
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the effect of oxide components on proton mobility in phosphate glasses using a statical analysis approach†
Takahisa Omata *a,
Issei Suzukia,
Aman Sharmaa,
Tomohiro Ishiyamab,
Junji Nishiic,
Toshiharu Yamashitad and
Hiroshi Kawazoed
*a,
Issei Suzukia,
Aman Sharmaa,
Tomohiro Ishiyamab,
Junji Nishiic,
Toshiharu Yamashitad and
Hiroshi Kawazoed
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Sendai 980-8577, Japan. E-mail: takahisa.omata.c2@tohoku.ac.jp; Fax: +81-22-217-5832; Tel: +81-22-217-5832
bFuel Cell Materials Group, Research Institute for Energy Conservation, National Institute of Advanced Industrial Science and Technology (AIST), AIST Central 5, Higashi 1-1-1, Tsukuba, Ibaraki 305-8565, Japan
cResearch Institute for Electronic Science, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo 001-0021, Japan
dKawazoe Frontier Technologies Corporation, Kuden-cho 931-113, Sakae-ku, Yokohama 247-0014, Japan
First published on 14th January 2021
Abstract
The models to describe the proton mobility (μH) together with the glass transition temperature (Tg) of proton conducting phosphate glasses employing the glass composition as descriptors have been developed using a statical analysis approach. According to the models, the effects of additional HO1/2, MgO, BaO, LaO3/2, WO3, NbO5/2, BO3/2 and GeO2 as alternative to PO5/2 were found as following. μH at Tg is determined first by concentrations of HO1/2 and PO5/2, and μH at Tg increases with increasing HO1/2 concentration and decreasing PO5/2. The component oxides are categorized into three groups according to the effects on μH at Tg and Tg. The group 1 oxides increase μH at Tg and decrease Tg, and HO1/2, MgO, BaO and LaO3/2 and BO3/2 are involved in this group. The group 2 oxides increase both μH at Tg and Tg, and WO3 and GeO2 are involved in this group. The group 3 oxides increase Tg but do not vary μH at Tg. Only NbO5/2 falls into the group 3 among the oxides examined in this study. The origin of the effect of respective oxide groups on μH at Tg and Tg were discussed.
Introduction
Inorganic glasses have been studied for decades as solid electrolytes because of their electrochemical stability and chemical durability, and various cationic conduction, such as Li+, Ag+ and H+ conduction, in oxide glass has been investigated extensively.1–3 Recent demands for highly proton conducting electrolytes in the temperature range between 250 and 500 °C that is operating temperatures of intermediate temperature fuel cells accelerate to explore proton conducting glasses.4–9 Our group developed a technique termed as alkali-proton substitution (APS) that injects high concentration of proton carriers, >1021 cm−3, into phosphate glasses10 and fabricated many proton conducting glasses by using APS.11–14 We studied characteristics of glasses that influence on proton conductivity, such as polymerization level of phosphate framework (ratio of the number of oxygen to phosphorous atoms; O/P ratio)15 and kinds of glass network modifier,16 and the effect of additional glass-network formers, such as GeO2, on the thermal stability.17 As a result, 2 × 10−3 S cm−1 of proton conductivity at 300 °C has been achieved by 34HO1/2–2NaO1/2–4NbO5/2–2BaO–4LaO3/2–4GeO2–1BO3/2–49PO5/2 glass (36H-glass) up to now.18 Based on the electromotive force and electrochemical hydrogen pump experiments, the phosphate glass electrolyte is confirmed that the mean transport number of proton is unity even under the oxidation atmosphere like an air electrode atmosphere in the fuel cell,19 suggesting that highly efficient operation of fuel cells and steam electrolysis cells is achievable owing to its no electronic leakage.20 In addition, fabrication of ultra-thin glass electrolytes with a thickness of 16 μm was recently demonstrated by the press forming.21 This will be a great advantage of the glass electrolyte in order to reduce electrolyte resistance (ohmic resistance) of the electrochemical cells. However, further increase of their proton conductivity >1 × 10−2 S cm−1 at the operating temperature is still required for practical applications.Very recently, we have found that the mobility of proton carriers (μH) at the glass transition temperature (Tg) in phosphate glasses converges in a small range between 2 × 10−9 and 2 × 10−7 cm2 V−1 s−1, whereas Tg of the glasses is in the wide range of 150 to 650 °C, proton conductivity at 200 °C is also wide range of 10−10 to 10−4 S cm−1, and proton carrier concentration is in the range of 1019 to 1022 cm−3.22 Because the μH at Tg of the 36H-glass is 5.4 × 10−8 cm2 V−1 s−1 that is the middle in the μH at Tg range from 2 × 10−9 to 2 × 10−7 cm2 V−1 s−1, it is suggested that its proton conductivity can be further increased by improving the μH at Tg. Because the determining factor of μH at Tg of proton conducting phosphate glasses has yet to be cleared, we unfortunately still do not understand how to improve μH at Tg.
The composition of glasses is continuatively controllable unlike the crystalline materials; therefore, various properties of glasses, have been empirically expressed by the mole fraction weighting mean of the respective components.23–26 Whereas to understand the effects of fundamental properties of glasses, such as O–H bonding, local structure surrounding protons and short range atomic structure of the glass framework, on μH at Tg are of course important to understand the proton conduction in phosphate glasses from the physical aspect, understanding the relationship between the glass composition and μH at Tg is also valuable in order to improve the electrolyte performance of proton conducting phosphate glasses. When the proton conductivity is successfully described by the glass composition, the proton conductivity of phosphate glasses will be easy to improve based on the obtained relationship between the glass composition and μH at Tg, and that will have a major impact on the electrochemical cells such as fuel cells and steam electrolysis cells working at intermediate temperatures. The proton conducting phosphate glasses prepared by using APS previously reported consists of many oxide components;22 for example, 36H-glass involves 8 oxides as HO1/2, NaO1/2, BaO, LaO3/2, NbO5/2, GeO2, BO3/2 and PO5/2; therefore, it is not easy to understand the role of the respective component oxides on μH at Tg and the relationship between the composition and μH at Tg.
Here, we have developed a model, using a statical analysis approach, to describe μH at Tg of phosphate glasses according to the glass composition, i.e., the mol% of respective component oxides were employed as descriptors. We also developed a model to describe Tg because the thermal stability of proton conducting glasses is another key property taking the working temperature of the electrochemical devices involving the glasses into account. The effect of respective component oxides on μH at Tg and Tg were discussed based on the model obtained.
Methodology
Dataset details
The dataset for μH at Tg and Tg of proton conducting phosphate glasses used as training data in this study is referenced from previous report (Table 1 in ref. 16). The dataset has originally 32 records, but for the 13 records in the original dataset, the proton carrier concentrations are smaller than 1 mol% because the proton carrier in those 13 glasses are originated from the residual water. Therefore, we used a dataset that consists of remaining 19 records as summarized in Table 1. Each record contains glass composition in mol% and experimentally determined μH at Tg and Tg.| No. | Mol% of component oxide | μH at Tg (cm2 V−1 s−1) | Tg (°C) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HO1/2 | NaO1/2 | WO3 | NbO5/2 | TaO5/2 | MgO | BaO | LaO3/2 | AlO3/2 | YO3/2 | GdO3/2 | GeO2 | BO3/2 | PO5/2 | |||
| 1 | 25 | 3 | 1 | 8 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 58 | 2.1 × 10−9 | 200 |
| 2 | 24 | 8 | 1 | 8 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 54 | 5.5 × 10−9 | 177 |
| 3 | 25 | 10 | 1 | 8 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 51 | 3.7 × 10−8 | 190 |
| 4 | 32 | 6 | 1 | 8 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 48 | 3.7 × 10−8 | 170 |
| 5 | 32 | 8 | 1 | 8 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 46 | 1.2 × 10−8 | 167 |
| 6 | 28 | 2 | 1 | 8 | 0 | 0 | 0 | 5 | 3 | 3 | 0 | 0 | 0 | 50 | 2.0 × 10−8 | 281 |
| 7 | 29 | 6 | 1 | 8 | 0 | 0 | 0 | 5 | 3 | 0 | 0 | 0 | 0 | 48 | 7.6 × 10−9 | 224 |
| 8 | 30 | 5 | 1 | 8 | 0 | 0 | 0 | 5 | 0 | 3 | 0 | 0 | 0 | 48 | 4.1 × 10−9 | 228 |
| 9 | 35 | 0 | 0 | 3 | 0 | 5 | 0 | 3 | 0 | 0 | 0 | 2 | 2 | 50 | 1.3 × 10−8 | 192 |
| 10 | 32 | 3 | 0 | 3 | 0 | 0 | 5 | 3 | 0 | 0 | 0 | 2 | 2 | 50 | 6.8 × 10−9 | 163 |
| 11 | 34 | 2 | 0 | 4 | 0 | 0 | 2 | 4 | 0 | 0 | 0 | 4 | 1 | 49 | 5.4 × 10−8 | 180 |
| 12 | 38 | 2 | 0 | 0 | 4 | 2 | 0 | 4 | 0 | 0 | 0 | 2 | 1 | 47 | 2.7 × 10−8 | 165 |
| 13 | 17 | 8 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 | 0 | 1 | 0 | 66 | 2.6 × 10−9 | 227 |
| 14 | 12 | 13 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 6 | 0 | 63 | 1.3 × 10−8 | 243 |
| 15 | 33 | 2 | 0 | 0 | 0 | 2 | 0 | 5 | 0 | 0 | 0 | 5 | 0 | 53 | 4.0 × 10−8 | 182 |
| 16 | 31 | 4 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 5 | 5 | 0 | 53 | 1.2 × 10−8 | 178 |
| 17 | 20 | 5 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 6 | 0 | 63 | 1.5 × 10−8 | 252 |
| 18 | 28 | 7 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 5 | 5 | 0 | 53 | 1.4 × 10−8 | 233 |
| 19 | 34 | 1 | 8 | 8 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 44 | 1.1 × 10−7 | 231 |
Regression models and method
A linear combination model, in which mol% of respective oxides are used as predictors, is employed for both log(μH at Tg) and Tg in this study. The regression algorithm used in this study is based on the linear regression as implemented in MATLAB (MathWorks, USA). When the general linear regression was preliminary performed for log(μH at Tg), the overtraining occurred maybe because of small number of training data; the predicted μH at Tg for the 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 296 glass compositions described later was unreasonable values in the range of 10−29 to 1017 cm2 V−1 s−1 (Fig. S1 and S2 in ESI†), although the range of the experimentally observed values is in the range of 2 × 10−9 to 2 × 10−7 cm2 V−1 s−1.22 Therefore, we employed the principal components analysis to fit a linear regression in order to avoid overtraining. Five principal components were employed to explain 95% of variance of original data. The mathematical model can be written as
296 glass compositions described later was unreasonable values in the range of 10−29 to 1017 cm2 V−1 s−1 (Fig. S1 and S2 in ESI†), although the range of the experimentally observed values is in the range of 2 × 10−9 to 2 × 10−7 cm2 V−1 s−1.22 Therefore, we employed the principal components analysis to fit a linear regression in order to avoid overtraining. Five principal components were employed to explain 95% of variance of original data. The mathematical model can be written as
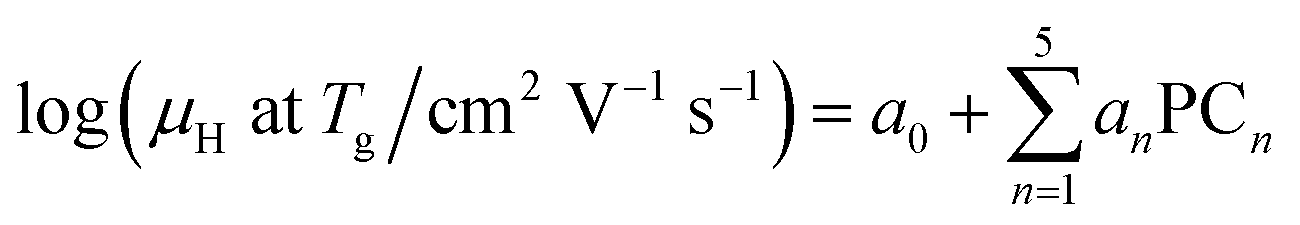 | (1) |
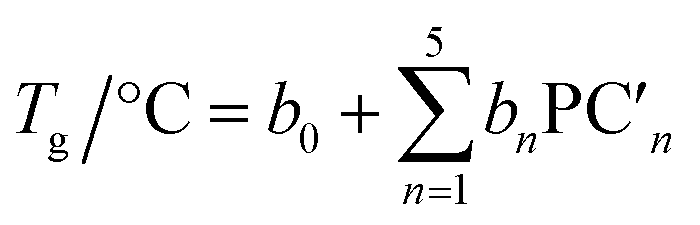 | (2) |
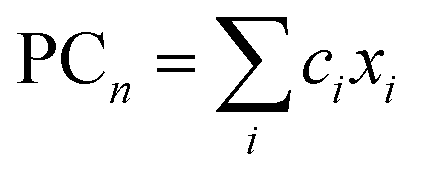 | (3) |
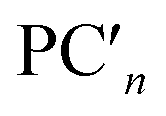 are nth principal component explaining the variance of experimentally observed log(μH at Tg) and Tg, respectively, a0 and b0 are intercepts, an and bn are coefficients of nth principal component, xi is the mol% of the oxide i, and ci is its coefficient.
are nth principal component explaining the variance of experimentally observed log(μH at Tg) and Tg, respectively, a0 and b0 are intercepts, an and bn are coefficients of nth principal component, xi is the mol% of the oxide i, and ci is its coefficient.
In order to check the validity of the models and to understand the effect of respective component oxides on μH at Tg and Tg, we performed to predict μH at Tg and Tg for 55296 glass compositions containing 30, 33 and 36 mol% of HO1/2, 0, 2 and 4 mol% of WO3, 0, 2, 4 and 6 mol% of NbO5/2, 0, 2, 4 and 6 mol% of MgO, 0, 2, 4 and 6 mol% of BaO, 0, 2, 4 and 6 mol% of LaO3/2, 0, 1, 2, 3, 4 and 5 mol% of GeO2, 0, 1, 2 and 3 mol% of BO3/2 and 28–70 mol% of PO5/2. In this prediction, all the compositions were assumed to form homogeneous glasses.
Results and discussion
Linear regression models for μH at Tg and Tg
The following relationships of log(μH at Tg) and Tg against the five principal components of glass composition were obtained after regression:| log(μH at Tg) = −7.8549 + 0.022233 × PC1 − 0.01167 × PC2 + 0.26874 × PC3 − 0.01727 × PC4 + 0.160456 × PC5, | (4) |
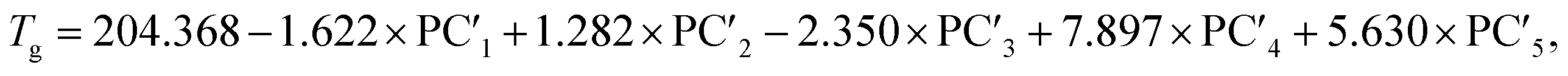 | (5) |
The principal components are summarized in Tables 2 and 3 for log(μH at Tg) and Tg, respectively. Fig. 1(a) and (b) show comparison of experimentally observed and predicted values of μH at Tg and Tg, respectively, for the 19 training data. The root mean square error (RMSE) was 0.2775 for log(μH at Tg) and was 23.6 °C for Tg. No systematic error was observed and the fitting were reasonably good for both log(μH at Tg) and Tg. Fig. 2(a) and (b) respectively show the predicted values of log(μH at Tg) and Tg for the 55296 phosphate glass compositions. The predicted values are ranging between 8.1 × 10−10 and 7.7 × 10−7 cm2 V−1 s−1 for μH at Tg and between 152 and 256 °C for Tg. As compared with experimentally determined μH at Tg,22 the range of the predicted values are very close to the range of the experimentally observed values from 2 × 10−9 to 2 × 10−7 cm2 V−1 s−1. These results indicate that the models obtained are quite reasonable and available to discuss the effects of respective component oxides on μH at Tg.
| Principal components | PC1 | PC2 | PC3 | PC4 | PC5 | |
| Proportion of variance | 0.659 | 0.183 | 0.061 | 0.026 | 0.021 | |
| Cumulative proportion | 0.659 | 0.842 | 0.903 | 0.929 | 0.950 | |
| Factor loading | x(HO1/2) | 0.69239 | −0.32693 | −0.07750 | −0.19571 | −0.15439 |
| x(NaO1/2) | −0.24549 | 0.28854 | 0.71623 | −0.35865 | −0.03489 | |
| x(WO3) | 0.06837 | 0.15986 | −0.11444 | 0.32222 | 0.77387 | |
| x(NbO5/2) | 0.16670 | 0.68417 | −0.16598 | 0.28247 | −0.30664 | |
| x(TaO5/2) | 0.02694 | −0.05991 | 0.00355 | −0.24336 | 0.04874 | |
| x(MgO) | 0.03954 | −0.18457 | 0.01768 | 0.06160 | −0.13778 | |
| x(BaO) | 0.02319 | −0.04547 | −0.02005 | −0.12335 | −0.09325 | |
| x(LaO3/2) | −0.08309 | 0.19943 | −0.33952 | −0.50726 | 0.26128 | |
| x(AlO3/2) | 0.01281 | 0.06277 | −0.04134 | 0.08739 | −0.09090 | |
| x(YO3/2) | 0.01467 | 0.05828 | −0.05950 | 0.09692 | −0.09979 | |
| x(GdO3/2) | −0.00319 | −0.15212 | 0.31784 | 0.52266 | −0.11954 | |
| x(GeO2) | −0.10164 | −0.37370 | 0.21746 | 0.08717 | 0.28694 | |
| x(BO3/2) | 0.02654 | −0.05917 | −0.04343 | −0.10435 | −0.08555 | |
| x(PO5/2) | −0.63774 | −0.25119 | −0.41100 | 0.07224 | −0.24811 | |
| Principal components | 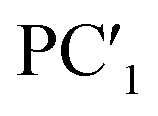 |
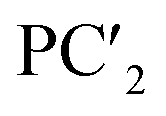 |
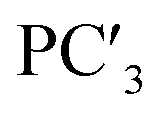 |
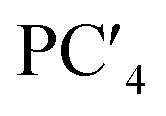 |
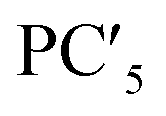 |
|
| Proportion of variance | 0.659 | 0.183 | 0.061 | 0.026 | 0.021 | |
| Cumulative proportion | 0.659 | 0.842 | 0.903 | 0.929 | 0.950 | |
| Factor loading | x(HO1/2) | 0.69239 | −0.32693 | −0.0775 | −0.19571 | −0.15439 |
| x(NaO1/2) | −0.24549 | 0.28854 | 0.71623 | −0.35865 | −0.03489 | |
| x(WO3) | 0.06837 | 0.15986 | −0.11444 | 0.32222 | 0.77387 | |
| x(NbO5/2) | 0.1667 | 0.68417 | −0.16598 | 0.28247 | −0.30664 | |
| x(TaO5/2) | 0.02694 | −0.05991 | 0.00355 | −0.24336 | 0.04874 | |
| x(MgO) | 0.03954 | −0.18457 | 0.01768 | 0.0616 | −0.13778 | |
| x(BaO) | 0.02319 | −0.04547 | −0.02005 | −0.12335 | −0.09325 | |
| x(LaO3/2) | −0.08309 | 0.19943 | −0.33952 | −0.50726 | 0.26128 | |
| x(AlO3/2) | 0.01281 | 0.06277 | −0.04134 | 0.08739 | −0.0909 | |
| x(YO3/2) | 0.01467 | 0.05828 | −0.0595 | 0.09692 | −0.09979 | |
| x(GdO3/2) | −0.00319 | −0.15212 | 0.31784 | 0.52266 | −0.11954 | |
| x(GeO2) | −0.10164 | −0.3737 | 0.21746 | 0.08717 | 0.28694 | |
| x(BO3/2) | 0.02654 | −0.05917 | −0.04343 | −0.10435 | −0.08555 | |
| x(PO5/2) | −0.63774 | −0.25119 | −0.411 | 0.07224 | −0.24811 | |
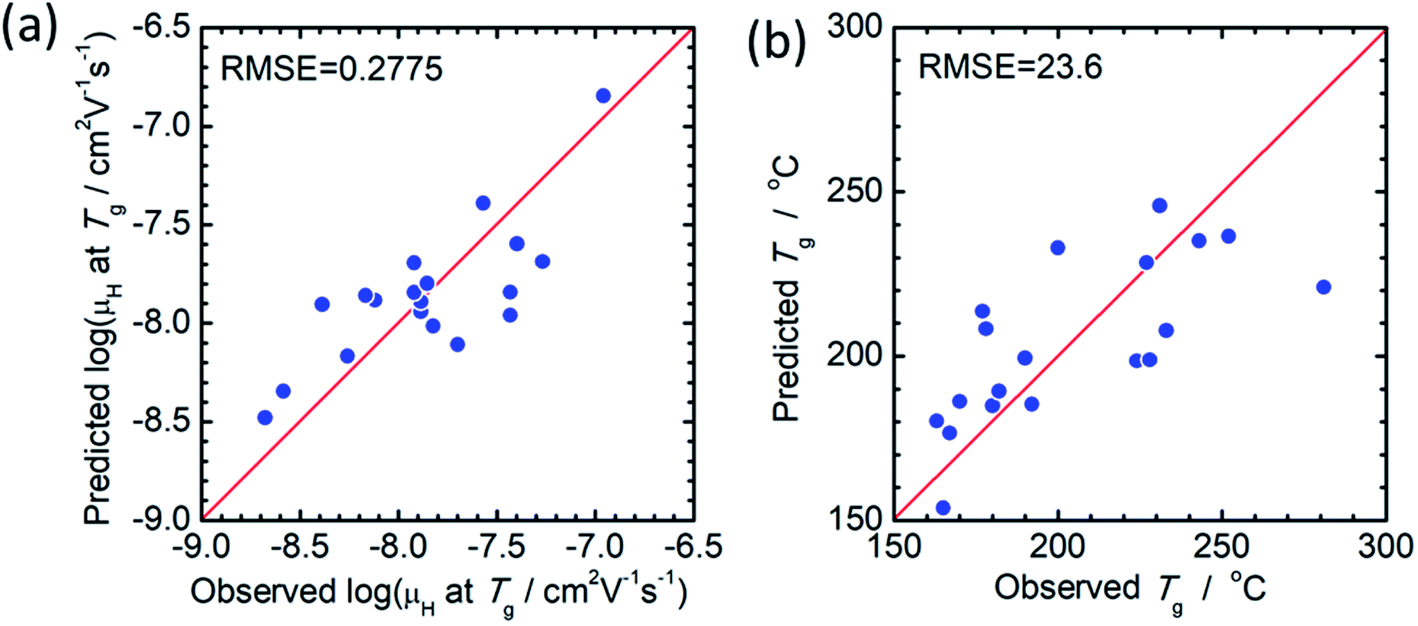 | ||
| Fig. 1 Comparison of experimentally observed and predicted values of (a) μH at Tg and (b) Tg. | ||
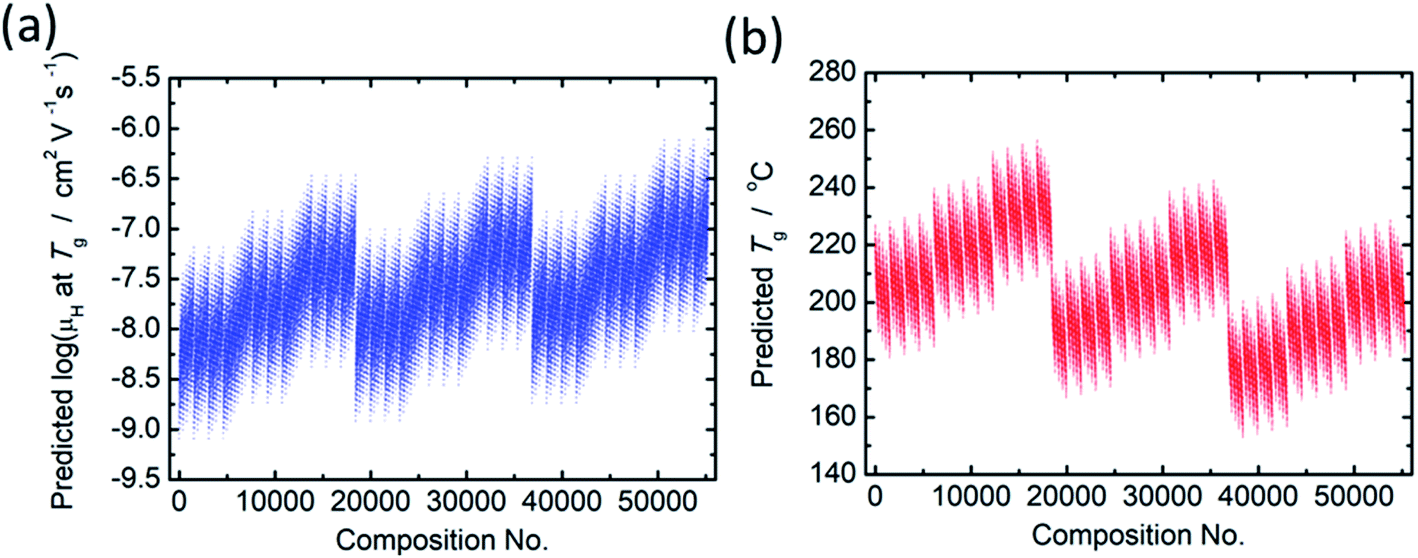 | ||
| Fig. 2 Predicted values of (a) log(μH at Tg) and (b) Tg for the 55296 phosphate glass compositions. | ||
As seen in Table 2, absolute values of the factor loading of HO1/2 and PO5/2 components are particularly larger than those of the other components, indicating that μH at Tg is first determined by the concentration of HO1/2 and PO5/2. Taking into account that the coefficient of PC1 in eqn (4) is positive, μH at Tg increases with the increasing HO1/2 concentration, and it reduces with the increasing PO5/2 concentration. In this respect, the experimental observation that the μH increases with the decreasing polymerization level of phosphate glass-network is reproduced well by the present model. μH turns into decrease at O/P ratio (ratio of the number of oxygen to phosphorous atoms) higher than 3.5–3.6;15 however, such a behavior cannot be reproduced using linear regression model. Consequently, applicable composition range of the present model is limited in a O/P ratio smaller than 3.5–3.6.
From comparison of the models of μH at Tg and Tg as summarized in Tables 2 and 3, the factor loadings of respective principal components for log(μH at Tg) and Tg are surprisingly found to be the same each other, i.e., the variance in both log(μH at Tg) and Tg are explained by the same principal components, clearly indicating that there should be some kind of relationship between log(μH at Tg) and Tg. This is quite consistent with our previously reported estimation that the motion of protons (proton diffusion or mobility) determines the motion of glass framework (Tg) in the proton conducting phosphate glasses.22 Fig. 3 shows log(μH at Tg) as a function of Tg of 55296 predicted values (black dots) together with the experimentally observed 19 values (red dots). A trend that log(μH at Tg) decreases linearly with the increasing Tg was clearly observed for the predicted values in Fig. 3. The observed relationship between log(μH at Tg) and Tg may be a key to understand physical factor to determine μH at Tg; however, we need additional information in order to go further this problem. Therefore, the origin of the relationship between log(μH at Tg) and Tg remains as an open question, and we do not discuss further in this paper.
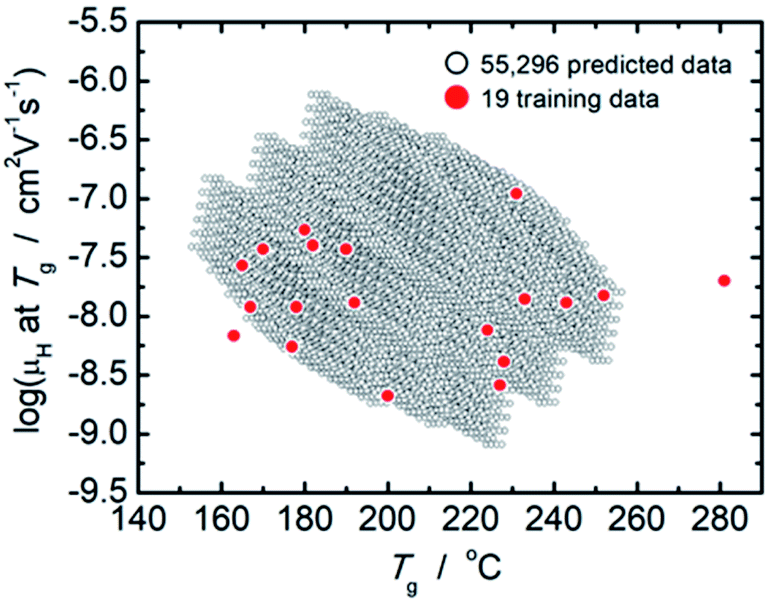 | ||
| Fig. 3 Plot of log(μH at Tg) as a function of Tg of 55296 predicted values (open black dots) together with the experimentally observed 19 values (closed red dots). | ||
Effects of respective component oxides on μH at Tg and Tg
As mentioned in the previous section, there is a clear relationship between log(μH at Tg) and Tg; therefore, the effect of each component oxide was studied in this regard. Fig. 4 shows the distribution of relationship between log(μH at Tg) and Tg depending on the concentration of respective component oxides. All data plotted in Fig. 4 are predicted values. In Fig. 4(a), 55296 predicted values are distinguished into three parts depending on the concentration of HO1/2. In Fig. 4(b), 18432 predicted values for the glasses with 30 mol% of HO1/2 are plotted and they are distinguished into three parts depending on the concentration of WO3. In Fig. 4(c), 6144 predicted values for the glasses with 30 mol% of HO1/2 and 0 mol% of WO3 are plotted and they are distinguished into four parts depending on the concentration of LaO3/2. In Fig. 4(d), (e), (f), (g) and (h), 1536 predicted values for the glasses with 30 mol% of HO1/2, 0 mol% of WO3 and 0 mol% of LaO3/2 are plotted and they are distinguished into four or six parts depending on the concentration of the oxide of interest (MgO, BaO, NbO5/2, BO3/2 and GeO2). The situation observed, when the component oxide of interest adds into the glass as alternative to PO5/2, is described as follows.
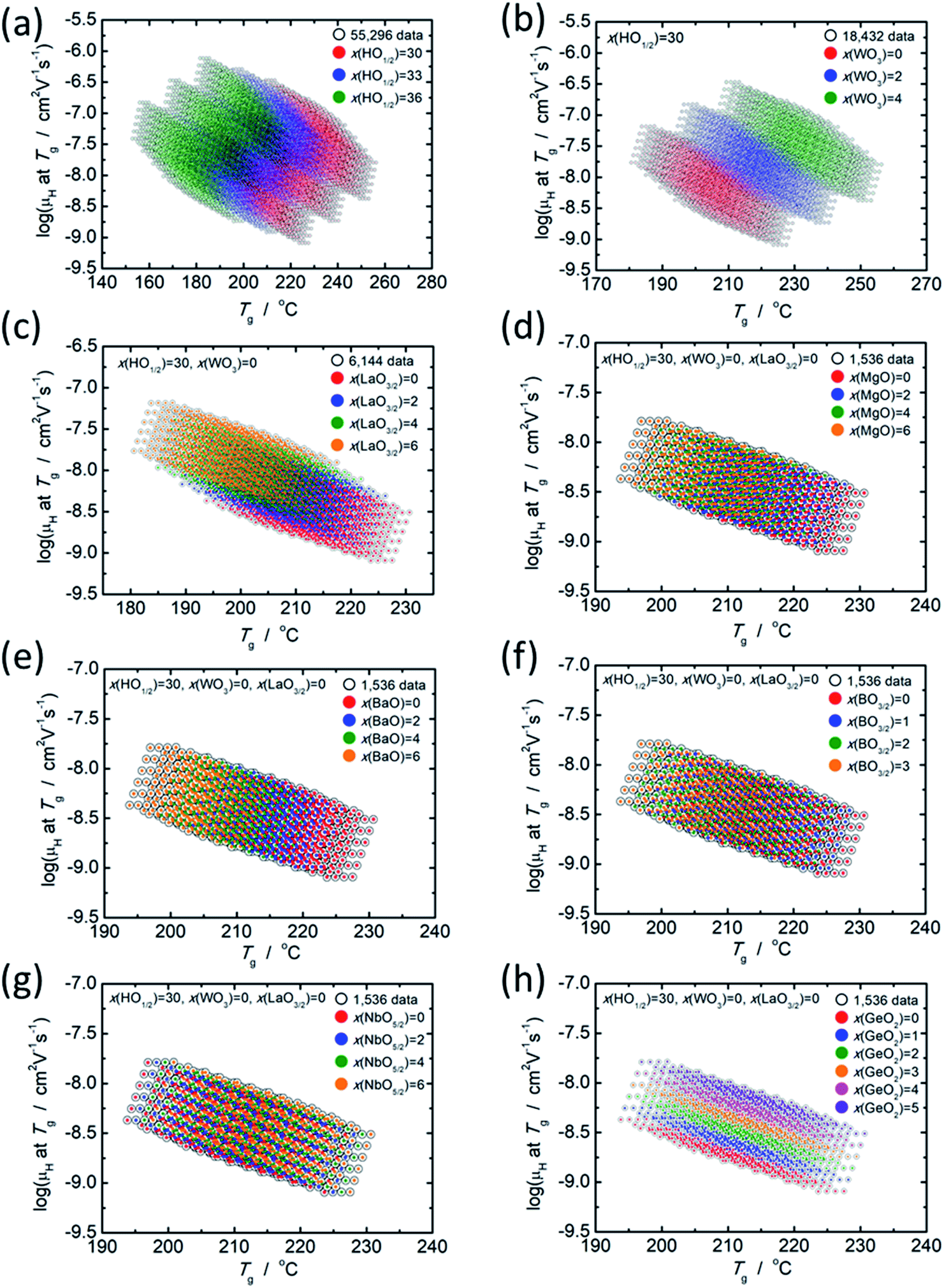 | ||
| Fig. 4 Distribution of the relationship between predicted values of log(μH at Tg) and Tg depending on the concentration of respective component oxides. (a) 55296 predicted values distinguished by the HO1/2 concentration (red dots = 30 mol% HO1/2, blue dots = 33 mol% HO1/2 and green dots = 36 mol% HO1/2), (b) 18432 predicted values for the glasses with 30 mol% of HO1/2 distinguished by the WO3 concentration (red dots = 0 mol% WO3, blue dots = 2 mol% WO3 and green dots = 4 mol% WO3), (c) 6144 predicted values for the glasses with 30 mol% of HO1/2 and 0 mol% of WO3 distinguished by the LaO3/2 concentration (red dots = 0 mol% LaO3/2, blue dots = 2 mol% LaO3/2, green dots = 4 mol% LaO3/2 and orange dots = 6 mol% LaO3/2). (d), (e), (f), (g) and (h) 1536 predicted values for the glasses with 30 mol% of HO1/2, 0 mol% of WO3 and 0 mol% of LaO3/2 respectively distinguished by the concentration of MgO, BaO, BO3/2, NbO5/2 and GeO2. | ||
With the increasing HO1/2 concentration (Fig. 4(a)), the Tg decreases by 5 °C per 1 mol% HO1/2 and log(μH at Tg) increases by 0.06 per 1 mol% of HO1/2. In contrast to the dependence of HO1/2 concentration, both Tg and log(μH at Tg) increases with the increasing WO3 concentration by 6.5 °C and 0.08 per 1 mol% of WO3, respectively (Fig. 4(b)). In the case of LaO3/2 shown in Fig. 4(c), Tg decreases with the increasing LaO3/2 concentration by 2.2 °C per 1 mol% of LaO3/2, and log(μH at Tg) increases with the increasing LaO3/2 concentration by 0.1 per 1 mol% of LaO3/2. In the cases for MgO, BaO and BO3/2 shown in Fig. 4(d), (e) and (f), respectively, the dependence are similar to the case of HO1/2 and LaO3/2; Tg decreases and log(μH at Tg) increases with the increasing concentration of the additional oxide. The variation in Tg and log(μH at Tg) are respectively −1.5 °C and 0.05 per 1 mol% of MgO, −2.4 °C and 0.05 per 1 mol% of BaO and −2.2 °C and 0.05 per 1 mol% of BO3/2. For NbO5/2, as clearly seen in Fig. 4(g), the relationship between log(μH at Tg) and Tg is little dependent on the NbO5/2 concentration, i.e., Tg increases by 0.7 °C per 1 mol% of NbO5/2 and log(μH at Tg) does not change regardless NbO5/2 concentration. In the case of GeO2 shown in Fig. 4(h), both Tg and log(μH at Tg) increases with the increasing GeO2 concentration similar to the case of WO3; however, increase in Tg, 0.6 °C per 1 mol% of GeO2, is much smaller than that of WO3 (6.5 °C per 1 mol% of WO3), while increase in log(μH at Tg), 0.12 per 1 mol% of GeO2, is slightly larger than that of WO3 (0.08 per 1 mol% of WO3). These situations are summarized in Table 4.
| Component oxide | Group 1 | Group 2 | Group 3 | |||||
|---|---|---|---|---|---|---|---|---|
| MgO | BaO | LaO3/2 | BO3/2 | WO3 | GeO2 | NbO5/2 | ||
| Variation per 1 mol% of oxide | log(μH at Tg) | 0.05 | 0.05 | 0.10 | 0.05 | 0.08 | 0.12 | 0.00 |
| Tg/°C | −1.5 | −2.4 | −2.2 | −2.2 | 6.5 | 0.6 | 0.7 | |
It is noticed that the component oxides are categorized into three groups in terms of the effect on the μH at Tg and Tg. The group 1 consists of HO1/2, MgO, BaO, LaO3/2 and BO3/2. They increase μH at Tg but decrease Tg, when their concentrations increase. The group 2 involves WO3 and GeO2 that increase both μH at Tg and Tg, when their concentrations increase. The group 3 consists of NbO5/2 only in the present study, and it increases Tg but does not changes μH at Tg, when its concentration increases. Such effects categorized into three groups could not be found in the experimentally observed data, i.e., 19 glass compositions that used as training data in this study. The information of the three groups is useful to obtain purpose-designed glasses.
The effect on Tg of respective group oxides is quite reasonable and explained according to the glass structural chemistry as following. The group 1 consists of the glass-modifiers except for BO3/2; therefore, the reduction of Tg with the increasing concentration of the group 1 oxide is reasonably understood as a result of breaking of the phosphate glass-network by introduction of the glass-modifier oxides. BO3/2 is a glass-former oxide, and it may exist in the glass as the trigonal planer BO3 in addition to the BO4 tetrahedron in the phosphate glasses assumed in the present study.27–29 When the trigonal planer BO3 is introduced into the glass as alternative to PO4 tetrahedra, the number of the bridging oxygens in the glass-network reduces as the concentration of the trigonal planer BO3 increases. Consequently, BO3/2 acts as almost glass-modifier, and its effect on Tg is similar to the other group 1 oxides that are glass-modifier oxides. The groups 2 and 3 consist of the oxides exhibiting high glass forming ability, i.e., GeO2 is a glass-former and WO3 and NbO5/2 are conditional glass-formers.30 When the groups 2 and 3 oxides are introduced into the glass as alternative to PO5/2, GeO2 tetrahedra and WO6 and NbO6 octahedra strengthen the phosphate glass-network, resulting in increasing Tg.
In contrast to the effect on Tg, the origin of the effect on μH at Tg is still an open question as already mentioned. However, the effect of the group 2 oxides, i.e., they increase μH at Tg with the increasing their concentration, may be explained phenomenologically as following. For the effect of WO3, we refer to the heteropoly acid of WO3 combined with PO5/2. It is well known that WO3 and PO5/2 form heteropoly acid, H3PW12O40·6H2O, and it exhibits strong acidity much stronger than H2SO4.31,32 The strong acidity, i.e., easy proton formation, is explained by dispersion of the negative charge over many atoms of the polyanion, PW12O403−, and the polarization of the outer W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.32 Of course, the molar ratio of WO3 over against PO5/2 is much smaller (4 mol% of WO3 is the highest, while 28 mol% of PO5/2 is the lowest) than that of PW12O403−; therefore, formation of PW12O403−-like polyanion should be excluded. However, the WO3 coexisting with PO5/2 may have an effect to enhance acidity of
O bond.32 Of course, the molar ratio of WO3 over against PO5/2 is much smaller (4 mol% of WO3 is the highest, while 28 mol% of PO5/2 is the lowest) than that of PW12O403−; therefore, formation of PW12O403−-like polyanion should be excluded. However, the WO3 coexisting with PO5/2 may have an effect to enhance acidity of ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P–O–H units. In this case, protons are easy to dissociate from P–O–H units; as a result, μH would be increased by the addition of WO3 into phosphate glasses.
P–O–H units. In this case, protons are easy to dissociate from P–O–H units; as a result, μH would be increased by the addition of WO3 into phosphate glasses.
In the case of GeO2, we refer to the silicophosphate gel that is prepared by reacting SiCl4 with anhydrous phosphoric acid (H3PO4).33 The silicophosphate gel that involves Si–O–P bondings exhibits evidently higher proton conductivity than H3PO4,33,34 although the increase in conductivity is not so large. Taking into account that the polymerization occurs in silicophosphate gel, the concentration of proton carriers in silicophosphate is smaller than that in phosphoric acid, indicating that the SiO2 addition enhances μH. Although the reason why SiO2 addition enhance proton conductivity has not been fully understood yet, the octahedrally coordinated SiO6 that appears in silicophosphate gel is pointed out as a key feature to explain the effect of SiO2 addition into phosphoric acid.33 While GeO2 exhibits similar feature to SiO2, i.e., both GeO2 and SiO2 are group 4 oxides and exhibit as glass-formers, preference of six-fold coordination of Ge4+ ion is higher than Si4+ ion. These imply that GeO2 would enhance μH, when it is added into the phosphoric acid. In this case, increase in μH by the addition of GeO2 to phosphate glass would be understood by the analogous to silicophosphate gel.
Conclusion
In summary, we developed a linear regression models for the compositional dependence of log(μH at Tg) and Tg for the proton conducting phosphate glass based on the approach of principal component analysis, and μH at Tg and Tg were predicted for 55296 of phosphate glasses involving 9 component oxide of HO1/2, MgO, BaO, LaO3/2, WO3, NbO5/2, BO3/2, GeO2 and PO5/2. The models themselves do not have any physical meaning of course, but they provide the following information about the effects of respective component oxides on μH at Tg and Tg: (i) the μH at Tg is determined first by concentrations of HO1/2 and PO5/2; μH at Tg increases with increasing HO1/2 concentration and decreasing PO5/2. (ii) There is a trend for log(μH at Tg) to increase linearly as Tg decreases. This is quite consistent with our estimation previously reported that the motion of protons determines the motion of glass framework in the proton conducting phosphate glasses. (iii) The component oxides are categorized into three groups according to the effects on μH at Tg and Tg. The group 1 oxides that behave as glass-modifiers increase μH at Tg and decrease Tg, and HO1/2, MgO, BaO and LaO3/2 and BO3/2 are involved in this group. The group 2 oxides increase both μH at Tg and Tg, and WO3 and GeO2 are involved in this group. The group 3 oxides increase Tg but do not vary μH at Tg. Only NbO5/2 falls into the group 3 among the oxides examined in this study. These information are very useful to obtain purpose-designed glasses; therefore, they will be applied to the future development of proton-conducting phosphate glasses. Especially, the effects of the additional glass-formers, such as GeO2 and WO3, are very important to design highly proton conducting phosphate glass at intermediate temperatures.
The enhance of μH at Tg by WO3 and GeO2 of group 2 oxide is phenomenologically understood by referring to the strong acidity of PW12O403− heteropoly acid and the enhancing μH of phosphoric acid by SiO2 addition, respectively. In contrast, the origin of the effect of groups 1 and 3 oxides on μH at Tg and the relationship between log(μH at Tg) and Tg still remain as open questions.
Author contributions
Takahisa Omata: conceptualization, methodology, formal analysis, software, writing – original draft and visualization, Issei Suzuki: validation, visualization and writing – review & editing, Aman Sharma: formal analysis and data curation, Tomohiro Ishiyama: writing – review & editing, Junji Nishii: conceptualization, funding acquisition and writing – review & editing, Toshiharu Yamashita: supervision and writing – review & editing, Hiroshi Kawazoe: supervision and writing – review & editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Prof. Junichi Kawamura (Tohoku University) for valuable comments. This work was supported in part by a Grant-in-Aid for Scientific Research (B) (Grant No. 20H02428). This work was partly performed under the Cooperative Research Program of the “Network Joint Research Center for Materials and Devices” (No. 20194020 and 20204012) and “Dynamic Alliance for Open Innovation Bridging Human, Environment, and Materials”.References
- A. Hayashi, A. Sakuda and M. Tatsumisago, Front. Energy Res., 2016, 4, 1–13 CrossRef.
- M. Nakayama, M. Hanaya, A. Hatate and M. Oguni, J. Non-Cryst. Solids, 1994, 172–174, 1252–1261 CrossRef CAS.
- Y. Abe, H. Hosono, O. Akita and L. L. Hench, J. Electrochem. Soc., 1994, 141, L64–L65 CrossRef CAS.
- Y. Daiko, J. Ceram. Soc. Jpn., 2013, 121, 539–543 CrossRef CAS.
- Y. Takamatsu, Y. Daiko, S. Kohara, K. Suzuya, A. Mineshige and T. Yazawa, Solid State Ionics, 2013, 245–246, 19–23 CrossRef CAS.
- H. Sumi, Y. Nakano, Y. Fujishiro and T. Kasuga, Solid State Sci., 2015, 45, 5–8 CrossRef CAS.
- Y. Huang, E. Christensen, Q. Shuai and Q. Li, Int. J. Hydrogen Energy, 2017, 42, 7235–7240 CrossRef CAS.
- H. Sumi, J. Ceram. Soc. Jpn., 2017, 125, 829–832 CrossRef CAS.
- S. H. Lee, S. B. Park and Y. Il Park, Solid State Ionics, 2020, 345, 115186 CrossRef CAS.
- T. Ishiyama, S. Suzuki, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, J. Electrochem. Soc., 2013, 160, E143–E147 CrossRef CAS.
- T. Ishiyama, S. Suzuki, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, Solid State Ionics, 2014, 262, 856–859 CrossRef CAS.
- T. Ishiyama, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, J. Mater. Chem. A, 2014, 2, 3940–3947 RSC.
- T. Yamaguchi, T. Ishiyama, K. Sakuragi, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, Solid State Ionics, 2015, 275, 22855–22861 CrossRef.
- T. Yamaguchi, T. Ishiyama, K. Sakuragi, J. Nishii, T. Yamashita, H. Kawazoe, N. Kuwata, J. Kawamura and T. Omata, Solid State Ionics, 2016, 288, 281–285 CrossRef CAS.
- T. Yamaguchi, T. Kataoka, S. Tsukuda, T. Ishiyama, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, Phys. Chem. Chem. Phys., 2017, 19, 29669–29675 RSC.
- T. Yamaguchi, Y. Saito, Y. Kuwahara, H. Yamashita, T. Ishiyama, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, J. Mater. Chem. A, 2017, 5, 12385–12392 RSC.
- K. Kawaguchi, T. Yamaguchi, T. Omata, T. Yamashita, H. Kawazoe and J. Nishii, Phys. Chem. Chem. Phys., 2015, 17, 22855–22861 RSC.
- T. Yamaguchi, S. Tsukuda, T. Ishiyama, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, J. Mater. Chem. A, 2018, 6, 23628–23637 RSC.
- T. Ishiyama, H. Kishimoto, K. Yamaji, T. Yamaguchi, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, Int. J. Hydrogen Energy, 2019, 44, 24977–24984 CrossRef CAS.
- T. Nakamura, S. Mizunuma, Y. Kimura, Y. Mikami, K. Yamauchi, T. Kuroha, N. Taniguchi, Y. Tsuji, Y. Okuyama and K. Amezawa, J. Mater. Chem. A, 2018, 6, 15771–15780 RSC.
- I. Suzuki, M. Tashiro, T. Yamaguchi, T. Ishiyama, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, Int. J. Hydrogen Energy, 2020, 45, 16690–16697 CrossRef CAS.
- T. Omata, T. Yamaguchi, S. Tsukuda, T. Ishiyama, J. Nishii, T. Yamashita and H. Kawazoe, Phys. Chem. Chem. Phys., 2019, 21, 10744–10749 RSC.
- T. Kentaro, J. Ceram. Assoc. Jpn., 1955, 63, 142–147 CrossRef.
- A. Makishima and J. D. Mackenzie, J. Non-Cryst. Solids, 1973, 12, 35–45 CrossRef CAS.
- B. Deng, J. Non-Cryst. Solids, 2020, 529, 119768 CrossRef CAS.
- S. Bishnoi, S. Singh, R. Ravinder, M. Bauchy, N. N. Gosvami, H. Kodamana and N. M. A. Krishnan, J. Non-Cryst. Solids, 2019, 524, 119643 CrossRef CAS.
- N. Mascaraque, A. Durán and F. Muñoz, J. Non-Cryst. Solids, 2011, 357, 3212–3220 CrossRef CAS.
- S. Le Roux, S. Martin, R. Christensen, Y. Ren and V. Petkov, J. Phys.: Condens. Matter, 2011, 23, 035403 CrossRef.
- R. K. Brow and D. R. Tallant, J. Non-Cryst. Solids, 1997, 222, 396–406 CrossRef CAS.
- N. Boubata, A. Roula and I. Moussaoui, Bull. Mater. Sci., 2013, 36, 457–460 CrossRef CAS.
- I. V. Kozhevnikov and K. I. Matveev, Appl. Catal., 1983, 5, 135–150 CrossRef CAS.
- M. Makoto, Mater. Chem. Phys., 2002, 75, 103–120 Search PubMed.
- Y. Ansari, T. G. Tucker and C. A. Angell, J. Power Sources, 2013, 237, 47–51 CrossRef CAS.
- Y. Ansari, T. G. Tucker, W. Huang, I. S. Klein, S. Y. Lee, J. L. Yarger and C. A. Angell, J. Power Sources, 2016, 303, 142–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10327f |
| This journal is © The Royal Society of Chemistry 2021 |