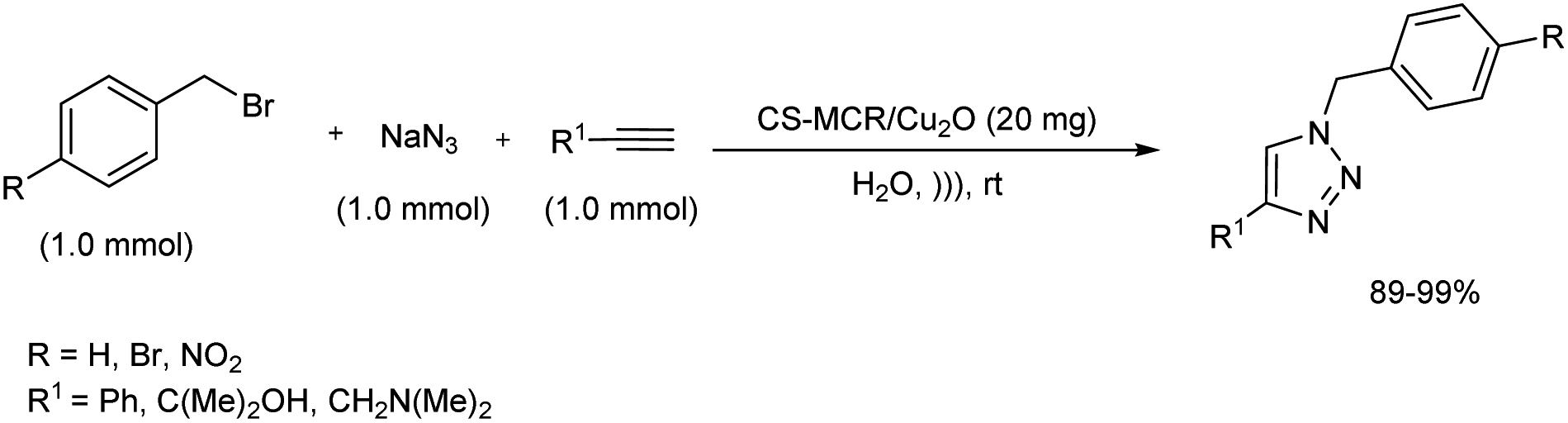DOI:
10.1039/D0RA10472H
(Review Article)
RSC Adv., 2021,
11, 3452-3469
Recent developments and perspectives in the copper-catalyzed multicomponent synthesis of heterocycles
Received
13th December 2020
, Accepted 8th January 2021
First published on 15th January 2021
Abstract
Heterocyclic compounds have become an inevitable part of organic chemistry due to their ubiquitous presence in bioactive compounds. Copper-catalyzed multicomponent synthesis of heterocycles has developed as the most convenient and facile synthetic route towards complex heterocyclic motifs. In this review, we discuss the advancements in the field of copper-catalyzed multicomponent reactions for the preparation of heterocycles since 2018.
 Jaleel Fairoosa | Jaleel Fairoosa was born in Wayanad, Kerala, India. She received her B.Sc. degree (2017) from the Department of Chemistry, Sir Syed College, Taliparamba and M.Sc. degree (2019) from the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. Her research interests revolve around the development of new catalytic methodologies in synthetic organic chemistry and employing this methodology to make medicinally relevant compounds. |
 Mohan Neetha | Mohan Neetha was born in 1996 in Ernakulam, Kerala, India. She pursued her B.Sc. Degree at St. Albert's College, Ernakulam in 2016 and her M.Sc. Degree at Sacred Heart College, Thevara in 2018. She acquired First rank in B.Sc. Chemistry Examination held in 2016 from Mahatma Gandhi University, Kottayam. She qualified KSCSTE Fellowship-2018 as well as CSIR-UGC National Eligibility Test – 2019 and is presently doing her doctoral research under the guidance of Dr G. Anilkumar at the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his PhD in 1996 at the Regional Research Laboratory (renamed as the National Institute for Interdisciplinary Science and Technology NIIST-CSIR), Trivandrum with Dr Vijay Nair. He did postdoctoral studies at the University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), the Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and the Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 100 papers in peer-reviewed journals, 7 patents, three book chapters and edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, in press). He has received a Dr S. Vasudev Award from the Govt. of Kerala, India for best research (2016) and an Evonik research proposal competition award (second prize 2016). His h-index is 30. |
1. Introduction
Synthesis of heterocycles is one among the most significant and widely explored chemical transformations. Efforts towards the development of new synthetic methodologies for the easy generation of heterocycles are constantly increasing due to their omnipresent nature in naturally occurring as well as pharmaceutically relevant compounds, biological processes (as vitamins, DNA, RNA, ATP, enzymes, coenzymes, and serotonin) and in industry (as additives and modifiers).1 Among the various heterocyclic compounds N-containing heterocycles are prominent and have widespread applications in pharmaceuticals,2 agrochemicals, catalysis, and coordination chemistry.3 Moreover, the high solubility and salt formation ability of N-heterocycles make them more significant in drug development.4 Oxygen5 and sulfur6-containing heterocycles are also widely synthesized and studied nowadays.
Among the various synthetic methods developed for heterocycle synthesis, transition metal-catalyzed multicomponent reactions (MCRs) are well established. Transition metals generally used in these reactions include palladium, copper, nickel, cobalt, zinc, iron and manganese. Copper-catalyzed reactions are having high acceptance in organic synthesis due to their natural abundance, low toxicity, sustainability and low cost, and copper has turned out as a practical alternative for expensive noble metal like palladium in catalysis.7 Numerous novel reactions catalyzed by copper reported in the last decade unveil the significance of copper in transition metal catalysis. Copper with different oxidation states are employed in multicomponent heterocycle formation and has emerged as the most applicable and effective catalyst among transition metals. Nowadays, copper nanoparticles are also efficiently used as catalysts in several chemical reactions and exhibit remarkable benefits.
Multicomponent heterocycle synthesis is an efficient and convenient method for the construction of complex heterocycles and in these one pot reactions three or more components react to form the target organic compounds. The first MCR accomplished was “benzoylazotide” synthesis from bitter almond oil and ammonia via benzaldehyde and hydrogen cyanide, done by Laurent and Gerhardt8 in 1838. In 1850, Strecker officially reported a multicomponent reaction for α-aminocyanide preparation. MCRs poses several advantages including higher product yield, reproducibility, simple reaction profiles and shorter reaction times.9 They also allow incorporation of numerous reactive functionalities into selected molecules by fewer steps which is often complicated in classical techniques. The main classifications of MCRs are consecutive reactions, domino-type reactions and sequential reactions.10 MCRs are frequently employed in drug development due to their competence in construction of multiple bonds and chemically flexible motifs. Simple execution, high atom economy, environmental-friendliness and simple purification made these reactions equally popular in academic and industrial research. Reviews on copper-catalyzed synthesis of aromatic heterocycles having single heteroatom11 and synthesis of nitrogen-containing heterocyclic compounds catalyzed by copper/L-proline12 have been reported. In 2018, our group has published a review on copper-catalyzed multicomponent synthesis of heterocycles.11 Owing to the high importance of copper-catalyzed synthesis of heterocycles, we hereby summarize the recent advancements in copper-catalyzed multicomponent synthesis of heterocycles covering literature since 2018.
2. Synthesis of heterocycles
For clarity, the topic is classified according to the size of the heterocycle formed.
2.1. Heterocyclic compounds
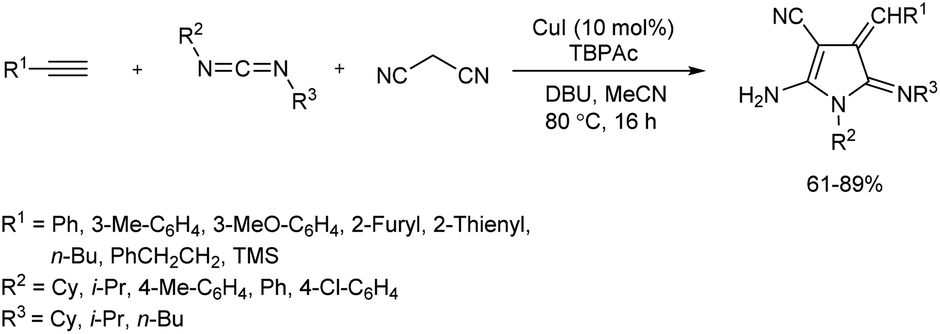
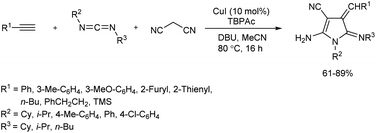
2.1.1. Five-membered heterocyclic compounds with one heteroatom. An efficient catalytic protocol towards pyrrole derivatives has been reported via a multicomponent reaction of terminal alkynes, malononitrile and carbodiimides in presence of CuI in anhydrous MeCN, which afforded the desired products in good yields (Scheme 1).13 DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) was the most suitable base for this reaction, and presence of copper(I) source and the additive tetrabutylphosphonium acetate (TBPAc) played a crucial role in the formation of the corresponding product in good amount. The substrate scope studies were carried out using diverse carbodiimides and terminal alkynes. Groups such as isopropyl and cyclohexyl on carbodiimide were well tolerated, and carbodiimides with a combination of aromatic and aliphatic substituents also showed good reactivity towards pyrrole synthesis. While methyl- and methoxy-substituted phenyl acetylenes gave good yield of target product, reaction using heteroaromatic alkynes resulted in excellent yields of product. Moderate yields of pyrroles were obtained when aliphatic alkynes were employed in the reaction.
 |
| | Scheme 1 Synthesis of pyrrole derivatives from terminal alkynes, carbodiimides and malononitrile. | |
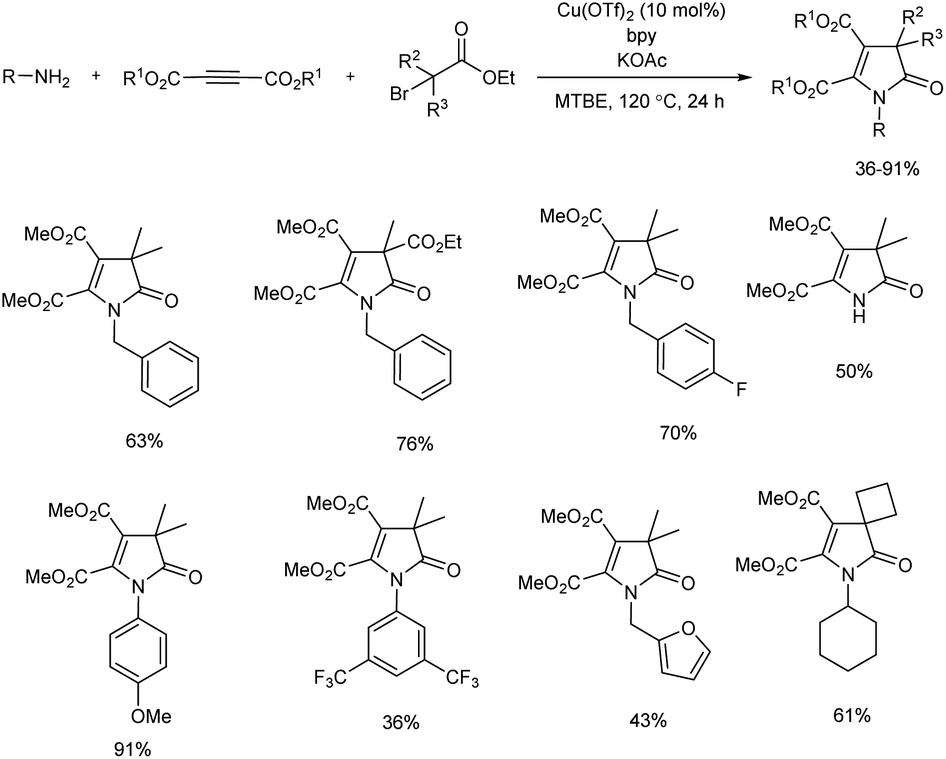
1,3-Dihydro-2H-pyrrol-2-one is widely found in bioactive compounds.14 A novel three-component reaction of acetylenedicarboxylates, alkylamines, and α-bromocarbonyls for the preparation of small library of fully-substituted 1,3-dihydro-2H-pyrrol-2-ones was discussed (Scheme 2).15 The team of researchers succeeded in realizing Heck-type coupling of in situ-generated trisubstituted alkene with a tertiary alkyl bromide for the first time. The multicomponent annulation gave better results with copper(II) triflate as catalyst in the presence of a ligand, and all the 2,2′-bipyridine ligands used gave good results. Moreover, a weak base was found more effective and KOAc was used in all reactions. Substrate scope of this cascade process was evaluated and a broad substrate scope was exhibited by the present protocol. The control experiments carried out using TEMPO (2,2,6,6-tetramethylpiperidine N-oxyl) and DPE (1,1-diphenylethylene) confirmed the radical transformation of this method.
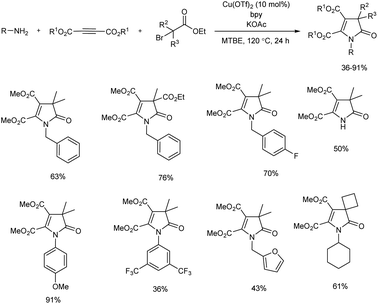 |
| | Scheme 2 Multicomponent synthesis of 1,3-dihydro-2H-pyrrol-2-one. | |
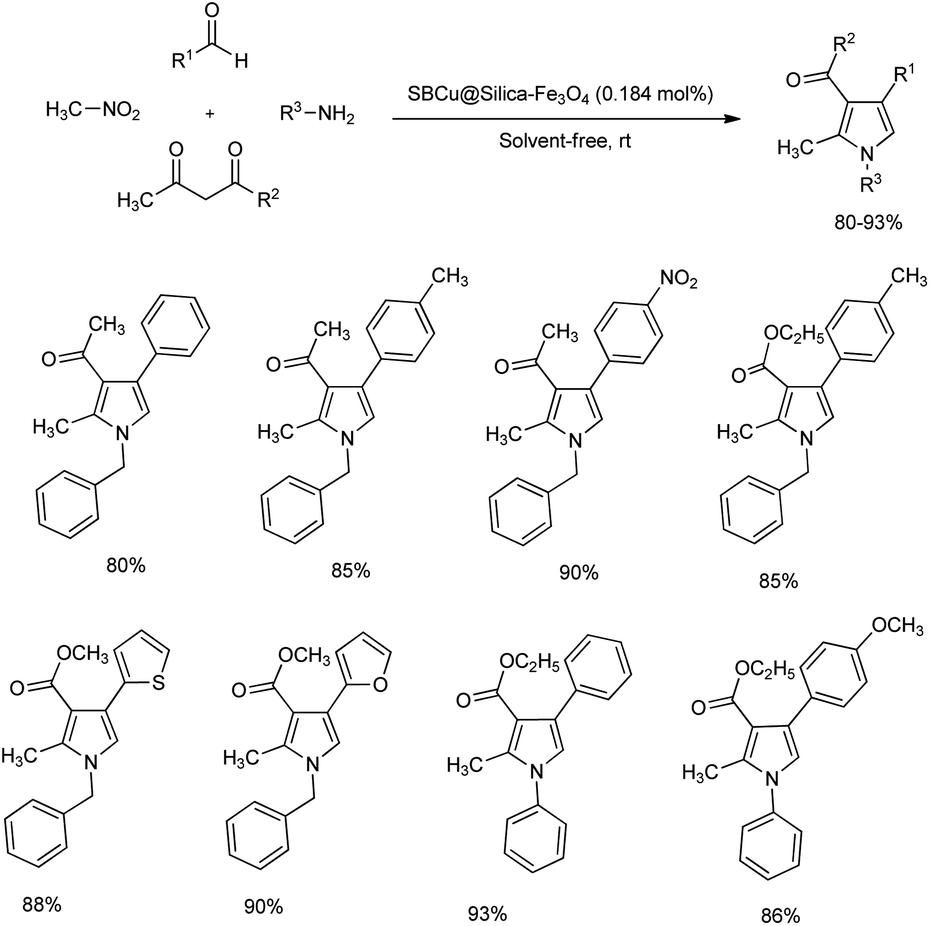
A copper Schiff base complex immobilized on silica-coated Fe3O4 nanoparticles for the synthesis of multi-substituted pyrroles under solvent-free condition was reported.16 One-pot reaction of aryl aldehyde, nitromethane, 1,3-dicarbonyl compound and amine at room temperature afforded poly-substituted pyrroles in high yield (Scheme 3). Catalyst complex was efficiently prepared by the mixing of SB@silica-Fe3O4 suspension with excess Cu(OAc)2 in ethanol. The mentioned protocol exhibited wide substrate scope, and aldehydes with electron-withdrawing group afforded a slightly higher yield of pyrroles than those having electron-donating group. Turn over frequency (TOF) and turn over number (TON) of the present catalyst was calculated and reusability of catalyst was found to be high throughout the reaction.
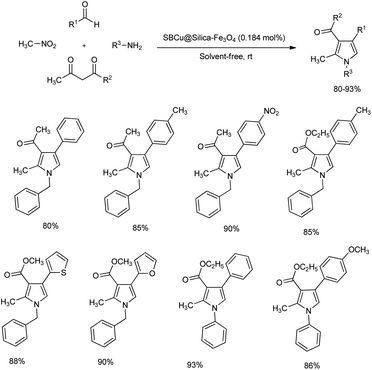 |
| | Scheme 3 One pot synthesis of poly-substituted pyrroles under solvent-free condition. | |
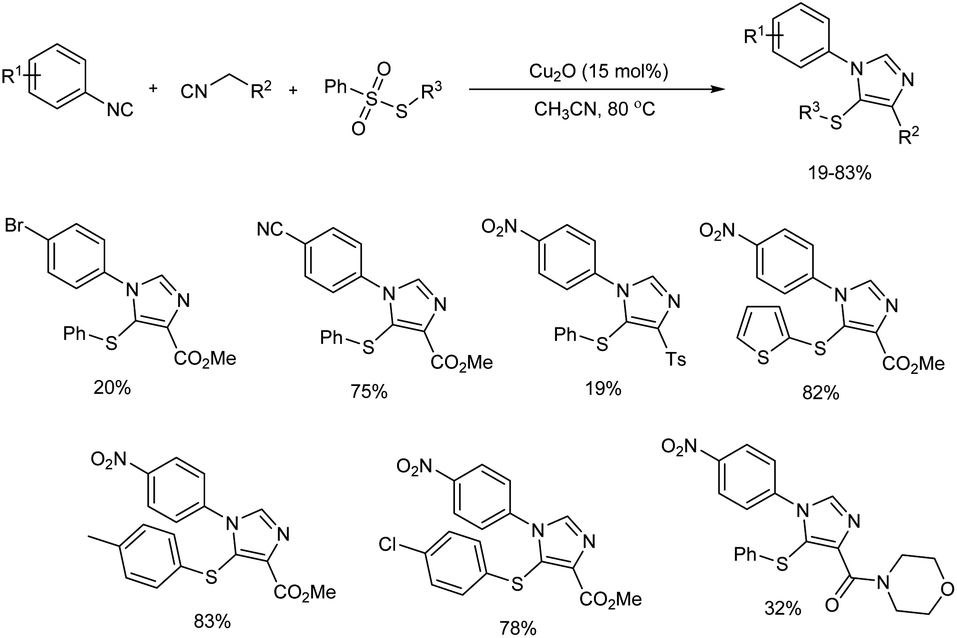
2.1.2. Five-membered heterocyclic compounds with two heteroatoms. Sulfur-containing trisubstituted imidazoles were successfully prepared via three-component reaction of aryl isocyanides with active methylene isocyanides and arylsulfonothioates in presence of Cu2O as catalyst.17 The trisubstituted imidazoles were constructed based on the isocyanide–isocyanide [3 + 2] cycloaddition and this Cu(I)-catalyzed reaction enabled formation of new C–C, C–S and C–N bonds in single step.Generality and viability of the reaction was analysed and moderate to good yield of final products were obtained (Scheme 4). When the reactions of aryl isocyanides with halogen group (Br, I) and other electron-withdrawing groups (CF3, CN, COMe, CO2Et) gave desired amount of products in moderate to good yield, 1-isocyano-4-methylbenzene failed to give the target product under standard conditions. Active methylene isocyanides such as 1-((isocyanomethyl)sulfonyl)-4-methylbenzene and 2-isocyano-1-morpholinoethan-1-one and several substituted arylsulfonothioates showed good reactivity towards the reaction.
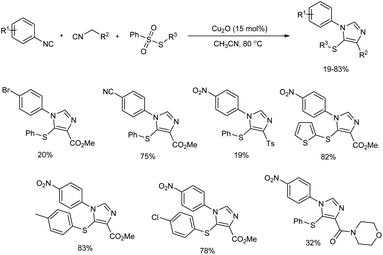 |
| | Scheme 4 Cu2O-catalyzed three-component reaction for trisubstituted imidazole preparation. | |
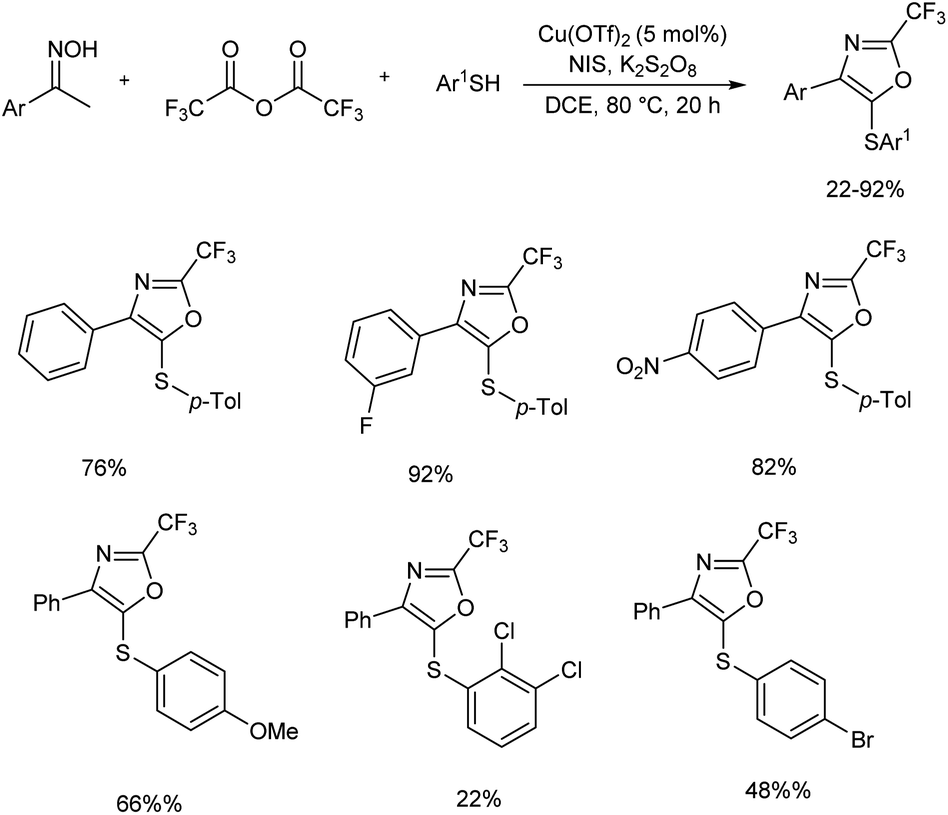
An efficient synthetic strategy for the incorporation of a trifluoromethyl group at oxazole ring was established by Xiao et al.18 This protocol involved oxidative cyclization of arylthiol, oxime and trifluoroacetic anhydride using a copper(II) catalyst, and a series of trisubstituted oxazoles was developed (Scheme 5). The reaction proceeded via N–O bond breaking, C–H functionalization, and an intramolecular annulation. Three-component reaction was initiated using acetophenone oxime, 4-methylbenzenethiol and trifluoroacetic anhydride as model substrates and along with Cu(OTf)2 catalyst. An iodine-containing additive N-iodosuccinimide (NIS) was found necessary to get better results.
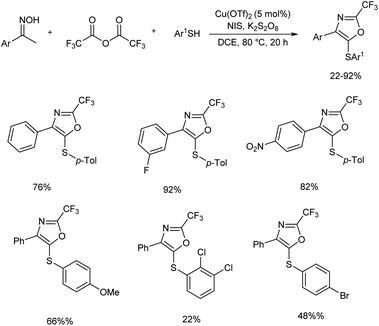 |
| | Scheme 5 Copper-catalyzed synthesis of trisubstituted oxazoles. | |
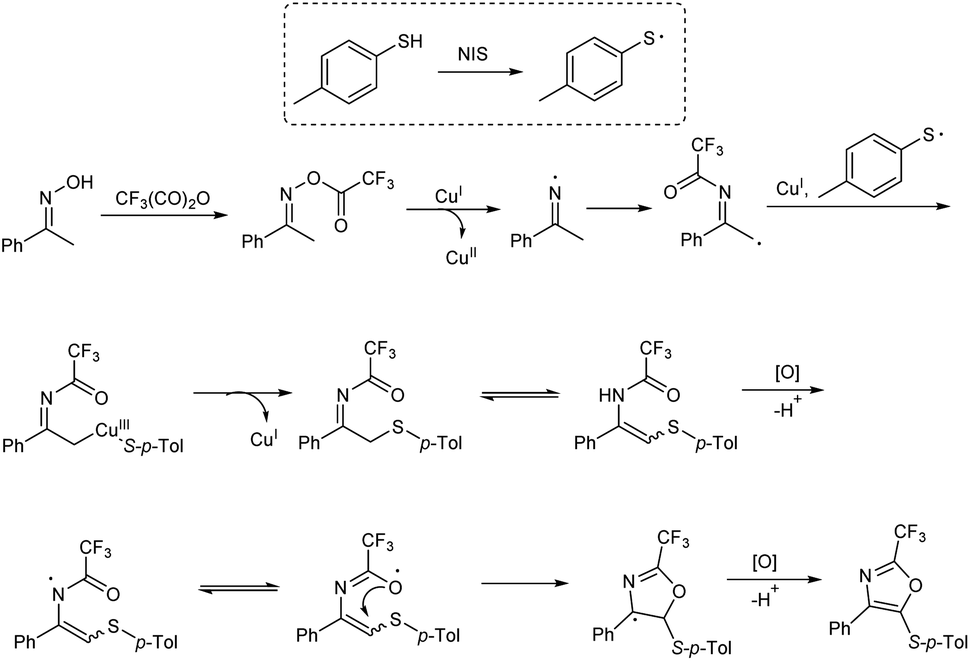
The functional group tolerance of the protocol was studied using diverse thiophenols, as well as ketoximes. Electron-donating groups at the para position of the benzene ring of thiophenols gave good yields of product and halogen substituents, such as fluoro, chloro and bromo also afforded the target products in moderate yields, without the cleavage of aryl-halide bond. A sharp decrease in product yield was observed when 2,3-dichlorobenzenethiol was used as the coupling reagent. Acetophenone oximes containing methyl, phenyl and isobutyl electron-donating groups were well tolerated and moderate to good yield of product was obtained. Ketoximes with halogen groups and electron-withdrawing groups like nitro and cyano also gave better results. The mechanistic studies performed by the team revealed that the reaction involved a three-component enamine formation as well as oxidative 5-endo cyclization (Scheme 6).
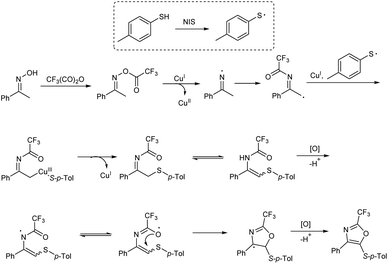 |
| | Scheme 6 Possible mechanism of trisubstituted oxazoles synthesis [this figure has been reproduced from ref. 18 with permission from American Chemical Society, copyright 2019]. | |
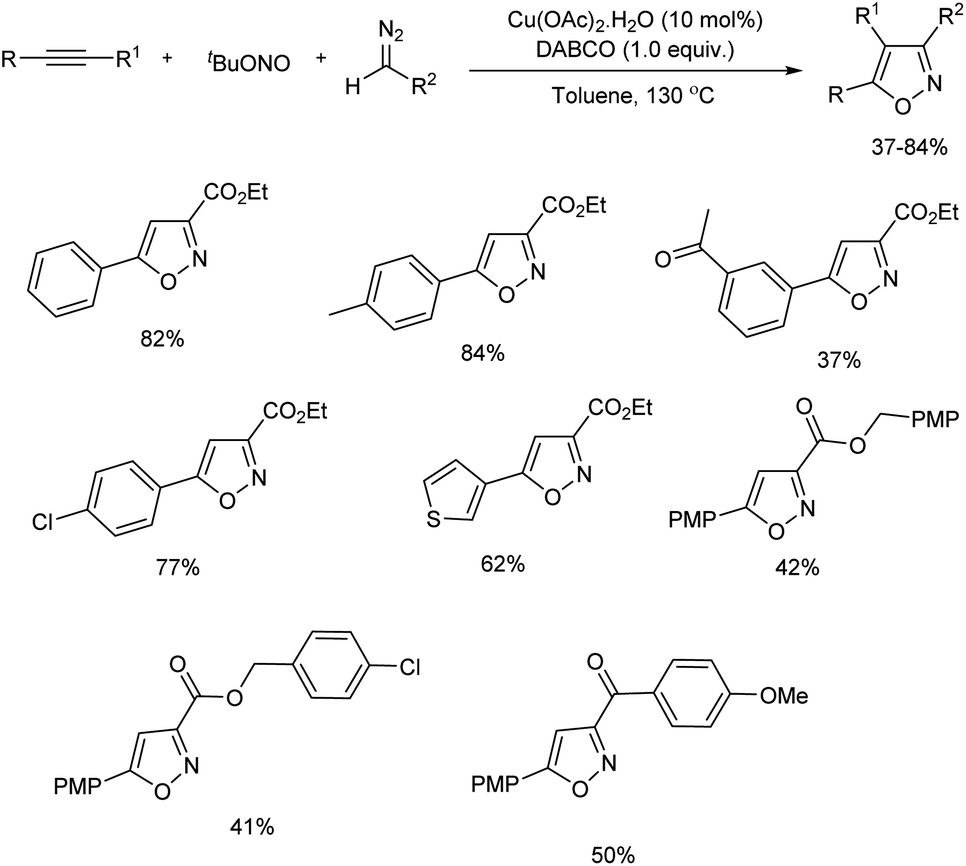
Zhao et al. reported a regioselective synthesis of isoxazoles from a copper-promoted [3 + 2] cycloaddition of alkynes with in situ formed nitrile oxides.19 Nitrile oxides were formed from the coupling reaction of copper carbene and nitroso radical. Initial reaction commenced with multi-component reaction of ethynylbenzene, tert-butyl nitrite and ethyl diazoacetate. Cu(OAc)2·H2O and 1,4-diazabicyclo[2.2.2]octane (DABCO) were the copper source and base chosen respectively for the reaction. The scope of the reaction was studied using various terminal alkynes and diazo compounds (Scheme 7). Terminal alkynes having diverse substitutions including phenyl, electron-rich and electron-deficient aryl, heteroaryl groups gave desired products in moderate to good yield. The reaction was tolerant towards aliphatic terminal alkynes and steric effect had no significant influence on the reaction. Electron-deficient internal alkyne also gave 37% yield of the desired isoxazole with excellent regioselectivity. α-Diazocarbonyl compounds containing alkoxy carbonyl as well as aroyl groups with diverse substitution also reacted well under optimal conditions. Several control experiments carried out by the team found a radical process was involved in this reaction and Cu(OAc)2·H2O plays a significant role in the conversion.
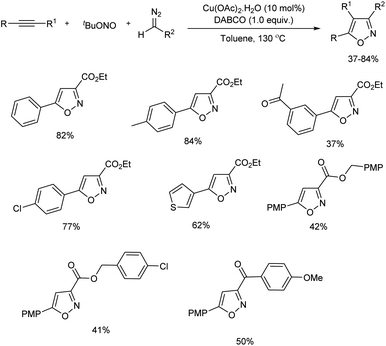 |
| | Scheme 7 Regioselective synthesis of isoxazoles from a copper-promoted [3 + 2] cycloaddition. | |
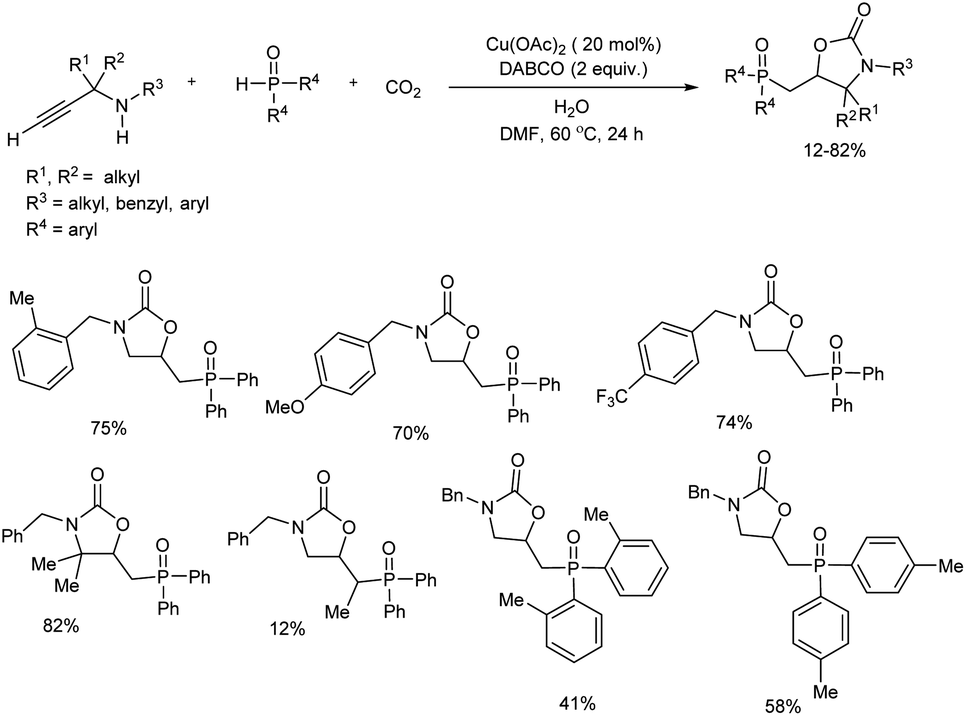
An efficient protocol towards a range of 5-((diarylphosphoryl)methyl)oxazolidin-2-ones via copper(II)-catalyzed phosphonocarboxylative cyclization of phosphine oxide and propargylic amines with CO2 was studied.20 N-Benzylprop-2-yn-1-amine reacted with diphenylphosphine oxide under CO2 balloon at 80 °C for 24 hours in the initial reaction. Among cupric and cuprous salts tested cupric acetate was the best catalyst and DABCO was the most efficient base for the reaction. The scope and limitations of the tandem reaction was explored and the target products were obtained in moderate to good yields (Scheme 8). Diversely substituted propargylic amines reacted smoothly in the reaction and showed no influence of steric and electronic effects of substituents. With respect to phosphine oxides, lower yield of final products offered by ortho-substituted substrates compared with para-substituted ones showed significant steric effect on ortho-position. Mechanistic studies conducted by the team suggested that the reaction proceeds via one-pot tandem cyclization/radical addition scheme, followed by phosphorylation/cyclization sequence.
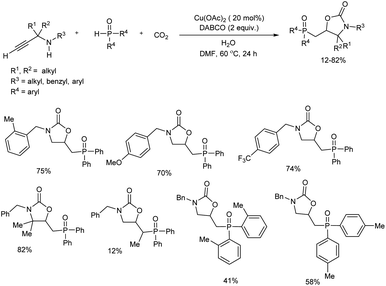 |
| | Scheme 8 Construction of 5-((diarylphosphoryl)methyl)oxazolidin-2-ones via the copper(II)-catalyzed phosphonocarboxylative cyclization reaction. | |
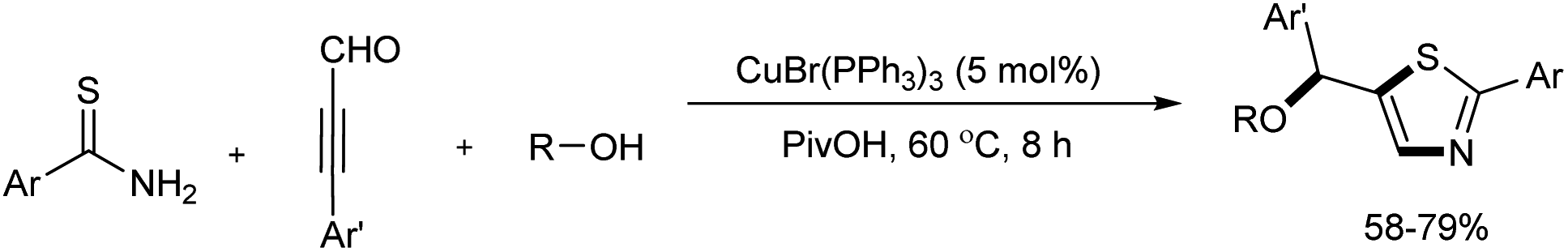
A series of functionalized thiazoles were efficiently prepared from thioamides, ynals, and alcohols through Cu(I)-catalyzed reaction.21 Multiple bonds including C–S, C–N, and C–O were formed in this one pot reaction and 5 mol% CuBr(PPh3)3 was used as the catalyst. Several acids including PivOH, MsOH, PhCO2H, TsOH were screened and the best results were offered by PivOH. The novel method exhibited wide substrate scope and good functional group compatibility (Scheme 9).
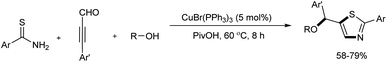 |
| | Scheme 9 Copper-mediated preparation of functionalized thiazoles. | |
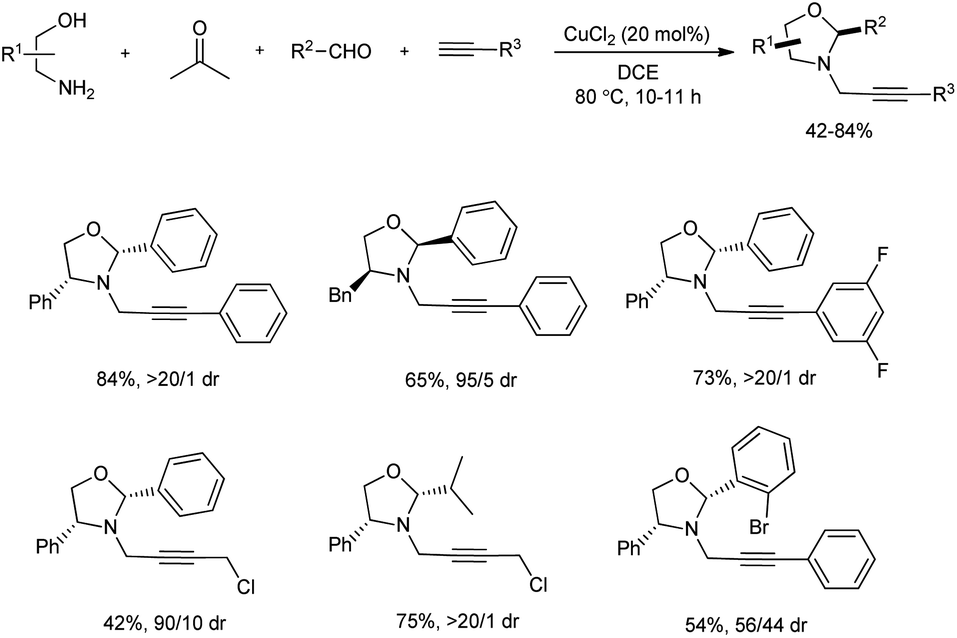
A significant chemoselective reaction of formaldehyde, amino alcohols, two types of alkynes and aldehydes using copper(II) catalysis was investigated by Feng and team (Scheme 10).22 In this A3-coupling/annulation reaction, chiral N-propargyl oxazolidines were prepared in acceptable yields with diastereoselectivities up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Among the several copper sources screened 20 mol% of CuCl2 was the most suitable, and it was noted that the reaction was highly sensitive to temperature. The scope of this protocol studied for amino alcohol and alkyne found that several substituents on these molecules were well-tolerated irrespective of the electronic behavior of substituents and a series of diverse oxazolidines were formed with good diastereomeric ratio in moderate to good yields. Similarly, reaction with various aldehydes also performed and similar results were attained. Moreover, the size of the group on ortho-position of the benzene ring had a direct influence on diastereoselectivity and a bulky substituent decreased diastereometric ratio to a great extent. A broad substrate scope was exhibited by this synthetic method.
:1. Among the several copper sources screened 20 mol% of CuCl2 was the most suitable, and it was noted that the reaction was highly sensitive to temperature. The scope of this protocol studied for amino alcohol and alkyne found that several substituents on these molecules were well-tolerated irrespective of the electronic behavior of substituents and a series of diverse oxazolidines were formed with good diastereomeric ratio in moderate to good yields. Similarly, reaction with various aldehydes also performed and similar results were attained. Moreover, the size of the group on ortho-position of the benzene ring had a direct influence on diastereoselectivity and a bulky substituent decreased diastereometric ratio to a great extent. A broad substrate scope was exhibited by this synthetic method.
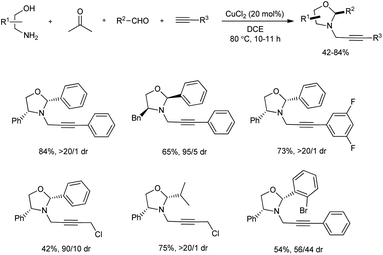 |
| | Scheme 10 Four-component synthesis of N-propargyl oxazolidines. | |
Mechanistic pathway of this reaction suggested by the team include generation of oxazolidine from the condensation of formaldehyde and amino alcohol. Reaction of in situ generated oxazolidine with aldehyde to form N-substituted oxazolidine. Subsequent treatment of N-substituted oxazolidine with the copper–alkyne complex (obtained from activation of terminal alkyne by the Cu(II) catalyst) and formation of propargylamine species A with regeneration of the catalyst used. Followed by the intramolecular cyclization of propargylamine species A to afford the chiral N-propargyl oxazolidines via another intermediate B (Scheme 11).
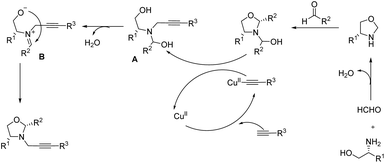 |
| | Scheme 11 Expected mechanism of N-propargyl oxazolidine formation [this figure has been reproduced from ref. 22 with permission from American Chemical Society, copyright 2019]. | |
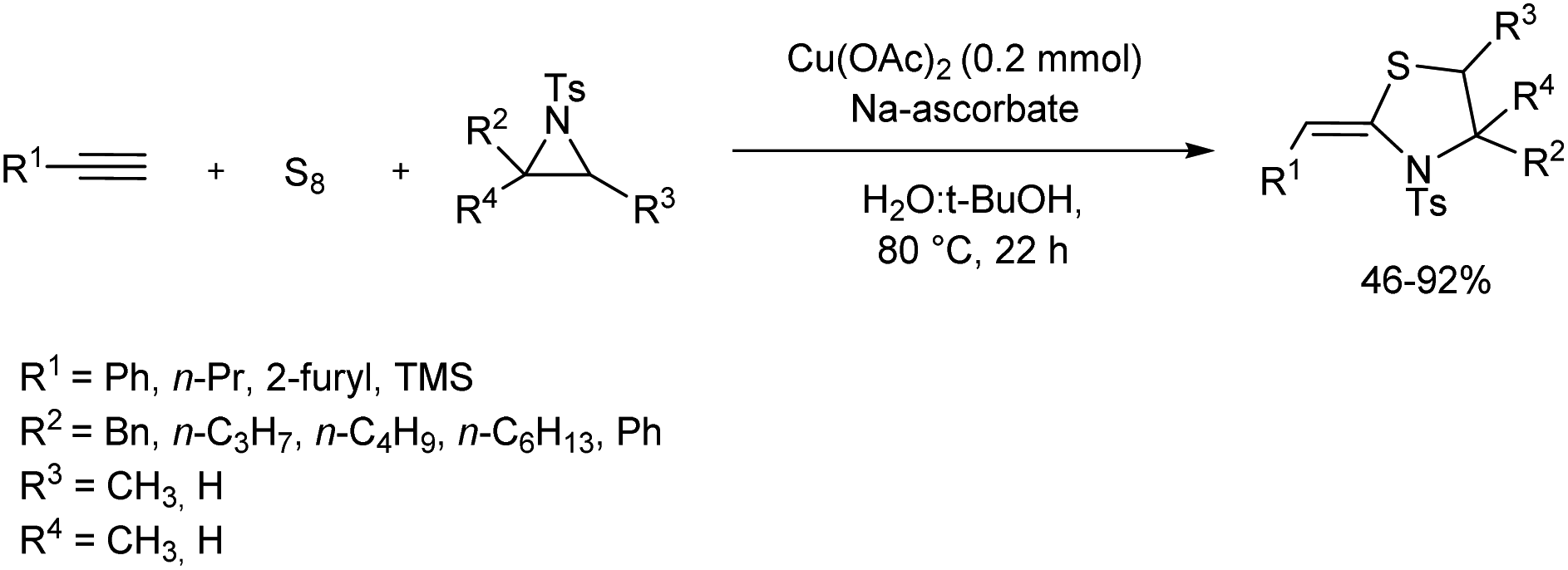
A small library of thiazolidine derivatives was constructed from copper-catalyzed reaction of aziridines, terminal alkynes, and elemental sulfur (Scheme 12).23 In this atom economic synthetic method alkyne thiolate formed from the reaction of elemental sulfur and terminal alkynes reacted with aziridines in presence of copper salts to offer thiazolidine derivatives efficiently. When the initial reaction carried out using phenyl acetylene, elemental sulfur and aziridine gave very low yield of the desired product, however, addition of Cu(II) (Cu(OAc)2) and sodium ascorbate to the system resulted profound increase in product yield.
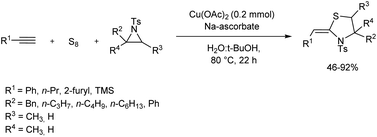 |
| | Scheme 12 Copper-catalyzed reaction of terminal alkynes, elemental sulfur and aziridines. | |
It is observed that benzyl-substituted aziridine gave good yield of the target product, and due to the steric issues of the alkyl groups gem-disubstituted aziridine afforded the desired product in moderate yield. Simple alkyl-substituted aziridines reacted smoothly throughout the synthesis. 1-Pentyne and heteroaromatic terminal alkyne engaged efficiently in the reaction. Good yield was obtained in the reaction of trimethylsilylacetylene which formed tosylthiazolidine skeleton having vinylsilane motif, which opened new avenues for the synthesis of organosilicon compounds.
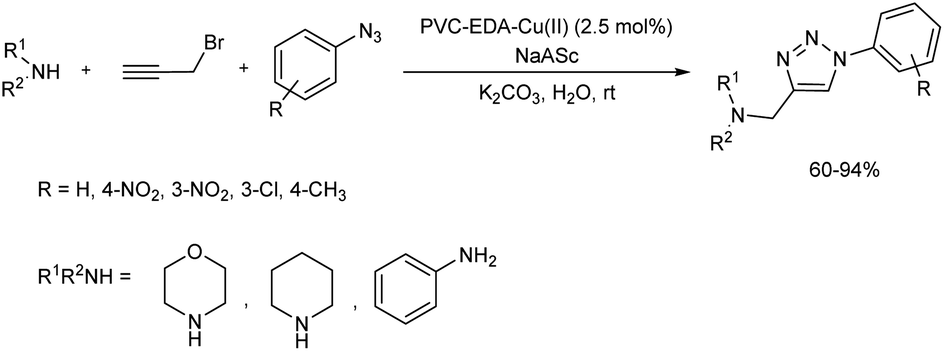
2.1.3. Five-membered heterocyclic compounds with three heteroatoms. A multicomponent synthesis in the presence of PVC-supported ethylenediamine–copper(II) complex was studied in 2018.24 Click reaction of aromatic azides, propargyl bromide and amines resulted in the formation of 1,4-disubstituted 1,2,3-triazoles in good yield (Scheme 13). Novel heterogeneous catalyst, PVC–EDA–Cu(II) was easily prepared by the reaction of ethylenediamine, PVC (polyvinyl chloride) and CuCl2·2H2O in water, and the catalyst structure was characterized through various analytical tools. The optimum reaction conditions selected for this reaction was 5 mol% of sodium ascorbate and 2.5 mol% of PVC–EDA–Cu(II) in the presence of K2CO3 base in water at room temperature. The scope of the reaction was studied using several aromatic azides as well as amines like morpholine, aniline and piperidine with propargyl bromide, and no significant effect of position of the substituent towards reaction yield was observed. Reusability of the catalyst was tested and found that the catalyst was reusable though a gradual decrease in activity was observed.
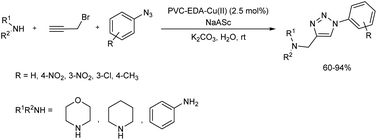 |
| | Scheme 13 PVC–EDA–Cu2+ complex catalyzed synthesis of 1,2,3-triazoles. | |
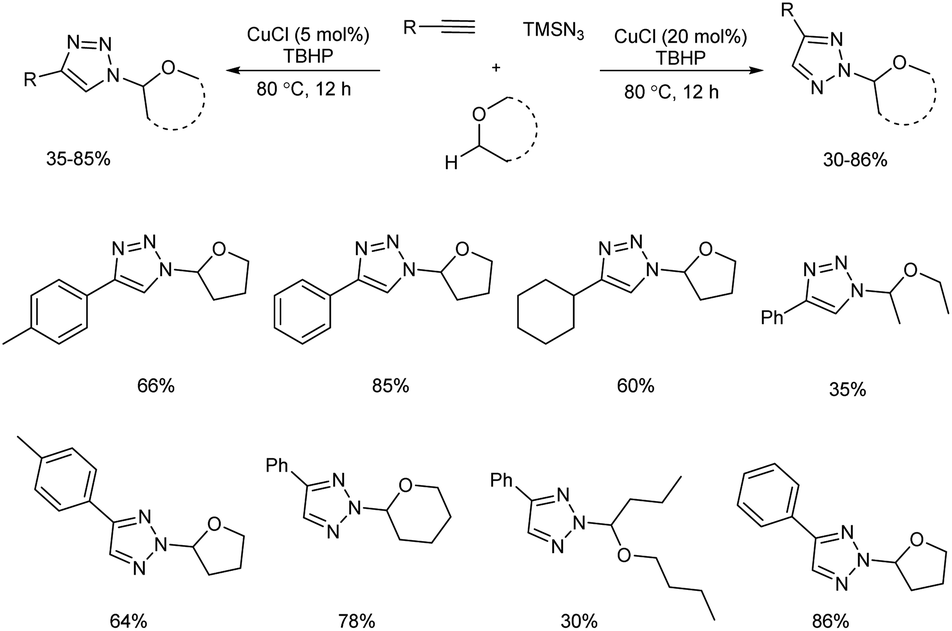
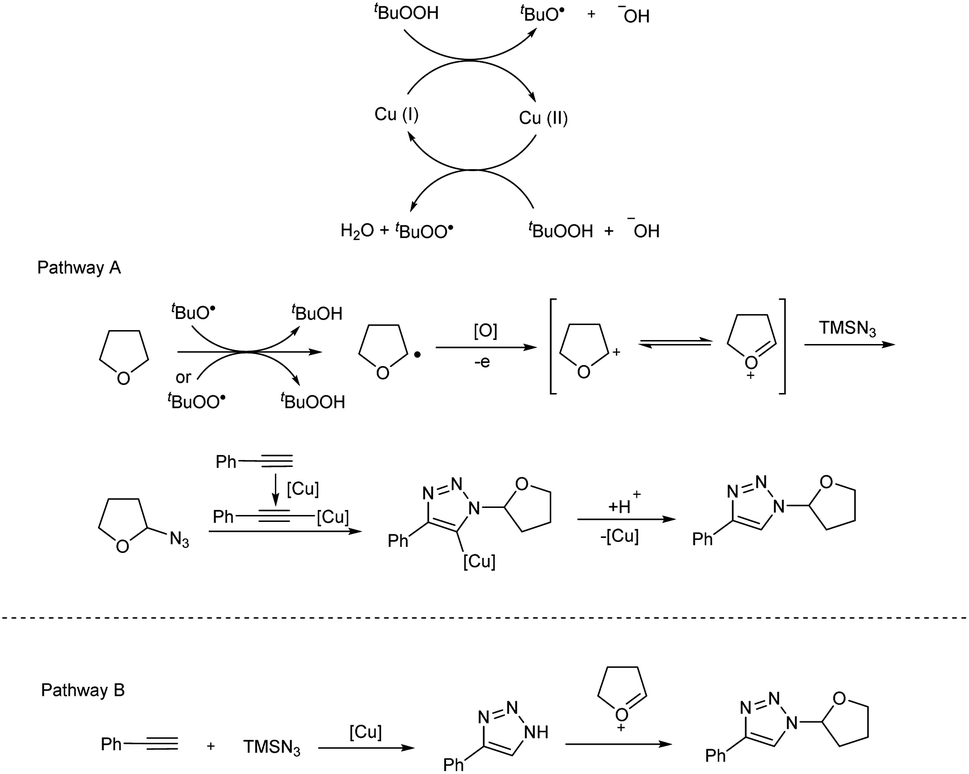
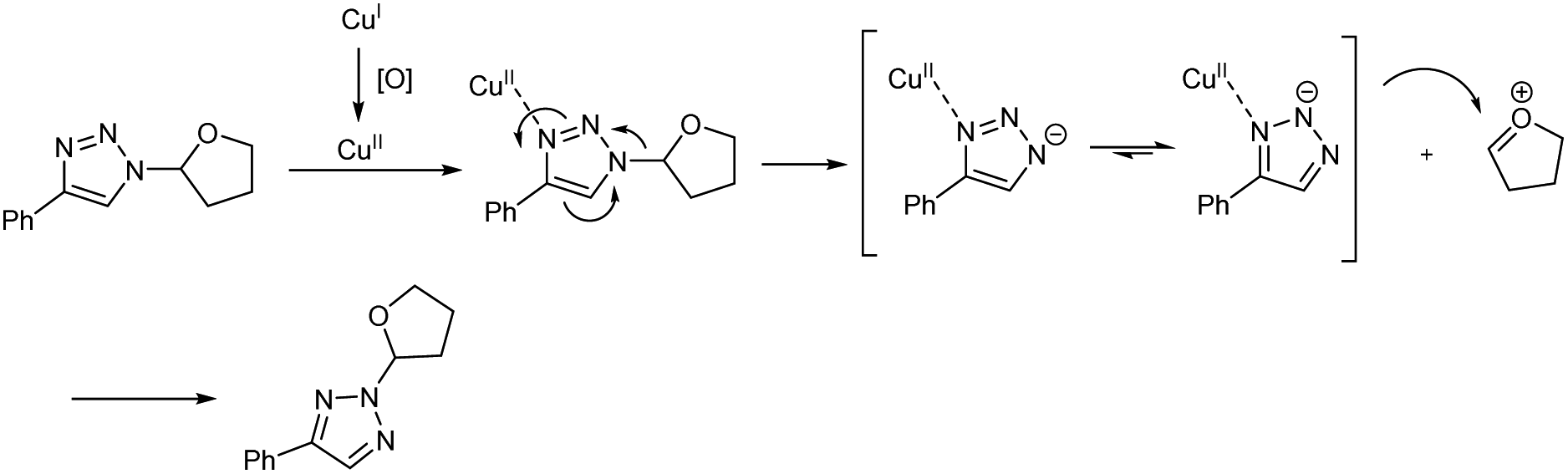
Copper(I)-catalyzed reaction of alkynes, ethers and TMSN3 for the preparation of N1- and N2-oxyalkylated 1,2,3-triazoles was revealed by Wei and co-workers.25 CuCl was the copper source used and usage of oxidant DTBP (di-tert-butyl peroxide) gave good results in this regioselective protocol (Scheme 14). Optimization of reaction conditions carried out concluded that 5 mol% and 20 mol% were the optimum amount of CuCl required for the synthesis of N1-substituted triazoles and N2-substituted triazoles respectively. The generality and limitations of the reaction was investigated separately for N1-substituted triazoles and N2-substituted triazoles. Aromatic alkynes with electron-withdrawing substituents and donating substituents and aliphatic alkynes like hex-1-yne and ethynylcyclohexane reacted smoothly under standard condition in the first case. The scope of ether tested under optimized conditions gave acceptable yield throughout the reaction. Aromatic and aliphatic alkynes used in the construction of N2-substituted triazoles showed good reactivity and better results were obtained when cyclic ethers were used in N2-substituted triazoles preparation compared to linear ethers. Authors described two separate pathways for N1-oxyalkylated 1,2,3-triazoles formation (Scheme 15). They also suggested an expected mechanism of N2-substituted triazoles synthesis which involved the copper metal promoted 1,2-shift of the ether group in N1-oxyalkylated 1,2,3-triazoles through a sequential electronic transfer process (Scheme 16).
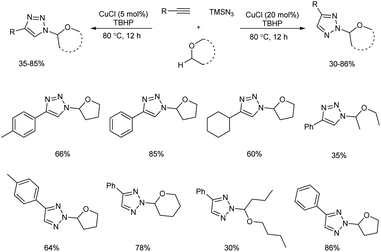 |
| | Scheme 14 Preparation of N1- and N2-oxyalkylated 1,2,3-triazoles using copper catalyst. | |
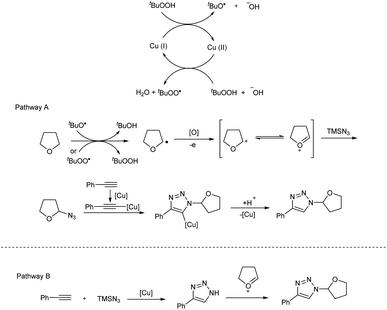 |
| | Scheme 15 Expected mechanism of N1-oxyalkylated 1,2,3-triazole formation [this figure has been reproduced from ref. 25 with permission from American Chemical Society, copyright 2019]. | |
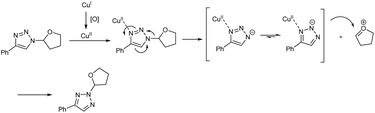 |
| | Scheme 16 Plausible mechanism of synthesis of N2-oxyalkylated 1,2,3-triazoles [this figure has been reproduced from ref. 25 with permission from American Chemical Society, copyright 2019]. | |
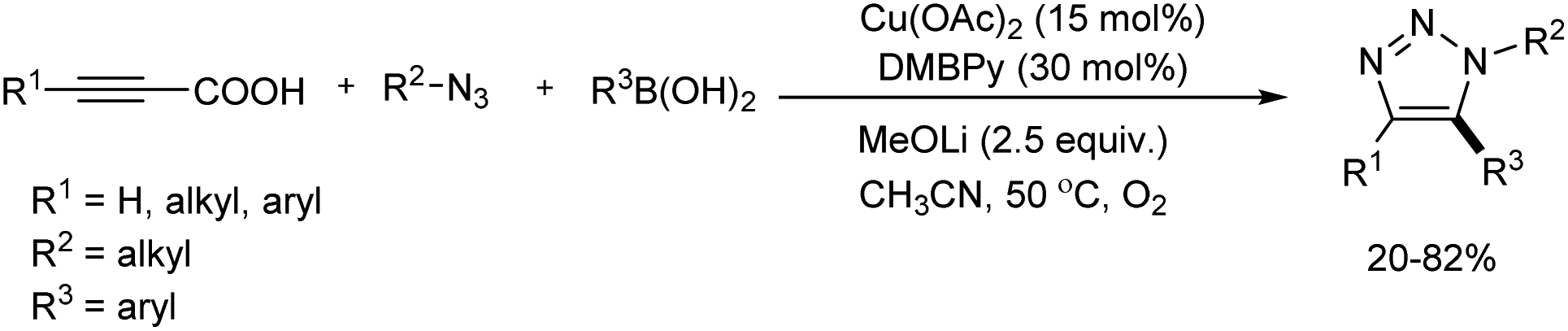
Li and co-workers put forward a convenient procedure for the construction of diverse fully-substituted 1,2,3-triazoles from easily available propiolic acids, azides, and arylboronic acids via decarboxylative cycloaddition reaction (Scheme 17).26 Several catalyst and ligand screened during optimization concluded that 15 mol% of Cu(OAc)2 and 30 mol% of 4,4′-dimethoxy-2,2′-bipyridine (DMBPy) were most suitable catalyst and ligand for the reaction. The role of MeOLi base and presence of O2 atmosphere were crucial in the reaction. Among various propiolic acids investigated phenylpropiolic acids bearing electron-donating and electron-withdrawing groups and halofunctional groups (F, Cl, Br) were compatible with this reaction. Ortho-substituted phenylpropiolic acids and several alkylpropiolic acids also reacted well in the reaction. With respect to arylboronic acids employed numerous functional groups (except hydroxyl and cyano groups) at ortho-, meta- or para-position gave good amount of 5-arylated triazoles. In the case of azides used, benzyl azide and various aliphatic azides gave the corresponding products in moderate yields.
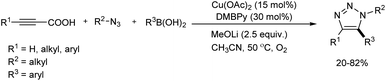 |
| | Scheme 17 Construction of fully-substituted 1,2,3-triazoles from propiolic acids, azides, and arylboronic. | |
Based on experimental results, authors proposed a plausible mechanism for the reaction which include; generation of copper(I) acetylide A from decarboxylation of propiolic acid in the presence of copper salt and base, followed by [3 + 2] cycloaddition of copper(I) acetylide with azide to form triazolyl-copper intermediate C, subsequent transmetallation with borate complex to generate intermediate D and reductive elimination of D to get the final product (Scheme 18).
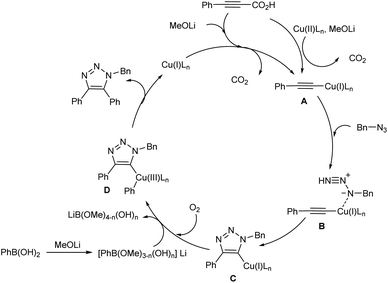 |
| | Scheme 18 Possible mechanism of substituted 1,2,3-triazoles synthesis [this figure has been reproduced from ref. 26 with permission from American Chemical Society, copyright 2020]. | |
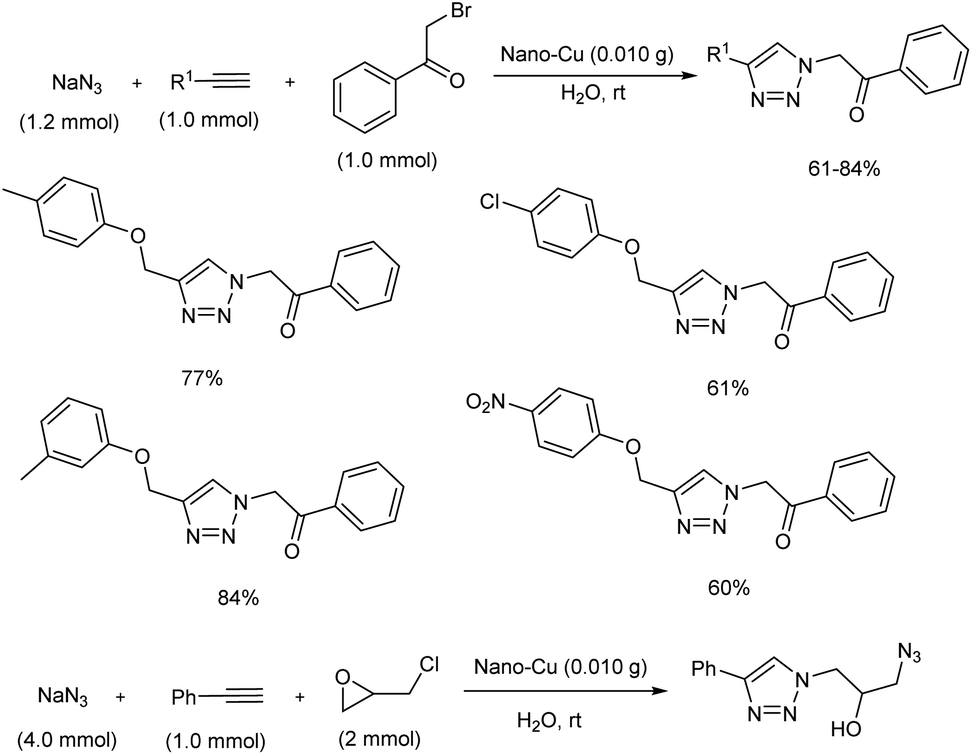
Inexpensive copper nanoparticles were efficiently used for the preparation of β-carbonyl 1,2,3-triazoles and triazole azido alcohols via Click reaction.27 The copper catalyst was prepared by the reduction of CuO in the presence of NaBH4 through ball milling, and the structure of the copper nanoparticles (Cu NPs) was confirmed using SEM, XRD, EDX and FTIR. Click reaction for β-carbonyl 1,2,3-triazoles and triazole azido alcohols was carried out using epichlorohydrin, sodium azide, phenacyl bromide and terminal alkynes (Scheme 19). Moderate to good yields of the target products were obtained using this strategy, and the catalyst was found to be smoothly recovered as well as reused several times.
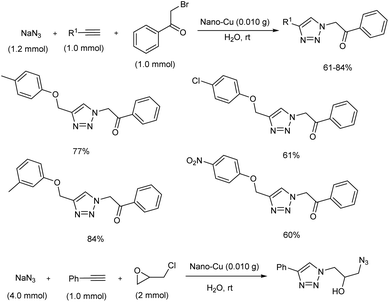 |
| | Scheme 19 Synthesis of β-carbonyl 1,2,3-triazoles and triazole azido alcohols in water. | |
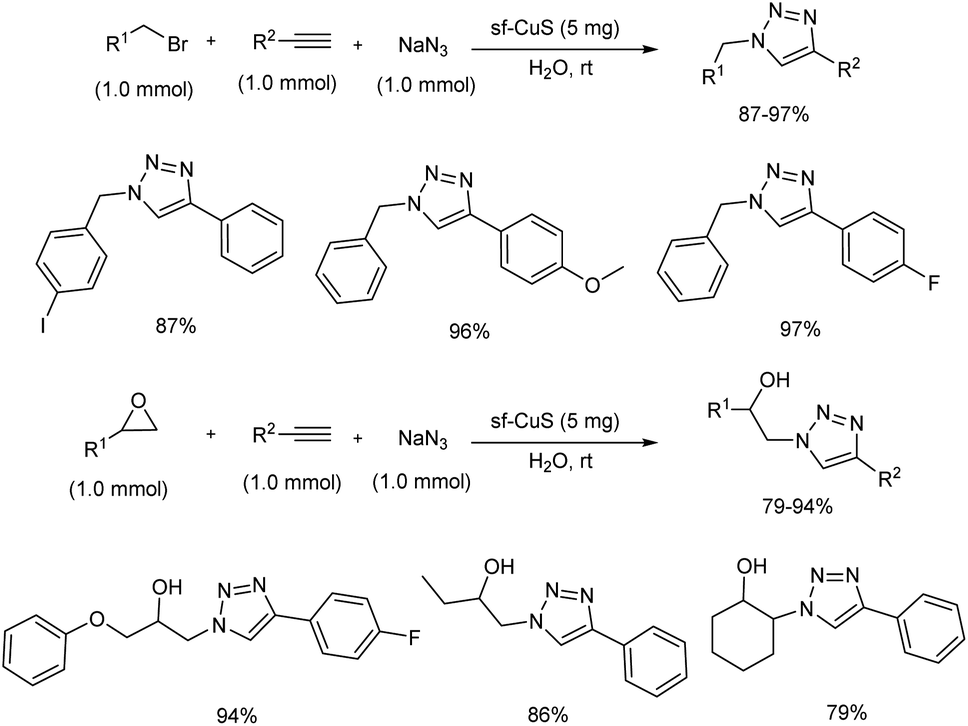
An important one-pot synthesis of triazoles (1,2,3-triazoles and β-hydroxy 1,2,3-triazoles) from benzyl halides, epoxide derivatives, sodium azide and phenylacetylene in water at room temperature was disclosed (Scheme 20).28 The reaction was done in the presence of organic surfactant-free copper sulfide (sf-CuS) nano/micro flower catalysts which were easily obtained by supersaturated condition by adding Cu(OAc)2·H2O to sulfur dissolved in THF. 1,2,3-Triazoles and β-hydroxy 1,2,3-triazoles were synthesized in high yields using this method. Triazole synthesis using very mild condition and reusability of the catalyst make this protocol more efficient.
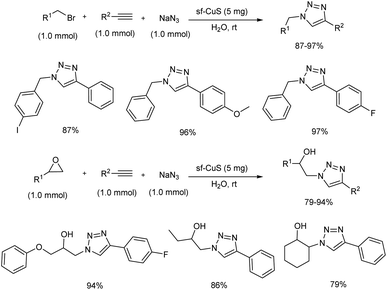 |
| | Scheme 20 One-pot synthesis of 1,2,3-triazoles and β-hydroxy 1,2,3-triazoles. | |
A systematic synthetic protocol towards an important A3 coupling as well as click reaction was reported in 2019.29 Chitosan complexed with Cu-nanoparticles (CS-MCR/Cu2O) was effectively used as catalyst for this reaction without metal leaching and loss of activity. CS-MCR/Cu2O catalyst was prepared by covalent functionalization of chitosan biopolymer via MCR method followed by complexing the in situ formed Cu2O nanoparticles with biopolymeric chitosan. Catalytic activity of the synthesized complex was initially tested on A3 coupling reactions. Propargylamines were smoothly prepared from a range of benzaldehyde with diverse substituents, phenylacetylene derivatives and several secondary amines under ultrasonic irradiation in high yield. Click reaction was also conducted using 20 mg catalyst for the preparation of 1,2,3-triazoles from different benzyl bromides, alkynes and sodium azide in water at 40 °C (Scheme 21).
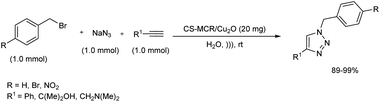 |
| | Scheme 21 Click reaction for the preparation of 1,2,3-triazoles under ultrasonic irradiation. | |
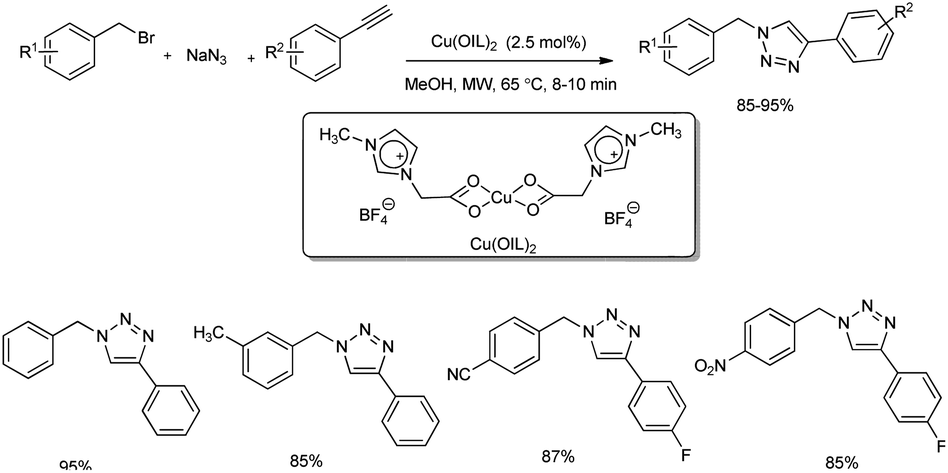
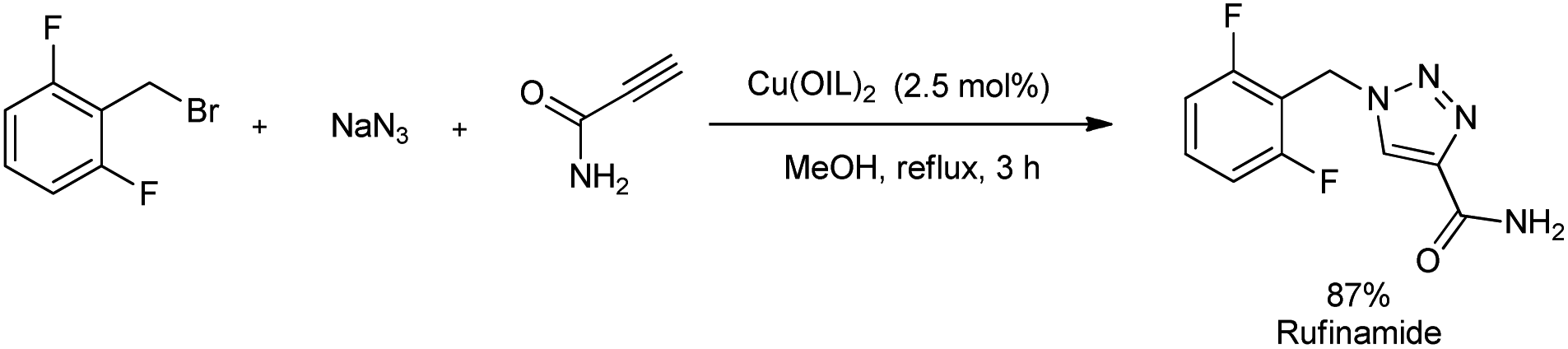
A microwave-assisted regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles by using ionic liquid supported Cu(II) precatalysts in methanol was reported.30 1,4-Disubstituted 1,2,3-triazoles were successfully synthesized from alkynes, benzyl bromide and NaN3 at 65 °C (Scheme 22). Several benzyl bromides and alkynes were used in the substrate scope study and good amount of 1,4-disubstituted 1,2,3-triazoles were formed. The product yield obtained using different benzyl bromides was independent of electronic nature of the substituents. The mentioned protocol was also extended to the preparation of rufinamide, an anti-epileptic drug, in 87% yield (Scheme 23). Previous reports on rufinamide synthesis showed that it was achieved through multistep process under high temperature (135 °C) and long reaction time (24 hours) compared to this method.31
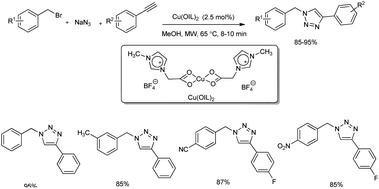 |
| | Scheme 22 Regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles. | |
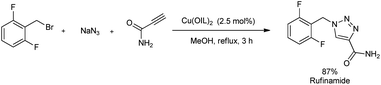 |
| | Scheme 23 Multicomponent synthesis of rufinamide from 2,6-difluorobenzyl bromide, NaN3, and propiolamide. | |
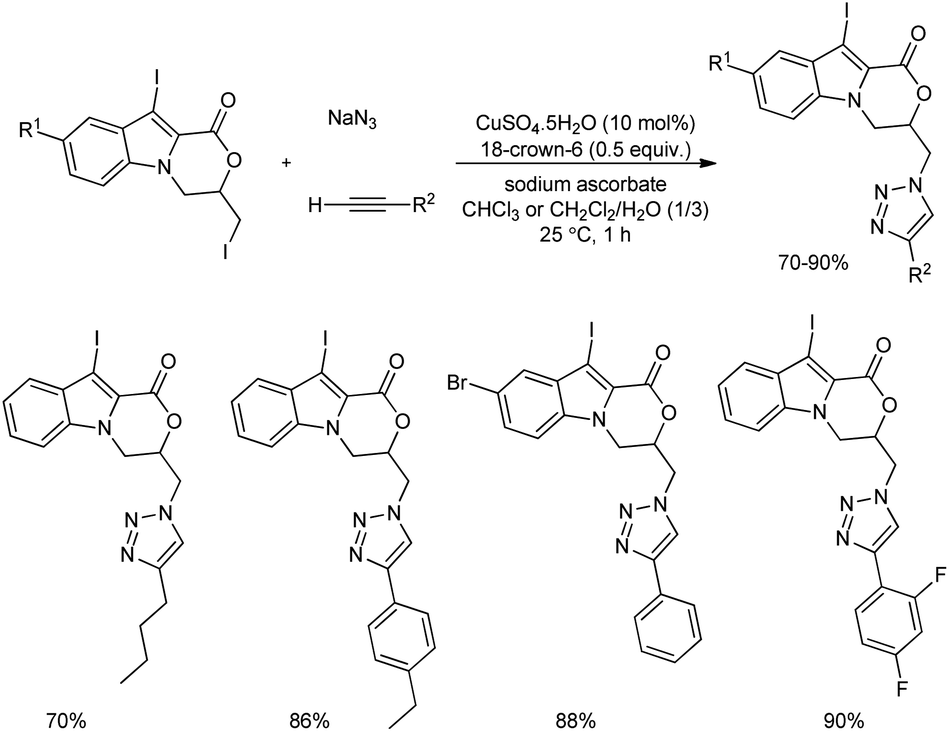
Inspired from the biological as well as pharmacological values of heterocyclic scaffolds, an efficient protocol for the formation novel heterocyclic compounds based on indole-fused oxazinone-1,2,3-triazole scaffolds was reported.32 1,2,3-Triazole and iodo-triazoles were installed on indole-fused oxazinones using CuAAC-based (copper-catalyzed azide–alkyne cycloaddition) multicomponent reaction in the presence of 18-crown-6. After the synthesis of di-iodinated indoles they were reacted with sodium azide and phenyl acetylene in the model reaction in the presence of 10 mol% of CuSO4·5H2O and a moderate yield of target product was obtained.
The scope of the reaction was explored using diverse alkynes and sodium azide, and the desired indole-fused oxazinone-1,2,3-triazole scaffolds were obtained in good to excellent yields (Scheme 24). It is observed that both electron-donating and electron-withdrawing groups can be easily incorporated in the alkyne reactant, and numerous examples of indole-fused oxazinone-1,2,3-triazole scaffolds were synthesized.
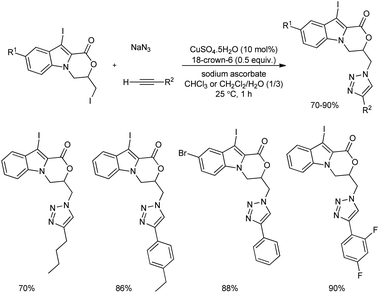 |
| | Scheme 24 Formation of indole-fused oxazinone-1,2,3-triazole scaffolds using CuAAC-based multicomponent reaction. | |
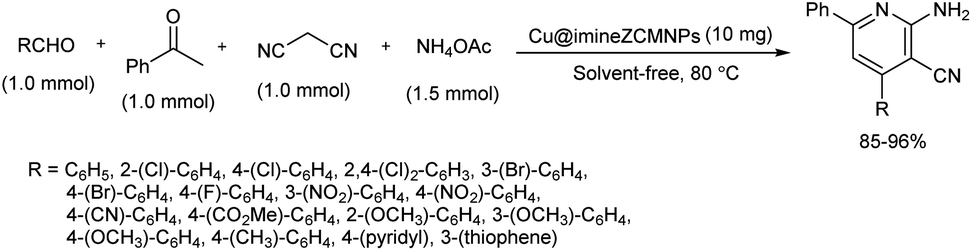
2.1.4. Six-membered heterocyclic compounds with one heteroatom. A four component reaction of various aldehydes, malononitrile, ammonium acetate and acetophenone using Cu@imineZCMNPs as catalyst was studied.33 Copper(II) chloride was the copper source used to prepare Cu@imineZCMNPs complex and a range of 2-amino-3-cyanopyridine derivatives were prepared using this one-pot strategy. After careful optimization studies, aldehydes were reacted with malononitrile, acetophenone and ammonium acetate at 80 °C in the presence of Cu@imineZCMNPs catalyst under solvent-free conditions and corresponding 2-amino-3-cyanopyridine derivatives were obtained in good to high yields (Scheme 25). Low reactivity showed by the aromatic aldehydes with electron-donating groups compared to the aldehydes with electron-withdrawing and heteroaromatic groups proved that electronic effect of substrate groups had a direct influence on the product formation. Moreover, magnetic nature of the catalyst allowed easy recovery of catalyst simply by an external magnetic field and they were reused several times.
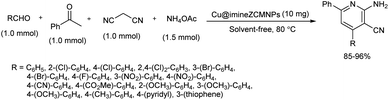 |
| | Scheme 25 Four component reaction of aldehyde, malononitrile, ammonium acetate and acetophenone. | |
Synthesis of functionalized tetrahydropyridine derivatives using Cu(OTf)2 catalyst and their anti-inflammatory activity was described.34 This simple protocol involved reaction of ethyl acetoacetate, substituted aromatic aldehyde and aniline in ethanol at room temperature. Good functional group tolerance and purification without the use of column chromatography was achieved by the method. The authors have successfully carried out in vitro anti-inflammatory activity of the synthesized compounds mainly against matrix metalloproteinases (MMPs) using gelatin zymography.
In 2018, Yao and team established a three-component reaction to form iminolactone from aryl acetylene, enaminone and sulfonyl azide through 6π electrocyclization (Scheme 26).35 Synthesis proceeds via reaction of in situ generated metalated ketenimine from copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) with enaminone followed by 6π electrocyclization. Model substrates chosen was p-tolyl enaminone, phenylacetylene and p-toluenesulfonyl azide. The reaction was carried out under mild conditions and CuI was the copper source employed. A range of aryl enaminones and aryl acetylenes were reacted with sulphonyl azide, and iminolactones were formed in moderate to good yield.
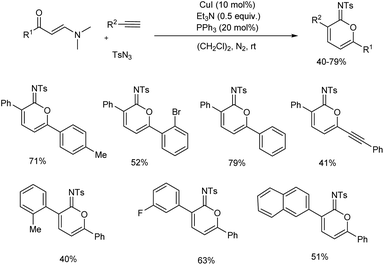 |
| | Scheme 26 Synthesis of iminolactone from aryl acetylene, enaminone and sulfonyl azide. | |
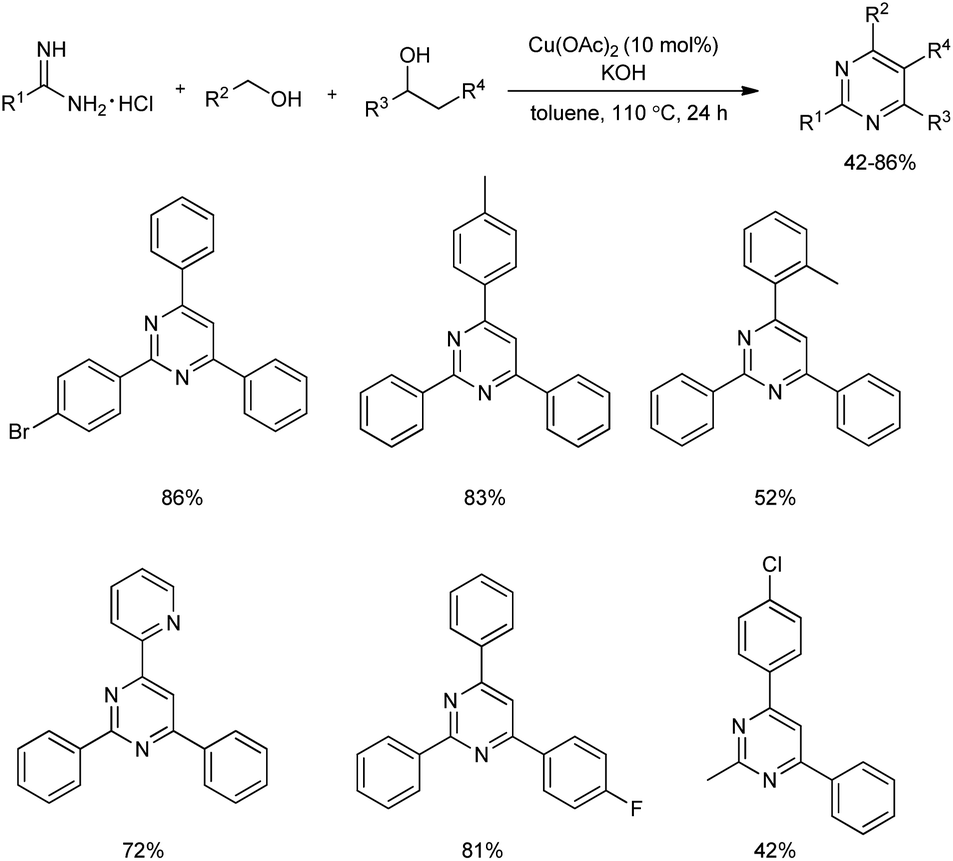
2.1.5. Six-membered heterocyclic compounds with two heteroatoms. Multi-substituted pyrimidines were efficiently synthesized from a three-component reaction of primary alcohols, secondary alcohols and amidines using 10 mol% of Cu(OAc)2 catalyst (Scheme 27).36 Initially, the reaction of benzamidine hydrochloride, benzyl alcohol, and 1-phenylethanol was carried out in the presence of 10 mol% CuBr as catalyst, 3 equiv. of KOH in toluene solvent at a temperature of 110 °C, which afforded 43% of the desired product. An increase of product yield upto 84% was observed when the catalyst CuBr was replaced by Cu(II) catalyst.
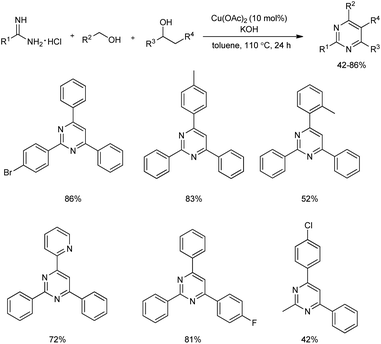 |
| | Scheme 27 Formation of multi-substituted pyrimidines using Cu(OAc)2 catalyst. | |
Aromatic alcohols used gave excellent yields irrespective of the electronic behavior of groups, and higher yields were observed for meta- and para-methyl substituted benzyl alcohols compared to the ortho-substituted groups which proved the significance of steric hindrance on this reaction. Alcohols such as 3-thiophenylmethanol and 2-pyridinylmethanol gave good results. However, reaction using primary aliphatic alcohols did not work. In the case of amidines and aryl amidines with electron-donating groups such as methyl and electron-deficient groups like F, Br, as well as CF3 afforded 2,4,6-trisubstituted pyrimidines in good yields. Interestingly, aliphatic amidine also participated in the reaction and gave a yield of 42%. Among the secondary alcohols used, 4-substituted phenylethanol having electron-withdrawing and electron-donating groups were well tolerated and good yields of corresponding products was offered by halogen-bearing substrates.
2.2. Fused heterocyclic compounds
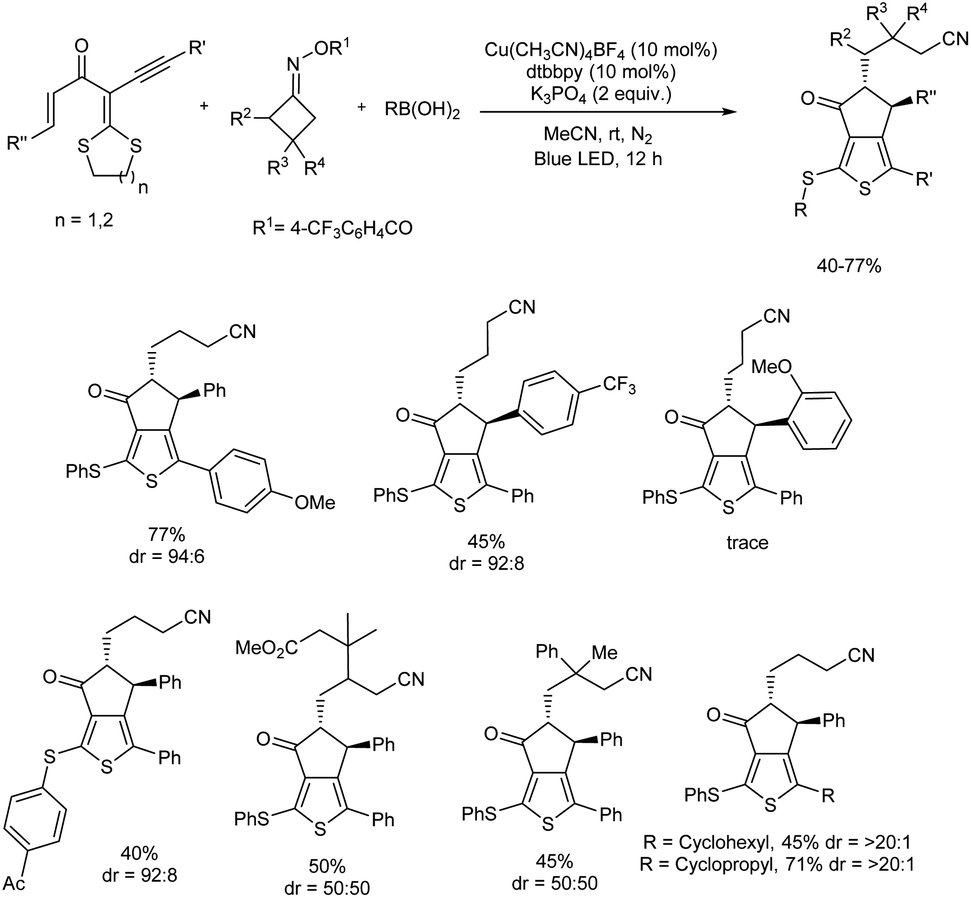
2.2.1. Fused five-membered heterocyclic compounds with one heteroatom. Highly functionalized aryl-2-thienyl sulfides were obtained with good chemo- and diastereoselectivities through a photoinduced, Cu(I)-catalyzed radical cross-coupling reaction of gem-dialkylthio 1,3-enynes, cycloketone oxime esters, and boronic acids.37 The major steps involved in the reaction are a domino sequence involving cyanoalkyl radical-mediated intramolecular annulation of gem-dialkylthio enyne, alkenyl radical-promoted C(sp3)–S bond cleavage, and sulfur-centered radical-trapped Cu(II)-facilitated C–S cross-coupling. The optimized reaction condition was 10 mol% of Cu(CH3CN)4BF4, 10 mol% of 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbbpy) as ligand, and 2 equiv. of K3PO4 in CH3CN under blue LED irradiation. Notably, the method enabled simultaneous establishment of thiophene, cyanoalkyl, and cyclopentanone moieties as well as a thioether C–S bond in one pot. Substrate scope analysis of the protocol studied concluded that steric and electronic effects of the substituents have a significant influence on the reaction and products were isolated with high diastereoselectivities (Scheme 28).
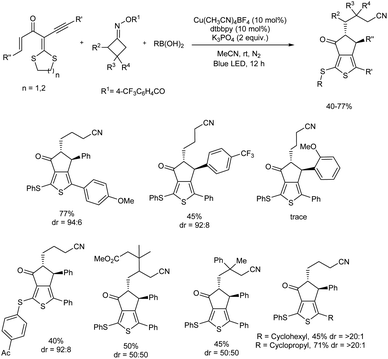 |
| | Scheme 28 Photoinduced, three-component radical synthesis of highly functionalized aryl thienyl sulfides. | |
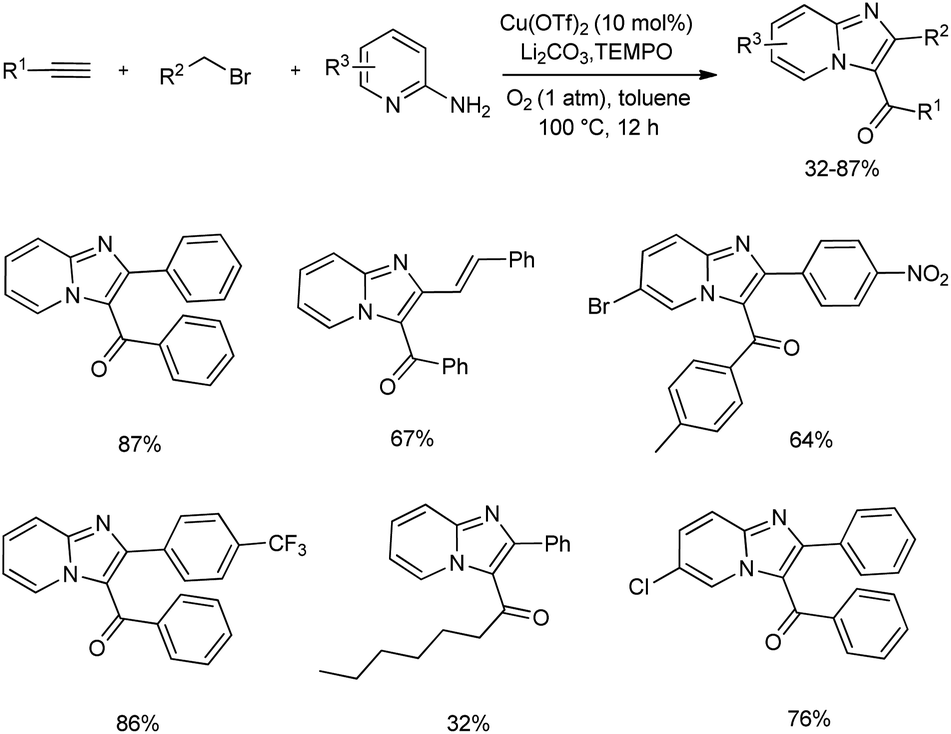
2.2.2. Fused five-membered heterocyclic compounds with two heteroatoms. An efficient synthetic method towards functionalized imidazo fused heterocycles from 2-amino N-heterocycle, terminal alkyne, allyl or benzyl bromide using molecular oxygen was unveiled (Scheme 29).38 The mentioned protocol consists of many advantages and is assumed to have the synthetic potential for the easy access to diverse bioactive molecules. The optimized reaction condition contained Cu(OTf)2 as the copper(II) source for catalysis, Li2CO3 as base, TEMPO as well as molecular oxygen as the suitable oxidant in toluene. The multicomponent cascade reaction exhibited good functional group tolerance, and moderate to good amount of the desired product was obtained.
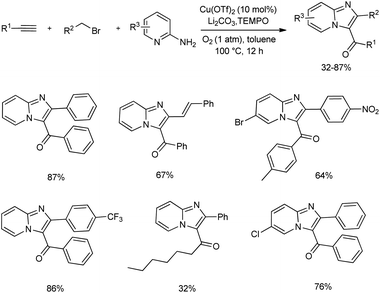 |
| | Scheme 29 Oxidative synthesis of imidazo fused heterocycles. | |
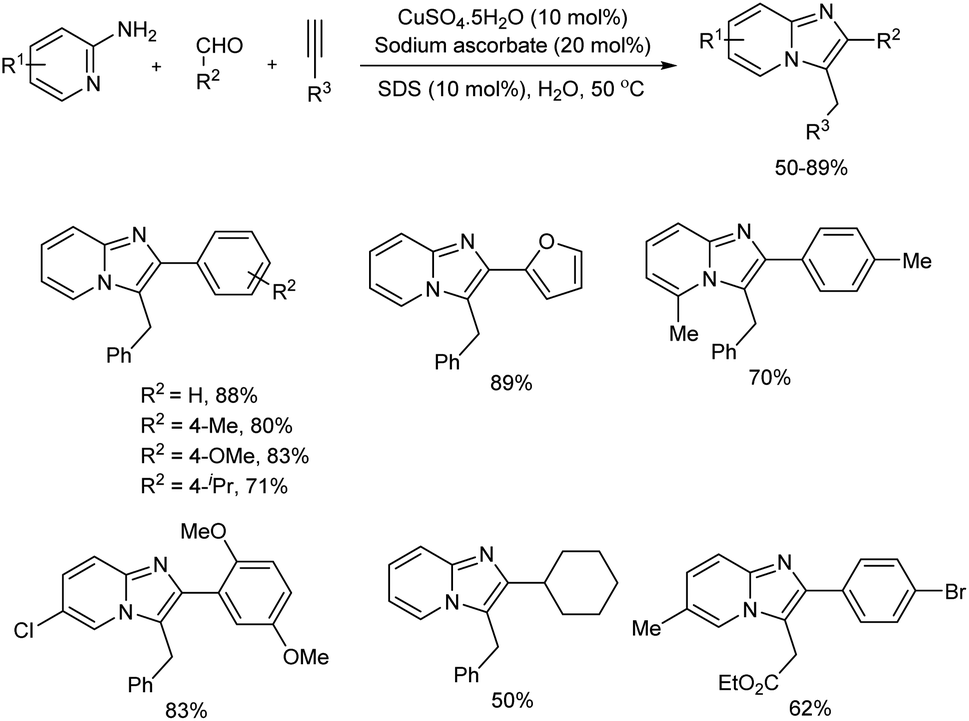
Imidazo[1,2-a]pyridine derivatives were prepared by Cu(II) ascorbate catalyzed domino A3-coupling reaction in aqueous micellar media in the presence of anionic surfactant, sodium dodecyl sulfate (SDS) (Scheme 30).39 In this eco-friendly procedure, the catalyst used was a dynamic combination of Cu(II)/Cu(I), which was generated in the reaction mixture in situ by mixing CuSO4 with sodium ascorbate. Imidazo[1,2-a]pyridines were synthesized in good amount from 5-exo-dig cycloisomerization of alkynes with the condensation products of aldehydes and 2-aminopyridines. The protocol worked well with 2-aminopyridine and benzaldehyde derivatives having electron-withdrawing and electron-donating substituents and aliphatic, aromatic as well as heteroaromatic aldehydes gave the corresponding products in good yields. The reaction was equally successful in gram-scale and present method is reported as highly efficient and useful than existing methods in many aspects including mild reaction condition, inexpensive catalyst, simple experimental setup and green reaction media.
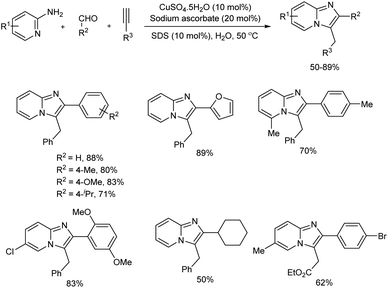 |
| | Scheme 30 Imidazo[1,2-a]pyridine derivatives synthesis in aqueous micellar media. | |
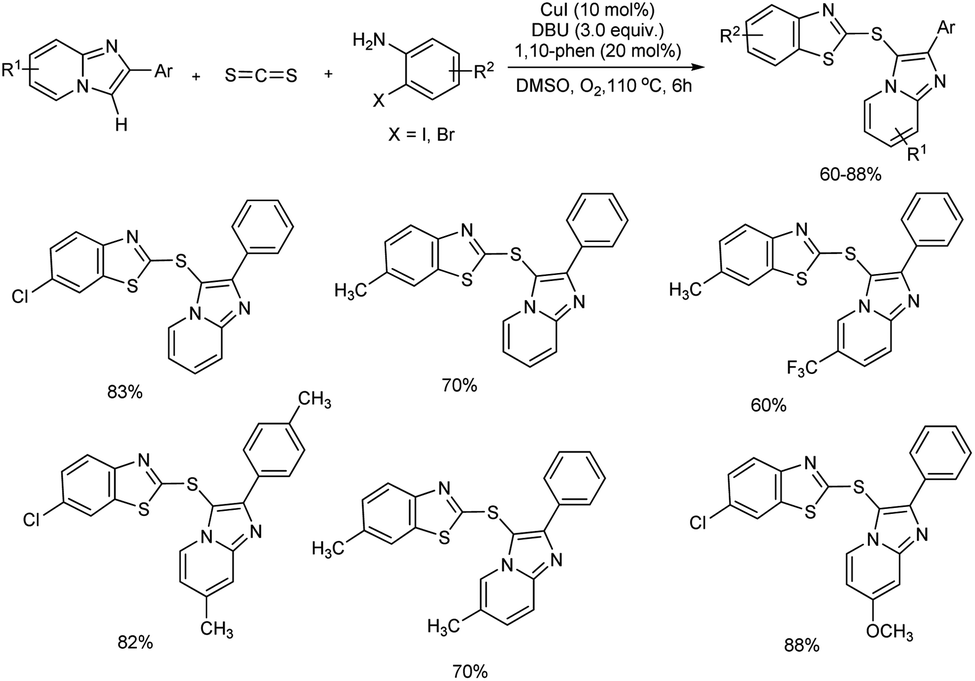
A convenient domino method for the C-3 sulfenylated imidazo[1,2-a]pyridine derivative construction using easily available carbon disulfide as a building block through Ullmann-type coupling and C–H functionalization was unveiled by Yang et al.40 This Cu(I)-catalyzed reaction of imidazo[1,2-a]pyridines and carbon disulfide with o-haloanilines was unsuccessful in the absence of copper source and the best results were obtained when CuI was used as catalyst and 1,10-phenanthroline (1,10-phen) as the ligand. Several bases and solvents evaluated found DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) and DMSO as the most efficient base and solvent for the reaction. The substrate scope studies were conducted using diversely substituted imidazo[1,2-a]pyridines and o-haloanilines. The corresponding products were obtained in moderate to good yields and the present strategy exhibited a moderate functional group tolerance (Scheme 31). Among o-haloanilides, o-iodoacetanilines and o-bromoaniline derivatives were compatible and among them o-haloanilides bearing electron-withdrawing groups were more reactive than those having electron-donating groups. However, substituted imidazo[1,2-a]pyridines with electron-withdrawing or electron-donating group showed no significant variation in reaction yield.
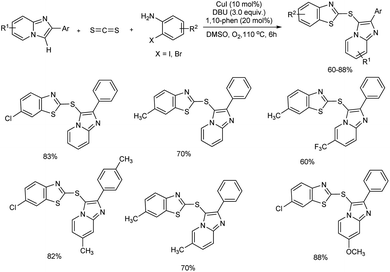 |
| | Scheme 31 Synthesis of C-3 sulfenylated imidazo[1,2-a]pyridine derivatives in the presence of CuI catalyst. | |
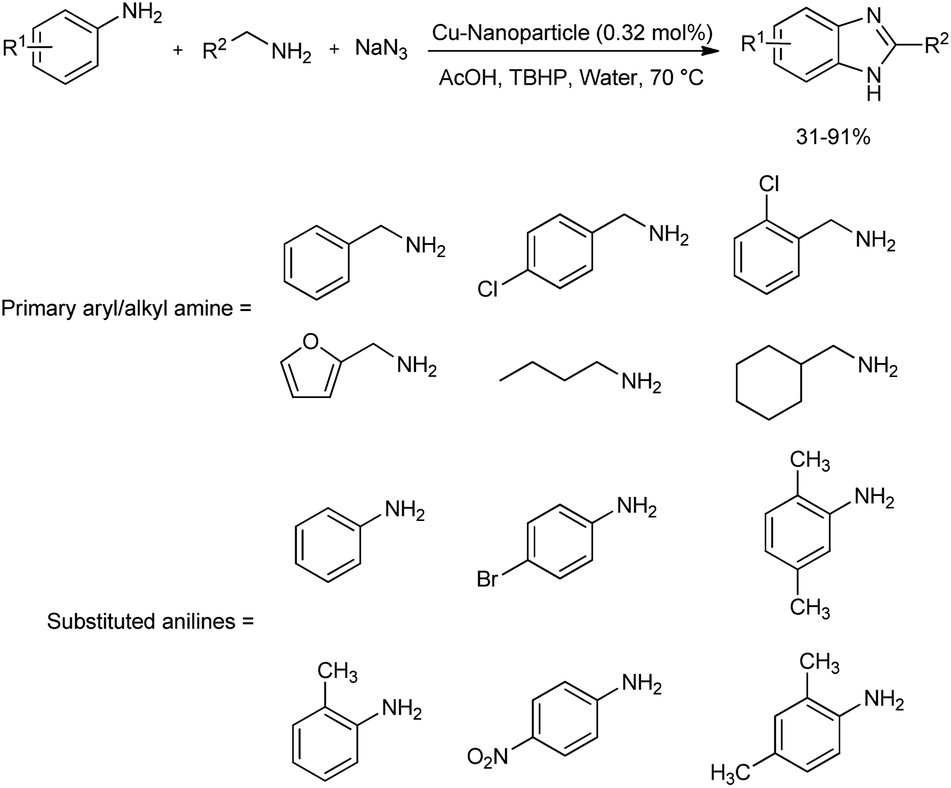
A magnetically recoverable Cu-isophthalate-based metal–organic framework (MOF) decorated with surface-modified cobalt ferrite (CoFe2O4) nanoparticles was prepared by chemical bonding method.41 This copper-based framework was successfully used as efficient catalyst for the preparation of a wide range of substituted benzimidazoles through oxidative cross coupling reaction (Scheme 32). Benzimidazoles were prepared by the multicomponent reaction of primary aryl/alkyl amine, substituted aniline and sodium azide in green solvent, water. tert-Butyl hydroperoxide (TBHP) was the suitable oxidizing agent found for the reaction, and a temperature of 70 °C was found optimum. The scope of primary amine as well as aniline functionalities was examined. Alkyl amines, amine with heterocyclic group like furfurylamine and aliphatic primary amines engaged in the reaction with good reactivity. Broad substrate applicability was reported in the case of aniline functionalities too. The catalyst was magnetically retrievable and could be used for eight successive times.
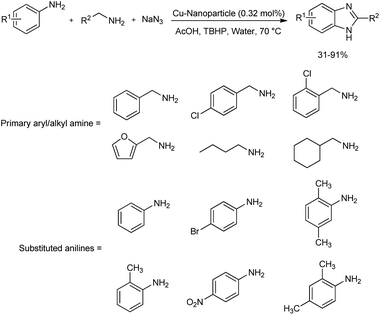 |
| | Scheme 32 Benzimidazole synthesis via oxidative cross-coupling. | |
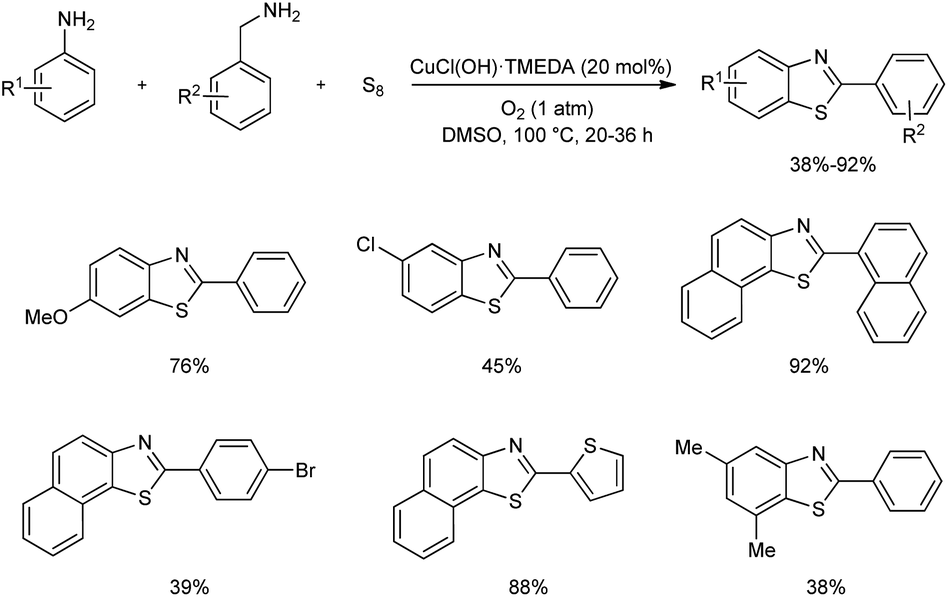
A one-pot three-component preparation of benzothiazoles using copper(II) catalyst was established very recently.42 Aerobic cross-coupling of amines and subsequent thiolation of arene by elemental sulfur gave a series of benzothiazoles in moderate to good yields (Scheme 33). Notably, undesirable oxidation pathways of benzylamines and anilines to the corresponding aldehydes and azobenzenes were avoided in this synthesis. Control experiments conducted by the team revealed that 20 mol% is the suitable amount of CuCl(OH)·TMEDA catalyst and molecular oxygen plays a crucial role in the catalytic cycle.
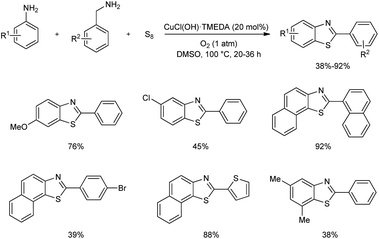 |
| | Scheme 33 Cu-based one-pot three-component synthesis of benzothiazoles. | |
Functional group tolerance on aniline derivatives accessed found alkyl groups at para-position of anilines gave the desired product in moderate to good yields, and electronic nature of groups on anilines showed a significant effect on product yield. Aniline without electron-donating group and anilines with halogen substituent gave low yields of benzothiazoles. Alkyl and allyl amines stopped at the aldehyde products and they were not employed in the reaction. In the case of benzylamines, groups such as chloro-, bromo-, and nitro- at the para-position gave lower yields of benzothiazoles. Moreover, 2-thiophenemethylamine reacted to form benzothiazoles in 88% yield.
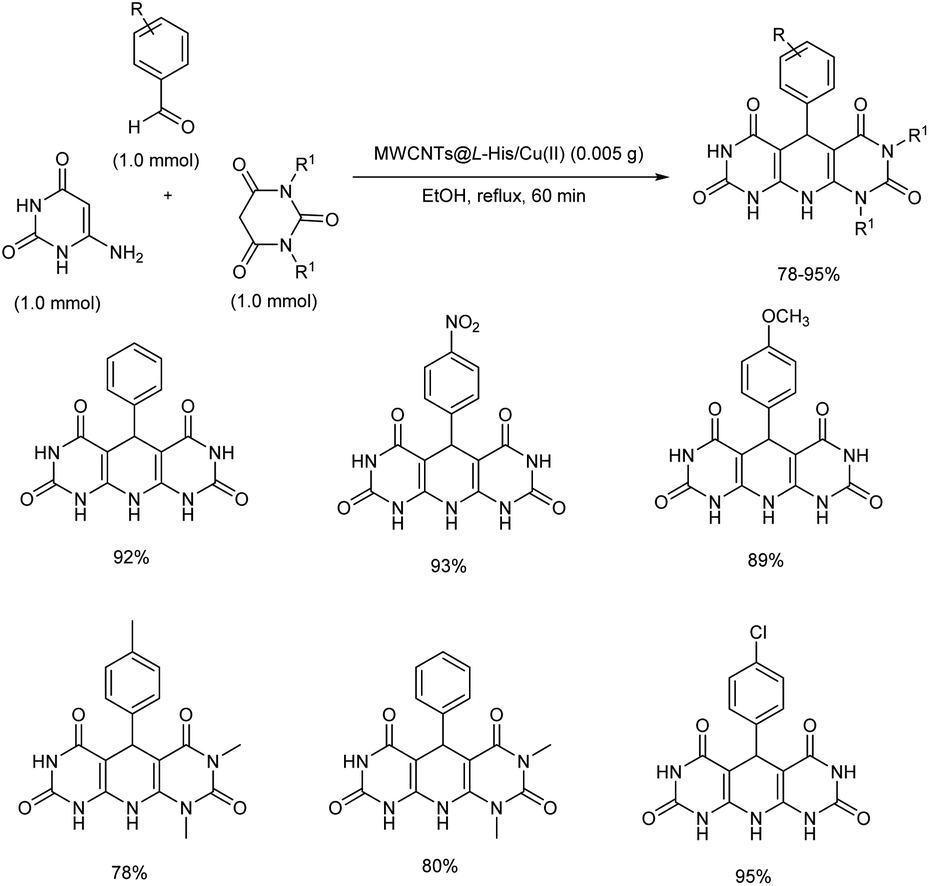
2.2.3. Fused six-membered heterocyclic compounds with one heteroatom. A novel heterogeneous organometallic catalyst was discovered from Cu(II) immobilization on L-histidine functionalized multi-walled carbon nanotubes (MWCNTs@L-His/Cu(II)).43 Simple one-pot condensation reaction of barbituric acid (1,3-dimethylbarbituric acid), aromatic aldehydes and 6-aminouracil using MWCNTs@L-His/Cu(II) afforded pyrido[2,3-d:5,6-d′]dipyrimidine derivatives in high to excellent yields within a relatively short reaction time (Scheme 34). Initially, reaction conditions were optimized using barbituric acid, 6-aminouracil and p-chlorobenzaldehyde as model substrates. Solvent screening conducted concluded that polar solvents were better, and the optimized reaction condition was use of MWCNTs@L-His/Cu(II) under reflux conditions for 60 minutes in ethanol. The aromatic aldehydes with electron-withdrawing substituents offered higher yields compared to those with electron-releasing ones. Gratifyingly, the catalyst synthesized was recoverable and reusable.
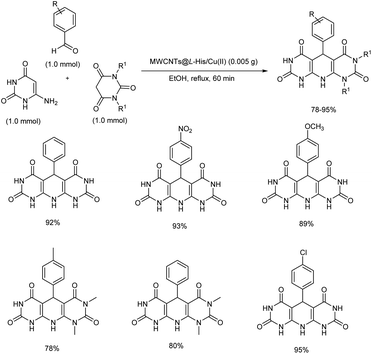 |
| | Scheme 34 MWCNTs@L-His/Cu(II) complex catalyzed synthesis of pyrido[2,3-d:5,6-d′]dipyrimidines. | |
Chitosan decorated copper nanoparticles (CS/CuNPs) were used as catalyst for the ultrasonic irradiated synthesis of quinoline derivatives.44 CS/CuNPs were prepared using reduction techniques via green protocols. Initial reaction was carried out using p-chlorobenzaldehyde, dimedone, ethyl cyanoacetate and ammonium acetate, and the optimal reaction conditions identified was 0.1 g of chitosan nanoparticles loaded Cu(II) complex in ethanol at 80 °C under ultrasonic irradiation for 15 minutes (Scheme 35). CS/CuNPs catalyst exhibited high turnover frequency and it was reusable up to five times.
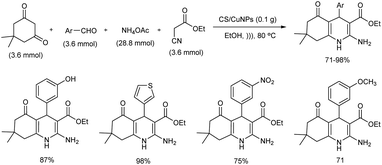 |
| | Scheme 35 CS/CuNPs catalyzed synthesis of quinoline derivatives. | |
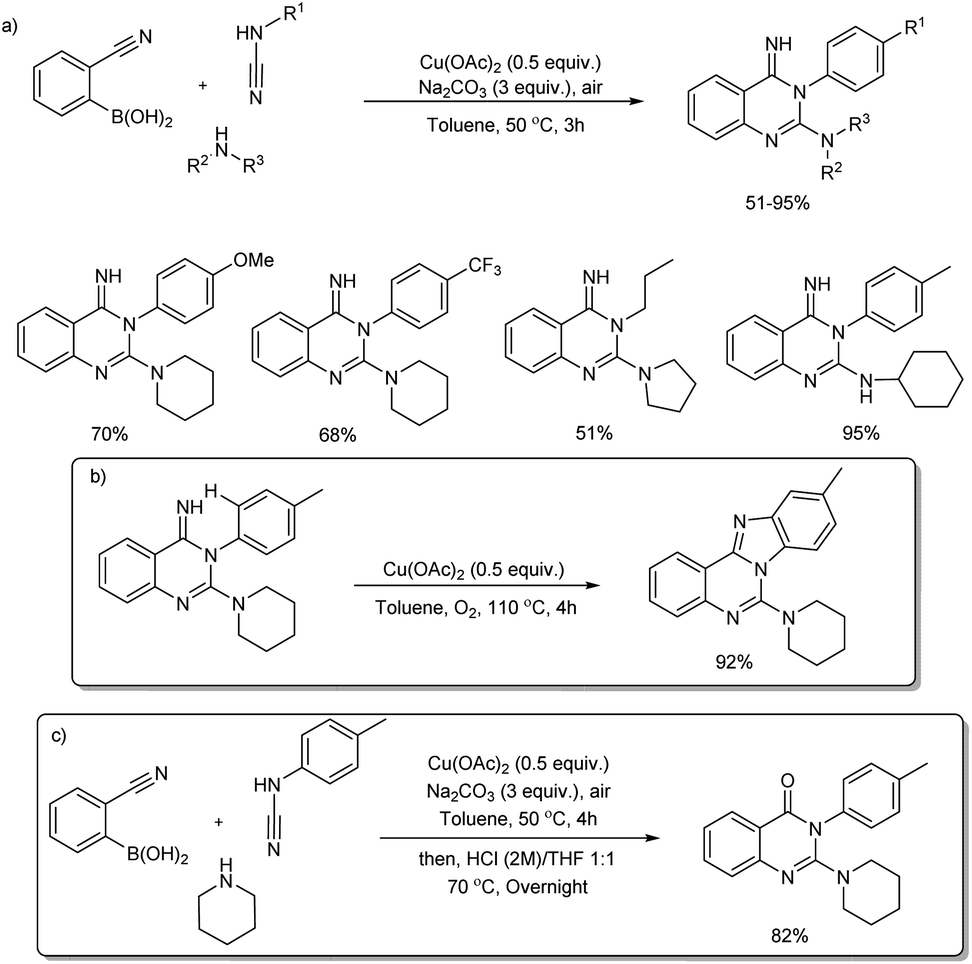
2.2.4. Fused six-membered heterocyclic compounds with two heteroatoms. A copper(II)-based three-component reaction for the synthesis of quinazolin-4(H)-imines and benzimidazo[1,2-c]quinazolines was reported by Neuville and team.45 Quinazolin-4(H)-imines were successfully prepared from 2-cyanoarylboronic acids, cyanamides and amines in the presence of Cu(OAc)2 and Na2CO3 at 50 °C for 3 hours (Scheme 36(a)) and cyclization of newly formed NH-imine delivered benzimidazo[1,2-c]quinazolines under oxidative copper catalysis (Scheme 36(b)). Under similar one pot hydrolytic conditions (more stronger conditions including Cu(OAc)2 and Na2CO3, HCl, in THF/water (1:1) at a temperature of 70 °C for 12 h) quinazolin-4(3H)-one was formed in 82% yield (Scheme 36(c)). The protocol exhibited wide substrate scope. Various aryl, alkyl and benzyl cyanamides and linear or branched primary alkyl amines, benzyl amines and cyclic as well as acyclic amines reacted very well in the reaction.
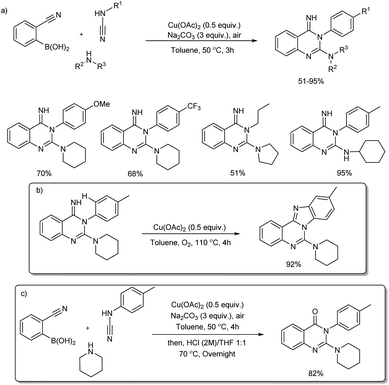 |
| | Scheme 36 Copper-catalyzed synthesis of (a) quinazolin-4(H)-imines, (b) benzimidazo[1,2-c]quinazolines and (c) quinazolin-4(3H)-one. | |
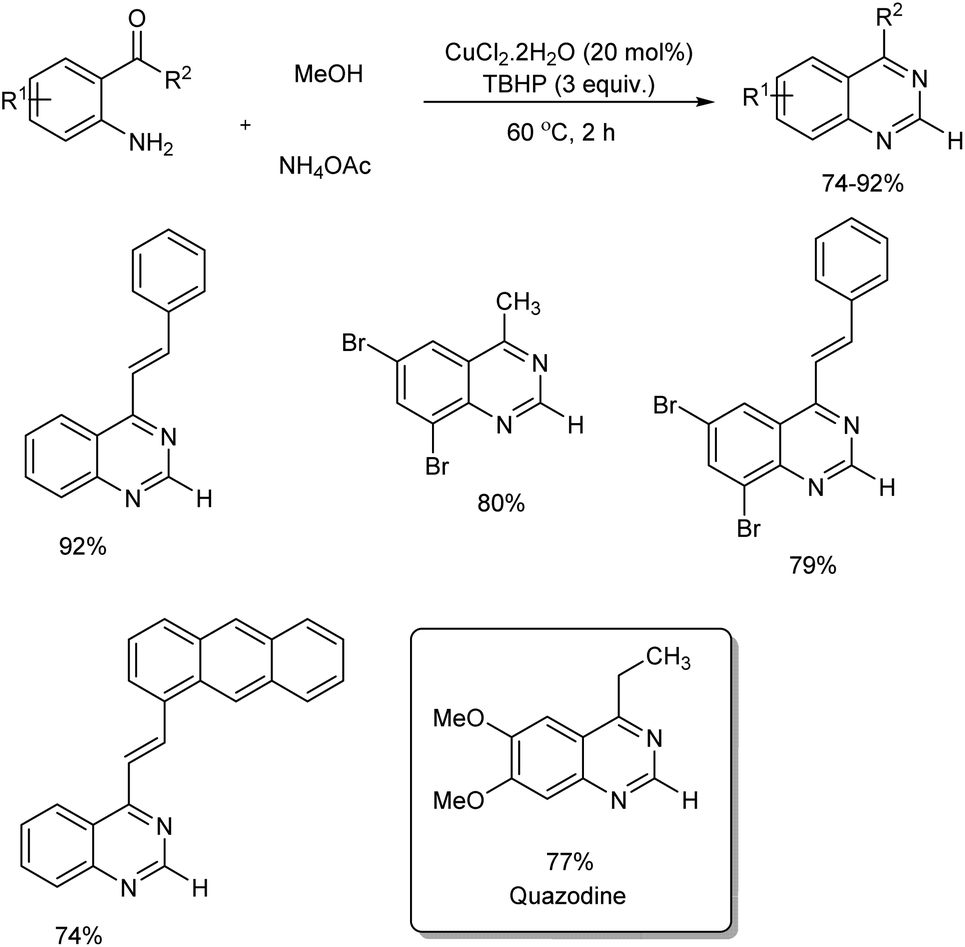
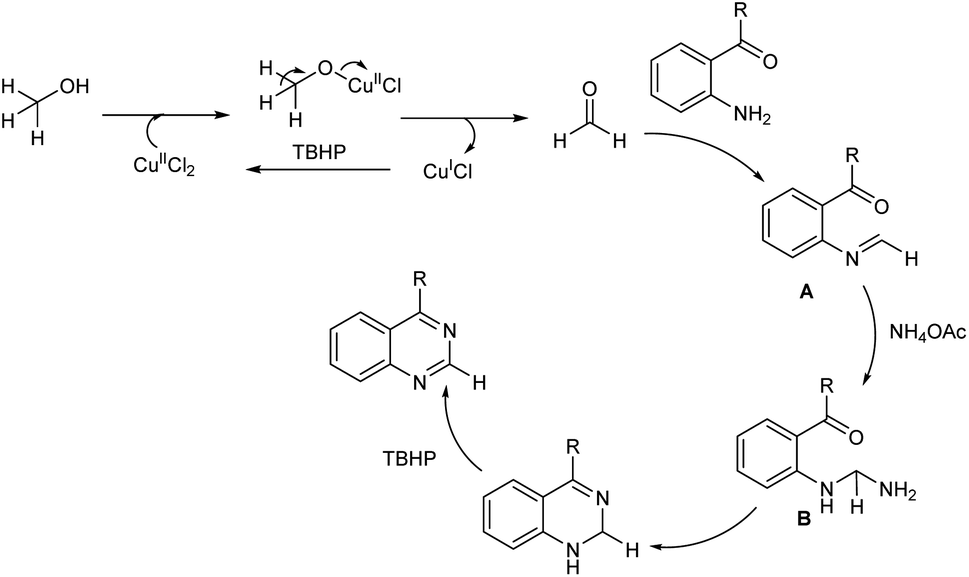
A facile and mild strategy for the construction of quinazolines via copper-based oxidative amination of 2′-aminoarylketones with naturally abundant methanol as C1 carbon source and ammonium acetate as amine source was implemented in 2019.46 The reaction was carried out in the presence of 0.2 mmol of CuCl2·2H2O, 3.0 mmol of TBHP in 3.0 mL MeOH at 60 °C (Scheme 37). Quazodine, a muscle relaxing drug was synthesized in 77% yield from dimethoxy 2′-aminopropiophenone under this condition. The procedure was applicable in multigram scale and exhibited broad functional group tolerance. The plausible mechanism suggested by the team involve, dehydrogenation of methanol to form the formaldehyde in the presence of copper(II) salt and oxidant. Condensation of in situ generated HCHO with aromatic primary amine of 2′-aminoarylketones forms imine A. The reaction of imine A with NH4OAc forms amine B. Subsequent intramolecular cyclization followed by oxidation facilitates the quinazoline in one pot (Scheme 38).
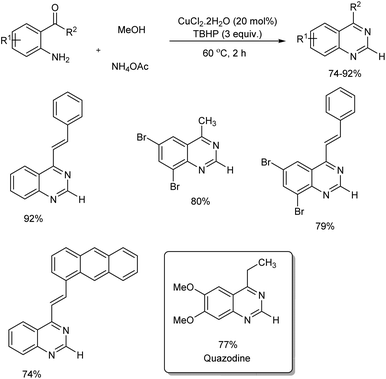 |
| | Scheme 37 One pot synthesis of quinazolines via oxidative amination of methanol. | |
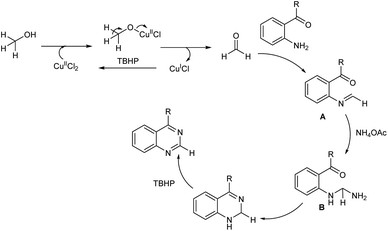 |
| | Scheme 38 Expected mechanism of quinazolines synthesis via oxidative amination of methanol [this figure has been reproduced from ref. 46 with permission from Royal Society of Chemistry, copyright 2019]. | |
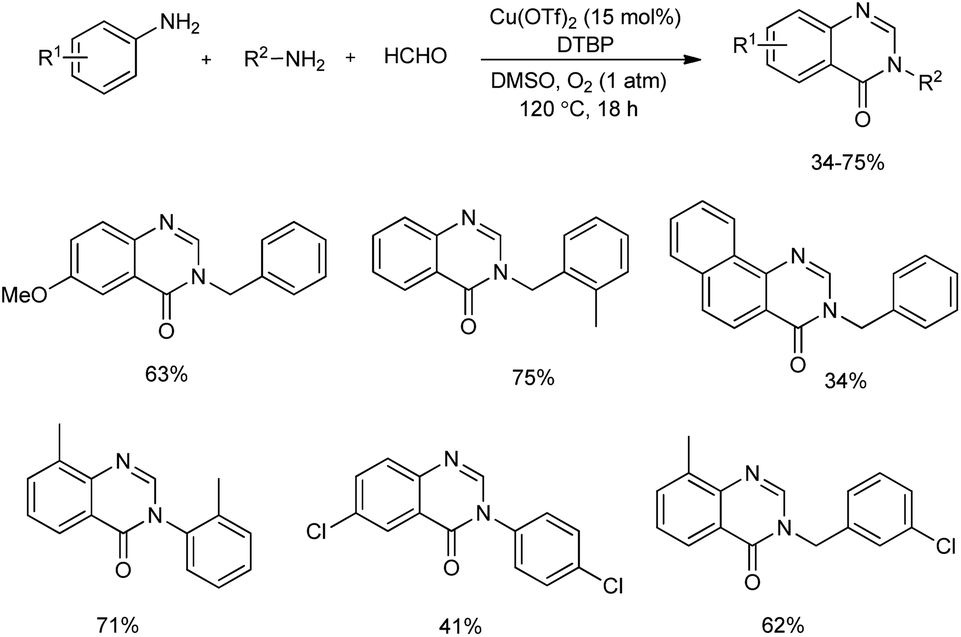
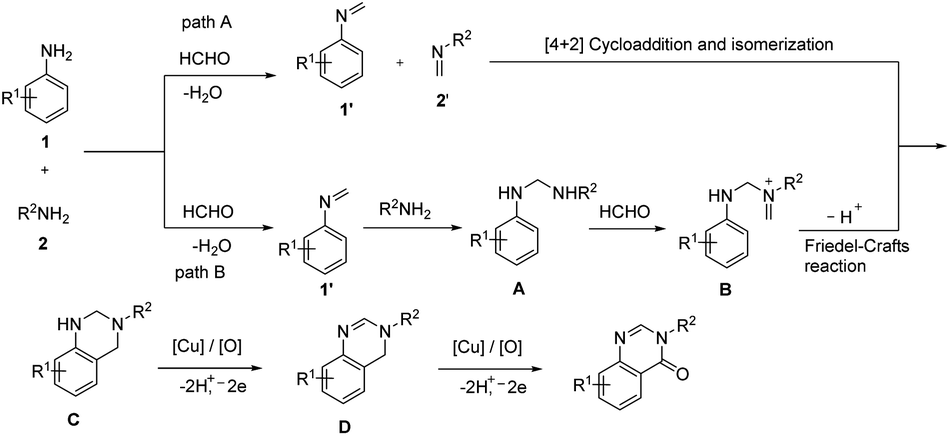
An unprecedented synthesis of quinazolinones through an imine-protection method using copper(II)-catalyzed oxidative multicomponent annulation was disclosed.47 Primary amines, anilines and formaldehyde reacted to yield quinazolinones by the formation of one C–C bond and three C–N bonds along with benzylic functionalization (Scheme 39). Optimization studies revealed that the use of Cu(OTf)2 as well as DMSO (dimethyl sulfoxide) gave the best results in the reaction. Several substituted anilines used revealed that anilines with electron-rich groups on aryl ring exhibited higher yields of quinazolinones than those with electron-withdrawing group. Moderate to good yield of target product was obtained in the case of different primary alkylamines used. Benzylamines with electron-donating group performed better in the reaction compared to benzylamine containing electron-withdrawing group. A detailed mechanistic investigation of the protocol resulted in two possible reaction pathways of the synthesis. In path A, the HCHO condenses with amines 1 and 2 and form imines 1′ and 2′. Then, the [4 + 2] cycloaddition between 1′ and 2′ and subsequent isomerization to give aminal C occurs. Under oxidative copper catalysis dihydroquinazoline D generates from the dehydrogenation of C, which is induced by single-electron oxidation. Finally, Cu(II)-catalyzed benzylic oxidation to give desired product (Scheme 40).
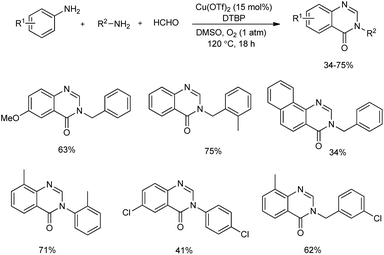 |
| | Scheme 39 Copper-catalyzed oxidative multicomponent annulation for the synthesis of quinazolinones. | |
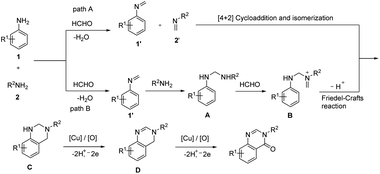 |
| | Scheme 40 Expected mechanism of quinazolinones synthesis [this figure has been reproduced from ref. 47 with permission from American Chemical Society, copyright 2019]. | |
2.3. Miscellaneous
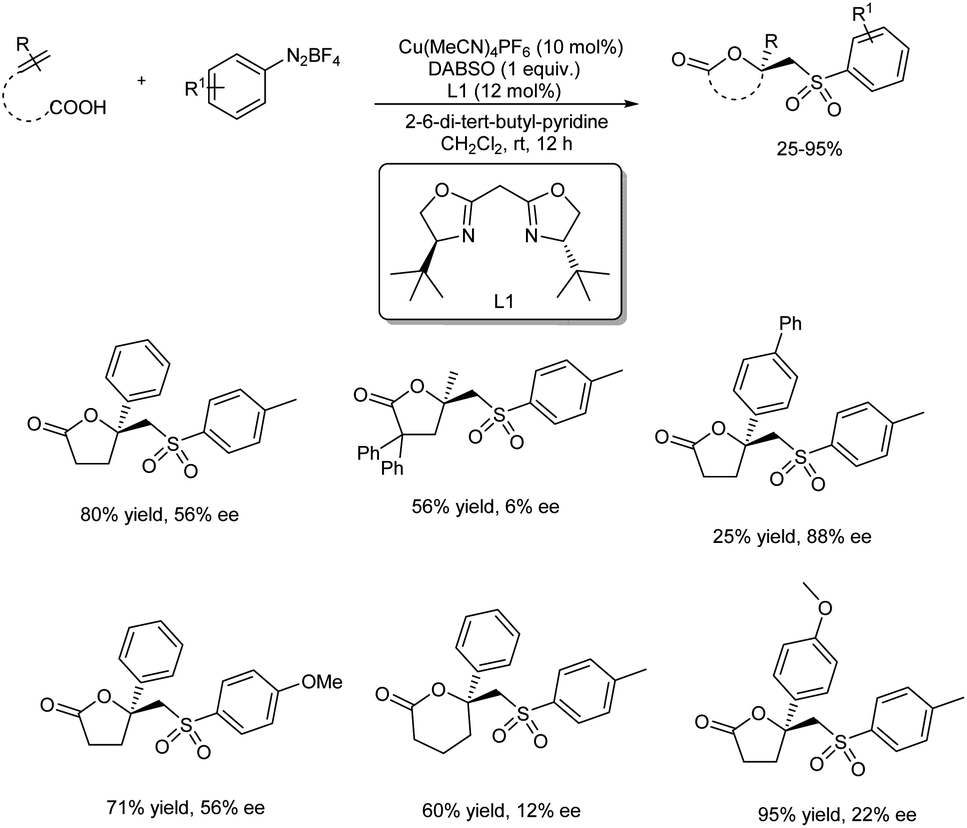
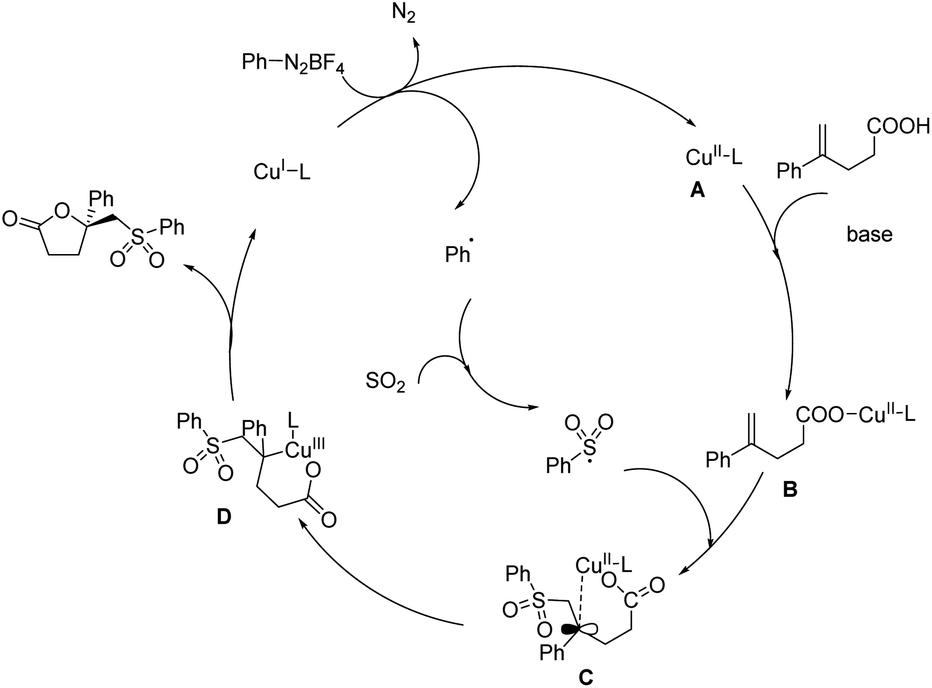
The first report on asymmetric copper-catalyzed radical multicomponent reaction for sulfonyl lactone formation from unsaturated carboxylic acids, DABCO·(SO2)2 (DABSO) and aryldiazonium tetrafluoroborates appeared in 2018 (Scheme 41).48 Model substrates chosen were 4-phenylpent-4-enoic acid, 4-methylbenzenediazonium tetrafluoroborate and DABSO. [Cu(MeCN)4]PF6 was the copper source and bis((S)-4-(tert-butyl)-4,5-dihydrooxazol-2-yl)methane (L1) was the chiral ligand selected for further reactions. The effect of substituents on unsaturated carboxylic acids was investigated, and most of the substrates with either electron-withdrawing or donating group gave good results. Reaction carried using 4-methyl-2,2-diphenylpent-4-enoic acid (substrate having two phenyl groups) to study the steric effect of the protocol gave the desired product with 6% ee and 56% chemical yield. The scope of aryl diazonium salts studied found that substrates with strong electron-withdrawing or electron-donating group worked well in this method giving the corresponding product up to 71% yield and also 71% ee. A plausible mechanism towards sulfonyl lactones was also proposed and it involve reduction of phenyldiazonium tetrafluoroborate by Cu(I) through SET (single electron transfer) to generate phenyl radical as well as Cu(II) complex A via nitrogen releasing. Benzene sulfonyl radical generation through trapping phenyl radical by DABCO·(SO2)2. Development of Cu(II)-unsaturated carboxylic acids complex B from the reaction of unsaturated carboxylic acid with A in the presence of base. Formation of intermediate C from coupling of benzene sulfonyl radical with complex. Followed by the chiral center and Cu(III) complex formation via single electron oxidation process of intermediate C and reductive elimination of Cu(III) complex D to afford corresponding sulfonyl lactones (Scheme 42).
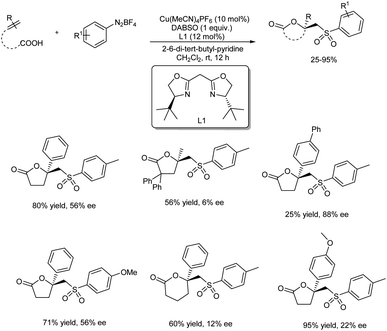 |
| | Scheme 41 Asymmetric copper-catalyzed multicomponent synthesis of sulfonyl lactones. | |
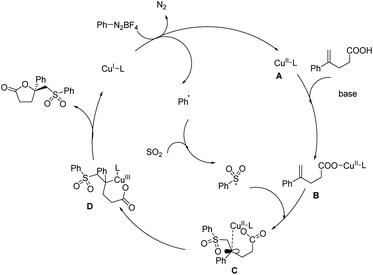 |
| | Scheme 42 Plausible mechanism of sulfonyl lactone synthesis [this figure has been reproduced from ref. 48 with permission from John Wiley & Sons, copyright 2018]. | |
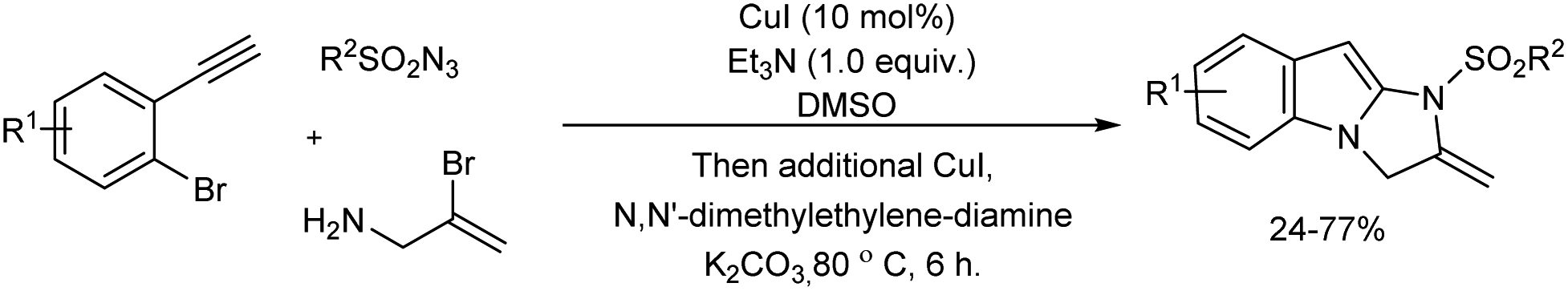
A Cu(I)-catalyzed three-component reaction of alkynes, sulfonyl azides and allylamines offering 2,3-dihydro-1H-imidazo-[1,2-a]indoles was reported (Scheme 43).49 2,3-Dihydro-1H-imidazo[1,2-a]indoles were obtained in moderate yields via sequential copper-catalyzed azide–alkyne cycloaddition (CuAAC) followed by a double copper-catalyzed C–N coupling reactions in one-pot.
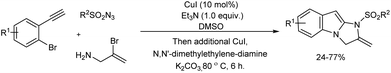 |
| | Scheme 43 Multi-component reaction of alkynes, sulfonyl azides and allylamines for the synthesis of 2,3-dihydro-1H-imidazo-[1,2-a]indoles. | |
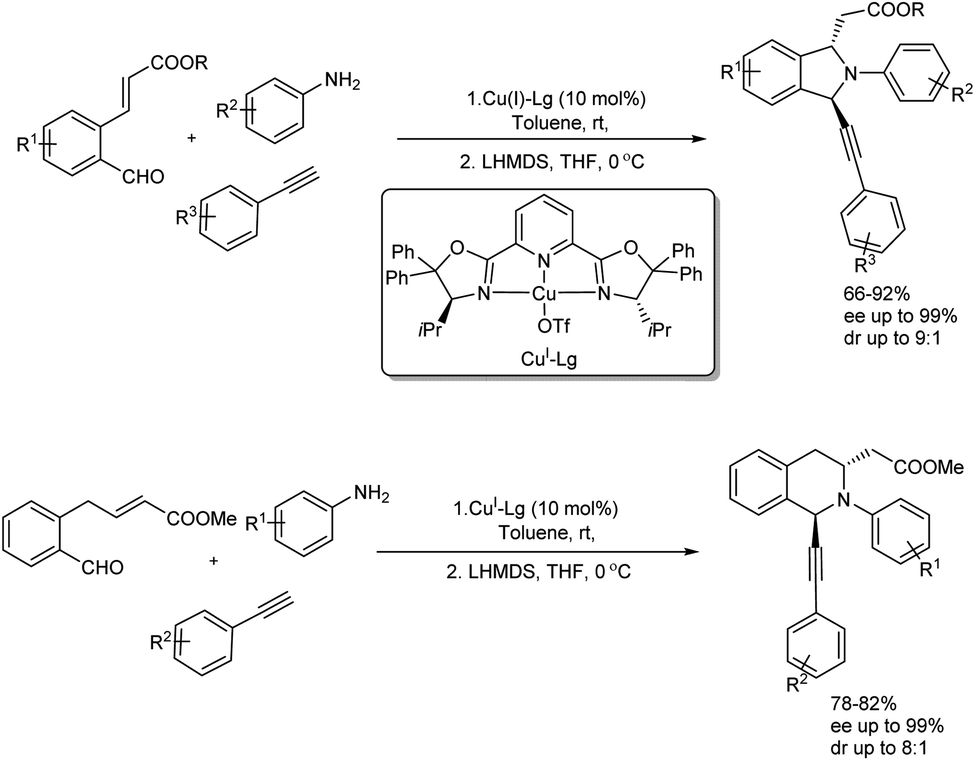
Singh et al. disclosed the enantio- and diastereoselective preparation of 1,3-disubstituted isoindolines and tetrahydroisoquinolines through Cu(I)-Pybox-diPh catalyzed one-pot imination–alkynylation–aza-Michael sequence.50 Among several catalysts screened Cu(I)-triflate and Cu(II)-triflate only gave the desired product in good amount and Cu(I)-Pybox-diPh were used in further reactions. One C–C and two C–N bonds were generated under the reaction and high yield of products with enantioselectivity and diastereoselectivity up to 99% and 9:1 was obtained respectively (Scheme 44). Substrate scope studies of isoindolines was carried using a variety of amines, terminal alkynes and aldehydes. Aromatic amines having an electron-donating group and halide substituted aniline gave high yield of the final product with increased enantioselectivity. Various terminal alkynes were also employed in the substrate scope analysis and electronic properties of substitution on the aromatic ring of alkyne showed no significant influence on the reaction yield. In the case of substituted aldehydes ester functionalities gave the desired isoindoline in very good yield and enantioselectivity. Comparatively lower yield was obtained for sterically bulkier tert-butyl ester. After synthesizing isoindolines successfully, 1,3-disubstituted tetrahydroisoquinolines (THIQ) were also prepared in high amount.
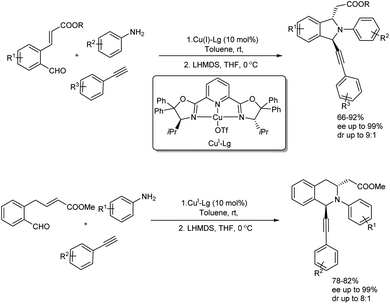 |
| | Scheme 44 Enantio- and diastereoselective synthesis of 1,3-disubstituted isoindolines and tetrahydroisoquinolines. | |
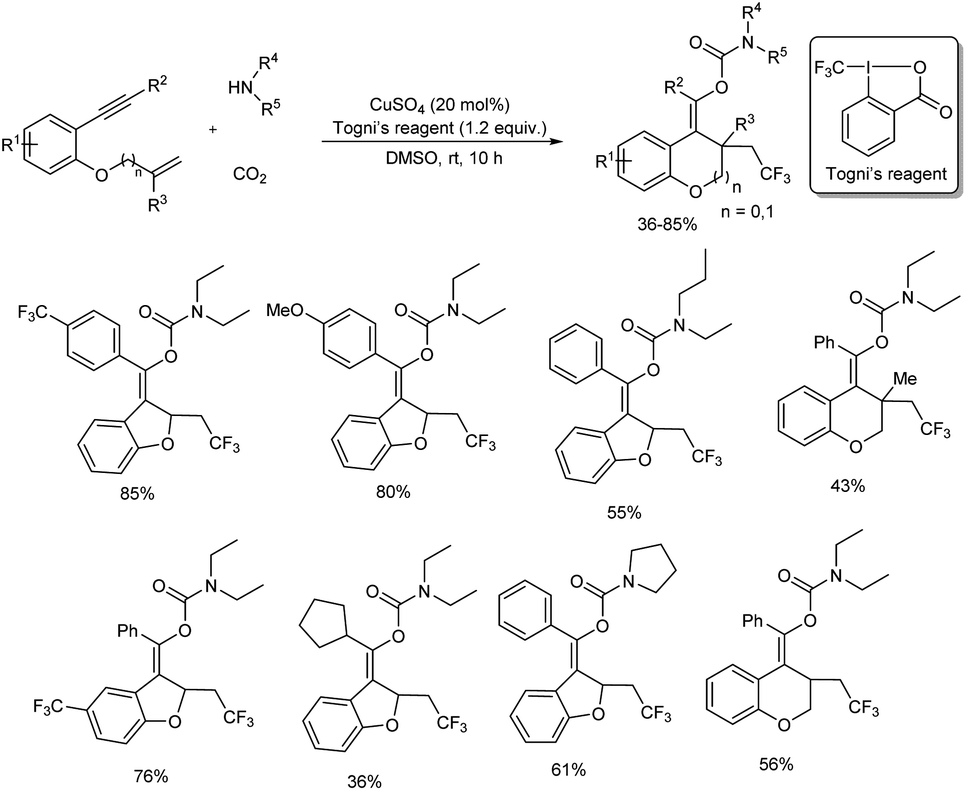
A novel and efficient method for the preparation of trifluoromethyl-substituted carbamates (which are difficult to prepare by other means) was achieved via an efficient copper(II)-catalyzed cascade cyclization of enynes with Togni's reagent, carbon dioxide, and amines.51 Copper source selected for the synthesis was CuSO4 and reaction was carried out in dry DMSO at room temperature for 10 hours. The catalytic method exhibited good functional group tolerance as well as broad substrate scope (Scheme 45). Diverse 1,6-enynes having substituted aryl groups at the terminal alkyne gave the corresponding products in moderate to high yields. Electron-donating as well as electron-withdrawing groups at the ortho, meta, or para positions of the benzene ring were well tolerated. Moreover, substrates with a heterocycle or fused aryl ring also reacted efficiently under this protocol. The scope of amines investigated found dialkylamines; both symmetric and unsymmetric ones and several cyclic secondary amines gave good yields of final product. Whereas, reaction using primary amines such as n-butylamine was unsuccessful.
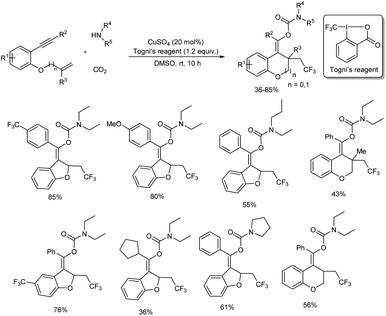 |
| | Scheme 45 Preparation of trifluoromethyl-substituted carbamates through copper-catalyzed cascade cyclization of enynes. | |
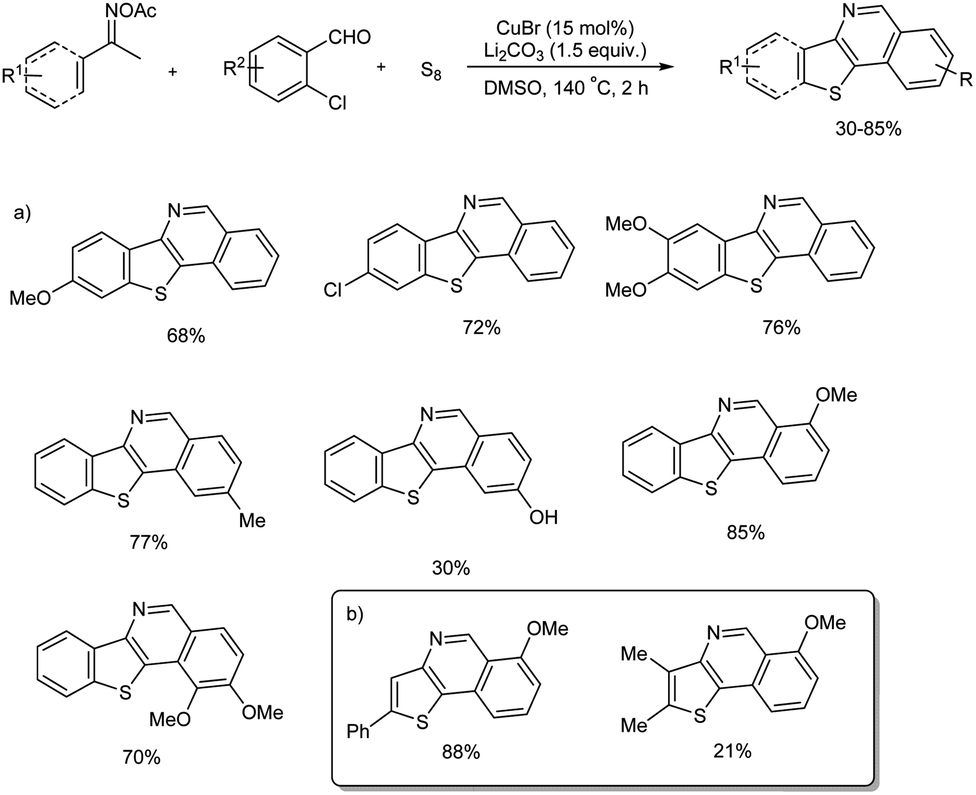
A range of benzo[4,5]thieno[3,2-c]isoquinoline and thieno[3,2-c]-isoquinoline compounds were prepared from a multi-component cascade bis-heteroannulation reaction of methylketoximes, o-halobenzaldehydes and elemental sulfur.52 Initial reaction commenced with acetophenone oxime ester, o-halo-substituted benzaldehydes and elemental sulfur in presence CuBr and Li2CO3 in DMSO. No desired product was obtained in the absence copper catalyst and mechanistic studies exhibited the significance of Cu(I) species in the conversion. Notably, when o-chlorobenzaldehyde was used instead of o-bromobenzaldehyde 52% yield of desired product was obtained. Substrate scope of the reaction was explored using a range of aromatic methyl ketoxime substrates and o-chlorobenzaldehydes (Scheme 46(a)). Several methyl ketone oximes with methoxy, alkyl, and halogen (Br, Cl, F) substituents were tolerated and thieno[3,2-c]isoquinolines were obtained in moderate to good yields. In the case of o-chlorobenzaldehydes electronic properties of the substituent attached to the benzaldehyde had clear influence on the amount of final products. While substrates with electron-donating group reacted smoothly, electron-withdrawing groups quenched the conversion. The vinyl ketoximes, tried for the reaction also worked well and thieno[3,2-c]isoquinoline products were obtained in 21–88% yield (Scheme 46(b)). The present protocol is an efficient as well as straightforward strategy towards benzo[4,5]thieno[3,2-c]-isoquinoline and thieno[3,2-c]isoquinoline compounds and the synthesis of these motifs are not much explored due to their structural complexity.
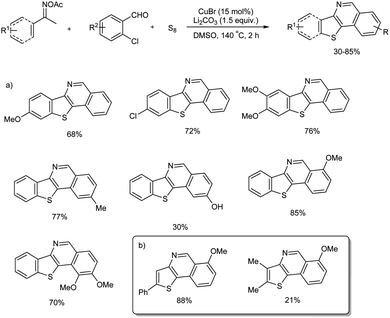 |
| | Scheme 46 Multi-component cascade bis-heteroannulation reaction using (a) aromatic methyl ketoximes, (b) vinyl ketoximes. | |
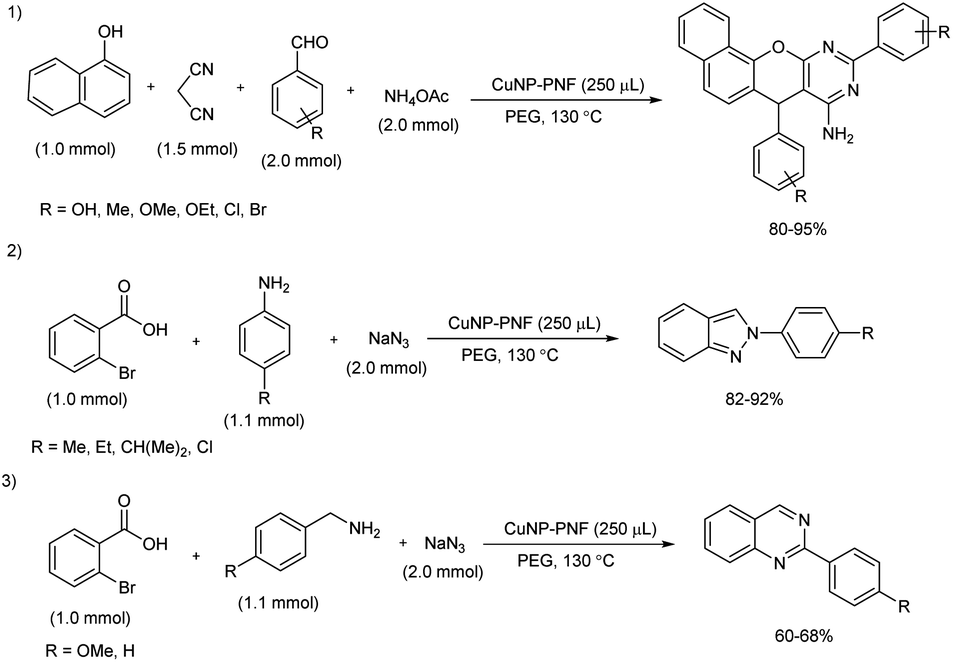
A facile and green protocol for the construction of 2H-indazole, 7,10-diaryl-7H-benzo[7,8]chromeno[2,3-d]pyrimidin-8-amine and quinazoline derivatives was achieved in the presence of decorated peptide nanofibers with copper nanoparticles (CuNP–PNF) (Scheme 47).53 These peptide nanofibers were prepared via self-assembly technique and using histidine as the building block. The role of succinic anhydride was accessed during preparation of peptide nanofiber in two ways, without adding succinic anhydride and by adding at pH 4 and at pH 5 respectively. During the synthesis of chromeno[2,3-d]pyrimidin-8-amine, α-naphthol, benzaldehyde, malononitrile and ammonium acetate were selected as model substrates and better results were obtained with 250 μL of CuNP–PNF catalyst in poly(ethylene glycol) (PEG) solvent at 130 °C. Several aldehydes with electron-donating and electron-withdrawing groups reacted well under the standard reaction condition and afforded good to excellent yield of the desired products. The efficiency of CuN–PNF catalysts was investigated for the preparation of 2H-indazoles as well as quinazoline derivatives from multicomponent reaction of aromatic amine or aliphatic amine, 2-bromobenzaldehydes and sodium azide, and the optimized condition of first synthesis was found suitable for 2H-indazoles as well as quinazolines synthesis. Both reaction exhibited satisfactory substrate scope and 2H-indazoles were formed in high amount than quinazolines.
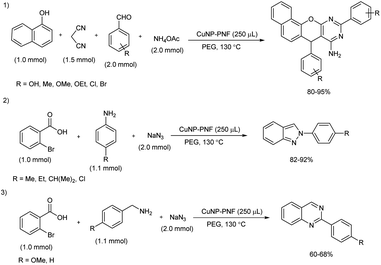 |
| | Scheme 47 Preparation of 7,10-diaryl-7H-benzo[7,8]chromeno[2,3-d]pyrimidin-8-amine, 2H-indazoles, and quinazolines. | |
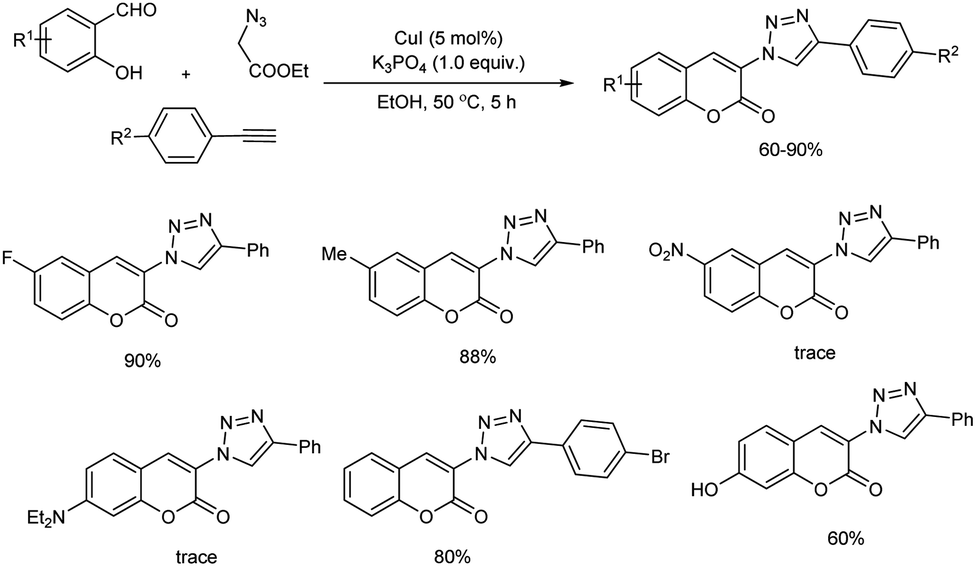
Shang and co-workers reported an environmentally sustainable one-pot, three-component reaction of ethyl 2-azidoacetate, salicylaldehydes and arylacetylenes for 3-triazolylcoumarins synthesis in the presence of copper(I) catalyst (Scheme 48).54 The reaction includes (1) copper-catalyzed azide–alkyne cycloaddition (2) Knoevenagel condensation (3) intramolecular transesterification sequence mediated by base. The reaction was performed in the presence of 5 mol% CuI, 1 equiv. of K3PO4 in ethanol at 50 °C for 5 hours. When salicylaldehydes having moderate electron-withdrawing or electron-donating group showed moderate to good reactivity towards the reaction. Whereas, strong electron-withdrawing and electron-donating groups gave only trace amount of corresponding product. For arylacetylenes, halide substituents were compatible and good amount of product was offered. Interestingly, fluorescence properties of 3-triazolylcoumarin derivatives evaluated and they exhibited broad range of fluorescence emission.
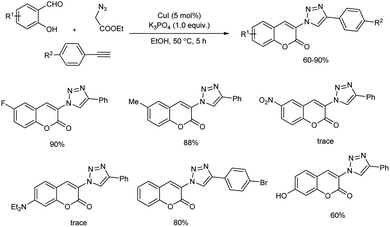 |
| | Scheme 48 Cu-based one-pot reaction cascade reaction of ethyl 2-azidoacetate, salicylaldehydes and arylacetylenes. | |
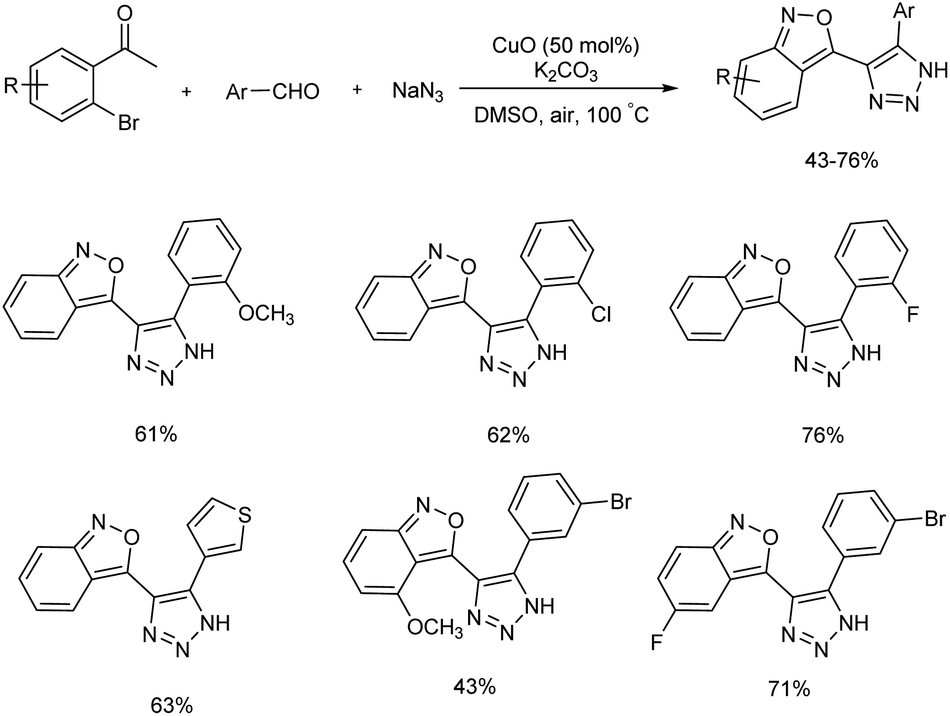
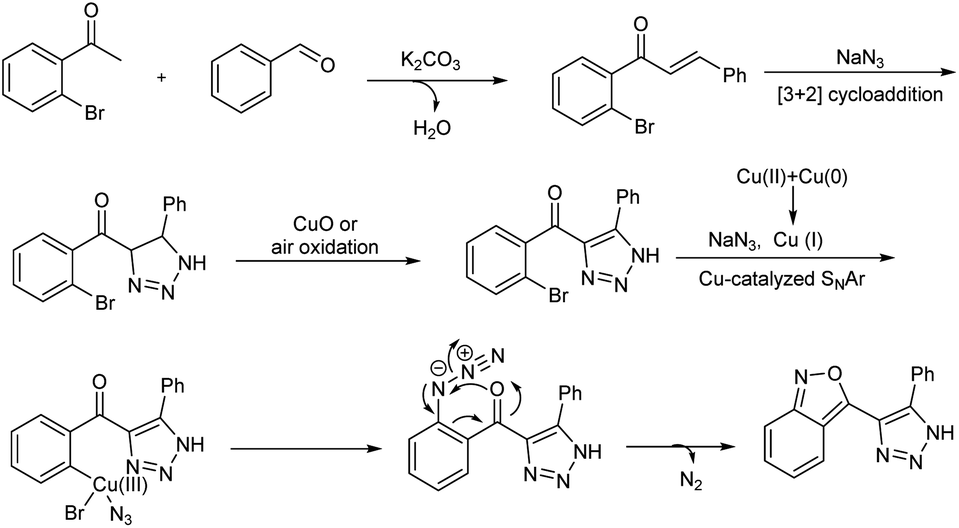
A domino protocol from easily available aldehydes, o-bromoacetophenones and sodium azide for the synthesis of 2,1-benzoisoxazole-containing 1,2,3-triazoles in the presence of Cu(II) catalyst was studied (Scheme 49).55 The model reaction involved o-bromoacetophenone, benzaldehyde, and sodium azide, and the optimized reaction condition was the reaction in presence of 50 mol% of CuO, 2.0 equiv. of K2CO3 in 3 mL DMSO at a temperature of 100 °C. Various aromatic aldehydes employed engaged in the reaction effectively irrespective of electronic effect and heteroaryl aldehydes like, thiophene-2-carbaldehyde, thiophene-3-carbaldehyde and furan-2-carbaldehyde also tolerated in this reaction. Substrate studies for o-bromoacetophenones conducted revealed that o-bromoacetophenones with electron-withdrawing substituents gave higher yield than o-bromoacetophenones with electron-releasing groups. Applicability of present protocol was studied by conducting further reactions with obtained products. A detailed mechanistic investigation of this synthesis explored found out various reactions involved in the process are aldol condensation, copper-catalyzed oxidative [3 + 2] cycloaddition, 1,2,3-triazole-assisted azidation and denitrogenative cyclization sequences (Scheme 50).
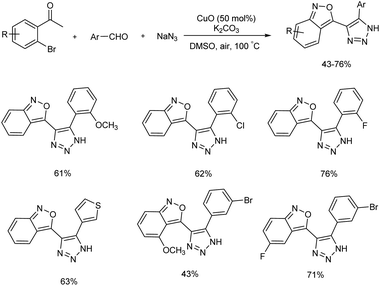 |
| | Scheme 49 Synthesis of 2,1-benzoisoxazole-containing 1,2,3-triazoles. | |
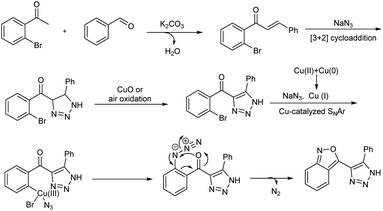 |
| | Scheme 50 Plausible mechanism for the synthesis of 2,1-benzoisoxazole-containing 1,2,3-triazoles synthesis [this figure has been reproduced from ref. 55 with permission from American Chemical Society, copyright 2020]. | |
3. Conclusions
Heterocycles are found extensively in biological as well as pharmaceutical compounds and a growing interest in heterocyclic synthesis is evident from the literature reports. Copper-catalyzed heterocycle synthesis is remarkably beneficial and are broadly studied at present time. In this highlight, we have discussed some of the developments in copper-catalyzed multicomponent synthesis of heterocycles in the past two years. Most of the reactions reviewed here have opened an economically favourable route towards large number of synthetically valuable molecules including potentially active biomolecules. Operational easiness and readily available starting materials make this procedure more acceptable than other existing heterocycle synthesis. Advantage of multicomponent reactions such as combining various reaction steps in single operation and incorporation of several reactive functionalities into molecules by fewer steps have enabled easy and efficient synthesis of structurally complex heterocycles. Incorporation of valuable and reactive functional groups in these methods pave way for further transformations. Profound advantages like, low catalyst loading, mild reaction conditions, high atom economy and wide functional group tolerance are widely seen in most of the methods reported here. This catalytic protocol has enabled easy preparation of numerous simple and fused 5- and 6-membered heterocycles with one, two or three heteroatoms. Solvent used varied in most of the reactions and heterocycles were also prepared in solvent-free conditions. Some reactions are carried out in water and reactions in aqueous media, which are environmentally sustainable have high significance in present as well as future research. It seen that copper(I) and copper(II) species are employed in the reactions and copper(I) is used in majority of the reactions. Even though, in some reactions electronic nature of substituent group on reactants exhibited a direct influence on the product yield, a general trend in this behaviour is not observed. Most of the reactions completed within relatively shorter reaction time, ligand-free conditions and at a temperature range of 25–130 °C. It is observed that comparatively higher yield of products was obtained when the reaction was carried under microwave or ultrasonic irradiation. Copper-catalyzed multicomponent synthesis of heterocycles is an active area with wide possibilities in organic synthesis. Amount of work published within this area portrays the significance of this heterocyclic synthesis. Constant developments in this field are seen and efficient as well as easy procedures for the synthesis of heterocycles is of high demand. Environmentally viable and economic reactions for heterocyclic synthesis is expected to be more explored in the coming years.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
MN thanks the University Grants Commission (UGC, New Delhi) for a research fellowship. GA thanks the Kerala State Council for Science, Technology and Environment (KSCSTE), India, for a research grant (No. 341/2013/KSCSTE dated 15.03.2013).
References
- P. Majumdar, A. Pati, M. Patra, R. K. Behera and A. Kumar Behera, Chem. Rev., 2014, 114, 2942 CrossRef CAS.
- C. Hu, L. Lu and J. P. Wan, Curr. Med. Chem., 2017, 24, 2241 CAS.
- L. Zhou, M. L. Hossain and T. Xiao, Chem. Rec., 2016, 16, 319 CrossRef CAS.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS.
- R. E. Ziegert, J. Toräng, K. Knepper and S. Bräse, J. Comb. Chem., 2005, 7, 147 CrossRef CAS.
- I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036 RSC.
- S. Thapa, B. Shrestha, S. K. Gurunga and R. Giri, Org. Biomol. Chem., 2015, 13, 4816 RSC.
- I. Ugi, Pure Appl. Chem., 2001, 73, 187 CAS.
- J. Fairoosa, S. Saranya, S. Radhika and G. Anilkumar, ChemistrySelect, 2020, 5, 5180 CrossRef CAS.
- R. M. Cherian, N. A. Harry, S. Saranya, K. R. Rohit and G. Anilkumar, Asian J. Org. Chem., 2019, 8, 197 CrossRef CAS.
- T. Pal, G. Kumar Lahiri and D. Maiti, Eur. J. Org. Chem., 2020, 2020, 6859 CAS.
- P. Shiri, Appl. Organomet. Chem., 2020, 34, e5600 CrossRef CAS.
- A. Samzadeh-Kermani and S. Ghasemi, J. Heterocycl. Chem., 2019, 56, 2202 CrossRef CAS.
- Q. Yuan, D. Liu and W. Zhang, Org. Lett., 2017, 19, 1144 CrossRef CAS.
- D. Ba, Y. Chen, W. Lv, S. Wen and G. Cheng, Org. Lett., 2019, 21, 8603 CrossRef CAS.
- S. A. Hamrahian, J. Rakhtshah, S. M. M. Davijani and S. Salehzadeh, Appl. Organomet. Chem., 2018, 32, 4501 CrossRef.
- P. Xu, Y.-M. Zhu, X.-J. Li, F. Wang, S.-Y. Wang and S.-J. Ji, Adv. Synth. Catal., 2019, 361, 1 CrossRef.
- F. Xiao, S. Yuan, H. Huang, F. Zhang and G. Deng, Org. Lett., 2019, 21, 8533 CrossRef CAS.
- X.-D. Wang, L.-H. Zhu, P. Liu, X.-Y. Wang, H.-Y. Yuan and Y.-L. Zhao, J. Org. Chem., 2019, 84, 16214 CrossRef CAS.
- W.-B. Huang, F.-Y. Ren, M.-W. Wang, L.-Q. Qiu, K.-H. Chen and L.-N. He, J. Org. Chem., 2020, 85, 14109 CrossRef CAS.
- Y. Wang, X. Liu, B. Zhu, P. Guo, Y. Pei, Q. He and H. Cao, J. Org. Chem., 2020, 85, 10118 CrossRef CAS.
- Y. Zhang, L. Huang, X. Li, L. Wang and H. Feng, J. Org. Chem., 2019, 84, 5046 CrossRef CAS.
- M. Khalaj, M. Sadeghpour, S. M. M. Safavi, A. Lalegani and S. M. Khatami, Monatsh. Chem., 2019, 150, 1085 CrossRef CAS.
- A. Keivanloo, M. Bakherad, M. Khosrojerdi and A. H. Amin, Res. Chem. Intermed., 2018, 44, 2571 CrossRef CAS.
- P. Bao, H. Yue, N. Meng, X. Zhao, J. Li and W. Wei, Org. Lett., 2019, 21, 7218 CrossRef CAS.
- X.-X. Wang, Y. Xin, Y. Li, W.-J. Xia, B. Zhou, R.-R. Ye and Y.-M. Li, J. Org. Chem., 2020, 85, 3576 CrossRef CAS.
- H. Esmaeili-Shahri, H. Eshghi, J. Lari, S. A. Rounaghi and E. Esmaeili-Shahri, Res. Chem. Intermed., 2019, 45, 2963 CrossRef CAS.
- V. R. Velpuri and K. Muralidharan, J. Organomet. Chem., 2019, 884, 59 CrossRef CAS.
- A. Shaabani, M. Shadi, R. Mohammadian, S. Javanbakht, M. T. Nazeri and F. Bahri, Appl. Organomet. Chem., 2019, 33, 5074 Search PubMed.
- A. A. Saikia, R. N. Rao, S. Das, S. Jena, S. Rej, B. Maiti and K. Chanda, Tetrahedron Lett., 2020, 61, 152273 CrossRef CAS.
- W. H. Mudd and E. P. Stevens, Tetrahedron Lett., 2010, 51, 3229 CrossRef CAS.
- A. Mayooufi, M. Romdhani-Younes, Y. Carcenac and J. Thibonnet, Synth. Commun., 2019, 49, 2168 CrossRef CAS.
- A. Yahyazadeh, E. Abbaspour-Gilandeh and M. Aghaei-Hashjin, Catal. Lett., 2018, 148, 1254 CrossRef CAS.
- K. N. Patil, R. A. Mane, S. B. Jadhav, M. M. Mane and V. B. Helavi, Chem. Data Collect., 2019, 21, 100233 CrossRef.
- J. Sun, X. Cheng, J. K. Mansaray, W. Fei, J. Wan and W. Yao, Chem. Commun., 2018, 54, 13953 RSC.
- T. Shi, F. Qin, Q. Li and W. Zhang, Org. Biomol. Chem., 2018, 16, 9487 RSC.
- J. Lou, J. Ma, B.-H. Xu, Y.-G. Zhou and Z. Yu, Org. Lett., 2020, 22, 5202 CrossRef CAS.
- X. Li, T. Wang, Y. Lu, S. Ji, Y. Huo and B. Liu, Org. Biomol. Chem., 2018, 16, 7143 RSC.
- Z. T. Bhutia, D. Das, A. Chatterjee and M. Banerjee, ACS Omega, 2019, 4, 4481 CrossRef CAS.
- Z. Gan, Q. Yan, G. Li, Q. Li, X. Dou, G.-Y. Li and D. Yang, Adv. Synth. Catal., 2019, 361, 4558 CrossRef CAS.
- R. K. Sharma, S. Yadav, S. Sharma, S. Dutta and A. Sharma, ACS Omega, 2018, 3, 15100 CrossRef CAS.
- J. Kim and K. Oha, Adv. Synth. Catal., 2020, 362, 3576 CrossRef CAS.
- H. Saeidiroshan and L. Moradi, J. Organomet. Chem., 2019, 893, 1 CrossRef CAS.
- K. S. Alghamdi, N. S. I. Ahmed, D. Bakhotmah and M. Mokhtar, J. Nanosci. Nanotechnol., 2020, 20, 890 CrossRef CAS.
- R. Rodrigues, L. Q. Tran, B. Darses, P. Daubana and L. Neuville, Adv. Synth. Catal., 2019, 361, 4454 CrossRef CAS.
- G. Satish, P. Ashok, L. Kota and A. Ilangovan, Org. Biomol. Chem., 2019, 17, 4774 RSC.
- Y. Liang, Z. Tan, H. Jiang, Z. Zhu and M. Zhang, Org. Lett., 2019, 21, 4725 CrossRef CAS.
- Y. Wang, L. Deng, J. Zhou, X. Wang, H. Mei, J. Han and Y. Pan, Adv. Synth. Catal., 2018, 360, 1060 CrossRef CAS.
- H. Jin, D. Liu, B. Zhou and Y. Liu, Synthesis, 2020, 52, 9 CrossRef.
- B. Gopal Das, S. Shah and V. K. Singh, Org. Lett., 2019, 21, 4981 CrossRef.
- L. Wang, C. Qi, R. Cheng, H. Liu, W. Xiong and H. Jiang, Org. Lett., 2019, 21, 7386 CrossRef CAS.
- Z. Xu, G.-J. Deng, F. Zhang, H. Chen and H. Huang, Org. Lett., 2019, 21, 8630 CrossRef CAS.
- Z. Taherinia, A. Ghorbani-Choghamarani and M. Hajjami, ChemistrySelect, 2019, 4, 2753 CrossRef CAS.
- X. He, R. Li, M. Xie, J. Duan, Q. Tang and Y. Shang, New J. Chem., 2020, 44, 12266 RSC.
- Y. Wang, P. Yu, Q. Ren, F. Jia, Y. Chen and A. Wu, J. Org. Chem., 2020, 85, 2688 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc