
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePseudo in situ construction of high-performance thermoelectric composites with a dioxothiopyrone-based D–A polymer coating on SWCNTs†
Wen-Qiang Quab,
Cai-Yan Gao *a,
Ping-Xia Zhangc,
Xin-Heng Fana and
Lian-Ming Yang*a
*a,
Ping-Xia Zhangc,
Xin-Heng Fana and
Lian-Ming Yang*a
aBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Green Printing, Institute of Chemistry, Chinese Academy of Sciences, Beijing, P. R. China. E-mail: gaocaiyan@iccas.ac.cn; yanglm@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, P. R. China
cKey Laboratory of Science and Technology on High-tech Polymer Materials, Institute of Chemistry, Chinese Academy of Sciences, Beijing, P. R. China
First published on 24th February 2021
Abstract
Organic polymer/inorganic particle composites with thermoelectric (TE) properties have witnessed rapid progress in recent years. Nevertheless, both development of novel polymers and optimization of compositing methods remain highly desirable. In this study, we first demonstrated a simulated in situ coagulation strategy for construction of high-performance thermoelectric materials by utilizing single-walled carbon nanotubes (SWCNTs) and a new D–A polymer TPO-TTP12 that was synthesized via incorporating dioxothiopyrone subunit into a polymeric chain. It was proven that the preparation methods have a significant influence on thermoelectric properties of the TPO-TTP12/SWCNT composites. The in situ prepared composite films tend to achieve much better thermoelectric performances than those prepared by simply mixing the corresponding polymer with SWCNTs. As a result, the in situ compositing obtains the highest Seebeck coefficient of 66.10 ± 0.05 μV K−1 at the TPO-TTP12-to-SWCNT mass ratio of 1/2, and the best electrical conductivity of up to 500.5 ± 53.3 S cm−1 at the polymer/SWCNT mass ratio of 1/20, respectively; moreover, the power factor for the in situ prepared composites reaches a maximum value of 141.94 ± 1.47 μW m−1 K−2, far higher than that of 104.68 ± 0.86 μW m−1 K−2 for the by-mixing produced composites. This indicates that the dioxothiopyrone moiety is a promising building block for constructing thermoelectric polymers, and the simulated in situ compositing strategy is a promising way to improve TE properties of composite materials.
Introduction
Thermoelectric materials, which can realize direct energy conversion between heat and electricity with no moving parts, no noise and a long operating lifetime even under very low temperature gradients relative to the environmental temperature, have been considered as promising candidates to meet current challenges of increasingly global energy shortages and environmental pollution.1,2 The thermoelectric performance is determined by the figure of merit (ZT = S2σT/k), where S, σ, T and k are the Seebeck coefficient, electrical conductivity, absolute temperature and thermal conductivity, respectively.3 Therefore, the high-performance thermoelectric materials are supposed to have a high electrical conductivity and high Seebeck coefficient, but low thermal conductivity. Given low thermal conductivities of organic materials or organic–inorganic composites, a simplified expression of power factor defined as PF = S2σ is most often used for evaluating their TE performances.4The organic–inorganic composites, composed of nano-sized inorganic semiconductors and organic conducting polymers, can produce dramatically enhanced TE performances through synergistic effects resulting from high electrical conductivities of inorganic components as well as large Seebeck coefficients and low thermal conductivities of organic constituents.5–12 In this respect, conducting polymers/carbon nanotube (CNT) composites have demonstrated tremendous progress.13–22 Currently, TE performance optimization of the polymer/CNT composites are concentrated on two aspects including the molecular design of polymers and the preparation technology.
The preparation methodology for organic/inorganic composites plays a vital role in determining performances of composites. Generally, two approaches are adopted to form the polymer/CNT TE composites. One is the in situ polymerization of monomers in a dispersion of SWCNTs, which is applicable to such classical conductive polymers as PANI, PPy, PTh, and PEDOT gotten easily by oxidative polymerization under simple conditions.13,14 The other is the drop-casting of a mixed suspension of the pre-prepared polymers and SWCNTs, which is suitable for those polymers that are structurally complicated and synthetically strict (such as water-free and oxygen-free conditions).16–22 Although many other film-forming technologies, such as dip-coating,23 spray-coating24 and spin-coating,25 were used for fabricating TE samples, almost all the recently-developed complicated TE polymer/CNT composites have been prepared through the direct drop-casting. However, the drop-casting method is difficult to form uniform interface coating layer of polymers on the surface of SWCNTs because of the rapid evaporation of solvents and static growth of polymers on the substrate, leading to conglutination and filling of the precipitated polymer in the gap of SWCNT networks. It is worth mentioning that a promising coagulation method has long been ignored in the preparation of thermoelectric materials, although it was widely used in nanotechnology and materials science because it can provide a better dispersion of inorganic particles in a polymer matrix26 and a uniform coating layer of polymers on the surface of inorganic particles.27
Another urgent and arduous task is to develop new types of conjugated polymers for TE polymer/CNT composites. In the past years, a number of new structural segments and special bonding modes were used for construction of more structurally complicated polymers than the classical polymers, and those conducting polymers were employed to form the composites with superior TE performances.15–22 In 2016, our group reported a kind of poly-Schiff base/SWCNT TE composites and the chelation effect of the Schiff base with transition metal ions on the TE performance.15 Subsequently, Wang et al. prepared a series of TE composite films based on SWCNTs and bipyridine-containing polyfluorene derivatives, discussing the variation trend of TE performance caused by chelation of the bipyridine unit with various metal ions.17 It is worth mentioning that Wang et al. made a great deal of work for developing new polymer/SWCNT composites with high TE performances. Notably, a benzodithiophene (BDT)-based conjugated polymer (PBDT-EDOT) was developed, and the thermoelectric behavior of its composite films with SWCNT was investigated elaborately, with a maximum power factor of 74.6 μW m−1 K−2 for the SWCNT content of 90% at 400 K.18 Making use of olefin metathesis, the cross-linking effect on thermoelectric properties of polymer/CNT composite films have been studied.19 Consecutively, a class of self-assembled alkyl chain-linked naphthalenediimide (NDI)/SWCNT composites were systematically examined, achieving the best power factor of 237.6 ± 20.8 μW m−1 K−2.20 Very recently, by controlling interfacial doping of 7,7,8,8-tetracyanoquinodimethane (TCNQ) deposited in vacuum, a type of high-performance thermoelectric composites of SWCNTs and 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene (C8BTBT), were developed.21 Almost simultaneously, Qiu et al. reported a new class of thermoelectric composites by combining SWCNTs and a BDT-based D–A polymer with carbazole segment as the side-chain. Both power factors reached about 60 μW m−1 K−2 before and after doping composites with F4TCNQ at room temperature.22
Despite these advances, the structural types of conjugated polymers for TE composites are still extremely limited and their TE performances remain far below the expected level. Apparently, it would be very meaningful to develop novel types of conjugated polymers and efficient compositing method for TE composite materials. Accordingly, 4-(dicyanomethylene)-2,6-diphenyl-4H-thiopyran-1,1-dioxide (Fig. 1a) and its precursor dioxothiopyranone (Fig. 1b) aroused our interest as a sulfone-containing analogue of tetracyanoquinodimethane (TCNQ).28 The dioxothiopyranone structure exhibits many advantages:29 first, the moiety is an approximately planar ring with only minor deviation, and a more rigid near-plane structure could be formed through intramolecular interaction between the ortho-hydrogen on the 2- and 6-phenyls and the oxygen atom of the sulfone group, being helpful to the improvement of the carrier transport and the electrical conductivity; second, its electron-deficient nature, reinforced synergistically by strongly electron-withdrawing carbonyl or dicyanomethylidene group and the sulfone group, offers the possibility for constructing a D–A type conjugated polymer, which may be beneficial to the enhancement of carrier mobility of the polymer on account of intramolecular charge transfer; and last but not least, the moiety is ready to undergo molecular modification or derivatization at its 2- and 6-positions, opening a pathway to enrich organic TE materials. In brief, dioxothiopyranone unit is a very promising building-block for construction of high-performance polymeric thermoelectric materials. However, there has yet been no report on the dioxothiopyranone-based polymers and their application in TE materials so far, although its small-molecular derivatives served as excellent electron-transporting materials in electrophotography in the mid of 1980s.30
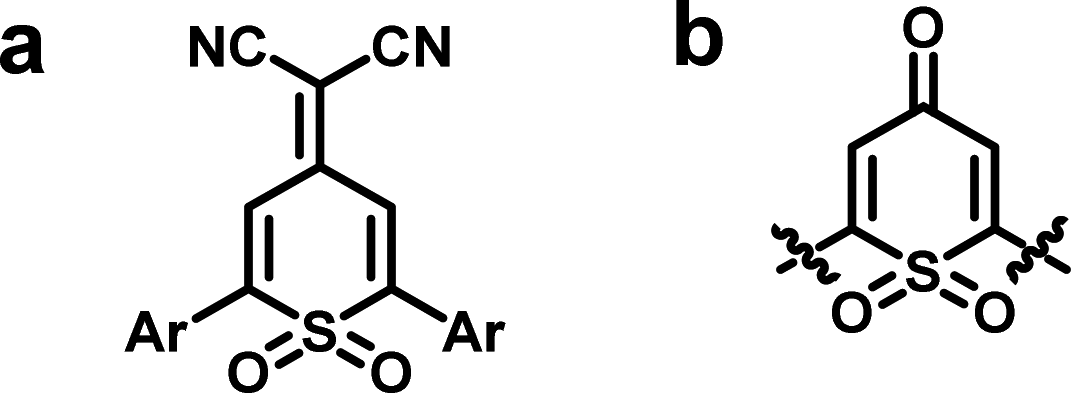 | ||
| Fig. 1 Typical structure of (a) dioxothiopyranone-based electron-transporting materials and (b) dioxothiopyranone substituted at the 2- and 6-positions. | ||
We designed and synthesized a novel thiopyranone-based D–A alternating conjugated polymer (coded as TPO-TTP12) by Stille coupling polymerization. Thereafter, two series of polymer/SWCNT composites were prepared and evaluated taking advantage of, respectively, the simulated in situ coagulation method (abbreviated as “in situ method”) and the directly-mixing preparation method (abbreviated as “by-mixing method”). The results show that introduction of dioxothiopyranone is effective to elevate the Seebeck coefficients of the composites. By comparison, the in situ method makes a well-balance between the electrical conductivity and the Seebeck coefficient of the composites, achieving significantly high Seebeck coefficients with a maximum value of 66.1 ± 0.05 μV K−1. As a result, the in situ prepared composite film exhibits the highest power factor of 141.9 ± 1.5 μW m−1 K−2, which is nearly 30% higher than that of the by-mixing prepared sample. To the best of our knowledge, it is the first time that the simulated in situ coagulation strategy was used for fabrication of polymer/CNT TE composites, and it may be expected to become a universally applicable means to develop high-performance polymer/CNT thermoelectric composites.
Results and discussion
Preparation of the polymer and its composite films with SWCNTs
The synthetic route to the polymer and the preparation process of the polymer/SWCNT composite films are shown in Schemes 1 and 2, respectively (the detailed synthesis procedure and characterization are described in the ESI Section†).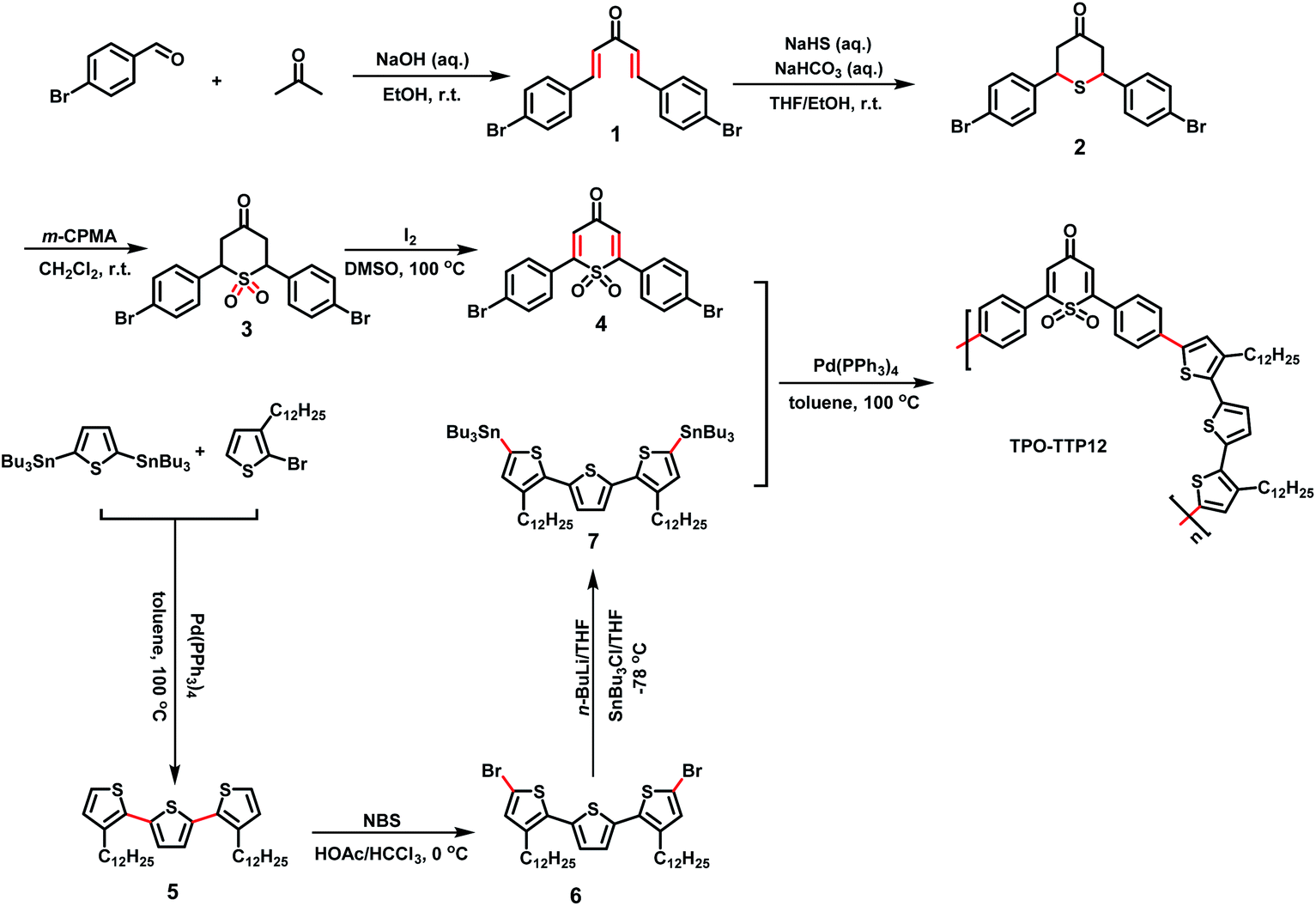 | ||
| Scheme 1 The synthetic route to the polymer TPO-TTP12. | ||
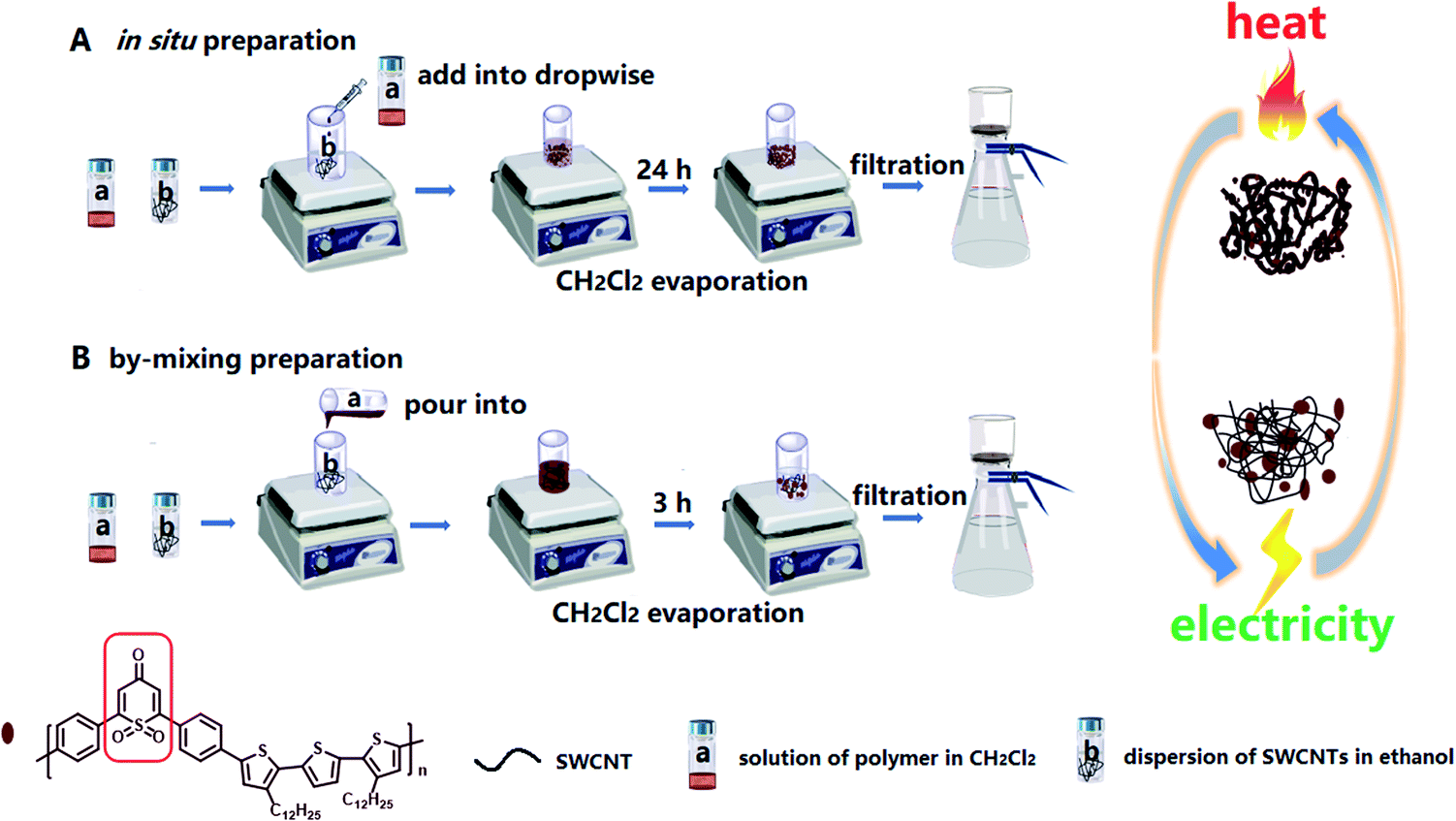 | ||
| Scheme 2 Two preparation processes of the TPO-TTP12/SWCNT composite films. | ||
Design and synthesis of the polymer TPO-TTP12
As demonstrated in Scheme 1, the polymer TPO-TTP12 was synthesized by the Stille coupling reaction between 1,1-dioxo-thiopyranone substituted by 4-bromophenyl (compound 4) and ditin reagent of terthiophene having two dodecyl side chains (compound 7). Following the previously reported reference with minor modification,30 the intermediate 4 as acceptor segment was achieved via a series of reactions from the condensation of 4-bromobenzaldehyde and acetone, to the addition of sodium hydrosulfide hydrate to dienone, the oxidation of tetrahydrothiopyran, and the dehydrogenation of sulfone. The intermediate 7 as donor fragment was obtained by the Stille coupling between the corresponding bromide and tin reagent. Apparently, the symmetry of dodecyl-substituted terthiophene simplifies the structural characterization of the polymer to some extent. Meanwhile, the incorporation of long alkyl chains increases significantly the solubility of polymers in common organic solvents. In particular, the combination of alternating dioxothiopyranone acceptor with terthiophene donor would be expected to enhance the carrier mobility due likely to strong intramolecular charge transfer effects.Fabrication of polymer/SWCNT composite films
Scheme 2 illustrates two different strategies for the fabrication of polymer/SWCNT composite films. A simulated in situ preparation procedure (the in situ method) is depicted in Scheme 2A: firstly, the solution (a) and the dispersion (b) were prepared respectively by dissolving the polymer TPO-TTP in anhydrous dichloromethane and dispersing homogeneously SWCNTs in ethanol; then the solution (a) was added drop-wise into the dispersion (b) with a syringe so that the polymer precipitated out slowly and wrapped on the surface of SWCNTs; and after slow evaporation of CH2Cl2 with stirring for 24 h, the residual suspension was filtrated by suction to give the composite films with a uniform surface. For comparison, a directly-mixing procedure (the by-mixing method) was performed as described in Scheme 2B. In contrast, such a by-mixing method led to the poor TE performance of composite films because of too quick precipitation of the polymer from solution (a) on the SWCNTs. However, all the composite films prepared by two methods show high thermal stability and good flexibility (as shown in Fig. S23 and S24 in the ESI†).Photophysical and electrochemical properties and theoretical simulation
To facilitate understanding, the structures of the polymer and its D and A units are displayed in Fig. 2a. The UV-vis absorption spectra of their CH2Cl2 solutions and solid films are shown in Fig. 2b. In the dilute dichloromethane solution, both TPO as acceptor (A unit) and TTP12 as donor (D unit) exhibit similar absorption bands in the range of 300–400 nm originating from the π–π* absorption. Compared to the spectra of individual fragments, the spectrum of TPO-TTP12, constructed from D and A units, shows a strong absorption in the broad region of from 300 to 700 nm, which can be ascribed to the strong intramolecular charge transfer (ICT) transition from the electron-donating unit (TTP12) to the electron-withdrawing part (TPO).31 By contrast, a red-shifted broadening of around 60 nm can be observed for the solid-state samples on account of the stronger molecule aggregations resulting from the enhanced intermolecular π–π interactions in the solid film. Simultaneously, the corresponding optical bandgap was calculated to be about 1.81 eV using the formula Eoptg = 1240/λfilmonset according to the onset absorption edge of the polymeric film at 685 nm.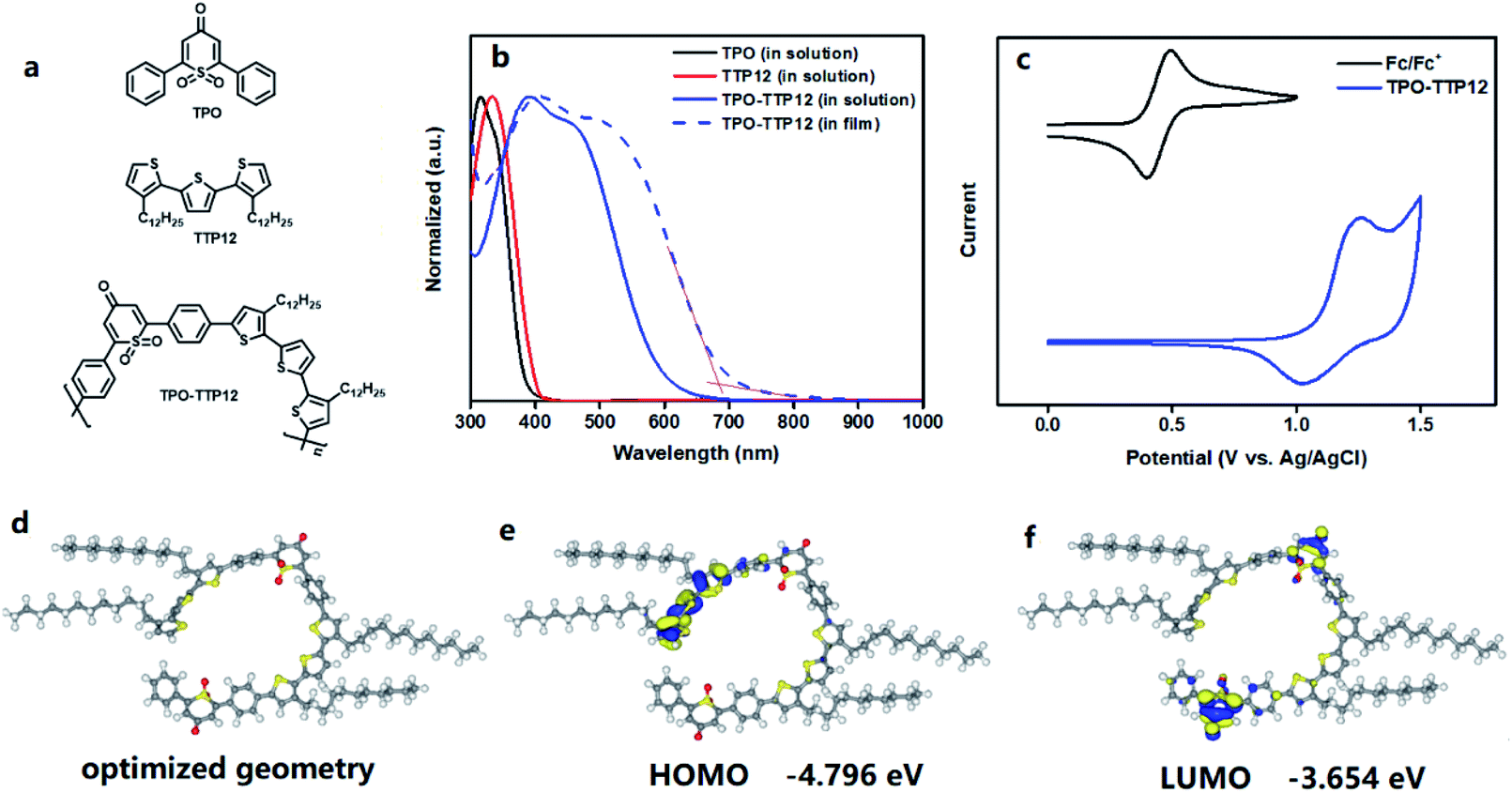 | ||
| Fig. 2 (a) The molecular structures of D and A units and the polymer TPO-TTP12; (b) UV-vis absorption spectra for the D and A units and the polymer TPO-TTP12 in CH2Cl2 solutions (solid lines) and solid films (dot line); (c) cyclic voltammetry curves of the polymer film with ferrocene as the internal standard substance; (d) the optimized geometry for the lowest energy conformation; (e and f) the DFT-calculated isosurfaces of the HOMO (e) and LUMO (f) orbital densities of the polymer at the GGA/BLYP level. | ||
The electronic energy levels of the polymer were investigated by cyclic voltammetry using the ferrocene/ferrocenium (Fc/Fc+) as internal calibration, which has an oxidation potential of 4.8 eV at vacuum level. As shown in Fig. 2c, the highest occupied molecular orbital (HOMO) level of the polymer is estimated to be −5.44 eV from the onset oxidation potential according to the equation EHOMO = −(4.80 eV − eφ1/2,Fc/Fc+ + eφoxonset), where φ1/2,Fc/Fc+ is the half wave potential of Fc/Fc+ versus Ag/AgCl in the measurement system (0.44 V). The lowest unoccupied molecular orbital (LUMO) energy level of the polymer can be calculated to be −3.63 eV from the equation of ELUMO = EHOMO + Eoptg. Evidently, a low LUMO energy level for polymers would be very beneficial to p-doping on SWCNTs.
Density functional theory (DFT) calculations were performed to investigate the molecular configuration and electronic structure of the polymer TPO-TTP12, which possesses two main conformations due to the rotation around the bonds that link the acceptor unit and donor unit. For ease of analysis, the dimer with two repeated units was chosen as a model system (Fig. S25†). Different from common linear and zigzag geometries, the lowest energy geometry is a nearly circular structure (Fig. 2d), which may probably be attributed to the unique geometric angle in the TPO unit. In addition, the calculated isosurfaces of the HOMO and LUMO orbital densities are shown in Fig. 2e and f, respectively. The HOMO orbital principally concentrates on the donor segment with three thiophene rings at one end of the dimer, but extends over the neighboring linking benzene ring, whereas with smaller contributions. In contrast, the LUMO orbital is mainly localized on two 1,1-dioxothiopyranone segments (TPO) with the electron-withdrawing nature, involving slightly the adjacent phenyl rings. From the distribution diagrams of wave functions, it can be deduced that the alkyl chains of the polymer have hardly any contribution to the frontier molecular orbits. As a result, the HOMO and LUMO levels were calculated to be −4.796 eV and −3.654 eV, respectively, and the corresponding band gap was acquired to be 1.142 eV from the equation ΔE = EHOMO − ELUMO. Besides, the calculated charge population shows that the oxygen atoms of sulfone and carbonyl in TPO carry negative charges, implying that these atoms can attract electrons from the polymer backbone, and thus promote the electron transfer from SWCNTs to the polymer.
IR and Raman spectra
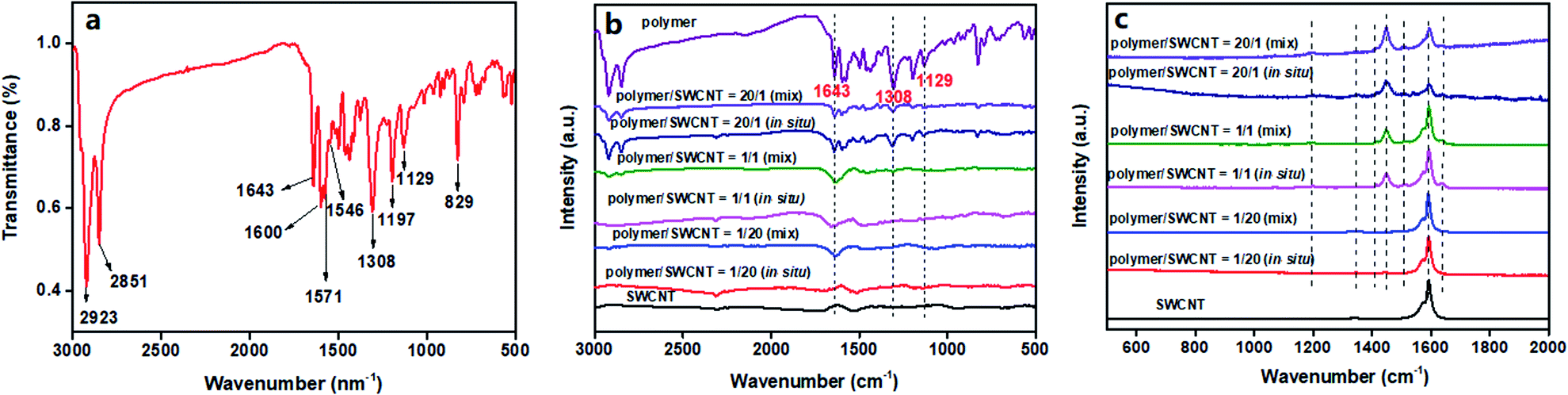
Fourier transform infrared spectroscopy (FT-IR) is a simple and effective means for analysis of polymeric structures. The FT-IR spectrum of the polymer TPO-TTP12 is shown in Fig. 3a. The strong absorption bands at 2923 cm−1 and 2851 cm−1 are assigned to the stretching vibration of saturated C–H bonds.32 The characteristic absorption arising from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration appears at a distinct lower wave number of 1643 cm−1 due to its conjugation with two adjacent CC bonds.33 A set of bands at 1600 cm−1, 1571 cm−1 and 1546 cm−1 are defined as the CC stretching vibration of the aromatic ring.34 The sharp peaks at 1308 cm−1 and 1129 cm−1 are corresponding to the characteristic absorption caused by the stretching vibration of SO in sulfone.28 Additionally, the bands at 1197 cm−1 and 829 cm−1 are identified as the in-plane and out-of-plane aromatic C–H bending vibrations for the 1,4-disubstituted aromatic groups.35 In a word, the FT-IR spectrum shows the characteristic absorptions of the important functional groups for the polymer, preliminarily confirming its successful synthesis.
O stretching vibration appears at a distinct lower wave number of 1643 cm−1 due to its conjugation with two adjacent CC bonds.33 A set of bands at 1600 cm−1, 1571 cm−1 and 1546 cm−1 are defined as the CC stretching vibration of the aromatic ring.34 The sharp peaks at 1308 cm−1 and 1129 cm−1 are corresponding to the characteristic absorption caused by the stretching vibration of SO in sulfone.28 Additionally, the bands at 1197 cm−1 and 829 cm−1 are identified as the in-plane and out-of-plane aromatic C–H bending vibrations for the 1,4-disubstituted aromatic groups.35 In a word, the FT-IR spectrum shows the characteristic absorptions of the important functional groups for the polymer, preliminarily confirming its successful synthesis.
 | ||
| Fig. 3 FT-IR spectra of (a) the polymer TPO-TTP12 and (b) its representative composite films; (c) Raman spectra of the representative composite films. | ||
In order to ensure the chemical structure of the polymer remain intact after compositing with SWCNTs, the IR spectra for pure SWCNTs and some representative composite films are also displayed in Fig. 3b with the reappearance of the IR spectrogram of the polymer. It is quite clear that all those composites exhibit spectral absorption patterns similar to the pristine polymer, without new peaks appearing. This suggests that the chemical structure of the polymer is relatively stable without the occurrence of structure damage during the combination with SWCNTs. However, it should be noted that the absorption bands, respectively, at 1643 cm−1 for CO and at 1308 cm−1 and 1129 cm−1 for SO, are obviously blue-shifted to higher wavenumbers. This should be a compromising consequence of two factors: on one hand, the intermolecular hydrogen bonding in the polymer weakened with decreasing the polymer concentration by introduction of SWCNTs, leading to a strong blue-shift of the absorption peaks; simultaneously, a modest red-shift occurs due to the reduced force constants for CO and SO, which are caused by the electron transfer from the SWCNT system to the electron-deficient polymeric backbone arising from the interfacial interactions between the polymer and SWCNTs. As a result, a relative blue shift of the absorption peaks can be observed since their blue-shifts are more prominent than the red-shifts. Furthermore, the degree of blue shift increases with the increase of SWCNT contents. Especially, compared to the by-mixing prepared composite films, a more remarkable blue-shift for the in situ prepared composites can be observed since the stronger π–π interfacial interaction is induced by the more uniform wrapping of the polymer on the surface of SWCNTs. The Raman spectra of the pure SWCNTs and several typical composite films are shown in Fig. 3c to study the interfacial interactions between the polymer and SWCNTs in composite films. The pure SWCNTs present strong characteristic G bands at 1573 cm−1 and 1593 cm−1, respectively, which arise from the stretching vibration of sp2-hybridized carbon atoms.36 In contrast, the polymer/SWCNT composite films reveal a gradual blue-shift of the G-band peaks with increasing polymeric contents, implying the effective charge transfer from the SWCNTs to the D–A type of polymeric molecules.37,38 In particular, the G peak at 1593 cm−1 for the pure SWCNTs shifts to by 5 cm−1 higher wave number, appearing at 1598 cm−1 in the composite film with polymer/SWCNT mass ratio of 20/1. Unfortunately, the ideal Raman spectrum of the pure polymer failed to be recorded due to the overlap of the strong fluorescence emission signals and the weak Raman scattering signals at an excitation wavelength of 514 nm. In the acquired spectra, because of the strong fluorescence interferences of the polymer, the background noises of the composite films were synchronously enhanced with the increase of polymeric contents, leading to the indiscernibility of some characteristic peaks; on the other hand, there remained several important vibration modes appearing in the spectra of some composite films. An extremely weak band at 1190 cm−1 is assigned to the characteristic peak of SO stretching vibration of the sulfone. The band at 1341 cm−1 originates from the C–C stretching vibration of the π–π conjugated backbone.19 The bands of the CC symmetrical stretching vibration appear at 1413 cm−1 and 1449 cm−1, respectively.19 Also, the asymmetric CC stretching mode is found at 1507 cm−1.18 In addition, the weak peak at 1634 cm−1 is definitely attributed to the stretching vibration of the CO conjugated with two adjacent CC bonds.39 At the same time, it should be noted that the in situ prepared composites show slightly obvious vibration signals in comparison to those of the by-mixing prepared ones. This is because their fluorescence interferences is attenuated more effectively, which is caused by the diluted concentration of the polymer resulting from the more effective interactions between polymeric molecules and SWCNTs.
Surface morphologies
As well known, the properties of materials depend on the structures, including their molecule-level structures and micromorphology. All the morphologies for the polymer, SWCNTs and the polymer/SWCNT composites were examined to understand the relationship between the micro-structure and performance of the composites through both the field-emission scanning electron microscope (FESEM) and transmission electron microscope (TEM).Several selected morphology images are shown in Fig. 4. Fig. 4a–f present respectively an adhesive sub-globose morphology formed via the agglomeration of scale-like granular particles for the pristine polymer and a randomly intertwined fibrillar morphology for the pure SWCNTs. For ease of viewing, the polymer/SWCNT composite film with mass ratio of 20/1 is taken as a typical example. The images for the in situ prepared composite film with different magnifications are displayed in Fig. 4b–d. From Fig. 4b, it can be seen that the morphology of the composite is apparently influenced by the introduction of SWCNTs, merging quasi-spheres of the polymer and fibers of the SWCNTs into a porous network structure with the pearl-necklace-like morphology, which is formed by the interfacial attractions from the π–π interactions and van der Waals forces between SWCNTs and the polymer. This morphology is strikingly different from the previously reported twining sphere-wire structure formed by the surface adhesion of SWCNTs on the spheres of polymers.15 Moreover, the unique morphological feature is further accentuated as shown in the TEM image of Fig. 4e, which is in sharp contrast to the thinner and smoother TEM morphology for the pure SWCNTs (Fig. S26†). With changing the enlargement factor from high to low magnifications, a small cellular structure with relative uniform pores is presented in Fig. 4c. Interestingly, a fairly smooth and homogenous morphology appears at smaller magnification, as demonstrated in Fig. 4d. For comparison, the images with the corresponding magnifications for the by-mixing produced polymer/SWCNT composite film with the mass ratio of 20/1 were shown in Fig. 4g–i. Comparing with Fig. 4b and g gives a more disorder network morphology with abundant bare SWCNTs, in which bulky grains of the polymer are sporadically scattered. As expected, a more uneven and irregular topography stands out at lower magnification, as shown in Fig. 4h and i. Especially, the morphology with large irregular bulges in Fig. 4i is in stark contrast to that of Fig. 4d. It is surprising that the morphology presented in Fig. 4e is obviously different from that in Fig. 4j in which some bulky grains are adhered to bare SWCNTs with almost no coating of the polymer on the surface of SWCNTs. Thus, it can be well understood that the preparation method for composite films has a huge impact on their microscopic morphologies.
 | ||
| Fig. 4 FESEM images of (a) the pristine polymer; (b–d) the in situ produced polymer/SWCNT composite film with a polymer/SWCNT mass ratio of 20/1 at different magnifications; (f) the pure SWCNTs and (g–i) the by-mixing produced polymer/SWCNT composite film with a polymer/SWCNT mass ratio of 20/1 at different magnifications; TEM images of (e) the in situ produced and (j) the by-mixing prepared polymer/SWCNT composite films with a polymer/SWCNT mass ratio of 20/1. | ||
XPS spectra
X-ray photoelectron spectroscopy (XPS) was adopted to investigate the compositions of materials and changes in the binding energies for the key elements. The polymer/SWCNT composites with the mass ratio of 20/1 were taken as representative examples with the pure SWCNTs and pristine polymer as controls. Notably, in comparison with the XPS spectrum of the SWCNT film, both the pristine polymer and its composites show a new energy peak at about 164 eV coming from the S 2p of the polymer, confirming the successful compositing of the polymer and SWCNTs (shown in Fig. 5a). From the deconvolution spectrum of S 2p of the pristine polymer provided by Fig. 5b, two set of S 2p doublets, originating from two species of S in the polymer, are clearly visible. One S 2p doublet at low binding energy is separated into S 2p3/2 at 163.65 eV and S 2p1/2 at 164.83 eV, which are derived from thienyls in the polymer; the other S 2p doublet located at higher binding energy is as well divided into two peaks at 167.87 eV and 169.05 eV respectively, corresponding to the S 2p3/2 and S 2p1/2 of the higher oxidation state of sulfur in the dioxothiopyrone unit.40,41 In the composite films, all of their XPS spectra display two set of similar S 2p doublets with only slight differences of both energy values and peak areas (as shown in Fig. S29 in the ESI†). In addition, a significant doping-induced change is discerned in both the C 1s and O 1s core levels. The deconvolution C 1s spectra of sp2-hybridized carbon for the pure SWCNTs and the composite films with the polymer/SWCNT mass ratio of 20/1 are displayed in Fig. 5c. It is observed that the peaks of the C 1s for the by-mixing and the in situ prepared composite films are downshifted, respectively, to lower binding energy levels of 284.32 eV and 284.25 eV relative to that of 284.41 eV for the pure SWCNTs, corresponding to a change in the Fermi level of SWCNTs. This indicates that the effective electron transfer occurs from SWCNTs to the polymer, namely that the polymer acts as an acceptor to attract electrons from the SWCNT backbone.42–44 And, the more distinct change in the binding energy for the in situ prepared composite reveals the more effective interaction between SWCNTs and polymer. Considering only two characteristic species of oxygen atoms in the polymer, the binding energies of O 1s for the polymer were also used for clarifying the charge transfer between the polymer and SWCNTs. From the deconvolution spectrum of O 1s for the pristine polymer in Fig. 5d, we can see that the original O 1s single peak is separated into two single peaks, including O 1s peak of carbonyl at 531.64 eV and O 1s peak of sulfone at 532.42 eV. Compared with the pristine polymer, the O 1s peaks of the by-mixing and the in situ prepared composite films are shifted to the lower bonding energies from 532.42 eV to 532.33 and 531.91 eV for sulfone (Fig. 5e) and from 531.64 eV to 531.52 and 531.09 eV for carbonyl (Fig. 5f), respectively, which may be explained for the enhanced electronic cloud density on the electronegative O atoms of carbonyl and sulfone resulting from the electron transfer from the SWCNT system to the polymer backbone.42–45 These results are exactly in concert with those of Raman spectra and simulated calculations, revealing the strong interfacial interactions between the polymer and SWCNTs.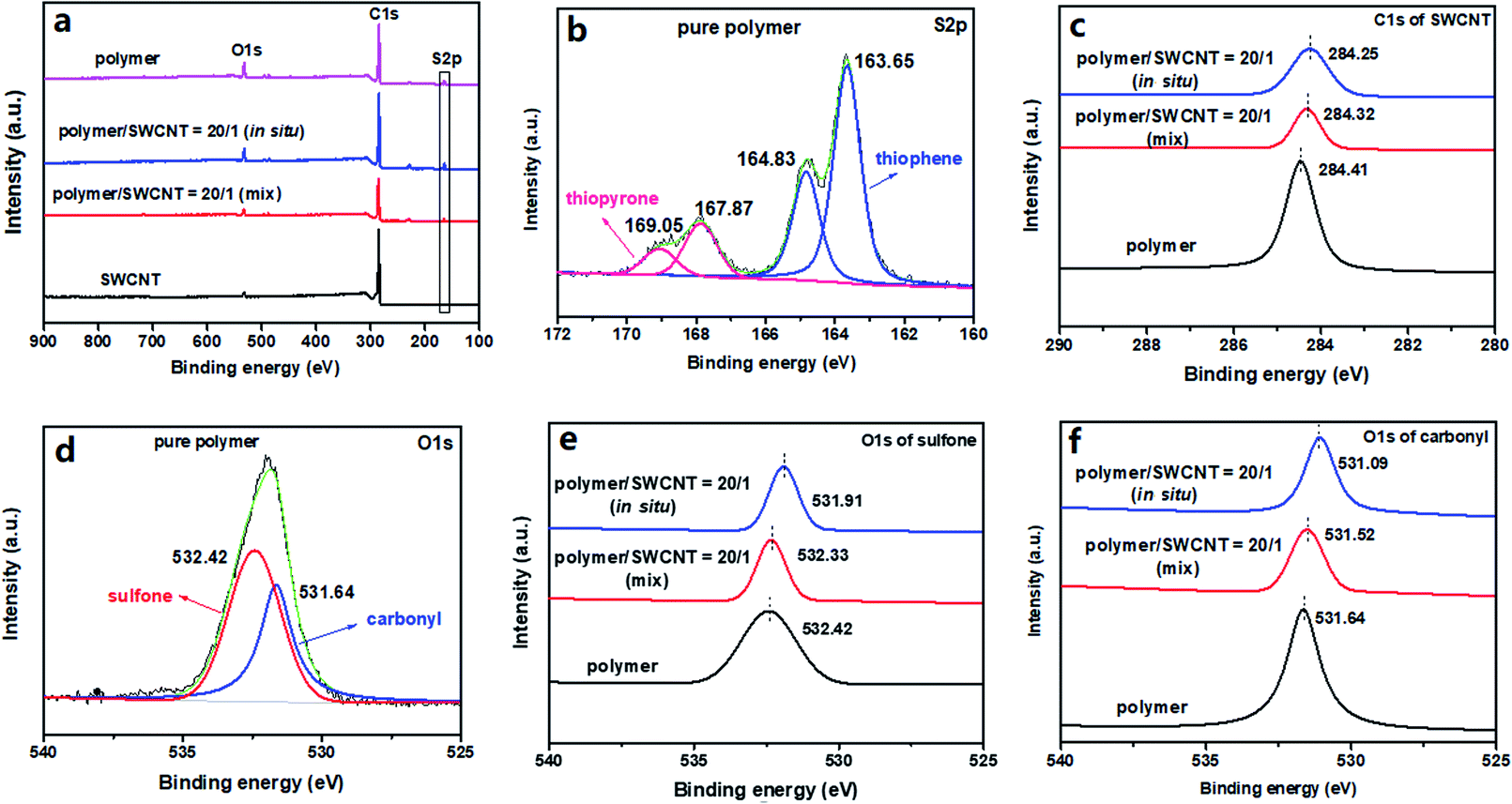 | ||
| Fig. 5 XPS spectra of (a) full-range for the pristine polymer, the pure SWCNT and the composite films prepared by two methods with a polymer/SWCNT mass ratio of 20/1; (b–f) the deconvolution of (b) S 2p for the pure polymer, (c) C 1s of Csp2 for the pure SWCNTs and the composite films prepared by two methods with a polymer/SWCNT mass ratio of 20/1, (d) O 1s for the pure polymer, (e) O 1s of sulfone and (f) O 1s of carbonyl for the pure SWCNTs and the composite films prepared by two methods with a polymer/SWCNT mass ratio of 20/1. | ||
Thermoelectric performance
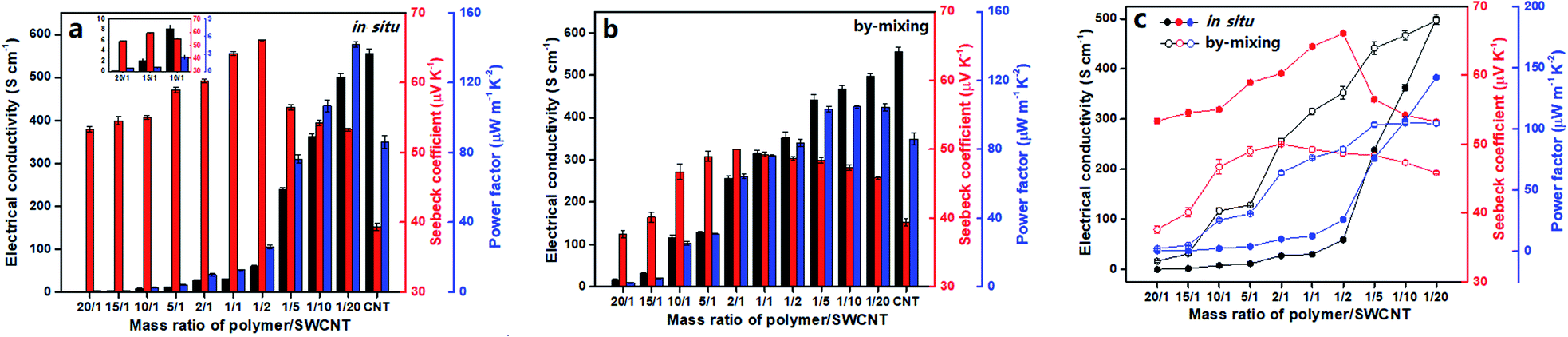
To evaluate the TE performance of the composites, the key parameters for organic thermoelectric materials, i.e., the electrical conductivity (σ), Seebeck coefficient (S) and power factor (PF), were investigated as a function of the polymer/SWCNT mass ratio. Note that the electrical conductivity and Seebeck coefficient of the pure polymer cannot be measured directly by the current four-probe method owing to its relatively poor conducting performance.Firstly, effects of polymer/SWCNT mass ratios on TE properties in the in situ prepared composite films are outlined in Fig. 6a. The electrical conductivity of the composite films increases monotonously with the increase of SWCNT doping contents, reaching a maximum value of 500.5 ± 53.3 S cm−1 after experiencing a first mild then sudden rise, which approaches the conductivity of 555.85 ± 10.1 S cm−1 for the pure SWCNTs. The reason should easily be understood. At a high ratio of polymers to SWCNTs, the low conductive polymer plays a leading role in limiting electrical conductivities of the composites due to its dense and thick wrapping on the SWCNTs' surface.46 But rather, the electrical conductivity of the composites is primarily dominated by highly conductive SWCNTs when the wrapping layer of the polymer on SWCNTs' surface turns thinner with declining polymer/SWCNT mass ratio, likely producing a rapid even steep rise. With regard to Seebeck coefficients, they go through a process of rising first and then descending with the increase of the SWCNT content, reaching a peak value of 66.10 ± 0.05 μV K−1 at the polymer/SWCNT mass ratio of 1/2, which is about 70% higher than that of the pure SWCNTs (39.36 ± 0.54 μV K−1). In principle, the composite with the smallest loading of SWCNTs should give the maximum Seebeck coefficient closest to that of the pristine polymer. Nevertheless, due to the strong correlations between the Seebeck coefficient and carrier mobility, low SWCNT loadings leads to low carrier mobilities and thereby low Seebeck coefficients. As anticipated, the Seebeck coefficients begins to smoothly fall after the peak value with the decrease of polymer contents, indicating a gradually weakening effect of the polymer on the Seebeck coefficient of the composites. The above results suggest that the polymer with dioxothiopyrone unit has a substantial contribution to the improvement of Seebeck coefficients of the composites. Additionally, the power factor of the composites also shows a monotonous increase with the increase of SWCNT content. It is mainly ascribed to a dramatic increase in the electrical conductivity and a relatively moderate fluctuation in Seebeck coefficient with the decrease of the polymer loadings. Unexpectedly, the best power factor of 141.94 ± 1.47 μW m−1 K−2 was obtained, which is far higher than that of the pure SWCNTs (86.04 ± 3.47 μW m−1 K−2) and approximately two and a half times as large as the maximum value of the poly-dodecylthiophene/SWCNT composites reported very recently by Chen et al. (60 μW m−1 K−2).47
 | ||
| Fig. 6 Effects of (a and b) polymer/SWCNT mass ratios on the TE performance for (a) the in situ prepared and (b) the by-mixing prepared polymer/SWCNT composite films; and (c) two compositing strategies on TE performances. | ||
As a comparison, Fig. 6b describes the variation tendency of TE properties for the by-mixing prepared polymer/SWCNT composites as the polymer/SWCNT mass ratio changes. Similarly, the curve of electrical conductivities for these by-mixing composites shows a monotonously upward trend, and maintains a steady linear growth within the entire range of mass ratios. As a result of the dispersion of almost totally bare SWCNTs in the polymer, Seebeck coefficients of the by-mixing prepared composites should be more approximate to that of SWCNTs because a large number of bare SWCNTs reinforce their contribution. As expected, the curve of the Seebeck coefficient goes up first and then down, with only a small fluctuation of around 10 μV K−1 from 37.64 ± 0.54 μV K−1 to 50.02 ± 0.04 μV K−1. Their power factors show a trend of rapid increase with the increase of SWCNT contents in the range of low SWCNT loadings, but keep almost unchanged after a mass ratio of 1/5. Finally, the highest power factor for such composites was calculated to be 104.68 ± 0.86 μW m−1 K−2, which is 18 μV m−1 K−2 or so higher than that of the pure SWCNTs (86.04 ± 3.47 μW m−1 K−2).
To more intuitively compare the influence of preparation methods on TE performances, Fig. 6c presents the TE performance of the composite films prepared by two different strategies at the same time. All Seebeck coefficients for the in situ prepared composite films are higher than those of the by-mixing prepared ones in the case of the corresponding polymer/SWCNT mass ratio, with a biggest difference of approximate 20 μV K−1. The most essential cause for this big gap between the Seebeck coefficients can be attributed to the difference of their electron energy filtering effects. For the in situ prepared composites, the uniform and dense coating of the nanostructured polymer on the surface of SWCNTs can effectively enhance the electron energy filtering effect between interfaces, allowing the high-energy carriers to pass but blocking the low-energy carriers simultaneously. The resulting mean carrier energy in electron transport increases, leading to the enhancement of the Seebeck coefficient.48,49 On the contrary, due to the aggregation of a great deal of bare SWCNTs resulting from inhomogeneous and discrete wrapping of the polymer on the surface of SWCNTs, the electron energy filtering effect for the by-mixing prepared composites becomes weak significantly or even disappears. Differing from the Seebeck coefficients, two curves of electrical conductivities from those composites prepared by two strategies intersect each other to form an atypical olive-like shape. This result may be explained as follows. The dense and thick enwrapping of polymer on the surface of SWCNTs for the in situ prepared composites and the crowded dispersion of polymer in the SWCNT network for the by-mixing prepared composites result in their respective approximate low electrical conductivity at the low loading of SWCNTs. With the increase of SWCNT contents, the contribution of the exposed SWCNTs in by-mixing prepared composites to the electrical conductivity gain gradually the upper hand over that of the SWCNTs wrapped heavily with polymer in the in situ prepared composites, causing two curves to diverge from each other. As the content of SWCNTs continues to increase, the thinning coating layer of polymer on the surface of SWCNTs for the in situ prepared composites leads to a dramatic increase of electrical conductivities, rendering two curves to get closer and closer and finally converge. Consequently, the curves of the power factors for two types of composites, regardless of preparation methods, show a variation trend similar to those of the electrical conductivity, suffering a reversal at the 1/10 mass ratio of polymer/SWCNT. Finally, the in situ prepared composite achieves the largest power factor of 141.94 ± 1.47 μW m−1 K−2 higher by about 37 μW m−1 K−2 than the maximum power factor the by-mixing prepared composite does.
Conclusions
In summary, we have designed and synthesized a new-type polymer TPO-TTP12 by integrating a dioxothiopyrone moiety as electron-withdrawing building-block into D–A polymers. By the simulated in situ compositing process utilizing the coagulation method, the flexible high-performance TPO-TTP12/SWCNT composite films with TPO-TTP12 coating layer on the SWCNTs' surface have been obtained. The effects of different methods for preparing the TPO-TTP/SWCNT composites on their thermoelectric properties were systematically examined. Compared to the by-mixing method, the in situ produced composites exhibit remarkably high maximum values in both the electrical conductivity and the Seebeck coefficient. The best power factor of the in situ prepared composites reached up to 141.94 ± 1.47 μW m−1 K−2, which is about 37 μW m−1 K−2 higher than that of the corresponding by-mixing prepared ones. This suggests that the in situ compositing method based on the coagulation process is a facile and effective strategy for constructing high-performance TE composites, and can be extended to other polymeric systems. In brief, this preliminary research not only enriches the family of TE composites but also opens a new avenue for improving TE performance of composites based on complicated polymers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank National Natural Science Foundation of China (Project No. 21572235, 21503234, and 21402209) for financial support of this work.Notes and references
- G. Tan, L. D. Zhao and M. G. Kanatzidis, Chem. Rev., 2016, 116, 12123–12149 CrossRef CAS.
- X. Shi and L. Chen, Nat. Mater., 2016, 15, 691–692 CrossRef CAS.
- M. S. Dresselhaus, G. Chen, M. Y. Tang, R. G. Yang, H. Lee, D. Z. Wang, Z. F. Ren, J.-P. Fleurial and P. Gogna, Adv. Mater., 2007, 19, 1043–1053 CrossRef CAS.
- T. M. Tritt and M. A. Subramanian, MRS Bull., 2006, 31, 188–198 CrossRef.
- Y. Du, S. Z. Shen, K. F. Cai and P. S. Casey, Prog. Polym. Sci., 2012, 37, 820–841 CrossRef CAS.
- M. He, F. Qiu and Z. Q. Lin, Energy Environ. Sci., 2013, 6, 1352–1361 RSC.
- B. T. McGrail, A. Sehirlioglu and E. Pentzer, Angew. Chem., Int. Ed., 2015, 54, 1710–1723 CrossRef CAS.
- K. Xu, G. Chen and D. Qiu, J. Mater. Chem. A, 2013, 1, 12395–12399 RSC.
- C. Gao and G. Chen, Compos. Sci. Technol., 2016, 124, 52–70 CrossRef CAS.
- G. Chen, W. Xu and D. Zhu, J. Mater. Chem. C, 2017, 5, 4350–4360 RSC.
- N. Feng, C. Gao, C. Guo and G. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 5603–5608 CrossRef CAS.
- C. Gao and G. Chen, Small, 2018, 1703453 CrossRef.
- C. Meng, C. Liu and S. Fan, Adv. Mater., 2010, 22, 535–539 CrossRef CAS.
- L. Liang, C. Gao, G. Chen and C.-Y. Guo, J. Mater. Chem. C, 2016, 4, 526–532 RSC.
- C. Gao and G. Chen, J. Mater. Chem. A, 2016, 4, 11299–11306 RSC.
- J. Jung, E. H. Suh, Y. J. Jeong, H. S. Yang, T. Lee and J. Jang, ACS Appl. Mater. Interfaces, 2019, 11, 47330–47339 CrossRef CAS.
- C. Pan, L. Wang, W. Zhou, L. Cai, D. Xie, Z. Chen and L. Wang, Polymers, 2019, 11, 278 CrossRef.
- X. Zhou, C. Pan, A. Liang, L. Wang and W.-Y. Wong, Compos. Sci. Technol., 2017, 145, 40–45 CrossRef CAS.
- T. Liu, A. Shinohara, G. Tan, C. Pan and L. Wang, Macromol. Mater. Eng., 2019, 304, 1800730 CrossRef.
- R. Niu, C. Pana, Z. Chen, L. Wang and L. Wang, Chem. Eng. J., 2020, 381, 122650 CrossRef CAS.
- J. Tan, Z. Chen, D. Wang, S. Qin, X. Xiao, D. Xie, D. Liu and L. Wang, J. Mater. Chem. A, 2019, 7, 24982–24991 RSC.
- Z. Chen, M. Lai, L. Cai, W. Zhou, D. Xie, C. Pan and Y. Qiu, Polymers, 2020, 12, 1447 CrossRef CAS.
- R. Amirabad, A. R. Saadatabadi and M. H. Siadati, Mater Renew Sustain Energy, 2020, 9, 1–12 CrossRef.
- E. Yvenou, M. Sandroni, A. Carella, M. N. Gueye, J. Faure-Vincent, S. Pouget, R. Demadrille and J.-P. Simonat, Mater. Chem. Front., 2020, 4, 2054–2063 RSC.
- C. T. Hong, W. Lee, Y. H. Kang, Y. Yoo, J. Ryu, S. Y. Cho and K.-S. Jang, J. Mater. Chem. A, 2015, 3, 12314–12319 RSC.
- F. Du, J. E. Fischer and K. I. Winey, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 3333–3338 CrossRef CAS.
- T. D. Prichard and B. D. Vogt, Polym. Eng. Sci., 2013, 53, 69–77 CrossRef CAS.
- C. H. Chen, G. A. Reynolds, H. R. Luss and J. H. Perlstein, J. Org. Chem., 1986, 51, 3282–3289 CrossRef CAS.
- M. R. Detty, R. S. Eachus, J. A. Sinicropi, J. R. Lenhard, M. McMillan, A. M. Lanzafame, H. R. Luss, R. Young and J. E. Eilers, J. Org. Chem., 1995, 60, 1674–1685 CrossRef CAS.
- M. Scozzafava, C. H. Chen, G. A. Reynolds and J. H. Perlstein, US Pat., 4514481, 1985 Search PubMed.
- G. D. Sharma, M. A. Reddy, K. Ganesh, S. P. Singh and M. Chandrasekharam, RSC Adv., 2014, 4, 732–742 RSC.
- Y. Zhang, B. Zhen, H. Li and Y. Feng, RSC Adv., 2018, 8, 36769–36774 RSC.
- Y. Wang, Y. Liu, W. Wang, L. Liu and C. Hu, ACS Omega, 2020, 5, 27709–27722 CrossRef CAS.
- M. Catauro and S. Pacifico, Materials, 2017, 10, 840 CrossRef.
- J. Li, C. Lai, X. Xiang and L. Wang, J. Mater. Chem. C, 2015, 3, 2693–2701 RSC.
- Y. Piao, J. R. Simpson, J. K. Streit, G. Ao, M. Zheng, J. A. Fagan and A. R. Hight Walker, ACS Nano, 2016, 10, 5252–5259 CrossRef CAS.
- D. Yu, K. Park, M. Durstock and L. Dai, J. Phys. Chem. Lett., 2011, 2, 1113–1118 CrossRef CAS.
- F. Liu, X. Zhou, C. Pan and L. Wang, J. Power Sources, 2019, 412, 153–159 CrossRef CAS.
- Y. Mukai, M. Abe, Y. Katsuta, S. Tomozoe, M. Ito and Y. Koyama, J. Phys. Chem., 1995, 99, 7160–7171 CrossRef CAS.
- H. Zhou, C. Gao, T. Liu, C. Pan and L. Wang, J. Mater. Chem. C, 2020, 8, 7096–7103 RSC.
- Y. Vaynzof, T. J. K. Brenner, D. Kabra, H. Sirringhaus and R. H. Friend, Adv. Funct. Mater., 2012, 22, 2418–2424 CrossRef CAS.
- Y. Nonoguchi, A. Tani, T. Ikeda, C. Goto, N. Tanifuji, R. M. Uda and T. Kawai, Small, 2017, 13, 1603420–1603425 CrossRef.
- K. Mistry, B. A. Larsen, J. D. Bergeson, T. M. Barnes, G. Teeter, C. Engtrakul and J. L. Blackburn, ACS Nano, 2011, 5, 3714–3723 CrossRef CAS.
- Y. Liu, Q. Dai, Y. Zhou, B. Li, X. Mao, C. Gao, Y. Gao, C. Pan, Q. Jiang, Y. Wu, Y. Xie and L. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 29320–29329 CrossRef CAS.
- C. Gao, Y. Liu, Y. Gao, Y. Zhou, X. Zhou, X. Yin, C. Pan, C. Yang, H. Wang, G. Chen and L. Wang, J. Mater. Chem. A, 2018, 6, 20161–20169 RSC.
- J. Liu, J. Sun and L. Gao, Nanoscale, 2011, 3, 3616–3619 RSC.
- M. Wang, Q. Yao, S. Qu, Y. Chen, H. Li and L. Chen, Polymers, 2020, 12, 2720 CrossRef CAS.
- Q. Yao, L. Chen, W. Zhang, S. Liufu and X. Chen, ACS Nano, 2010, 4, 2445–2451 CrossRef CAS.
- R. C. Y. King, F. Roussel, J.-F. Brun and C. Gors, Synth. Met., 2012, 162, 1348–1356 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10625a |
| This journal is © The Royal Society of Chemistry 2021 |