
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCe3+-enriched spherical porous ceria with an enhanced oxygen storage capacity†
Ayano Taniguchia,
Yoshitaka Kumabea,
Kai Kan abc,
Masataka Ohtani*abc and
Kazuya Kobiro*abc
abc,
Masataka Ohtani*abc and
Kazuya Kobiro*abc
aSchool of Environmental Science and Engineering, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami, Kochi 782-8502, Japan. E-mail: ohtani.masataka@kochi-tech.ac.jp; kobiro.kazuya@kochi-tech.ac.jp
bLaboratory for Structural Nanochemistry, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami, Kochi 782-8502, Japan
cResearch Center for Molecular Design, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami, Kochi 782-8502, Japan
First published on 1st February 2021
Abstract
Porous ceria was obtained using a unique solvothermal reaction in acetonitrile, applying high temperature and pressure. The resulting material comprised homogeneous and monodisperse spheres and exhibited an extremely large surface area of 152 m2 g−1. From catalytic performance evaluation by vapor- and liquid-phase reactions, the synthesized porous ceria showed superior and different reaction activity compared with commercial CeO2. To examine the origin of the reaction activity of the present porous ceria, synchrotron hard X-ray photoelectron spectroscopy (HAXPES) measurements were carried out. The systematic study of HAXPES measurements revealed that the obtained porous ceria with the present solvothermal method contained a very high concentration of Ce3+. Moreover, O2-pulse adsorption analyses demonstrated a significant oxygen adsorption capacity exceeding 268 μmol-O g−1 at 400 °C owing to its high contents of Ce3+ species.
Introduction
Ceria is well known as a catalyst or a catalyst support with applications in many different reactions.1 Especially in the case of oxidation reactions, the presence of Ce3+ ions (ascribed to defects in CeO2 crystals) and the Ce3+/Ce4+ redox cycle are believed to be important in terms of establishing the catalytic effects of ceria.2–4 Additionally, the oxygen storage capacity (OSC) of this material is also directly affected by oxygen defects due to the presence of neighbouring Ce3+,5 and so there has been a significant effort to develop methods of synthesizing ceria with a high Ce3+ content. However, it is challenging to obtain ceria with a high proportion of stable Ce3+ such that this lower valence state is maintained over time, because Ce3+ is easily oxidized to the more thermodynamically stable Ce4+ state in air.6,7One preparation method that has been successfully demonstrated is the reduction of CeO2 by Ce metal at high temperatures in the vicinity of 1500 °C,8 although sintering of the ceria particles inevitably leads to a small surface area. The reduction of CeO2 nanosheets, which have relatively large surface areas, is an alternative approach, but involves a more complex two-step process.9 Mesoporous CeO2 with an increased OSC has also been obtained using hard templates such as SBA-15, although the template must subsequently be removed.10 There are several reports related on high OSC values of un-doped CeO2 determined by several different methods, for example 156 μmol-O2 g−1 (determined by thermogravimetry at 200 °C),11 461 μmol-O g−1 (determined by thermogravimetry at 300 °C)12 and 554 μmol-O g−1 (determined by CO-pulse at 400 °C).13 Finally, the doping of CeO2 with different elements, including Si, Ti, Y, Zr, La and Cr, has been employed as a means of introducing stable defects based on lattice strain into ceria crystals to improve the OSC.14–16
Recently, Hao et al. reported a unique procedure yielding ceria nanocrystals with a high concentration of Ce3+ based on an improved hydrothermal method.17 Their work showed that smaller nanocrystals contained higher levels of Ce3+ defects and that these defects were primarily situated at the surfaces of the nanoparticles. Additionally, the proportion of defects (that is, unsaturated coordination sites) was found to increase as the particle size decreased, and these defects were primarily present at the corners and edges of the nanocrystals.
As can be seen from the above examples, the concentration of Ce3+ in ceria is highly dependent on both the synthetic methodology and the reaction environment. Our group previously established a simple one-step solvothermal method for the preparation of submicron-sized, porous spheres made of oxides such as SiO2, TiO2, ZrO2 and CeO2 as well as composites of these oxides. These materials exhibited large surface areas and unique concave-convex surface structures on the nanoscale.18–20 These oxides were termed micro/mesoporously architected roundly integrated metal oxides (MARIMOs). In prior work, an Au metal nanoparticle catalyst deposited on a TiO2 MARIMO substrate showed excellent resistance to temperature stress during the CO oxidation reaction, without sintering of the Au nanoparticles.21 The submicron spherical porous morphology of these MARIMOs also favors heterogeneous catalysis applications, because these spheres are more readily dispersed than standard nanoparticles, which tend to agglomerate.
We considered that a solvothermal reaction performed under a reductive atmosphere over a very short time span would produce an appreciable concentration of Ce3+ defects in ceria nanocrystals, based on previous syntheses of metal oxide nanoparticles.22–24 Therefore, in the present work, we applied this technique to produce a ceria MARIMO comprising nanocrystals rich in Ce3+ by optimizing the solvothermal reaction conditions (including the solvent, additives, temperature and time). The unique properties of the resulting ceria MARIMO were examined and are discussed herein.
Experimental details
Materials
Methanol, acetonitrile, toluene, formic acid, acetic acid, benzoic acid, phthalic acid, ethylene glycol, diethylene glycol, cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O), potassium chloroplatinate(II) (K2PtCl4) and aniline were purchased from FUJIFILM Wako Pure Chemical Corporation. Triethylene glycol and n-tridecane was purchased from Tokyo Chemical Industry Co., Ltd. Benzyl alcohol was purchased from Nacalai Tesque Inc. Reference ceria catalyst, JRC-CEO-5, was received from Daiichi Kigenso Kagaku Kogyo Co., Ltd. All chemicals were used as received without further purification.Preparation of CeO2-AN-tEG
The ceria MARIMO (CeO2-AN-tEG) was synthesized according to a previously developed procedure for the preparation of a TiO2 MARIMO.18 An acetonitrile solution (140 mL) including Ce(NO3)3·6H2O (6.08 g, 14.0 mmol) and triethylene glycol (66.7 g, 444 mmol) was transferred to an SUS316 autoclave 500 mL volume high pressure micro reactor (MMJ-500, OM Labtech Co., Ltd. Available maximum temperature and pressure are 300 °C and 20 MPa, respectively.). The reactor was heated to 300 °C at a rate of 5.4 °C min−1. The temperature was kept at 300 °C for 10 min. Then, the reactor was cooled to room temperature to quench the reaction. The obtained product was centrifuged, washed with methanol, and then dried under vacuum for overnight at room temperature to give a powder. Preparation methods of other samples are described in ESI.†Catalyst preparation
CeO2-AN-tEG supported 1 wt% Pt nanoparticles catalyst was prepared by using a standard precipitation–deposition method.25 Ceria support (CeO2-AN-tEG, 1 g) was dispersed in 100 mL of reverse osmosis water. K2PtCl4 aqueous solution (5.18 mL, 10.0 mmol L−1) was added to the suspension with vigorous stirring at room temperature. After 2 h, the mixture was centrifuged to collect precipitate. The precipitate was washed with reverse osmosis water, and centrifuged. The obtained product was dried under vacuum overnight (ca. 15 h), and calcined in air at 300 °C for 4 h to get a powder, namely, 1 wt% Pt/CeO2-AN-tEG catalyst. 1 wt% Pt/JRC-CEO-5 catalyst was prepared in the similar procedure.Characterization
Transmission electron microscope (TEM) and scanning electron microscope (SEM) images were taken on a JEOL JEM-2100F microscope and a Hitachi SU8020 microscope, respectively. Energy-dispersive X-ray spectroscopy (EDX) were performed on an Oxford INCA X-Max 80 EDX spectrometer equipped on the TEM instrument. X-ray diffraction (XRD) patterns were measured using a Rigaku SmartLab diffractometer with graphite-monochromatized Cu-Kα radiation (X-ray wavelength: 1.5418 Å). Nitrogen adsorption/desorption isotherm measurements were carried out using a BEL BELSORP Mini II. Specific surface area and average pore size were calculated by the Brunauer–Emmett–Teller (BET) method and the Barrett–Joyner–Halenda (BJH) calculation, respectively. Thermogravimetry (TG) was carried out using a HITACHI STA7200RV. Hard X-ray photoelectron spectroscopy (HAXPES) spectra were measured at the SPring-8 BL15XU beam line. CO oxidation test, O2-pulse absorption measurement and H2 temperature-programmed reduction (H2-TPR) measurement were carried out using a MicrotracBEL BELCAT II.Nitrogen adsorption/desorption isotherm
Before performing measurement, CeO2 (as-synthesized CeO2-AN-tEG and JRC-CEO-5) were pre-treated by vacuum heating at 250 °C for 4 h to remove organic residue and absorbed moisture on surface.TG analysis
TG of CeO2-AN-tEG was performed to investigate burning or decomposing temperature of organic residue in the product and its amount. As-synthesized CeO2-AN-tEG (ca. 5 mg) was heated up to 800 °C from 25 °C ramp rate of 2 °C min−1 in air flow (100 mL min−1). Similarly, calcined CeO2-AN-tEG (300 °C, 1 h, in air) was also analysed to check amounts of organic residues.Analysis of HAXPES spectra
Peak fitting of Ce 3d HAXPES spectra was carried out by using KolXPD software. A background signal was fitted using well known Shirley method. All multiple peaks were fitted as Gaussian/Lorentzian peak. The multiple peaks were fitted to 10 peaks correspond to the spin–orbit split 3d5/2 (= v0, v, v′, v′′, v′′′) and 3d3/2 (= u0, u, u′, u′′, u′′′). The spin–orbit splitting was fixed to 18.4 eV. The peak intensity ratio of 3d5/2/3d3/2 was fixed to 1.5. The peaks of v0, v′, u0 and u′ and v, v′′, v′′′, u, u′′ and u′′′ correspond to the Ce(III) and Ce(IV) species, respectively. The Ce3+ concentration was calculated by following equation:Here, Ai shows the peak area of the i peak.
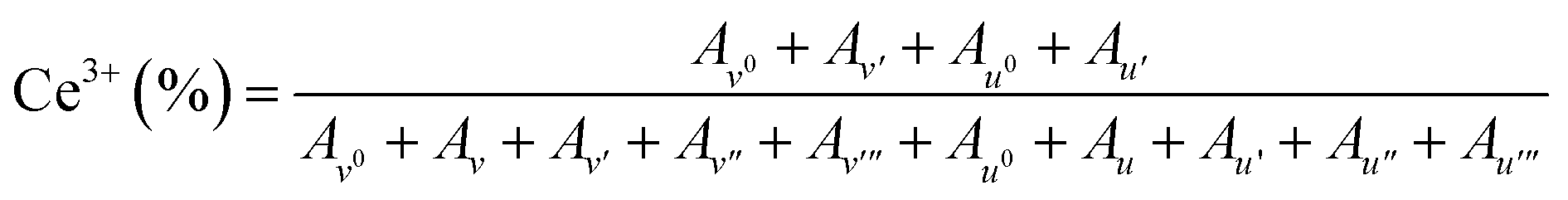
Vapor-phase catalytic reactions
Before performing CO oxidation, CeO2 (as-synthesized CeO2-AN-tEG and JRC-CEO-5) were calcined at 300 °C for 1 h in air to remove organic residue. Calcination temperature was decided by TG profile of as-synthesized CeO2-AN-tEG (Fig. S1†). The calcined CeO2 (50 mg) or prepared Pt/CeO2 catalyst (30 mg) was pre-treated at 300 °C for 60 min in O2 flow at a flow rate of 30 sccm (100 vol%) before reaction. Then, CO oxidation was performed using CO (4 sccm, 13 vol%), O2 (2 sccm, 7 vol%) and Ar (24 sccm, 80 vol%) in the temperature range of 150–350 °C for the test of CeO2 or in a temperature range of 50–300 °C for the test of Pt/CeO2. The outlet gas was analyzed by a GL Science GC-3200 gas chromatograph with an active carbon column and a thermal conductivity detector (TCD) using Ar as a carrier gas.Liquid-phase catalytic reaction
To a solution of benzyl alcohol (517 μL, 5.00 mmol), aniline (912 μL, 10.0 mmol) and n-tridecane (608 μL, 2.50 mmol) as an internal standard for GC in 10.0 mL toluene was added as-synthesized CeO2-AN-tEG (250 mg) under O2 atmosphere. The mixture was heated at 60 °C. Then, an aliquot amount of reaction mixture was withdrawn by syringe through a membrane filter. The product distribution was determined by a Shimadzu GC-17A gas chromatograph equipped with an Agilent Technologies DB-1 column (30 m × 0.25 mm × 0.25 μm), flame ionization detector (FID), and helium as a carrier gas. Only a negligible amount of benzaldehyde was detected during the reaction. Similarly, the reaction was also performed in the cases of JRC-CEO-5 and calcined CeO2-AN-tEG (300 °C, 1 h, in air) as catalysts. In pre-treatment, as-synthesized CeO2-AN-tEG was stirred in toluene (7.00 mL) at 60 °C for 48 h under O2 atmosphere prior to an addition of a mixture of a toluene solution (3.00 mL) of aniline (10.0 mmol), benzyl alcohol (5.00 mmol) and n-tridecane (2.50 mmol, internal standard for GC analysis). Reaction time was recorded after 48 h in the case of pre-treatment in Fig. S6a.†H2-TPR
Before H2-TPR measurement, CeO2 (as-synthesized CeO2-AN-tEG and JRC-CEO-5) were pre-treated for 60 min each at 300 °C and 50 °C in 20 vol% O2/Ar flow at flow rate of 30 sccm to remove organic residue and oxidation of CeO2. Then, H2-TPR measurement was performed using 6 vol% H2/Ar (30 sccm) in the temperature range of 50–700 °C (ramp rate was 10 °C min−1). The outlet gas was analyzed by a BELCAT II equipped with a TCD using Ar as a carrier gas.Cyclic O2-pulse adsorption
Prior to pulse measurement, CeO2 (as-synthesized CeO2-AN-tEG or JRC-CEO-5) was calcined at 300 or 400 °C for 60 min in air to remove organic residue, and the calcined CeO2 (50 mg) was pre-treated at 200 or 400 °C for 60 min in 10 vol% H2/He (30 sccm). Then, O2-pulse adsorption measurement was performed using 10 vol% O2/He (30 sccm) at 200 or 400 °C. These procedures were repeated several times. The outlet gas was analyzed by BELCAT II equipped with a TCD using Ar as a carrier gas.Results and discussion
Synthesis of porous CeO2 spheres
Porous CeO2 were synthesized by simple procedure involved heating a mixture of Ce(NO3)3·6H2O and an additive in a solvent to 300 °C (Table S1†). The particular combination of solvent and additive was found to modify the morphology of the product (Fig. 1). Interestingly, a combination of methanol and formic acid or acetic acid, which was previously employed in the synthesis of the TiO2 MARIMO,18,26 did not yield a monodisperse CeO2 MARIMO. Rather, irregularly aggregated porous spheres having a cubic crystal phase were obtained (Fig. 1a, b and 2a, b). Using benzoic acid as an additive, primary CeO2 nanoparticles with a cubic crystal phase were generated but did not aggregate to form secondary structures (Fig. 1c and 2c). Moreover, the usage of phthalic acid resulted in thin flakes (Fig. 1d) that generated an XRD pattern suggesting a crystal phase different from that of cubic ceria (Fig. 2d). Additional trials were performed using oxyethylene compounds as additives because these are well known chelating reagents based on the coordination of oxygen atoms to the central metal ions of metal salts.27,28 These trials produced cubic CeO2 aggregates with improved porous spherical morphologies in accordance with the number of oxyethylene units (Fig. 1e–g and 2e–g). However, the products were not monodispersed but rather showed significant aggregation of the spheres.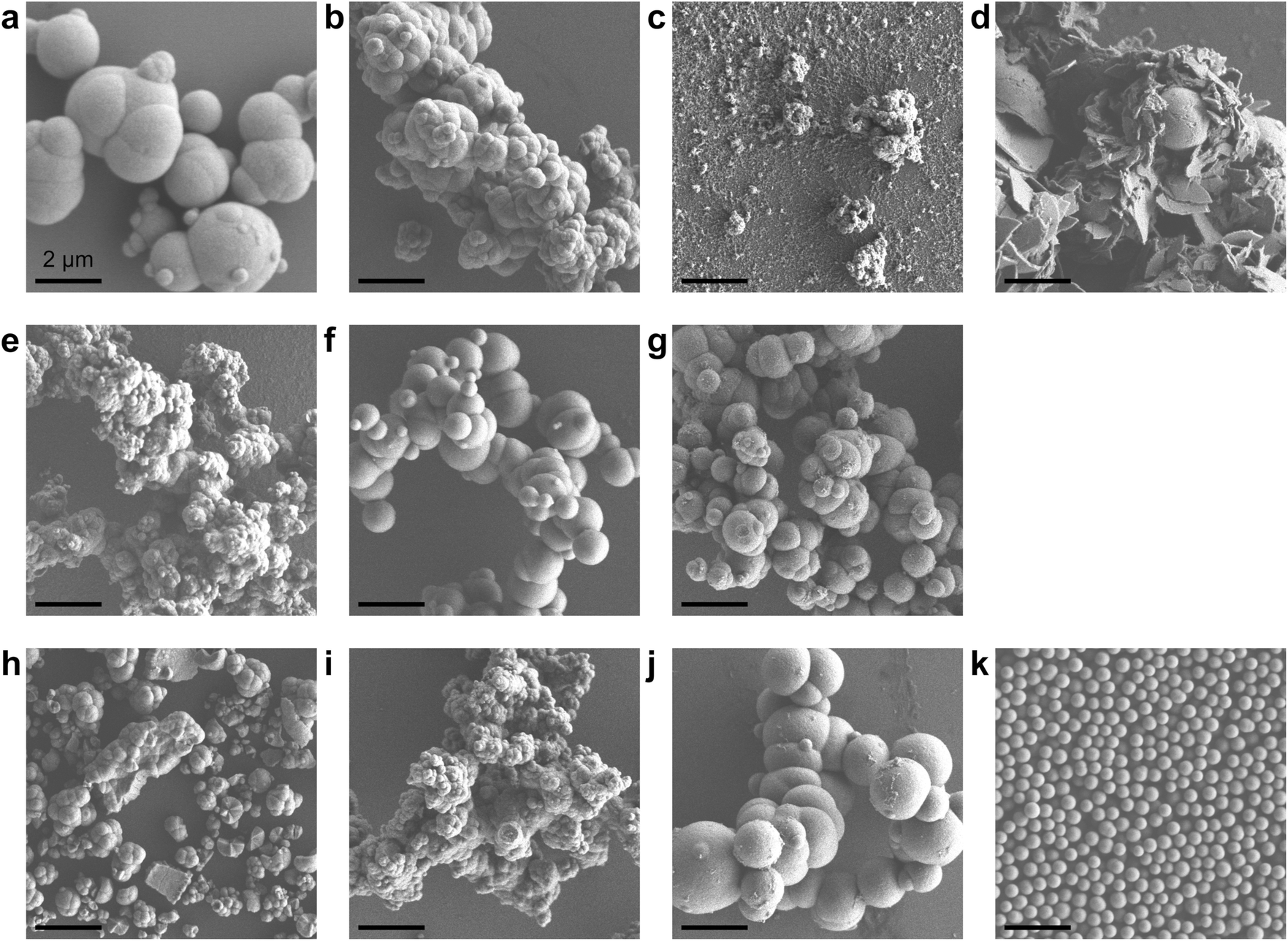 | ||
| Fig. 1 SEM images of the prepared ceria (scale bar: 2 μm). (a) CeO2-ME-FA, (b) CeO2-ME-AA, (c) CeO2-ME-BA, (d) CeO2-ME-PA, (e) CeO2-ME-EG, (f) CeO2-ME-dEG, (g) CeO2-ME-tEG, (h) CeO2-AN, (i) CeO2-AN-EG, (j) CeO2-AN-dEG and (k) CeO2-AN-tEG, where short abbreviations ME, FA, AA, BA, PA, EG, dEG, tEG and AN represent methanol, formic acid, acetic acid, benzoic acid, o-phthalic acid, ethylene glycol, diethylene glycol, triethylene glycol and acetonitrile, respectively. | ||
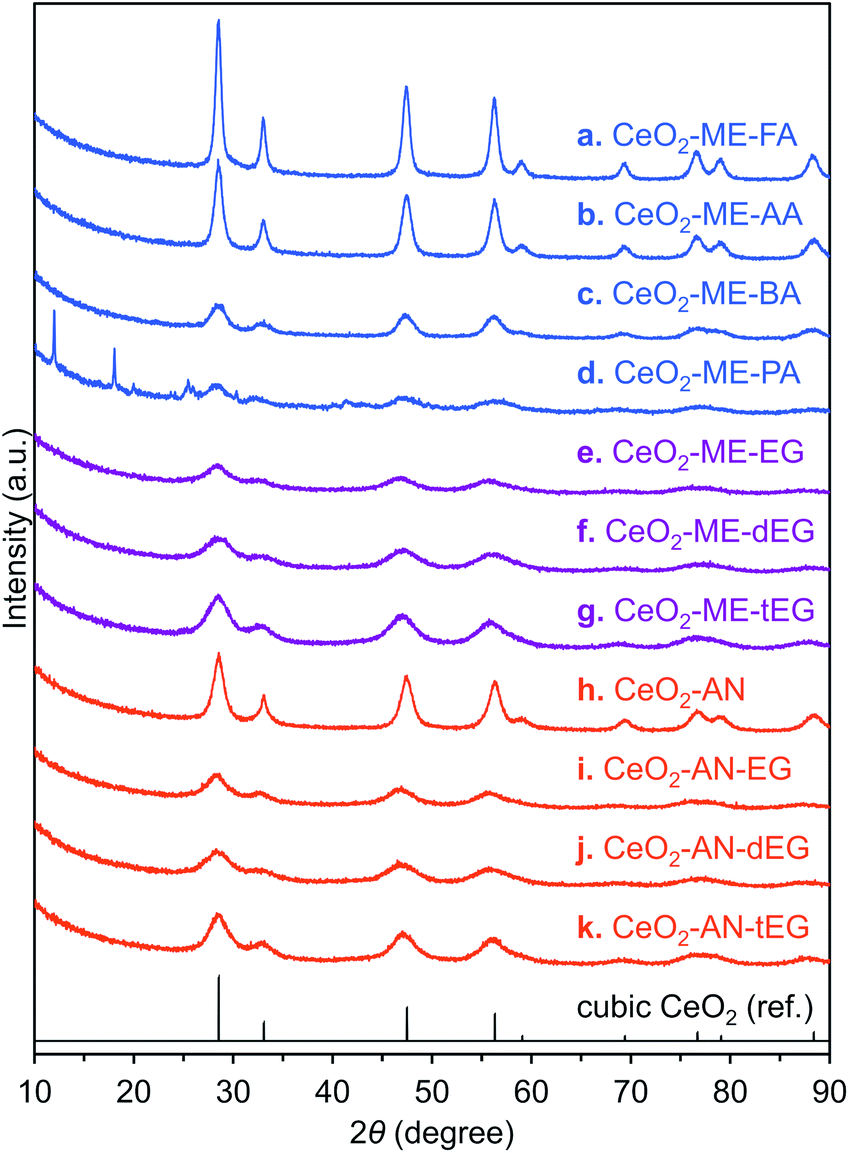 | ||
| Fig. 2 XRD patterns of ceria porous spheres. (a) CeO2-ME-FA, (b) CeO2-ME-AA, (c) CeO2-ME-BA, (d) CeO2-ME-PA, (e) CeO2-ME-EG, (f) CeO2-ME-dEG, (g) CeO2-ME-tEG, (h) CeO2-AN, (i) CeO2-AN-EG, (j) CeO2-AN-dEG and (k) CeO2-AN-tEG. Black line represents reference XRD pattern of cubic CeO2. | ||
Subsequently, we selected acetonitrile as an alternative solvent because it is a representative polar solvent being capable of dissolving a number of different inorganic compounds.29 In trials without the use of additives, non-uniform aggregates were obtained (Fig. 1h), while improved morphologies were observed in accordance with the number of oxyethylene units in additives (Fig. 1i–k). Surprisingly, the use of triethylene glycol resulted in a completely monodisperse and almost perfectly spherical morphology (sample CeO2-AN-tEG, Fig. 1k and 3). Such beautiful spherical morphology can be derived from minimizing contact surface area with outer atmosphere (solvent) in the reaction, as can be seen in emulsification polymerization, which could be one reason.26 This product had a crystallite size of 3.3 nm as determined using Scherrer's equation in conjunction with the XRD pattern for the material (Fig. 2k). The diameters of the spheres were determined to be 440 ± 40 nm based on SEM images (Fig. 1k). Comparing to XRD patterns of four samples CeO2-AN, CeO2-AN-EG, CeO2-AN-dEG and CeO2-AN-tEG, crystallinity of CeO2-AN was significantly higher than that of other three samples. Therefore, it is considered that oxyethylene compounds suppress crystal growth of the primary particles by chelate effect to precursor Ce ion and/or generated primary particles. Furthermore, judging from the monodisperse state of sample CeO2-AN-tEG, it was suggested that the oxyethylene compounds suppressed the aggregation of secondary particles, too. Thus, triethylene glycol performed the best additive in the three oxyethylene compounds, which can be ascribed to effective coordination through three oxyethylene units. The BET specific surface area was large at 152 m2 g−1 (Fig. S2a†) while the BJH pore size distribution peak was less than 2.43 nm (the lower limit of the instrument, Fig. S2b†). Concave-convex surface structures on the nanoscale are clearly evident in TEM images of the MARIMO surface (Fig. 3b), which would be expected to provide a heat-tolerant catalyst support based on previous work.21,30 Judging from these physical properties, it was anticipated that this CeO2 MARIMO (CeO2-AN-tEG) could potentially serve as a high-performance catalyst support for metal nanoparticle catalysts.
 | ||
| Fig. 3 Morphology of porous ceria sphere (CeO2-AN-tEG). (a) TEM image and (b) high resolution TEM image. | ||
Evaluation of the catalytic performance
The performance of the CeO2-AN-tEG as a catalyst or a catalyst support was examined by performing a highly exothermic CO oxidation reaction as a probe reaction. In these trials, a JRC-CEO-5 (Japan Reference Catalyst) standard with a specific surface area of 92.0 m2 g−1 and an average crystallite size of 9.9 nm was employed as a reference material.31 The TEM image in Fig. S3b† clearly shows several Pt nanoparticles on the JRC-CEO-5 catalyst, while no Pt particles are evident in the case of the Pt/CeO2-AN-tEG catalyst (Fig. S3a†). However, based on EDX analyses, the JRC-CEO-5 and CeO2-AN-tEG contained 1.1 and 0.9 wt% Pt, respectively, thus, indicating that the Pt nanoparticles would be highly dispersed on the CeO2-AN-tEG.The catalytic CO oxidation reaction was examined using both the porous ceria spheres and the reference ceria, both with Pt nanoparticles. Fig. 4a plots the CO2 yields as functions of reaction temperature. As expected, the Pt/CeO2-AN-tEG catalyst exhibited superior low temperature catalytic activity. Although the oxidation reaction started at 150 °C in the case of the Pt/JRC-CEO-5, a substantially lower onset temperature of 50 °C was observed when using the Pt/CeO2-AN-tEG. Notably, the CO2 yield reached almost 100% at 100 °C with the latter material, at which temperature the Pt/JRC-CEO-5 reaction had just begun. The higher oxidation ability of Pt/CeO2-AN-tEG catalyst than that of reference Pt/JRC-CEO-5 can be explained as follows: it is well known that the ceria–metal interface activates oxygen in CO oxidation reactions at the ceria–metal interface with smooth oxygen transfer between Pt and ceria support.32 The large surface area and convex-concave surface structure would disperse Pt particles well and serve larger amounts of ceria–metal interface.
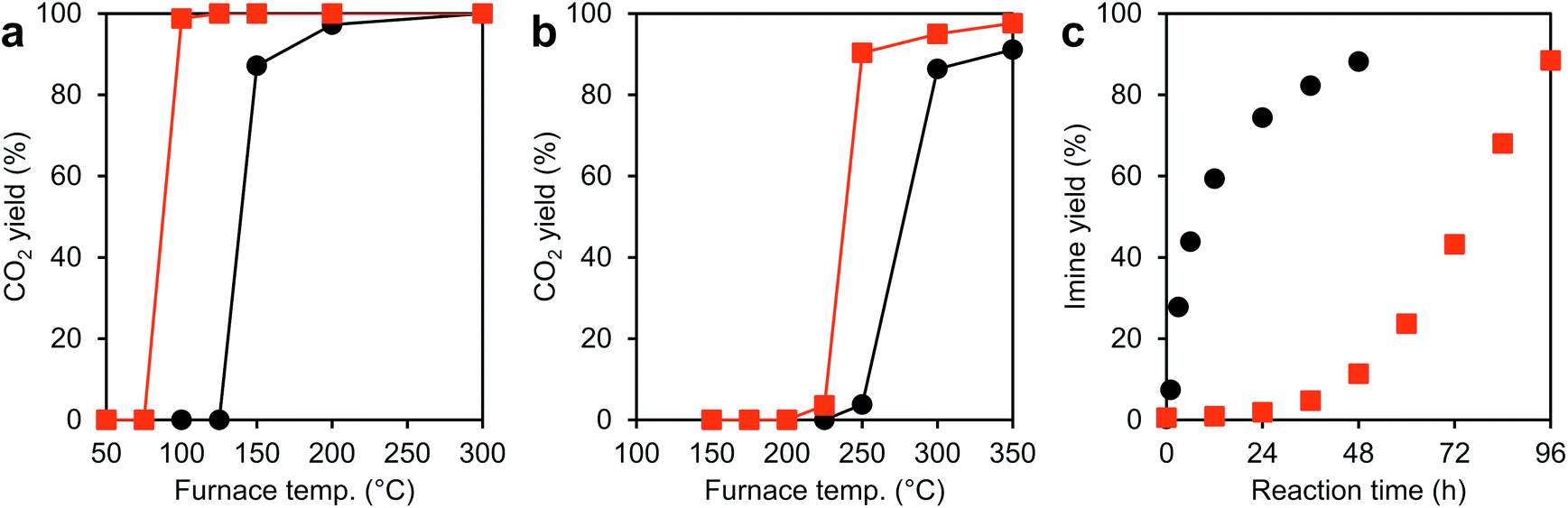 | ||
| Fig. 4 Results of catalytic reactions with Pt/CeO2 and CeO2 catalysts. (a) The vapor-phase CO oxidation reaction catalyzed by Pt/CeO2-AN-tEG (red) and Pt/JRC-CEO-5 (black). (b) The vapor-phase CO oxidation reaction catalyzed by CeO2-AN-tEG (red) and JRC-CEO-5 (black). (c) The liquid-phase oxidation reaction of benzyl alcohol catalyzed by CeO2-AN-tEG (red) and JRC-CEO-5 (black) followed by condensation with aniline. | ||
In trials using the CeO2-AN-tEG without Pt during the CO oxidation reaction, the reaction was found to start at 225 °C with 90% conversion at 250 °C, as can be seen in Fig. 4b. In contrast, the reaction started at 250 °C when using the JRC-CEO-5 and 86% conversion was achieved at 300 °C. Thus, the bare CeO2-AN-tEG exhibited higher CO conversion as well as superior low temperature activity. As discussed above, the higher specific surface area of the CeO2-AN-tEG (127 m2 g−1: after calcined at 300 °C for 1 h in air) compared with that of the JRC-CEO-5 (87.6 m2 g−1: after calcined at 300 °C for 1 h in air) may be one reason for its superior catalytic CO conversion properties. However, it is still difficult to fully explain the reason for the higher catalytic activity of this material at low temperatures, unless differences in another factor of the porous ceria are taken into account. Evaluation in other catalytic reaction should be performed to understand the superior low temperature catalytic activity of CeO2-AN-tEG.
In other trials, an unexpected difference in reactivity was observed in liquid-phase reactions. CeO2 is known to act as a redox catalyst in liquid-phase reactions and so the CeO2-AN-tEG was applied to the catalytic oxidation reaction of benzyl alcohol to benzaldehyde under O2. This reaction was followed by conversion to N-benzylideneaniline via condensation with aniline in situ as shown in Fig. S4.†33 In this reaction, the oxidation of benzyl alcohol is promoted by redox sites on the ceria, while the subsequent condensation reaction between benzaldehyde and aniline is facilitated by the Lewis acid sites on the material.34–37 The associated reaction profiles are presented in Fig. 4c. Based on the specific surface areas of the porous ceria and of the reference compound (JRC-CEO-5), it was predicted that the porous ceria would be more active. However, the reaction profile using CeO2-AN-tEG was found to be very different from that of JRC-CEO-5, such that the former required a long induction period (ca. 48 h) to initiate the reaction. Thus, it seems that other factors, such as structure, electronic state, high oxygen diffusivity and lower activation energy by high lattice strain, relate to catalytic activity of CeO2, which could compensate smaller specific surface area in the case of this liquid-phase reaction.
HAXPES analysis
To establish the origin of the unexpected catalytic activity of this material, the valences of the Ce ions in the ceria were assessed using HAXPES at the SPring-8 facility (Fig. S5†), where photoelectrons released from ca. 7 nm depth from the particle surface can be detected because of strong synchrotron X-ray (5949 eV) at the SPring-8 synchrotron radiation facility.38 Judging from the primary particle size of the CeO2 MARIMOs (ca. 4 nm), entire inside electronic structure can be detected. This was done because the concentration of point defects in CeO2, which directly affects oxygen uptake/release efficiencies, is an important factor controlling catalytic activity. The peak fitting curves (Fig. 5) clearly demonstrates that the reference ceria JRC-CEO-5 generated small peaks at the positions attributed to Ce3+ (880.9, 885.2, 899.1 and 903.4 eV).39 However, the CeO2-AN-tEG prepared by the solvothermal reaction in acetonitrile exhibited obviously large peaks at these positions.7,40–42 These results clearly demonstrate that the JRC-CEO-5 contained exclusively Ce4+ while the CeO2-AN-tEG had a high proportion of Ce3+, where concentration of Ce3+ in JRC-CEO-5 was determined to 22.5 at% by peak fitting of HAXPES profiles. On the other hand, Ce3+ proportion of CeO2-AN-tEG turned out extremely high, to be 57.4 at%. Hao et al. reported that the amount of Ce3+ in CeO2 nano-sized crystals increased as the CeO2 particle size decreased.17 The crystallite size in the CeO2-AN-tEG was estimated to be 3.3 nm and so was comparable to the value previously reported for CeO2 nanocrystals (5.4 nm) including high proportions of Ce3+.17 Thus, the small crystallite size of the CeO2-AN-tEG could be one reason for the existence of an appreciable amount of Ce3+ in the CeO2-AN-tEG nanocrystals.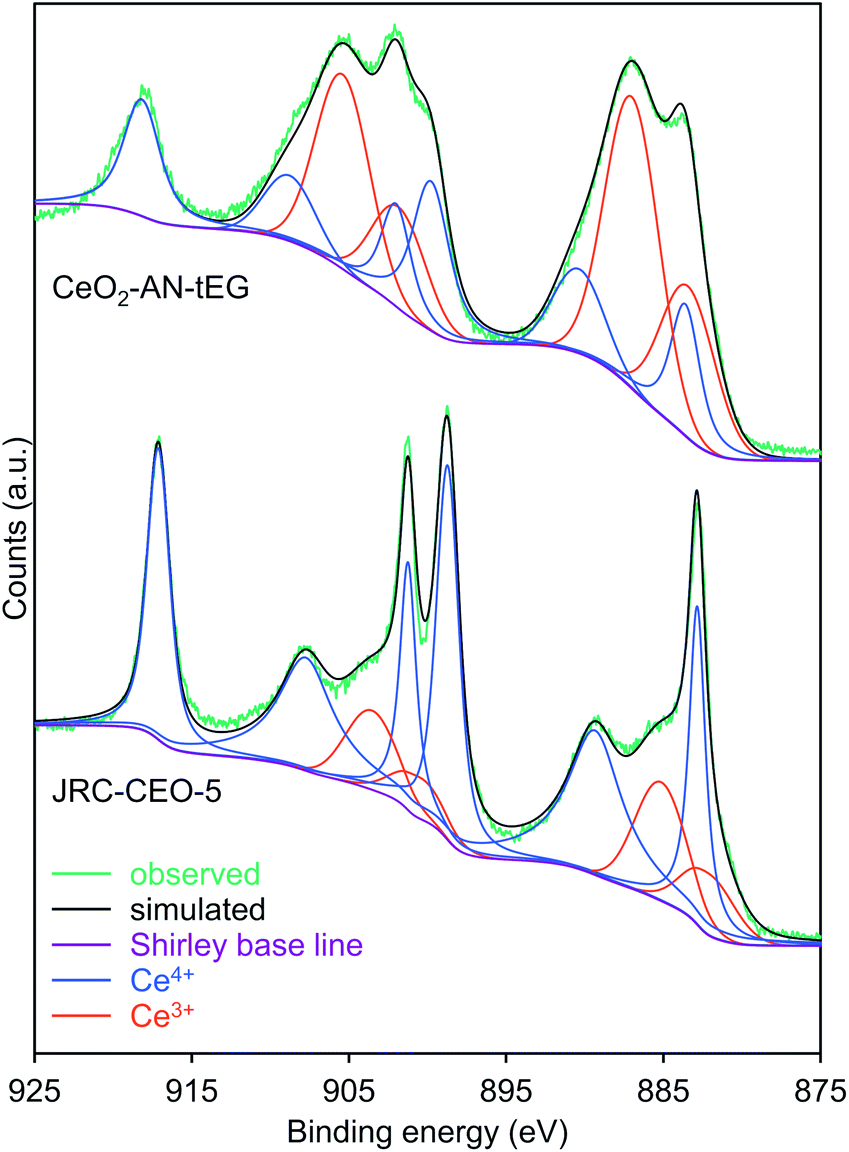 | ||
| Fig. 5 Ce 3d HAXPES spectra and peak fitting curves of CeO2-AN-tEG and JRC-CEO-5. Observed (green), Shirley base line (violet), deconvolution peaks of Ce4+ (blue), deconvolution peaks of Ce3+ (red) and simulated curve (black). | ||
The Ce3+ in the original Ce(NO3)3·6H2O would be expected to be oxidized to Ce4+ during the solvothermal reaction that yielded CeO2. However, the reductive nature of the solvothermal system could be another reason for the large amount of Ce3+ in the porous CeO2 spheres. That is, the hydroxy groups of the triethylene glycol could have acted as reducing agents, while the acetonitrile would have no oxidation ability. The relatively short heating time of 10 min could also partly explain the high Ce3+ content in the product. In general, Ce3+ species in CeO2−x are easily oxidized to Ce4+ species in oxidative atmosphere.6,7 So, prolonged heating at high temperatures (such as 300 °C) would be expected to promote the formation of the more thermodynamically stable Ce(IV)O2. Thus, we succeeded in synthesizing a CeO2 MARIMO rich in Ce3+ with a very high specific surface area using a facile one-step solvothermal reaction with acetonitrile as the solvent.
According to the literature,43 the presence of Ce3+ in the CeO2 improves the low temperature activity of this material in the vapor-phase CO oxidation reaction. Consequently, the superior low temperature activity of the CeO2-AN-tEG compared with the JRC-CEO-5 in the vapor phase (Fig. 4b) can be ascribed to the larger amount of Ce3+ in the former, as confirmed by the HAXPES analysis. However, in the case of the liquid-phase benzyl alcohol oxidation (Fig. 4c), Ce4+ on the surfaces of the CeO2 nanocrystals played a crucial role in catalyzing the reaction by oxidizing the alcohol with the concurrent reduction of Ce4+ to Ce3+. The valence change from Ce3+ to Ce4+ in this catalytic cycle is not as pronounced when using the CeO2-AN-tEG, because it has more Ce3+ contents. Thus, a longer time span is required for Ce3+ in this material to be oxidized by atmospheric O2 to Ce4+ prior to the benzyl alcohol oxidation. In fact, the induction period was reduced by 48 h pre-treatment of CeO2-AN-tEG under O2 prior to liquid-phase catalytic reaction (Fig. S6a†). In addition, the induction period was found to disappear when calcined CeO2-AN-tEG was used as a catalyst (Fig. S6a†), which mostly composed of Ce4+ species (Fig. S6b†). The BET specific surface area of 127 m2 g−1 and the BJH pore size less than 2.43 nm (the lower limit of the instrument) in the calcined CeO2-AN-tEG were almost same to those of as-synthesized CeO2-AN-tEG. Therefore, absence of the induction period was due to difference of an electronic state of CeO2 instead of its structure.
As for the solvents for CeO2 synthesis, when a methanol solution is heated to 300 °C, the condensation of methanol would be expected to produce dimethyl ether and H2O. The resulting H2O would accelerate the hydrolysis of Ce(NO3)3·6H2O to yield CeO2 via Ce(OH)4, and the increased crystal size would in turn increase the Ce4+ concentration in the CeO2 nanocrystals. Consequently, we propose that the suppression of H2O formation from methanol during the solvothermal reaction could be an additional reason for the high Ce3+ concentration in the CeO2. Then, we selected an aprotic polar solvent (acetonitrile) instead of methanol in expectation of no H2O formation at elevated temperature. To test our hypothesis, mixtures of acetonitrile and H2O were applied as the reaction media in solvothermal reaction trials (Table 1). The crystallite sizes were found to increase in proportion to the amount of water in the reaction solution (Table S3† and Fig. 6a), which indicates that water does indeed accelerate crystal growth. Fig. 6b presents the HAXPES profiles normalized by the peak intensities at the position of a Ce4+ peak (883.0 eV). As expected, the peak intensities at 885.2 and 903.4 eV ascribed to Ce3+ species strengthened as the water level in the reaction solvent decreased. Thus, the Ce3+/Ce4+ ratio in the CeO2 MARIMO could be readily adjusted by varying the water/acetonitrile ratio. In addition, XRD peak shifted to lower angles with increasing acetonitrile ratio in acetonitrile/water mixed solvents (Fig. S9†). The result indicates lattice expansion of cubic CeO2, where increasing amounts of Ce3+ cation with larger ionic radius than that of Ce4+ cation would expand lattice size of CeO2 crystallites.17,44 The peak fitting curves of HAXPES spectra were showed in Fig. S10,† and the details of the crystallite sizes, the calculated Ce3+ concentrations in the CeO2 and lattice parameter are summarized in Tables S2 and S3.†
| Sample namea | Solvent | Additive | Temperature (°C) |
|---|---|---|---|
| a AN, tEG and W indicate acetonitrile, triethylene glycol and water, respectively.b Diffraction peaks ascribed to a non-cubic ceria phase were recognized in the XRD patterns of the product obtained at 300 °C (Fig. S7), so that the reaction temperature was lowered to 250 °C (Fig. S8). | |||
| CeO2-AN-tEG | CH3CN | Triethylene glycol | 300 |
| CeO2-AN7-W3-tEG | CH3CN/H2O = 7/3 (v/v) | Triethylene glycol | 250b |
| CeO2-AN5-W5-tEG | CH3CN/H2O = 5/5 (v/v) | Triethylene glycol | 250b |
| CeO2-W-tEG | H2O | Triethylene glycol | 250b |
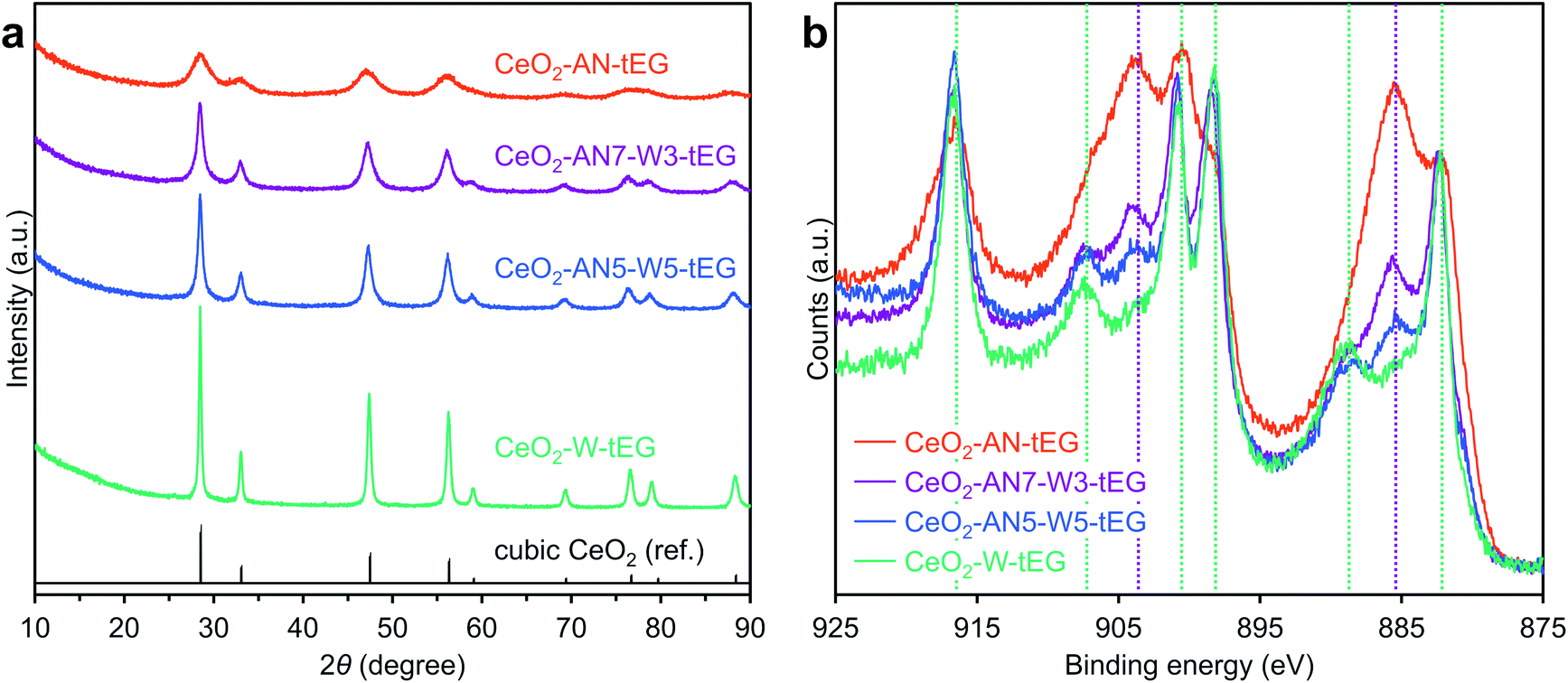 | ||
| Fig. 6 (a) XRD patterns of CeO2-AN-tEG, CeO2-AN7-W3-tEG, CeO2-AN5-W5-tEG and CeO2-W-tEG. Black line represents reference XRD pattern of cubic CeO2. (b) Ce 3d HAXPES spectra of CeO2-AN-tEG, CeO2-AN7-W3-tEG, CeO2-AN5-W5-tEG and CeO2-W-tEG. Green and purple lines show peak positions of Ce4+ and Ce3+, respectively. | ||
Oxygen storage capacity
Ceria has been recognized as one of the best materials for oxygen storage and transfer in catalytic systems.5,11–13 As an example, the Ce3+/Ce4+ cycling phenomenon has been applied to the catalytic treatment of automobile exhaust gases. There has also been much effort devoted to obtaining higher concentrations of Ce3+ in CeO2 catalysts so as to enhance the oxygen transfer process. Based on the HAXPES data, the CeO2-AN-tEG was expected to exhibit exceptional OSC, and this was evaluated by cyclic O2-pulse measurements at 200 and 400 °C (Fig. 7). The reference material JRC-CEO-5 demonstrated an OSC of approximately 22 μmol-O g−1 during five cycles at 200 °C (Fig. 7a), whereas the CeO2-AN-tEG showed a much larger value of 232 μmol-O g−1 during the first cycle at 200 °C. However, this high OSC abruptly decreased to quite a low value of approximately 11 μmol-O g−1 on subsequently cycling. The initial large OSC is attributed to the high concentration of oxygen defects originally associated with Ce3+ ions in the material. The subsequent drop in the OSC value is believed to have resulted from the insufficient reduction of the CeO2 by H2 at the low temperature of 200 °C. For this reason, reduction temperatures of CeO2-AN-tEG and JRC-CEO-5 were investigated by H2-TPR. Fig. S11† showed that reduction of CeO2-AN-tEG and JRC-CEO-5 started at 250 °C and 350 °C, respectively. Then, additional cyclic O2-pulse measurements were performed at 400 °C, and the OSC values for the JRC-CEO-5 and CeO2-AN-tEG were found to be 114 and 268 μmol-O g−1 , respectively (Fig. 7b). Unlike the result obtained at 200 °C, small changes (less than 12.4% decreasing) of these OSC values were observed during 5 cycles. The enhanced durability of the OSC value can be explained by a larger amount of oxygen defect generated by higher temperature reduction at 400 °C as illustrated in Fig. S12.† Importantly, the OSC value for the CeO2-AN-tEG was two times larger than that for the JRC-CEO-5. Interestingly, the specific surface areas of the JRC-CEO-5 and CeO2-AN-tEG before cyclic O2-pulse measurements at 400 °C (after calcination at 400 °C for 1 h in air) were 86 and 78 m2 g−1, respectively. Thus, although the specific surface area of CeO2-AN-tEG was smaller a little bit than that of JRC-CEO-5, this material exhibited a high OSC, and so these results are ascribed to the improved diffusion of oxygen in the CeO2-AN-tEG nanocrystals.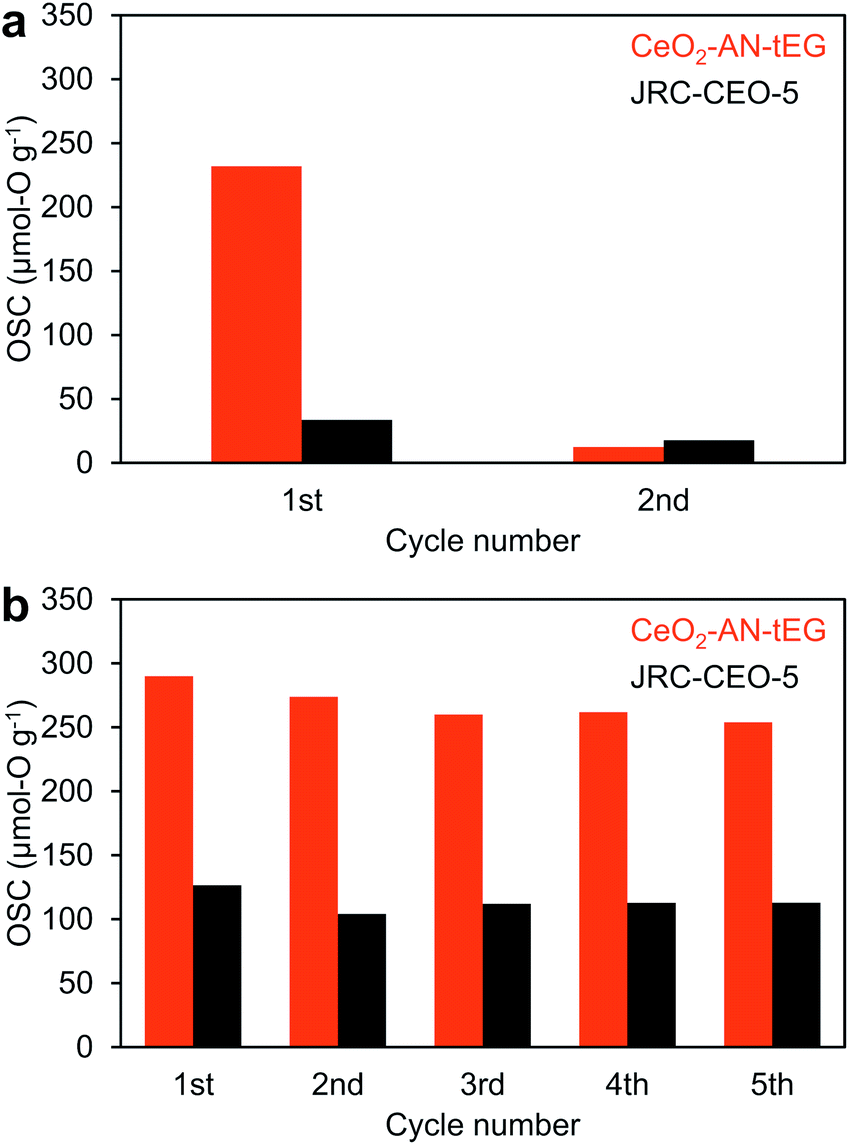 | ||
| Fig. 7 Oxygen storage capacity of CeO2-AN-tEG (red) and JRC-CEO-5 (black) measured by cyclic O2-pulse measurements at (a) 200 °C and (b) 400 °C. | ||
Conclusions
A simple one-pot single-step solvothermal method using acetonitrile as a solvent and triethylene glycol as an additive produced porous ceria with a monodisperse spherical morphology. Compared with reference CeO2, the porous spheres generated in this work showed high performance not only as a catalyst support but also as a catalyst in the CO oxidation reaction. A high Ce3+ concentration in this material was confirmed by HAXPES at the SPring-8 facility. The superior performance of these porous CeO2 spheres during the CO oxidation reaction and their OSC of 268 μmol-O g−1 g at 400 °C are both attributed to the high concentration of Ce3+ species in this material.Author contributions
Ayano Taniguchi: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing. Yoshitaka Kumabe: data curation, formal analysis, investigation, writing – review & editing. Kai Kan: investigation, supervision, writing – review & editing. Masataka Ohtani: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing. Kazuya Kobiro: funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the Creation of New Business and Industry Program of the Kochi Prefectural Industry-Academia-Government Collaboration Research Promotion Operation and funding from JSPS KAKENHI Grant Number 15K06560, 17K14858 and 19K05143. The HAXPES measurements were performed with the approval of the NIMS Synchrotron X-ray Station (proposal numbers 2018A4910, 2019A4906 and 2020A4904). The authors are also grateful to HiSOR, Hiroshima University, and JAEA/SPring-8 for the development of the HAXPES unit at the BL15XU beamline of SPring-8. The authors thank Dr Shigenori Ueda of the NIMS Synchrotron X-ray Station for his support during the HAXPES work and Professor Hisao Makino of the Kochi University of Technology for helpful discussions related to the HAXPES analyses. The authors also thank the Daiichi Kigenso Kagaku Kogyo Co., Ltd. for providing the CeO2 nanoparticles. We thank Michael Judge, MSc, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.References
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS.
- H.-H. Liu, Y. Wang, A.-P. Jia, S.-Y. Wang, M.-F. Luo and J.-Q. Lu, Appl. Surf. Sci., 2014, 314, 725–734 CrossRef CAS.
- Q. Xie, H. Zhang, J. Kang, J. Cheng, Q. Zhang and Y. Wang, ACS Catal., 2018, 8, 4902–4916 CrossRef CAS.
- J. Xu, Y. Zhang, Y. Liu, X. Fang, X. Xu, W. Liu, R. Zheng and X. Wang, Eur. J. Inorg. Chem., 2019, 183–194 CrossRef.
- P. Li, X. Chen, Y. Li and J. W. Schwank, Catal. Today, 2019, 327, 90–115 CrossRef CAS.
- Y. Namai, K. Fukui and Y. Iwasawa, J. Phys. Chem. B, 2003, 107, 11666–11673 CrossRef CAS.
- J. P. Holgado, G. Munuera, J. P. Espino and A. R. Gonzalez-Elipe, Appl. Surf. Sci., 2000, 158, 164–171 CrossRef CAS.
- C. M. Hamm, L. Alff and B. Albert, Z. Anorg. Allg. Chem., 2014, 640, 1050–1053 CrossRef CAS.
- T. Naganuma and E. Traversa, Nanoscale, 2012, 4, 4950–4953 RSC.
- J. Lu, S. Asahina, S. Takami, A. Yoko, G. Seong, T. Tomai and T. Adschiri, ACS Appl. Nano Mater., 2020, 3, 2346–2353 CrossRef CAS.
- Y. Ishikawa, M. Takeda, S. Tsukimoto, K. S. Nakayama and N. Asao, Adv. Mater., 2016, 28, 1467–1471 CrossRef CAS.
- D. Wang, Y. Kang, V. Doan-Nguyen, J. Chen, R. Küngas, N. L. Wieder, K. Bakhmutsky, R. J. Gorte and C. B. Murray, Angew. Chem., Int. Ed., 2011, 50, 4378–4381 CrossRef CAS.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS.
- D. Devaiah, T. Tsuzuki, C. U. Aniz and B. M. Reddy, Catal. Lett., 2015, 145, 1206–1216 CrossRef CAS.
- B. M. Reddy and A. Khan, Catal. Surv. Asia, 2005, 9, 155–171 CrossRef CAS.
- Y. Zhu, G. Seong, T. Noguchi, A. Yoko, T. Tomai, S. Takami and T. Adschiri, ACS Appl. Energy Mater., 2020, 3, 4305–4319 CrossRef CAS.
- X. Hao, A. Yoko, C. Chen, K. Inoue, M. Saito, G. Seong, S. Takami, T. Adschiri and Y. Ikuhara, Small, 2018, 14, 1802915 CrossRef.
- P. Wang, K. Ueno, H. Takigawa and K. Kobiro, J. Supercrit. Fluids, 2013, 78, 124–131 CrossRef CAS.
- E. K. C. Pradeep, T. Habu, H. Tooriyama, M. Ohtani and K. Kobiro, J. Supercrit. Fluids, 2015, 97, 217–223 CrossRef CAS.
- H. T. T. Nguyen, T. Habu, M. Ohtani and K. Kobiro, Eur. J. Inorg. Chem., 2017, 3017–3023 CrossRef CAS.
- F. Duriyasart, A. Irizawa, K. Hayashi, M. Ohtani and K. Kobiro, ChemCatChem, 2018, 10, 3392–3396 CrossRef CAS.
- B. Liu, B. Liu, Q. Li, Z. Li, R. Liu, X. Zou, W. Wu, W. Cui, Z. Liu, D. Li, B. Zou, T. Cui and G. Zou, J. Alloys Compd., 2010, 503, 519–524 CrossRef CAS.
- D. Sun, M. Gu, R. Li, S. Yin, X. Song, B. Zhao, C. Li, J. Li, Z. Feng and T. Sato, Appl. Surf. Sci., 2013, 280, 693–697 CrossRef CAS.
- C. Slostowski, S. Marre, O. Babot, T. Toupance and C. Aymonier, Langmuir, 2014, 30, 5965–5972 CrossRef CAS.
- J. Papavasiliou, A. Paxinou, G. Słowik, S. Neophytides and G. Avgouropoulos, Catal, 2018, 8, 544–563 CrossRef.
- P. Wang and K. Kobiro, Chem. Lett., 2012, 41, 264–266 CrossRef CAS.
- C. Ho, J. C. Yu, T. Kwong, A. C. Mak and S. Lai, Chem. Mater., 2005, 17, 4514–4522 CrossRef CAS.
- M. Ohtani, T. Muraoka, Y. Okimoto and K. Kobiro, Inorg. Chem., 2017, 56, 11546–11551 CrossRef CAS.
- E. K. C. Pradeep, M. Ohtani, T. Kawaharamura and K. Kobiro, Chem. Lett., 2017, 46, 940–943 CrossRef CAS.
- H. T. T. Nguyen, Y. Kumabe, S. Ueda, K. Kan, M. Ohtani and K. Kobiro, Appl. Catal., A, 2019, 577, 35–43 CrossRef CAS.
- Japan Reference Catalyst, https://www.shokubai.org/com/sansyo/ceria.html, accessed June 2020 Search PubMed.
- S. Song, X. Wang and H. Zhang, NPG Asia Mater., 2015, 7, 179–196 CrossRef.
- M. Tamura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 864–867 CrossRef CAS.
- Z. Zhang, Y. Wang, M. Wang, J. Lü, L. Li, Z. Zhang, M. Li, J. Jiang and F. Wang, Chin. J. Catal., 2015, 36, 1623–1630 CrossRef CAS.
- J. Zhang, J. Yang, J. Wang, H. Ding, Q. Liu, U. Schubert, Y. Rui and J. Xu, Mol. Catal., 2017, 443, 131–138 CrossRef CAS.
- H. Ding, J. Yang, S. Ma, N. Yigit, J. Xu, G. Rupprechter and J. Wang, ChemCatChem, 2018, 10, 4100–4108 CrossRef CAS.
- X. Ren, Z. Zhang, Y. Wang, J. Lu, J. An, J. Zhang, M. Wang, X. Wang and Y. Luo, RSC Adv., 2019, 9, 15229–15237 RSC.
- S. Tanuma, C. J. Powellb and D. R. Penn, Surf. Interface Anal., 2011, 43, 689–713 CrossRef CAS.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264–267 CrossRef.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J.-C. Volta, J. Phys. Chem. B, 2003, 107, 11475–11484 CrossRef CAS.
- L. Qiu, F. Liu, L. Zhao, Y. Ma and J. Yao, Appl. Surf. Sci., 2006, 252, 4931–4935 CrossRef CAS.
- K. I. Maslakov, Y. A. Teterin, A. J. Popel, A. Y. Teterin, K. Y. Ivanov, S. N. Kalmykov, V. G. Petrov, P. K. Petrov and I. Farnan, Appl. Surf. Sci., 2018, 448, 154–162 CrossRef CAS.
- J. Li, Z. Zhang, Z. Tian, X. Zhou, Z. Zheng, Y. Ma and Y. Qu, J. Mater. Chem. A, 2014, 2, 16459–16466 RSC.
- L. Chen, P. Fleming, V. Morris, J. D. Holmes and M. A. Morris, J. Phys. Chem. C, 2010, 114, 12909–12919 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10186a |
| This journal is © The Royal Society of Chemistry 2021 |