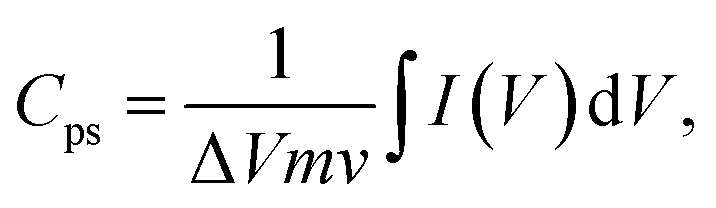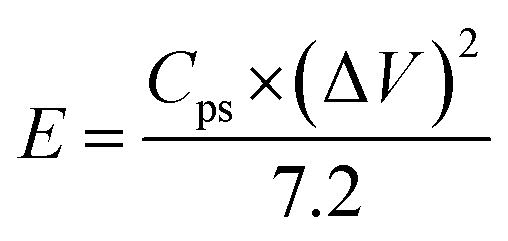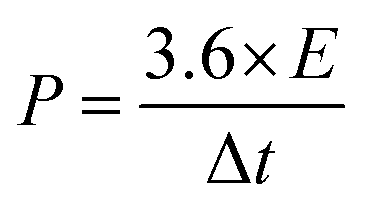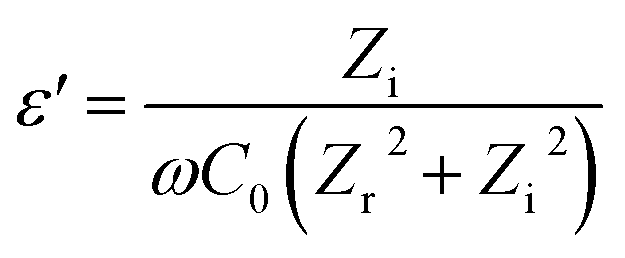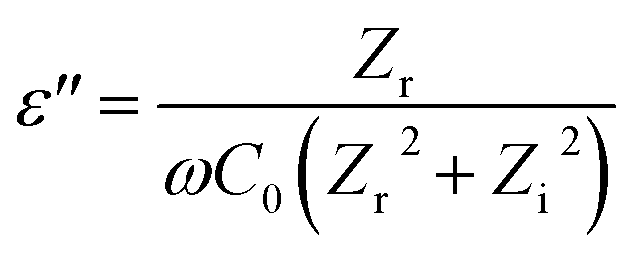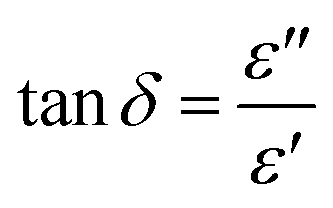DOI:
10.1039/D0RA10257A
(Paper)
RSC Adv., 2021,
11, 11233-11243
Physicochemical properties and performance of graphene oxide/polyacrylonitrile composite fibers as supercapacitor electrode materials
Received
4th December 2020
, Accepted 10th March 2021
First published on 17th March 2021
Abstract
Graphene oxide derived from palm kernel shells (rGOPKS) and polyacrylonitrile (PAN) were electrospun into composite fiber mats and evaluated as supercapacitor electrode materials. Their morphologies and crystalline properties were examined, and chemical interactions between rGOPKS and PAN were investigated. The diameters of individual fibers in the rGOPKS/PAN composite mats ranged from 1.351 to 1506 μm and increased with increasing rGOPKS content. A broad peak centered near 23° in the X-ray diffraction (XRD) pattern of rGOPKS corresponded to the (002) planes in graphitic carbon. Characteristic rGOPKS and PAN peaks were observed in the XRD patterns of all the composite fibers, and their Fourier-transform infrared (FTIR) spectra indicated hydrogen bond formation between rGOPKS and PAN. The composite fiber mats had smooth and homogeneous surfaces, and they exhibited excellent flexibility and durability. Their electrochemical performance as electrodes was assessed, and a maximum specific capacitance of 203 F g−1 was achieved. The cycling stability of this electrode was excellent, and it retained over 90% of its capacitance after 5000 cycles. The electrode had an energy density of 17 W h kg−1 at a power density of 3000 W kg−1. Dielectric results showed a nanofiber composite dielectric constant of 72.3 with minor leakage current (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) i.e., 0.33 at 51 Hz. These results indicate that the rGOPKS/PAN composite fibers have great promise as supercapacitor electrode materials.
δ) i.e., 0.33 at 51 Hz. These results indicate that the rGOPKS/PAN composite fibers have great promise as supercapacitor electrode materials.
1. Introduction
Interest in commercially available portable electronics, such as flexible screens, television screens, and cellphones, has grown among consumers over the last decade.1–3 The rapid development of portable electronics has increased the demand for flexible and efficient energy storage devices. Supercapacitors (SCs) are among the most promising energy storage devices. They are more efficient than conventional batteries due to their extraordinary storage capacity and effective release of electrical energy.4 The electrochemical characteristics of SCs include a rapid charging process, high power density, excellent cycling stability, and a long service life.5,6
Conventional SCs include two-electrode systems with planar structures. In this configuration, the positive and negative electrodes are separated by an electrical insulator that is impregnated with an electrolyte to allow ionic conduction.7,8 Currently available supercapacitors are not suitable for portable electronic devices, because they are heavy, thick, and large.7 A great deal of effort has gone into overcoming these limitations, including the development of nanofiber-based supercapacitors. Nanofibers are flexible, lightweight, and they can be easily incorporated into portable electronic devices.9–11 Nanofiber membranes are small and have high surface-to-volume ratios. They provide efficient ion diffusion pathways, which enable rapid charging and extend service life.12,13
The selection of electroactive materials for supercapacitor devices has an important impact on their performance. Activated carbon,14,15 graphene,16,17 carbon nanocubes,9,18 and carbon nanofibers are often used as electrode materials.19 Graphene has many advantageous properties, which include large surface area, high thermal conductivity, good electrochemical stability, and excellent charge carrier mobility. The efficient separation of electrons and ionic charge carriers at the interfaces between graphene-based electrodes and electrolytes is advantageous for energy storage and release, and graphene can improve the physicochemical properties of SC electrodes.7,20,21 The specific capacitances of graphene-based SC electrodes range from 270 to 310 F g−1.22,23 Electrodes with specific capacitances ranging from 214 to 374 F g−1 have been successfully fabricated using graphene derived from biomass, including coconut shells, wood, rice husks, walnut shells, banana peels, bagasse, tea leaves, bamboo, palm oil waste, and palm kernel shells.19,24–27 However, few nanofiber-based rGOPKS electrodes have been developed.
Palm oil is obtained from the fruit of African oil palm (Elaeis guineensis). African oil palm is widely cultivated in Malaysia, Thailand, and Indonesia, and oil palm plantations cover a total of 15 million hectares.28 Asian countries are the most important producers and exporters of palm oil in the world. Over ∼21.94 tons of fresh fruit bunches per hectare are produced each year, 70% of which is discarded as waste.25,29 Asian factories will generate approximately 350 million tons of biomass waste in 2020, 8% of which is expected to be palm kernel shell (PKS) waste.29 PKS waste is the solid product obtained after palm oil extraction. Given the large amount of PKS waste generated annually, using PKS waste to fabricate value-added products is highly desirable.
Electrically conductive polymers have been intensively studied as binding materials, and polyacrylonitrile (PAN) is frequently used as an electrode precursor.30 The electrochemical performance of PAN can be enhanced by loading it with graphene derived from PKS waste and incorporating it into composite nanofibers. The synergistic effects of carbon-based materials and conductive polymers in composite nanofibers have recently been shown to enhance the capacitance and stability of supercapacitors.9,31 Therefore, rGOPKS/PAN composite nanofibers are promising materials for flexible, high-performance SC electrodes. In this study, rGOPKS/PAN composite fiber mats with varying rGOPKS contents were fabricated. Their physicochemical properties, including morphology, fiber diameter, crystallinity, and chemical interactions, were examined. The electrochemical performance of the flexible fiber mats as SC electrodes was then investigated.
2. Experimental
2.1 Materials
Polyacrylonitrile with a molecular weight of 150000 g mol−1 was obtained from Sigma-Aldrich (Singapore). Dimethylformamide (DMF) was obtained from Merck (USA). PKS waste was donated by PT Sumber Waras Banyuasin in South Sumatra (Indonesia). Distilled water, HCl (6 N), urea, iron(III) chloride (FeCl3), and N2 gas were obtained from Bratachem (Indonesia). All reagents were of analytical grade and used as received without further purification.
2.2 Graphitization and activation of the PKS waste
The PKS waste was dried under sunlight for approximately six days and ground into a 180–200 mesh powder in a bulk mill. Carbonization and activation were achieved in two steps32 using FeCl3 as a pyrolysis catalyst, ZnCl2 as an activating agent, and urea as a nitrogen source. A mixture of the raw PKS powder, ZnCl2, and urea in 1 M FeCl3 was stirred at 80 °C on a hot plate for 2 h. The solid material was separated by freeze drying in an oven for 1 h. The dried material was placed in a tube furnace, and the furnace was charged with nitrogen. The furnace was heated to 250 °C over a period of 45 min, and the material was heated at this temperature for 1 h. The temperature was then increased to 900 °C, and the material was graphitized for 90 minutes under flowing nitrogen. Complete pyrolysis yielded graphene with Fe impurities. The impurities were removed by immersing the crude product in 2 M HCl and stirring the mixture for 2 h. The reduced graphene oxide (rGO) was rinsed with distilled water, dried to a constant mass at 105 °C, and designated rGOPKS.
2.3 Electrospinning process
rGOPKS/PAN composite nanofiber mats were prepared by first adding the rGOPKS powder to 10% (w/w) solutions of PAN in DMF. The precursor mixtures were designated CNF1, CNF2, and CNF3, which contained 0.06 g, 0.16 g, and 0.26 g rGOPKS, respectively. A solution without rGOPKS was designated CNF0. The rGOPKS/PAN precursor mixtures were then stirred for approximately 20 h at 80 °C to obtain homogeneous black suspensions.
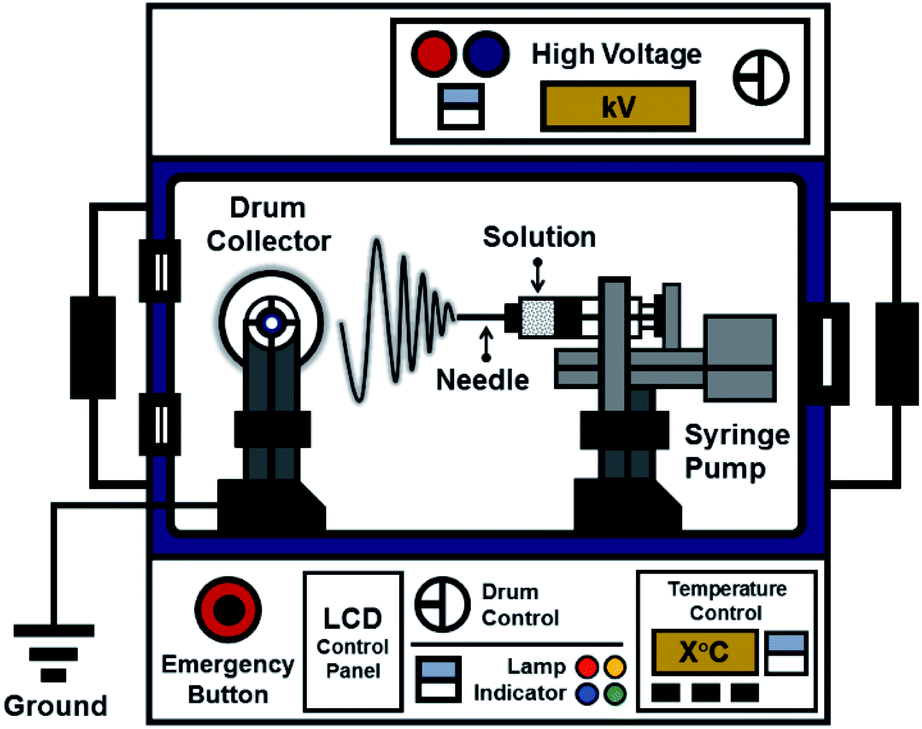
The ES106 electrospinning apparatus (Nanolab, Malaysia) used to fabricate the nanofiber mats is shown in Fig. 1. It consisted of a high-voltage power supply, a drum collector, and a syringe pump equipped with a 10 mL syringe. The precursor solutions were ejected through the syringe needle at the positive pole, and the fibers were collected on the drum at the negative pole. The tip of the 0.45 mm (ϕ (inner diameter)) needle was placed 8 cm from the collector. Electrospinning was performed at 10 kV with a syringe pump flow rate of 56 μL h−1.
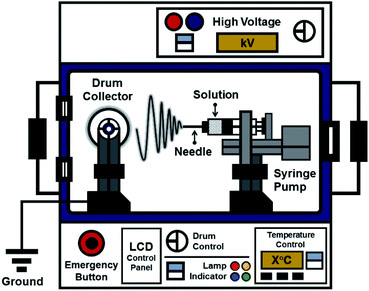 |
| | Fig. 1 Schematic diagram of the electrospinning apparatus used in the study. | |
2.4 Scanning electron microscopy (SEM) and energy dispersive X-ray (EDX)
SEM and EDX analysis were performed using a JSM-6510 SEM (JEOL, Japan) to examine the morphologies of the composite nanofiber mats. Each sample was coated with a conductive layer prior to SEM imaging, and images of the fibers at 10000× magnification were collected at 10 kV. The size distributions of the fibers were determined using the Origin version 9 software package. Transmission Electron Microscope (TEM HT7700, USA).
2.5 Fourier-transform infrared spectroscopy (FTIR)
FTIR spectroscopy was performed using the Alpha II spectrometer equipped with a A220/D-01 Platinum ATR sampling module (Bruker, USA) to investigate potential chemical interactions between rGOPKS and PAN in the composite nanofibers. The FTIR spectra of the nanofiber mats encompassed the range from 500 to 4000 cm−1.
2.6 X-ray diffractometer (XRD) and X-ray photoelectron spectroscopy (XPS)
The XRD patterns of rGOPKS, PAN, and the composite nanofiber mats were obtained using a MiniFlex 600 X-ray diffractometer (Rigaku, Japan). The samples were placed in a standard Cu tube with an operating voltage of 40 kV and irradiated with CuKα radiation (λ = 1.5405 Å) at 35 mA. XRD patterns were recorded from (2θ) 5° to 70°. The XPS data were obtained using an Kratos AXIS Ultra DLD; USA.
2.7 Tensile tests
The tensile strength and Young's modulus of each mat were measured using a Favigraph tensile tester (Textechno, Germany). Nanofiber mat samples of equal thickness were cut into rectangles with dimensions of approximately 3 × 20 mm2. A 1 N cell load was applied, and each sample was stretched at a rate of 20 mm min−1. Measurements were performed over 10 mm lengths as described in a previous report.33 The maximum tensile strength (MTS) of each sample was determined from the highest point in its stress curve, and the Young's modulus was obtained by locating the highest point in its stress–strain curve.
2.8 Electrochemical characteristics of the rGOPKS/PAN composite fiber mats
The electrochemical characteristics of the composite fiber mats were evaluated by performing cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) analysis, and electrochemical impedance spectroscopy (EIS). All experiments were conducted at room temperature using an Autolab potentiostat/galvanostat (Metrohm, Switzerland). CV curves with a −0.2 to 0.4 V voltage window were obtained at scan rates of 10 mV s−1, 20 mV s−1, 30 mV s−1, 40 mV s−1, 50 mV s−1, and 100 mV s−1. GCD measurements were performed at 0 and 1.0 V and current densities ranging from 0.5 to 5 mA cm2. EIS was conducted in the frequency range from 100 kHz to 10 mHz at an applied voltage of 5 mV relative to the open circuit voltage. The rGOPKS/PAN nanofibers served as working electrodes in a three-electrode system in 4 M KOH electrolyte at room temperature with an Ag/AgCl reference electrode and a Pt counter electrode. The specific capacitance (Cps) values of the PAN/rGOPKS composite nanofibers were calculated using eqn (1).34| |
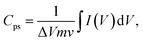 | (1) |
where Cps has units of F g−1, ΔV is the potential window, m is the mass of the supercapacitor electrode (g), v is the scan rate (mV s−1), and the integrated value is the area under the CV curve. The energy density (E) and power density (P) of each electrode were calculated using eqn (2) and (3), respectively.| |
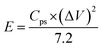 | (2) |
| |
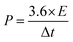 | (3) |
2.9 Dielectric properties of the rGOPKS/PAN composite fiber mats
Dielectric properties are measured using an impedance analyzer (HIOKI 3532-50 LCR HiTESTER-Japan) with a nanofiber membrane size of approximately 8 × 8 × 4 mm placed between two parallel electrodes. The measurement parameters are carried out at room temperature, voltage amplitude 1.4 V and over frequency range 50–500 kHz. The dielectric properties of the nanofiber are calculated according to the eqn (4)–(7):35,36| |
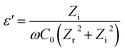 | (4) |
| |
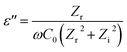 | (5) |
where ε′ is dielectric constant, ε′′ is dielectric loss, Zi is imaginary impedance (Ω), Zr is real impedance (Ω), C0 is the geometrical capacitance and ω is angular frequency (s−1). Furthermore, the energy dissipation and AC conductivity of nanofiber PAN/rGOPKS are calculated according to eqn (6) and (7), respectively:| |
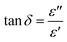 | (6) |
| | |
σAC = ε0ε′ωtanδ
| (7) |
where tanδ is energy dissipation, σAC is AC conductivity (S m−1), and ε0 is absolute dielectric permittivity (8.85 × 10–12 F m−1).
3. Result and discussion
3.1 rGOPKS morphology
SEM and TEM images of rGOPKS are shown in Fig. 2. The rGOPKS nanosheets formed at temperatures between 700 °C and 900 °C. As Fe3+ in the FeCl3 catalyst was reduced to Fe, carbon atoms diffused from the FeCl3 to form rGOPKS. Graphene derivatives were also generated as the pyrolysis temperature increased from 700 °C to 900 °C.37 Widiatmoko et al. (2019) reported that activated carbon from palm fruit transformed into graphene as the pyrolysis temperature increased from 700 °C to 800 °C.32 The corrugated morphology of rGOPKS was due to bending of the 2D sheets, which generated more thermodynamically stable rumpled structures.38
 |
| | Fig. 2 SEM (a and b) and TEM (c and d) of rGOPKS. | |
3.2 rGOPKS composition
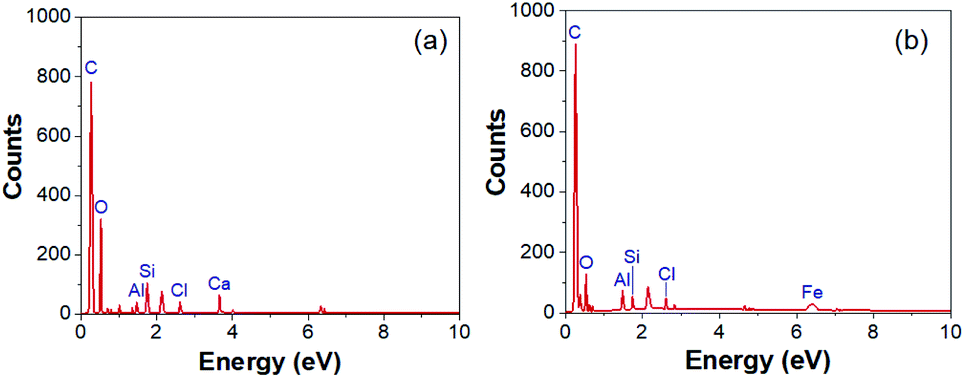
The EDX spectra of rGOPKS and a CNF3 are shown in Fig. 3. As expected, carbon and oxygen peaks in the rGOPKS spectrum indicated the presence of graphene oxide (GO). Other elements are also found as impurities on the surface of sample. Kharkwal et al. (2016) observed peaks in the EDX spectrum of a biocompatible nanofilm that were attributed to impurities in GO.39 The oxygen and carbon content in rGOPKS were 19.69% and 78.06%, so the rGOPKS had a C/O ratio of 3.96. This value did not differ significantly from the C/O ratios of pure GO.40 The proportion of carbon increased as the rGOPKS was incorporated into CNFs, and the oxygen content decreased to 6.47%. This was because the high voltage applied during the electrospinning process caused the solvent and oxygen in the rGOPKS to evaporate.
 |
| | Fig. 3 Energy dispersive X-ray (EDX) spectra of (a) rGOPKS and (b) CNF. | |
3.3 Electrospun PAN/rGOPKS nanofibers
Photographs of electrospun PAN and a rGOPKS/PAN composite nanofiber mat are shown in Fig. 4a. The PAN/rGOPKS composite nanofiber mats were smooth, homogeneous, and flexible. The PAN fiber sample (CNF0) was white. The PAN/rGOPKS composite fibers were black, which indicated that PAN and the rGOPKS mixed completely. Three forces acted on the mixtures during the electrospinning process, which contributed to the smoothness and homogeneity of the flexible rGOPKS/PAN nanofiber composites. A high voltage was applied to the tip of the needle to charge the polymer, and coulombic force (FC) drew it toward the grounded collector. The polymer formed droplets at the tip of the needle due to the force of surface tension (Fγ) at the air-polymer interface. The formation of polymer droplets for spinning was due to the loading force (Fd) of the syringe pump.41,42 Flexible PAN and PAN/rGOPKS composite fibers with an 8 cm radius of curvature is shown in Fig. 4b.
 |
| | Fig. 4 Photographs of (a) PAN and PAN/rGOPKS mats and (b) electrodes. | |
3.4 SEM analysis
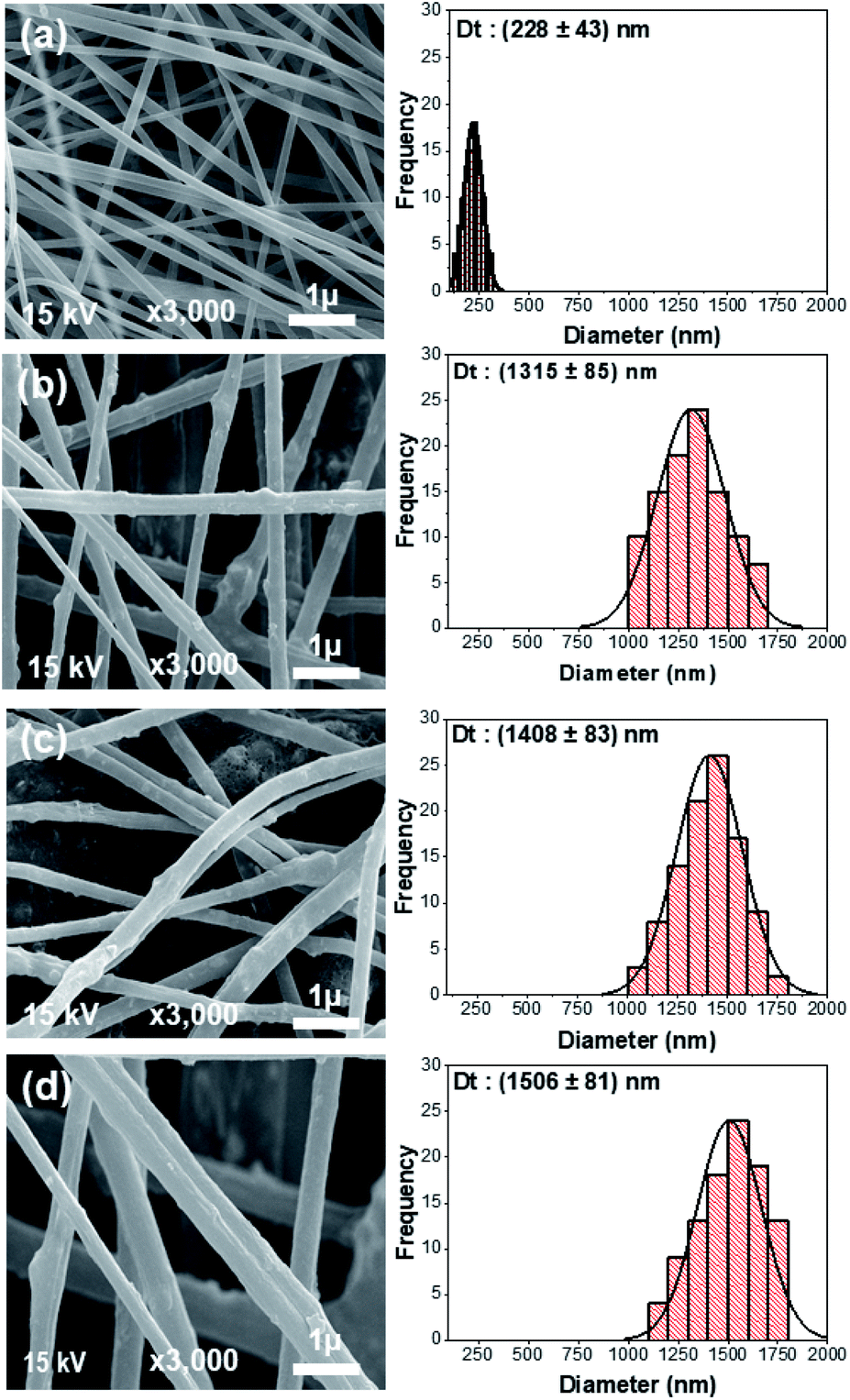
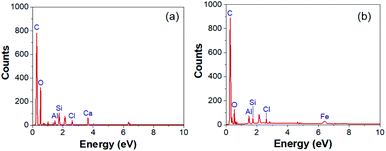
The morphologies of the electrospun PAN sample (CNF0) and the CNF1, CNF2, and CNF3 rGOPKS/PAN composite nanofiber mats can be seen in the SEM images in Fig. 5. Electrospinning usually produces fine fibers, fibers with a bead-like morphology, or simple beads.33 Differences between the rGOPKS concentrations in the precursor mixtures did not affect fiber morphology. The bead-free PAN and rGOPKS/PAN fibers differed only in terms of diameter. This was consistent with the results of a previous study on fine PVA/graphene nanofibers, in which graphene in a PVA matrix did not affect nanofiber morphology.21
 |
| | Fig. 5 SEM images of the nanofiber composite mats and their fiber size distributions. (a) CNF0, (b) CNF1, (c) CNF2, and (d) CNF3. | |
The size distributions of the fibers in the CNF0, CNF1, CNF2, and CNF3 samples are plotted next to the corresponding SEM images in Fig. 5a–d. Nanofibers in the CNF0 sample ranged from 100 to 300 nm in diameter. They had an average diameter (D) of 228 nm with a standard deviation (SD) of 43 nm. Nanofibers in the CNF1 sample ranged from 1000 to 2000 nm in diameter with an average diameter of 1351 ± 169 nm. The diameters of nanofibers in the CNF2 and CNF3 samples had very similar distributions to that of the CNF1 nanofibers. The average diameters of the fibers in the CNF2 and CNF3 samples were 1408 ± 165 nm and 1506 nm ± 161 nm, respectively. Mixing rGOPKS into the PAN polymer solution clearly increased the fiber diameter, and the mixture containing the largest quantity of rGOPKS (CNF3) yielded the widest nanofibers. That was because the rGOPKS powder increased the viscosity of the PAN precursor mixture. It is more difficult to stretch the jet of a highly viscous precursor solution in an electric field, so the resulting nanofibers generally have larger diameters.33,43,44
The homogeneity of the PAN/rGOPKS fibers was evaluated by calculating the coefficient of variation (CF) of the fibers in each PAN/rGOPKS sample. The CF is defined as the ratio of the SD to the average fiber diameter, and homogeneous fibers have CFs below 0.3.33,43,45 The results indicated that the composite fibers were homogeneous. High voltage did not destabilize the jet or generate many side jets, so the fibers had similar diameters. While previous studies have shown that the distribution of nanofibers is not homogeneous, which is due to the influence of the high voltage used in the electrospinning process.44
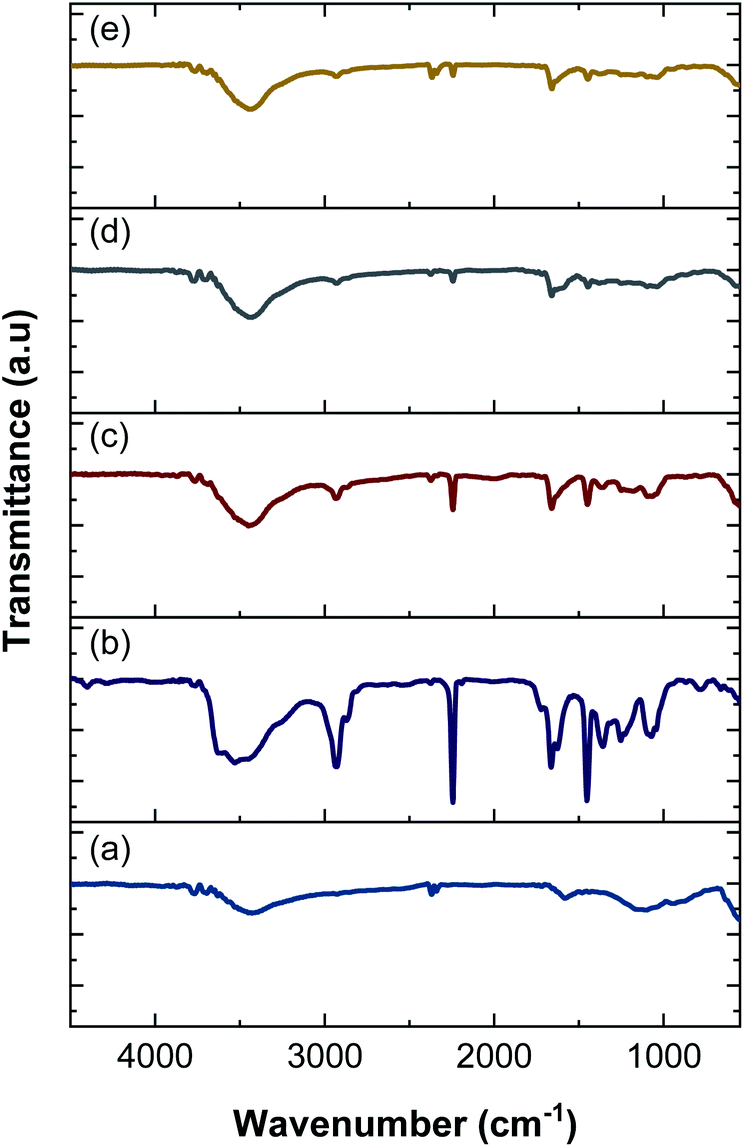
3.5 FTIR analysis
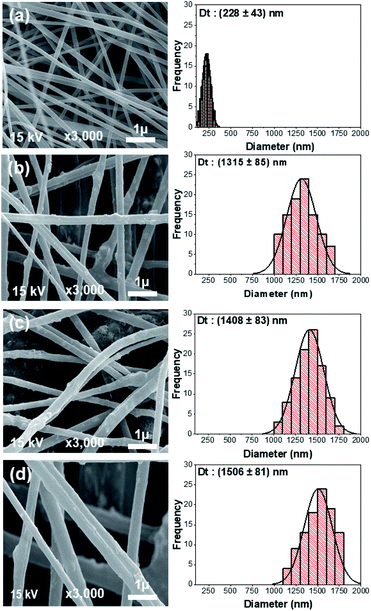
FTIR spectroscopy was performed to identify functional groups in the nanofiber electrodes and rGOPKS. The spectra are shown in Fig. 6. The spectrum of the PAN nanofiber mat (CNF0) contained peaks at 3527 cm−1, 2928 cm−1, 2243 cm−1, 1665 cm−1, 1451 cm−1, and 1070 cm−1. The peak at 3527 cm−1 was ascribed to –OH stretching vibrations,46 and the peak at 2928 cm−1 was assigned to –CH2 stretching vibrations. The peaks at 2243 cm−1 and 1451 cm−1 were attributed to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (nitrile) stretching and –CH vibrations in PAN, respectively.47 The peak at 1665 cm−1 was assigned to the stretching vibrations of C
N (nitrile) stretching and –CH vibrations in PAN, respectively.47 The peak at 1665 cm−1 was assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds in amidoxime groups,46–48 and the peak at 1070 cm−1 was attributed to C–O–C stretching in ether groups.46 Peaks were observed at 3428 cm−1, 2366 cm−1, 1601 cm−1, and 1198 cm−1 in the FTIR spectrum of the rGOPKS powder. The broad peak at 3428 cm−1 was ascribed to O–H stretching vibrations. The peak at 1662 cm−1 was attributed to CC stretching in aromatic groups, while the peak at 1198 cm−1 was due to carboxyl C–O stretching vibrations.49 The hydroxyl and carboxyl peaks in the rGOPKS FTIR spectrum indicated that oxidation occurred. The introduction of oxygen-bearing functional groups during rGOPKS formation demonstrated that surface modification with acidic and oxidative chemicals was successful.49
N bonds in amidoxime groups,46–48 and the peak at 1070 cm−1 was attributed to C–O–C stretching in ether groups.46 Peaks were observed at 3428 cm−1, 2366 cm−1, 1601 cm−1, and 1198 cm−1 in the FTIR spectrum of the rGOPKS powder. The broad peak at 3428 cm−1 was ascribed to O–H stretching vibrations. The peak at 1662 cm−1 was attributed to CC stretching in aromatic groups, while the peak at 1198 cm−1 was due to carboxyl C–O stretching vibrations.49 The hydroxyl and carboxyl peaks in the rGOPKS FTIR spectrum indicated that oxidation occurred. The introduction of oxygen-bearing functional groups during rGOPKS formation demonstrated that surface modification with acidic and oxidative chemicals was successful.49
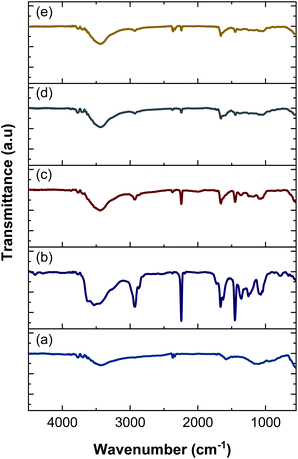 |
| | Fig. 6 FTIR spectra of rGOPKS and the nanofiber mats. (a) rGOPKS, (b) CNF0, (c) CNF1, (d) CNF2, and (e) CNF3. | |
The addition of rGOPKS to the PAN nanofibers caused changes in the intensity and positions of some peaks in the spectra of the CNF1, CNF2, and CNF3 samples. A high rGOPKS content resulted in sharper hydroxyl peaks in the region between 3700 and 3000 cm−1 and less intense amidoxime peaks from PAN in the region from 1000 to 2000 cm−1. A high RGOPKS content also caused the hydroxyl and amidoxime peaks of PAN to shift to lower wavenumbers. The PAN hydroxyl peak at 3527 cm−1 was shifted to 3439 cm−1 in the spectrum of the CNF1 nanofibers. Shifts to 3435 cm−1 and 3433 cm−1 were observed in the spectra of the CNF2 and CNF3 nanofibers, respectively. The amidoxime peak shifted to wavenumbers below 1665 cm−1 in the spectra of CNF1 (1662 cm−1), CNF2 (1660 cm−1), and CNF3 (1657 cm−1). Changes in the positions and intensities of peaks in the CNF1, CNF2, and CNF3 spectra were attributed to the interactions of the hydrogen bond-mediated intermolecular bonds of the hydroxyl in rGOPKS and the nitrile bonds in PAN.
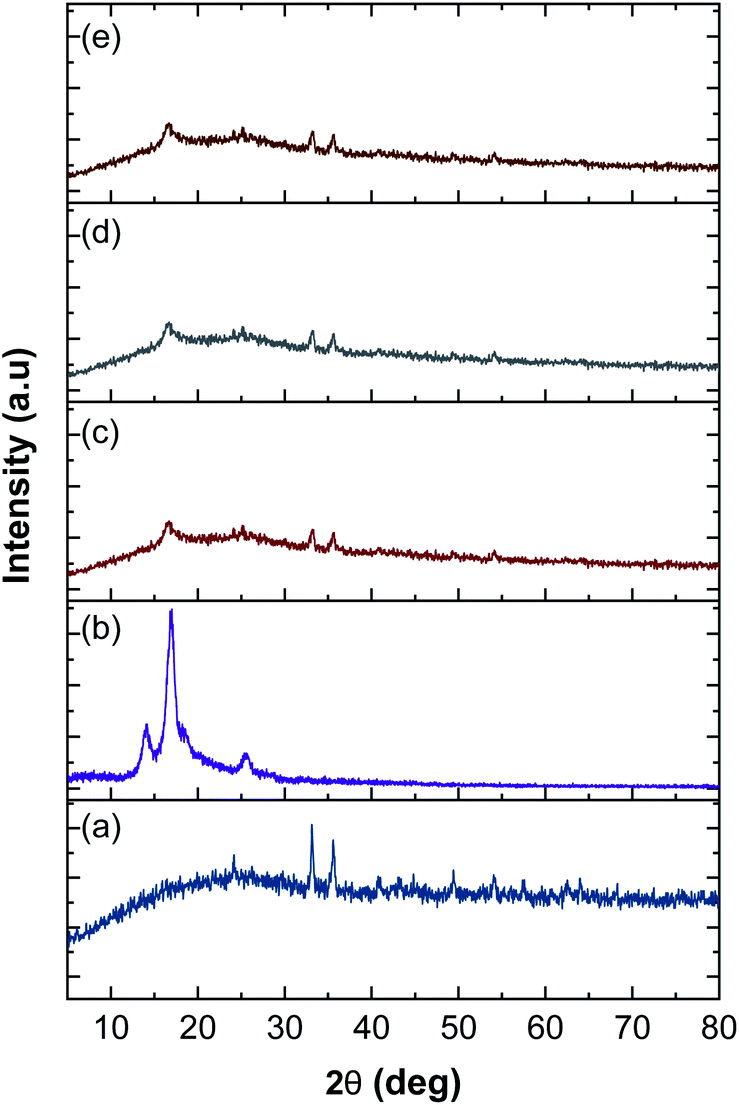
3.6 XRD analysis
The XRD patterns of rGOPKS, CNF0, and the CNF1, CNF2, and CNF3 composite nanofiber mats are shown in Fig. 7. The pattern of PKS in rGOPKS contained two sharp diffraction peaks at 33.30° and 36.2°. The broad peak centered near 23° corresponded to the (002) planes in amorphous graphitic carbon, and it along with the peak at 49.32° were characteristic of GO in biomass.29,34,49–51
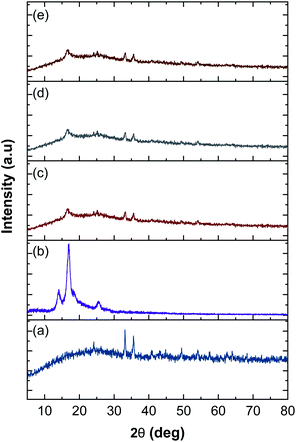 |
| | Fig. 7 X-ray diffraction patterns of rGOPKS and the nanofiber mats. (a) rGOPKS, (b) CNF0, (c) CNF1, (d) CNF2, and (e) CNF3. | |
The diffraction pattern of the PAN nanofiber mat (CNF0) contained a sharp peak at 17°, which corresponded to the (010) planes in crystalline PAN.47 A sharp peak appears near 28° in the XRD pattern of pure PAN.52 The peak centered near 28° in the PAN nanofiber pattern was weak, which may have been due to a decrease in crystallinity during the electrospinning process. The high applied voltage caused the PAN polymer solution to travel rapidly from the tip of the needle to the collector. The polymer was elongated and solidified into fibers as the solvent evaporated,42,44 which rapidly changed the three-dimensional arrangement of the PAN molecules. As a result, the PAN nanofibers did not have a highly crystalline structure.
The patterns of the CNF1, CNF2, and CNF3 composite nanofiber mats were similar to each other and contained broad halo peaks that were characteristic of rGOPKS. Peaks attributed to PKS in rGOPKS appeared at 23°, 33.5°, and 36.32°. The patterns of all the composite nanofiber mats contained the characteristic PAN peak at 17°, which was consistent with the formation of covalent bonds between rGOPKS and PAN molecules.
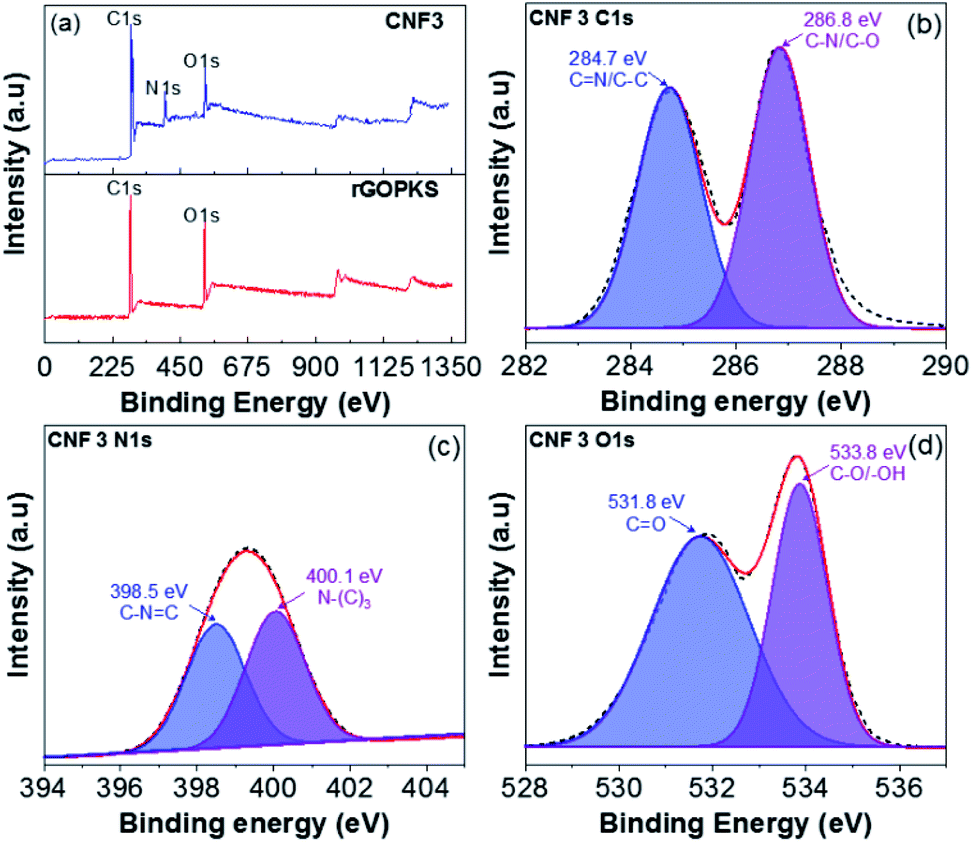
3.7 XPS analysis
The XPS of rGOPKS and CNF3 composite nanofiber mats are shown in Fig. 8a, some of the samples contain O, C, and N elements. The rGOPKS spectrum shows two clearly visible peaks centered on C1s (285.3 eV) and O1s (531.2 eV), these peaks correspond with graphene peaks reported by Yu et al. (2016).53 Meanwhile, the CNF3 spectrum offers three peaks at C1s (286.6 eV), N1s (400.6 eV), and O1s (532.9 eV). After rGOPKS and PAN were composited to form CNF3, there was a decrease in the peak intensity of C1s and O1s. It was due to the presence of surface conjunctions in CNF3 through π-stacking which is in turn due to the increased interaction between PAN and rGOPKS, this effect can facilitate the electron transfer for electrochemical properties.54
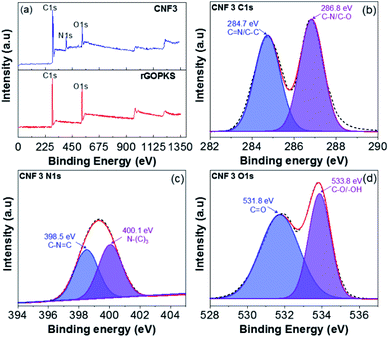 |
| | Fig. 8 XPS spectra of rGOPKS and CNF3 (a) survey spectrum, (b) C1s, (c) N1s and (d) O1s. | |
Furthermore, the C1s spectrum (CNF3-Fig. 8b) has two peak binding energies of 284.7 eV and 286.8 eV originating from the CN and C–N/C–O functional groups, and while the N1s spectrum (CNF3-Fig. 8c) has two peaks derived from the primary pyridinic N (N-6) functional group observed at 398.5 eV along with the pyrrolic/pyridone N (N-5) and quaternary N (N-Q) peaks at 400.1 eV.53,55,56 Moreover, the O1s spectrum (CNF3-Fig. 8d) has two peaks concentrated in the binding energy 531.8 eV associated with the carbonyl functional group (CO) and 533.8 eV associated with the hydroxyl functional group (C–O/–OH).57,58 The presence of N-Q can enlarge the electron transfer so that it has an impact on increasing the specific capacity performance. Meanwhile, the surface functional groups O and N can enhance conductivity of the material thereby increasing the performance of capacitive material.59,60
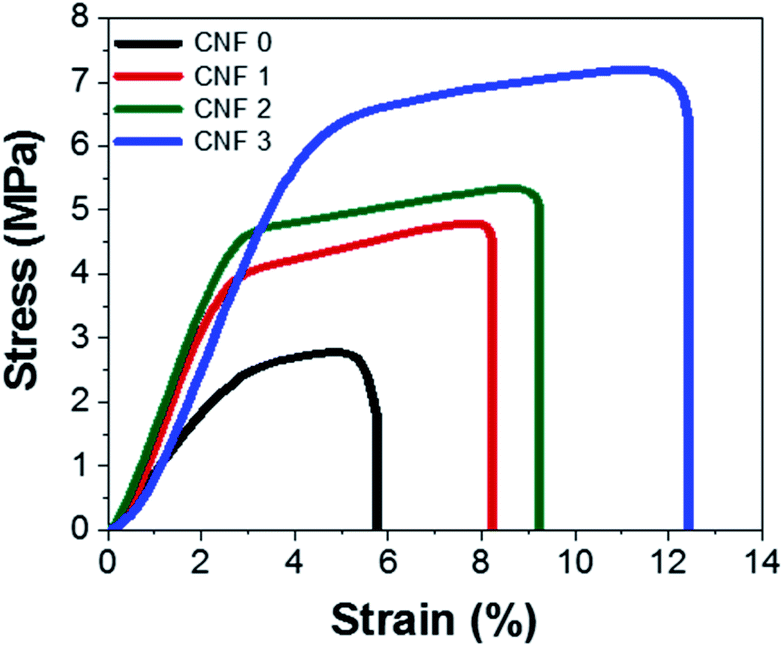
3.8 Mechanical test
Mechanical tests were performed to determine how flexible the nanofiber composite mats were. The stress–strain curves of the mats are shown in Fig. 9. Their shapes resembled that of a polymer stress–strain curve61 and indicated that the composite nanofiber mats underwent straightening, elastic, and plastic deformation. The SEM images and nanofiber size distributions showed that PAN nanofibers prepared with rGOPKS had larger diameters. The addition of rGOPKS also increased the Young's modulus, because it formed many bonds with the large number of parallel chains.33,62 The measured strains of the CNF0, CNF1, CNF2, and CNF3 rGOPKS/PAN nanofiber mats at breakage were 5%, 8%, 9%, and 11%, respectively. All of the strain values exceeded that of typical CNFs (∼2.5%).9 The flexible tensile strengths of the nanofiber composite mats ranged from 2.8 MPa to 7.2 MPa, which exceeded that of typical CNFs (∼0.48 MPa). These results indicated that the flexible nanofiber mat electrodes could serve as SC electrodes.
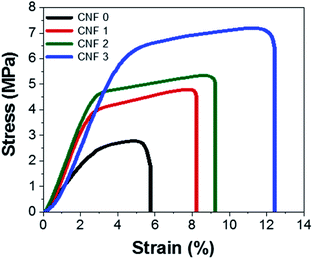 |
| | Fig. 9 Stress–strain curves of the fibrous mats. | |
3.9 Electrochemical properties of the rGOPKS/PAN composite nanofiber mats as supercapacitor electrodes
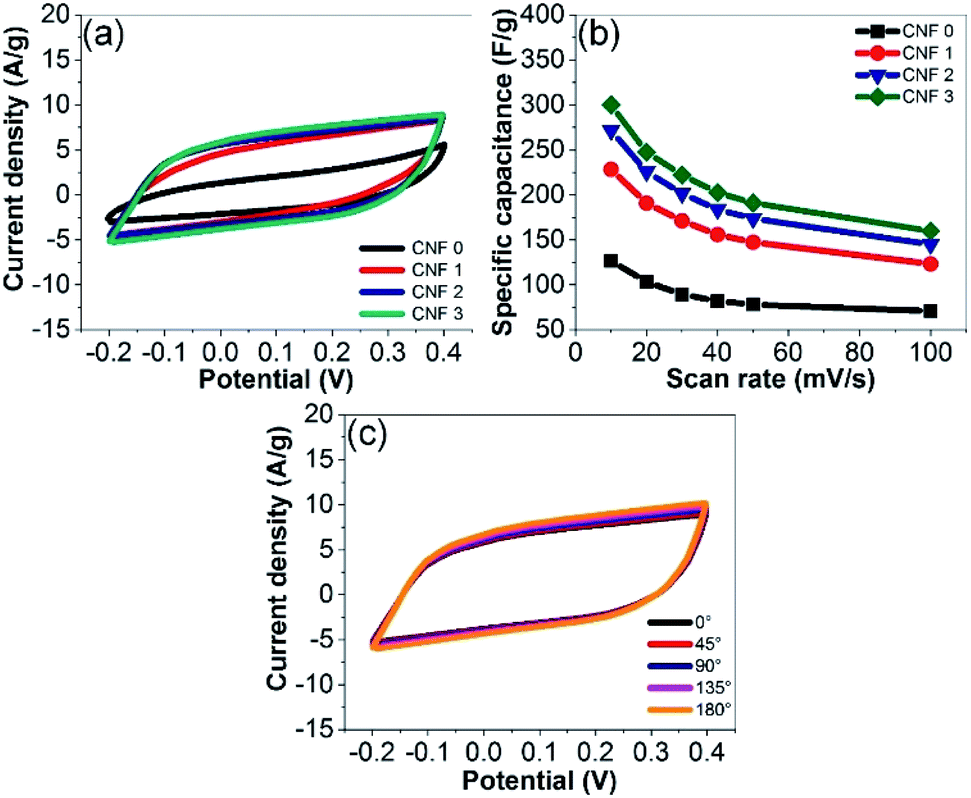
The electrochemical performance of the rGOPKS/PAN composite nanofiber mats as supercapacitor electrodes was investigated by performing CV and GCD measurements. The CV curves of the samples in a 4 M KOH aqueous electrolyte solution recorded at a scan rate of 40 mV s−1 are shown in Fig. 10a. The CV curves of the CNF1, CNF2, and CNF3 rGOPKS/PAN electrodes were quasi-rectangular and contained no peaks in the potential range of −0.2 to 0.4 V. The electrodes exhibited the ideal double-layer capacitance of carbon,12 which indicated that specific capacitance (Cs) could not be attributed to reduction or oxidation on the surfaces of the electrodes. Rather, the electrostatic discharge release response from the pure cell.63,64 The Cs values of the rGOPKS/PAN composite nanofiber electrodes were calculated using eqn (1).34 The Cs of the CNF3 electrode (203 F g−1) was higher than those of the CNF2 (184 F g−1) and CNF1 (156 F g−1) electrodes at a 40 mV s−1 scan rate. All of the rGOPKS/PAN nanofiber electrodes had higher Cs values than electrodes prepared from wartel and banana biomass.65 The Cs values decreased as the scan rate was increased from 10 mV s−1 to 100 mV s−1 (Fig. 10b). However, the CV curve of the CNF3 nanofiber electrode remained quasi-rectangular at a scan rate of 100 mV s−1 (Fig. 9b), which was indicative of good performance. At the highest scan rate, collisions between ions were so frequent that ion exchange on the electrode surfaces was inhibited.34 Similar behavior has been observed on electrodes fabricated using a PVA/GO composite and a polymer composite that contained waste carbon from tires.66,67
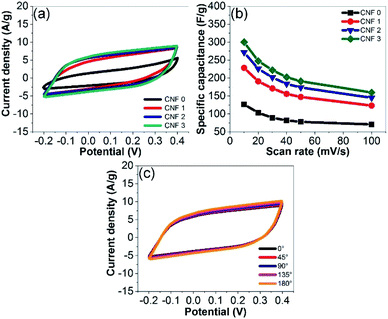 |
| | Fig. 10 (a) CV curves of the fibrous mats recorded at a scan rate of 40 mV s−1. (b) CV curves recorded at various scan rates. (c) CV curves of CNF3 bent at various angles. | |
Flexibility was evaluated by recording CV curves while the devices were bent at various angles (Fig. 10c). The CV curves were identical regardless of the flexural angle. These results were consistent with those of the tensile tests and confirmed that the SC electrodes could be used for flexible electronic device applications. Similar findings were reported by Gupta et al. (2015).68
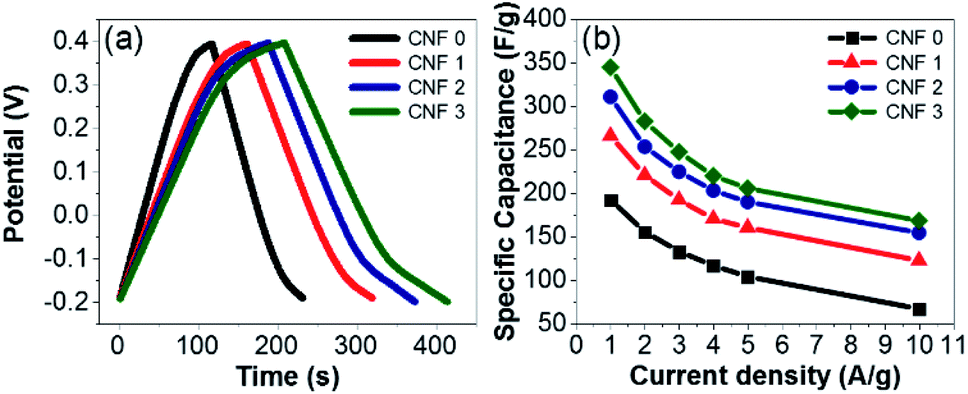
The GCD profiles of the electrodes were symmetric and triangular at current densities ranging from 1 to 10 A g−1 (Fig. 11a). The high degree of reversibility indicated ideal electrical double-layer capacitance. The CNF3 electrode displayed the best capacity retention, excellent cycling stability, and outstanding electrochemical performance. The Cs values of the samples were calculated using eqn (1). The decrease in Cs as the current density increased (Fig. 11b) was attributed to inhibited diffusion. At higher current densities, ions in the electrolyte did not have sufficient time to diffuse into the inner pores of the electrodes.68
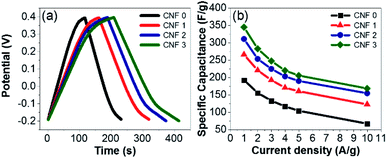 |
| | Fig. 11 (a) CGD profiles of the mats at a current density of 2 A g−1. (b) Specific capacitance values of the composite fiber mat electrodes at various current densities. | |
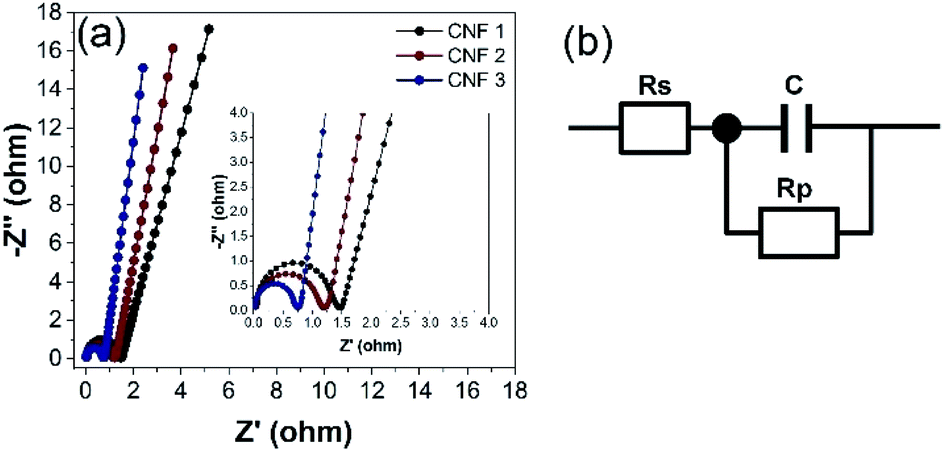
EIS was performed to investigate the behavior of the electrodes at frequencies from 10 kHz to 10 mHz. The Nyquist plots of the CNF1, CNF2, and CNF3 electrodes are shown in Fig. 12a. The equivalent circuit diagram used to fit the EIS data is shown in Fig. 12b. Rs is the equivalent series resistance (ESR); Rp is the charge transfer resistance; and C is the double-layer capacitance value.12 Two regions were observed in the Nyquist plots. Each contained a semicircle in the high-frequency range and a linear portion in the low-frequency region. At high frequencies, the x-axis intercept corresponded to the sum of contact resistance at the current collector and ohmic resistance at the electrode–electrolyte interface (Rs).29 The rGOPKS/PAN composite devices had Rs values of 0.05 Ω (CNF1), 0.04 Ω (CNF2), and 0.03 Ω. As expected, CNF3 provided the lowest ESR. This indicated good contact between the electrolyte solution and the active components in the interfacial. The semicircles in the high- and mid-frequency regions of the plots were indicative of interfacial charge transfer resistance (Rp).12,59 The semicircle in the plot of CNF3 had a smaller radius than those in the plots of CNF2 and CNF1 (Fig. 10a, inset), which meant that Rp was lower in CNF3. The recorded Rp values were 1.47 Ω (CNF1), 1.21 Ω (CNF2), and 0.76 Ω (CNF3). These results confirmed that contact resistance was low, and the distribution of charge in the interfacial regions was ideal. The charge transfer capability and electrical conductivity of the CNF3 electrode were superior,59 which was consistent with its Cs value based on the CV and GCD measurements. These results were consistent with the charge storage and distribution mechanisms of an electrode with a large surface area. A large surface area enables ions to rapidly penetrate into the micropores and mesopores, which enhances capacitive performance.59,69 The advantages of the CNF3 electrode were largely due to two factors. The large proportion of rGOPKS in the PAN nanofibers enhanced the conductivity of PAN,70 and the porous structure of the CNFs provided ample surface area for ions to participate in electrochemical reactions at the electrolyte/electrode interface.12,71
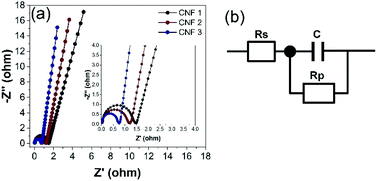 |
| | Fig. 12 (a) Nyquist plots of the electrospun composite nanofibers. Inset: magnification of the region between 0.0 and 4.0 Ω. (b) Equivalent circuit diagram derived from the Nyquist plots. | |
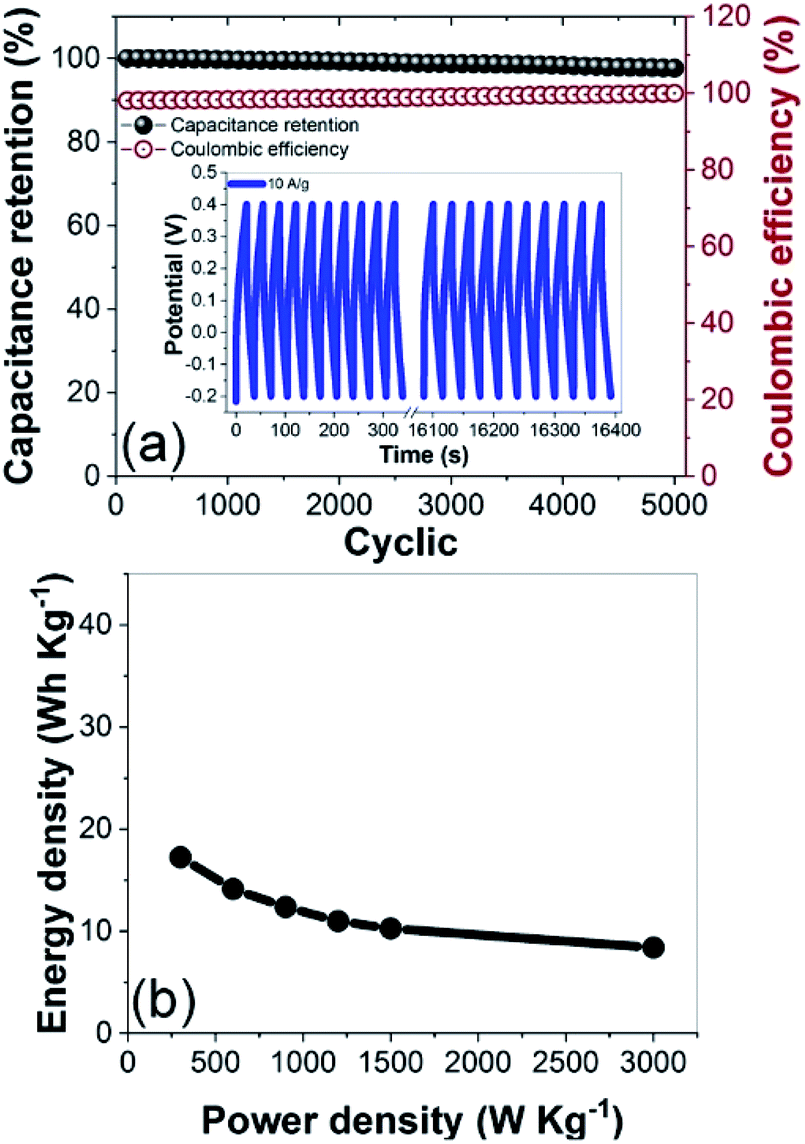
The CNF3 electrode had a long cycle life at a current density of 10 A g−1 (Fig. 13a). Its capacitance retention was expected to exceed 90%, and it retained 98.96% of its initial capacitance after 5000 cycles.66 These results demonstrated that the cycling stability and charge–discharge reversibility of the CNF3 electrode were excellent,60 and its coulombic efficiency was stable at 98.85%. A maximum energy density of 17 W h kg−1 was attained at 3000 W kg−1 (Fig. 13b). This was comparable to the energy density of commercially available ELDCs (5 W h kg−1) at 5000 W kg−1 and lithium-ion batteries (10 W h kg−1) at 1000 W kg−1.72,73 These results confirmed that CNF3 efficiently stored energy, and that its rate of utilization was high. Thus, the rGOPKS/PAN-based CNFs demonstrated great potential as supercapacitor electrode materials.
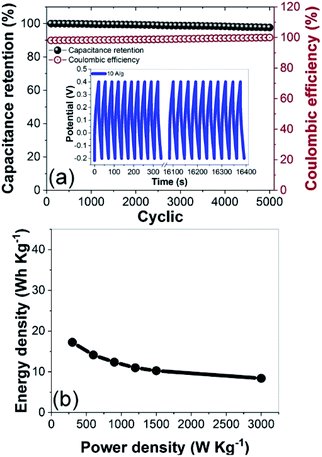 |
| | Fig. 13 (a) Cycling stability and coulombic efficiency of the CNF3 electrode over 5000 charge–discharge cycles at a current density of 10 A g−1. (b) Ragone plot of the CNF3 electrode. | |
3.10 Dielectric studies of the rGOPKS/PAN composite nanofiber mats
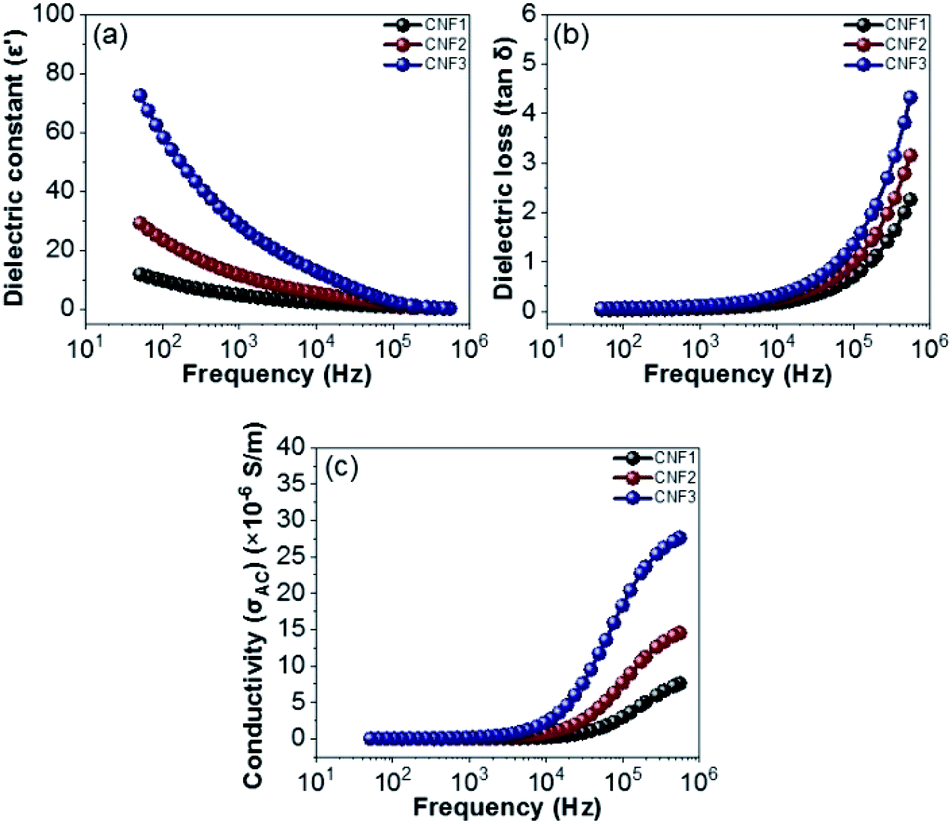
The dielectric properties for all the nanofiber composites are further investigated in the 50 Hz–500 kHz range. Fig. 14a shows the relationship between the frequency and the dielectric constant of the CNF1, CNF2, and CNF3 nanofiber composites. All nanofiber composites decrease dramatically with a higher frequency, this is due to the interfacial polarization relaxation and dipole.74 At low frequencies, the time required by the dipole is more than sufficient to reach the maximum value limit at which the charge carrier is formed, which is responsible for the magnitude of the polarization formed.75 Whereas at high frequency the field changes direction very quickly, as a result polarization does not occur so that the carrier cannot reach the grain boundaries.76 The highest dielectric constant is generated at point 72.3 (CNF3). This is the effect of adding rGOPKS to the nanofibers, therefore it results more defects. The broad surface area of the nanofiber composite can accommodate most of the defects to produce rotational direction polarization (RDP)77 Meanwhile, the dielectric loss of CNF1, CNF2, and composite CNF3 is shown in Fig. 14b. Dielectric loss increases with increasing frequency. Dielectric loss for all samples is very small with a value less than 0.10 (frequency below 1000 Hz). Gradual increase in dielectric loss begins at frequencies of 10000–100000 Hz, with the dielectric samples of CNF1, CNF2 and CNF3 increasing from 0.16, 0.22, and 0.33 to 0.70, 0.98, and 1.35, respectively. The increase is continued until at a frequency of 500000 Hz, the dielectric loss samples CNF1, CNF2 and CNF3 reach their maximum points at 2.26, 3.15 and 4.32, respectively.
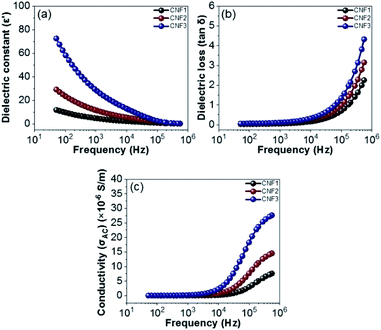 |
| | Fig. 14 Frequency dependency of (a) dielectric constant (ε′), (b) dielectric loos (tanδ), and (c) electrical conductivity (σAC). | |
The increase in dielectric loss is due to: (1) dipole polarization relaxation, the establishment of dipole polarization cannot follow the electric field, so the relaxation leads to enhanced loss;78 and (2) the addition of rGOPKS inhibits the movement of electrons in the polymer chain, which increases the dielectric loss of dipole polarization relaxation. Fig. 14c shows the conductivity vs. frequency of the nanofiber composites (CNF1, CNF2, and CNF3). When it is below 1000 Hz, the conductivity of all samples does not depend on its frequency. Within this range, the low frequency applied causes the charge to flow randomly because the electric field does not have sufficient energy to discharge the charge in the unit direction. Conductivity increases gradually from 7.6 × 10−6 S m−1 to 27.6 × 10−6 S m−1 which is observed in the frequency range above 10000–500000 Hz. In this condition (moderate frequency) makes more energy produced so that the carrying charge begins to flow into the field, further this flow increases exponentially at much higher frequencies via the loop mechanic. The increase in the surface area of the nanofiber composites, results in the trapping of charges in a large number, and coupled with high frequency conditions cause the charge to be released which in turn increases the maximum compound conductivity.74,79 Thus, each sample meets the requirements for dielectric properties, where CNF1, CNF2, and CNF3 samples have dielectric constant values of 11.8, 29.0, and 72.3, respectively. Under the same conditions dielectric losses become independent of frequency. Furthermore, the conductivity reachs a maximum value of 27.6 × 10−6 S m−1 at high frequencies.
4. Conclusions
rGOPKS/PAN composite fiber mats were fabricated using an electrospinning technique and applied as supercapacitor electrode materials. The fibers were homogeneous, and the rGOPKS/PAN composite fiber mats were flexible. Changes in the intensities and positions of peaks in the FTIR spectra of the composite nanofiber mats were attributed to molecular interactions between PAN and rGOPKS. XRD analysis indicated that crystalline PAN became amorphous when it was electrospun into fibers. The specific capacitance of a CNF3 composite nanofiber electrode, which contained the largest quantity of rGOPKS, was 203 F g−1 at a scan rate of 40 mV s−1. The cycling stability of the electrode was excellent, and it retained over 90% of its initial capacitance after 5000 cycles. The Ragone plot this electrode showed an energy density of 17 W h kg−1 at a power density of 3000 W kg−1. The CNF1, CNF2 and CNF3 samples have dielectric constant values of 11.8, 29.0 and 72.3 and the conductivity is successfully reaches a maximum value of 27.6 × 10−6 S m−1 at high frequencies. The good mechanical flexibility and superior electrochemical performance of the composite nanofiber mat electrodes make them promising candidates as supercapacitor electrodes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by Sriwijaya University, Republik of Indonesia, under the University's Excellence Research (PUPT) Grant for the fiscal year of 2019–2020 (0179.093/UN9/SB3.LPPM.PT/2020) and by BPDPKS, Republik of Indonesia, under the Research and Development of Palm Oil for the fiscal year of 2019–2020.
References
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao and G. Yu, J. Mater. Chem. A, 2014, 2, 6086–6091 RSC.
- W. K. Chee, H. N. Lim, Z. Zainal, I. Harrison, Y. Andou, N. M. Huang, M. Altarawneh and Z. T. Jiang, Mater. Lett., 2017, 199, 200–203 CrossRef CAS.
- K. Gao, Z. Shao, J. Li, X. Wang, X. Peng, W. Wang and F. Wang, J. Mater. Chem. A, 2013, 1, 63–67 RSC.
- Z. Htut, M. Kim, E. Lee, G. Lee, S. H. Baeck and S. E. Shim, Synth. Met., 2017, 227, 61–70 CrossRef.
- M. Horn, B. Gupta, J. MacLeod, J. Liu and N. Motta, Current Opinion in Green and Sustainable Chemistry, 2019, 17, 42–48 CrossRef.
- T. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899–6905 CrossRef CAS PubMed.
- Y. Shao, M. F. El-Kady, L. J. Wang, Q. Zhang, Y. Li, H. Wang, M. F. Mousavi and R. B. Kaner, Chem. Soc. Rev., 2015, 44, 3639–3665 RSC.
- S. K. Simotwo, C. Delre and V. Kalra, ACS Appl. Mater. Interfaces, 2016, 8, 21261–21269 CrossRef CAS PubMed.
- M. Yanilmaz, M. Dirican, A. M. Asiri and X. Zhang, Journal of Energy Storage, 2019, 24, 100766 CrossRef.
- X. Lu, M. Yu, G. Wang, Y. Tong and Y. Li, Energy Environ. Sci., 2014, 7, 2160–2181 RSC.
- C. Ma, X. Wang, Y. Ma, J. Sheng, Y. Li, S. Li and J. Shi, Mater. Lett., 2015, 145, 197–200 CrossRef CAS.
- M. A. A. Mohd Abdah, N. A. Zubair, N. H. N. Azman and Y. Sulaiman, Mater. Chem. Phys., 2017, 192, 161–169 CrossRef CAS.
- D. Wang and J. Wang, Chem. Eng. J., 2017, 314, 714–726 CrossRef CAS.
- R. Farzana, R. Rajarao, B. R. Bhat and V. Sahajwalla, J. Ind. Eng. Chem., 2018, 65, 387–396 CrossRef CAS.
- J. Zeng, L. Wei and X. Guo, J. Mater. Chem. A, 2017, 5, 25282–25292 RSC.
- J. Xiao, W. Lv, Y. Song and Q. Zheng, Chem. Eng. J., 2018, 338, 202–210 CrossRef CAS.
- J. Huang, H. Deng, D. Song and H. Xu, Anal. Chim. Acta, 2015, 878, 102–108 CrossRef CAS PubMed.
- E. Azwar, W. A. Wan Mahari, J. H. Chuah, D. V. N. Vo, N. L. Ma, W. H. Lam and S. S. Lam, Int. J. Hydrogen Energy, 2018, 43, 20811–20821 CrossRef CAS.
- J. E. Omoriyekomwan, A. Tahmasebi, J. Zhang and J. Yu, Energy Convers. Manage., 2017, 148, 583–592 CrossRef CAS.
- H. A. Abdul Bashid, H. N. Lim, S. Kamaruzaman, S. Abdul Rashid, R. Yunus, N. M. Huang, C. Y. Yin, M. M. Rahman, M. Altarawneh, Z. T. Jiang and P. Alagarsamy, Nanoscale Res. Lett., 2017, 12, 1–10 CrossRef CAS PubMed.
- K. J. Ramalingam, N. R. Dhineshbabu, S. R. Srither, B. Saravanakumar, R. Yuvakkumar and V. Rajendran, Synth. Met., 2014, 191, 113–119 CrossRef CAS.
- Y. Shao, H. Wang, Q. Zhang and Y. Li, NPG Asia Mater., 2014, 6, e119 CrossRef CAS.
- P. K. Jha, S. K. Singh, V. Kumar, S. Rana, S. Kurungot and N. Ballav, Chem, 2017, 3, 846–860 CAS.
- A. Dhar, N. S. Kumar, M. Khimani, A. S. Al-Fatesh, A. A. Ibrahim, A. H. Fakeeha, P. Bhadja and R. L. Vekariya, Int. J. Energy Res., 2020, 44, 913–924 CrossRef CAS.
- A. M. Abioye and F. N. Ani, Renewable Sustainable Energy Rev., 2015, 52, 1282–1293 CrossRef CAS.
- A. R. Tobi, J. O. Dennis, H. M. Zaid, A. A. Adekoya, A. Yar and U. Fahad, J. Mater. Res. Technol., 2019, 8, 3688–3695 CrossRef CAS.
- M. Shanmuga Priya, P. Divya and R. Rajalakshmi, Sustainable Chem. Pharm., 2020, 16, 100243 CrossRef.
- J. Pirker, A. Mosnier, F. Kraxner, P. Havlík and M. Obersteiner, Global Environ. Change, 2016, 40, 73–81 CrossRef.
- I. I. Misnon, N. K. M. Zain, R. A. Aziz, B. Vidyadharan and R. Jose, Electrochim. Acta, 2015, 174, 78–86 CrossRef CAS.
- J. Yan, J. H. Choi and Y. G. Jeong, Mater. Des., 2018, 139, 72–80 CrossRef CAS.
- M. Zhi, S. Liu, Z. Hong and N. Wu, RSC Adv., 2014, 4, 43619–43623 RSC.
- P. Widiatmoko, I. F. Sukmana, I. Nurdin, T. Prakoso and H. Devianto, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 543, 12032 CrossRef CAS.
- D. Edikresnha, T. Suciati, M. M. Munir and K. Khairurrijal, RSC Adv., 2019, 9, 26351–26363 RSC.
- C. Zequine, C. K. Ranaweera, Z. Wang, P. R. Dvornic, P. K. Kahol, S. Singh, P. Tripathi, O. N. Srivastava, S. Singh, B. K. Gupta, G. Gupta and R. K. Gupta, Sci. Rep., 2017, 7, 1–12 CrossRef CAS PubMed.
- S. Ishaq, F. Kanwal, S. Atiq, M. Moussa, U. Azhar and D. Losic, Materials, 2020, 13, 1–11 CrossRef PubMed.
- M. Y. Chong, Y. C. Y. Evyan, N. Zebardastan, K. Kumar, K. Ramesh and S. Ramesh, International Journal of Nanoelectronics and Materials, 2020, 13, 119–128 Search PubMed.
- S. Zhang, M. Zeng, J. Li, J. Li, J. Xu and X. Wang, J. Mater. Chem. A, 2014, 2, 4391–4397 RSC.
- S. Nasir, M. Z. Hussein, N. A. Yusof and Z. Zainal, Nanomaterials, 2017, 7, 1–18 CrossRef PubMed.
- H. Kharkwal, H. C. Joshi and K. P. Singh, Int. J. Biochem. Biophys., 2018, 6, 1–19 CAS.
- H. Kalil, S. Maher, T. Bose and M. Bayachou, J. Electrochem.
Soc., 2018, 165, G3133–G3140 CrossRef CAS.
- J. Jauhari, S. Wiranata, A. Rahma, Z. Nawawi and I. Sriyanti, Mater. Res. Express, 2019, 6, 064002 CrossRef CAS.
- S. Ramakrishna, K. Fujihara, W. E. Teo, T. C. Lim and Z. Ma, An introduction to electrospinning and nanofibers, 2005 Search PubMed.
- I. Sriyanti, D. Edikresnha, A. Rahma, M. M. Munir, H. Rachmawati and K. Khairurrijal, Int. J. Nanomed., 2018, 13, 4927–4941 CrossRef CAS PubMed.
- I. Sriyanti, D. Edikresnha, A. Rahma, M. M. Munir, H. Rachmawati and K. Khairurrijal, J. Nanomater., 2017, 1–10 Search PubMed.
- J. Matulevicius, L. Kliucininkas, T. Prasauskas, D. Buivydiene and D. Martuzevicius, J. Aerosol Sci., 2016, 92, 27–37 CrossRef CAS.
- X. Zhao, Y. Dong, B. Cheng and W. Kang, Int. J. Photoenergy, 2013, 2013, 1–9 CrossRef.
- S. Tas, O. Kaynan, E. Ozden-Yenigun and K. Nijmeijer, RSC Adv., 2016, 6, 3608–3616 RSC.
- Y. Dong, F. Li, X. Zhao, B. Cheng, W. Kang and G. Cui, J. Ind. Text., 2018, 48, 146–161 CrossRef CAS.
- S. Nasir, M. Z. Hussein, Z. Zainal and N. A. Yusof, J. Nanomater., 2019, 1–13 CrossRef CAS.
- M. Rafi, B. Samiey and C. H. Cheng, Materials, 2018, 11, 1–24 CrossRef PubMed.
- K. Jurkiewicz, M. Pawlyta and A. Burian, C, 2018, 4, 68 CAS.
- S. W. Choi, J. R. Kim, S. M. Jo, W. S. Lee and Y.-R. Kim, J. Electrochem. Soc., 2005, 152, A989 CrossRef CAS.
- H. Yu, B. Zhang, C. Bulin, R. Li and R. Xing, Sci. Rep., 2016, 6, 1–7 CrossRef CAS PubMed.
- J. Li, D. Xiao, Y. Ren, H. Liu, Z. Chen and J. Xiao, Electrochim. Acta, 2019, 300, 193–201 CrossRef CAS.
- K. Alam, Y. Sim, J.-H. Yu, J. Gnanaprakasam, H. Choi, Y. Chae, U. Sim and H. Cho, Materials, 2019, 13, 12 CrossRef PubMed.
- G. He, Y. Song, S. Chen and L. Wang, J. Mater. Sci., 2018, 53, 9721–9730 CrossRef CAS.
- H. Fu, X. Zhang, J. Fu, G. Shen, Y. Ding, Z. Chen and H. Du, J. Alloys Compd., 2020, 829, 154557 CrossRef CAS.
- S. B. Kulkarni, U. M. Patil, I. Shackery, J. S. Sohn, S. Lee, B. Park and S. Jun, J. Mater. Chem. A, 2014, 2, 4989–4998 RSC.
- H. Yin, H. Zheng, L. Yang, S. Wang and L. Liu, RSC Adv., 2020, 10, 5666–5672 RSC.
- W. Wu, L. Zhang, C. Wang, J. Wang, J. Qian, S. Song and Z. Yue, RSC Adv., 2020, 10, 5436–5442 RSC.
- G. K. Meenashisundaram, M. H. Nai, A. Almajid and M. Gupta, Materials, 2016, 9, 1–21 CrossRef PubMed.
- J. Yao, C. W. M. Bastiaansen and T. Peijs, Fibers, 2014, 2, 158–187 CrossRef.
- C. Costentin, T. R. Porter and J. M. Savéant, ACS Appl. Mater. Interfaces, 2017, 9, 8649–8658 CrossRef CAS PubMed.
- E. Lee, M. Kim, J. Ju, S. Jang, S. H. Baeck and S. E. Shim, Synth. Met., 2017, 226, 195–206 CrossRef CAS.
- S. Ahmed, A. Ahmed and M. Rafat, J. Saudi Chem. Soc., 2018, 22, 993–1002 CrossRef CAS.
- S. Nabati Shoghl, J. Jamali and M. Keshavarz Moraveji, Exp. Therm. Fluid Sci., 2016, 74, 339–346 CrossRef CAS.
- M. Boota, M. P. Paranthaman, A. K. Naskar, Y. Li, K. Akato and Y. Gogotsi, ChemSusChem, 2015, 8, 3576–3581 CrossRef CAS PubMed.
- R. K. Gupta, J. Candler, S. Palchoudhury, K. Ramasamy and B. K. Gupta, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- X. Zheng, X. Yan, Y. Sun, Y. Yu, G. Zhang, Y. Shen, Q. Liang, Q. Liao and Y. Zhang, J. Colloid Interface Sci., 2016, 466, 291–296 CrossRef CAS PubMed.
- F. Miao, C. Shao, X. Li, N. Lu, K. Wang, X. Zhang and Y. Liu, Energy, 2016, 95, 233–241 CrossRef CAS.
- L. Wang, M. Huang, S. Chen, L. Kang, X. He, Z. Lei, F. Shi, H. Xu and Z. H. Liu, J. Mater. Chem. A, 2017, 5, 19107–19115 RSC.
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623–1632 RSC.
- B. Xu, F. Wu, S. Chen, Z. Zhou, G. Cao and Y. Yang, Electrochim. Acta, 2009, 54, 2185–2189 CrossRef CAS.
- Y. Feng, M. L. Li, W. L. Li, T. D. Zhang, Y. Zhao and W. D. Fei, Appl. Phys. Lett., 2018, 112, 2–7 Search PubMed.
- X. Zhao, Y. Dong, B. Cheng and W. Kang, Int. J. Photoenergy, 2013, 2013, 10005–10012 CrossRef.
- S. Mishra, R. Sahoo, L. Unnikrishnan, A. Ramadoss, S. Mohanty and S. K. Nayak, Mater. Res. Bull., 2020, 124, 110732 CrossRef CAS.
- P. Li, P. Li, J. Yu, J. Yu, S. Jiang, H. Fang, K. Liu, H. Hou and H. Hou, e-Polym., 2020, 20, 226–232 CAS.
- Y. Kou, W. Zhou, L. Xu, H. Cai, G. Wang, X. Liu, Q. Chen and Z. M. Dang, High Perform. Polym., 2019, 31, 1183–1194 CrossRef CAS.
- S. Wageh, L. He, A. A. Al-Ghamdi, Y. A. Al-Turki and S. C. Tjong, RSC Adv., 2014, 4, 28426–28431 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
M. Rama Almafieb,
Leni Marlinab,
Zainuddin Nawawi*c and
Ida Sriyanti
a,
M. Rama Almafieb,
Leni Marlinab,
Zainuddin Nawawi*c and
Ida Sriyanti