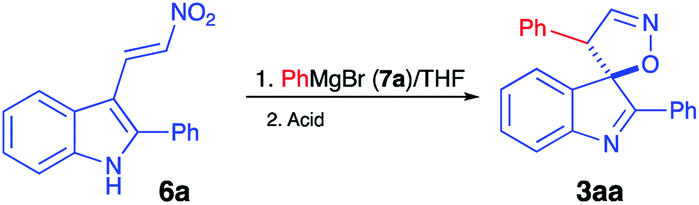DOI:
10.1039/D0RA10219A
(Paper)
RSC Adv., 2021,
11, 1783-1793
Preparation of spiro[indole-3,5′-isoxazoles] via Grignard conjugate addition/spirocyclization sequence†
Received
3rd December 2020
, Accepted 21st December 2020
First published on 6th January 2021
Abstract
A highly efficient one-pot procedure combining conjugate addition of Grignard reagents to (2-nitroalkenyl)indoles and sub-sequent Brønsted acid-assisted spirocyclization allowed for preparation of 4′H-spiro[indole-3,5′-isoxazoles] in a diastereomerically selective fashion. Utilization of alkyl Grignard reagents provided an easy access to 4′-alkylsubstituted derivatives hardly available by other means.
Introduction
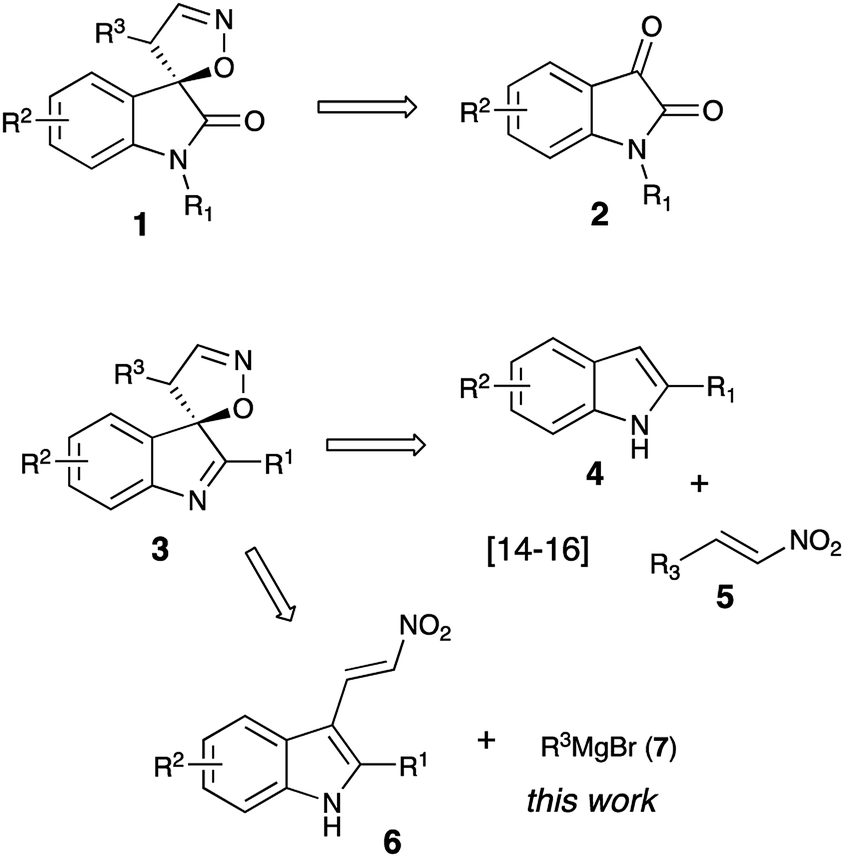
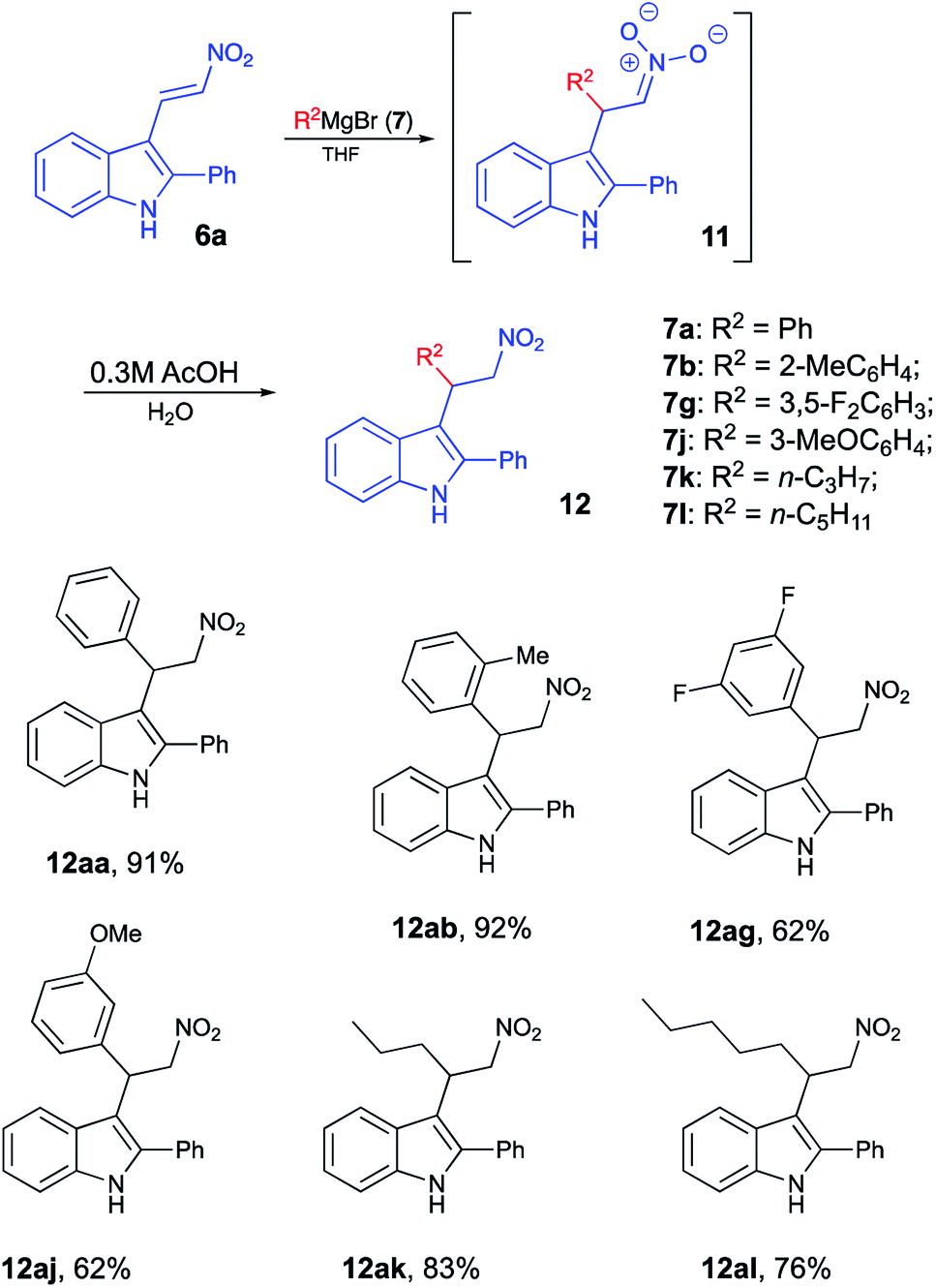
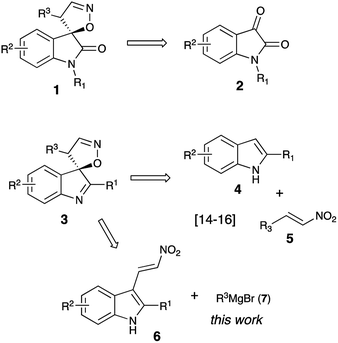
Due to their diverse biological profiles and important pharmaceutical activities, natural and synthetic spiroheterocycles have been the constant focus of attention of many research groups as synthetic targets.1–3 Although the subjects of the present study, 4′H-spiro[indoline-3,5′-isoxazoles] and 4′H-spiro[indole-3,5′-isoxazoles], have not been found in nature, they share a privileged polycyclic framework with an array of highly potent natural products.1,2,4–8 The chemistry of related 4′H-spiro[indoline-3,5′-isoxazol]-2-ones (1) is much better known, and quite a few methods toward their synthesis have been developed to date. Most of these employ readily available isatin precursors 2 as a synthetic platform (Scheme 1), and typically involve an acid-assisted 1,5-spirocyclization of mono-oximes of 3-ene-2,5-diones,9,10 [3 + 2]-cycloaddition of nitrile oxides to 3-methyleneoxindoles,11–13 or metal-catalyzed selective vinylation of isatin oximes with vinylboronic acids.14 However, any subsequent manipulations at the carbonyl moiety, should they be required, would be limited by the vulnerability of the α-spirocyclic fragment in 1. We have recently reported a highly efficient and diastereoselective formal [4 + 1]-spirocyclization of indoles with nitroolefins serving as unusual 1,4-CCNO dipoles (Scheme 1).15–17 This reaction proceeded in the presence of phosphorous acid, affording very good yields of spiroindoles 3, hardly accessible by other means. For a series of these compounds promising antitumor activity against neuroblastoma cells was discovered.16 Implementing a synthesis of focused libraries for SAR studies, we stumbled upon a limitation of this previously published method, which proved to work great for preparation of compounds possessing an aryl substituent at C-4′ (R3 = Ar). Primary alkyl groups, however, could not be efficiently introduced this way as the corresponding nitroalkenes 5 (R3 = 1°-Alk) seldom produce spirocyclic products 3 in acceptable yields. Herein we disclose an improved design of an alternative and more general approach featuring a diversity-oriented late-stage introduction of substituent R3 via the nucleophilic addition of Grignard reagents 7 to nitroolefins 6. At this point we did not set a goal to pursue the library synthesis, but wanted to evaluate the new synthetic protocol.
 |
| | Scheme 1 | |
Results and discussion
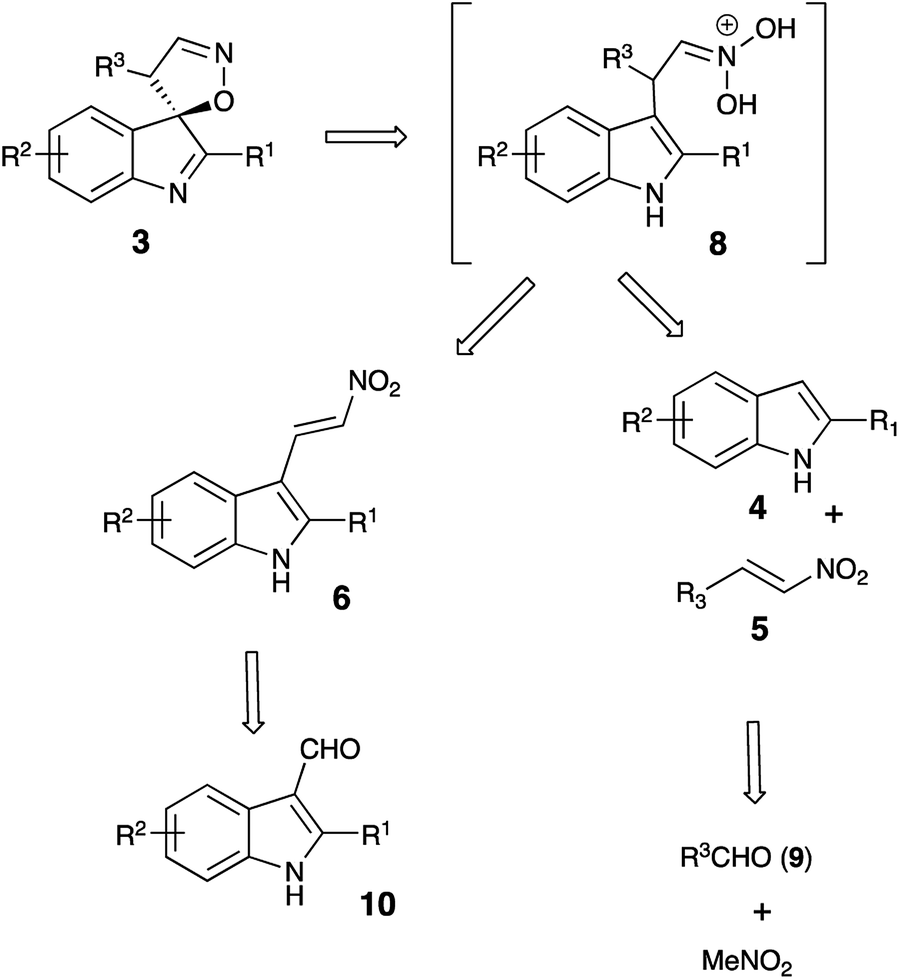
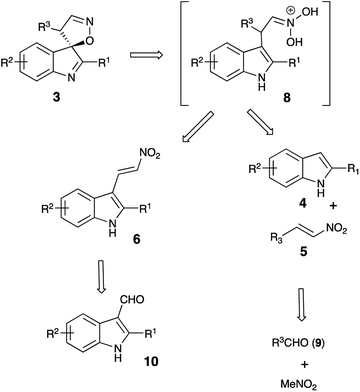
It should be pointed out that the featured spirocyclization is believed to proceed via formation of nitronic acid 8 (Scheme 2). According to the first-generation method, this key intermediate is generated via nucleophilic addition of indoles 4 to nitroalkenes 5 which, in turn, are accessible via Henry reaction between nitromethane and aldehydes 9. Thus, this method greatly depends on the availability of the corresponding nitroalkenes, and an attempt at diversification of the R3 substituent would require a whole array of Henry reactions to be carried out in order to gain access to different nitroalkenes 5. To avoid this bottleneck, we penciled down an alternative design, in which nitronic acid 8 would be generated in a one-pot fashion after conjugate nucleophilic addition of Grignard reagents to nitroolefin 6.18–26 When only the diversification of the R3 substituent is pursued, structure 6 could serve as a common intermediate, prepared in a single scaled-up experiment via Henry reaction of readily available 3-indolylaldehyde 10 (Scheme 2). To test this idea, (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) was subjected to a reaction with phenylmagnesium bromide (7a) in dry THF. The first step of the reaction, involving conjugate nucleophilic addition proceeded smoothly, but the acid-assisted spirocyclization step required some optimization. Originally, a mixture of phosphorous and formic acids was employed as an optimal medium for this step.16 This mixture, however, provided marginal results when used for work up of the nitronate generated in the reaction with a Grignard reagent. A complex mixture of products was formed with partial resinification, and the content of the spirocyclic product 3aa was 51% (Table 1, entry 1). The situation was improved slightly when the acidic work up was carried out at 0 °C. Spirane 3aa was formed in 67% yield and isolated along with ca. 6% of an unidentified impurity (entry 2). The same impurity was present in notable amounts in the crude product, isolated from the mixture quenched with sulfuric acid in acetic acid (entry 3). Unfortunately, this impurity proved to be inseparable, so further exercises with these quenching reagents were counterproductive, so we started to look for alternatives. Among many other strong acids tested, we found that the quench with orthophosphoric and methanesulfonic acids afforded only 45% and 51% of 3aa, respectively, but in both cases, these were pure samples (entries 4 and 5). The best yield of 72% of spirane was obtained after work up in ice-cold concentrated hydrochloric acid (entry 6). Diastereomeric purity of this material was ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
 |
| | Scheme 2 | |
Table 1 Optimization of spirocyclization towards 2aa
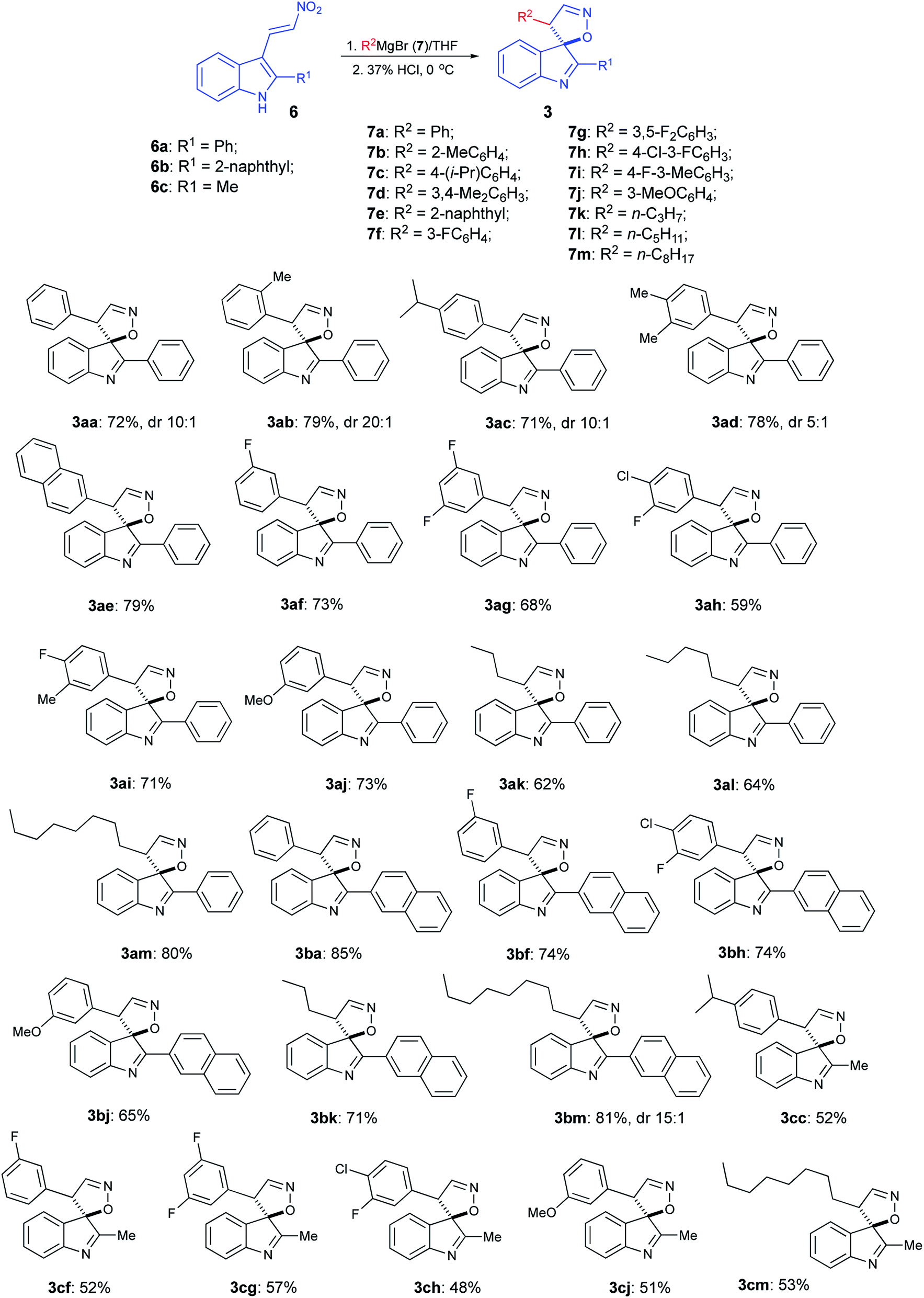
With optimized reaction conditions in hand, we proceeded with the synthesis of several arrays of spirocyclic structures in order to investigate the scope and limitations of the featured transformation. In the first series of experiments, nitroalkene 6a was treated with phenyl- (7a), o-tolyl- (7b), p-cumyl- (7c), 3,4-xylyl- (7d), and 2-naphthyl- (7e) magnesium bromides. All of these reactions proceeded smoothly, affording, after acidic quench, the corresponding spiranes 3aa–3ae in good yields (Scheme 3). Next, the same substrate was subjected in the reaction with a series of Grignard reagents (7f–7i), derived from fluorinated aromatic hydrocarbons. Again, spiranes 3af–3ai were formed smoothly, albeit in somewhat lower yields. Reaction with m-anisylmagnesium bromide (7j) provided spirocyclic product 3aj, thus showcasing the tolerance of the method to the presence of the ether function (Scheme 3). Most importantly, reactions with 1°-alkyl Grignard reagents 7k–m also worked well, yielding the corresponding spiranes alkyl-substituted at C-4′, which were not easily available via previous version of the spirocyclization.16 We also tested reactions of two other nitroalkenes, 6b and 6c, bearing the bulky 2-naphthyl and small methyl group at C-2 of indole core, respectively. It seems that the steric hindrance at this position has a positive effect on the featured transformation. Indeed, yields of naphthyl-substituted products 3ba–3bm were consistently higher than their methyl-substituted analogs 3cc–3cm (Scheme 3). It should be pointed out that formation of spirocyclic core was unambiguously confirmed by single crystal X-ray diffraction of compound 3ag (CCDC #2024756, see ESI for details†).
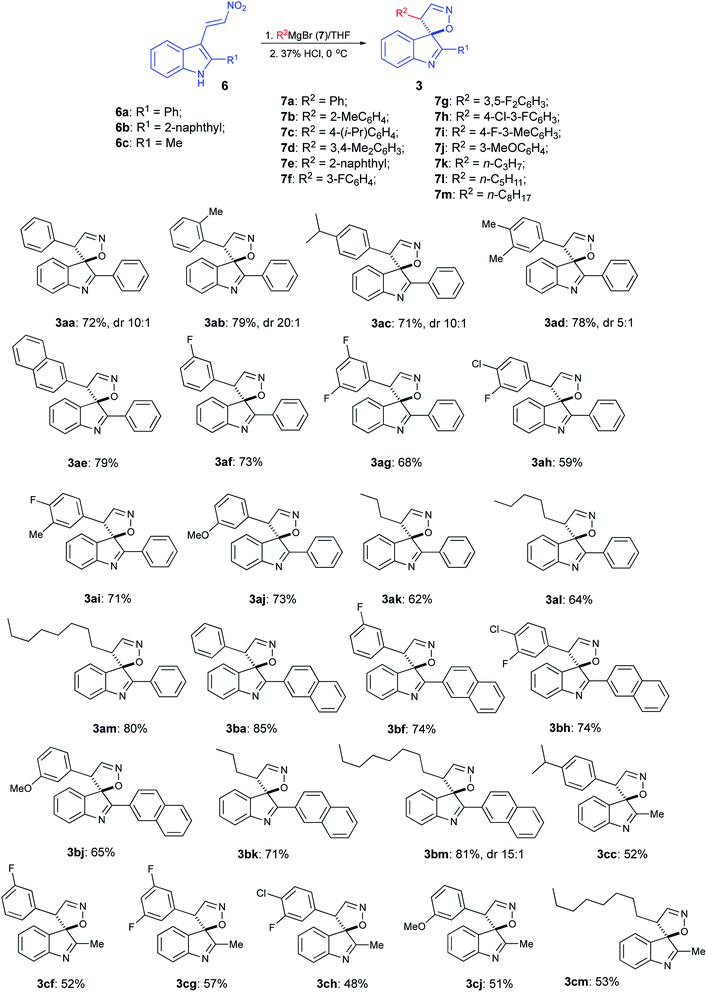 |
| | Scheme 3 | |
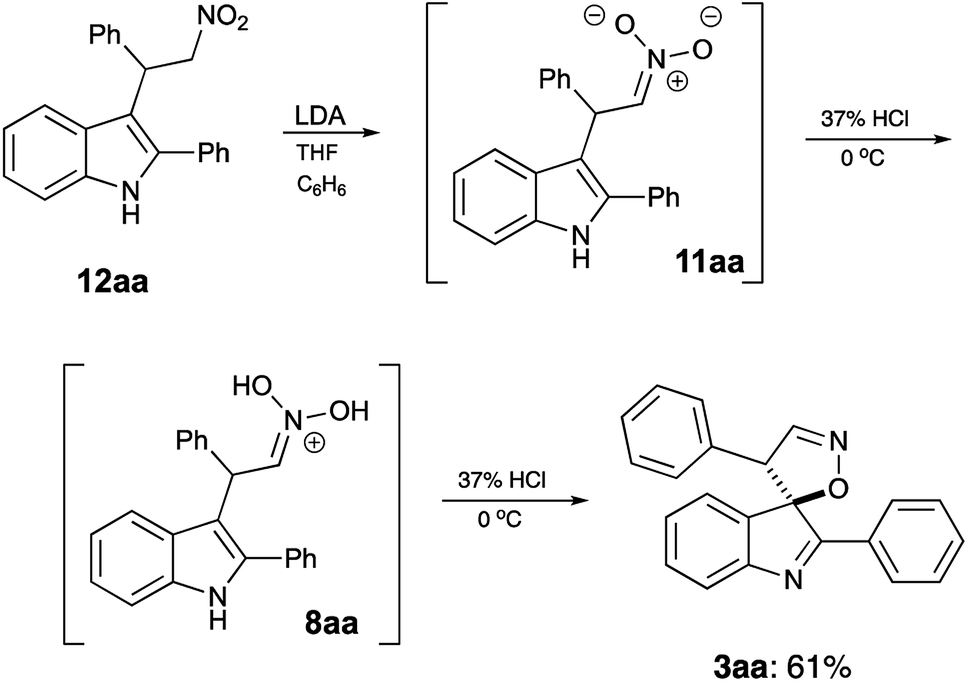
It should be stressed, that formation of spirocyclic product 3 occurs during the work up with cold hydrochloric acid, as it relies on formation of nitronic acid 8, which requires quite a low pH level. If this treatment is replaced with a regular quench with water or an aqueous solution of weak acid, 8 does not form, as mono-protonation of nitronate 11 affords nitroalkane 12 instead (Scheme 4). It should be understood, however, that nitro-aci tautomerization of nitroalkanes proceeds very slowly under acidic conditions, so simple treatment of 12 with concentrated hydrochloric acid did not produce spirocyclic products in detectable amounts. Under basic conditions, however, tautomeric equilibrium is efficient and shifted towards formation of the aci-form. Thus, upon treatment of 12aa with LDA nitronate 11aa was produced. Subsequent acidification of this species with cold concentrated HCl affords nitronic acid 8aa under conditions suitable for spirocyclization. Indeed, after such treatment, spirane 3aa was isolated in 61% yield (Scheme 5).
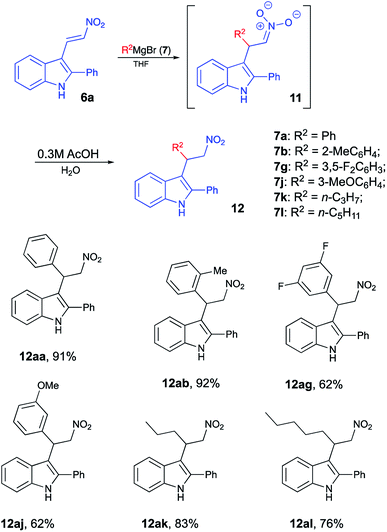 |
| | Scheme 4 | |
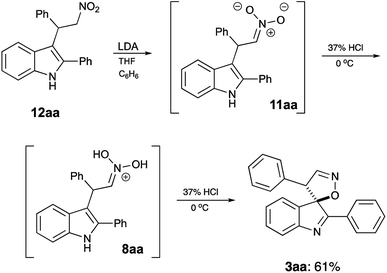 |
| | Scheme 5 | |
Conclusion
An improved protocol combining conjugate addition of Grignard reagents to 3-(2-nitrovinyl)-1H-indoles 6 and subsequent treatment with concentrated HCl allowed for efficient and highly diastereoselective preparation of 4′H-spiro[indole-3,5′-isoxazoles] 3. These structures can be obtained in good yields, including hardly available compounds with primary alkyl substituents at C-4′. Very low pH during the acidic work up proved to be highly essential for spirocyclization step. Quench with water or weak acids provides linear nitroalkane products 12, which cannot be converted into 3 in the following acidification. However, conversion of 12 into nitronate species under strongly basic conditions and subsequent treatment with concentrated HCl allowed to obtain spirocyclic product 3, thus proving the participation of nitronic acid intermediates.
Experimental part
General information
1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 101 MHz, respectively) equipped with BBO probe in CDCl3 or DMSO-d6, using TMS as internal standard. High-resolution mass spectra were registered with a Bruker Maxis spectrometer (electrospray ionization, in MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart SMP30 apparatus. Reaction progress and purity of isolated compounds were controlled by TLC on Silufol UV-254 plates. All other reagents and solvents were purchased from commercial vendors and used as received.
(3R*,4′S*)-4′-(3,5-Difluorophenyl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3ag), typical procedure A
A 10 mL two necked round bottomed flask equipped with magnetic spin bar, rubber septum, and reflux condenser was charged with fine magnesium shavings (96 mg, 4.0 mmol). The apparatus was dried with heat gun (at 300 °C) in vacuum and refilled with dry argon, and this operation was repeated three times. The glassware was allowed to cool to room temperature and 1-bromo-3,5-difluorobenzene (96 mg, 0.50 mmol) was injected via syringe, and the mixture was stirred for 2 min. Then, an additional amount of 1-bromo-3,5-difluorobenzene (288 mg, 1.50 mmol) in dry THF (1 mL) was added dropwise, which should bring the content of the flask to the boiling. If this did not happen, the mixture was carefully heated to 55 °C, to initiate the reaction and boiling of the solvent. When the exothermic effect seized, the dark-brown mixture was stirred for 2 h at room temperature, and (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) 132 mg (0.50 mmol) was added. This typically leads to discoloration or color change, and heating of the mixture to the boiling point once again. The stirring was continued for 10 min, then the mixture was cooled to 0 °C in ice bath and transferred via cannula to a cold concentrated HCl (2 mL). To achieve the quantitative transfer the apparatus was rinsed with small portions of ethyl acetate, which was combined with the acid solution. The mixture was digested for 1–2 h at 0 °C, then water was added (20 mL). The product was extracted with ethyl acetate (4 × 25 mL), concentrated in vacuum and purified by preparative column chromatography. The titled material was obtained as colorless crystals, mp 168.8–172.9 °C (EtOH/hexane). Rf 0.44 (EtOAc/hexane, 1:4), Rf 0.53 (benzene). Yield 122 mg (0.34 mmol, 68%). 1H NMR (400 MHz, CDCl3) δ 8.17–8.09 (m, 2H), 7.66 (d, J = 1.6 Hz, 1H), 7.58–7.49 (m, 3H), 7.46 (d, J = 7.7 Hz, 1H), 7.22 (ddd, J = 7.7, 6.6, 2.2 Hz, 1H), 7.00–6.92 (m, 2H), 6.55 (tt, J = 8.8, 2.2 Hz, 1H), 6.43–6.36 (m, 2H), 5.03 (d, J = 1.4 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 176.8, 163.0 (dd, J = 250.6, 12.8 Hz, 2C), 152.9, 148.1, 136.9 (t, J = 9.2 Hz), 135.1, 132.0, 131.4, 130.6, 129.3 (2C), 128.3 (2C), 126.4, 124.3, 121.6, 110.9 (dd, J = 19.2, 6.8 Hz, 2C), 103.9 (t, J = 25.1 Hz), 97.5, 60.9 (t, J = 1.7 Hz); 19F NMR (377 MHz, CDCl3) δ −108.3 (s). FT IR (ZnSe, cm−1): 3088, 2095, 1700, 1623, 1586, 1586, 1449, 1323, 1279, 1138, 1116, 993; HRMS (ES TOF) m/z: (M + Na)+ calcd for C22H14F2N2NaO 383.0966; found 383.0964.
(3R*,4′S*)-2,4′-Diphenyl-4′H-spiro[indole-3,5′-isoxazole] (3aa)16
This compound was prepared via typical procedure A starting with (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and phenylmagnesium bromide (7a) generated from bromobenzene (312 mg, 2.0 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless oil. Yield 117 mg (0.36 mmol, 72%) (method A), dr 10:1, a mixture of inseparable (3R*,4′S*)- and (3S*,4′S*)-diastereomers. 1H NMR (400 MHz, CDCl3) δ 8.21–8.14 (m, 2H), 7.70 (d, J = 1.6 Hz, 1H), 7.59–7.50 (m, 3H), 7.44–7.37 (m, 1H), 7.19–7.05 (m, 4H), 6.99–6.80 (m, 4H), 5.10 (d, J = 1.7 Hz, 1H).
(3R*,4′S*)-2-Phenyl-4′-(o-tolyl)-4′H-spiro[indole-3,5′-isoxazole] (3ab)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and o-tolylmagnesium bromide (7b), generated from 1-bromo-2-methylbenzene (340 mg, 2.00 mmol). Eluent for column chromatography: benzene/hexane, 1:1. The titled material was obtained as colorless oil. Rf 0.46 (EtOAc/hexane, 1:4), Rf 0.78 (benzene). Yield 133 mg (0.39 mmol, 79%). 1H NMR (400 MHz, CDCl3) δ 8.10 (dd, J = 8.1, 1.4 Hz, 2H), 7.61–7.49 (m, 4H), 7.46 (d, J = 7.7 Hz, 1H), 7.24–7.13 (m, 3H), 7.08–7.02 (m, 1H), 6.85 (ddd, J = 11.6, 5.9, 2.1 Hz, 2H), 6.77 (d, J = 7.3 Hz, 1H), 5.14 (d, J = 1.6 Hz, 1H), 1.73 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 178.3, 153.7, 149.8, 136.9, 135.2, 132.5, 132.2, 131.7, 130.8, 130.1, 129.2 (2C), 128.3, 128.22 (2C), 128.19, 126.04, 125.96, 125.4, 121.3, 96.5, 58.3, 19.1; FT IR (ZnSe, cm−1): 3074, 2927, 2857, 1989, 1941, 1735, 1528, 1458, 1444, 1381, 1264, 1074; HRMS (ES TOF) m/z: (M + Na)+ calcd for C23H18N2NaO 361.1311; found 361.1308.
(3R*,4′S*)-4′-(4-Isopropylphenyl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3ac)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and (4-isopropylphenyl)magnesium bromide (7c) generated from 1-bromo-4-isopropylbenzene (396 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless oil. Rf 0.36 (EtOAc/hexane, 1:4), 0.38 (benzene). Yield 130 mg (0.36 mmol, 71%). 1H NMR (400 MHz, CDCl3) δ 8.22–8.12 (m, 2H), 7.67 (d, J = 1.6 Hz, 1H), 7.57–7.49 (m, 3H), 7.40 (d, J = 7.7 Hz, 1H), 7.14 (td, J = 7.5, 1.4 Hz, 1H), 6.95 (d, J = 8.1 Hz, 2H), 6.91 (dd, J = 7.4, 1.0 Hz, 1H), 6.86 (td, J = 7.4, 0.8 Hz, 1H), 6.76 (d, J = 8.1 Hz, 2H), 5.04 (d, J = 1.4 Hz, 1H), 2.81–2.65 (m, 1H), 1.10 (d, J = 6.9 Hz, 3H), 1.10 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.5, 153.0, 149.7, 149.0, 135.8, 131.8, 131.7, 130.5, 129.9, 129.2 (2C), 128.4 (2C), 127.8 (2C), 126.7 (2C), 125.9, 124.8, 121.2, 97.8, 61.5, 33.8, 23.9, 23.9; FT IR (ZnSe, cm−1): 3055, 2960, 2872, 1958, 1909, 1610, 1537, 1460, 1447, 1268, 1197, 1019; HRMS (ES TOF) m/z: (M + H)+ calcd for C25H23N2O 367.1805; found 367.1806.
(3R*,4′S*)-4′-(3,4-Dimethylphenyl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3ad)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and (3,4-dimethylphenyl)magnesium bromide (7d) generated from 4-bromo-1,2-dimethylbenzene (368 mg, 2.0 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish oil. Rf 0.55 (benzene). Yield 137 mg (0.39 mmol, 78%), dr 5:1, a mixture of inseparable (3R*,4′S*)- and (3S*,4′S*)-diastereomers. Individual (3R*,4′S*)-diastereomer can be obtained after crystallization of this material from benzene.1 H NMR (400 MHz, CDCl3) δ 8.20–8.12 (m, 2H), 7.65 (d, J = 1.3 Hz, 1H), 7.59–7.49 (m, 3H), 7.41 (d, J = 7.7 Hz, 1H), 7.16 (t, J = 7.7 Hz, 1H), 6.98–6.94 (m, 1H), 6.90 (t, J = 7.3 Hz, 1H), 6.85 (d, J = 7.9 Hz, 1H), 6.59 (d, J = 6.8 Hz, 2H), 5.01 (s, 1H), 2.08 (s, 3H), 2.07 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.6, 152.9, 149.9, 136.9, 136.6, 135.8, 131.8, 131.7, 130.3, 130.0, 129.8, 129.2 (2C), 129.1, 128.4 (2C), 126.0, 125.3, 124.8, 121.1, 97.7, 61.5, 19.7, 19.5; FT IR (ZnSe, cm−1): 3065, 2929, 2852, 1956, 1904, 1726, 1614, 1537, 1444, 1264; HRMS (ES TOF) m/z: (M + Na)+ calcd for C24H20N2NaO 375.1268; found 375.1473.
(3R*,4′S*)-4′-(Naphthalen-2-yl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3ae)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and naphthalen-2-ylmagnesium bromide (7e) generated from 2-bromonaphthalene (412 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless solid, mp 198.6–202.5 °C (acetone/hexanes). Rf 0.69 (acetone/hexanes 1:5), Rf 0.53 (benzene). Yield 148 mg (0.4 mmol, 79%). 1H NMR (400 MHz, CDCl3) δ 8.28–8.23 (m, 2H), 7.76–7.73 (m, 1H), 7.70–7.55 (m, 5H), 7.48–7.37 (m, 2H), 7.31 (t, J = 7.3 Hz, 1H), 7.25–7.16 (m, 3H), 6.94 (t, J = 7.4 Hz, 1H), 6.70 (d, J = 7.1 Hz, 1H), 6.65 (t, J = 7.4 Hz, 1H), 5.67 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.5, 153.4, 149.7, 135.1, 133.6, 132.1, 131.9, 131.6, 130.7, 129.9, 129.3 (2C), 129.1, 128.6, 128.3 (2C), 126.7, 126.1, 125.8, 125.6, 124.8, 124.7, 122.6, 121.2, 96.8, 57.6; FTIR (ZnSe, film, cm−1): 3070, 2909, 1965, 1746, 1614, 1546, 1451, 1240, 1193, 1059; HRMS (ES TOF) m/z: (M + Na)+ calcd for C26H18N2NaO 397.1311; found 397.1306.
(3R*,4′S*)-4′-(3-Fluorophenyl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3af)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and (3-fluorophenyl)magnesium bromide (7f) generated from 1-bromo-3-fluorobenzene (348 mg, 2.0 mmol). Eluent for column chromatography: benzene/hexane, 1:1. The titled material was obtained as colorless oil. Rf 0.68 (benzene). Yield 125 mg (0.37 mmol, 73%) 1H NMR (400 MHz, CDCl3) δ 8.19–8.11 (m, 2H), 7.68 (d, J = 1.7 Hz, 1H), 7.60–7.50 (m, 3H), 7.42 (d, J = 7.7 Hz, 1H), 7.18 (td, J = 7.9, 6.6, 2.3 Hz, 1H), 7.13–7.02 (m, 1H), 6.96–6.88 (m, 2H), 6.80 (td, J = 8.4, 2.6 Hz, 1H), 6.65–6.61 (m, 1H), 6.60–6.55 (m, 1H), 5.08 (d, J = 1.6 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.1, 162.7 (d, J = 247.7 Hz), 153.0, 148.8, 135.5 (d, J = 7.2 Hz), 135.4, 131.9, 131.6, 130.32 (d, J = 8.5 Hz), 130.28, 129.3 (2C), 128.3 (2C), 126.2, 124.5, 123.5 (d, J = 3.0 Hz), 121.4, 115.3 (d, J = 21.1 Hz), 114.8 (d, J = 22.1 Hz), 97.6, 61.2 (d, J = 1.9 Hz); 19F NMR (376 MHz, chloroform-d) δ −112.0 (s); HRMS (ES TOF) m/z: (M + Na)+ calcd for C22H15FN2NaO 365.1061; found 365.1064.
(3R*,4′S*)-4′-(4-Chloro-3-fluorophenyl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3ah)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and (4-chloro-3-fluorophenyl)magnesium bromide (7h) generated from 4-bromo-1-chloro-2-fluorobenzene (418 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless oil. Rf 0.45 (EtOAc/hexane, 1:4), Rf 0.35 (benzene). Yield 112 mg (0.30 mmol, 59%). 1H NMR (400 MHz, CDCl3) δ 8.17–8.08 (m, 2H), 7.65 (d, J = 1.6 Hz, 1H), 7.60–7.49 (m, 3H), 7.44 (d, J = 7.7 Hz, 1H), 7.24–7.19 (m, 1H), 7.13 (t, J = 7.9 Hz, 1H), 7.01–6.89 (m, 2H), 6.66 (dd, J = 9.5, 2.0 Hz, 1H), 6.58 (dd, J = 8.2, 1.7 Hz, 1H), 5.04 (d, J = 1.4 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 176.9, 158.0 (d, J = 250.7 Hz), 152.9, 148.4, 135.2, 133.9 (d, J = 6.2 Hz), 132.0, 131.4, 130.9, 130.6, 129.3 (2C), 128.3 (2C), 126.4, 124.3, 124.2 (d, J = 3.5 Hz), 121.6, 121.0 (d, J = 17.6 Hz), 116.0 (d, J = 21.8 Hz), 97.6, 60.8; 19F NMR (377 MHz, CDCl3) δ −113.9 (s). FT IR (ZnSe, cm−1): 3063, 2912, 2854, 2586, 1747, 1733, 1539, 1488, 1455, 1427, 1374, 1244, 1061, 1008; HRMS (ES TOF) m/z: (M + Na)+ calcd for C22H14ClFN2NaO1 399.0671; found 399.0675.
(3R*,4′S*)-4′-(4-Fluoro-3-methylphenyl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3ai)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and (4-fluoro-3-methylphenyl)magnesium bromide (7i) generated from 4-bromo-1-fluoro-2-methylbenzene (376 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless oil. Rf 0.44 (EtOAc/hexane, 1:4), Rf 0.43 (benzene). Yield 126 mg (0.35 mmol, 71%). 1H NMR (400 MHz, CDCl3) δ 8.22–8.08 (m, 2H), 7.65 (d, J = 1.6 Hz, 1H), 7.59–7.48 (m, 3H), 7.42 (d, J = 7.7 Hz, 1H), 7.22–7.11 (m, 1H), 6.96–6.87 (m, 2H), 6.76–6.70 (m, 1H), 6.65 (d, J = 5.8 Hz, 2H), 5.02 (d, J = 1.2 Hz, 1H), 2.09 (d, J = 1.7 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.4, 160.9 (d, J = 246.3 Hz), 153.0, 149.4, 135.7, 131.8, 131.7, 130.9 (d, J = 5.4 Hz), 130.2, 129.2 (2C), 128.6 (d, J = 3.8 Hz), 128.3 (2C), 126.7 (d, J = 8.3 Hz), 126.1, 125.3 (d, J = 17.8 Hz), 124.6, 121.3, 115.3 (d, J = 22.8 Hz), 97.7, 61.1, 14.5 (d, J = 3.5 Hz); 19F NMR (377 MHz, CDCl3) δ −117.7 (s). FT IR (ZnSe, cm−1): 3055, 2927, 2846, 1962, 1735, 1601, 1535, 1502, 1444, 1242, 1208, 1123, 1023; HRMS (ES TOF) m/z: (M + H)+ calcd for C23H18FN2O 357.1398; found 357.1391.
(3R*,4′S*)-4′-(3-Methoxyphenyl)-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3aj)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and (3-methoxyphenyl)magnesium bromide (7j) generated from 1-bromo-3-methoxybenzene (372 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless solid, mp 114.9–119.1 °C (benzene/hexanes). Rf 0.56 (EtOAc/hexanes 1:4), Rf 0.37 (benzene). Yield 129 mg (0.37 mmol, 73%). 1H NMR (400 MHz, DMSO) δ 8.30 (s, 1H), 8.07 (d, J = 7.4 Hz, 2H), 7.62 (d, J = 7.3 Hz, 3H), 7.41 (d, J = 7.6 Hz, 1H), 7.20 (t, J = 7.5 Hz, 1H), 7.07–6.98 (m, 2H), 6.95 (t, J = 7.4 Hz, 1H), 6.65 (d, J = 7.9 Hz, 1H), 6.43 (s, 2H), 5.21 (s, 1H), 3.60 (s, 3H); 13C{1H} NMR (101 MHz, DMSO) δ 177.4, 159.7, 153.1, 149.4, 135.7, 134.6 (2C), 131.8, 130.1, 129.7, 129.2 (2C), 128.3 (2C), 126.1, 124.6, 121.2, 120.1, 113.7, 113.5, 97.7, 61.6, 55.3; FTIR (ZnSe, film, cm−1): 3432, 2956, 2931, 2854, 1725, 1601, 1460, 1270, 1120, 1068; HRMS (ES TOF) m/z: (M + Na)+ calcd for C23H18N2NaO2 377.1260; found 377.1262.
(3R*,4′S*)-2-Phenyl-4′-propyl-4′H-spiro[indole-3,5′-isoxazole] (3ak)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and n-propylmagnesium bromide (7k) generated from 1-bromopropane (244 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish oil. Rf 0.35 (benzene). Yield 90 mg (0.31 mmol, 62%). 1H NMR (400 MHz, CDCl3) δ 8.11–7.99 (m, 2H), 7.62 (d, J = 7.7 Hz, 1H), 7.56–7.44 (m, 3H), 7.44–7.38 (m, 2H), 7.36 (d, J = 7.2 Hz, 1H), 7.25–7.16 (m, 1H), 3.78–3.68 (m, 1H), 1.58–1.42 (m, 1H), 1.31–1.14 (m, 1H), 1.14–1.00 (m, 1H), 1.00–0.78 (m, 1H), 0.64 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.8, 153.4, 151.1, 135.9, 131.7, 131.5, 130.5, 129.0 (2C), 128.2 (2C), 126.4, 124.1, 121.8, 96.5, 56.1, 30.5, 21.1, 13.8; FT IR (ZnSe, cm−1): 3058, 2928, 1958, 1911, 1710, 1608, 1535, 1446, 1264; HRMS (ES TOF) m/z: (M + Na)+ calcd for C19H18N2NaO 313.1311; found 313.1308.
(3R*,4′S*)-4′-Pentyl-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3al)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and n-pentylmagnesium bromide generated from 1-bromopentane (7l) (300 mg, 2.00 mmol). Eluent for column chromatography: benzene/hexane, 1:1. The titled material was obtained as yellowish oil. Rf 0.41 (EtOAc/hexane, 1:4), Rf 0.57 (benzene). Yield 102 mg (0.32 mmol, 64%) 1H NMR (400 MHz, chloroform-d) δ 8.08–8.02 (m, 2H), 7.61 (d, J = 7.7 Hz, 1H), 7.55–7.44 (m, 3H), 7.44–7.37 (m, 2H), 7.35 (d, J = 7.1 Hz, 1H), 7.22 (td, J = 7.5, 0.9 Hz, 1H), 3.73 (td, J = 7.9, 1.3 Hz, 1H), 1.56–1.44 (m, 1H), 1.26–1.18 (m, 1H), 1.11–0.79 (m, 6H), 0.69 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.8, 153.6, 151.2, 136.0, 131.7, 131.6, 130.5, 129.0 (2C), 128.2 (2C), 126.3, 124.1, 121.8, 96.6, 56.3, 31.4, 28.4, 27.4, 22.2, 13.9; FT IR (ZnSe, cm−1): 3059, 2923, 1963, 1537, 1453, 1374, 1273, 1195, 1028; HRMS (ES TOF) m/z: (M + Na)+ calcd for C21H22N2NaO 341.1624; found 341.1617.
(3R*,4′S*)-4′-Octyl-2-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3am)
This compound was prepared via typical procedure A from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and n-octylmagnesium bromide (7m) generated from 1-bromooctane (384 mg, 2.00 mmol). Eluent for column chromatography: benzene/hexane, 1:1. The titled material was obtained as yellowish oil. Rf 0.44 (EtOAc/hexane, 1:4), Rf 0.62 (benzene/hexane, 1:1). Yield 144 mg (0.40 mmol, 80%). 1H NMR (400 MHz, chloroform-d) δ 8.10–8.00 (m, 2H), 7.61 (d, J = 7.7 Hz, 1H), 7.54–7.44 (m, 3H), 7.43–7.38 (m, 2H), 7.35 (d, J = 7.3 Hz, 1H), 7.22 (t, J = 7.4 Hz, 1H), 3.76–3.70 (m, 1H), 1.54–1.43 (m, 1H), 1.30–0.91 (m, 13H), 0.83 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.8, 153.6, 151.2, 136.0, 131.7, 131.6, 130.6, 129.0 (2C), 128.2 (2C), 126.3, 124.1, 121.8, 96.6, 56.3, 31.8, 29.2, 29.1, 29.0, 28.5, 27.7, 22.7, 14.2; FT IR (ZnSe, cm−1): 3066, 2923, 2850, 2670, 1938, 1738, 1608, 1537, 1455, 1359, 1237, 1025; HRMS (ES TOF) m/z: (M + Na)+ calcd for C24H28N2NaO 383.2094; found 383.2095.
(3R*,4′S*)-2-(Naphthalen-2-yl)-4′-phenyl-4′H-spiro[indole-3,5′-isoxazole] (3ba)16
This compound was prepared via typical procedure A from (E)-2-(naphthalen-2-yl)-3-(2-nitrovinyl)-1H-indole (6b) (157 mg, 0.50 mmol) and phenylmagnesium bromide (7a) generated from bromobenzene (312 mg, 2.0 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish oil. Yield 159 mg (0.42 mmol, 85%); 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.33 (dd, J = 8.7, 1.7 Hz, 1H), 8.03–7.96 (m, 2H), 7.94–7.89 (m, 1H), 7.76 (d, J = 1.6 Hz, 1H), 7.64–7.53 (m, 2H), 7.45 (d, J = 7.7 Hz, 1H), 7.17 (td, J = 7.6, 1.3 Hz, 1H), 7.14–7.07 (m, 3H), 6.99 (d, J = 6.8 Hz, 1H), 6.93–6.84 (m, 3H), 5.19 (d, J = 1.4 Hz, 1H); HRMS (ES TOF) m/z: (M + H)+ calcd for C26H19N2O 375.1492; found 375.1490.
(3R*,4′S*)-4′-(3-Fluorophenyl)-2-(naphthalen-2-yl)-4′H-spiro[indole-3,5′-isoxazole] (3bf)
This compound was prepared via typical procedure A from (E)-2-(naphthalen-2-yl)-3-(2-nitrovinyl)-1H-indole (6b) (157 mg, 0.50 mmol) and 3-fluorophenylmagnesium bromide (7f) generated from 1-bromo-3-fluorobenzene (348 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless solid, mp 138.5–140.0 °C (benzene/hexanes), Rf 0.34 (EtOAc/hexanes 1:4), Rf 0.53 (benzene). Yield 145 mg (0.37 mmol, 74%). 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.31 (dd, J = 8.7, 1.6 Hz, 1H), 7.99 (t, J = 7.2 Hz, 2H), 7.91 (d, J = 7.5 Hz, 1H), 7.74 (d, J = 1.5 Hz, 1H), 7.65–7.53 (m, 2H), 7.47 (d, J = 7.7 Hz, 1H), 7.24–7.15 (m, 1H), 7.12–7.03 (m, 1H), 7.02–6.91 (m, 2H), 6.85–6.74 (m, 1H), 6.64 (d, J = 7.7 Hz, 1H), 6.62–6.57 (m, 1H), 5.16 (d, J = 1.1 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.1, 162.7 (d, J = 247.9 Hz), 153.0, 148.9, 135.53, 135.46 (d, J = 7.5 Hz), 135.0 (2C), 133.2, 130.4, 130.3 (d, J = 8.9 Hz), 129.5, 129.1, 128.9, 128.2, 128.0, 127.0, 126.2, 124.9, 124.5, 123.5 (d, J = 3.0 Hz), 121.4, 115.3 (d, J = 21.2 Hz), 114.8 (d, J = 22.4 Hz), 97.8, 61.7 (d, J = 1.3 Hz). 19F NMR (377 MHz, CDCl3) δ −111.9 (s). FTIR (ZnSe, film, cm−1): 3044, 2923, 1854, 1742, 1550, 1456, 1376, 1235, 1189, 1145; HRMS (ES TOF) m/z: (M + H)+ calcd for C26H18FN2O 393.1398; found 393.1400.
(3R*,4′S*)-4′-(4-Chloro-3-fluorophenyl)-2-(naphthalen-2-yl)-4′H-spiro[indole-3,5′-isoxazole] (3bh)
This compound was prepared via typical procedure A from (E)-2-(naphthalen-2-yl)-3-(2-nitrovinyl)-1H-indole (6b) (157 mg, 0.50 mmol) and (4-chloro-3-fluorophenyl)magnesium bromide (7h) generated from 4-bromo-1-chloro-2-fluorobenzene (416 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish solid, mp 154.9–158.3 °C (benzene/hexanes), Rf 0.61 (EtOAc/hexanes 1:4), Rf 0.34 (benzene). Yield 157 mg (0.37 mmol, 74%). 1H NMR (400 MHz, CDCl3) δ 8.55 (s, 1H), 8.30 (dd, J = 8.7, 1.3 Hz, 1H), 7.98 (t, J = 8.2 Hz, 2H), 7.91 (d, J = 7.7 Hz, 1H), 7.73–7.68 (m, 1H), 7.65–7.53 (m, 2H), 7.48 (d, J = 7.7 Hz, 1H), 7.26–7.18 (m, 1H), 7.13 (t, J = 7.9 Hz, 1H), 6.98 (d, J = 4.2 Hz, 2H), 6.67 (dd, J = 9.5, 1.6 Hz, 1H), 6.59 (d, J = 8.2 Hz, 1H), 5.12 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3) δ 176.9, 158.0 (d, J = 250.8 Hz), 153.0, 148.5, 135.3, 135.0, 133.9 (d, J = 6.5 Hz), 133.2, 130.9, 130.6, 129.5, 129.2, 128.9, 128.7, 128.2, 128.0, 127.0, 126.4, 124.9, 124.3, 124.2 (d, J = 3.7 Hz), 121.6, 121.0 (d, J = 17.6 Hz), 116.0 (d, J = 21.9 Hz), 97.7, 61.3; 19F NMR (377 MHz, CDCl3) δ −113.9 (s); FTIR (ZnSe, film, cm−1): 3055, 2927, 2579, 1941, 1749, 1733, 1536, 1506, 1419, 1240, 1195, 1169, 1063; HRMS (ES TOF) m/z: (M + Na)+ calcd for C26H16ClFN2NaO 449.0827; found 449.0822.
(3R*,4′S*)-4′-(3-Methoxyphenyl)-2-(naphthalen-2-yl)-4′H-spiro[indole-3,5′-isoxazole] (3bj)
This compound was prepared via typical procedure A from (E)-2-(naphthalen-2-yl)-3-(2-nitrovinyl)-1H-indole (6b) (157 mg, 0.50 mmol) and (3-methoxyphenyl)magnesium bromide (7j) generated from 1-bromo-3-methoxybenzene (372 mg, 2.0 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish solid, mp 172.1–174.4 °C (benzene/hexanes), Rf 0.50 (EtOAc/hexanes 1:4), Rf 0.76 (benzene/hexanes 1:1). Yield 132 mg (0.33 mmol, 65%). 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 1.8 Hz, 1H), 8.31 (dd, J = 8.6, 1.8 Hz, 1H), 8.04–7.96 (m, 2H), 7.94–7.87 (m, 1H), 7.74 (d, J = 1.6 Hz, 1H), 7.65–7.52 (m, 2H), 7.46 (d, J = 7.7 Hz, 1H), 7.19 (td, J = 7.6, 1.3 Hz, 1H), 7.07–6.98 (m, 2H), 6.96–6.88 (m, 1H), 6.62 (dd, J = 8.3, 2.5 Hz, 1H), 6.47 (d, J = 7.6 Hz, 1H), 6.37 (t, J = 2.1 Hz, 1H), 5.14 (d, J = 1.6 Hz, 1H), 3.64 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.4, 159.7, 153.1, 149.5, 135.8, 134.9, 134.6, 133.2, 130.2, 129.7, 129.5, 129.1, 129.0, 128.9, 128.1, 128.0, 126.9, 126.1, 125.0, 124.7, 121.3, 120.1, 113.7, 113.5, 97.8, 62.1, 55.3; FTIR (ZnSe, film, cm−1): 3070, 2931, 2561, 1747, 1535, 1508, 1451, 1272, 1242, 1043; HRMS (ES TOF) m/z: (M + Na)+ calcd for C27H20N2NaO2 427.1417; found 427.1405.
(3R*,4′S*)-2-(Naphthalen-2-yl)-4′-propyl-4′H-spiro[indole-3,5′-isoxazole] (3bk)
This compound was prepared via typical procedure A from (E)-2-(naphthalen-2-yl)-3-(2-nitrovinyl)-1H-indole (6b) (157 mg, 0.50 mmol) and n-propylmagnesium bromide (7k) generated from 1-bromopropane (244 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish oil. Rf 0.44 (EtOAc/hexane, 1:4), Rf 0.51 (benzene). Yield 121 mg (0.36 mmol, 71%). 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 8.24 (dd, J = 8.7, 1.6 Hz, 1H), 7.94 (d, J = 8.7 Hz, 2H), 7.91–7.84 (m, 1H), 7.66 (d, J = 7.7 Hz, 1H), 7.56 (pd, J = 7.0, 1.4 Hz, 2H), 7.47–7.35 (m, 3H), 7.28–7.21 (m, 1H), 3.87–3.78 (m, 1H), 1.57–1.43 (m, 1H), 1.30–1.18 (m, 1H), 1.13–1.00 (m, 1H), 0.97–0.86 (m, 1H), 0.63 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.8, 153.5, 151.2, 136.1, 134.9, 133.1, 130.6, 129.5, 128.9, 128.8 (2C), 128.0, 127.9, 126.8, 126.4, 124.9, 124.2, 121.8, 96.7, 56.6, 30.5, 21.2, 13.8; FT IR (ZnSe, cm−1): 3058, 2964, 1736, 1549, 1529, 1456, 1273, 1238, 1196; HRMS (ES TOF) m/z: (M + H)+ calcd for C23H21N2O 341.1648; found 341.1650.
(3R*,4′S*)-2-(Naphthalen-2-yl)-4′-octyl-4′H-spiro[indole-3,5′-isoxazole] (3bm)
This compound was prepared via typical procedure A from (E)-2-(naphthalen-2-yl)-3-(2-nitrovinyl)-1H-indole (6b) (157 mg, 0.50 mmol) and n-octylmagnesium bromide (7m) generated from 1-bromooctane (384 mg, 2.00 mmol). Eluent for column chromatography: benzene/hexane, 1:1. The titled material was obtained as yellow oil. Rf 0.43 (EtOAc/hexane, 1:4), Rf 0.60 (benzene/hexane, 1:1). Yield 166 mg (0.41 mmol, 81%), dr 15:1; 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.23 (dd, J = 8.6, 1.7 Hz, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.88 (dd, J = 7.8, 1.7 Hz, 1H), 7.66 (d, J = 7.7 Hz, 1H), 7.55 (pd, J = 6.9, 1.5 Hz, 2H), 7.47–7.36 (m, 3H), 7.26–7.21 (m, 2H), 3.81 (td, J = 7.8, 1.5 Hz, 1H), 1.60–1.45 (m, 1H), 1.37–1.14 (m, 3H), 1.12–0.86 (m, 9H), 0.81 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 177.9, 153.6, 151.3, 136.1, 134.9, 133.1, 130.6, 129.5, 129.0, 128.8 (2C), 128.0, 127.9, 126.8, 126.4, 124.9, 124.2, 121.9, 96.7, 56.8, 31.8, 29.2, 29.1, 29.0, 28.5, 27.7, 22.7, 14.2; FT IR (ZnSe, cm−1): 3063, 2930, 2854, 1945, 1914, 1733, 1601, 1537, 1453, 1358, 1277, 1233, 1128, 1012; HRMS (ES TOF) m/z: (M + H)+ calcd for C28H31N2O 411.2431; found 411.2429.
(3R*,4′S*)-4′-(3-Isopropylphenyl)-2-methyl-4′H-spiro[indole-3,5′-isoxazole] (3cc)
This compound was prepared via typical procedure A from (E)-2-methyl-3-(2-nitrovinyl)-1H-indole (6c) (101 mg, 0.50 mmol) and (4-isopropylphenyl)magnesium bromide (7c) generated from 1-bromo-4-isopropylbenzene (396 mg, 2.00 mmol). Eluent for column chromatography: EtOAc/hexane, 1:1. The titled material was obtained as dark yellow amorphous solid. Rf 0.41 (EtOAc/hexanes 1:1), Rf 0.56 (EtOAc/benzene 1:4). Yield 79 mg (0.26 mmol, 52%). 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 1.5 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.16 (t, J = 7.6 Hz, 1H), 7.10 (d, J = 8.0 Hz, 2H), 6.84 (d, J = 8.1 Hz, 2H), 6.72 (t, J = 7.5 Hz, 1H), 6.36 (d, J = 7.4 Hz, 1H), 4.57 (d, J = 1.2 Hz, 1H), 2.88–2.80 (m, 1H), 2.35 (s, 3H), 1.20 (d, J = 2.1 Hz, 3H), 1.18 (d, J = 2.1 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 181.1, 154.5, 149.6, 149.4, 133.0, 130.4, 130.2, 128.5 (2C), 127.0 (2C), 125.4 (2C), 120.3, 96.4, 58.4, 33.9, 24.03, 23.98, 15.5; FTIR (ZnSe, film, cm−1): 3062, 2953, 2586, 1742, 1599, 1458, 1429, 1242, 1118, 1052, 1023; HRMS (ES TOF) m/z: (M + Na)+ calcd for C20H20N2NaO 327.1468; found 327.1462.
(3R*,4′S*)-4′-(3-Fluorophenyl)-2-methyl-4′H-spiro[indole-3,5′-isoxazole] (3cf)
This compound was prepared via typical procedure A from (E)-2-methyl-3-(2-nitrovinyl)-1H-indole (6c) (101 mg, 0.50 mmol) and 3-fluorophenylmagnesium bromide (7f) generated from 1-bromo-3-fluorobenzene (348 mg, 2.0 mmol). Eluent for column chromatography: EtOAc/hexane, 1:1. The titled material was obtained as dark yellow amorphous solid. Rf 0.72 (EtOAc/hexanes 1:1), Rf 0.70 (EtOAc/benzene 1:4). Yield 73 mg (0.26 mmol, 52%). 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 1.3 Hz, 1H), 7.35 (d, J = 7.4 Hz, 1H), 7.25–7.15 (m, 2H), 6.95 (td, J = 8.3, 2.0 Hz, 1H), 6.80 (t, J = 7.5 Hz, 1H), 6.70 (d, J = 7.8 Hz, 1H), 6.69–6.62 (m, 1H), 6.44 (d, J = 7.4 Hz, 1H), 4.60 (d, J = 1.0 Hz, 1H), 2.36 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 180.7, 163.0 (d, J = 248.2 Hz), 154.5, 148.6, 135.6 (d, J = 7.1 Hz), 132.6, 130.6 (d, J = 8.3 Hz), 130.5, 125.7, 125.1, 124.2 (d, J = 3.0 Hz), 120.6, 115.7 (d, J = 12.3 Hz), 115.5 (d, J = 13.4 Hz), 96.4, 58.2 (d, J = 1.7 Hz), 15.4; 19F NMR (377 MHz, CDCl3) δ −111.6 (s). FTIR (ZnSe, film, cm−1): 3073, 2930, 2846, 2612, 1731, 1585, 1458, 1381, 1248, 1142, 1079; HRMS (ES TOF) m/z: (M + Na)+ calcd for C17H13FN2NaO 303.0904; found 303.0897.
(3R*,4′S*)-4′-(3,5-Difluorophenyl)-2-methyl-4′H-spiro[indole-3,5′-isoxazole] (3cg)
This compound was prepared via typical procedure A from (E)-2-methyl-3-(2-nitrovinyl)-1H-indole (6c) (101 mg, 0.50 mmol) and 3,5-difluorophenylmagnesium bromide (7g) generated from 1-bromo-3,5-difluorobenzene (384 mg, 2.00 mmol). Eluent for column chromatography: EtOAc/hexane, 1:1. The titled material was obtained as colorless solid, mp 136.9–139.3 °C (benzene/hexanes). Rf 0.68 (EtOAc/hexanes 1:1), Rf 0.72 (EtOAc/benzene 1:4). Yield 85 mg (0.29 mmol, 57%). 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 1.7 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 7.25–7.20 (m, 1H), 6.85 (t, J = 7.6 Hz, 1H), 6.71 (tt, J = 8.7, 2.2 Hz, 1H), 6.50–6.46 (m, 3H), 4.56 (d, J = 1.6 Hz, 1H), 2.34 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 180.4, 163.3 (dd, J = 251.0, 12.7 Hz, 2C), 154.4, 148.0, 137.0 (t, J = 9.0 Hz), 132.3, 130.8, 125.9, 124.9, 120.8, 111.6 (dd, J = 18.8, 7.5 Hz, 2C), 104.3 (t, J = 25.1 Hz), 96.3, 57.9 (t, J = 1.9 Hz), 15.3; 19F NMR (377 MHz, CDCl3) δ −107.9 (s). FTIR (ZnSe, film, cm−1): 3081, 2945, 2579, 2429, 1738, 1594, 1460, 1310, 1248, 1120, 997; HRMS (ES TOF) m/z: (M + Na)+ calcd for C17H12F2N2NaO 321.0810; found 321.0805.
(3R*,4′S*)-4′-(4-Chloro-3-fluorophenyl)-2-methyl-4′H-spiro[indole-3,5′-isoxazole] (3ch)
This compound was prepared via typical procedure A from (E)-2-methyl-3-(2-nitrovinyl)-1H-indole (6c) (101 mg, 0.50 mmol) and (4-chloro-3-fluorophenyl)magnesium bromide (7h) generated from 4-bromo-1-chloro-2-fluorobenzene (416 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish oil. Rf 0.86 (EtOAc/benzene, 1:4), Rf 0.25 (benzene). Yield 75 mg (0.24 mmol, 48%). 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 1.8 Hz, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.22 (td, J = 7.6, 1.1 Hz, 1H), 6.84 (td, J = 7.5, 0.8 Hz, 1H), 6.73 (dd, J = 9.4, 2.0 Hz, 1H), 6.65 (dd, J = 8.2, 1.6 Hz, 1H), 6.45 (d, J = 7.4 Hz, 1H), 4.56 (d, J = 1.7 Hz, 1H), 2.34 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 180.6, 158.2 (d, J = 251.2 Hz), 154.4, 148.3, 133.9 (d, J = 6.3 Hz), 132.4, 131.2, 130.7, 125.9, 125.0, 124.9 (d, J = 3.7 Hz), 121.4 (d, J = 17.6 Hz), 120.7, 116.7 (d, J = 21.8 Hz), 96.3, 57.7, 15.3; 19F NMR (377 MHz, CDCl3) δ −113.5 (s). FT IR (ZnSe, cm−1): 3070, 2927, 2857, 2308, 1751, 1606, 1489, 1453, 1424, 1242, 1068; HRMS (ES TOF) m/z: (M + Na)+ calcd for C17H12ClFN2NaO 337.0514; found 337.0513.
(3R*,4′S*)-4′-(3-Methoxyphenyl)-2-methyl-4′H-spiro[indole-3,5′-isoxazole] (3cj)
This compound was prepared via typical procedure A from (E)-2-methyl-3-(2-nitrovinyl)-1H-indole (6c) (101 mg, 0.50 mmol) and (3-methoxyphenyl)magnesium bromide (7j) generated from 1-bromo-3-methoxybenzene (372 mg, 2.0 mmol). Eluent for column chromatography: EtOAc/hexane, 1:1. The titled material was obtained as colorless solid, mp 84.2–87.9 °C (benzene/hexanes). Rf 0.66 (EtOAc/hexanes 1:1), Rf 0.68 (EtOAc/benzene 1:3). Yield 74 mg (0.25 mmol, 51%). 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 1.4 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 7.19 (d, J = 7.7 Hz, 1H), 7.15 (d, J = 8.0 Hz, 1H), 6.88–6.70 (m, 2H), 6.52 (d, J = 7.5 Hz, 1H), 6.46 (d, J = 7.4 Hz, 1H), 6.41 (t, J = 2.1 Hz, 1H), 4.58 (d, J = 1.0 Hz, 1H), 3.69 (s, 3H), 2.36 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 181.0, 160.0, 154.5, 149.1, 134.6, 132.9, 130.3, 130.0, 125.6, 125.3, 120.8, 120.3, 114.2, 114.1, 96.4, 58.6, 55.4, 15.4. FTIR (ZnSe, film, cm−1): 3077, 2600, 1740, 1597, 1240, 1120, 1039; HRMS (ES TOF) m/z: (M + Na)+ calcd for C18H16N2NaO2 315.1104; found 315.1102.
(3R*,4′S*)-2-Methyl-4′-octyl-4′H-spiro[indole-3,5′-isoxazole] (3cm)
This compound was prepared via typical procedure A from (E)-2-methyl-3-(2-nitrovinyl)-1H-indole (6c) (101 mg, 0.50 mmol) and n-octylmagnesium bromide (7m) generated from 1-bromooctane (384 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish oil. Rf 0.69 (EtOAc/hexane, 1:1), Rf 0.43 (benzene). Yield 79 mg (0.27 mmol, 53%) 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.7 Hz, 1H), 7.41–7.29 (m, 3H), 7.17 (t, J = 7.4 Hz, 1H), 3.50–3.41 (m, 1H), 2.29 (s, 3H), 1.64–1.51 (m, 1H), 1.40–1.02 (m, 13H), 0.85 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (400 MHz, CDCl3) δ 181.1, 153.6, 150.0, 133.8, 129.9, 125.2, 124.1, 120.2, 95.3, 52.8, 31.2, 28.7, 28.6, 28.5 (2C), 27.3, 22.1, 14.5, 13.6; FT IR (ZnSe, cm−1): 3052, 2920, 2854, 1608, 1458, 1378, 1255, 1242, 1100, 1015; HRMS (ES TOF) m/z: (M + H)+ calcd for C19H27N2O 299.2118; found 299.2111.
3-(1-(3,5-Difluorophenyl)-2-nitroethyl)-2-phenyl-1H-indole (12ag), typical procedure B
Grignard reagent 7g was prepared form 1-bromo-3,5-difluorobenzene (384 mg, 2.00 mmol) in THF and its reaction with (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) 132 mg (0.50 mmol) was carried out in the same manner as described in typical procedure A. Then, the reaction mixture was quenched with water (30 mL), and the resulting suspension was acidified with dilute acetic acid (0.3 M, 10 mL). The product was extracted with ethyl acetate (4 × 25 mL), concentrated in vacuum and purified by preparative column chromatography. The titled material was obtained as yellowish oil. Rf 0.65 (EtOAc/hexanes 1:6), Rf 0.46 (benzene/hexanes 2:1). Yield 117 mg (0.31 mmol, 62%). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.55–7.39 (m, 7H), 7.29–7.22 (m, 1H), 7.18–7.11 (m, 1H), 6.90–6.81 (m, 2H), 6.69 (tt, J = 8.8, 2.2 Hz, 1H), 5.29 (t, J = 7.8 Hz, 1H), 5.20–5.06 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 163.4 (dd, J = 249.4, 12.9 Hz, 2C), 144.1 (t, J = 8.5 Hz), 137.4, 136.1, 131.9, 129.3 (2C), 129.1, 128.9 (2C), 126.7, 123.0, 120.8, 119.6, 111.7, 110.7 (dd, J = 18.7, 7.2 Hz, 2C), 108.6, 103.0 (t, J = 25.3 Hz), 78.5, 40.5 (t, J = 2.2 Hz); 19F NMR (376 MHz, chloroform-d) δ −108.5 (s); FTIR (ZnSe, film, cm−1): 3411, 2916, 1725, 1621, 1596, 1552, 1455, 1374, 1312, 1244, 1116, 1041; HRMS (ES TOF) m/z: (M + Na)+ calcd for C22H16F2N2NaO2 401.1072; found 401.1082.
3-(2-Nitro-1-phenylethyl)-2-phenyl-1H-indole (12aa)27
This compound was prepared via typical procedure B from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and 1-bromobenzene (7a) (312 mg, 2.0 mmol). Eluent for column chromatography: EtOAc/hexane, 1:4. The titled material was obtained as colorless solid. Rf 0.63 (EtOAc/hexane, 1:1). Yield 154 mg (0.45 mmol, 91%). 1H NMR (400 MHz, chloroform-d) δ 8.05 (s, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.37–7.28 (m, 5H), 7.27–7.08 (m, 7H), 7.01 (t, J = 7.5 Hz, 1H), 5.22 (t, J = 8.0 Hz, 1H), 5.14–4.95 (m, 2H).
3-(2-Nitro-1-(o-tolyl)ethyl)-2-phenyl-1H-indole (12ab)
This compound was prepared via typical procedure B from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and o-tolylmagnesium bromide (7b), generated from 1-bromo-2-methylbenzene (340 mg, 2.0 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless crystals, mp 178.3–181.4 °C (benzene). Rf 0.69 (EtOAc/hexane, 1:4), Rf 0.63 (benzene/hexane, 1:1). Yield 163 mg (0.46 mmol, 92%). 1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H), 7.82 (d, J = 8.1 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.55 (d, J = 6.4 Hz, 4H), 7.47 (td, J = 6.0, 2.6 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.21–7.08 (m, 4H), 7.02 (t, J = 7.5 Hz, 1H), 5.58–5.43 (m, 2H), 5.32 (t, J = 8.2 Hz, 1H), 1.98 (s, 3H); 13C{1H} NMR (101 MHz, DMSO-d6) δ 138.1, 136.6, 136.3, 135.8, 132.6, 130.9, 128.8 (2C), 128.7 (2C), 128.2, 126.9, 126.8, 126.7, 126.0, 121.4, 120.1, 119.3, 111.7, 107.8, 77.3, 37.9, 18.7; FT IR (ZnSe, cm−1): 3396, 3055, 1951, 1764, 1550, 1455, 1380, 1317, 1237, 1120; HRMS (ES TOF) m/z: (M + H)+ calcd for C23H20N2O2 357.1417; found 357.1410.
3-(1-(4-Methoxyphenyl)-2-nitroethyl)-2-phenyl-1H-indole (12aj)
This compound was prepared via typical procedure B from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and (3-methoxyphenyl)magnesium bromide (7j) generated from 1-bromo-3-methoxybenzene (372 mg, 2.0 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellowish amorphous solid. Rf 0.39 (EtOAc/hexanes 1:4), Rf 0.48 (benzene). Yield 153 mg (0.41 mmol, 82%). 1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.47–7.42 (m, 5H), 7.37 (d, J = 8.1 Hz, 1H), 7.29–7.19 (m, 2H), 7.13 (t, J = 7.5 Hz, 1H), 6.96 (d, J = 7.7 Hz, 1H), 6.91 (s, 1H), 6.80 (dd, J = 8.2, 1.7 Hz, 1H), 5.31 (t, J = 7.9 Hz, 1H), 5.21–5.10 (m, 2H), 3.73 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 160.0, 141.7, 137.1, 136.2, 132.3, 130.0, 129.0 (2C), 128.9 (2C), 128.7, 127.1, 122.6, 120.4, 120.0, 119.8, 113.9, 112.3, 111.5, 109.6, 79.1, 55.3, 40.9; FTIR (ZnSe, film, cm−1): 3407, 2923, 1956, 1596, 1455, 1380, 1264, 1147, 1034; HRMS (ES TOF) m/z: (M + Na)+ calcd for C23H20N2NaO3 395.1366; found 395.1376.
3-(1-Nitropentan-2-yl)-2-phenyl-1H-indole (12ak, ROS 542)
This compound was prepared via typical procedure B from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and n-propylmagnesium bromide (7k) generated from 1-bromopropane (244 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as colorless amorphous solid, mp 133.0–134.6 °C (benzene). Rf 0.71 (EtOAc/hexane, 1:4), Rf 0.69 (benzene/hexane, 1:1). Yield 127 mg (0.41 mmol, 83%). 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.48 (dd, J = 20.9, 4.4 Hz, 5H), 7.39 (t, J = 4.0 Hz, 1H), 7.28–7.22 (m, 1H), 7.18 (td, J = 8.0, 7.1, 0.9 Hz, 1H), 4.90–4.76 (m, 2H), 4.04–3.87 (m, 1H), 2.05 (dtd, J = 13.3, 10.2, 5.2 Hz, 1H), 1.81–1.64 (m, 1H), 1.28–1.07 (m, 2H), 0.77 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 136.9, 136.1, 132.5, 128.9 (2C), 128.8 (2C), 128.4, 126.5, 122.3, 119.9, 119.5, 111.4, 109.9, 80.0, 36.1, 34.2, 20.6, 13.7; FT IR (ZnSe, cm−1): 3400, 3059, 2923, 2853, 1746, 1738, 1552, 1456, 1370, 1244, 1200, 1046; HRMS (ES TOF) m/z: (M + Na)+ calcd for C19H20N2NaO2 331.1417; found 331.1408.
3-(1-Nitroheptan-2-yl)-2-phenyl-1H-indole (12al)
This compound was prepared via typical procedure B from (E)-3-(2-nitrovinyl)-2-phenyl-1H-indole (6a) (132 mg, 0.50 mmol) and n-pentylmagnesium bromide (7l) generated from 1-bromopentane (300 mg, 2.00 mmol). Eluent for column chromatography: benzene. The titled material was obtained as yellow oil. Rf 0.36 (EtOAc/hexane, 1:4), Rf 0.64 (benzene/hexane, 1:1). Yield 128 mg (0.38 mmol, 76%). 1H NMR (400 MHz, chloroform-d) δ 8.10 (s, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.52–7.42 (m, 5H), 7.41–7.37 (m, 1H), 7.24 (ddd, J = 8.2, 7.1, 1.2 Hz, 1H), 7.17 (ddd, J = 8.1, 7.1, 1.1 Hz, 1H), 4.90–4.75 (m, 2H), 3.99–3.85 (m, 1H), 2.12–1.98 (m, 1H), 1.79–1.67 (m, 1H), 1.24–1.05 (m, 6H), 0.76 (t, J = 6.9 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 136.7, 135.9, 132.3, 128.7 (2C), 128.7 (2C), 128.2, 126.4, 122.1, 119.7, 119.3, 111.2, 109.8, 79.9, 36.1, 31.8, 31.2, 26.8, 22.2, 13.7; FT IR (ZnSe, cm−1): 3400, 3055, 2923, 2849, 1546, 1455, 1447, 1425, 1380, 1306, 1240, 1072; HRMS (ES TOF) m/z: (M + Na)+ calcd for C21H24N2NaO2 (M + Na)+ 359.1730; found 359.1722.
(3R*,4′S*)-2,4′-Diphenyl-4′H-spiro[indole-3,5′-isoxazole] (3aa) from nitroalkane (12aa)
A 10 mL two necked round bottomed flask was equipped with magnetic spin bar, rubber septum, and reflux condenser. The apparatus was dried with heat gun (at 300 °C) in vacuum and refilled with dry argon, and this operation was repeated three times. The glassware was allowed to cool to room temperature and LDA (2 M in THF, 1.00 mL, 2.00 mmol) was added via syringe, followed by 0.5 mL of benzene. 3-(2-Nitro-1-phenylethyl)-2-phenyl-1H-indole (12aa) (171 mg, 0.50 mmol) was introduced, and the reaction mixture was stirred at room temperature for 2 h. The work up with cold concentrated HCl was performed, followed by aqueous workup, extraction and isolation in the same manner as described in typical procedure A. The titled compound was isolated as colorless oil, yield 99 mg (0.31 mmol, 61%).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Synthetic studies performed in the frame of this project were supported by grants from the Russian Science Foundation (grant number 18-13-00238).
Notes and references
- A. Singh and G. P. Roth, Org. Lett., 2011, 13, 2118–2121 CrossRef CAS.
- A. Singh and G. P. Roth, Tetrahedron Lett., 2012, 53, 4889–4891 CrossRef CAS.
- S.-Y. Wu, W.-L. Chen, X.-P. Ma, C. Liang, G.-F. Su and D.-L. Mo, Adv. Synth. Catal., 2019, 361, 965–970 CrossRef CAS.
- C. J. A. Ribeiro, J. D. Amaral, C. M. P. Rodrigues, R. Moreira and M. M. M. Santos, Bioorg. Med. Chem., 2014, 22, 577–584 CrossRef CAS.
- A. V. Velikorodov, V. A. Ionova, O. V. Degtyarev and L. T. Sukhenko, Pharm. Chem. J., 2013, 46, 715–719 CrossRef CAS.
- H. M. Refat, J. Heterocycl. Chem., 2015, 52, 1488–1495 CrossRef CAS.
- A. A. El-Gendy and A. M. Ahmedy, Arch. Pharmacal Res., 2000, 23, 310–314 CrossRef CAS.
- M. S. K. Youssef and A. A. O. Abeed, Heterocycl. Commun., 2014, 20, 25–31 CAS.
- I. A. Khan, V. M. Balaramnavar and A. K. Saxena, Tetrahedron, 2012, 68, 10122–10129 CrossRef CAS.
- A. Dandia, R. Singh, G. Kumar, K. Arya and H. Sachdeva, Heterocycl. Commun., 2001, 7, 571–576 CAS.
- F. Risitano, G. Grassi, F. Foti, G. Bruno and A. Rotondo, Heterocycles, 2003, 60, 857–863 CrossRef CAS.
- C. J. A. Ribeiro, S. Praveen Kumar, R. Moreira and M. M. M. Santos, Tetrahedron Lett., 2012, 53, 281–284 CrossRef CAS.
- A. V. Velikorodov, O. Y. Poddubnyi, A. K. Kuanchalieva and O. O. Krivosheev, Russ. J. Org. Chem., 2010, 46, 1826–1829 CrossRef CAS.
- C.-H. Chen, Q.-Q. Liu, X.-P. Ma, Y. Feng, C. Liang, C.-X. Pan, G.-F. Su and D.-L. Mo, J. Org. Chem., 2017, 82, 6417–6425 CrossRef CAS.
- A. V. Aksenov, D. A. Aksenov, N. A. Aksenov, E. V. Aleksandrova and M. Rubin, J. Org. Chem., 2019, 84, 12420–12429 CrossRef CAS.
- A. V. Aksenov, D. A. Aksenov, N. A. Arutiunov, N. A. Aksenov, E. V. Aleksandrova, Z. Zhao, L. Du, A. Kornienko and M. Rubin, J. Org. Chem., 2019, 84, 7123–7137 CAS.
- A. V. Aksenov, N. A. Aksenov, D. A. Aksenov, V. F. Khamraev and M. Rubin, Chem. Commun., 2018, 54, 13260–13263 RSC.
- T. Betke, P. Rommelmann, K. Oike, Y. Asano and H. Groeger, Angew. Chem., Int. Ed., 2017, 56, 12361–12366 CrossRef CAS.
- I. Deb, S. John and I. N. N. Namboothiri, Tetrahedron, 2007, 63, 11991–11997 CrossRef CAS.
- R. C. Dhakal and R. K. Dieter, Org. Lett., 2014, 16, 1362–1365 CrossRef CAS.
- S.-C. Lu, P.-R. Zheng and G. Liu, J. Org. Chem., 2012, 77, 7711–7717 CrossRef CAS.
- P. Reddy and R. Bandichhor, Tetrahedron Lett., 2013, 54, 3911–3915 CrossRef CAS.
- M. Tissot and A. Alexakis, Chem.–Eur. J., 2013, 19, 11352–11363 CrossRef CAS.
- K. Wood, D. S. C. Black and N. Kumar, Tetrahedron, 2011, 67, 4093–4102 CrossRef CAS.
- K. Wood, D. S. C. Black, I. N. N. Namboothiri and N. Kumar, Tetrahedron Lett., 2010, 51, 1606–1608 CrossRef CAS.
- C.-F. Yao, K.-H. Kao, J.-T. Liu, C.-M. Chu, Y. Wang, W.-C. Chen, Y.-M. Lin, W.-W. Lin, M.-C. Yan, J.-Y. Liu, M.-C. Chuang and J.-L. Shiue, Tetrahedron, 1998, 54, 791–822 CrossRef CAS.
- C. Praveen, K. Karthikeyan and P. T. Perumal, Tetrahedron, 2009, 65, 9244–9255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2024756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10219a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Dmitrii A. Aksenov
*a,
Dmitrii A. Aksenov