DOI:
10.1039/D0RA10409D
(Paper)
RSC Adv., 2021,
11, 5529-5536
Production and characterization of chitooligosaccharides by the fungal chitinase Chit42 immobilized on magnetic nanoparticles and chitosan beads: selectivity, specificity and improved operational utility†
Received
10th December 2020
, Accepted 25th January 2021
First published on 29th January 2021
Abstract
Chitin-active enzymes are of great biotechnological interest due to the wide industrial application of chitinolytic materials. Non-stability and high cost are among limitations that hinder industrial application of soluble enzymes. Here we report the production and characterization of chitooligosaccharides (COS) using the fungal exo-chitinase Chit42 immobilized on magnetic nanoparticles and food-grade chitosan beads with an immobilization yield of about 60% using glutaraldehyde and genipin linkers. The immobilized enzyme gained operational stability with increasing temperature and acidic pH values, especially when using chitosan beads-genipin that retained more than 80% activity at pH 3. Biocatalysts generated COS from colloidal chitin and different chitosan types. The immobilized enzyme showed higher hydrolytic activity than free enzyme on chitosan, and produced COS mixtures with higher variability of size and acetylation degree. In addition, biocatalysts were reusable, easy to handle and to separate from the reaction mixture.
Introduction
Chitin is a renewable and biodegradable semi crystalline biopolymer mainly formed by N-acetyl-β-D-glucosamine (GlcNAc) units. It is the most abundant polysaccharide of the marine environment and the second (after cellulose) on the earth. This ubiquitous material offers strength to the exoskeleton of insects and arthropods and it is an essential component of fungi cell walls. The deacetylated form of chitin is chitosan, polysaccharide composed of fully or partial deacetylated β-(1-4)-linked GlcNAc units. Both chitin and chitosan are large biopolymers, with molecular size depending on the producing organism and the method of their production.1 Sustainability, renewability, availability and other benefits of chitinolytic polymers seemed to be the solution for the critical problem faced worldwide on waste disposal and management by reducing hundreds of millions of tons of synthetic polymers produced annually.1,2 The hydrolysis of chitin and chitosan to produce chitooligosaccharides (COS) (MW 2–30 kDa) can be carried out chemically, physically, or enzymatically, the first being the most used strategy and the latter the most specific, controllable and environmentally friendly option.3 COS are soluble in aqueous solutions and show biocompatibility and biodegradability properties. Furthermore, they show more prominent antiviral, antibacterial, antitumor and antioxidant activities than chitin and high molecular weight chitosan making them gain value in the biotechnological sector. The biological activity of COS depending on their size, degree and pattern of acetylation.4–6 Specific enzymes such as chitinases and chitosanases, or unspecific such as carbohydrases and proteases have been employed in conversion of chitin and chitosan to COS.7,8 Chitinases (E.C 3.2.2.14) are extensively distributed Glycoside Hydrolases (GH), which cleave at internal or terminal β-(1-4) glycosidic linkages of chitin. These enzymes are structurally included in GH families 18, 19 and 20 (http://www.cazy.org) that are present in all organism kingdoms.8,9 Exo-chitinases attack the polysaccharide from the non-reducing end of the polymer chain releasing the disaccharide chitobiose (di-acetyl-glucosamine (GlcNAc)2) as major product, whereas endo-enzymes attack randomly from internal points along the polysaccharide producing tetrasaccharides and higher size sugars as majority products.9,10 As mentioned above, enzymatic methods for production of COS are the most interesting alternative for the industrial sector, but undoubtedly, yields of the products obtained in hydrolytic reactions are still far from being suitable. In fact, only less than 25% of the substrate is usually transformed into products by free chitinases, which makes industries to use less environmentally friendly strategies to produce COS.6 Immobilization of enzymes constitutes a powerful tool to improve some biocatalyst limitations such as their stability, high cost production cycle and low productivity levels.11–13 Over other types of support systems used in immobilization, the use of Magnetic Nanoparticles (MNPs) of 10 to 20 nm shows advantages like non-toxicity, large surface area and surface volume ratio or the simplicity in separating the biocatalyst after use. In addition, the MNPs surface can be modified coating with different compounds such as (3-aminopropyl) triethoxysilane (APTES). In this way, the amino group on their surface allows the attack of linker such as glutaraldehyde (GA), to facilitate the protein binding. Attachment of enzymes on chitosan beads is also a widely used immobilization method and different chitosan-based supports have been reported given the biodegradable and environmentally friendly nature of this material. The presence of active amino groups in deacetylated GlcNAc units of chitosan also enables the binding of GA and that of proteins. In addition, Chitosan beads/Macro-Spheres (CMS) employ centrifugation or filtration for separation of the catalysts from the reaction mixture. Therefore, some chitosanases, lipases, amylases and laccases (among others) have been already immobilized using MNPs and/or CMS with successful results.11,14–18 In contrast, most chitinases previously immobilized using both types of supports lost their activity after being reused in consecutive cycles or no data concerning production and characterization of COS were reported.19–21 Chitinase Chit42 from the fungus Trichoderma harzianum was previously expressed in the yeast Pichia pastoris with about 3 g L−1 of the protein produced in this heterologous system. This fungal exo-chitinase of the family GH18 hydrolyzed chitin oligomers with a minimal degree of polymerization (DP) of 3 units, with chitobiose being the main hydrolysis product.8 Chitosan and COS are produced and commercialized by different companies as safe dietary supplements.22 They are Generally Recognized As Safe (GRAS) compounds also at high dietary concentrations in animals and humans. However, the extraordinary biological activity of COS requires to identify new methods for production, purification and characterization. The goal of this work is to produce and characterize COS with the immobilized biocatalyst for the bioconversion of colloidal chitin and chitosan polymers. In addition, the reusability of the immobilized biocatalyst has also been evaluated.
Results and discussion
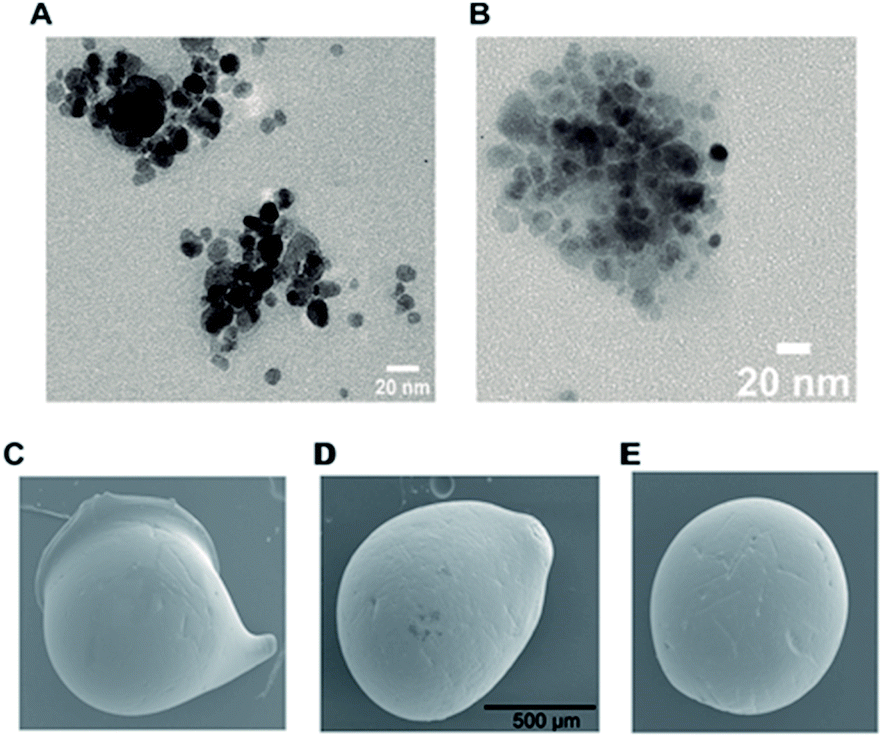
Morphology and size of MNPs, before and after immobilization of Chit42, were analyzed using TEM. The MNPs were rough spheres with an average diameter of about 10 nm that formed scattered groups due to the forces of attraction between them, and which remain even after sonication (Fig. 1A). The nanoparticles with the supported enzyme, MNPs-GA-Chit42, are visualized in Fig. 1B. All complexes apparently showed the same size and shape to each other and to that previously obtained in other works23,24 even if particles larger than 20 nm have also been reported before.16
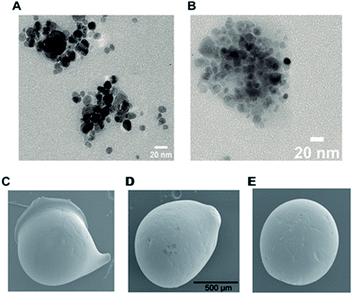 |
| | Fig. 1 TEM and SEM analyses of generated supports. TEM images of magnetic nanoparticles. (A) MNPs-GA and (B) MNPs-GA-Chit42. SEM images of dry chitosan macrospheres of (C) CMS-GA, (D) CMS-GA-Chit42 and (E) CMS-Gpn-Chit42. GA 0.5% and Gpn 0.125% were used. Scale bars are showed; same scale in (C) and (E) as in (D). | |
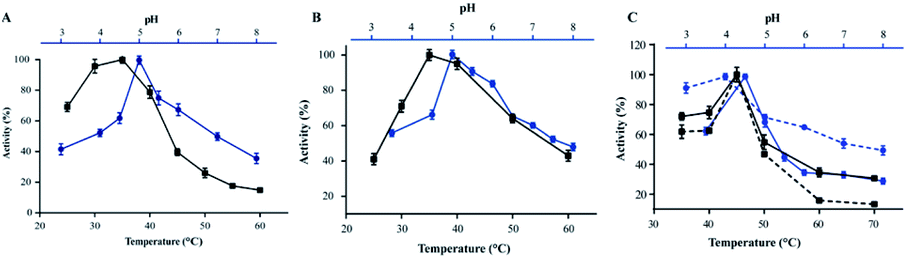
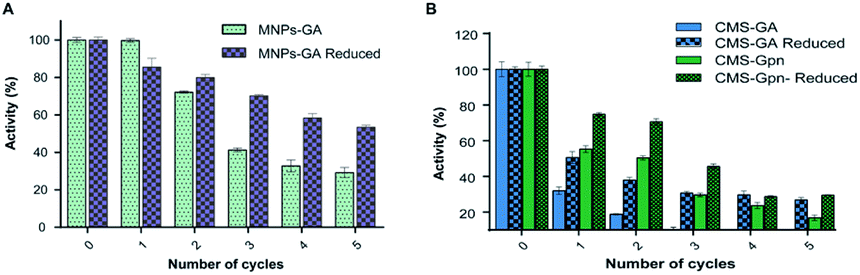
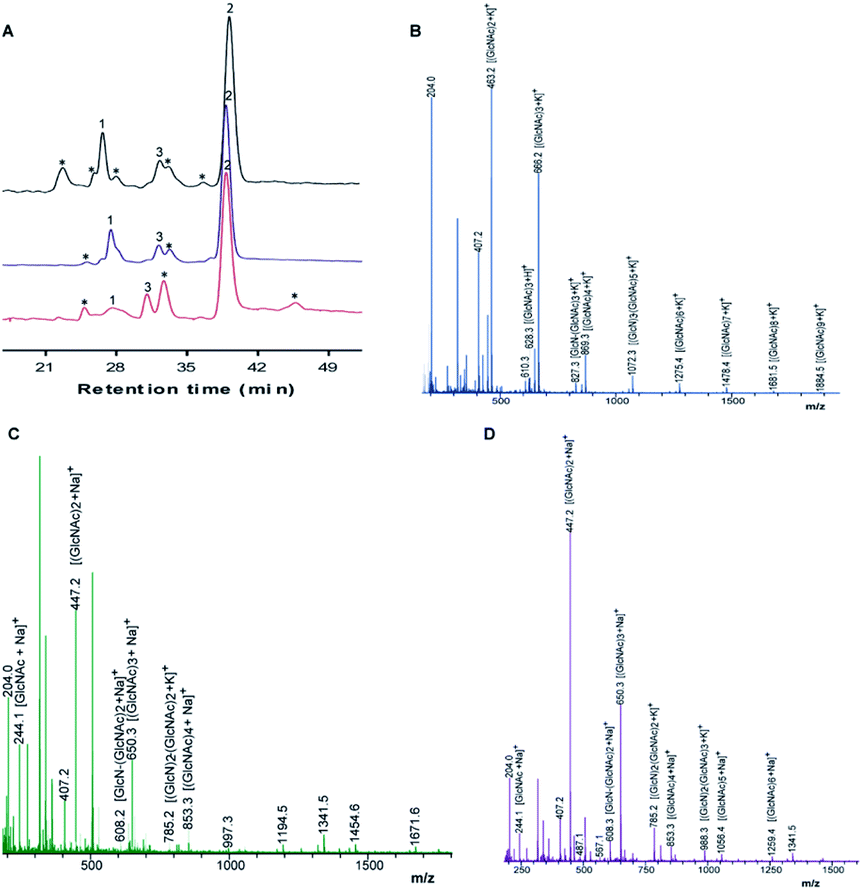
Chitosan beads produced in this study were clear macrospheres that proved to be stable in conditions used in enzymatic assays. The size of wet and dry chitosan beads was of about 2 mm and apparently 1 mm, respectively, by using millimeter paper (data not shown). Dry spheres were also analyzed using SEM and showed a size of 0.99 mm. Initially, 0.5% GA or 0.125% Gpn and different ratios of protein (Chit42) per gram of support were used. Best results of immobilization on MNPs were obtained using 6.2 mg of protein, where the immobilization yield and the recovered hydrolytic chitinase activity were both of around 62–67% (Table 1), and with less than 50% of recovery activity when using higher or lower amounts of proteins (see information in the Table 1 footnote). When chitinase Chit42 was immobilized on chitosan beads using GA or Gpn, best results were obtained using 6.2 and 2 mg of protein/g of CMS beads, with a recovery activity of 71% and 62%, respectively (Table 1), and less than 40% and 60% of recovery activity for CMS-GA and CMS-Gpn, respectively, when other conditions were used (Table 1). The selection of MNPs and CMS beads as supporting materials were motivated by the fact that in the first case the products can be easily separated from the medium while CMS beads are more sustainable to be used for food purposes. Furthermore with CMS, different linkers, GA and Gpn, have been tested for their different structure and chain length and because the latter is a natural and completely atoxic linker. To our knowledge, partial information is available on immobilized chitinases from different sources using MNPs and CMS beads in literature but no information on the production and detailed characterization of the produced COS by immobilized enzymes are reported. Thus, a recombinant chitinase was already immobilized using carboxyl functional MNPs with immobilization yield of 64%, which showed activity against the fungi Botrytis cinerea25 and a commercial chitinase from Trichoderma viride on superparamagnetic particles using a rotational magnetic field with no activity/immobilization yields reported.26 Chitosan beads and GA have also been used to immobilize chitinases such as that from Streptomyces griseus and Paenibacillus illinoisensis, with an immobilization yield of about 42%,19 and a thermostable enzyme from Thermomyces lanuginosus with almost 100% of immobilization yield but without data of the activity recovery reported.20 In addition, different polymers have also been previously used to immobilize chitinolytic enzymes. Among them, hydroxypropyl methylcellulose acetate succinate for a commercial chitinase from Serratia marcescens, with an immobilization yield of 78% and recovery activity of 41%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 and k-carrageenan-alginate gel for chitinase from Aspergillus awamori with immobilization and recovery activity yields of 93% and 77%, respectively.28 A lower immobilization yield (25%) was also obtained by covalent immobilization of a chitinolytic activity of Bacillus amyloliquefaciens in glyoxal agarose beads.29 Similar to free Chit42, the immobilized enzyme on MNPs displayed maximum activity at 35–40 °C but a priori could be more thermostable because retained more than 60% of its activity after 1 h at 50 °C (Fig. 2B) instead of about 20% retained by the non-immobilized enzyme (Fig. 2A). Immobilization on chitosan beads also increases the thermal activity profile of Chit42, which showed an optimal temperature of 45 °C and retained 50% of its activity at 50 °C (Fig. 2B). This stabilization effect is well-reported in covalent immobilization.30 Concerning the pH dependence, free enzyme showed optimal activity at pH 5.0–6.0 (Fig. 2A). However, the activity of the immobilized Chit42 increased remarkably at acidic pH values, and specially, when using chitosan beads and Gpn, which showed maxima activity at pH 4 and retained more than 80% of the activity at pH 3 (Fig. 2C). The formation of covalent bond through amino groups of the immobilized Chit42 can cause imbalanced partition of H+ and OH− concentrations resulting in the protein higher acidic stability.31 In any case, the higher apparent acidic stability of the immobilized Chit42 gives this biocatalyst the advantage to act more efficiently on chitin and chitosan, which are more soluble at low pH values. The reuse of enzymes in industrial processes is of enormous economic relevance. The reusability of chitinase Chit42 immobilized on MNPS and CMS was investigated using batch reactions (Fig. 3). After each cycle of reaction (1 h), catalysts MNPs-GA-Chit42 and CMS-GA/Gpn-Chit42 were separated from their reaction mixtures using external magnetic field and centrifugation, respectively. Before every one of the new reused cycles, all catalysts were washed in distilled water and phosphate buffer. The catalyst MNPs-GA-Chit42 retained higher than 70% of its initial chitinolytic activity after 2 cycles but only 30% after 5 cycles (Fig. 3A). However the catalyst CMS-GA-Chit42 completely lost its activity after the second cycle, which made us to test also Gpn as linker, but CMS-Gpn-Chit42 retained only about 30% of the initial activity (Fig. 3B), a value still clearly lower than that obtained with MNPs. In addition, the loss of activity during recovery steps due to possible outflow of the enzyme from the support was also investigated, but no free enzyme was detected on the washing buffer. Loss of chitinase activity after immobilization on MNPs and CMS has been previously reported using commercial chitinases, which retained less of 20% of their initial activity after only 2 reuse cycles.19,26 Nevertheless to stabilize bonds between enzymes and supports, the reduction with sodium borohydride, which converts weak Schiff bases into stable secondary amino bonds32 was attempted and evaluated (Fig. 3). The reduction of the immobilized catalyst caused a clear decrease of the initial Chit42 hydrolytic activity, of about 50% (from 1.0 to 0.52 units), 70% (from 1.5 to 0.42 U) and 33% (from 1.3 to 0.86 U) in the catalysts MNPs-GA-Chit42, CMS-GA-Chit42 and CMS-Gpn-Chit42, respectively. However, the reduction clearly improved the reusability of the immobilized Chit42, the MNPs-GA-Chit42 catalyst being the most favoured and retaining more than 50% of its activity after at least 5 cycles of reuse (Fig. 3). This makes the linkers stabilization an important aspect for the operational activity of the biocatalyst. Regardless of the support used, Chit42 hydrolyzed colloidal chitin and all the chitosan of different size and DD tested (Table 2). Both free and immobilized chitinase showed maximum hydrolytic activity on colloidal chitin and with more activity on small chitosan with DD 77–81 than in larger ones with DD > 90. In addition, immobilized enzyme had at least 3 times higher hydrolytic activity than free enzyme on any of the chitosan tested. Curiously, Chit42 immobilized on CMS-GA hydrolyzed commercial chitosan CHIT50 and colloidal chitin with the same effectiveness. The application of immobilized Chit42 on the production of COS was evaluated using HPAEC-PAD chromatography and mass spectrometry analyses. Independently of the type of support used, and as happened previously by using free Chit42,8 chitobiose ((GlcNAc)2) was the main product obtained from colloidal chitin, followed by (GlcNAc)3 and GlcNAc when using the immobilized enzyme (Fig. 4A). In addition, a large number of masses corresponding to different series of fully acetylated and partially acetylated COS (faCOS and paCOS, respectively) were detected using mass spectrometry analyses (Fig. 4B–D).
27 and k-carrageenan-alginate gel for chitinase from Aspergillus awamori with immobilization and recovery activity yields of 93% and 77%, respectively.28 A lower immobilization yield (25%) was also obtained by covalent immobilization of a chitinolytic activity of Bacillus amyloliquefaciens in glyoxal agarose beads.29 Similar to free Chit42, the immobilized enzyme on MNPs displayed maximum activity at 35–40 °C but a priori could be more thermostable because retained more than 60% of its activity after 1 h at 50 °C (Fig. 2B) instead of about 20% retained by the non-immobilized enzyme (Fig. 2A). Immobilization on chitosan beads also increases the thermal activity profile of Chit42, which showed an optimal temperature of 45 °C and retained 50% of its activity at 50 °C (Fig. 2B). This stabilization effect is well-reported in covalent immobilization.30 Concerning the pH dependence, free enzyme showed optimal activity at pH 5.0–6.0 (Fig. 2A). However, the activity of the immobilized Chit42 increased remarkably at acidic pH values, and specially, when using chitosan beads and Gpn, which showed maxima activity at pH 4 and retained more than 80% of the activity at pH 3 (Fig. 2C). The formation of covalent bond through amino groups of the immobilized Chit42 can cause imbalanced partition of H+ and OH− concentrations resulting in the protein higher acidic stability.31 In any case, the higher apparent acidic stability of the immobilized Chit42 gives this biocatalyst the advantage to act more efficiently on chitin and chitosan, which are more soluble at low pH values. The reuse of enzymes in industrial processes is of enormous economic relevance. The reusability of chitinase Chit42 immobilized on MNPS and CMS was investigated using batch reactions (Fig. 3). After each cycle of reaction (1 h), catalysts MNPs-GA-Chit42 and CMS-GA/Gpn-Chit42 were separated from their reaction mixtures using external magnetic field and centrifugation, respectively. Before every one of the new reused cycles, all catalysts were washed in distilled water and phosphate buffer. The catalyst MNPs-GA-Chit42 retained higher than 70% of its initial chitinolytic activity after 2 cycles but only 30% after 5 cycles (Fig. 3A). However the catalyst CMS-GA-Chit42 completely lost its activity after the second cycle, which made us to test also Gpn as linker, but CMS-Gpn-Chit42 retained only about 30% of the initial activity (Fig. 3B), a value still clearly lower than that obtained with MNPs. In addition, the loss of activity during recovery steps due to possible outflow of the enzyme from the support was also investigated, but no free enzyme was detected on the washing buffer. Loss of chitinase activity after immobilization on MNPs and CMS has been previously reported using commercial chitinases, which retained less of 20% of their initial activity after only 2 reuse cycles.19,26 Nevertheless to stabilize bonds between enzymes and supports, the reduction with sodium borohydride, which converts weak Schiff bases into stable secondary amino bonds32 was attempted and evaluated (Fig. 3). The reduction of the immobilized catalyst caused a clear decrease of the initial Chit42 hydrolytic activity, of about 50% (from 1.0 to 0.52 units), 70% (from 1.5 to 0.42 U) and 33% (from 1.3 to 0.86 U) in the catalysts MNPs-GA-Chit42, CMS-GA-Chit42 and CMS-Gpn-Chit42, respectively. However, the reduction clearly improved the reusability of the immobilized Chit42, the MNPs-GA-Chit42 catalyst being the most favoured and retaining more than 50% of its activity after at least 5 cycles of reuse (Fig. 3). This makes the linkers stabilization an important aspect for the operational activity of the biocatalyst. Regardless of the support used, Chit42 hydrolyzed colloidal chitin and all the chitosan of different size and DD tested (Table 2). Both free and immobilized chitinase showed maximum hydrolytic activity on colloidal chitin and with more activity on small chitosan with DD 77–81 than in larger ones with DD > 90. In addition, immobilized enzyme had at least 3 times higher hydrolytic activity than free enzyme on any of the chitosan tested. Curiously, Chit42 immobilized on CMS-GA hydrolyzed commercial chitosan CHIT50 and colloidal chitin with the same effectiveness. The application of immobilized Chit42 on the production of COS was evaluated using HPAEC-PAD chromatography and mass spectrometry analyses. Independently of the type of support used, and as happened previously by using free Chit42,8 chitobiose ((GlcNAc)2) was the main product obtained from colloidal chitin, followed by (GlcNAc)3 and GlcNAc when using the immobilized enzyme (Fig. 4A). In addition, a large number of masses corresponding to different series of fully acetylated and partially acetylated COS (faCOS and paCOS, respectively) were detected using mass spectrometry analyses (Fig. 4B–D).
Table 1 Optimal immobilisation condition for chitinase Chit42a
| Support type |
Chit42 (mg g−1 support) |
Immobilization yield (%) |
Recovery of activity (%) |
| 100% activity: 32.2, 32.2 and 10.4 U g−1 of biocatalyst for MNPs-GA, CMS-GA and CMS-Gpn, respectively. GA 0.5% and Gpn 0.125%. For: 3.12, 6.2, 12.4, 24.8 mg g−1 MNPs-GA, immobilization yields: 56.1, 62.3, 43.2, 23% and recovery activity: 46.5, 66.7, 38.3, 19%. For 0.78, 1.55, 3.1, 6.2 mg g−1 CMS-GA, immobilization yields: 17.4, 32.4, 53.7, 57% and recovery activity: 4.7, 13.2, 34.6, 71%. For 0.5, 1.5, 2, 2.5 mg g−1 CMS-Gpn, immobilization yields: 28, 70.7, 86.6, 64.5% and recovery activity: 43.4, 50.1, 62.3, 55.7%; all respectively. |
| MNPS-GA |
6.2 |
62.3 |
66.7 |
| CMS-GA |
6.2 |
57.0 |
71.0 |
| CMS-GPN |
2.0 |
86.5 |
62.3 |
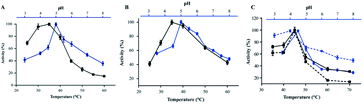 |
| | Fig. 2 Temperature and pH dependence profiles of the free and immobilized chitinase activity. (A and B) Temperature (black) and pH (blue) profiles for the free and the linked to MNPs-GA enzyme, respectively. (C) Temperature (black) and pH (blue) when CMS-GA (continuous line) and CMS-Gpn (discontinuous line) supports were used. Data are means of three independent values. Standard errors are indicated. | |
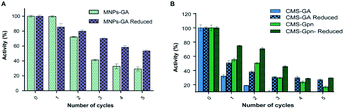 |
| | Fig. 3 Reusability of immobilized Chit42 on (A) MNPs and (B) CMS supports. Relative activity was evaluated on colloidal chitin. The 100% activity was 33.3, 28.9, 66.7, 23.3, 72.2 and 48.3 units per g of biocatalyst for MNPs-GA, MNPs-GA-reduced, CMS-GA, CMS-GA-reduced, CMS-Gpn and CMS-Gpn-Reduced, respectively. GA/Gpn-reduced with NaBH4. Assays were conducted in triplicate and data are means of three parallel measurements. Standard errors are indicated. | |
Table 2 Hydrolytic activity of the immobilized Chit42 on the referred substratesa
| Substrate |
MW (kDa) |
DD (%) |
Hydrolytic activity of Chit42 (%) |
| Free |
MNPs-GA |
CMS-GA |
CMS-Gpn |
| 100% activity: 5.3 U mg−1 of the protein, 5.6, 6.5, 6.4 U mg−1 of the immobilized enzyme for MNPs-GA-Chit42, CMS-GA-Chit42 and CMS-Gpn-Chit42, respectively. Data are means of three independent experiments and standard errors are indicated. |
| Colloidal chitin |
n.d. |
≤8 |
100.0 ± 2.9 |
100.0 ± 4.2 |
100.0 ± 2.4 |
100.0 ± 8.2 |
| QS1 |
98 |
81 |
14.8 ± 3.5 |
46.5 ± 6.1 |
69.0 ± 1.6 |
44.9 ± 4.0 |
| QS2 |
31 |
77 |
13.5 ± 0.8 |
50.8 ± 8.0 |
60.7 ± 5.3 |
49.2 ± 7.5 |
| CHIT100 |
100–300 |
>90 |
9.8 ± 3.5 |
34.2 ± 1.9 |
43.8 ± 1.4 |
34.5 ± 7.2 |
| CHIT600 |
600–800 |
>90 |
4.2 ± 3.6 |
3.1 ± 2.8 |
14.6 ± 1.9 |
16.3 ± 4.5 |
| CHIT50 |
50–190 |
77 |
16.0 ± 0.8 |
72.2 ± 4.2 |
98.0 ± 3.0 |
43.5 ± 8.6 |
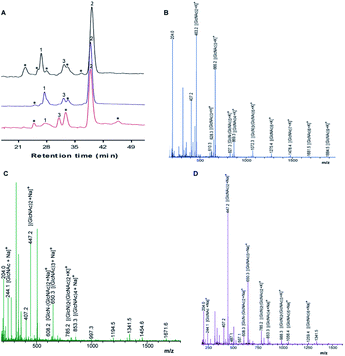 |
| | Fig. 4 Analysis of COS obtained from colloidal chitin. (A) High-performance anion exchange chromatography (HPAEC-PAD) analyses. The 24 h reactions catalyzed by immobilized Chit42 on MNPs (red) and CMS using GA linker (blue) and Gpn (black). Peaks: (1) GlcNAc; (2) (GlcNAc)2; (3) (GlcNAc)3; (*) Unidentified. MALDI-TOF MS analysis of the COS mixtures formed with the enzyme immobilized on (B) MNPs-GA, (C) CMS-GA and (D) CMS-Gpn. Data obtained with free Chit42 were previously indicated.8 The peaks in the spectra correspond to the monoisotopic masses of hydrogen adducts [M + H]+, [M + K]+ and [Ma + Na]+ of the COS. Only the major identified products were marked. | |
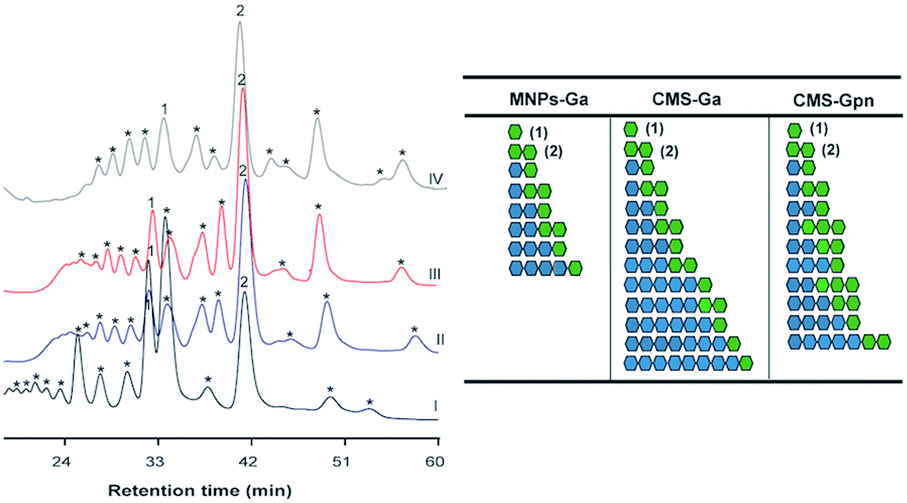
Specifically, masses corresponding to faCOS of (GlcNAc)1–9 units and the paCOS GlcN-(GlcNAc)1–3,5–7, (GlcN)2-(GlcNAc)1–3, (GlcN)3-(GlcNAc)2,4, (GlcN)4-(GlcNAc)5 were detected (Fig. 4 and Table S1†). As expected, masses corresponding to (GlcNAc)2 followed by (GlcNAc)3 being the majority. Concerning paCOS, masses corresponding to GlcN-(GlcNAc)2 followed of GlcN-(GlcNAc)3 were the majority detected when using MNPs (Table S1†) but that of GlcN-GlcNAc followed by GlcN-(GlcNAc)2 when using CMS (Table S2†). Pattern of products obtained could have been slightly altered due to the support used, which could alter the structure/specificity of the protein. Especially, considering that when free Chit42 was previously used in the hydrolysis of colloidal chitin only masses corresponding to the faCOS of (GlcNAc)1–4 units and the paCOS GlcN-(GlcNAc)2,4 and (GlcN)2-(GlcNAc)3 were detected. However, despite the high number of compounds detected in mass spectrometric analyses, only the faCOS of (GlcNAc)1–3 units could be clearly identified due to the availability of the corresponding commercial standards. The data points to that immobilized Chit42 shows a higher range of product variability from colloidal chitin than the free enzyme. The possibility that immobilization could affect the adsorption of substrates in the enzyme active site modifying enzymatic activity, selectivity or even the final product profile was previously suggested.31,33 Thus this was observed with glycosidases, such as the β-galactosidase from Aspergillus oryzae, and lipases from Rhizopus oryzae and Candida antarctica, which improved its selectivity and specificity after immobilization.34–37 Structure of Chit42 was previously determined, and showed the expected folding described for chitinases included in the GH18 family with a characteristic groove shaped substrate-binding size able to accommodate at least six sugars units.8 The unusual substrate-assisted catalytic mechanism of chitinases-GH18 requires a glutamic residue in the protein chain providing the acid protonating the glycosidic bond to be hydrolyzed and a mandatory GlcNAc residue in the substrate (after which to cut), which provides the oxygen (of the N-acetyl group) acting as nucleophile. Therefore, to the greater substrate acetylation degree, in principle, correspond both the greater hydrolysis and the diversity of products formed as previous reported.8,37 This, together with the non-availability of commercial paCOS that could be used in the identification of products, led us to use the smallest available substrate with the highest degree of acetylation (chitosan QS2) in this section (Fig. 5). To our knowledge no paCOS produced by immobilized chitinases have been previously reported. It is conceivable that the use of supported enzymes (1 h contact time with substrate in a thermal shaker before products analysis) reduces substrate crystallinity making the crystalline regions more accessible to the hydrolytic enzyme activity, which would increase yield of low molecular weight products (see Table 2). This represents an interesting achievement for the industrial bioconversion of chitinolytic polymers, as with the use of supported enzymes the pretreatment of substrate could be avoided. Furthermore, Gpn is a better promising linker for immobilized chitinase in terms of enzymatic activity recover. This might be due to a greater mobility of the covalent bonded enzyme. Concerning the COS properties, and although there is no broad consensus on the results previously obtained, the size, degree and pattern of acetylation exert a notable influence on the physicochemical and biological activity of these molecules.22,38–42 Thus, in general the lower MW COS the higher water solubility and lower viscosity, but also the lower antimicrobial activity and the higher antioxidant properties.22,38 In addition, paCOS are excellent plant elicitors39 and exhibit better antibacterial activity toward Escherichia coli and Listeria monocytogenes than COS fully acetylated.43 This increases the biotechnological interest of the immobilized catalysts obtained in this work. However a deeper understanding on how the immobilized enzyme produces paCOS and the structural aspects involved in this process, as well as the development of improved methods for separation, characterization and quantification of products, will be essential to improve the enzyme activity/specificity to produce COS with different characteristics and, a priori, properties.
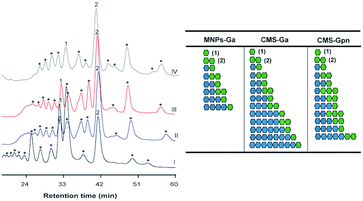 |
| | Fig. 5 HPAEC-PAD analysis of COS produced by the immobilized Chit42 with chitosan QS2 as substrate. On the right, a schematic representation of the polymerization degree and composition of reaction products predicted from mass spectrometry data (Tables S3 and S4†) is presented. Blue circles: GlcN. Green circles: GlcNAc. Identified peaks: (1) GlcNAc; (2) (GlcNAc)2. Chromatograms obtained with (I) free enzyme; (II) MNPs-GA-Chit42; (III) CMS-GA-Chit42; (IV) CMS-Gpn-Chit42. | |
Conclusions
Chitinase Chit42 was successfully immobilized on magnetic nanoparticles and chitosan beads using GA and Gpn as intermediate linker, with a recovery of activity above 60%. The immobilized enzyme presented higher activity in acidic conditions and improved thermal resistance than free enzyme. Generated biocatalysts could be reused and have potential application in environmentally friendly production of fully acetylated and partially acetylated COS from colloidal chitin and chitosan wastes. The acidic and higher temperature operational stability of the immobilized chitinase Chit42 could be useful for the potential industrial application of this biocatalyst. The results show that the immobilized enzyme is more active than the free counterpart for COS production with widely variable size distribution depending on the type of chitosan used. These results envisage the use of immobilized chitinase for more efficient production of low molecular chitosan oligomers for biotechnological applications.
Experimental
Materials
Iron(III) chloride hexahydrated (FeCl3·6H2O), ammonium iron(II) sulfate hexahydrate ((NH4)2Fe(SO4)2·6H2O), (3-aminopropyl)triethoxysilane (APTES), glutaraldehyde (GA; 25%) and sodium borohydride (NaBH4) were from Sigma-Aldrich (St. Louis, America). Sodium hydroxide, ethanol and hydrochloric acid were from Merck (Germany). Genipin (Gpn), di-acetyl-glucosamine ((GlcNAc)2), N,N′,N′′-tri-acetyl-glucosamine ((GlcNAc)3) were from Carbosynth Ltd (Berkshire, UK). Chitosan with different degree of deacetylation (DD) and polymerization (DP) were used, CHIT100 (100–300 kDa) and CHIT600 (600–800 kDa) all from shrimp shells (DD > 90%) were from Acros Organics (Geel, Belgium), chitosan QS1 from Paralomis granulosa (98 kDa, DD 81%) and chitosan QS2 from Pandalus borealis (31 kDa, DD 77%) from InFiQus (Madrid, Spain). Chitin (coarse flakes, DD ≤ 8%) from shrimp shells, chitosan CHIT50 (50–190 kDa, DD 77%), chitosan low molecular weight CHITLMW (50–190 kDa, DD ≥ 92%), N-acetyl-glucosamine and glucosamine (GlcN) were from Sigma-Aldrich (St. Louis, America).
Production of Chit42 in fed-batch fermenter
The chitinase Chit42 from Trichoderma harzianum CECT2413 fused to the Saccharomyces cerevisiae MFα1 secretion signal was previously cloned in plasmid pIB4 and expressed in Pichia pastoris as referred.8 The P. pastoris strain expressing chitinase Chit42 was cultivated to high cell density (for about 24 h) in 500 mL of BMG-F (13.4 mg mL−1 YNB, 4 mg mL−1 biotin, 1% (w/v) glycerol, 100 mM potassium phosphate, pH 6.0). Then this culture was grown in a 5 L bioreactor (Biostart BPluss Sartorius Ltd., Gottingen, Germany) containing 3.5 L of batch medium (40 g L−1 glycerol, 26.7 mL H3PO4 85%, 0.93 g L−1 CaSO4, 18.2 g L−1 K2SO4, 14.9 g L−1 MgSO4, 4.13 g L−1 KOH, 2 mL biotin (0.2 g L−1), and 4.35 mL of PTM1 trace salts). The fermentation parameters were maintained at 30 °C, 600 rpm agitation, 20% dissolved oxygen and pH was controlled at 5.0 units with NH4OH 28% (v/v) during 24 h (∼40 OD600 units). Then 100% methanol was added continuously during 4 days at 20 μL min−1 L−1 of fermentation volume to induce the expression of protein Chit42 (final ∼290 OD600 units). Culture growth was monitored spectrophotometrically at 600 nm (OD600) and protein concentration using NanoDrop at 280 nm. To obtain the pure protein from the expression medium, the cells were removed by centrifuging at 6000 × g for 15 min, then the extracellular fraction was concentrated using 30000 MWCO PES membranes in a Vivaflow 50 system (Sartorius, Gottingen, Germany).
Enzymatic activity analyses
Chitinase activity was evaluated by detection of reducing sugars produced from colloidal chitin and chitosan. Colloidal chitin preparation was obtained as previously reported.8 For 1% (w/v) chitosan preparation, 1 g of solid materials was dissolved in 100 mL of 0.1 M acetic acid and then pH was adjusted to 5.0 with 1 M sodium acetate pH 5.5. For free enzyme, reactions were performed in 1.5 mL Eppendorf tubes by addition of 100 μL of the enzymatic solution (previously diluted in 70 mM potassium phosphate pH 5.5, if required) to 400 μL of 1% (w/v) colloidal chitin and other substrates. Tubes were incubated at 35 °C and 900 rpm in a Termo Shaker TS-100 (Boeco, Hamburg, Germany) during 1 h. Reactions were boiled for 10 min and one volume of 0.2 M NaOH was added. Polysaccharides were removed by centrifugation at 12000 × g for 5 min. The quantification of reducing sugars in the supernatant was carried out using 3,5-dinitrosalicylic acid (DNS) method adapted to a 96-well microplate scale as described before.8 A calibration curve of D-glucosamine (0–3 mg mL−1) was used. The unit of chitinase activity (U) was defined as that corresponding to the release of 1 μmol of reducing sugar per minute (μmol min−1).
Synthesis of magnetic nanoparticles (MNPs) and chitosan beads
Magnetic nanoparticles were prepared by the co-precipitation method according to Yamaura with some modifications.23 Briefly, using a reflux condenser reactor 0.24 M of ammonium iron(II) sulfate hexahydrate solution was added to 0.25 M of iron(III) chloride hexahydrate solution at 60 °C under nitrogen gas (N2) to suppress oxygen to avoid the iron(III) to undergo oxidation.23 Then 1 M sodium hydroxide was added to obtain a final concentration of 0.5 M and the reaction was left for 1 h at room temperature. Particles were magnetically separated from the soluble phase and washed with distilled water until pH 7.0 was reached. MNPs were functionalized with amino groups by coating them with APTES according the procedure reported in.44 Briefly, 1 g of nanoparticles were dispersed in 332 mL of absolute ethanol. A black powder was obtained after drying and grinding this reaction product. Then, 8 mL of APTES and 34.6 mL of 1 M HCl 1 M was added under agitation that was maintained for 16 hours. Particles were sequentially washed with ethanol (twice), water (one time), and finally dried with nitrogen flux.
Chitosan macro-spheres (CMS) were prepared from 2% (w/v) chitosan CHITLMW in 0.2 M acetic acid maintained with agitation for 16 hours at room temperature. Using a Cole Parmer™ Masterflex™ Brushless Pump System (Thermo Fisher Scientific), drop wise of viscous solution was coagulated using 0.5 M NaOH and 13% ethanol solution. The formed chitosan beads were washed with an excess of water until neutral pH was reached. The beads obtained were golden yellow, the same physical aspects as the ones already obtained in previous works.32,45 When chitosan beads reacted with GA or Gpn their color changed to warm yellow or greenish blue, respectively, because the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–imine bonds act as chromophore as reported when using GA.46
N–imine bonds act as chromophore as reported when using GA.46
Characterization of magnetic nanoparticles and chitosan beads
The size and morphology of magnetic nanoparticles before and after the protein immobilization were analyzed by transmission electron microscopy (TEM Seiko JEOL Japan). A few micrograms of nanoparticles were suspended in water and then were sonicated. Dry chitosan macrospheres were coated with gold under vacuum before observation using scanning electron microscopy (SEM) for imaging at 11.500 μA and 15 kV accelerating voltage, using Zeiss DSM-950 (Zeiss, Oberkochen, Germany). The working magnification range was ×20–80 and higher magnification was not possible due to the bead's preparation instability.
Immobilization of Chit42
To immobilize the protein Chit42 to the generated MNPs and CMS particles, 0.05 g of nanoparticles and 0.2 g of wet chitosan beads, in separate Eppendorf tubes were used. Then 1 mL of 0.5% (v/v) GA in 100 mM potassium phosphate pH 7.0 was added. Tubes were incubated at 25 °C with agitation for 2 h. Particles were washed with the phosphate buffer to remove the excess of GA and then different concentrations of the protein Chit42 in the range of 3.12–24.8 mg g−1 of MNP or 0.78–6.2 mg g−1 of CMS were added. Tubes were maintained at 25 °C for 16 h with agitation. The MNPs-GA-Chit42 and CMS-GA-Chit42 immobilized catalysts were washed with 1 mL potassium phosphate two times to remove the unbound protein. Similar protocols were used to obtain the CMS-Gpn-Chit42 biocatalyst with Gpn (0.5% (w/v)) and CMS but using protein concentrations in the range 0.5–2.5 mg g−1. Furthermore, the use of Gpn as linker for CMS ensures to have a fully non-toxic system for food grade applications of chitinases. The amount of the enzyme in the solutions before and after the immobilization process were measured by NanoDrop at 280 nm. The immobilization yield and activity recovery, both in percentage, were calculated as follows: immobilization yield (%) = 100 × (amount of protein immobilized/starting amount of protein) and the activity recovered yield (%) = 100 × (Ui/(Ua − Uw)). Where, amount of protein immobilized = amount of protein added for its immobilization − total amount of protein in washing buffer. Ui = activity (units) of Chit42 immobilized, Ua = activity (units) of Chit42 added and Uw = activity (units) of Chit42 in washing buffer. Different amounts of GA (0.5, 1 and 2% of GA solution) and Gpn (0.063, 0.125, 0.25, 0.312 and 0.5%; w/v) were also used to evaluate the optimal conditions for immobilization. GA 0.5% and Gpn 0.125% were selected as optimal conditions. The reduction of the Schiff base was done as previously referred.41,47 In briefly, after washing the immobilized catalysts, 1 mg mL−1 of NaBH4 in potassium phosphate pH 7.0 was added. Catalysts were maintained for 30 minutes at 25 °C with gentle stirring and then were washed three times with the referred phosphate buffer.
Biochemical characterization and reuse assay for the immobilized Chit42
Optimal pH of free and immobilized Chit42 was analyzed using colloidal chitin 1% (w/v) at different pH values, using 70 mM of sodium citrate (pH 3.0–5.0) or potassium phosphate (pH 5.0–8.0). Samples were incubated at 35 °C and 900 rpm in a Thermo Shaker TS-100 (Boeco, Hamburg, Germany) for 1 h. For optimal temperature assays, reactions including 70 mM potassium phosphate pH 5.0 and 1% (w/v) colloidal chitin were incubated in the range of 35–70 °C. For MNPs, biocatalysts were separated from the reaction mixture by applying magnetic field, and for CMS using centrifugation at 12000 × g for 5 min. Then all reaction mixtures were boiled for 10 min. In reuse assays, 30 mg of chitosan beads, already attached to Chit42, and 0.6 mL of 1% (w/v) colloidal chitin pH 5.0 were incubated as explained above. Biocatalysts were precipitated at 1000 rpm 5 min and then the supernatant (200 μL) was mixed with DNS. For MNPs, 50 mg of support were used and reactions were developed as before but stopped by applying magnetic field to separate the catalysts from the mixture. Reducing sugars were analyzed by spectrophotometry at 540 nm after treatment at 100 °C for 10 min. MNPs and CMS were washed with 100 mM potassium phosphate pH 7.0 between each one of the reuse cycles.
COS production, characterization and quantification by HPAEC-PAD and mass spectrometry
Analysis of the products obtained by the hydrolytic activity of Chit42 immobilized to MNPs or CMS was carried out using 1% substrate (colloidal chitin and chitosan), incubation at 35 °C and 900 rpm as referred in previous section. Aliquots of 0.3 mL of reactions were mixed with an equal volume of 0.2 M NaOH, and centrifuged as referred. The supernatant was analyzed by HPAEC-PAD as described before.8 The chromatography equipment was a Dionex ICS3000 system (Dionex, Thermo Fisher Scientific Inc., Waltham, MA) consisting of an SP gradient pump, an electrochemical detector with a gold working electrode and Ag/AgCl as reference electrode, and an auto sampler (model AS-HV). An anion-exchange 4 × 250 mm Carbo-Pack PA-100 column (Dionex) connected to a 4 × 50 mm CarboPac PA-100 guard column was used at 30 °C. The initial mobile phase was 4 mM NaOH at 0.3 mL min−1 for 30 min. Then, column was washed for 20 min at 0.5 mL min−1 with a solution containing 100 mM sodium acetate and 100 mM NaOH and further equilibrated with 4 mM NaOH. Standards of fully acetylated COS with DP ranging from 1 to 4 were used for identification of the reaction products. The molecular weight of COS was assessed by MALDI-TOF mass spectrometry using a mass spectrometer with Ultraflex III TOF/TOF (Bruker, Billerica, MA, USA) and an NdYAG laser. Registers were taken in positive reflector mode within the mass interval 40–5000 Da, with external calibration and with 20 mg mL−1 2,5-dihydroxybenzoic in acetonitrile (3:7) (v/v) as matrix. Samples were mixed with the matrix in a 4:1 proportion and 0.5 μL were analyzed.
Funding
This work was supported by the EU EMFF-Blue Economy-2018-FISH4FISH-863697 project, the Spanish Ministry of Economy and Competitiveness [BIO2016-76601-C3-1/-2], the Spanish Ministry of Science and Innovation PID2019-105838RB-C32/-31, Fundación Ramón Areces [XIX Call of Research Grants in Life and Material Sciences], and by an institutional grant from Fundación Ramón Areces to the Centro de Biología Molecular. CSGI (Consorzio per lo Sviluppo dei Sistemi a Grande Interfase), Florence, Italy and MIUR for the Dipartimento di Eccellenza 2018-2022 grant are gratefully acknowledged.
Abbreviations
| COS | Chitooligosaccharides |
| paCOS | Partially acetylated COS |
| GlcN | D-Glucosamine |
| GlcNAc | N-Acetyl-D-glucosamine |
| GH | Glycoside hydrolase |
| DP | Degree of polymerization |
| DD | Degree of deacetylation |
| MNPs | Magnetic nanoparticles |
| CMS | Chitosan macro-spheres |
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors thank Asunción Martín-Redondo and Fadia Cervantes for technical support and advice on the use of equipment.
Notes and references
- M. M. Abo Elsoud and E. M. El Kady, Bull. Natl. Res. Cent., 2019, 43, 59–70 CrossRef.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- J. Zhou, X. Liu, F. Yuan, B. Deng and X. Yu, ACS Sustainable Chem. Eng., 2020, 8, 4781–4791 CrossRef CAS.
- M. Jiang, Z. Guo, C. Wang, Y. Yang, X. Liang and F. Ding, Neurosci. Lett., 2014, 581, 32–36 CrossRef CAS PubMed.
- P. Santos-Moriano, P. Kidibule, N. Míguez, L. Fernández-Arrojo, A. O. Ballesteros, M. Fernández-Lobato and F. J. Plou, Catalysts, 2019, 9, 405–416 CrossRef CAS.
- M. B. Kaczmarek, K. Struszczyk-Swita, X. Li, M. Szczęsna-Antczak and M. Daroch, Front. Bioeng. Biotechnol., 2019, 7, 243–268 CrossRef PubMed.
- P. Santos-Moriano, P. E. Kidibule, E. Alleyne, A. O. Ballesteros, A. Heras, M. Fernandez-Lobato and F. J. Plou, Process Biochem., 2018, 73, 102–108 CrossRef CAS.
- P. E. Kidibule, P. Santos-Moriano, E. Jiménez-Ortega, M. Ramírez-Escudero, M. C. Limón, M. Remacha, F. J. Plou, J. Sanz-Aparicio and M. Fernández-Lobato, Microb. Cell Fact., 2018, 17, 1–13 CrossRef PubMed.
- I. A. Hoell, G. Vaaje-Kolstad and V. G. H. Eijsink, Biotechnol. Genet. Eng. Rev., 2010, 27, 331–366 CrossRef CAS PubMed.
- A. Oyeleye and Y. M. Normi, Biosci. Rep., 2018, 38, 1–21 CrossRef PubMed.
- P. Torres-Salas, A. Del Monte-Martinez, B. Cutiño-Avila, B. Rodriguez-Colinas, M. Alcalde, A. O. Ballesteros and F. J. Plou, Adv. Mater., 2011, 23, 5275–5282 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- K. Wlizło, J. Polak, J. Kapral-Piotrowska, M. Graz, R. Paduch and A. Jarosz-Wilkołazka, Catalysts, 2020, 10, 1–21 CrossRef.
- M. Bilal, Y. Zhao, T. Rasheed and H. M. N. Iqbal, Int. J. Biol. Macromol., 2018, 120, 2530–2544 CrossRef CAS PubMed.
- W. Wang, N. Guo, W. Huang, Z. Zhang and X. Mao, Catalysts, 2018, 8, 1–10 Search PubMed.
- S. Rouhani, A. Rostami and A. Salimi, RSC Adv., 2016, 6, 26709–26718 RSC.
- T. J. Raharjo, L. Febrina, F. A. Wardoyo and R. T. Swasono, Asian J. Biochem., 2016, 11, 127–134 CrossRef CAS.
- C. A. Gasser, E. M. Ammann, A. Schäffer, P. Shahgaldian and P. F. X. Corvini, Appl. Microbiol. Biotechnol., 2016, 100, 7281–7296 CrossRef CAS PubMed.
- D. J. Seo, Y. H. Jang, R. D. Park and W. J. Jung, Carbohydr. Polym., 2012, 88, 391–394 CrossRef CAS.
- M. Prasad and P. Palanivelu, Biotechnol. Appl. Biochem., 2015, 62, 523–529 CrossRef CAS PubMed.
- S. Rathan and T. Thayumanavan, J. Glob. Biosci., 2017, 6, 5032–5045 Search PubMed.
- F. Liaqat and R. Eltem, Carbohydr. Polym., 2018, 184, 243–259 CrossRef CAS PubMed.
- M. Yamaura, R. L. Camilo, L. C. Sampaio, M. A. Macêdo, M. Nakamura and H. E. Toma, J. Magn. Magn. Mater., 2004, 279, 210–217 CrossRef CAS.
- F. Assa, H. Jafarizadeh-Malmiri, H. Ajamein, N. Anarjan, H. Vaghari, Z. Sayyar and A. Berenjian, Nano Res., 2016, 9, 2203–2225 CrossRef CAS.
- N. Zhang, R. X. Yan and W. Q. Guan, Adv. Mater. Res., 2014, 936, 674–680 Search PubMed.
- T. Mizuki, M. Sawai, Y. Nagaoka, H. Morimoto and T. Maekawa, PLoS One, 2013, 8, 6–9 CrossRef PubMed.
- J. -P. Chen and K. -C. Chang, J. Chem. Technol. Biotechnol., 1994, 60, 133–140 CrossRef CAS PubMed.
- M. A. Esawy, A. A. Gamal, M. M. I. Helal, M. E. Hassan, N. M. Hassanein and A. M. Hashem, Int. J. Biol. Macromol., 2016, 92, 56–62 CrossRef CAS PubMed.
- P. Santos-Moriano, J. M. Woodley and F. J. Plou, J. Mol. Catal. B: Enzym., 2016, 133, 211–217 CrossRef CAS.
- I. Eş, J. D. G. Vieira and A. C. Amaral, Appl. Microbiol. Biotechnol., 2015, 99, 2065–2082 CrossRef PubMed.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- T. Charoenwongpaiboon, K. Wangpaiboon, R. Pichyangkura and M. H. Prousoontorn, RSC Adv., 2018, 8, 17008–17016 RSC.
- J. Palomo, Curr. Org. Synth., 2009, 6, 1–14 CrossRef CAS.
- C. Guerrero, C. Aburto, S. Suárez, C. Vera and A. Illanes, Biocatal. Agric. Biotechnol., 2018, 16, 353–363 CrossRef.
- M. Ashjari, M. Mohammadi and R. Badri, J. Mol. Catal. B: Enzym., 2015, 115, 128–134 CrossRef CAS.
- O. Barbosa, R. Torres, C. Ortiz and R. Fernandez-Lafuente, Process Biochem., 2012, 47, 1220–1227 CrossRef CAS.
- A. Sørbotten, S. J. Horn, V. G. H. Eijsink and K. M. Vårum, FEBS J., 2005, 272, 538–549 CrossRef PubMed.
- T. Laokuldilok, T. Potivas, N. Kanha, S. Surawang, P. Seesuriyachan, S. Wangtueai, Y. Phimolsiripol and J. M. Regenstein, Food Biosci., 2017, 18, 28–33 CrossRef CAS.
- S. Basa, M. Nampally, T. Honorato, S. N. Das, A. R. Podile, N. E. El Gueddari and B. M. Moerschbacher, J. Am. Chem. Soc., 2020, 142, 1975–1986 CrossRef CAS PubMed.
- L. Phil, M. Naveed, I. S. Mohammad, L. Bo and D. Bin, Biomed. Pharmacother., 2018, 102, 438–451 CrossRef CAS PubMed.
- H. Wu, B. B. Aam, W. Wang, A. L. Norberg, M. Sørlie, V. G. H. Eijsink and Y. Du, Carbohydr. Polym., 2012, 89, 511–518 CrossRef CAS PubMed.
- M. Mengíbar, I. Mateos-Aparicio, B. Miralles and Á. Heras, Carbohydr. Polym., 2013, 97, 776–782 CrossRef PubMed.
- Á. Sánchez, M. Mengíbar, G. Rivera-Rodríguez, B. Moerchbacher, N. Acosta and A. Heras, Carbohydr. Polym., 2017, 157, 251–257 CrossRef PubMed.
- R. Barbucci, D. Pasqui, G. Giani, M. De Cagna, M. Fini, R. Giardino and A. Atrei, Soft Matter, 2011, 7, 5558–5565 RSC.
- E. Biró, A. S. Németh, C. Sisak, T. Feczkó and J. Gyenis, J. Biochem. Biophys. Methods, 2008, 70, 1240–1246 CrossRef PubMed.
- L. Poon, L. D. Wilson and J. V. Headley, Carbohydr. Polym., 2014, 109, 92–101 CrossRef CAS PubMed.
- A. H. Orrego, M. Romero-Fernández, M. D. C. Millán-Linares, M. D. M. Yust, J. M. Guisán and J. Rocha-Martin, Catalysts, 2018, 8, 1–15 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. MALDI-TOF MS analysis of COS produced from chitosan CHIT600. Table S1. Main peaks and intensities of the mass spectrum corresponding to the reaction mixture obtained with colloidal chitin as substrate and MNPs-Ga-Chit42. Table S2. Main peaks and intensities of the mass spectrum corresponding to the reaction mixture obtained with colloidal chitin as substrate and CMS-Ga-Chit42. Table S3. Main peaks and intensities of the mass spectrum corresponding to the reaction mixture obtained with chitosan QS2 as substrate and MNPs-Ga-Chit42. Table S4. Main peaks and intensities of the mass spectrum corresponding to the reaction mixture obtained with chitosan QS2 as substrate and CMS-Ga-Chit42. See DOI: 10.1039/d0ra10409d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b