phosphodiesterase-4 (PDE4) represents phosphodiesterase (PDE) superfamily of enzymes that regulate epithelial stability and inflammation. Four types of PDE4 namely PDE4A, PDE4B, PDE4C, and PDE4D have been recognized, which are principally found in immune cells, brain cells, and epithelial cells. PDE4 enzymes are responsible for the hydrolysis of cyclic adenosine monophosphate (cAMP) or 3′-5′-cyclic adenosine monophosphate, an intracellular second messenger that regulates a system of anti-inflammatory and proinflammatory mediators. Inhibition of PDE4 has been confirmed as an efficient curative approach for inflammatory disorders such as psoriasis, chronic obstructive pulmonary disease (COPD), asthma, neuroinflammation, inflammatory bowel diseases (IBD), lupus, rheumatic arthritis (RA), and atopic dermatitis (AD). PDE4 inhibitors suppress the breakdown of cAMP to 5′-adenosine monophosphate (5′-AMP), causing an increase in the amount of cAMP which then loosens smooth muscle and prevents inflammation and fibrosis. In the past few decades, several PDE4 inhibitors were synthesized, among which apremilast, crisaborole, and roflumilast have been recommended for the treatment of psoriatic arthritis, atopic dermatitis, and inflammatory airway diseases, respectively.140–142Lipoxygenases (LOXs) are non-heme iron-containing enzymes that catalyze the breakdown of arachidonic acid into leukotrienes which facilitate the phenomenon of inflammation. For instance, suppression of leukotrienes generation can reduce the number of pro-inflammatory cells, in addition to relieving the harmful effects of inflammation.143,144 Depending on the ability of LOXs to insert oxygen atoms into the appropriate site of arachidonic acid, they are divided into four types i.e. 5-LOX, 8-LOX, 12-LOX, and 15-LOX. Furthermore, 5-LOX, 12-LOX, and 15-LOX are associated with particular disorders, such as atherosclerosis, asthma, and even cancer.145 The excessive production of leukotrienes is one of the main reasons for inflammatory disorders such as glomerulonephritis, acute lung injury, inflammatory bowel diseases, psoriasis, and asthma. LOX inhibitors slow down or block the activity of the LOXs to prevent the overproduction of leukotrienes. Consequently, it is imperative to design potential selective LOX inhibitors for the treatment of inflammatory diseases.146,147
Based on the mechanism of actions described above for PDE4 and LOX, several research groups attempted to synthesize novel pyrimidine derivatives for their potential inhibitory effects against the two enzymes.
Synthesis of several nitraquazone derivatives with a pyrimidindione core has been reported for their potential application as PDE4 inhibitors. For the majority of the analogs, an ethyl group at position 3 of the pyrimidindione nucleus was introduced to meet the requirement for nitraquazone derivatives. PFE4 activity assay was performed in undifferentiated human U937 cells. Preliminary results showed that four analogs (169–172) (Fig. 51) had the most potent inhibitory activity against PFE4 with their IC50 values reported to be 6.54 ± 1.38, 0.62 ± 9.92, 5.72 ± 0.80, and 4.87 ± 1.37 μM, respectively. SAR analysis revealed that derivatives in which the phenyl ring at position 1 is substituted at para position either with chlorine (169 and 170) or nitro group (171), exhibited significant PDE4 inhibitory activity. Furthermore, compound (172) with a cyclopropylmethyl moiety at position 3 also displayed considerable inhibitory activity.148
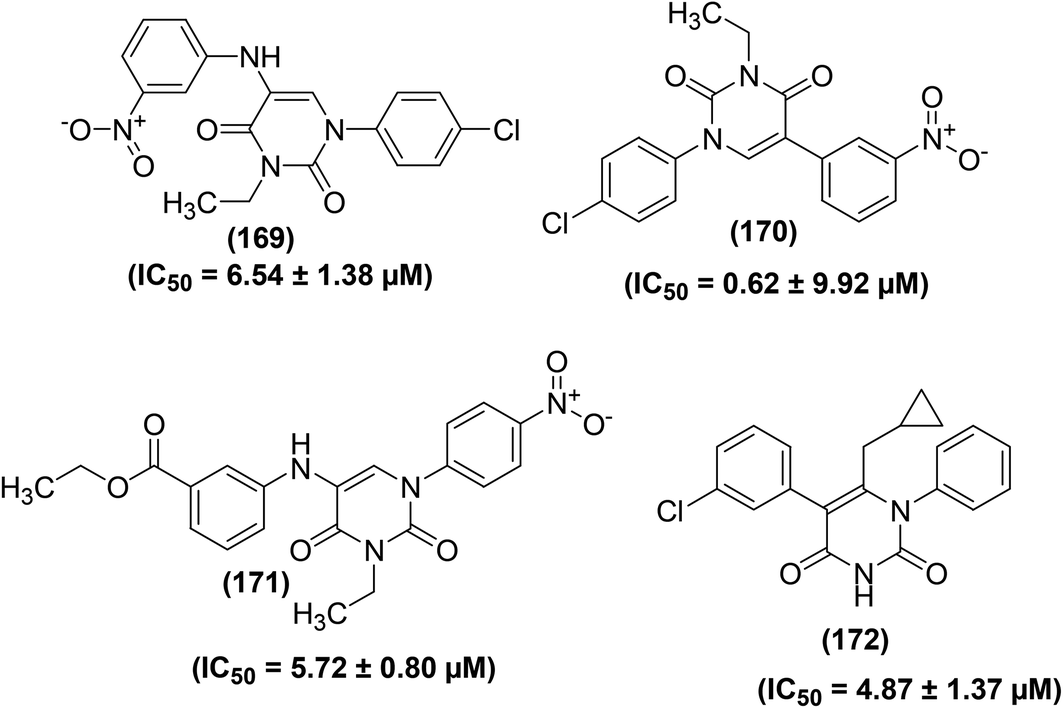 | ||
| Fig. 51 Chemical structures of selected pyrimidinedione derivatives (169–172). | ||
Adepu et al. reported the synthesis of several thieno[2,3-d]pyrimidine derivatives carrying a cyclohexane ring linked to a five- or six-membered heterocyclic scaffold together with a benzylic nitrile. The target derivatives were subsequently screened for their PDE4-inhibitory effects in vitro. Preliminary results revealed that some of the synthesized compounds had significant PDE4 inhibitory activities at a concentration of 30 μM. Compound (173) was observed to be the most potent inhibitor with its IC50 values calculated to be 2.0 ± 0.41 and 3.14 ± 1.02 μM against PDE4B and PDE4D, respectively. The noteworthy PDE4-inhibitory effect of compound (173) (Fig. 52) was attributed to the presence of 4,5,6,7-tetrahydro-1H-indazole-5-carbonitrile moiety. Moreover, the docking results of some selected derivatives exhibited decent overall similarity to their experimental PDE4B inhibitory activities in vitro.149
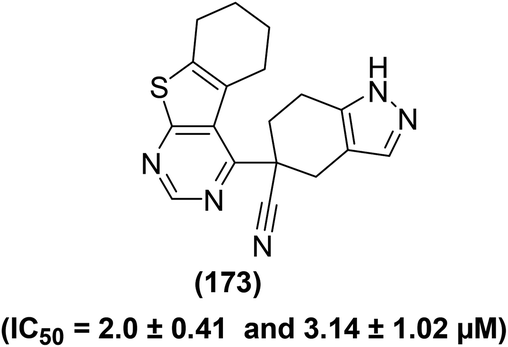 | ||
| Fig. 52 Chemical structure of selected thieno[2,3-d]pyrimidine derivative (173). | ||
Synthesis of pyranones linked to a pyrazolopyrimidine scaffold has been reported through the regioselective fabrication of pyranone ring utilizing coupling–iodocyclization together with Sonogashira/Heck/Suzuki reactions. The target compounds were investigated in vitro for their inhibitory action using PDE4 enzyme separated from Sf9 cells. Experimental data revealed that compound (174) (Fig. 53) exhibited the most potent inhibitory activity with its IC50 values reported being 1.33 ± 0.64 and 2.84 ± 0.64 μM against PDE4B and PDE4D, respectively. SAR analysis suggested that the existence of an aryl moiety at C-3 of the pyranone ring was vital, and a –CONH2 functionality connected to this aryl ring contributed to PDE4B inhibition. Further experiments revealed that compound (174) had fewer side effects and was better tolerated than rolipram (reference drug) in zebrafish embryos under the same experimental conditions. Overall, the results suggested that a novel and safer PDE4B inhibitor was identified for the possible therapy of asthma and COPD.150
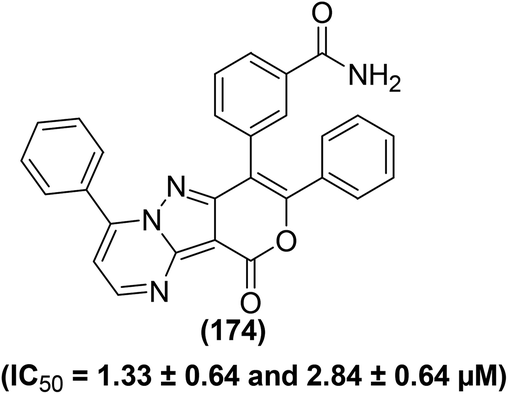 | ||
| Fig. 53 Chemical structure of selected pyrazolopyrimidine derivative (174). | ||
Goto et al. synthesized several 4-amino-2-phenylpyrimidines bonded to sulfur-containing rings as new and efficient PDE4 inhibitors. Preliminary results showed that the target compounds had potent PDE4B inhibitory action in vitro. Among the synthesized derivatives, four compounds (175a–d) (Fig. 54) showed the most potent inhibitory activities against PDE4B with their IC50 values calculated to be 7.5, 6.6, 11, and 4.7 nm, respectively. Furthermore, compound (175c) also displayed a nice in vivo anti-inflammatory effect in the LPS-stimulated lung inflammation model in mice (ID50 = 18 mg kg−1). Studies on the X-ray crystal structure of (175c) in PDE4B showed that the nitrogen atom of pyrimidine bonds to Glu443 and the oxygen atoms of the sulfone establish hydrogen bonds with Asn395 and Tyr233 through water molecules. The aliphatic part of the sulfone ring captures a lipophilic cavity analogous to the difluoromethoxy scaffold of Roflumilast. Additionally, the lateral chain of (175c) makes a bonding conformation and the tertiary butyl moiety captures a lipophilic cavity consisting of Phe414, Phe506, and Met431.151
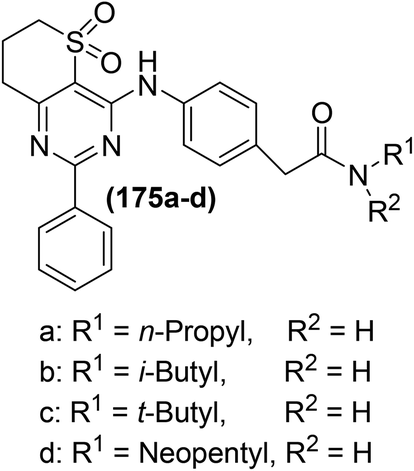 | ||
| Fig. 54 Chemical structures of selected 4-amino-2-phenylpyrimidines (175a–d). | ||
Goto et al. also reported the synthesis of novel 5-carbamoyl-2-phenylpyrimidine derivatives as efficient PDE4 inhibitors. The PDE4 inhibitory action of the target derivatives was assessed through the human PDE4B enzyme. Experimental data revealed that derivative (176) had modest PDE4B inhibitory effect (IC50 = 200 nM). However, chemical transformation of compound (176) afforded N-neopentylacetamide derivative (177), which exhibited potent inhibitory effect (IC50 = 8.3 nM) against PDE4 as well as high in vivo potency against LPS-stimulated pulmonary neutrophilia in mice. SAR analysis revealed that incorporation of methylthio and methyl moieties at position-6 of the pyrimidine skeleton reduced the PDE4B inhibitory activity. Similarly, substitution on the amide functionality also caused a decrease in the potency. The result of the X-ray crystallography analysis indicated that 5-carbamoyl group of compound (177) establishes water-bridged hydrogen bonds with Gln443 and Asn395. Moreover, the methylene group of the neopentyl moiety might be a useful binder between the amide bond and terminal t-butyl group, which possibly has contributed to the enhanced PDE4B inhibitory activity of the compound (177). Chemical structures of compounds (176 and 177) are shown (Fig. 55).152
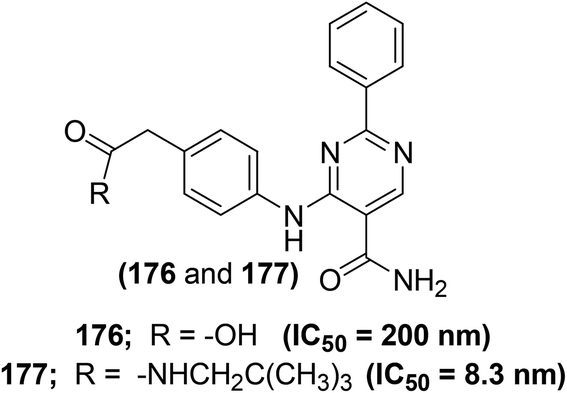 | ||
| Fig. 55 Chemical structures of selected 5-carbamoyl-2-phenylpyrimidines (176 and 177). | ||
Synthesis of several catecholopyrimidines as potential PDE4 inhibitors has been reported to cure atopic dermatitis. Catechol derivatized pyrimidines were screened for dose-dependent PDE4B enzyme suppression at various doses (1–5000 nM) utilizing rolipram as a controlled drug via PDE4 enzyme assay. Experimental data revealed that compound (178) (Fig. 56) carrying pyrimidine nucleus derivatized with pyridine, catechol, and trifluoromethyl moieties potently suppressed (IC50 = 15 ± 0.4 nM) PDE4 enzyme. SAR analysis revealed that the presence of catechol moiety at positions −4 and −5 of pyrimidine scaffold contributed to enhanced PDE4-inhibitory effects. Furthermore, compound (178) selectively suppressed PDE4B as compared to other PDE4s. The molecular docking analysis indicated that compound (178) increases the chances of binding interactions in the catalytic zone of PDE4B. In vivo studies proved that compound (178) efficiently reduced the symptoms of atopic dermatitis in DNCB-treated Balb/c mice by inhibiting the generation of TNF-α, IL-4, IL-5, and IL-17. Overall, the results suggested that compound (178) could prove a promising candidate for the treatment of atopic dermatitis.153,154
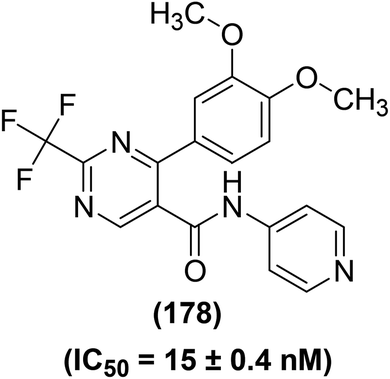 | ||
| Fig. 56 Chemical structure of selected catecholopyrimidine derivative (178). | ||
Kumar et al. reported the synthesis of pyrazolopyrimidines through a novel new 3-component reaction. The target compounds were then evaluated for their potential PDE4-inhibitory potential in vitro using rolipram as a reference compound. At a concentration of 30 nM, compounds (179 and 180) (Fig. 57) exhibited 33 and 67% suppression of PDE4, respectively. These results were further verified by docking analysis of compound (180) with PDE4B enzyme (binding energy = 10.2 kcal mol−1), which indicated hydrogen bonding between the –OH moiety of (180) and THR345 residue of PDE4B.155
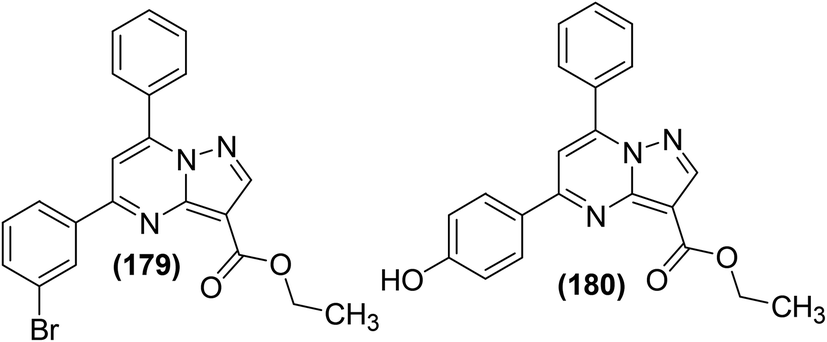 | ||
| Fig. 57 Chemical structures of selected pyrazolopyrimidine derivatives (179 and 180). | ||
Several new 7-(hetero)aryl-substituted pyrazolo[1, 5-a]pyrimidines have been synthesized for their potential use as PDE4 inhibitors. All the target derivatives (at 10 μM concentration) were evaluated for their inhibitory ability with reference to rolipram through cell-based cAMP reporter assay in HEK 293 cell lines. Observed results indicated that compounds (181 and 182) (Fig. 58) caused noteworthy enhancement of cAMP (90 and 76%, respectively) at 10 μM with reference to rolipram (100%). It is worth mentioning that though the compound (181) looked to be more potent than the compound (182) at 10 μM concentration, however, it exhibited lower inhibitory potential at lower concentrations. Results further indicated that compound (182) suppressed PDE4B in vitro with its EC50 of 5.01 μM. SAR analysis indicated that the existence of a heteroaryl moiety (possessing acidic hydrogen) or bi-cyclic aryl at the carbon-7 of the pyrazolopyrimidine skeleton was key for the inhibitory potential of this group of derivatives.156
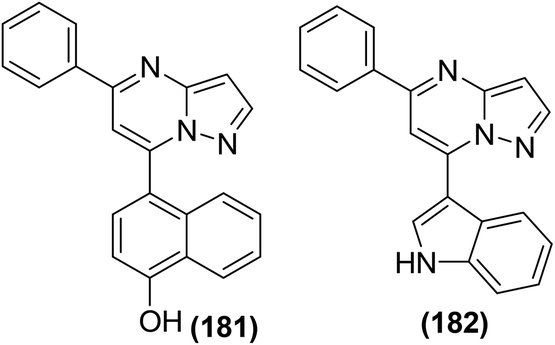 | ||
| Fig. 58 Chemical structures of selected 7-(hetero)aryl-substituted pyrazolo[1,5-a]pyrimidines (181 and 182). | ||
Based on the structure of 2-[(4,6-diphenethoxypyrimidin-2-yl)thio]hexanoic acid (183), Hieke et al. discovered a new family of strong 5-LOX inhibitors. All the target compounds were characterized for their influence on the activity of human 5-LOX. Compounds (184 and 185) were observed to be the most promising 5-LOX inhibitors (at a concentration of 10 μM) with their IC50 values calculated to be 0.4 and 0.5 μM, respectively via cell-based 5-LOX inhibition assay. Whereas, IC50 values of the two compounds (184 and 185) were calculated to 2.8 and 3.1 μM, respectively via cell-free 5-LOX inhibition assay. SAR studies revealed that lipophilic bulky substituents in the α-position of the carboxylic moiety control potency. Elongation of the aliphatic chain in α-position to the carboxylic acid caused a potency increase of the synthesized derivatives. Furthermore, the introduction of electron-releasing groups on para position of the phenyl moieties of compound (183) also enhanced the 5-LOX inhibitory activity. Chemical structures of compounds (183–185) are shown in (Fig. 59).157
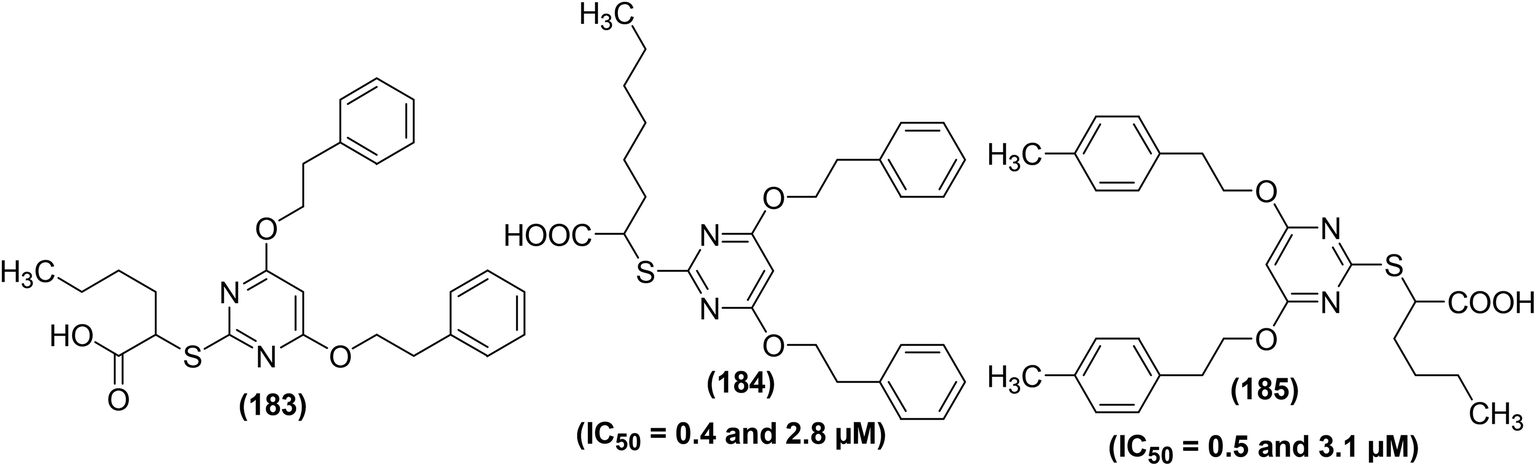 | ||
| Fig. 59 Chemical structures 2-[(4,6-diphenethoxypyrimidin-2-yl)thio]hexanoic acid (183) and its selected derivatives (184 and 185). | ||
Nam et al. evaluated pyrimidine derivatives of acetaminophen as potential LOX inhibitors. The LOX-inhibitory potential of the target derivatives was evaluated using the soybean lipoxygenase-1 (sLOX-1) enzyme. sLOX was selected instead of the mammalian LOX because of its greater availability and stability. sLOX-1 activity was evaluated with its natural substratum, linoleic acid, under characteristic conditions with variable quantities of pyrimidine derivatives of acetaminophen. The results showed that two derivatives (186 and 187) (Fig. 60) exhibited inhibitory activity against sLOX-1 with their IC50 values calculated to be 602 and 593 μM, respectively. SAR analysis revealed that the incorporation of nitrogen into the phenolic ring of acetaminophen enhances the efficacy of the target pyrimidine analogs of acetaminophen as sLOX-1 inhibitors, suggesting a different mode of suppression reliant on the acidity of the phenolic O–H.158
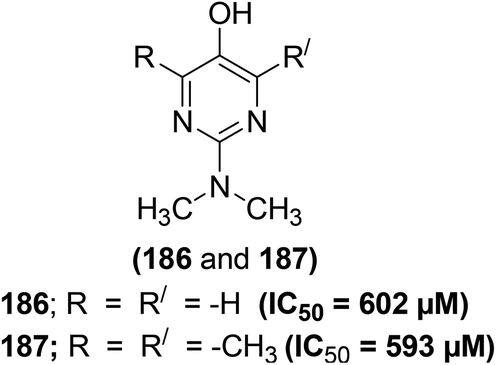 | ||
| Fig. 60 Chemical structures of selected pyrimidine derivatives of acetaminophen (186 and 187). | ||
Synthesis of several novel pyrazolopyrimidines as anti-soybean-5-LOX agents has been reported. The majority of the synthesized derivatives were tested for their soybean 5-LOX inhibitory activities. The results indicated that two derivatives (188 and 189) (Fig. 61) exhibited the most potent inhibitory effect against soybean 5-LOX with their (%) inhibition of 68.0% and 72.2% at 100 μM, respectively. The marginally greater potency of compound (189) compared to that of compound (188) proposes that the presence of a longer alkyl chain of the ether group at position 3 of the aryl ring contributes to better inhibitory activity. SAR results revealed the effect of the nature of the substituent(s) attached to the aryl system on the anti-5-LOX activity. Overall, the existence of the hydroxyl functionality and the ether group (-ethoxy or -methoxy) at positions 4 and 3 of the phenyl ring, respectively enhanced the anti-soybean 5-LOX activity.159
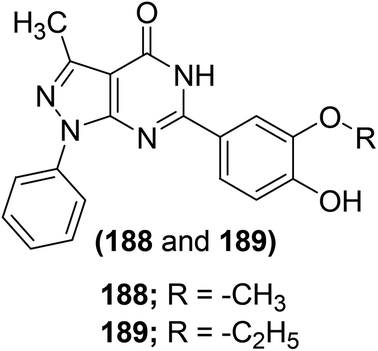 | ||
| Fig. 61 Chemical structures of selected pyrazolopyrimidines (188 and 189). | ||
Shehab et al. reported the synthesis and anti-5-LOX activities of some isolated and fused pyrimidine derivatives. The synthesized analogs were evaluated for their inhibitory potential with reference to a standard drug meclofenamate sodium via bnova 5-LOX inhibitor screening assay. Two pyrimidine derivatives (190 and 191) (Fig. 62) were noted to be the most potent inhibitors of 5-LOX with their IC50 values calculated to be 4.91 and 6.98 μM, respectively as compared to the standard drug meclofenamate sodium (IC50 = 6.15 μM). Observed results further indicated that the two derivatives possessed promising anti-inflammatory activities both in vitro and in vivo.160
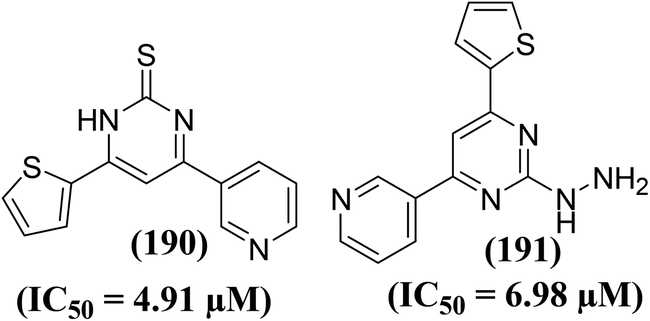 | ||
| Fig. 62 Chemical structures of selected pyrimidines coupled to pyridine and thiophene scaffolds (190 and 191). | ||
The potency of the selected pyrimidine derivatives versus PDE4 and LOX enzymes is provided in Table SI-4.†
5. Complete SAR analysis of pyrimidines
Overall SAR analysis of pyrimidine analogs can be summed up in the following key points (Fig. 63).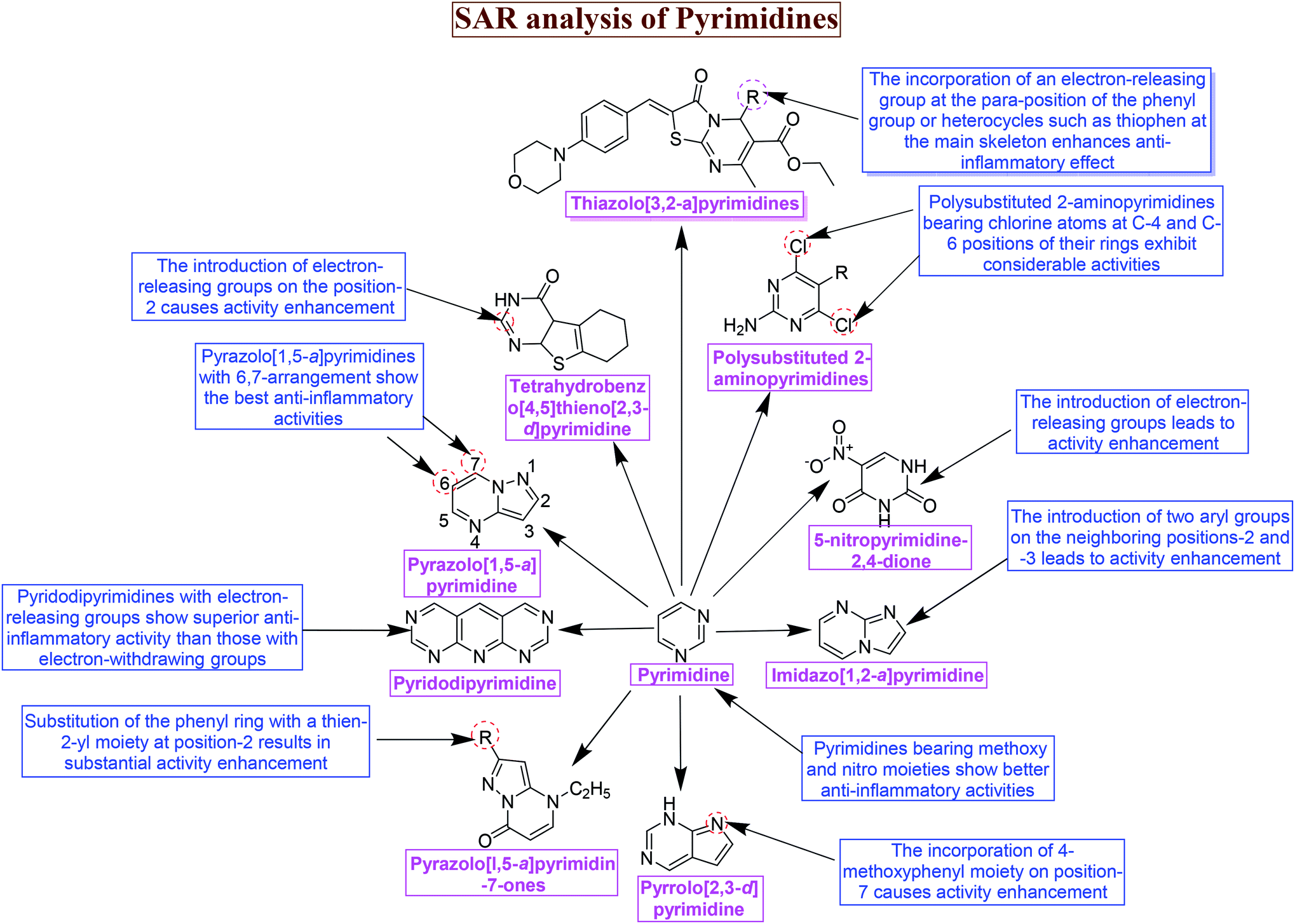 | ||
| Fig. 63 Overall SAR analysis of pyrimidines. | ||
(a) The introduction of electron-releasing moieties on position-2 of the core pyrimidine in tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine causes activity enhancement.79
(b) The incorporation of 4-methoxyphenyl moiety on position-7 of pyrrolo[2,3-d]pyrimidine results in activity enhancement.80
(c) Pyridodipyrimidine derivatives with electron-releasing moieties show superior anti-inflammatory potential than the derivatives of the same type with electron-withdrawing moieties.82
(d) Both monoaryl- and bisarylsubstituted 2-aminopyrimidines exhibit improved activities regardless of the length of the C-5 substituents in polysubstituted pyrimidines.83
(e) The introduction of two aryl moieties on the neighboring positions-2 and -3 in imidazo[1,2-a]pyrimidines leads to activity enhancement.86
(f) The existence of five-member thiophene moiety at positions-4,6 and electron-releasing chlorine atom at position-2 of tricyclic pyrimidines improve the anti-inflammatory potential of the target derivatives.87
(g) Substitution of the phenyl ring with a thien-2-yl moiety at position-2 in pyrazolo[l,5-a]pyrimidin-7-ones results in substantial activity enhancement.88
(h) Introduction of 3-chlorophenyl and carboxamide groups on positions-1 and -4 of pyrazolone ring, respectively causes substantial enhancement in anti-inflammatory activities of pyrazolediazenylpyrimidines.89
(i) Pyrimidines bearing a pyrazolyl group in a hybrid configuration with the pyrazolo[3,4-d]pyrimidine moiety exhibit significant anti-inflammatory activities.91
(j) Pyrazolo[1,5-a]pyrimidines with 6,7-arrangement show the best anti-inflammatory activities.93
(k) Pyrimidines containing cycloheptenes fused to their rings display better activities than the derivatives containing cyclohexenes fused to their rings.94
(l) The introduction of p-sulfonamide or p-methylsulfone on one of the two aromatic rings in diarylheterocycle encompassing pyrimidines contributes to activity enhancement.95
(m) Quinazolinone–pyrimidine hybrid bearing an anilino moiety at position-4 of the pyrimidine ring displays the most potent activity.96
(n) Pyrimidines bearing methoxy and nitro moieties show better anti-inflammatory activities.97
(o) 3-Aroylpyrido[1,2-a]pyrimidines carrying a benzyloxybenzoyl, biphenyloyl or naphthoyl moiety show superior activities, which can further be improved by the incorporation of a methyl moiety on position-8 of the pyrido[1,2-a]pyrimidine.112
(p) Polysubstituted 2-aminopyrimidines bearing chlorine at C-4 and C-6 positions of their rings exhibit considerable activities, and 4,6-dichloro analogs are more potent than monochloro analogs.115
(q) The introduction of electron-releasing substituents in 5-nitropyrimidine-2,4-dione derivatives leads to activity enhancement.116
(r) The introduction of electron–releasing moieties on the position-2 of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines causes activity enhancement.117
(s) 1,6,8-Trisubstituted tetrahydro-2H-pyrazino[1,2-a]pyrimidine-4,7-diones carrying the fluorobenzyl and methoxybenzyl substituents at carbon 1 and carbon 8, respectively exhibit potent activities.123
(t) The presence of a chlorine atom on position-2 in 2-chloro-4-(trifluoromethyl)pyrimidine-5-N-(3′,5′-bis(trifluoromethyl)phenyl) carboxamides is necessary for a good effect. The existence of carboxamide moiety at position-5 of these pyrimidines is crucial for the anti-inflammatory effect. Moreover, the introduction of a substituted aromatic moiety at the amide nitrogen in this class of pyrimidines results in activity enhancement.127
(u) Introduction of 3-morpholinopropan-1-amine moiety into pyrazolo[4,3-d]pyrimidine can enhance the activity.135
(v) The incorporation of an electron-releasing group at the para-position of the phenyl group or heterocycles such as thiophen at the main skeleton of thiazolo[3,2-a]pyrimidines enhances the anti-inflammatory effect.137
6. Conclusions
Inflammation is biological feedback of the immune system that is initiated by several toxic stimuli, namely, microorganisms, chemical irritants, damaged cells, and noxious compounds. Inflammation acts via the elimination of all these harmful stimuli, and thus triggers the process of tissue repair. Nevertheless, chronic inflammations can cause severe disorders, for example, diabetes, hepatitis, asthma, cardiovascular diseases, and rheumatoid arthritis. Presently, several NSAIDs are utilized to cure inflammatory disorders. Pyrimidines represent a group of aromatic heterocyclic compounds, enclosing two nitrogen atoms at positions-1 and -3 of the main six-member ring. They occur abundantly in natural as well as synthetic products and demonstrate a variety of pharmacological effects namely antibacterial, anti-inflammatory, antiviral, antifungal, and antitubercular activities. Several procedures for the synthesis of pyrimidines are described. Pyrimidines act as anti-inflammatory agents by suppressing the expressions and activities of inflammatory mediators. The inhibitory action of pyrimidine derivatives versus inflammatory mediators namely PGE2, NO, NF-κB, IL-1β, IL-6, TNF-α, and leukotrienes are explained. The potential use of various pyrimidine derivatives as anti-inflammatory agents is expected owing to their high potency and minimum toxicity. SAR studies of various pyrimidine derivatives are discussed in detail. The introduction of electron-releasing groups on position-2 of pyrimidine ring increases the anti-inflammatory activity. Similarly, pyrimidines with methoxy and nitro groups also exhibit improved anti-inflammatory activities. Pyridodipyrimidines carrying electron-releasing groups display superior anti-inflammatory activities than pyrimidines of the same type with electron-withdrawing groups.7. Prospects
Long-term consumption of NSAIDs (to alleviate the inflammatory pain) causes some adverse effects such as gastrointestinal toxicity, renal toxicity, hepatotoxicity, and cardiovascular disorders. The FDA also recommends the consumption of a minimum effective dosage of NSAIDs.161 Consequently, the prime focus of future research must be on the safety of novel pyrimidines-based anti-inflammatory agents. Harmful effects of novel pyrimidine-based anti-inflammatory agents can be kept to a minimum by enhancing their inhibitory potency against proinflammatory mediators, as low dosages of such drugs will be required to cure inflammation. Several approaches can be adopted to improve the anti-inflammatory potency of novel pyrimidines in the future.Drug synergy might be a useful strategy to enhance the anti-inflammatory potency of novel pyrimidines. Drug synergism involves the interaction between two or more drugs that leads to a larger overall remedial effect of the drugs as compared to the total of the separate effects of every single drug. It targets various constituents of complicated diseases including inflammatory disorders.162 The anti-inflammatory potency of new pyrimidines can be synergistically increased by taking them in combination with other anti-inflammatory drugs. It will potentially allow for improving the anti-inflammatory effects, whereas decreasing the toxicity, or side effects of pyrimidines.
Several natural products such as resveratrol, curcumin, capsaicin, boswellic acid, green tea polyphenols, colchicine, quercetin, epigallocatechin-3-gallate (EGCG), and cucurbitacins exhibit significant anti-inflammatory effects.163,164 Therefore, a synthetic methodology involving the coupling of the anti-inflammatory pyrimidines with the aforementioned natural products might be useful for the preparation of new anti-inflammatory candidates with better potency and minimum toxicity.
Similarly, the preparation of pyrimidines-NSAIDs hybrids might also improve the anti-inflammatory potency of the target compounds.161 Such anti-inflammatory hybrids can be prepared by the coupling reactions of active pyrimidine derivatives with suitable NSAIDs namely aspirin, ibuprofen, diclofenac, indomethacin, oxaprozin, etodolac, diflunisal, etc. Nevertheless, the safety of pyrimidines-NSAIDs hybrids has to be ensured through in vivo and in vitro studies.
Another approach to improve the anti-inflammatory potency of pyrimidines is through the enhancement of their absorption and bioavailability. For this purpose, several compounds of plant-origin namely naringin, genistein, glycyrrhizin, sinomenine, piperine, quercetin, and nitrile glycoside can be applied as possible bioavailability enhancers of new pyrimidines-based anti-inflammatory agents.165,166
Modern manufacturing techniques can be used to improve the absorption and dissolution of novel pyrimidine-based anti-inflammatory agents.167 Due to enhanced absorption and dissolution of novel pyrimidines, minimum doses will be administered, which might cause better drug tolerability without reducing their pain-relieving and anti-inflammatory potency. Similarly, the pyrimidine fabrications targeted exactly to the spot of inflammation are likely to decrease the systemic drug exposure, and thus reduce the threat of systemic side effects.
Novel drug delivery systems (NDDSs) such as liposomes, nanoparticles, dendrimers, microspheres, and transdermal delivery are greatly encouraging as they offer advantages in terms of enhanced bioavailability, target specificity, less dosage frequency, prevention from decomposition especially versus the severe gastric conditions, and minimum toxicity. NDDSs have been known to be more effective than conventional drug transport systems. The shortcomings (toxicity, poor water solubility, and low bioavailability) associated with the use of current NSAIDs can also be overcome by replacing the conventional drug delivery system with NDDSs.168,169 Novel pyrimidine-based anti-inflammatory drugs will be efficiently transported precisely to the desired tissues through the use of NDDSs. Therefore, the use of NDDSs for pyrimidine-based anti-inflammatory agents will improve drug efficacy, and control drug release to produce a constant anti-inflammatory effect besides offering better safety.
The computational chemistry technique is a useful approach that can expedite the progress of the traditional methods applied for drug discovery and development. In this approach, arithmetical operations and computer-based modeling are applied for chemical phenomena along with calculating the structures and chemical properties of molecular species. Specific computational chemistry approaches are utilized to evaluate and estimate the occurrences such as the drug coupling to its target and the chemical features for designing promising new drug candidates.170 Thus, this important method is time-saving and cost-efficient for the identification and synthesis of novel pyrimidine-based anti-inflammatory drug candidates.
Furthermore, clinical studies must pursue significant positive impacts for patients to gain real benefits from novel pyrimidine-based anti-inflammatory agents. These agents must be more effective than the present anti-inflammatory drugs or any other existing treatment. If there is no progress in terms of the potency of the novel anti-inflammatory pyrimidines, they should be minimally more tolerant, less harmful, convenient to use, or more economical than the current standard anti-inflammatory drugs. Generally, the synthesis and discovery of new pyrimidines are crucial, attributed to their promising potential to behave as prominent anti-inflammatory candidates.
List of abbreviations
| COPD | Chronic obstructive pulmonary disease |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| RNA | Ribonucleic acid |
| DNA | Deoxyribonucleic acid |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| N-PMB | N-p-Methoxybenzyl |
| PGE2 | Prostaglandins E2 |
| IEDDA | Inverse electron demand Diels–Alder |
| COX | Cyclooxygenase |
| TFA | Trifluoroacetic acid |
| FDA | Food and drug administration |
| rDA | Retro-Diels–Alder |
| GI | Gastrointestinal |
| iNOS | Inducible nitric oxide synthase |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| ED50 | 50% effective dose |
| SI | Selectivity indexes |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| mRNA | Messenger ribonucleic acid |
| EIA | Enzyme immunoassay |
| HWB | Human whole blood |
| PE | Purified enzyme |
| ELISA | Enzyme-linked immunosorbent assay |
| eNOS | Endothelium nitric oxide synthase |
| nNOS | Neural nitric oxide synthase |
| L-NNA | Nω-Nitro-L-arginine |
| SEITU | S-Ethyliso-thiourea |
| 7-NI | 7-Nitroindazole |
| LPS | Lipopolysaccharide |
| IL-1 | Interleukin-1 |
| IFNγ | Interferon-gamma |
| TNF-α | Tumor necrosis factor-α |
| CoMFA | Comparative molecular field analysis |
| AP-1 | Activator protein 1 |
| 3D-QSAR | Three-dimensional quantitative structure–activity relationship |
| SAR | Structure–activity relationship |
| PBMC | Peripheral blood mononuclear cells |
| MAPK | Mitogen-activated protein kinase p38 |
| STAT3 | Signal transducer and activator of transcription 3 |
| ERKs | Extracellular-signal regulated kinases |
| EGCG | Epigallocatechin-3-gallate |
| NDDSs | Novel drug delivery systems |
| MAPKs | Mitogen-activated protein kinase |
| 5-LOX | Arachidonate 5-lipoxygenase |
Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The authors would like to express their thanks to the Universidade Federal de Mato Grosso do Sul. This study was funded in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001, and CAPES-PrInt, Finance Code 88881.311799/2018-01. The authors are also grateful to the Conselho Nacional de Desenvolvimento Científico e Tecnológico—Brasil (CNPq)—Finance Code 420852/2018-2 and Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul—Brasil (FUNDECT-MS)—grants 036/2017 PPSUS-MS (59/300.074/2017) providing financial support for undertaking this project.References
- H. ur Rashid, Y. Xu, N. Ahmad, Y. Muhammad and L. Wang, Bioorg. Chem., 2019, 87, 335–365 CrossRef CAS.
- L. Chen, H. Deng, H. Cui, J. Fang, Z. Zuo, J. Deng, Y. Li, X. Wang and L. Zhao, Oncotarget, 2018, 9, 7204–7218 CrossRef.
- C. Nordqvist, Everything you need to know about inflammation, Medical News Today, 24 November 2017, https://www.medicalnewstoday.com/articles/248423 Search PubMed.
- P. A. Ward, Acute and Chronic Inflammation, In: Fundamentals of Inflammation, ed. C. N. Serhan, P. A. Ward and D. W. Gilroy, Cambridge University Press, New York, 2010, pp. 1–16 Search PubMed.
- E. O. Iwalewa, L. J. McGaw, V. Naidoo and J. N. Eloff, Afr. J. Biotechnol., 2007, 6, 2868–2885 CrossRef CAS.
- D. Wakefield and R. K. Kumar, Inflammation: chronic, in Encycl. Life Sci. (eLS), Nature Publishing Group, London, 2001, pp. 1–6 Search PubMed.
- J. C. Maroon, J. W. Bost and A. Maroon, Afr. J. Biotechnol., 2010, 1, 80 Search PubMed.
- J. R. Vane, Nature New Biol., 1971, 231, 232–235 CrossRef CAS.
- S. Chaiamnuay, J. J. Allison and J. R. Curtis, Am. J. Health-Syst. Pharm., 2006, 63, 1837–1851 CrossRef CAS.
- C. Litalien and P. Beaulieu, Molecular Mechanisms of Drug Actions: From Receptors to Effectors, in Pediatric Critical Care, ed. B. P. Fuhrman and J. J. Zimmerman, Elsevier, 4th edn, 2011. pp. 1553–1568 Search PubMed.
- R. O. Day and G. G. Graham, Aust. Prescr., 2004, 27, 142–145 CrossRef.
- W. A. Carvalho, R. D. S. Carvalho and F. Rios-Santos, Rev. Bras. Anestesiol., 2004, 54, 448–464 CrossRef CAS.
- K. U. Tufekci, R. Meuwissen, S. Genc and K. Genc, Inflammation in parkinson's disease, in Advances in Protein Chemistry and Structural Biology, ed. R. Donev, Elsevier Inc., 1st edn, 2012, vol. 88. pp. 69–132 Search PubMed.
- I. Ghlichloo and V. Gerriets, Anti-inflammatory Drugs (NSAIDs) [updated 2020 May 18], in StatPearls, StatPearls Publishing, Treasure Island, FL, 2020, https://www.ncbi.nlm.nih.gov/books/NBK547742/ Search PubMed.
- C. Sostres, C. J. Gargallo, M. T. Arroyo and A. Lanas, Best Pract. Res., Clin. Gastroenterol., 2010, 24, 121–132 CrossRef CAS.
- S. Harirforoosh, W. Asghar and F. Jamali, J. Pharm. Pharm. Sci., 2013, 16, 821–847 Search PubMed.
- G. Yuan, M. L. Wahlqvist, G. He, M. Yang and D. Li, Asia Pac. J. Clin. Nutr., 2006, 15, 143–152 CAS.
- Y. Ju and R. S. Varma, J. Org. Chem., 2006, 71, 135–141 CrossRef CAS.
- Y. Ju, D. Kumar and R. S. Varma, J. Org. Chem., 2006, 71, 6697–6700 CrossRef CAS.
- P. D. Lokhande, B. Y. Waghamare and S. S. Sakate, Indian J. Chem., 2005, 44, 2338–2342 Search PubMed.
- J. Jampilek, Molecules, 2019, 24, 10–13 CrossRef.
- M. Garcí-Valverde and T. Torroba, Molecules, 2005, 10, 318–320 CrossRef.
- D. J. Brown, Pyrimidines and their Benzo Derivatives, in Comprehensive Heterocyclic Chemistry, ed. C. Katritzky and A. R. Rees, Pergamon Press, Oxford, UK, vol. 3, 1984, pp. 57–155 Search PubMed.
- O. O. Ajani, J. T. Isaac, T. F. Owoeye and A. A. Akinsiku, Int. J. Biol. Chem., 2015, 9, 148–177 CrossRef CAS.
- V. Sharma, N. Chitranshi and A. K. Agarwal, Int. J. Med. Chem., 2014, 2014, 1–31 Search PubMed.
- A. E. A. Porter, Diazines and Benzodiazines, in Comprehensive Organic Chemistry, ed. D. H. R. Barton and W. D. Ollis, Pergamon Press, Oxford, UK, 1979, vol. 4, pp. 122–124 Search PubMed.
- N. Agarwal, S. K. Raghuwanshi, D. N. Upadhyay, P. K. Shukla and V. J. Ram, Bioorg. Med. Chem. Lett., 2000, 10, 703–706 CrossRef CAS.
- H. S. Basavaraja, G. M. Sreenivasa and E. Jayachandran, Indian J. Heterocycl. Chem., 2005, 15, 69 Search PubMed.
- Y. Nakagawa, S. Bobrov, C. R. Semer, T. A. Kucharek and M. Harmoto, Fungicidal pyrimidine derivatives, US Pat., 6, 818, 631 B1, 2004 Search PubMed.
- S. Ito, K. Masuda, S. Kusano, T. Nagata, Y. Kojima, N. Sawai and S. I. Maeno, Pyrimidine derivative, process for preparing same and agricultural or horticultural fungicidal composition containing same, US Pat., 4,814,338, 1989 Search PubMed.
- S. Marquais-Bienewald, W. Holzl, A. Preuss and A. Mehlin, Use of substituted 2,4-bis(alkylamino) pyrimidines, US Pat., 2006/0188453 A1, 2006 Search PubMed.
- J. Cieplik, M. Stolarczyk, J. Pluta, O. Gubrynowicz, I. Bryndal, T. Lis and M. Mikulewicz, Acta Pol. Pharm., 2015, 72, 53–64 Search PubMed.
- M. Botta, M. Artico, S. Massa, A. Gambacorta, M. E. Marongiu, A. Pani and P. L. Colla, Eur. J. Med. Chem., 1992, 27, 251–257 CrossRef CAS.
- O. Prakash, V. Bhardwaj, R. Kumar, P. Tyagi and K. R. Aneja, Eur. J. Med. Chem., 2004, 39, 1073–1077 CrossRef CAS.
- P. Sharma, N. Rane and V. K. Gurram, Bioorg. Med. Chem. Lett., 2004, 14, 4185–4190 CrossRef CAS.
- S. Vega, J. Alonso, J. A. Diaz and F. Junquera, J. Heterocycl. Chem., 1990, 27, 269–273 CrossRef CAS.
- V. J. Ram, N. Haque and P. Y. Guru, Eur. J. Med. Chem., 1992, 27, 851–855 CrossRef CAS.
- A. M. Farghaly, O. M. AboulWafa, Y. A. M. Elshaier, W. A. Badawi, H. H. Haridy and H. A. E. Mubarak, Med. Chem. Res., 2019, 28, 360–379 CrossRef CAS.
- K. Rana, B. Kaur and B. Kumar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2004, 43, 1553–1557 Search PubMed.
- A. Holý, I. Votruba, M. Masojídková, G. Andrei, R. Snoeck, L. Naesens, E. De Clercq and J. Balzarini, J. Med. Chem., 2002, 45, 1918–1929 CrossRef.
- M. M. M. Ramiz, W. A. El-Sayed, E. Hagag and A. A.-H. Abdel-Rahman, J. Heterocycl. Chem., 2011, 48, 1028–1038 CrossRef CAS.
- J. Balzarini, J. Antimicrob. Chemother., 2002, 50, 5–9 CrossRef CAS.
- P. A. S. Smith and R. O. Kan, J. Org. Chem., 1964, 29, 2261–2265 CrossRef CAS.
- H. W. Lee, B. Y. Kim, J. B. Ahn, S. K. Kang, J. H. Lee, J. S. Shin, S. K. Ahn, S. J. Lee and S. S. Yoon, Eur. J. Med. Chem., 2005, 40, 862–874 CrossRef CAS.
- A. A. Abu-Hashem, M. M. Youssef and H. A. R. Hussein, J. Chin. Chem. Soc., 2011, 58, 41–48 CrossRef CAS.
- A. K. Gupta, Sanjay, H. P. Kayath, S. Ajit, S. Geeta and K. C. Mishra, Indian J. Pharmacol., 1994, 26, 227–228 CAS.
- S. A. Rahaman, Y. Rajendra Pasad, P. Kumar and B. Kumar, Saudi Pharm. J., 2009, 17, 255–258 CrossRef.
- S. M. Sondhi, S. Jain, A. D. Dwivedi, R. Shukla and R. Raghubir, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2008, 47, 136–143 Search PubMed.
- T. Sasada, F. Kobayashi, N. Sakai and T. Konakahara, Org. Lett., 2009, 11, 2161–2164 CrossRef CAS.
- S. D. Jadhav and A. Singh, Org. Lett., 2017, 19, 5673–5676 CrossRef CAS.
- L. Su, K. Sun, N. Pan, L. Liu, M. Sun, J. Dong, Y. Zhou and S. F. Yin, Org. Lett., 2018, 20, 3399–3402 CrossRef CAS.
- X. Q. Chu, W. B. Cao, X. P. Xu and S. J. Ji, J. Org. Chem., 2017, 82, 1145–1154 CrossRef CAS.
- W. Guo, C. Li, J. Liao, F. Ji, D. Liu, W. Wu and H. Jiang, J. Org. Chem., 2016, 81, 5538–5546 CrossRef CAS.
- J. L. Zhan, M. W. Wu, F. Chen and B. Han, J. Org. Chem., 2016, 81, 11994–12000 CrossRef CAS.
- N. Deibl, K. Ament and R. Kempe, J. Am. Chem. Soc., 2015, 137, 12804–12807 CrossRef CAS.
- P. Zhichkin, D. J. Fairfax and S. A. Eisenbeis, Synthesis, 2002, 720–722 CrossRef CAS.
- M. D. Hill and M. Movassaghi, Synthesis, 2008, 823–827 CAS.
- O. K. Ahmad, M. D. Hill and M. Movassaghi, J. Org. Chem., 2009, 74, 8460–8463 CrossRef CAS.
- E. Gayon, M. Szymczyk, H. Gérard, E. Vrancken and J. M. Campagne, J. Org. Chem., 2012, 77, 9205–9220 CrossRef CAS.
- A. A. Estrada, J. P. Lyssikatos, F. St-Jean and P. Bergeron, Synlett, 2011, 2387–2391 CrossRef CAS.
- M. G. Barthakur, M. Borthakur, P. Devi, C. J. Saikia, A. Saikia, U. Bora, A. Chetia and R. C. Boruah, Synlett, 2007, 223–226 CAS.
- A. S. Karpov and T. J. J. Müller, Synthesis, 2003, 5, 2815–2826 Search PubMed.
- M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 14254–14255 CrossRef CAS.
- K. Yang, Q. Dang, P. J. Cai, Y. Gao, Z. X. Yu and X. Bai, J. Org. Chem., 2017, 82, 2336–2344 CrossRef CAS.
- M. Vidal, M. Garcí-Arriagada, M. C. Rezende and M. Domínguez, Synthesis, 2016, 48, 4246–4252 CrossRef CAS.
- K. S. Vadagaonkar, H. P. Kalmode, S. Prakash and A. C. Chaskar, New J. Chem., 2015, 39, 3639–3645 RSC.
- D. Tejedor, S. López-Tosco and F. Garcí-Tellado, J. Org. Chem., 2013, 78, 3457–3463 CrossRef CAS.
- K. A. Chkhikvadze, N. I. Koretskaya, N. S. Rodnyanskaya and O. Yu. Magidson, Chem. Heterocycl. Compd., 1969, 5, 108–111 CrossRef.
- M. Amir, S. A. Javed and H. Kumar, Indian J. Pharm. Sci., 2007, 68, 337–343 CrossRef.
- M. J. Alam, O. Alam, N. Shrivastava, M. J. Naim and P. Alam, Int. J. Pharmacol., 2015, 2, 55–69 CAS.
- A. M. Omar, H. A. Abd El Razik, A. A. Hazzaa, M. A. Z. El-Attar, M. A. El Demellawy, A. E. Abdel Wahab and S. A. M. El Hawash, Future Med. Chem., 2019, 11, 1585–1603 Search PubMed.
- G. Weingarten and G. Bravi, Pyrimidine derivative as selective COX-2 inhibitors, US Pat., 7,235,560 B2, 2007, pp. 1–13 Search PubMed.
- J. R. Vane and R. M. Botting, Inflammation Res., 1998, 47, 78–87 CrossRef.
- J. N. Cashman, Drugs, 1996, 52, 13–23 CrossRef CAS.
- N. Atatreh, A. M. Youssef, M. A. Ghattas, M. Al Sorkhy, S. Alrawashdeh, K. B. Al-Harbi, I. M. El-Ashmawy, T. I. Almundarij, A. A. Abdelghani and A. S. Abd-El-Aziz, Bioorg. Chem., 2019, 86, 393–400 CrossRef CAS.
- M. H. M. AbdEl-Azim, M. A. Aziz, S. M. Mouneir, A. F. EL-Farargy and W. S. Shehab, Arch. Pharm., 2020, 353, 1–13 CrossRef.
- A. A. Bekhit, H. T. Y. Fahmy, S. A. F. Rostom and A. M. Baraka, Eur. J. Med. Chem., 2003, 38, 27–36 CrossRef CAS.
- G. N. Tageldin, S. M. Fahmy, H. M. Ashour, M. A. Khalil, R. A. Nassra and I. M. Labouta, Bioorg. Chem., 2018, 78, 358–371 CrossRef CAS.
- Y. Zhang, L. Luo, C. Han, H. Lv, D. Chen, G. Shen, K. Wu, S. Pan and F. Ye, Molecules, 2017, 22, 1–20 Search PubMed.
- M. S. Mohamed, R. Kamel and R. H. Abd El-Hameed, Med. Chem. Res., 2013, 22, 2244–2252 CrossRef CAS.
- M. A. Abdelgawad, R. B. Bakr, W. Ahmad, M. M. Al-Sanea and H. A. H. Elshemy, Bioorg. Chem., 2019, 92, 103218 CrossRef.
- M. A. Abdelgawad, R. B. Bakr and A. A. Azouz, Bioorg. Chem., 2018, 77, 339–348 CrossRef CAS.
- V. Kolman, F. Kalčic, P. Jansa, Z. Zídek and Z. Janeba, Eur. J. Med. Chem., 2018, 156, 295–301 CrossRef CAS.
- D. Raffa, B. Maggio, F. Plescia, S. Cascioferro, M. V. Raimondi, S. Plescia and M. G. Cusimano, Arch. Pharm., 2009, 342, 321–326 CrossRef CAS.
- R. B. Bakr, A. A. Ghoneim and A. A. Azouz, Bioorg. Chem., 2019, 88, 102964 CrossRef CAS.
- J. P. Zhou, Y. W. Ding, H. B. Zhang, L. Xu and Y. Dai, Chin. Chem. Lett., 2008, 19, 669–672 CrossRef CAS.
- P. K. Singour, A. Khare, H. Dewangan and R. S. Pawar, J. Pharm. Res., 2012, 5, 4853–4855 CAS.
- F. Bruni, A. Costanzo, S. Selleri, G. Guerrini, R. Fantozzi, R. Pirisino and S. Brunelleschi, J. Pharm. Sci., 1993, 82, 480–486 CrossRef CAS.
- Z. M. Nofal, H. H. Fahmy, E. S. Zarea and W. El-Eraky, Acta Pol. Pharm., 2011, 68, 507–517 CAS.
- O. I. Abd El-Salam, M. E. A. Zaki, M. M. Said and M. Abdulla, Egypt. J. Chem., 2012, 55, 529–547 CrossRef.
- R. B. Bakr, A. A. Azouz and K. R. A. Abdellatif, J. Enzyme Inhib. Med. Chem., 2016, 31, 6–12 CrossRef CAS.
- P. J. Beswick, A. P. Blackaby, C. Bountra, T. Brown, K. Browning, I. B. Campbell, J. Corfield, R. J. Gleave, S. B. Guntrip, R. M. Hall, S. Hindley, P. F. Lambeth, F. Lucas, N. Mathews, A. Naylor, H. Player, H. S. Price, P. J. Sidebottom, N. L. Taylor, G. Webb and J. Wiseman, Bioorg. Med. Chem. Lett., 2009, 19, 4509–4514 CrossRef CAS.
- C. Almansa, A. F. De Arriba, F. L. Cavalcanti, L. A. Gómez, A. Miralles, M. Merlos, J. Garcí-Rafanell and J. Forn, J. Med. Chem., 2001, 44, 350–361 CrossRef CAS.
- A. E. G. E. Amr, M. A. Al-Omar and M. M. Abdalla, Int. J. Pharmacol., 2016, 12, 86–91 CAS.
- A. Orjales, R. Mosquera, B. López, R. Olivera, L. Labeaga and M. T. Núñez, Bioorg. Med. Chem., 2008, 16, 2183–2199 CrossRef CAS.
- S. E. Abbas, F. M. Awadallah, N. A. Ibrahin, E. G. Said and G. M. Kamel, Eur. J. Med. Chem., 2012, 53, 141–149 CrossRef CAS.
- S. N. A. Bukhari, W. Ahmad, A. M. Butt, N. Ahmad, M. W. B. Amjad, M. A. Hussain, V. H. Shah and A. R. Trivedi, Afr. J. Pharm. Pharmacol., 2012, 6, 1064–1068 CAS.
- A. A. El-Malah and A. E. Kassab, Anti-Inflammatory Anti-Allergy Agents Med. Chem., 2015, 14, 204–214 CrossRef CAS.
- P. Molina, E. Aller, A. Lorenzo, P. López-Cremades, I. Rioja, A. Ubeda, M. C. Terencio and M. J. Alcaraz, J. Med. Chem., 2001, 44, 1011–1014 CrossRef CAS.
- O. Tietz, J. Kaur, A. Bhardwaj and F. R. Wuest, Org. Biomol. Chem., 2016, 14, 7250–7257 RSC.
- O. Tietz, S. K. Sharma, J. Kaur, J. Way, A. Marshall, M. Wuest and F. Wuest, Org. Biomol. Chem., 2013, 11, 8052–8064 RSC.
- D. Lokwani, R. Azad, A. Sarkate, P. Reddanna and D. Shinde, Bioorg. Med. Chem., 2015, 23, 4533–4543 CrossRef CAS.
- E. K. A. Abdelall, P. F. Lamie, A. K. M. Ahmed and E. S. EL-Nahass, Bioorg. Chem., 2019, 86, 235–253 CrossRef CAS.
- W. Seebacher, J. Faist, A. Presser, R. Weis, R. Saf, T. Kaserer, V. Temml, D. Schuster, S. Ortmann, N. Otto and R. Bauer, Eur. J. Med. Chem., 2015, 101, 552–559 CrossRef CAS.
- F. Kalčic, V. Kolman, H. Ajani, Z. Zídek and Z. Janeba, ChemMedChem, 2020, 15, 1398–1407 CrossRef.
- N. M. Ahmed, S. Nofal and S. M. Awad, J. Pharm. Res. Int., 2020, 32, 49–67 CrossRef.
- W. Akhtar, L. M. Nainwal, M. F. Khan, G. Verma, G. Chashoo, A. Bakht, M. Iqbal, M. Akhtar, M. Shaquiquzzaman and M. M. Alam, J. Fluorine Chem., 2020, 236, 109579 CrossRef CAS.
- R. Korhonen, A. Lahti, H. Kankaanranta and E. Moilanen, Curr. Drug Targets: Inflammation Allergy, 2005, 4, 471–479 CrossRef CAS.
- C. R. Lyons, Adv. Immunol., 1995, 60, 323–371 CAS.
- G. B. Lenon, C. G. Li, C. C. Xue, F. C. K. Thien and D. F. Story, J. Ethnopharmacol., 2008, 116, 547–553 CrossRef.
- D. K. Mahapatra, S. K. Bharti and V. Asati, Curr. Top. Med. Chem., 2017, 17, 3146–3169 CrossRef CAS.
- U. Bluhm, J. L. Boucher, U. Buss, B. Clement, F. Friedrich, U. Girreser, D. Heber, T. Lam, M. Lepoivre, M. Rostaie-Gerylow and U. Wolschendorf, Eur. J. Med. Chem., 2009, 44, 2877–2887 CrossRef CAS.
- P. Jansa, A. Holý, M. Dračínský, V. Kolman, Z. Janeba, P. Kostecká, E. Kmoníčková and Z. Zídek, Med. Chem. Res., 2014, 23, 4482–4490 CrossRef CAS.
- P. Jansa, A. Holý, M. Dračínský, V. Kolman, Z. Janeba, E. Kmoníčková and Z. Zídek, Med. Chem. Res., 2015, 24, 2154–2166 CrossRef CAS.
- Z. Zídek, M. Kverka, A. Dusilová, E. Kmoníčková and P. Jansa, Nitric Oxide, 2016, 57, 48–56 CrossRef.
- L. Ma, L. He, L. Lei, X. Liang, K. Lei, R. Zhang, Z. Yang and L. Chen, Chem. Biol. Drug Des., 2015, 85, 296–299 CrossRef CAS.
- Y. Zhang, L. Luo, C. Han, H. Lv, D. Chen, G. Shen, K. Wu, S. Pan and F. Ye, Molecules, 2017, 22, 1960 CrossRef.
- J. B. Shi, L. Z. Chen, B. S. Wang, X. Huang, M. M. Jiao, M. M. Liu, W. J. Tang and X. H. Liu, J. Med. Chem., 2019, 62, 4013–4031 CrossRef CAS.
- K. Taniguchi and M. Karin, Nat. Rev. Immunol., 2018, 18, 309–324 CrossRef CAS.
- T. Lawrence, Cold Spring Harbor Perspect. Biol., 2009, 1, a001651 Search PubMed.
- T. Liu, L. Zhang, D. Joo and S.-C. Sun, Signal Transduction Targeted Ther., 2017, 2, 17023 CrossRef.
- K. Resch, Cytokine Inhibitors, In Encyclopedic Reference of Immunotoxicology, ed. H.-W. Vohr, Springer Berlin Heidelberg, 2005, pp. 170–173 Search PubMed.
- J. W. Kim, J. H. Cho, T. Han, J. Lee and C. H. Oh, Bull. Korean Chem. Soc., 2006, 27, 484–488 CrossRef CAS.
- L. Qian, S. Y. Liao, Z. L. Huang, Y. Shen and K. C. Zheng, J. Mol. Model., 2010, 16, 1139–1150 CrossRef CAS.
- R. W. Sullivan, C. G. Bigam, P. E. Erdman, M. S. S. Palanki, D. W. Anderson, M. E. Goldman, L. J. Ransone and M. J. Suto, J. Med. Chem., 1998, 41, 413–419 CrossRef CAS.
- D. M. Gerlag, L. Ransone, P. P. Tak, Z. Han, M. Palanki, M. S. Barbosa, D. Boyle, A. M. Manning and G. S. Firestein, J. Immunol., 2000, 165, 1652–1658 CrossRef CAS.
- M. S. S. Palanki, P. E. Erdman, L. M. Gayo-Fung, G. I. Shevlin, M. E. Goldman, L. J. Ransone, B. L. Bennett, A. M. Manning and M. J. Suto, J. Med. Chem., 2000, 43, 3995–4004 CrossRef CAS.
- M. S. S. Palanki, L. M. Gayo-Fung, G. I. Shevlin, P. Erdman, M. Sato, M. Goldman, L. J. Ransone and C. Spooner, Bioorg. Med. Chem. Lett., 2002, 12, 2573–2577 CrossRef CAS.
- H. H. Ha, J. S. Kim and B. M. Kim, Bioorg. Med. Chem. Lett., 2008, 18, 653–656 CrossRef CAS.
- T. A. Brugel, J. A. Maier, M. P. Clark, M. Sabat, A. Golebiowski, R. G. Bookland, M. J. Laufersweiler, S. K. Laughlin, J. C. VanRens, B. De, L. C. Hsieh, M. J. Mekel and M. J. Janusz, Bioorg. Med. Chem. Lett., 2006, 16, 3510–3513 CrossRef CAS.
- S. A. Laufer and G. K. Wagner, J. Med. Chem., 2002, 45, 2733–2740 CrossRef CAS.
- K. C. Rupert, J. R. Henry, J. H. Dodd, S. A. Wadsworth, D. E. Cavender, G. C. Olini, B. Fahmy and J. J. Siekierka, Bioorg. Med. Chem. Lett., 2003, 13, 347–350 CrossRef CAS.
- A. H. Gerards, S. de Lathouder, E. R. de Groot, B. A. C. Dijkmans and L. A. Aarden, Rheumatology, 2003, 42, 1189–1196 CrossRef CAS.
- H. J. Jang, S. M. Kim, M. C. Rho, S. W. Lee and Y. H. Song, J. Microbiol. Biotechnol., 2019, 29, 856–862 CrossRef CAS.
- B. S. Wang, X. Huang, L. Z. Chen, M. M. Liu and J. B. Shi, J. Enzyme Inhib. Med. Chem., 2019, 34, 1121–1130 CrossRef CAS.
- K. Somakala, S. Tariq and M. Amir, Bioorg. Chem., 2019, 87, 550–559 CrossRef CAS.
- L. Chen, Y. Jin, W. Fu, S. Xiao, C. Feng, B. Fang, Y. Gu, C. Li, Y. Zhao, Z. Liu and G. Liang, ChemMedChem, 2017, 12, 1022–1032 CrossRef CAS.
- T. Y. Zhang, C. S. Li, P. Li, X. Q. Bai, S. Y. Guo, Y. Jin and S. J. Piao, Mol. Diversity, 2020 DOI:10.1007/s11030-020-10154-7.
- S. Patel, P. Modi, V. Ranjan and M. Chhabria, Bioorg. Chem., 2018, 78, 258–268 CrossRef CAS.
- H. Li, J. Zuo and W. Tang, Front. Pharmacol., 2018, 9, 1–21 CrossRef.
- M. J. Sanz, J. Cortijo and E. J. Morcillo, Pharmacol. Ther., 2005, 106, 269–297 CrossRef CAS.
- A. M. Mulhall, C. A. Droege, N. E. Ernst, R. J. Panos and M. A. Zafar, Expert Opin. Invest. Drugs, 2015, 24, 1597–1611 CrossRef CAS.
- X. Z. Ding, P. Iversen, M. W. Cluck, J. A. Knezetic and T. E. Adrian, Biochem. Biophys. Res. Commun., 1999, 261, 218–223 CrossRef CAS.
- R. Natarajan and J. L. Nadler, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 1542–1548 CrossRef CAS.
- C. Hu and S. Ma, MedChemComm, 2018, 9, 212–225 RSC.
- O. Rådmark, O. Werz, D. Steinhilber and B. Samuelsson, Trends Biochem. Sci., 2007, 32, 332–341 CrossRef.
- H. Hedi and G. Norbert, J. Biomed. Biotechnol., 2004, 2004, 99–105 CrossRef.
- M. P. Giovannoni, A. Graziano, R. Matucci, M. Nesi, N. Cesari, C. Vergelli, C. Biancalani, L. Crocetti, A. Cilibrizzi and V. D. Piaz, Drug Dev. Res., 2011, 72, 274–288 CrossRef CAS.
- R. Adepu, D. Rambabu, B. Prasad, C. L. T. Meda, A. Kandale, G. R. Krishna, C. M. Reddy, L. N. Chennuru, K. V. L. Parsa and M. Pal, Org. Biomol. Chem., 2012, 10, 5554–5569 RSC.
- P. M. Kumar, K. S. Kumar, C. L. T. Meda, G. R. Reddy, P. K. Mohakhud, K. Mukkanti, G. R. Krishna, C. M. Reddy, D. Rambabu, K. S. Kumar, K. K. Priya, K. S. Chennubhotla, R. K. Banote, P. Kulkarni, K. V. L. Parsa and M. Pal, MedChemComm, 2012, 3, 667–672 RSC.
- T. Goto, A. Shiina, T. Yoshino, K. Mizukami, K. Hirahara, O. Suzuki, Y. Sogawa, T. Takahashi, T. Mikkaichi, N. Nakao, M. Takahashi, M. Hasegawa and S. Sasaki, Bioorg. Med. Chem. Lett., 2013, 23, 3325–3328 CrossRef CAS.
- T. Goto, A. Shiina, T. Yoshino, K. Mizukami, K. Hirahara, O. Suzuki, Y. Sogawa, T. Takahashi, T. Mikkaichi, N. Nakao, M. Takahashi, M. Hasegawa and S. Sasaki, Bioorg. Med. Chem., 2013, 21, 7025–7037 CrossRef CAS.
- B. Purushothaman, P. Arumugam, G. Kulsi and J. M. Song, Eur. J. Med. Chem., 2018, 145, 673–690 CrossRef CAS.
- B. Purushothaman, P. Arumugam and J. M. Song, Front. Pharmacol., 2018, 9, 1–11 CrossRef.
- P. M. Kumar, K. S. Kumar, P. K. Mohakhud, K. Mukkanti, R. Kapavarapu, K. V. L. Parsa and M. Pal, Chem. Commun., 2012, 48, 431–433 RSC.
- A. Kodimuthali, R. Gupta, K. V. L. Parsa, P. L. Prasunamba and M. Pal, Lett. Drug Des. Discovery, 2010, 7, 402–408 CrossRef CAS.
- M. Hieke, C. Greiner, M. Dittrich, F. Reisen, G. Schneider, M. Schubert-Zsilavecz and O. Werz, J. Med. Chem., 2011, 54, 4490–4507 CrossRef CAS.
- T. Nam, S. J. Nara, I. Zagol-Ikapitte, T. Cooper, L. Valgimigli, J. A. Oates, N. A. Porter, O. Boutaud and D. A. Pratt, Org. Biomol. Chem., 2009, 7, 5103–5112 RSC.
- A. Rahmouni, S. Souiei, M. A. Belkacem, A. Romdhane, J. Bouajila and H. B. Jannet, Bioorg. Chem., 2016, 66, 160–168 CrossRef CAS.
- W. S. Shehab, M. H. Abdellattif and S. M. Mouneir, Chem. Cent. J., 2018, 12, 1–8 CrossRef.
- N. U. A. Mohsin and M. Ahmad, Turk. J. Chem., 2018, 42, 1–20 CrossRef.
- K. R. Roell, D. M. Reif and A. A. Motsinger-Reif, Front. Pharmacol., 2017, 8, 1–11 Search PubMed.
- M. C. Recio, I. Andujar and J. L. Rios, Curr. Med. Chem., 2012, 19, 2088–2103 CrossRef CAS.
- R. Fürst and I. Zündorf, Mediators Inflammation, 2014, 2014, 146832 Search PubMed.
- B. Peterson, M. Weyers, J. H. Steenekamp, J. D. Steyn, C. Gouws and J. H. Hamman, Pharmaceutics, 2019, 11(33), 1–46 Search PubMed.
- K. Kesarwani and R. Gupta, Asian Pac. J. Trop. Biomed., 2013, 3, 253–266 CrossRef CAS.
- B. H. McCarberg and B. Cryer, Am. J. Ther., 2015, 22, e167–e178 CrossRef.
- A. M. Faheem and D. H. Abdelkader, Novel drug delivery systems, in Engineering Drug Delivery Systems, ed. A. Seyfoddin, S. M. Dezfooli and C. A. Greene, Elsevier, 2020, pp. 1–16 Search PubMed.
- S. Thakur, B. Riyaz, A. Patil, A. Kaur, B. Kapoor and V. Mishra, Biomed. Pharmacother., 2018, 106, 1011–1023 CrossRef CAS.
- G. Palermo and M. D. Vivo, Computational Chemistry for Drug Discovery, in Encyclopedia of Nanotechnology, ed. B. Bhushan, Springer, Dordrecht, Netherlands, 2015, pp. 1–15 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10657g |
| This journal is © The Royal Society of Chemistry 2021 |