
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFullerene soot and a fullerene nanodispersion as recyclable heterogeneous off-the-shelf photocatalysts†
Augustina Jozeliūnaitėa,
Domantas Valčeckasa and
Edvinas Orentas *ab
*ab
aDepartment of Organic Chemistry, Naugarduko 24, Vilnius, LT-03225, Lithuania. E-mail: edvinas.orentas@chf.vu.lt
bCenter for Physical Sciences and Technology, Saulėtekio Av. 3, LT-10257 Vilnius, Lithuania
First published on 21st January 2021
Abstract
Metal-free heterogeneous photocatalysis, which requires no prior catalyst immobilization or chemical modification and can operate in green solvents, represents a highly-sought after, yet currently still underdeveloped, synthetic method. In this report we present a comparative study which aims to evaluate the use of unmodified fullerene soot and a fullerene nanodispersion as non-soluble and quasi-soluble carbon-based photocatalysts, respectively, for sulfide oxidation and other transformations using oxygen as an oxidant in ethanol. A wide range of sulfoxides were successfully prepared with good yields and chemoselectivity using a very low catalyst loading. The fullerene soot photocatalyst is easily recovered and shows excellent stability of the catalytic properties. The reaction was shown to proceed via a singlet oxygen pathway and has a high selectivity for aliphatic sulfides, whereas the oxidation of thioanisoles can be accomplished using an amine mediated electron transfer mechanism. The applicability of the fullerene nanodispersion as a general purpose photocatalyst was demonstrated in radical cyclization, boronic acid oxidation and imine formation reactions.
1. Introduction
Catalytic chemical transformations utilizing heterogeneous catalysts are essential to the modern chemical industry and range in scale from the refinery to the pharmaceutical industry.1 According to recent estimates, 80–90% of industrial processes use catalysis, reaching global sales of 1.5 trillion US$ and generating 35% of the world's global domestic product (GDP).2 Although bulk or supported metal (metal oxides) and porous zeolite catalysts dominate the industrial fields, partially organic compounds that are amenable to the rational tailoring of catalytic sites and the porosity, such as metal organic frameworks, have become increasingly important players.3 Moreover, various nanostructured catalysts, mostly in the form of nanoparticles, allow unprecedented activities and chemoselectivities to be achieved.4 Owing to the separation of reactants/products and the catalyst in different phases and the complicated chemical properties of surfaces, heterogeneous catalysis is much more challenging to approach analytically or mechanistically; however, it provides the added advantage of easy separation from the products, thus enabling efficient recycling and environmentally more sustainable processes.Among the various catalytic systems, visible-light mediated photoredox catalysis has emerged as a functional group tolerant, mild, chemoselective and environmentally benign synthetic method for free radical generation from various starting materials. The high diversity of the starting materials and compatible reaction partners with these new catalytic methods has allowed chemists to harvest otherwise inaccessible chemical space.5 This particular field of catalysis is now considered to be one of the most rapidly developing and promising research areas in organic chemistry. In contrast, metal-free heterogeneous catalysts still remain largely underdeveloped, especially the ones operating in the most convenient visible region of the spectrum.6
In our quest for efficient environmentally friendly and sustainable heterogeneous catalysts, we turned our attention to carbon-based materials,7 in particular fullerenes, as possible candidates to enable the oxidation of sulfides to the corresponding sulfoxides using air or oxygen as the sole oxidant. Based on the well-known strong electron acceptor properties of fullerenes, the oxidation of organic substrates, initiated by an electron transfer and the ensuing formation of the radical cation–anion pair can be envisioned.8 Moreover, the electron transfer reaction can be further driven using visible light, increasing the oxidation potency of the catalyst. Alternatively, fullerene, as a highly stable oxygen photosensitizer with an almost quantitative quantum yield over a broad range of wavelengths, can be utilized in singlet oxygen mediated reactions.9 Indeed, some work in this direction has been reported previously, using C60 derivatives10 or solid support immobilized forms.11 The wider application of C60 in the pure form is limited by its high price, whereas modified catalysts require additional functionalization steps using commercial materials. We hypothesized that the cheap raw material used for fullerene production, fullerene soot (FS), could also serve as a heterogeneous oxidation photocatalyst and that it may be comparable with pure C60 in terms of the substrate scope, chemoselectivity and catalytic activity. FS is a low-cost nanomaterial produced by vaporizing the pure carbon in an inert atmosphere and condensing the vapor to produce a wide range of structures that include fullerenes, mostly C60 and C70. For comparison, the photocatalytic activity of the quasi-heterogeneous C60 nanodispersion was also probed.
The oxidation of sulfides to sulfoxides has been selected as a model reaction to assess the catalytic activity of FS. Sulfoxides are valuable chemical products which have widespread uses in medicinal chemistry and are also found in the structure of bioactive natural products.12,13 The main challenge associated with the oxidation of sulfides is related to chemoselectivity issues, in which over-oxidation to sulfones must be prevented. In a broader context, the oxidation of noxious (mustard gas or derivatives) or foul-smelling low-valent organosulfur derivatives has practical implications in wastewater treatment or the disposal of chemical warfare agents. To date, the oxidation of sulfides has been achieved using both homogeneous14 and heterogeneous photocatalysis;15 however, all the reported examples of heterogeneous catalysis involve additional steps to prepare the catalyst.
In this report we utilized FS as an off-the-shelf photocatalyst for sulfide oxidation in ethanol and also applied it to other oxidative transformations, such as the oxidation of boronic acids, amines or radical cascade cyclization reactions. In addition to the batch process, a flow reactor with a non-immobilized heterogeneous catalyst has been developed for larger scale reactions. A C60 nanodispersion in ethanol has also been successfully applied for the above described transformations and in most cases showed an enhanced catalytic performance.
2. Results and discussion
2.1 Reaction scope and catalyst screening
The list of sulfoxides prepared in this study is outlined in Fig. 1. In addition to simple dialkyl sulfides, compounds possessing a bulky tert-butyl group (2a, 2c, 2e, 2l, 2m–n) or oxidation sensitive alkene functionalities (2e–f) were also included for better investigation of the reaction scope. The heterogeneous catalysts were used in three different forms: untreated FS ([FS]), a suspension of FS in toluene ([FS in Tol]) or a saturated solution of C60 in toluene ([C60 in Tol]). The latter solution was used to prepare the C60 nanodispersion in poor solvents, such as ethanol or acetonitrile by injecting a saturated solution of C60 in toluene.16 The as-obtained yellow solution (quasi-heterogeneous system) was stable for some time, however, in all cases presented, it eventually converted into an amorphous precipitate of C60 after the reaction, enabling easy separation of the catalyst. Typically, on the 0.2 mmol reaction scale, 50 μL of the saturated C60 toluene solution (c = 4.2 mM) was used to prepare the nanodispersion, resulting in a substrate to catalyst molar ratio of approximately 1400. For the FS suspension in toluene, 100 μL of stock suspension (2 mg of FS in 4 mL of toluene) was added to 2 mL of ethanol. For the FS powder, 1 mg of FS was used for 0.2 mmol of substrate. Taking into account that commercial FS contains up to 7% w/w of extractable fullerenes,17 this translates into substrate to catalyst molar ratios of approximately 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 2000 for the FS suspension and FS powder, respectively (assuming that all fullerenes in FS are C60). Ethanol (95/5 v/v mixture with water) was used as a green solvent, which is also capable of stabilizing one of the key reaction intermediates (reaction mechanisms are discussed below). Another polar, but aprotic solvent, acetonitrile was chosen as a control in which, similar to ethanol, fullerenes are virtually insoluble. The reactions were performed using solvents saturated with O2 (bubbling) in closed borosilicate culture tubes under irradiation with a 100 W 450 nm blue LED (at 10 cm distance) with external air cooling.
000 and 2000 for the FS suspension and FS powder, respectively (assuming that all fullerenes in FS are C60). Ethanol (95/5 v/v mixture with water) was used as a green solvent, which is also capable of stabilizing one of the key reaction intermediates (reaction mechanisms are discussed below). Another polar, but aprotic solvent, acetonitrile was chosen as a control in which, similar to ethanol, fullerenes are virtually insoluble. The reactions were performed using solvents saturated with O2 (bubbling) in closed borosilicate culture tubes under irradiation with a 100 W 450 nm blue LED (at 10 cm distance) with external air cooling.
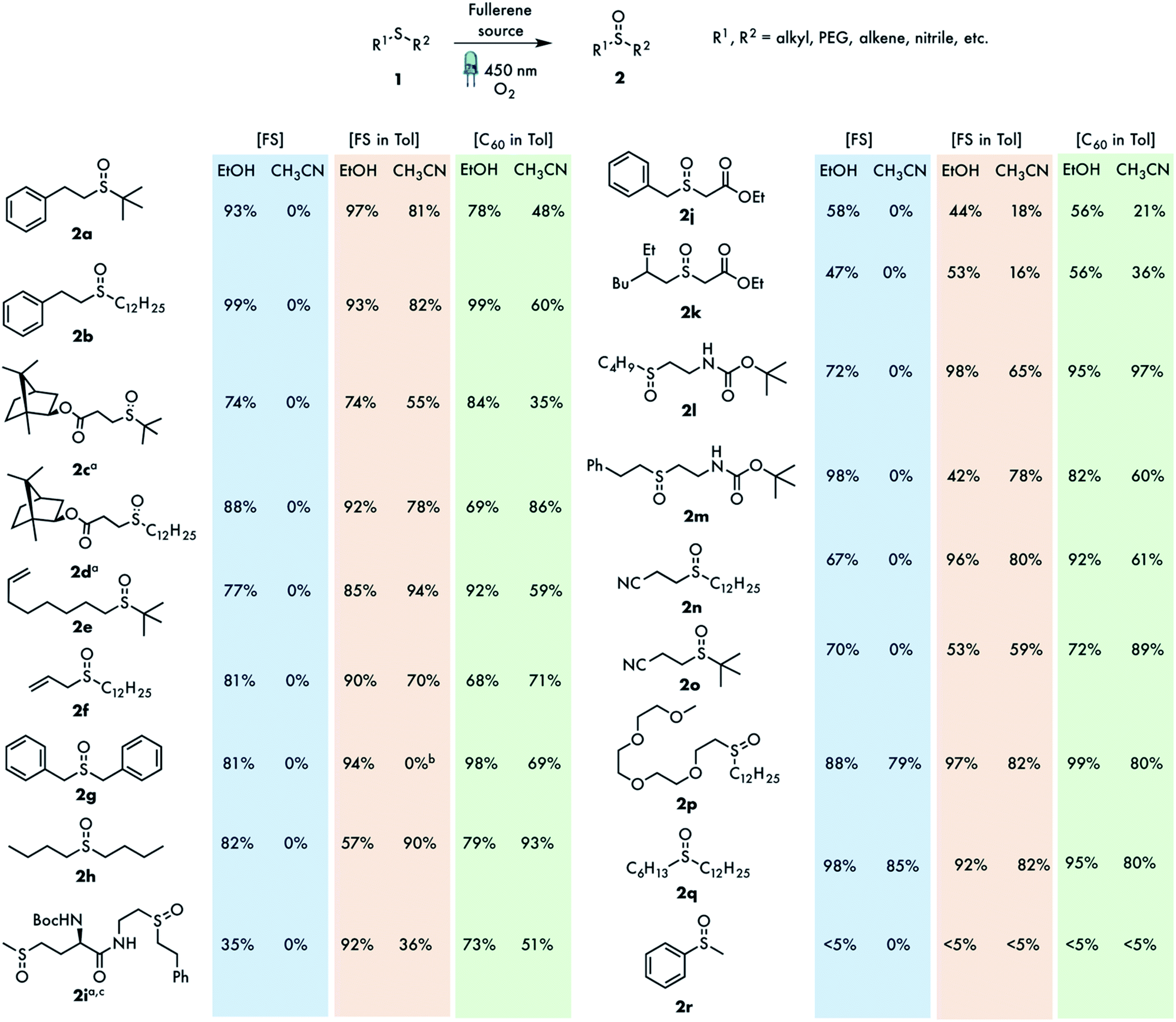 | ||
| Fig. 1 Exploration of the substrate scope. FS: fullerene soot; Tol: toluene. For experimental details see the ESI.† aMixture of diastereomers; bcomplex mixture; cdouble oxidation product. | ||
As shown in Fig. 1, the oxidation efficiency varies depending on the solvent or catalyst used. In most cases, FS performed well, delivering sulfoxide products in good yields. On average, the reaction was completed in 48 and 24 h using the FS or C60 nanodispersion, respectively. Sulfides with tert-butyl groups were also successfully oxidized, indicating the high tolerance to steric hindrance of this catalytic system. Remarkably, the challenging substrate, dibenzyl sulfide 2g, known for its propensity to fragment into benzaldehyde during oxidation, was smoothly oxidized to the expected sulfoxide. The lowest yields were obtained with substrates 2j and 2k. It should be noted, however, that these substrates, similar to dibenzylsulfide, are also sensitive to fragmentation via enolization owing to the increased acidity of the CH2 protons between the sulfur atom and ester group (vide infra). In our case the moderate yields obtained were still higher than those obtained using other reported oxidation methods.18 The alkene functionality in 2e–f was compatible with the reaction conditions. In the few cases tested, the sulfone by-product was not detected in significant amounts, based on the NMR analysis of the crude product. Control reactions without light, oxygen or catalyst did not deliver tangible amounts of the products. As expected, fullerene depleted FS, obtained after continuous extraction with 1,2-dichlorobenzene was notably less efficient, providing 2b in only a 20% yield. Non extractable carbon nanotubes, that are probably present in FS might be responsible for the small amount of the product formed. A control experiment using single walled carbon nanotubes did indeed afford a 13% yield of 2b, whereas powdered activated charcoal provided no oxidation product.
Comparison of the three different catalysts revealed some notable trends. In the majority of cases, FS delivered as a suspension in toluene provided higher yields of the products in both solvents. At first glance, these results can be easily rationalized by assuming the possible extraction of fullerene components from carbon black. This assumption is also corroborated by the slightly enhanced performance of the C60 nanodispersion compared to FS. In FS, the active fullerenes are less accessible by the substrate and are competitively adsorbed on carbon black. The C60 in nanodispersion, on the other hand, is more accessible and has a greater effective surface area. On the other hand, in quite a few cases FS performed better than the C60 nanodispersion, which might be attributed to the combined action of C60 and C70. For instance, a slight increase in the yield (∼5%) was observed for substrate 1f using the nanodispersion of C60/C70 (1:1 molar ratio). The change in morphology of FS upon treatment with toluene is an alternative explanation for the enhancement of the catalytic properties. Indeed, notable changes were observed in the scanning electron microscopy (SEM) images obtained after stirring FS into toluene (Fig. S2†). Stirring with toluene resulted in a much finer powder (0.80 μm) compared to the untreated FS (1.43 μm) which might contribute to the enhanced performance of this system. Strikingly, in contrast to our study, the previously reported related photocatalyst, fullerene-modified carbon nitride, produced sulfone as the main product in ethanol and was not able to discriminate between different types of sulfides.11b
2.2 Development of the flow reactor
The notorious drawback of all photochemical reactions is the short penetration length of light into the reaction mixture. This limitation severely reduces the scale of batch processes. To overcome this obstacle, larger scale reactions are typically carried out using a flow system which is easily implemented for homogeneous processes. As anticipated, our attempt to scale up the reaction with substrate 2b to the gram scale under standard conditions was not successful, resulting in incomplete conversion, even after weeks of irradiation. Although heterogeneous reactions are routinely performed under flow, insoluble reagents or catalysts are typically immobilized in the stationary phase, which is not always feasible for photochemical reactions.19 We sought to develop a flow process with a heterogeneous catalyst within the mobile phase. For this, a flow loop was assembled, consisting of a mini-peristaltic pump connected to 3 mm Teflon tubing and passing through a reservoir of FS suspension in ethanol and a high surface area irradiation section (Fig. 2a and b). The reservoir was connected to either the cylinder of oxygen or another mini-peristaltic pump that acted as an adjustable air feed. We found that the flow of the well mixed suspension could be maintained with a flow rate of 30 mL min−1 without sedimentation of the catalyst powder, even at the tube turning points (Fig. 2c and d). If necessary, the irradiated part can be refrigerated with either water or a fan. Using this setup, gram quantities of 2b were obtained in a 94% yield within 5 d with a turnover number of 11000.
 | ||
| Fig. 2 (a) Schematic representation of the flow system; (b) working unit assembly; (c) FS dispersion flowing through the irradiated section; (d) snapshot of the FS dispersion under flow in the tubing; (e) FS dispersion in ethanol before the flow was initiated; and (f) flow reaction under irradiation. | ||
2.3 Recyclability studies
The long-term stability is one of the most important characteristics of a heterogeneous catalyst. Owing to the extremely low solubility of FS in ethanol, the catalyst can be quantitively recovered after the reaction by simple filtration or centrifugation. The chemical stability of FS was evaluated by performing the oxidation reaction of 2b for 10 reaction cycles. Essentially, no drop in catalytic activity was noted (Fig. 3a). | ||
| Fig. 3 (a) Recyclability of the FS catalyst. (b) SEM image of the FS powder before the reaction (top) and after 10 cycles (bottom). | ||
In order to check whether the chemical nature of FS had changed during the reaction, Raman measurements on fresh and recycled FS were carried out. After the reaction, the FS was filtered, washed with ethanol and dried. The spectra of both samples were almost identical, demonstrating the structural and chemical integrity of the catalyst. The SEM measurements showed that in contrast to the FS in toluene, the size of the particles increased significantly during cycling of the catalyst, but surprisingly did not compromise its catalytic activity (Fig. 3b). The formation of fullerene oxides by the action of singlet oxygen has been demonstrated previously; however, even small quantities of alcohol almost fully suppresses this reaction.10b
2.4 Reaction mechanism
The mechanism of sulfide oxidation with oxygen has been a subject of many studies over the years. Based on the data obtained from the pioneering work performed by Foote20 and Clennan21 and the later extensive studies reported by Baciocchi,22,18b Albini23 and others,24 it is now generally accepted that oxidation of sulfides with oxygen can proceed either through energy transfer or electron transfer pathways. In the energy transfer mechanism, an excited sensitizer (in our case fullerene) undergoes intersystem crossing to produce a triplet state, which via energy transfer generates singlet oxygen, 1O2 (Scheme 1a). Singlet oxygen reacts with sulfide 1 to produce a key intermediate, persulfoxide 3. The reaction of the latter with another molecule of 1 yields two molecules of sulfoxide 2. In another mechanism, the excited photosensitizer acts as an oxidant and abstracts an electron from sulfide 1 resulting in a pair, a radical cation 5 and a radical anion 4 (Scheme 1b). After the electron transfer from 4 to O2, a superoxide radical anion is formed which can combine with 5 to generate the same persulfoxide 3, as in the case of the singlet oxygen mechanism. It is worth noting that 3 is a strongly nucleophilic species and its reaction with another nucleophile, sulfide 1 is expected to be very slow. In fact, persulfoxide 3 can be trapped with electrophilic diphenylsulfoxide and the product formed, diphenylsulfone, can be used as a marker for the presence of 3. Therefore, thiadioxirane 6 has also been proposed as a possible source of electrophilic oxygen in some cases, although some computational studies reported by Albini and co-workers suggest that this intermediate can probably form only from the high energy radical cation 5, but not from 1 and singlet oxygen, that is only in the electron transfer channel.23b Alternatively, for the electron transfer mechanism, direct reaction of the radical cation 5 with oxygen to produce 7 was also proposed to account for the notable difference between these two mechanisms. For substrates having labile hydrogen atoms in the α-position to sulfur, especially benzylic sulfides (2g) or thioacetic acid derivatives (2j–k), the formation of an electrophilic hydrosulfoperoxide 8 is possible via proton transfer (Scheme 1c). Unfortunately, 8 is prone to Pummerer rearrangement to form hydroperoxide 9, which typically further decomposes into an aldehyde and various S-oxidized products. For this reason, this type of substrate often suffers from low yields in oxidation reactions. To account for the fact that the singlet oxygen oxidation of sulfides proceeds much faster in the presence of protic solvents, such as alcohols or water despite the significantly shorter lifetime of 1O2 in these solvents, the formation of H-bond stabilized persulfoxide 10 or alcohol adduct 11 was postulated and experimentally confirmed.25 These stabilized intermediates also possess the electrophilic oxygen required for the oxidation of another sulfide molecule, as opposed to the nucleophilic oxygen in 3. It should be noted that in many cases sulfide oxidation is taking place via the combination of both mechanisms depending on the structure of sulfide, the sensitizer and the solvent. | ||
| Scheme 1 Mechanisms for sensitized sulfide oxidation (a) and (b) and the commonly observed side products (c). | ||
The high efficiency of the FS mediated oxidation in ethanol provides a strong hint that the reaction proceeds via the singlet oxygen mechanism. The poor results obtained with thioanisole 1m, known to be a poor substrate in 1O2 oxidations also corroborate this assumption. To evaluate the relative contribution of the energy and electron transfer mechanisms, quenching experiments with various additives were performed using dialkylsulfide 2a as a control (Table S7†). The presence of a radical species was confirmed by strong inhibition with TEMPO. Selective singlet oxygen quenchers, Co(acac)3 (ref. 14g) and DABCO also significantly reduced the reaction rate. In contrast, 1,4-dimethoxybenzene, capable of reducing the radical cation 5 has little effect, indicating the negligible fraction of the electron transfer process. Likewise, 1,3-dinitrobenzene had very little impact on the reaction yield. The superoxide radical anion scavenger, 1,4-benzoquinone, also reduced the oxidation yield, indicating the contribution of the electron transfer mechanism. This result, however, should be taken with caution as some degradation of the scavenger was observed, presumably owing to its reaction with persulfoxide 3 or water to generate the hydroxylated quinones among other products.26 The possible intermediacy of thiadioxirane 6 was ruled out with the unsaturated substrates 2e and 2f, as the formation of epoxide was not observed, in line with the proposed energy transfer mechanism.27 Taken together, the results obtained corroborate the involvement of singlet oxygen as the main oxidant for the dialkyl sulfides.
As seen from Fig. 1, the oxidation of sulfides with FS in acetonitrile did not proceed at all, except for two substrates (2p–q). Taking into account the sluggish singlet oxygen mediated reaction in this solvent, one may assume a mechanism occurs in which the electron transfer process to fullerene requires tight contact between the sulfide and fullerene surface.28 The substrates, having a long, sterically non-demanding alkyl or polyethylene glycol chain, can interact with the fullerene surface via dispersive forces facilitating the electron transfer event.29 This interaction might be too weak for other substrates owing to the bulky or too short alkyl chains. The significant contribution of the electron transfer in the oxidation of 1q in acetonitrile was also evident from the drastic reduction in yield from 85% to 35% that was observed upon using the sulfide radical cation quencher 1,4-dimethoxybenzene (Table S7†).
The photooxidation of thioanisoles with FS via the singlet oxygen mechanism was very sluggish, limiting the scope of our method to dialkyl sulfides. On the other hand, systems operating under a charge transfer regime are well-documented in the literature and deliver the corresponding alkyl aryl sulfoxides in a high yield. We reasoned that the direct electron transfer from thioanisole to fullerene was inefficient and therefore a sub-stoichiometric amount of triethylamine was introduced as a mediator. Indeed, under these modified conditions thioanisole 2r and its derivatives 2s–u were oxidized in good yields (Fig. 4a and Tables S2, S3†). Most likely the very efficient electron transfer from triethylamine to 3C60 generates an amine radical cation which then oxidizes thioanisole into the reactive compound 5, regenerating the starting amine.30
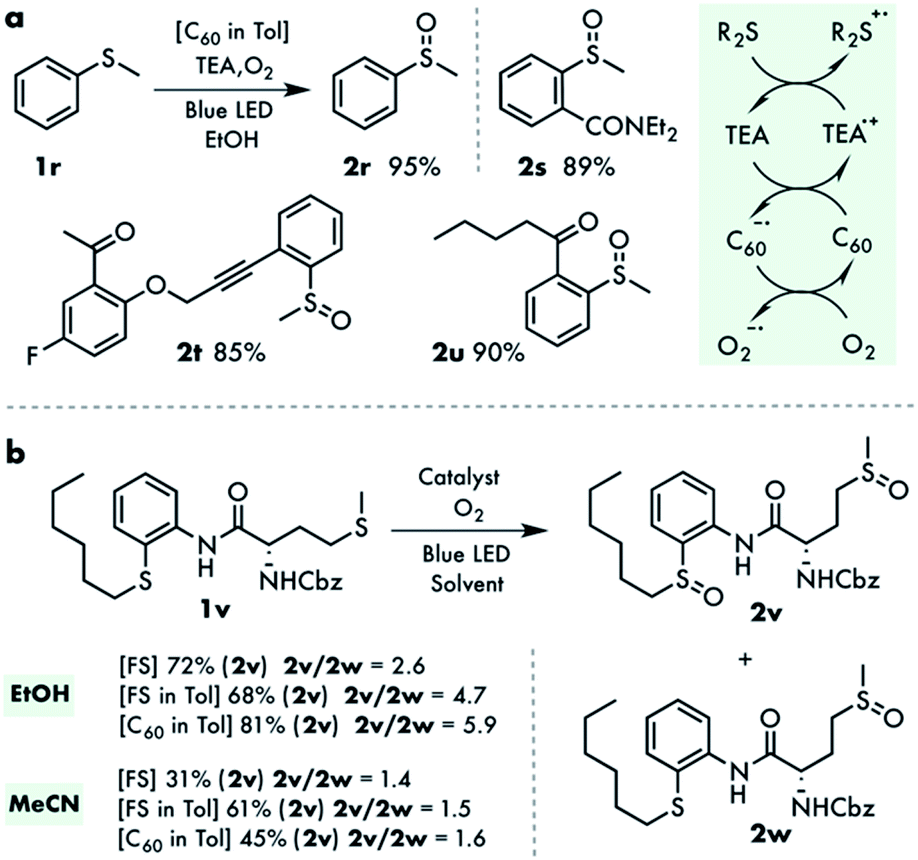 | ||
| Fig. 4 (a) The amine mediated oxidation of the thioanisoles. (b) Probing of the intramolecular oxygen transfer in bisulfide. | ||
Given the high selectivity of the current method for dialkyl sulfides, that is not observed in other reported systems, an experiment to perform the chemoselective oxidation of the model bisulfide 1v, possessing both dialkyl and arylalkyl sulfide moieties, was attempted. This chemoselective oxidation is not trivial as the persulfoxide formed at the reactive site can indirectly oxidize the arylalkyl thiol sulfur via oxygen transfer. This was indeed observed in compound 1v with all catalysts in ethanol, in which disulfoxide was obtained in a 68–81% yield (Fig. 4b and Table S1†). The yield is remarkable considering that the only source of oxygen for the arylalkyl sulfide part of the molecule is an intramolecular oxygen transfer. The efficiency of the intramolecular oxidation was also demonstrated with an external nucleophilic quencher, triphenylphosphite (P(OPh)3).31 Despite being a much better electrophilic oxygen acceptor compared to sulfide, the amount of monooxidized product increased only marginally using 5 equivalents of P(OPh)3. In addition, no significant co-oxidation of diphenylsulfoxide Ph2SO was observed in this model system, highlighting the electrophilic nature of the intermediate involved. The reaction in acetonitrile was less efficient, as observed in all previous cases. Moreover, the ratio of the di- and monooxidized products was significantly smaller, in accordance with the nucleophilic nature of the non-stabilized persulfoxide intermediate and the more sluggish oxygen transfer reaction (Fig. 4b).
2.5 Other synthetic applications
Having established the FS and C60 nanodispersion as an efficient, selective and recyclable heterogeneous photocatalyst for sulfide oxidation, we attempted to probe whether its use can be extended beyond this particular application while maintaining green reaction conditions. Encouraged by the enabling effect of the amine to promote the electron transfer mechanism, the addition–cyclization cascade reaction between N,N-dimethylaniline 12 and maleimide 13 was tested first, using the C60 nanodispersion as a photocatalyst in an air atmosphere (Table S4†).32 In this reaction, the amine radical cation 12a produced upon electron transfer is converted into the neutral radical 12b during deprotonation (Fig. 5a). Subsequent addition to maleimide, cyclization into an aromatic ring and a hydrogen atom and proton transfer to the superoxide radical anion affords the cyclization product and hydrogen peroxide.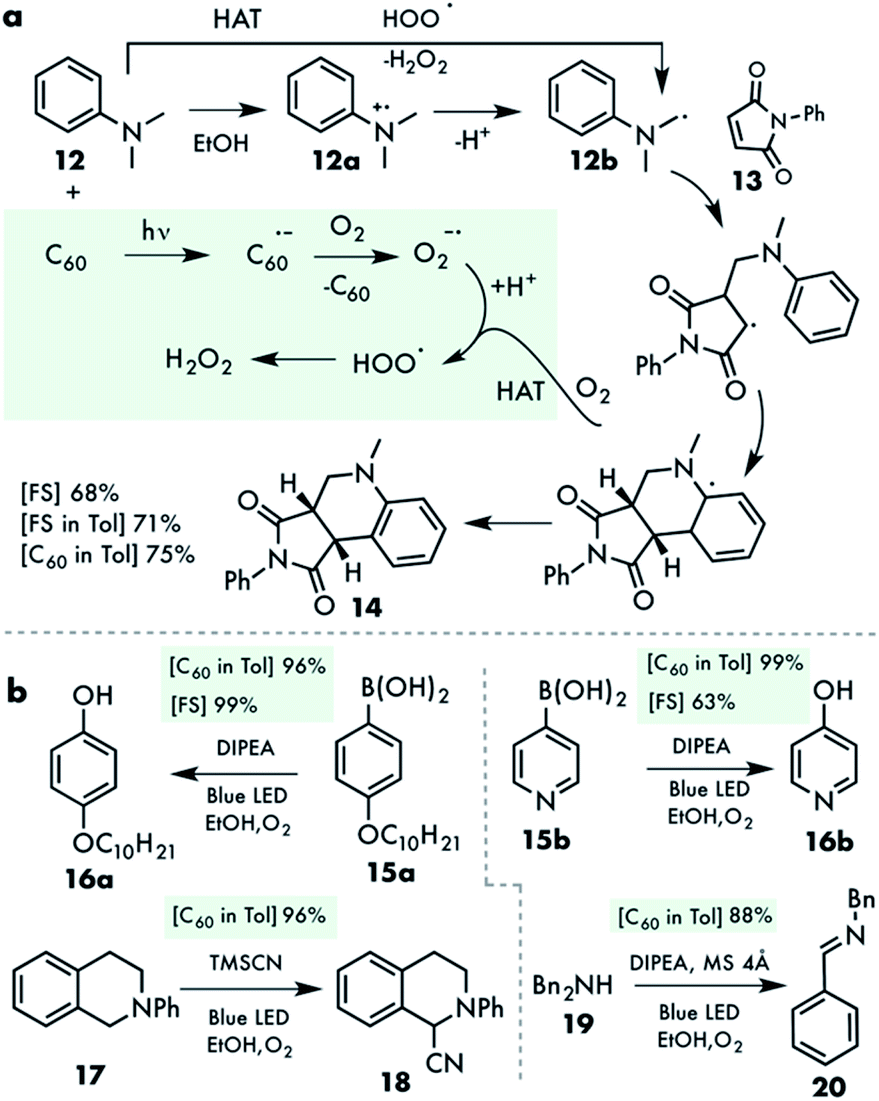 | ||
| Fig. 5 (a) Radical cyclization of N,N-dimethylaniline and N-phenyl maleimide (HAT: hydrogen atom transfer). (b) Oxidation of aryl boronic acids and amines. | ||
In another application, the superoxide radical anion was generated using the C60 nanosuspension in ethanol and the amines for arylboronic acid ipso-hydroxylation to the corresponding phenols.33 Almost quantitative yields were obtained for the aryl- and pyridyl boronic acids 15 (Fig. 5b). Sterically more hindered diisopropylethylamine (DIPEA) was more effective than triethylamine in this case. The oxidative transformation of amines to the corresponding imines by singlet oxygen was also possible with this photocatalytic system. Thus, 1,2,3,4-tetrahydroisoquinoline 17 was subjected to the C60 nanodispersion and TMSCN in an O2 saturated solution. The one-pot imine formation and trapping with TMSCN afforded product 18 in an excellent yield (Fig. 5b and Table S5†). Likewise, imine 20 derived from dibenzylamine 19 was quantitatively obtained under identical conditions (Table S6†).34
3. Conclusions
In summary, we have successfully introduced a new entry, fullerene soot, into the family of heterogeneous photocatalysts for oxygen activation. The commercially available inexpensive mixture, composed solely of carbon, was shown to be a competent catalyst for singlet oxygen-based oxidation, whereas the C60 nanodispersion can also be successfully applied for superoxide mediated oxidation or cyclization reactions. The catalysts described herein are very efficient (substrate/catalyst molar ratio up to 20000), robust and can be easily recycled. In addition, the feasibility of large scale photooxidation with the heterogeneous catalyst dispersed within the mobile phase of a flow reactor has been demonstrated. The results disclosed in this report set the stage for many other conceivable applications of carbon-based photocatalysts to advance the field of green chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Research Council of Lithuania (grant no. S-MIP-17-46). We are grateful to Dr Lina Mikoliūnaitė and Andrius Pakalniškis from Vilnius University for Raman spectroscopy and SEM measurements, respectively.Notes and references
- (a) G. Ertl, H. Knozinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, VCH, Weinheim, 1997 CrossRef; (b) N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS; (c) C. Lucarelli and A. Vaccari, Green Chem., 2011, 13, 1941 RSC.
- Z. Ma and F. Zaera, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd., 2014 Search PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468 CrossRef CAS.
- (a) P. W. N. M. van Leeuwen and C. Claver, Recent Advances in Nanoparticle Catalysis, Springer Nature, Cham, 2020 CrossRef; (b) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS; (b) B. König, Eur. J. Org. Chem., 2017, 3358 Search PubMed.
- (a) X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC; (b) G. Marcì and L. Palmisano, Heterogeneous Photocatalysis, Elsevier, 2019 Search PubMed.
- (a) C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275 CrossRef CAS; (b) Y. Pan, X. Liu, W. Zhang, Z. Liu, G. Zeng, B. Shao, Q. Liang, Q. He, X. Yuan, D. Huang and M. Chen, Appl. Catal., B, 2020, 265, 118579 CrossRef; (c) S. Yao, X. Yuan, L. Jiang, T. Xiong and J. Zhang, Materials, 2020, 13, 2924 CrossRef CAS; (d) N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307 CrossRef CAS; (e) N. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, J. Catal., 2013, 303, 60–69 CrossRef CAS.
- (a) R. Haddon, Philos. Trans. R. Soc., A, 1993, 343, 53 CrossRef CAS; (b) J. W. Arbogast, C. S. Foote and M. Kao, J. Am. Chem. Soc., 2002, 114, 2277 CrossRef; (c) Y. Yamakoshi, N. Umezawa, A. Ryu, K. Arakane, N. Miyata, Y. Goda, T. Masumizu and T. Nagano, J. Am. Chem. Soc., 2003, 125, 12803 CrossRef CAS.
- (a) J. W. Arbogast, A. P. Darmanyan, C. S. Foote, F. N. Diederich, R. L. Whetten, Y. Rubin, M. M. Alvarez and S. J. Anz, J. Phys. Chem., 1991, 95, 11 CrossRef CAS; (b) H. Tokuyama and E. Nakamura, J. Org. Chem., 1994, 59, 1135 CrossRef CAS.
- (a) Y. Takaguchi, Y. Yanagimoto, S. Fujima and S. Tsuboi, Chem. Lett., 2004, 33, 1142 CrossRef CAS; (b) Y. Maeda, Y. Niino, T. Kondo, M. Yamada, T. Hasegawa and T. Akasaka, Chem. Lett., 2011, 40, 1431 CrossRef CAS.
- (a) D. Latassa, O. Enger, C. Thilgen, T. Habicher, H. Offermanns and F. Diederich, J. Mater. Chem., 2002, 12, 1993 RSC; (b) X. Chen, K. Deng, P. Zhou and Z. Zhang, ChemSusChem, 2018, 11, 1 CrossRef; (c) T. Hino, T. Anzai and N. Kuramoto, Tetrahedron Lett., 2006, 47, 1429 CrossRef CAS; (d) D.-Y. Zheng, E.-X. Chen, C.-R. Ye and X.-C. Huang, J. Mater. Chem. A, 2019, 7, 22084 RSC; (e) K. J. Moor and J. H. Kim, Environ. Sci. Technol., 2014, 48, 2785 CrossRef CAS.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS.
- (a) C. Jacob, Nat. Prod. Rep., 2006, 23, 851 RSC; (b) E. Block, Angew. Chem., Int. Ed., 1992, 31, 1135 CrossRef.
- (a) A. Casado-Sánchez, R. Gómez-Ballesteros, F. Tato, F. J. Soriano, G. Pascual-Coca, S. Cabrera and J. Alemán, Chem. Commun., 2016, 52, 9137 RSC; (b) Y. Gao, H. Xu, S. Zhang, Y. Zhang, C. Tang and W. Fan, Org. Biomol. Chem., 2019, 17, 7144 RSC; (c) A. Guerrero-Corella, A. M. Martinez-Gualda, F. Ahmadi, E. Ming, A. Fraile and J. Alemán, Chem. Commun., 2017, 53, 10463 RSC; (d) X. Liang, Z. Guo, H. Wei, X. Liu, H. Lv and H. Xing, Chem. Commun., 2018, 54, 13002 RSC; (e) Y. Li, S. A.-E.-A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499 CrossRef CAS; (f) X. Gu, X. Li, Y. Chai, Q. Yang, P. Li and Y. Yao, Green Chem., 2013, 15, 357 RSC; (g) T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654 CrossRef; (h) W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417 RSC; (i) J. Dad'ova, E. Svobodova, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620 CrossRef.
- (a) A. Sridhar, R. Rangasamy and M. Selvaraj, New J. Chem., 2019, 43, 17974 RSC; (b) K. Suzuki, J. Jeong, K. Yamaguchi and N. Mizuno, New J. Chem., 2016, 40, 1014 RSC; (c) Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001 CrossRef CAS; (d) X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697 CrossRef CAS; (e) T. Uematsu, Y. Miyamoto, Y. Ogasawara, K. Suzuki, K. Yamaguchi and N. Mizuno, Catal. Sci. Technol., 2016, 6, 222 RSC; (f) J. Jiang, R. Luo, X. Zhou, Y. Chen and H. Ji, Adv. Synth. Catal., 2018, 360, 4402 CrossRef CAS; (g) T. Pigot, T. Arbitre, H. Martinez and S. Lacombe, Tetrahedron Lett., 2004, 45, 4047 CrossRef CAS; (h) J. Jiang, R. Luo, X. Zhou, Y. Chen and H. Ji, Adv. Synth. Catal., 2018, 360, 4402 CrossRef CAS.
- G. R. Alargova, S. Deguchi and K. Tsujii, J. Am. Chem. Soc., 2001, 123, 10460 CrossRef.
- H. Keypour, M. Noroozi and A. Rashidi, J. Nanostruct. Chem., 2013, 3, 45 CrossRef.
- (a) A. Toutchkine and E. L. Clennan, J. Am. Chem. Soc., 2000, 122, 1834 CrossRef CAS; (b) E. Baciocchi, C. Crescenzi and O. Lanzalunga, Tetrahedron, 1997, 53, 4469 CrossRef CAS.
- For some examples of open flow system for fast photochemical heterogeneous reactions, see: (a) T. Carofiglio, P. Donnola, M. Maggini, M. Rossetto and E. Rossi, Adv. Synth. Catal., 2008, 350, 2815 CrossRef CAS; (b) M. Woźnica, N. Chaoui, S. Taabache and S. Blechert, Chem.–Eur. J., 2014, 20, 14624 CrossRef.
- (a) J. Eriksen, C. S. Foote and T. L. Parker, J. Am. Chem. Soc., 1977, 99, 6455 CrossRef CAS; (b) J. J. Liang, C. L. Gu, M. L. Kacher and C. S. Foote, J. Am. Chem. Soc., 1983, 105, 4717 CrossRef CAS.
- (a) E. L. Clennan, Acc. Chem. Res., 2001, 34, 875 CrossRef CAS; (b) E. L. Clennan, Sulfur Rep., 1996, 19, 171 CrossRef CAS; (c) E. L. Clennan, Tetrahedron, 2006, 62, 10724 CrossRef CAS; (d) E. L. Clennan, S. E. Hightower and A. Greer, J. Am. Chem. Soc., 2005, 127, 11819 CrossRef CAS; (e) F. Jensen, A. Greer and E. L. Clennan, J. Am. Chem. Soc., 1998, 120, 4439 CrossRef CAS.
- (a) E. Baciocchi, T. Del Giacco, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444 CrossRef CAS; (b) E. Baciocchi, T. Del Giacco, O. Lanzalunga and A. Lapi, J. Org. Chem., 2007, 72, 9582 CrossRef CAS.
- (a) S. M. Bonesi, M. Fagnoni and A. Albini, J. Org. Chem., 2004, 69, 928 CrossRef CAS; (b) S. M. Bonesi, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem.–Eur. J., 2006, 12, 4844 CrossRef CAS; (c) S. M. Bonesi, M. Fagnoni and A. Albini, Eur. J. Org. Chem., 2008, 2612 CrossRef CAS.
- (a) S. Lacombe, H. Cardy, M. Simon, A. Khoukh, J. P. Soumillion and M. Ayadim, Photochem. Photobiol. Sci., 2002, 1, 347 RSC; (b) Y. Che, W. Ma, Y. Ren, C. Chen, X. Zhang, J. Zhao and L. Zang, J. Phys. Chem. B, 2005, 109, 8270 CrossRef CAS.
- (a) E. L. Clennan and A. Greer, J. Org. Chem., 1996, 61, 4793 CrossRef CAS; (b) S. M. Bonesi and A. Albini, J. Org. Chem., 2000, 65, 4532 CrossRef CAS.
- For possible degradation of benzoquinone, see: H. Görner, Photochem. Photobiol., 2006, 82, 71 CrossRef.
- Alkenes were shown to react with thiadioxirane: T. Akasaka, A. Sakurai and W. Ando, J. Am. Chem. Soc., 1991, 113, 2696 CrossRef CAS.
- In our unpublished studies on supramolecular fullerene receptors bearing sulfide side chains nearby the recognition site, we indeed observe very fast oxidation upon C60 complexation, even in the dark.
- Polyethylene glycols (PEGs) are well known solubilizing agents for C60, see: H. Hungerbuhler, D. M. Guldi and K. D. Asmus, J. Am. Chem. Soc., 1993, 115, 3386 CrossRef.
- (a) M. Zou, L. Feng, T. Thomas and M. Yang, Catal. Sci. Technol., 2017, 7, 4182 RSC; (b) X. Lang, W. Hao, W. R. Leow, S. Li, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 5000 RSC.
- (a) K. Nahm and C. S. Foote, J. Am. Chem. Soc., 1989, 111, 1909 CrossRef CAS; (b) Y. Sawaki and Y. Ogata, J. Am. Chem. Soc., 1981, 103, 5947 CrossRef CAS.
- For homogeneous photochemical variant of this reaction, see: (a) X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561 CrossRef CAS; (b) Z. Liang, S. Xu, W. Tian and R. Zhang, Beilstein J. Org. Chem., 2015, 11, 425 CrossRef CAS.
- I. Kumar, R. Sharma, R. Kumar, R. Kumar and U. Sharma, Adv. Synth. Catal., 2018, 360, 2013 CrossRef CAS.
- R. Kumar, E. H. Gleißner, E. V. Tiu and Y. Yamakoshi, Org. Lett., 2016, 18, 184 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10147h |
| This journal is © The Royal Society of Chemistry 2021 |