DOI:
10.1039/D0RA10178H
(Paper)
RSC Adv., 2021,
11, 6804-6817
Kinetics and reaction mechanism of photochemical degradation of diclofenac by UV-activated peroxymonosulfate
Received
2nd December 2020
, Accepted 31st January 2021
First published on 10th February 2021
Abstract
Diclofenac (DCF) is a common non-steroidal anti-inflammatory drug, which is frequently detected in different environmental media such as surface water, groundwater, domestic sewage, and sediment. In this study, UV-activated peroxymonosulfate (PMS) was used to degrade DCF by generating active radicals (i.e., SO4˙− and HO˙) with strong oxidizing properties. The effects of PMS dosage, pH, initial DCF concentration and common water constituents on the removal of DCF as well as its degradation mechanism in UV/PMS system were investigated. Compared to UV alone and PMS alone systems, DCF was removed more efficiently in the UV/PMS system at pH 7.0 due to the contribution of SO4˙− and HO˙, and its degradation followed the pseudo-first order kinetic model. As the dosage of PMS or solution pH increased, the degradation efficiency of DCF was gradually enhanced. The highest DCF degradation was obtained at pH 11.0 in this study, because the molar absorption coefficient of PMS increased with increasing pH at 254 nm resulting in generation of more reactive radicals at high pH. Removal efficiency of DCF was decreased significantly with the increase in its initial concentration due to the insufficient concentration of radicals. The presence of HCO3− and NO3− could promote the degradation of DCF because of the role of carbonate radicals and extra HO˙ formed, respectively, while NOM inhibited DCF degradation due to its competition with DCF for reactive radicals. No obvious influence on DCF degradation was observed in the UV/PMS system with the addition of Cl− and SO42−. The degradation of DCF by UV/PMS in real waters was slightly suppressed compared with its removal in ultrapure water. Seven transformation products were detected using UPLC-QTOF/MS, and the potential degradation mechanism of DCF was thus proposed showing six reaction pathways including hydroxylation, decarboxylation, dechlorination–cyclization, formylation, dehydrogenation and dechlorination–hydrogenation.
1. Introduction
In recent decades, pharmaceutical and personal care products (PPCPs) have been widely used in many fields and have attracted growing attention.1,2 PPCPs that can resist pathogens in human and animal medicine have gradually increased with the continuous development of science and the medical industry.3,4 Since PPCPs cannot be fully absorbed by organisms, they are mainly discharged into water and soil through feces and urine, causing potential pollution and unpredictable threats to the environment and human health.5 Diclofenac (DCF) is a type of common PPCPs which is used as a nonsteroidal anti-inflammatory drug (NSAID) to reduce inflammation and pain associated with arthritis, osteoarthritis, rheumatoid arthritis, and ankylosing spondylitis.6 However, the removal of DCF in conventional wastewater treatment is incomplete due to its toxicity and recalcitrant performance.7–9 Hence, DCF has been widely detected in effluent of wastewater treatment plants (WWTPs), surface water, groundwater and even tap water due to its widespread use and low removal.10–12 Furthermore, in the drinking water treatment process, the harmful disinfection byproducts (DBPs) produced by the reaction between disinfectants and DCF may pose a serious threat to human health.13 Therefore, it is indispensable to develop efficient treatment methods for DCF removal in aquatic environment.
Advanced oxidation processes (AOPs) are a kind of methods which rely on the production of radical species to treat refractory organic contaminants.14 Hydroxyl radical (HO˙)-based AOPs have been widely studied for the removal of emerging contaminants, because HO˙ is a non-selective and powerful oxidant.15–19 Except hydroxyl radical, sulfate radical (SO4˙−) has received increasing attention due to its higher redox potential (E0 = 2.5–3.1 V) compared with hydroxyl radical (E0 = 1.9–2.7 V).20,21 Moreover, the selectivity and half-life of sulfate radical (30–40 μs) are higher and longer than those of hydroxyl radical (1 μs).22–24 Thus, sulfate radical-based AOPs (SR-AOPs) have a good application prospect in removing organic pollution.25
Currently, peroxymonosulfate (PMS) and peroxydisulfate (PDS) are the common oxidants utilized to generate SO4˙−.26 Their redox potentials are 1.82 V27 and 2.01 V,28 respectively. The main difference between these two oxidants is that PMS is a type of an S-inorganic hydroperoxide while the peroxide group in PDS bridges two sulfur atoms. According to the activation methods of PMS and PDS, the (hydro) peroxide bond (O–O) can be broken by homolytic or heterolytic cleavage.26 Transition metals (e.g., Fe2+, Cu2+, Mn2+ and Co2+) are efficient activators for PMS and PDS, but they may cause the risk of secondary pollution. Although PMS can undergo self-decomposition under slightly alkaline condition, singlet oxygen (1O2) is mainly produced,29 and this process increases the dosage of alkalis. High energy input such as heat and ultraviolet (UV) are other types of common methods which split PMS or PDS into SO4˙−. Compared with thermal activation, UV is widely used in the actual water treatment processes for disinfection. Hence, UV-activated PMS or PDS is easier to be accepted due to its environmentally friendly and effective properties, and its activation processes are shown in eqn (1) and (2). At present, UV/PMS and UV/PDS are widely utilized to remove pollutants in wastewater, but some studies have reported the degradation of DCF by UV/PDS, while there is very limited research report on the application of UV/PMS to degrade DCF in water. Consequently, based on the high efficiency, easy operation and moderate cost of the UV/PMS system, this technology is used to degrade DCF in this study.
| | |
HSO5− + hν → SO4˙− + HO˙
| (1) |
The objective of this study is to systematically investigate the photochemical degradation of DCF by UV/PMS. The effects of pH, PMS dosage, initial DCF concentration and common water constituents including inorganic anions and natural organic matter (NOM) on DCF removal were assessed. The removal of DCF in real waters and its mineralization were also explored. Eventually, the potential degradation mechanism of DCF was proposed based on the detected transformation products.
2. Materials and methods
2.1. Materials
Diclofenac (DCF, ≥99%) and fulvic acid (FA) were purchased from Aladdin (China). Potassium peroxymonosulfate (KHSO5), sodium nitrate (NaNO3), sodium sulfate (NaSO4), sodium chloride (NaCl), sodium bicarbonate (NaHCO3), sulfuric acid (H2SO4), sodium hydroxide (NaOH), dipotassium hydrogen phosphate (K2HPO4), monobasic potassium phosphate (KH2PO4), sodium thiosulfate (Na2S2O3), tertiary butanol (C4H10O), formic acid (CH2O2) and acetic acid (C2H4O2) were all analytical grade and bought from Chengdu Kelong Chemical Reagent Co. Ltd. Methyl alcohol (CH4O) and acetonitrile (CH3CN) were both chromatographic grade and obtained from Fisher Scientific. All the solutions were prepared by ultrapure water (18 MΩ cm).
2.2. Experimental procedure
The photochemical experiments were conducted in a self-made parallel light emitting device, as shown in Fig. 1. The light source used was two low-pressure mercury lamps (15 W, Philips), which mainly emitted ultraviolet light with a wavelength of 254 nm. The intensity of the emitted ultraviolet light was 0.24 mW cm−2 measured by an UV radiometer. Before experiments, UV lamps were turned on for preheating and stabilizing for 30 min. A total volume of 50 mL of the reaction solution was added to the reactor (glass Petri dish with a quartz cover), which was then put on the magnetic stirrer. After pulling the visor, the UV light was irradiated into the reactor and the reaction was started. At the specified time intervals, 0.5 mL of the reaction solution was sampled and added to 0.5 mL of 100 mM sodium thiosulfate (to quench radicals and residual oxidant) in the liquid phase vial. The samples were mixed quickly and placed in the refrigerator to avoid light and wait for testing. All the experiments were carried out under constant temperature (25 °C) and repeated at least three times.
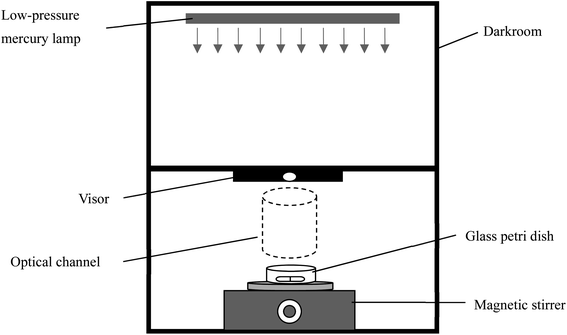 |
| | Fig. 1 Self-made parallel light emitting device. | |
2.3. Analysis
The concentration of DCF was determined by a high-performance liquid chromatograph (HPLC, Waters 2695) equipped with a C18 column (4.6 × 150 mm, 5 μm). The column temperature was 30 °C with the injection volume of 20 μL. The mobile phase consisted of 0.1% acetic acid aqueous solution and methanol (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75, v/v) at a flow rate of 1 mL min−1 and the detection wavelength was set at 276 nm. DCF degradation products were detected by an ultra-high performance liquid chromatograph-quadrupole time-of-flight tandem mass spectrometer (UPLC-QTOF/MS). The C18 column (2.1 × 100 mm, 1.7 μm) was used as a stationary phase with the column temperature of 30 °C. The mobile phase was a mixture of 0.1% formic acid water solution (A) and acetonitrile (B) at a flow rate of 0.3 mL min−1. The gradient elution procedure was 90% A for 1 min constantly, linearly decreasing to 0% A (100% B) for 12 min, keeping for 3 min and slowly increasing to 90% A for 18 min. The injection volume was 10 μL. The electrospray ionization (ESI) was used in the mass spectrometer to scan in the positive ion mode, and the scan range was m/z 50–500. All the mass spectrum data were analyzed by Masslynx 4.1 software. The mineralization of DCF was assessed through measuring the total organic carbon (TOC) of the reaction solution using a TOC analyzer (Elementar). The solution pH was monitored by a pH meter (PHS-3C, Leici).
:75, v/v) at a flow rate of 1 mL min−1 and the detection wavelength was set at 276 nm. DCF degradation products were detected by an ultra-high performance liquid chromatograph-quadrupole time-of-flight tandem mass spectrometer (UPLC-QTOF/MS). The C18 column (2.1 × 100 mm, 1.7 μm) was used as a stationary phase with the column temperature of 30 °C. The mobile phase was a mixture of 0.1% formic acid water solution (A) and acetonitrile (B) at a flow rate of 0.3 mL min−1. The gradient elution procedure was 90% A for 1 min constantly, linearly decreasing to 0% A (100% B) for 12 min, keeping for 3 min and slowly increasing to 90% A for 18 min. The injection volume was 10 μL. The electrospray ionization (ESI) was used in the mass spectrometer to scan in the positive ion mode, and the scan range was m/z 50–500. All the mass spectrum data were analyzed by Masslynx 4.1 software. The mineralization of DCF was assessed through measuring the total organic carbon (TOC) of the reaction solution using a TOC analyzer (Elementar). The solution pH was monitored by a pH meter (PHS-3C, Leici).
3. Results and discussion
3.1. Degradation of DCF by UV, PMS and UV/PMS
It is reported that 1O2 can be produced through PMS self-decomposition, while its generation is highly dependent on solution pH. Yang et al.29 found that this reaction is mainly occurred under slightly alkaline pH (pH = 9.4), while it is hardly occurred at pH 7.0. The experiment in this study was carried out in a phosphate buffer at pH 7.0, thus 1O2 was barely produced in current conditions in this study. As shown in Fig. 2, PMS alone could hardly remove DCF after 20 min of reaction, indicating that PMS could not effectively oxidize DCF and the role of 1O2 on DCF degradation could be negligible. Under UV irradiation, DCF could undergo photolysis, and about 81.2% of DCF was degraded after 20 min. When PMS was added to UV alone system, the removal of DCF was significantly accelerated, which might be attributed to the role of SO4˙− and HO˙ generated by UV activated PMS. In order to clarify the contribution of these two radicals to the degradation of DCF, two commonly used radical quenchers, namely tert-butanol (TBA) and isopropanol (i-PrOH), were added to the UV/PMS system. The results are shown in Fig. 2. The inhibition on DCF removal with the addition of TBA might be caused by HO˙ oxidation, because TBA can only inhibit HO˙ (KHO˙/TBA = 7.6 × 108 M−1 s−130) rather than SO4˙− (KSO4˙−/TBA = 8.4 × 105 M−1 s−130). The addition of i-PrOH further inhibited the removal of DCF, because it can react with both HO˙ and SO4˙− (KHO˙/i-PrOH = 1.9 × 109 M−1 s−1, KSO4˙−/i-PrOH = 8.2 × 107 M−1 s−1).30,31 In the UV/PMS/i-PrOH system, the degradation of DCF was basically similar to its direct UV photolysis, suggesting that the addition of i-PrOH might completely quench SO4˙− and HO˙ in UV/PMS system. These results showed that under current experimental conditions (pH = 7.0), both SO4˙− and HO˙ were existed in UV/PMS system, and they both played a certain role in the removal of DCF. The degradation of DCF in UV alone and UV/PMS systems both followed the pseudo-first order kinetic model, and their apparent rate constants (kobs) were 0.083 min−1 and 0.109 min−1, respectively.
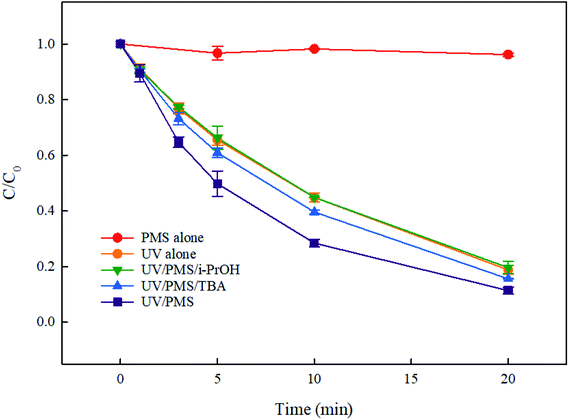 |
| | Fig. 2 Degradation of DCF by different reaction systems. Experimental conditions: [DCF]0 = 1 μM, [PMS]0 = 50 μM, [i-PrOH]0 = [TBA]0 = 10 mM, T = 25 °C, pH 7.0 phosphate buffer (10 mM). | |
3.2. Effect of pH
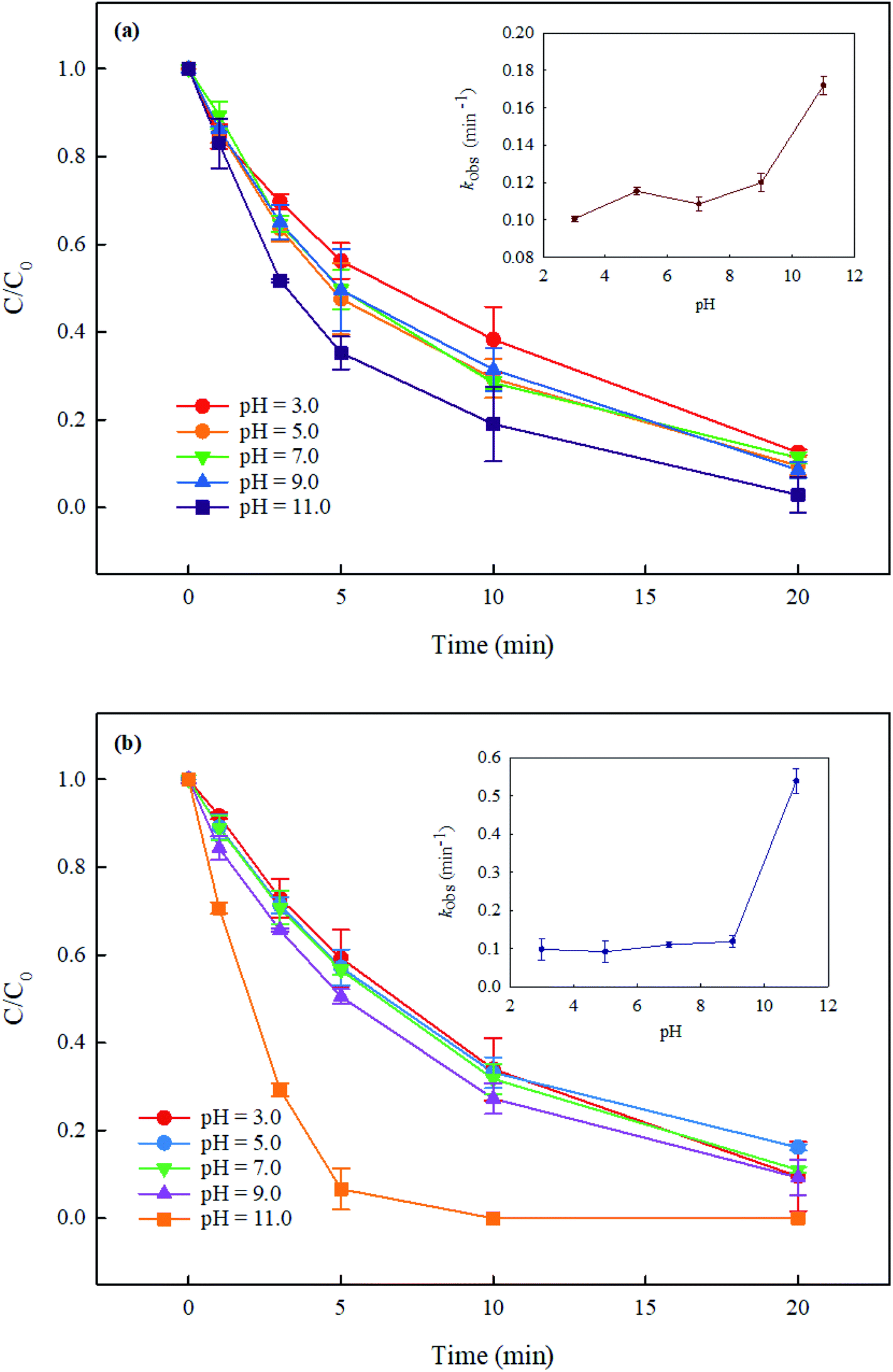
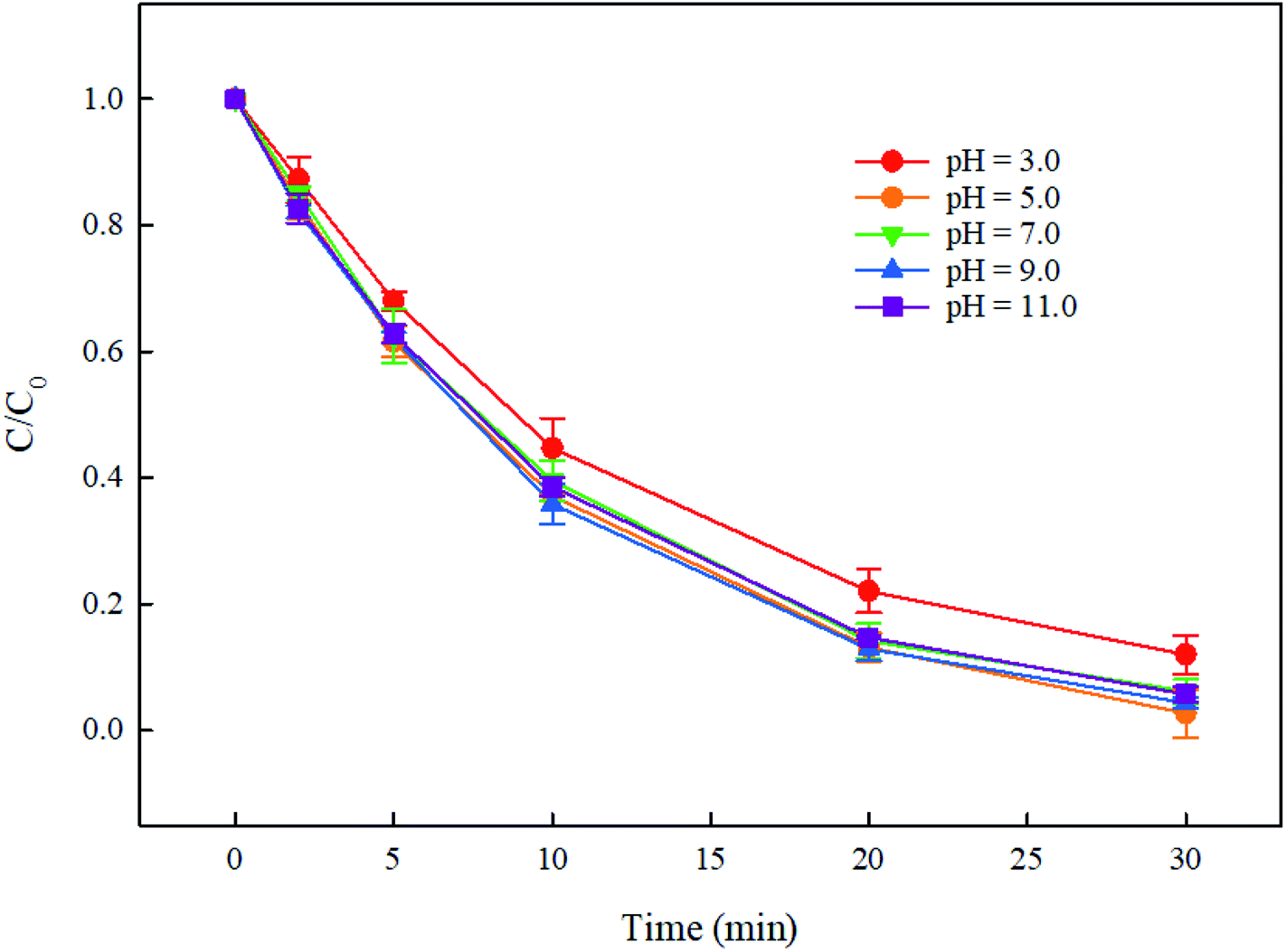
In the UV/PMS system, the pH of the solution is an important parameter, which can affect the concentration and distribution of active radicals SO4˙− and HO˙, and further may affect the degradation of pollutants. In addition, at high pH, PMS can be activated by alkali to generate active species. Therefore, it is necessary to explore the effect of solution pH on the degradation of DCF by UV/PMS. In this study, the degradation of DCF in UV/PMS system at pH of 3, 5, 7, 9 and 11 with phosphate buffer and ultrapure water were investigated, respectively. As shown in Fig. 3, the degradation efficiency of DCF in phosphate buffer and ultrapure water were both gradually increased as the pH of the solution increased. Since the pKa of DCF is 4.15,32 it was mainly existed in the form of protonation at pH 3; when pH > 5, it was mainly existed in a deprotonated form. Hence, its photolysis efficiency by UV alone might be different at different pHs. To prove the supposition above, the degradation of DCF in UV alone system at different pH conditions was studied. As shown in Fig. 4, the photolysis of DCF at pH 3 was lower than those at other pH values, indicating that the deprotonated form of DCF was more prone to undergo photolysis. At pH 5–11, DCF was existed in the same form, and therefore its direct photolysis was almost the same. Thus, in UV/PMS system, the lower removal efficiency of DCF at pH 3 might be attributed to its poor photolysis. Although the concentration of OH− in UV/PMS system would gradually increase with the increase of pH, it would react with SO4˙− to generate HO˙ again, as presented in eqn (3).33 These two kinds of radicals have similar reactivities toward DCF (KSO4˙−/DCF = 9.2 × 109 M−1 s−1,22 KHO˙/DCF = 7.5 × 109 M−1 s−134), as a result, the degradation of DCF was almost the same under near-neutral pH conditions. When the pH was 11, the degradation efficiency of DCF reached the highest. Since the molar absorption coefficient of PMS (εPMS) increased with increasing pH in the range of 6–12, its photolysis rate under UV irradiation was thus enhanced with pH, which would further lead to the increase in the steady-state concentration of HO˙ and SO4˙− produced through UV-activated PMS.35 Therefore, the fastest degradation of DCF at pH 11.0 in this work might be ascribed to the increased steady-state concentration of reactive radicals.
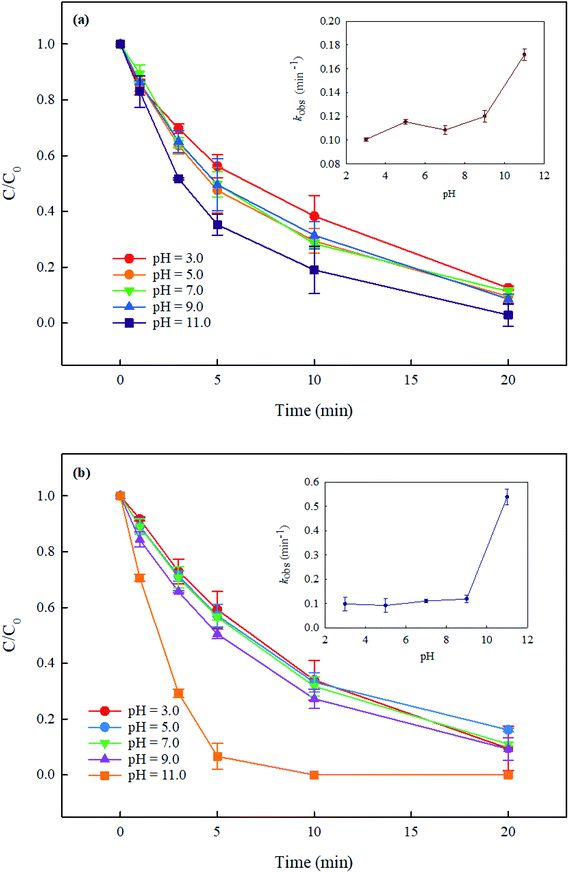 |
| | Fig. 3 Effect of pH on DCF degradation in UV/PMS system in phosphate buffer (a) and ultrapure water (b). Experimental conditions: [DCF]0 = 1 μM, [PMS]0 = 50 μM, T = 25 °C, phosphate buffer (10 mM), ultrapure water (18 MΩ cm). | |
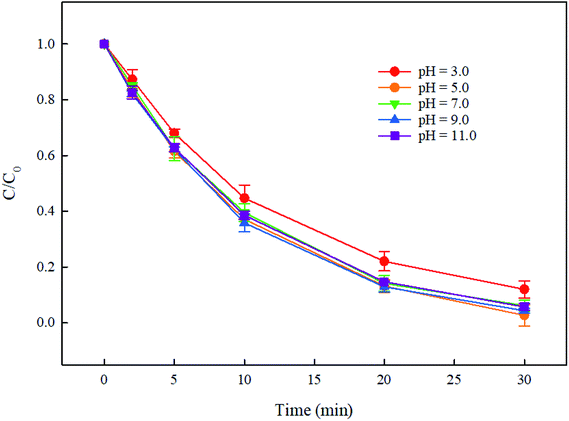 |
| | Fig. 4 UV photolysis of DCF at different pH values. Experimental conditions: [DCF]0 = 1 μM, T = 25 °C, phosphate buffer (10 mM). | |
Compared with DCF removal in phosphate buffer (Fig. 3(a)), its degradation rate in ultrapure water (Fig. 3(b)) was comparable at pH 3.0–9.0 but much faster at pH 11.0. The inhibited degradation of DCF in phosphate buffer at pH 11.0 was probably due to the competition of phosphate (HPO42−) with DCF for HO˙ and SO4˙− with the rate constants of 1.5 × 105 and 1.2 × 106 M−1 s−1, respectively, at a higher steady-state concentration of reactive radicals. PMS activation by phosphate buffer in this study was not observed, although it was reported by Lou et al.36 Yang et al.29 also did not find the PMS activation by phosphate in the degradation of furfuryl alcohol in PMS system. These results indicated that there was almost no influence of phosphate buffer on DCF removal in UV/PMS system at pH 7.0.
| | |
SO4˙− + OH− → SO42− + HO˙, K = 7.3 × 107 M−1 s−1
| (3) |
3.3. Effect of PMS dosage
The dosage of PMS is a key parameter in the UV/PMS system, because it will not only affect the removal efficiency of pollutants, but also influence the operating cost of the reaction system. Consequently, this study investigated the effect of PMS dosage (10–200 μM) on the degradation of DCF by UV/PMS. As presented in Fig. 5, the removal of DCF was gradually increased with the increase of PMS concentration, and its degradation rate constant was gradually enhanced from 0.095 min−1 to 0.219 min−1 when PMS dosage changed from 10 μM to 200 μM. This might be due to the increase in the steady-state concentration of reactive radicals (SO4˙− and HO˙) with the increase in the dosage of PMS. However, excessive PMS might consume SO4˙− and HO˙, as shown in eqn (3), (4) and (5),37 and SO4˙− and HO˙ might also undergo recombination (eqn (6)).38 Hence, the growth trend of DCF degradation would slow down at high concentration of PMS. However, this phenomenon was not observed in this study, indicating that 200 μM of PMS has not reached the critical value of inhibition under current experimental conditions.| | |
HSO5− + SO4˙− → HSO4− + SO5˙−, K < 1.0 × 105 M−1 s−1
| (4) |
| | |
HSO5− + HO˙ → SO5˙− + H2O, K = 1.7 × 107 M−1 s−1
| (5) |
| | |
SO4˙− + HO˙ → HSO4− + 0.5 O2, K = 1–10 × 109 M−1 s−1
| (6) |
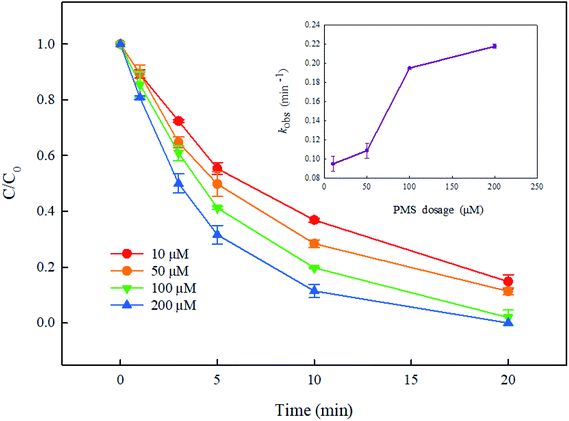 |
| | Fig. 5 Effect of PMS dosage on DCF degradation in UV/PMS system. Experimental conditions: [DCF]0 = 1 μM, T = 25 °C, pH 7.0 phosphate buffer (10 mM). | |
3.4. Effect of initial concentration of DCF
Besides the solution pH and PMS dosage, the degradation rate of DCF might also be affected by its initial concentration in UV/PMS system. Fig. 6 shows the degradation of DCF with different initial concentrations when the dosage of PMS was 50 μM. The removal rate of DCF was gradually slowed down with its increasing concentration, and the kobs was decreased from 0.133 min−1 to 0.088 min−1 when the initial concentration of DCF increased from 0.2 μM to 5 μM. Possible reasons include the following two aspects: (1) the increase in DCF concentration might affect the absorption of UV by the oxidant PMS, reducing the production of active radicals SO4˙− and HO˙ accordingly; (2) the concentration of reaction products would also increase with the increase of DCF concentration, and they would compete with DCF for reactive radicals. Similar conclusions were also obtained in other studies. Shah et al.39 found that the degradation rate of the pesticide endosulfan was gradually decreased with its increasing concentration in UV/PDS system.
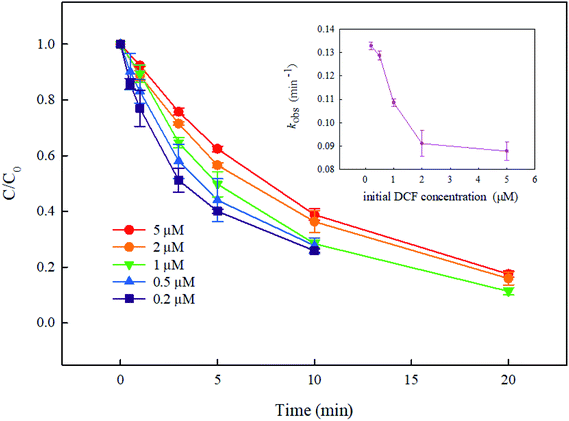 |
| | Fig. 6 Effect of initial DCF concentration on its degradation in UV/PMS system. Experimental conditions: [PMS]0 = 50 μM, T = 25 °C, pH 7.0 phosphate buffer (10 mM). | |
3.5. Effect of common water constituents
3.5.1. Effect of inorganic anions. Inorganic anions (e.g., HCO3−, Cl−, NO3− and SO42−) are common constituents in natural water bodies. The research on their influence on the degradation of DCF by UV/PMS is helpful in determining the application of this technology in the treatment of organic pollutants in real waters. As shown in Fig. 7(a), the presence of Cl− could inhibit the elimination of DCF in the first 10 min of the reaction in UV/PMS system. This was because the added Cl− could react with SO4˙− and HO˙ to generate Cl˙ and Cl2˙− (eqn (7)–(11)) which were less oxidative,40,41 and thereby hindering the degradation of DCF. As the reaction time increased, the steady-state concentration of the formed Cl˙ and Cl2˙− accumulated in the system would increase, and they might be able to degrade DCF. Therefore, the removal efficiency of DCF was almost the same after the reaction even if with different concentrations of Cl−. Since SO42− nearly did not react with SO4˙− and HO˙, its presence in the reaction system basically did not affect the removal of DCF, as shown in Fig. 7(b). The presence of NO3− could promote the degradation of DCF by UV/PMS, and the higher the NO3− concentration, the more significant the promotion effect, as depicted in Fig. 7(c). This was mainly because NO3− could be excited to produce HO˙ under UV irradiation as shown in eqn (12)–(15).42 Huang et al.43 also found that compared with UV alone, UV/NO3− significantly promoted the elimination of DCF, which was due to the contribution of HO˙. The presence of HCO3− enhanced the removal of DCF, and the promotion effect was gradually increased with the increase of HCO3− concentration, as presented in Fig. 7(d). The addition of HCO3− could change the pH of the solution, and it was varied from 8.8 to 9.2 when the concentration of HCO3− increased from 1 mM to 20 mM. In this pH range, the degradation of DCF in UV/PMS system was almost the same, as discussed in Section 3.2. Thus, the pH change caused by the addition of HCO3− had almost no effect on the degradation of DCF. The promotion effect of HCO3− on DCF removal might be attributed to the carbonate radical (CO3˙−) generated through the reactions of HCO3− with SO4˙− and HO˙, as shown in eqn (16) and (17).44 Liu et al.45,46 also found this enhancement effect in the degradation of oxytetracycline by UV/NO3−, UV/H2O2 and UV/PDS in the presence of HCO3−, which was ascribed to the role of generated CO3˙−.| | |
SO4˙− + Cl− → Cl˙ + SO42−, K = 3.1 × 108 M−1 s−1
| (7) |
| | |
Cl− + HO˙ → ClOH˙−, K = (4.3 ± 0.4) × 109 M−1 s−1
| (8) |
| | |
ClOH˙− → Cl− + HO˙, K = (6.1 ± 0.8) × 109 M−1 s−1
| (9) |
| | |
ClOH˙− + H+ → Cl˙ + H2O, K = (2.1 ± 0.7) × 1010 M−1 s−1
| (10) |
| | |
Cl˙ + Cl− → Cl2˙−, K = 2.1 × 1010 M−1 s−1
| (11) |
| | |
[NO3−]* → NO2− + O(3P)
| (13) |
| | |
[NO3−]* → NO2˙ + O˙−
| (14) |
| | |
O˙− + H2O → HO˙ + OH−
| (15) |
| | |
HCO3− + SO4˙− → CO3˙− + SO42− + H+, K = 9.1 × 106 M−1 s−1
| (16) |
| | |
HCO3− + HO˙ → CO3˙− + H2O, K = 8.5 × 106 M−1 s−1
| (17) |
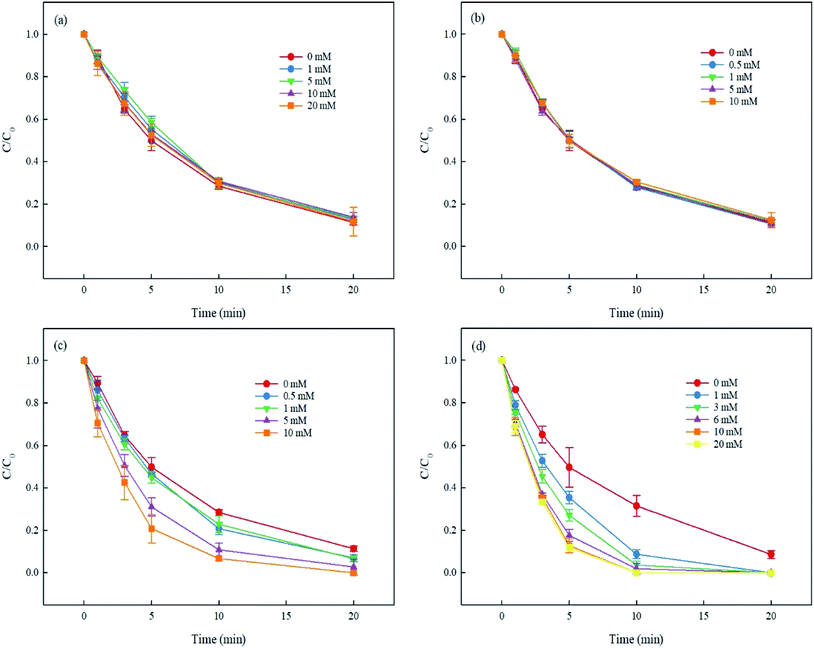 |
| | Fig. 7 Effect of Cl− (a), SO42− (b), NO3− (c) and HCO3− (d) on DCF degradation in UV/PMS system. Experimental conditions: [DCF]0 = 1 μM, [PMS]0 = 50 μM, T = 25 °C, except for (d) which is pure water, all others are pH 7.0 phosphate buffer (10 mM). | |
3.5.2. Effect of NOM. NOM is a common type of photosensitive materials, which can be excited to produce reactive oxygen species (ROS, such as 1O2, O2˙− and HO˙) under UV irradiation, and then promoting the removal of pollutants.47,48 Meanwhile, NOM can compete with the target pollutants for incident UV, inhibiting the direct photolysis of pollutants accordingly. Furthermore, they can also react with radicals such as HO˙ and SO4˙−, thereby inhibiting the degradation of the target contaminants. In summary, in the UV-based advanced oxidation systems, NOM has a dual effect and their impact on the degradation of pollutants depends on their own nature and the reaction conditions. In this study, fulvic acid was chosen to represent NOM to investigate the effect of NOM on DCF degradation by UV/PMS. As shown in Fig. 8, the presence of FA hindered the degradation of DCF, and the higher the FA concentration, the more obvious the inhibition effect. This result showed that in UV/PMS system, the inhibition effect of FA on DCF degradation exceeded its promotion effect, that is, the inhibition effect caused by the competition in incident UV, HO˙ and SO4˙− between NOM and DCF was greater than the promotion effect caused by ROS generated by UV-activated NOM.
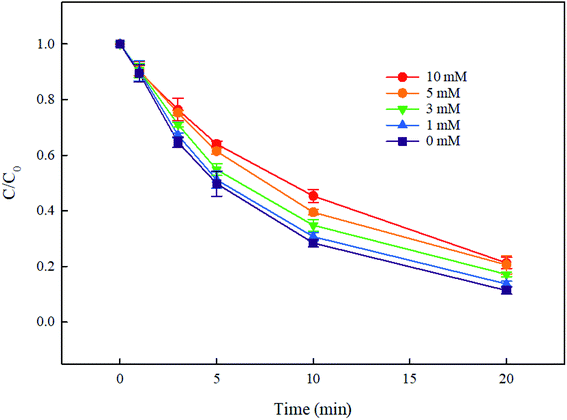 |
| | Fig. 8 Effect of FA on DCF degradation in UV/PMS system. Experimental conditions: [DCF]0 = 1 μM, [PMS]0 = 50 μM, T = 25 °C, pH 7.0 phosphate buffer (10 mM). | |
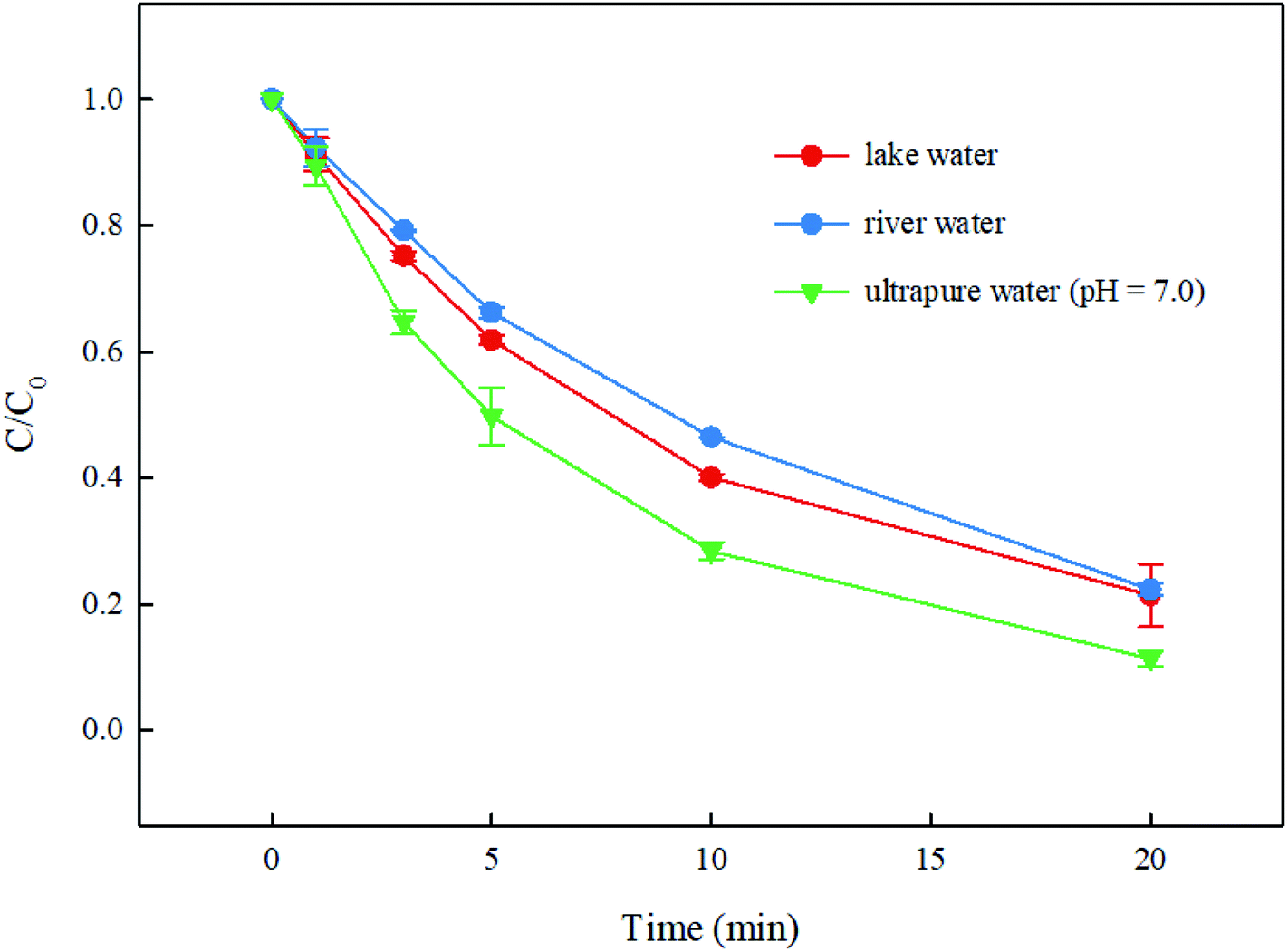
3.6. Degradation of DCF by UV/PMS in real waters
The degradation of DCF in real waters from a lake and a river by UV/PMS was conducted. The water quality parameters of these two real waters are presented in Table 1. As shown in Fig. 9, compared with the degradation of DCF in ultrapure water (pH = 7.0 adjusted by 10 mM phosphate buffer), its removal in lake water and river water were both inhibited slightly. According to the analysis in Section 3.5, it is clear that Cl− and SO42− almost had no effect on DCF removal in this system while HCO3− and NO3− could promote the degradation of DCF. Moreover, NOM could suppress DCF degradation due to its competition with DCF for reactive radicals. The promotion of HCO3− and NO3− in real waters might be inferior to the inhibition of NOM, which might result in a slight inhibition on DCF degradation in real waters in this system.
Table 1 Water quality parameters of real waters
| Real waters |
pH |
UV254 |
CODcr (mg L−1) |
NH3-N (mg L−1) |
Total alkalinity (mg CaCO3 per L) |
Cl− (mg L−1) |
NO3− (mg L−1) |
SO42− (mg L−1) |
| ND: not detected. |
| River water |
7.6 |
0.0672 |
22.4 |
3.87 |
662.11 |
0.2368 |
9.1605 |
0.00854 |
| Lake water |
7.2 |
0.0365 |
20.8 |
0.93 |
470.36 |
0.1302 |
8.7245 |
NDa |
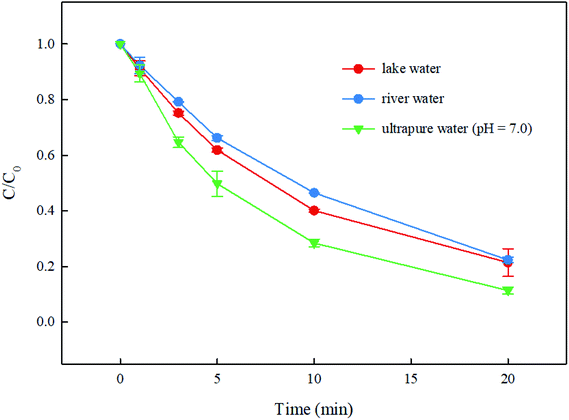 |
| | Fig. 9 Degradation of DCF by UV/PMS in real waters. Experimental conditions: [DCF]0 = 1 μM, [PMS]0 = 50 μM, T = 25 °C. | |
3.7. Mineralization of DCF
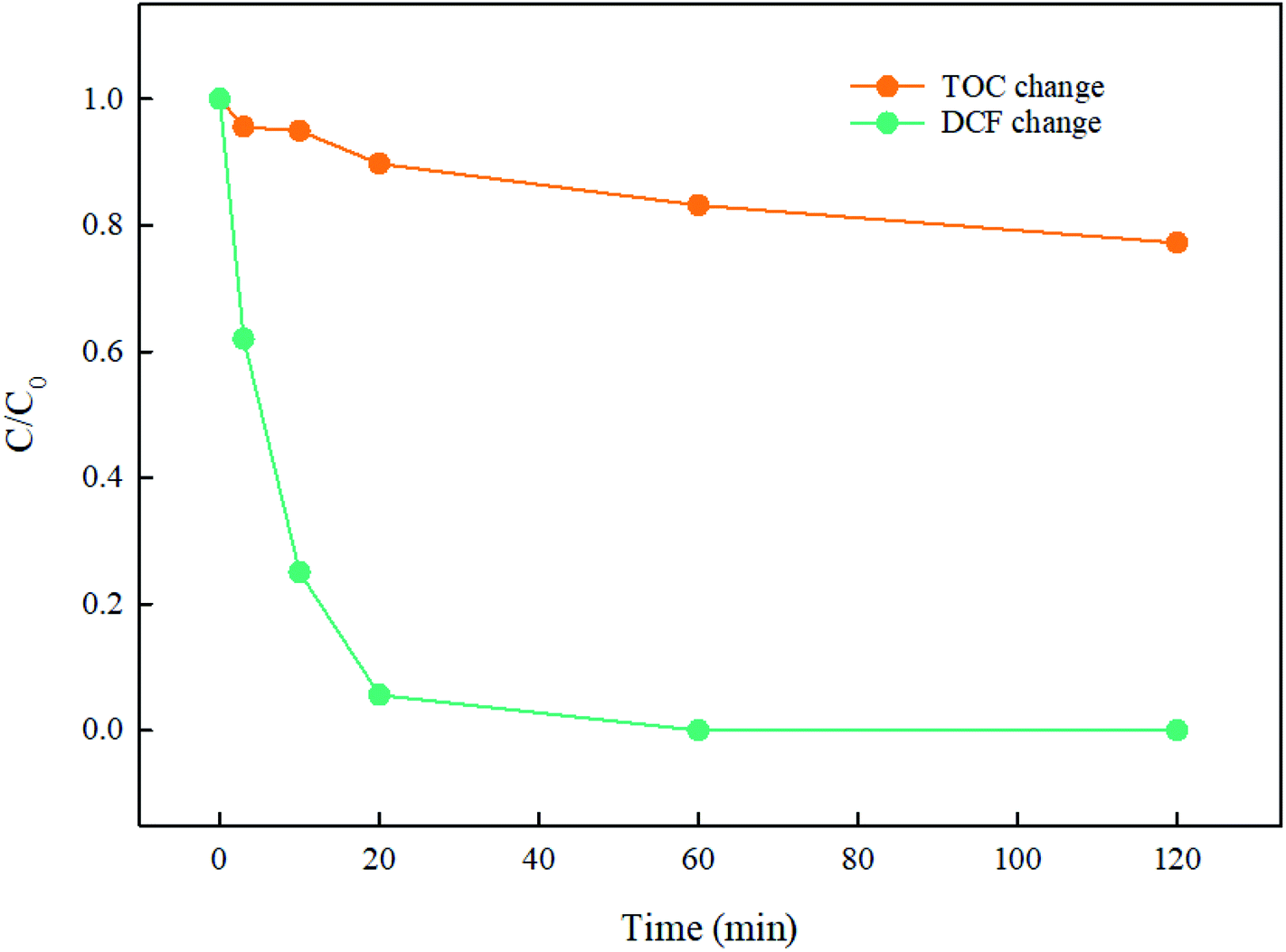
The mineralization of DCF by UV/PMS was performed and the result is seen in Fig. 10. In first 20 min, 94.4% of DCF was degraded rapidly, while the total organic carbon was merely removed 10.2%. As the reaction went on, DCF was completely degraded and TOC was gradually eliminated, indicating that this system was feasible for the degradation and mineralization of DCF. Since DCF was firstly decomposed to intermediates as stated in Section 3.8, which was then degraded to other products such as CO2 and H2O, the mineralization of DCF was much slower than its degradation, and a prolonged reaction time might be needed for obtaining a good TOC elimination.
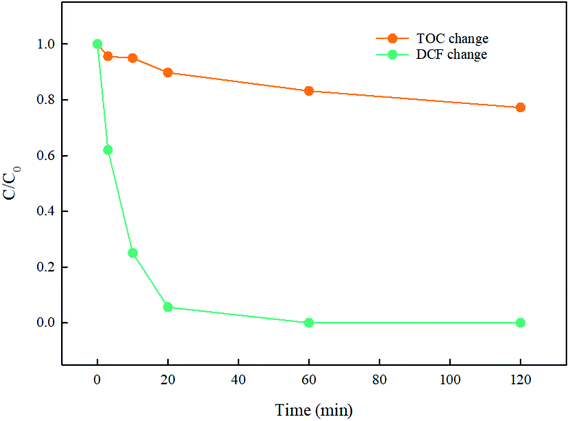 |
| | Fig. 10 Mineralization of DCF in UV/PMS system. Experimental conditions: [DCF]0 = 10 μM, [PMS]0 = 500 μM, T = 25 °C, pH 7.0 phosphate buffer (10 mM). | |
3.8. Identification of degradation products and transformation pathways of DCF
In the degradation of DCF by UV/PMS, seven reaction products were detected. Their molecular mass, mass to charge ratio (m/z), formula and possible structures are presented in Table 2. Based on these identified transformation products, the probable degradation mechanism of DCF in UV/PMS system was proposed including six different reaction pathways, namely hydroxylation, decarboxylation, dechlorination–cyclization, formylation, dehydrogenation and dechlorination–hydrogenation, as shown in Fig. 11. In current experimental conditions, both SO4˙− and HO˙ were existed in UV/PMS system. Under the electrophilic attack of these two active radicals, DCF could undergo hydroxylation and decarboxylation to generate hydroxylated product m/z 312 and decarboxylation product m/z 252. The formed product m/z 312 could further undergo formylation reaction under the oxidation of SO4˙− and HO˙ to produce product m/z 282, which could be further converted to product m/z 280 through dehydrogenation, as described in Fig. 11.
Table 2 Degradation products of DCF in UV/PMS system
| Serial number |
Molecular mass |
Mass-to-charge ratio |
Formula |
Proposed structure |
| DCF |
295 |
296 |
C14H11Cl2NO2 |
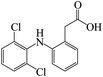 |
| 1 |
311 |
312 |
C14H11Cl2NO3 |
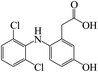 |
| 2 |
281 |
282 |
C13H9Cl2NO2 |
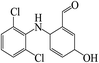 |
| 3 |
279 |
280 |
C13H7Cl2NO2 |
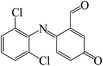 |
| 4 |
259 |
260 |
C14H10ClNO2 |
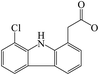 |
| 5 |
251 |
252 |
C13H11Cl2N |
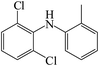 |
| 6 |
225 |
226 |
C14H11NO2 |
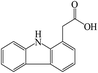 |
| 7 |
215 |
216 |
C13H10ClN |
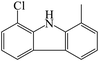 |
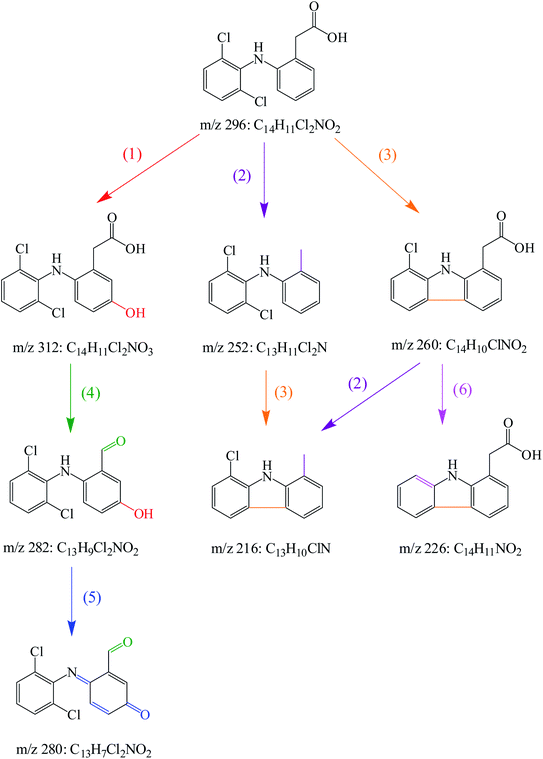 |
| | Fig. 11 Probable degradation pathways of DCF by UV/PMS: (1) hydroxylation, (2) decarboxylation, (3) dechlorination–cyclization, (4) formylation, (5) dehydrogenation, (6) dechlorination–hydrogenation. | |
A total of three dechlorination products of DCF were detected in this system, i.e., m/z 260, m/z 226 and m/z 216. The generation of these three products might be due to the cleavage of C–Cl bonds induced by UV photolysis and the oxidation of active radicals. Since the low-pressure mercury lamp mainly emitted ultraviolet light with a wavelength of 254 nm (the corresponding energy was 112 kcal mol−1), the C–Cl bonds of DCF might undergo bond cleavage under high-energy UV irradiation. As shown in Fig. 11, under UV radiation, DCF underwent dechlorination–cyclization reaction to generate product m/z 260, which could further undergo dechlorination–hydrogenation or decarboxylation to form products m/z 226 and m/z 216, respectively. The formed product m/z 216 might also be converted from the decarboxylation product m/z 252 through a dechlorination–cyclization reaction. Lekkerkerker-Teunissen et al.49 also detected dechlorination products m/z 260 and m/z 216 during the UV photolysis of DCF, and the other three photolysis products m/z 256 (C14H9NO4), m/z 242 (C14H11NO3) and m/z 212 (C13H9NO2) were not detected in this study.
4. Conclusions
In this study, the removal of DCF by the UV/PMS system was investigated systematically. Compared with UV alone, UV/PMS could significantly promote the degradation of DCF. Radical quenching experiments proved that the promotion effect was attributed to the role of SO4˙− and HO˙ produced by UV activation of PMS. The degradation efficiency of DCF was gradually increased with increasing pH, and its degradation rate was the fastest at pH 11, which was probably due to the increased steady-state concentration of reactive radicals produced through UV-activated PMS. The degradation of DCF was gradually enhanced with the increase in PMS dosage, but was gradually slowed down with the increase of its own initial concentration. The presence of Cl− could inhibit the removal of DCF in the first 10 min of the reaction, and then the inhibition effect was disappeared after reaction. NO3− and HCO3− could promote the degradation of DCF because of the contribution of additional HO˙ and CO3˙− generated, respectively. The presence of SO42− could hardly affect the elimination of DCF, while NOM could restrain DCF removal, and the inhibition effect was gradually increased with the increase of NOM concentration. Compared with DCF degradation in ultrapure water, its removal in real waters was slightly suppressed. The mineralization of DCF could be obtained by UV/PMS, but its efficiency was much slower than DCF degradation. Based on the detected seven degradation products, the probable reaction mechanism of DCF in UV/PMS system was proposed exhibiting six different transformation pathways including hydroxylation, decarboxylation, dechlorination–cyclization, formylation, dehydrogenation and dechlorination–hydrogenation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by Sichuan Science and Technology Program (2018SZDZX0026). We also gratefully appreciate financial support from the Fundamental Research Funds for the Central Universities (2682018CX32).
References
- P. Kovalakova, L. Cizmas, T. J. McDonald, B. Marsalek, M. Feng and V. K. Sharma, Occurrence and toxicity of antibiotics in the aquatic environment: a review, Chemosphere, 2020, 251, 1–15 CrossRef.
- J. Wang and S. Wang, Removal of pharmaceuticals and personal care products (PPCPs) from wastewater: a review, J. Environ. Manage., 2016, 182, 620–640 CrossRef CAS.
- M. Qiao, G. G. Ying, A. C. Singer and Y. G. Zhu, Review of antibiotic resistance in China and its environment, Environ. Int., 2018, 110, 160–172 CrossRef CAS.
- X. Wang, R. Yin, L. Zeng and M. Zhu, A review of graphene-based nanomaterials for removal of antibiotics from aqueous environments, Environ. Pollut., 2019, 253, 100–110 CrossRef CAS.
- R. Zhuan and J. Wang, Degradation of diclofenac in aqueous solution by ionizing radiation in the presence of humic acid, Sep. Purif. Technol., 2020, 234, 1–8 CrossRef.
- M. Parolini, Toxicity of the Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) acetylsalicylic acid, paracetamol, diclofenac, ibuprofen and naproxen towards freshwater invertebrates: A review, Sci. Total Environ., 2020, 740, 1–13 CrossRef.
- A. Ameri, M. Shakibaie, M. Pournamdari, A. Ameri, A. Foroutanfar, M. Doostmohammadi and H. Forootanfar, Degradation of diclofenac sodium using UV/biogenic selenium nanoparticles/H2O2: optimization of process parameters, J. Photochem. Photobiol., A, 2020, 392, 1–10 CrossRef.
- F. Yu, Y. Wang, H. Ma and M. Zhou, Hydrothermal synthesis of FeS2 as a highly efficient heterogeneous electroFenton catalyst to degrade diclofenac via molecular oxygen effects for Fe(II)/Fe(III) cycle, Sep. Purif. Technol., 2020, 248, 1–12 Search PubMed.
- X. Li, M. Zhou and Y. Pan, Degradation of diclofenac by H2O2 activated with pre-magnetization Fe(0): influencing factors and degradation pathways, Chemosphere, 2018, 212, 853–862 CrossRef CAS.
- M. Sadeghi, M. H. Mehdinejad, N. Mengelizadeh, Y. Mahdavi, H. Pourzamani, Y. Hajizadeh and M. R. Zare, Degradation of diclofenac by heterogeneous electro-Fenton process using magnetic single-walled carbon nanotubes as a catalyst, J. Water Process. Eng., 2019, 31, 1–12 Search PubMed.
- A. Aguinaco, F. J. Beltrán, J. F. García-Araya and A. Oropesa, Photocatalytic ozonation to remove the pharmaceutical diclofenac from water: influence of variables, Chem. Eng. J., 2012, 189–190, 275–282 CrossRef CAS.
- M. Huguet, M. Deborde, S. Papot and H. Gallard, Oxidative decarboxylation of diclofenac by manganese oxide bed filter, Water Res., 2013, 47, 5400–5408 CrossRef CAS.
- X. Lu, Y. Shao, N. Gao, J. Chen, Y. Zhang, H. Xiang and Y. Guo, Degradation of diclofenac by UV-activated persulfate process: kinetic studies, degradation pathways and toxicity assessments, Ecotoxicol. Environ. Saf., 2017, 141, 139–147 CrossRef CAS.
- K. Kowalska, G. Maniakova, M. Carotenuto, O. Sacco, V. Vaiano, G. Lofrano and L. Rizzo, Removal of carbamazepine, diclofenac and trimethoprim by solar driven advanced oxidation processes in a compound triangular collector based reactor: a comparison between homogeneous and heterogeneous processes, Chemosphere, 2020, 238, 1–8 CrossRef.
- J. Wang and S. Wang, Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- E. Rosales, S. Diaz, M. Pazos and M. A. Sanromán, Comprehensive strategy for the degradation of anti-inflammatory drug diclofenac by different advanced oxidation processes, Sep. Purif. Technol., 2019, 208, 130–141 CrossRef CAS.
- H. Yu, E. Nie, J. Xu, S. Yan, W. J. Cooper and W. Song, Degradation of diclofenac by advanced oxidation and reduction processes: kinetic studies, degradation pathways and toxicity assessments, Water Res., 2013, 47, 1909–1918 CrossRef CAS.
- J. Wang and Z. Bai, Fe-based catalysts for heterogeneous catalytic ozonation of emerging contaminants in water and wastewater, Chem. Eng. J., 2017, 312, 79–98 CrossRef CAS.
- K. V. Plakas, A. Mantza, S. D. Sklari, V. T. Zaspalis and A. J. Karabelas, Heterogeneous Fenton-like oxidation of pharmaceutical diclofenac by a catalytic iron-oxide ceramic microfiltration membrane, Chem. Eng. J., 2019, 373, 700–708 CrossRef CAS.
- A. Shad, J. Chen, R. Qu, A. A. Dar, M. Bin-Jumah, A. A. Allam and Z. Wang, Degradation of sulfadimethoxine in phosphate buffer solution by UV alone, UV/PMS and UV/H2O2: kinetics, degradation products, and reaction pathways, Chem. Eng. J., 2020, 398, 1–14 CrossRef.
- C. Tan, Y. Dong, D. Fu, N. Gao, J. Ma and X. Liu, Chloramphenicol removal by zero valent iron activated peroxymonosulfate system: kinetics and mechanism of radical generation, Chem. Eng. J., 2018, 334, 1006–1015 CrossRef CAS.
- M. Mahdi Ahmed, S. Barbati, P. Doumenq and S. Chiron, Sulfate radical anion oxidation of diclofenac and sulfamethoxazole for water decontamination, Chem. Eng. J., 2012, 197, 440–447 CrossRef CAS.
- U. Ushani, X. Lu, J. Wang, Z. Zhang, J. Dai, Y. Tan, S. Wang, W. Li, C. Niu, T. Cai, N. Wang and G. Zhen, Sulfate radicals-based advanced oxidation technology in various environmental remediation: a state-of-the–art review, Chem. Eng. J., 2020, 402, 1–19 CrossRef.
- H. V. Lutze, N. Kerlin and T. C. Schmidt, Sulfate radical-based water treatment in presence of chloride: formation of chlorate, inter-conversion of sulfate radicals into hydroxyl radicals and influence of bicarbonate, Water Res., 2015, 72, 349–360 CrossRef CAS.
- S. Giannakis, K.-Y. A. Lin and F. Ghanbari, A review of the recent advances on the treatment of industrial wastewaters by Sulfate Radical-based Advanced Oxidation Processes (SR-AOPs), Chem. Eng. J., 2021, 406, 1–20 CrossRef.
- X. Duan, S. Yang, S. Wacławek, G. Fang, R. Xiao and D. D. Dionysiou, Limitations and prospects of sulfate-radical based advanced oxidation processes, J. Environ. Chem. Eng., 2020, 8, 1–5 Search PubMed.
- F. Ghanbari and M. Moradi, Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: review, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- S. Wacławek, H. V. Lutze, K. Grübel, V. V. T. Padil, M. Černík and D. D. Dionysiou, Chemistry of persulfates in water and wastewater treatment: a review, Chem. Eng. J., 2017, 330, 44–62 CrossRef.
- Y. Yang, G. Banerjee, G. W. Brudvig, J.-H. Kim and J. J. Pignatello, Oxidation of Organic Compounds in Water by Unactivated Peroxymonosulfate, Environ. Sci. Technol., 2018, 52, 5911–5919 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (HO˙/˙O−) in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- C. L. Clifton and R. E. Huie, Rate Constants for Hydrogen Abstraction Reactions of the Sulfate Radical, SO4˙−, Alcohols, Int. J. Chem. Kinet., 1989, 21, 677–687 CrossRef CAS.
- N. M. Vieno, H. Härkki, T. Tuhkanen and L. Kronberg, Occurrence of Pharmaceuticals in River Water and Their Elimination in a Pilot-Scale Drinking Water Treatment Plant, Environ. Sci. Technol., 2007, 41, 5077–5084 CrossRef CAS.
- D. Yuan, C. Zhang, S. Tang, M. Sun, Y. Zhang, Y. Rao, Z. Wang and J. Ke, Fe3+-sulfite complexation enhanced persulfate Fenton-like process for antibiotic degradation based on response surface optimization, Sci. Total Environ., 2020, 727, 1–10 Search PubMed.
- M. M. Huber, S. Canonica, G.-Y. Park and U. V. Gunten, Oxidation of pharmaceuticals during ozonation, Environ. Sci. Technol., 2003, 37, 1016–1024 CrossRef CAS.
- Y. Guan, J. Ma, X. Li, J. Fang and L. Chen, Influence of pH on the Formation of Sulfate and Hydroxyl Radicals in the UV/Peroxymonosulfate System, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS.
- X. Lou, L. Wu, Y. Guo, C. Chen, Z. Wanga, D. Xiao, C. Fang, J. Liu, J. Zhao and S. Lu, Peroxymonosulfate activation by phosphate anion for organics degradation in water, Chemosphere, 2014, 117, 582–585 CrossRef CAS.
- P. Maruthamuthu and P. Neta, Radiolytic chain decomposition of peroxomonophosphoric and peroxomonosulfuric acids, J. Phys. Chem., 1977, 81, 937–940 CrossRef CAS.
- Y. Liu, X. He, Y. Fu and D. D. Dionysiou, Kinetics and mechanism investigation on the destruction of oxytetracycline by UV-254 nm activation of persulfate, J. Hazard. Mater., 2016, 305, 229–239 CrossRef CAS.
- N. S. Shah, X. He, H. M. Khan, J. A. Khan, K. E. O'Shea, D. L. Boccelli and D. D. Dionysiou, Efficient removal of endosulfan from aqueous solution by UV-C/peroxides: a comparative study, J. Hazard. Mater., 2013, 263, 584–592 CrossRef CAS.
- Y. Liu, X. He, Y. Fu and D. D. Dionysiou, Degradation kinetics and mechanism of oxytetracycline by hydroxyl radical-based advanced oxidation processes, Chem. Eng. J., 2016, 284, 1317–1327 CrossRef CAS.
- K. H. Chan and W. Chu, Degradation of atrazine by cobalt-mediated activation of peroxymonosulfate: different cobalt counteranions in homogenous process and cobalt oxide catalysts in photolytic heterogeneous process, Water Res., 2009, 43, 2513–2521 CrossRef CAS.
- G. Mark, H.-G. Korth, H.-P. Schuchmann and C. v. Sonntag, The photochemistry of aqueous nitrate ion revisited, J. Photochem. Photobiol., A, 1996, 101, 89–103 CrossRef CAS.
- Y. Huang, M. Kong, D. Westerman, E. G. Xu, S. Coffin, K. H. Cochran, Y. Liu, S. D. Richardson, D. Schlenk and D. D. Dionysiou, Effects of HCO3− on Degradation of Toxic Contaminants of Emerging Concern by UV/NO3−, Environ. Sci. Technol., 2018, 52, 12697–12707 CrossRef CAS.
- S. Canonica, T. Kohn, M. Mac, F. Real, J. Wirz and U. V. Gunten, Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds, Environ. Sci. Technol., 2005, 39, 9182–9188 CrossRef CAS.
- Y. Liu, X. He, X. Duan, Y. Fu and D. D. Dionysiou, Photochemical degradation of oxytetracycline: influence of pH and role of carbonate radical, Chem. Eng. J., 2015, 276, 113–121 CrossRef CAS.
- Y. Liu, X. He, X. Duan, Y. Fu, D. Fatta-Kassinos and D. D. Dionysiou, Significant role of UV and carbonate radical on the degradation of oxytetracycline in UV-AOPs: kinetics and mechanism, Water Res., 2016, 95, 195–204 CrossRef CAS.
- H. Xu, W. J. Cooper, J. Jung and W. Song, Photosensitized degradation of amoxicillin in natural organic matter isolate solutions, Water Res., 2011, 45, 632–638 CrossRef CAS.
- D. Zhang, S. Yan and W. Song, Photochemically induced formation of reactive oxygen species (ROS) from effluent organic matter, Environ. Sci. Technol., 2014, 48, 12645–12653 CrossRef CAS.
- K. Lekkerkerker-Teunissen, M. J. Benotti, S. A. Snyder and H. C. van Dijk, Transformation of atrazine, carbamazepine, diclofenac and sulfamethoxazole by low and medium pressure UV and UV/H2O2 treatment, Sep. Purif. Technol., 2012, 96, 33–43 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*