
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of substituted 3,4-dihydroquinazolinones via a metal free Leuckart–Wallach type reaction†
Suvarna Bokale-Shivalea,
Mohammad A. Amin a,
Rajiv T. Sawanta,
Marc Y. Stevensa,
Lewend Turanlia,
Adam Hallberga,
Suresh B. Waghmodeb and
Luke R. Odell*a
a,
Rajiv T. Sawanta,
Marc Y. Stevensa,
Lewend Turanlia,
Adam Hallberga,
Suresh B. Waghmodeb and
Luke R. Odell*a
aDepartment of Medicinal Chemistry, Uppsala Biomedical Center, Uppsala University, P. O. Box 574, SE-751 23 Uppsala, Sweden. E-mail: luke.odell@ilk.uu.se
bDepartment of Chemistry, Savitribai Phule Pune University (formerly Pune University), Ganeshkhind, Pune 411 007, India
First published on 23rd December 2020
Abstract
The 3,4-dihydroquinazolinone (DHQ) moiety is a highly valued scaffold in medicinal chemistry due to the vast number of biologically-active compounds based on this core structure. Current synthetic methods to access these compounds are limited in terms of diversity and flexibility and often require the use of toxic reagents or expensive transition-metal catalysts. Herein, we describe the discovery and development of a novel cascade cyclization/Leuckart–Wallach type strategy to prepare substituted DHQs in a modular and efficient process using readily-available starting materials. Notably, the reaction requires only the addition of formic acid or acetic acid/formic acid and produces H2O, CO2 and methanol as the sole reaction byproducts. Overall, the reaction provides an attractive entry point into this important class of compounds and could even be extended to isotopic labelling via the site-selective incorporation of a deuterium atom.
Introduction
The 3,4-dihydroquinazolinone (DHQ) moiety is a privileged scaffold in medicinal chemistry, as such compounds containing this unit have been reported to exhibit biological activity against a wide range of therapeutic targets. The most significant compound in this class, in terms of clinical utility, is the calcitonin gene-related peptide receptor antagonist olcegepant (Fig. 1), which demonstrated efficacy and safety in phase II trials as an anti-migraine agent.1,2 In addition, DHQs with potent anti-HIV,3 anti-psychotic,4,5 anti-cancer6,7 and anti-microbial8 activities as well as potential for the treatment of cardiovascular9 and anti-inflammatory disorders10 have been disclosed. Recently PFI-1, a potent and selective inhibitor of the bromo- and extra C-terminal domain (BET) family of bromodomains was developed using a fragment based approach starting from small DHQ-containing hit.11,12 As consequence of their pervading biological importance, numerous synthetic approaches to access this ring system are available. These include the cyclization of o-acyl/o-aminoanilines13–17 or o-nitrobenzylamines18 with a carbonyl donor or the nucleophilic annulation of o-functionalized aniline derivatives19 in addition to more recent methodologies relying on the use of expensive transition metal catalysts20–24 or toxic selenium and carbon monoxide.25 In 2015, we disclosed a novel multicomponent strategy to assemble diversely substituted DHQs via an N-acyliminium ion cyclization cascade.26 This is a simple, highly attractive approach for accessing novel and densely substituted DHQ analogues, based on an array of different chemistries (Scheme 1).27–31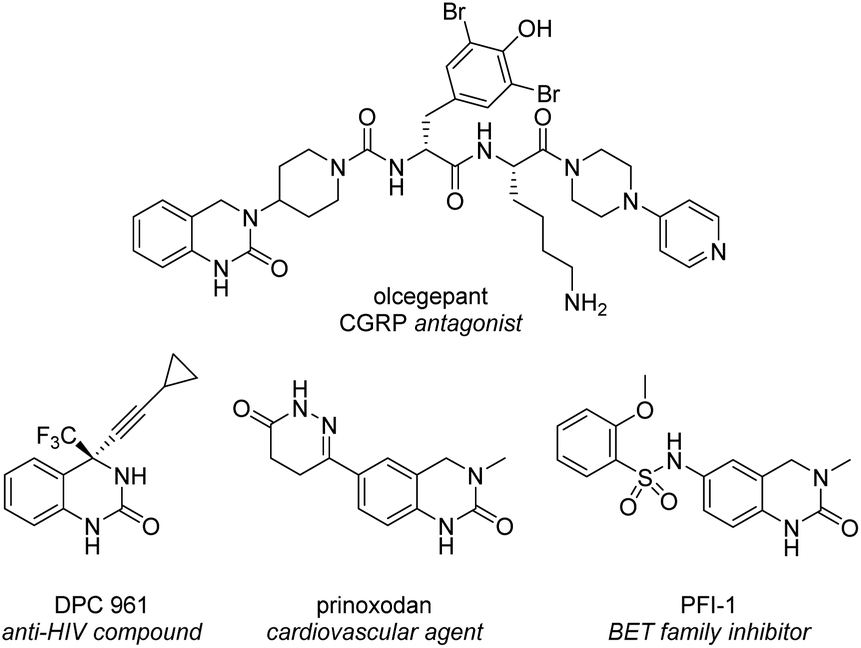 | ||
| Fig. 1 Representative biologically-active 3,4-dihydroquinazolinones. | ||
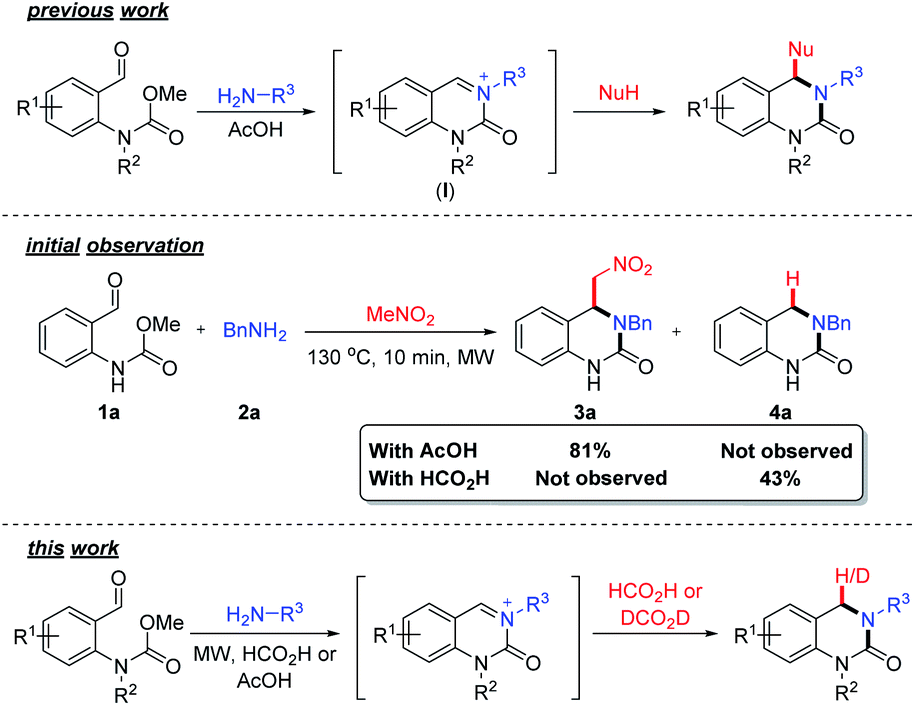 | ||
| Scheme 1 Overview of previous work on the formation of substituted DHQs via carbamate induced cyclization cascade. Initial observation using formic acid and an overview of this work. | ||
During the course of our investigations on the reactivity of N-acyliminium ions (I), we observed the formation of an unknown side-product (4a) when formic acid was used as a solvent, in an aza-Henry based cyclization cascade, in lieu of acetic acid (Scheme 1). Subsequent characterization studies revealed that this compound retained the DHQ core but lacked the expected C4-substituent. The most plausible explanation for the formation of 4a is the in situ reduction of the iminium ion intermediate and indeed, the catalyst-free formic acid (or derivative) mediated reduction of iminium ions was first reported over a century ago32,33 and is known as the Leuckart–Wallach reaction.
Although the use of formic acid as a green and renewable reductant in transfer hydrogen chemistry34 has received significant attention, there are surprisingly few examples utilizing the Leuckart–Wallach reaction manifold in the literature.35–43 Realizing that this would provide an efficient, sustainable and straightforward entry point into DHQ scaffolds structurally similar to olcegepant and PFI-1, we set about further exploring the formation of 4a. Herein, we describe the discovery and development of novel metal-free Leuckart–Wallach type reductive cyclization cascade of o-formyl methylcarbamates for the preparation of biologically relevant DHQs.
Results and discussion
Our study commenced with a survey of reaction conditions with the aim of increasing the yield of DHQ 4a (Table 1). Simple removal of nitromethane led to a marked increase in yield (64%, entry 1) most likely by suppression of competing Henry-type side reactions. An increase in the reaction temperature and time (150 °C and 30 min) afforded full consumption of 1a and the desired product was isolated in 77% yield (entry 2). Although the amount of amine 2a could be reduced to 1.5 equiv. without affecting the yield (entry 4) further decreases were found to be detrimental to the reaction outcome (entry 5). In cases where a larger excess (>1.5 equiv.) of 2a was used, purification was troublesome due to the formation of benzylformamide as a side-product and in entry 5 unreacted starting material 1a was observed (LCMS analysis). Finally, the reaction time was probed with shorter times leading to the presence of unreacted 1a and N-acyliminium ion I and extending the reaction time afforded a lower yield (entry 6). Accordingly, the conditions from entry 4 were chosen for further evaluation.
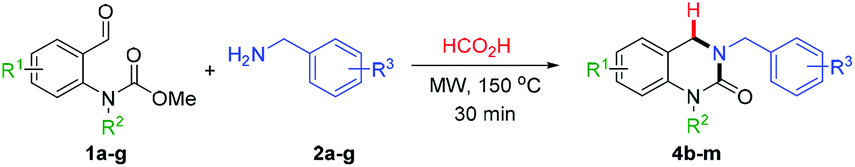
With the optimized reaction conditions in hand, the substrate scope was evaluated using various o-formyl methylcarbamate and benzyl amine derivatives (Table 2). In general, the reaction was compatible with a wide range of substrates, affording moderate to excellent yields of the desired DHQ products. Carbamates bearing electron donating (4b, c) or electron withdrawing (4d–h) substituents were well tolerated with the latter returning slightly lower yields. The presence of an o-substituent (4c) or an N-benzyl group (4i) was also compatible with the reaction indicating a tolerance towards steric bulk around the carbamate center. Similarly, the amine nucleophile scope was found to be broad with sterically and electronically diverse substrates all delivering the target DHQ products in good to excellent yields. Notably, the reaction was also successfully extended to heterocyclic amines to afford the thiophene and pyridine derivatives 4n and 4o, respectively. Finally, to investigate the potential for scalability, the reaction was performed on a 2 mmol scale resulting in an 86% yield of 4a.
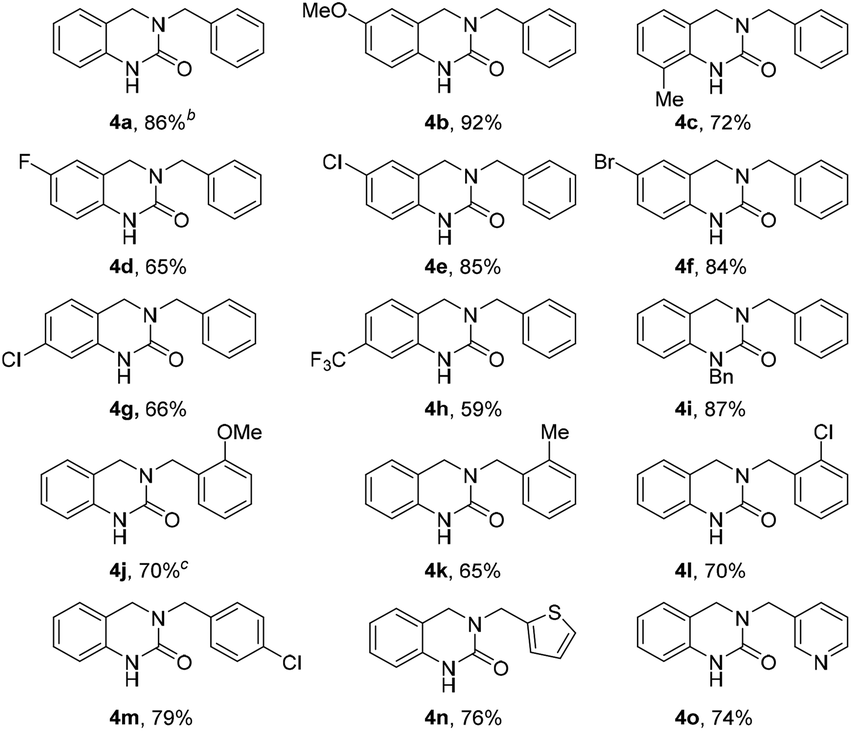
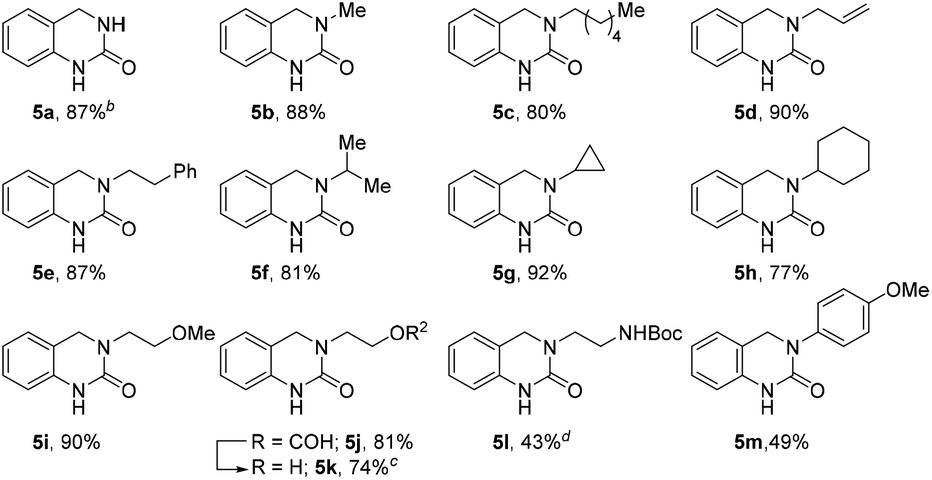
To further expand the reaction scope, we sought to explore the use of aliphatic amine nucleophiles in lieu of substituted benzyl amines. However, during our initial scouting reactions we noted a marked difference in reactivity, with only low levels of conversion observed (LCMS analysis) even after prolonged heating times. We reasoned that this could be the result of the higher basicity of these substrates, leading to an increase in the proportion of unreactive ammonium cations, under the acidic reaction conditions. To overcome this issue, we investigated a telescoped one-pot protocol where the key N-acyliminium ion intermediate was first generated using the less acidic acetic acid26 and subsequently reduced through the addition of formic acid. As shown in Table 3, the reaction performed well with wide range of amine nucleophiles to afford the corresponding DHQ products 5a–5m in up to 92% yield. Pleasingly, linear, branched and cyclic primary amines were all found to be suitable substrates and the use of NH4OAc was also successful. When ethanolamine was employed as the nucleophile, the corresponding formate ester 5j was isolated in 81% yield and this was readily hydrolyzed to afford alcohol 5k in an overall yield of 74%. The reaction scope could even be extended to acid-sensitive and less-reactive nucleophiles to afford moderate yields of the corresponding products 5l and 5m, respectively. The synthesis of 5l is particularly noteworthy given the labile nature of the Boc group and the elevated temperature and acidic reaction conditions.
|
|---|
| a Isolated yield. Unless otherwise stated, reactions were performed with 1 equiv. aldehyde (0.28 mmol scale), 1.5 equiv. amine in AcOH (1 mL) and HCO2H (1 mL).b Heated at 130 °C in step 2.c Treated with NaOAc (10 equiv.) in EtOH at reflux for 5 h.d Heated at 100 °C with 10 equiv. HCO2H for 30 min in step 2. |
 |
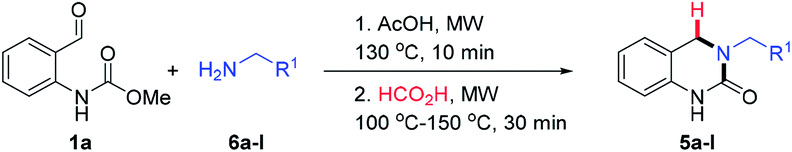
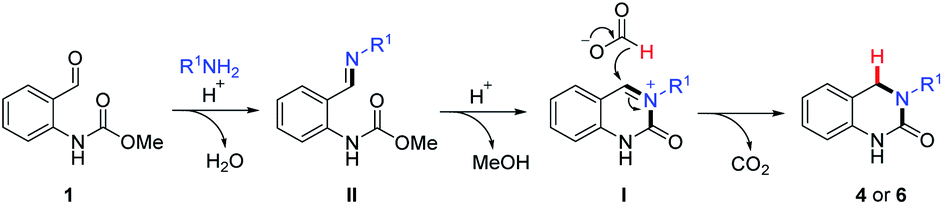
Based on the above results and our previous studies,26 the reaction is believed to occur via a cascade imine/iminium ion/Leuckart–Wallach type reaction process (Scheme 2). The reaction begins with the acid-mediated formation of imine II followed by annulation onto the pendant carbamate resulting in the formation of cyclic N-acyl iminium ion I. Finally, reduction of this highly electrophilic intermediate by formic acid leads to the target DHQ products 4 and 6, in an effective and efficient process, generating H2O, CO2 and MeOH as the only reaction byproducts. This is the most likely set of events in the two-step reaction (Table 3), as complete conversion to I was routinely observed (LCMS analysis) prior to the addition of formic acid. However, when the reaction is conducted solely in formic acid (Tables 1 and 2) an alternative scenario where reduction of II occurs prior to cyclization is also plausible. To further investigate this possibility, we set about synthesizing an imine of type-II and its corresponding reduced form (Scheme 3) to assess their relevance in our reaction system.
 | ||
| Scheme 2 Proposed reaction pathway. | ||
 | ||
| Scheme 3 Control experiments probing possible reaction intermediates. | ||
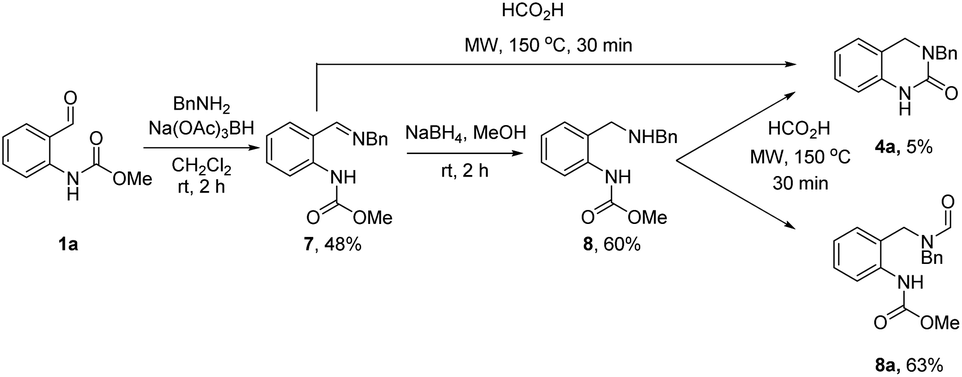
To this end, o-formyl carbamate (1a) and benzylamine (2a) were first reacted under standard reductive amination conditions using Na(OAc)3BH (Scheme 3). Surprisingly, formation of amine 8 was not observed (LCMS analysis) and instead imine 7 was isolated in 48% yield. Subsequent treatment of 7 with the more reactive NaBH4 led to formation of the desired amine product 8. We next subjected 7 and 8 to the optimized reaction conditions from Table 2. Interestingly, full conversion of 7 to 4a was observed (LCMS analysis) whereas the reaction with 8 gave the N-formyl derivative 8a as the major product (63%) and 4a was isolated in only 5% yield. These results strongly support the intermediacy of a cyclic N-acyl iminium ion species under both sets of reaction conditions.
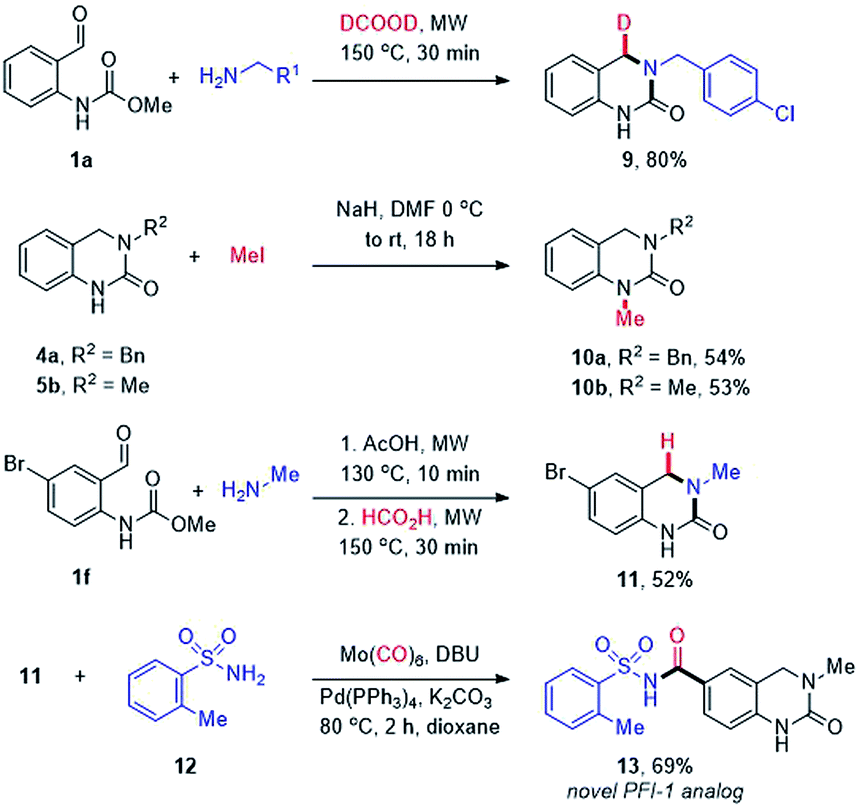
Finally, the utility of our methodology was demonstrated through the synthesis of additional, more elaborate DHQ derivatives (Scheme 4). Firstly, we were intrigued by the possibility of using deuterated formic acid to potentially enable the site-selective introduction of a deuterium atom. The H/D isotopic replacement is an important tool to modulate the PK/PD properties of drug candidates44 and provides a potential handle for mechanistic studies. Thus, 1a was reacted with 4-chlorobenzylamine (2m) using formic acid-d2 to afford the mono deuterated DHQ compound 9 in 80% yield. The site-selective incorporation of the deuterium atom at the benzylic position was confirmed by 1H and 13C NMR analysis. Next, selective N1-methylation was conducted using NaH and methyl iodide to afford moderate yields of 10a and 10b. Lastly, a novel analog of the BET bromodomain inhibitor PFI-1 (13) was synthesized via an efficient two-step route starting with the cascade cyclization of 1f and methylamine (4l), followed by a palladium-catalyzed aminocarbonylation with o-toluenesulfonamide (12).
 | ||
| Scheme 4 Synthetic utility of the cascade DHQ methodology. | ||
Conclusions
In summary, we have developed a straightforward and high-yielding protocol for the synthesis of 3,4-dihydroquinazolinone by a novel imine/cyclization/Leuckart–Wallach type cascade process. Notably, the reaction is metal-free and requires only the addition of formic acid as a dual Brønsted acid/reductant or a combination of acetic acid and formic acid for aliphatic amine substrates. Moreover, the only byproducts formed during the reaction are H2O, MeOH and CO2 making the overall process highly efficient and sustainable. Mechanistic studies supported the formation of a cyclic N-acyl iminium ion intermediate prior to reduction by formic acid. Finally, the methodology was exemplified on a range of different substrates and was even extended to deuterium incorporation and synthesis of a novel analog of the BET bromodomain inhibitor PFI-1. We hope that this work will encourage others to explore the underutilized Leuckart–Wallach reaction as a green synthetic manifold to prepare biologically important compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by Uppsala University and the Swedish Research Council (Vetenskapsrådet 2018-05133).Notes and references
- A. Recober and A. F. Russo, IDrugs, 2007, 10, 566–574 CAS.
- I. M. Bell, J. Med. Chem., 2014, 57, 7838–7858 CrossRef CAS.
- J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, K. Logue, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, J. Med. Chem., 2000, 43, 2019–2030 CrossRef CAS.
- Y. Uruno, Y. Konishi, A. Suwa, K. Takai, K. Tojo, T. Nakako, M. Sakai, T. Enomoto, H. Matsuda, A. Kitamura and T. Sumiyoshi, Bioorg. Med. Chem. Lett., 2015, 25, 5357–5361 CrossRef CAS.
- Y. Sugiyama, M. Hazama and N. Iwakami, U.S. Pat., US 9,199,969 B, 2005 Search PubMed.
- E. J. E. Freyne, L. A. Mevellec, J. E. Vialard, C. Meyer, E. T. J. Pasquier, X. M. Bourdrez and P. R. Angibaud, WO2009118384, 2009.
- W. Dohle, F. L. Jourdan, G. Menchon, A. E. Prota, P. A. Foster, P. Mannion, E. Hamel, M. P. Thomas, P. G. Kasprzyk, E. Ferrandis, M. O. Steinmetz, M. P. Leese and B. V. L. Potter, J. Med. Chem., 2018, 61, 1031–1044 CrossRef CAS.
- A. K. Tiwari, A. K. Mishra, A. Bajpai, P. Mishra, R. K. Sharma, V. K. Pandey and V. K. Singh, Bioorg. Med. Chem. Lett., 2006, 16, 4581–4585 CrossRef CAS.
- J. A. Barrett, R. F. Woltmann, R. S. Swillo, C. Kasiewski, W. C. Faith, H. F. Campbell and M. H. Perrone, J. Cardiovasc. Pharmacol., 1990, 16, 537–545 CrossRef CAS.
- J. E. Stelmach, L. Liu, S. B. Patel, J. V. Pivnichny, G. Scapin, S. Singh, C. E. C. A. Hop, Z. Wang, J. R. Strauss, P. M. Cameron, E. A. Nichols, S. J. O'Keefe, E. A. O'Neill, D. M. Schmatz, C. D. Schwartz, C. M. Thompson, D. M. Zaller and J. B. Doherty, Bioorg. Med. Chem. Lett., 2003, 13, 277–280 CrossRef CAS.
- S. Picaud, D. Da Costa, A. Thanasopoulou, P. Filippakopoulos, P. V. Fish, M. Philpott, O. Fedorov, P. Brennan, M. E. Bunnage, D. R. Owen, J. E. Bradner, P. Taniere, B. O'Sullivan, S. Müller, J. Schwaller, T. Stankovic and S. Knapp, Cancer Res., 2013, 73, 3336–3346 CrossRef CAS.
- P. V. Fish, P. Filippakopoulos, G. Bish, P. E. Brennan, M. E. Bunnage, A. S. Cook, O. Federov, B. S. Gerstenberger, H. Jones, S. Knapp, B. Marsden, K. Nocka, D. R. Owen, M. Philpott, S. Picaud, M. J. Primiano, M. J. Ralph, N. Sciammetta and J. D. Trzupek, J. Med. Chem., 2012, 55, 9831–9837 CrossRef CAS.
- J. C. Barrow, K. E. Rittle, T. S. Reger, Z. Q. Yang, P. Bondiskey, G. B. McGaughey, M. G. Bock, G. D. Hartman, C. Tang, J. Ballard, Y. Kuo, T. Prueksaritanont, C. E. Nuss, S. M. Doran, S. V. Fox, S. L. Garson, R. L. Kraus, Y. Li, M. J. Marino, V. Kuzmick Graufelds, V. N. Uebele and J. J. Renger, ACS Med. Chem. Lett., 2010, 1, 75–79 CrossRef CAS.
- H. Takai, H. Obase, M. Teranishi, A. Karasawa, K. Kubo, K. Shuto, Y. Kasuya and K. Shigenobu, Chem. Pharm. Bull., 1986, 34, 1907–1916 CrossRef CAS.
- W. Seitz, H. Geneste, G. Backfisch, J. Delzer, C. Graef, W. Hornberger, A. Kling, T. Subkowski and N. Zimmermann, Bioorg. Med. Chem. Lett., 2008, 18, 527–531 CrossRef CAS.
- M. E. Camacho, M. Chayah, M. E. García, N. Fernández-Sáez, F. Arias, M. A. Gallo and M. D. Carrión, Arch. Pharm., 2016, 349, 638–650 CrossRef CAS.
- M. Wang, J. Han, X. Si, Y. Hu, J. Zhu and X. Sun, Tetrahedron Lett., 2018, 59, 1614–1618 CrossRef CAS.
- D. Shi, G. Dou and Z.-Y. Li, J. Chem. Res., 2007, 2007, 545–547 CrossRef.
- J. Bergman, A. Brynolf, B. Elman and E. Vuorinen, Tetrahedron, 1986, 42, 3697–3706 CrossRef CAS.
- R. Ferraccioli and D. Carenzi, Synthesis, 2003, 9, 1383–1386 CrossRef.
- J. Willwacher, S. Rakshit and F. Glorius, Org. Biomol. Chem., 2011, 9, 4736–4740 RSC.
- R. K. Saunthwal, M. Patel, A. K. Danodia and A. K. Verma, Org. Biomol. Chem., 2015, 13, 1521–1530 RSC.
- G. B. Frost, M. N. Mittelstaedt and C. J. Douglas, Chem.–Eur. J., 2019, 25, 1727–1732 CrossRef CAS.
- Q. Wang, J. An, H. Alper, W. J. Xiao and A. M. Beauchemin, Chem. Commun., 2017, 53, 13055–13058 RSC.
- R. Zhou, X. Qi and X.-F. Wu, ACS Comb. Sci., 2019, 21, 573–577 CrossRef CAS.
- M. Y. Stevens, K. Wieckowski, P. Wu, R. T. Sawant and L. R. Odell, Org. Biomol. Chem., 2015, 13, 2044–2054 RSC.
- R. T. Sawant, M. Y. Stevens and L. R. Odell, Molbank, 2015, M866 CrossRef.
- R. T. Sawant, M. Y. Stevens, C. Sköld and L. R. Odell, Org. Lett., 2016, 18, 5392–5395 CrossRef CAS.
- R. T. Sawant, M. Y. Stevens and L. R. Odell, Chem. Commun., 2017, 53, 2110–2113 RSC.
- D. V. Ramana, B. Vinayak, V. Dileepkumar, U. S. N. Murty, L. R. Chowhan and M. Chandrasekharam, RSC Adv., 2016, 6, 21789–21794 RSC.
- R. T. Sawant, M. Y. Stevens and L. R. Odell, ACS Omega, 2018, 3, 14258–14265 CrossRef CAS.
- R. Leuckart, Ber. Dtsch. Chem. Ges., 1885, 18, 2341–2344 CrossRef.
- O. Wallach, Ber. Dtsch. Chem. Ges., 1891, 24, 3992–3993 CrossRef.
- W. Supronowicz, I. A. Ignatyev, G. Lolli, A. Wolf, L. Zhao and L. Mleczko, Green Chem., 2015, 17, 2904–2911 RSC.
- A. Loupy, D. Monteux, A. Petit, J. M. Aizpurua, E. Domínguez and C. Palomo, Tetrahedron Lett., 1996, 37, 8177–8180 CrossRef CAS.
- S. C. Lee and S. B. Park, Chem. Commun., 2007, 3714–3716 RSC.
- D. O'Connor, A. Lauria, S. P. Bondi and S. Saba, Tetrahedron Lett., 2011, 52, 129–132 CrossRef.
- F. Barba, J. Recio and B. Batanero, Tetrahedron Lett., 2013, 54, 1835–1838 CrossRef CAS.
- Y. Wei, C. Wang, X. Jiang, D. Xue, Z. T. Liu and J. Xiao, Green Chem., 2014, 16, 1093–1096 RSC.
- M. O. Frederick and D. P. Kjell, Tetrahedron Lett., 2015, 56, 949–951 CrossRef CAS.
- M. O. Frederick, M. A. Pietz, D. P. Kjell, R. N. Richey, G. A. Tharp, T. Touge, N. Yokoyama, M. Kida and T. Matsuo, Org. Process Res. Dev., 2017, 21, 1447–1451 CrossRef CAS.
- S. Y. Skachilova, N. K. Zheltukhin, V. N. Sergeev and N. K. Davydova, Pharm. Chem. J., 2018, 52, 545–549 CrossRef CAS.
- A. De, N. C. Ghosal, S. Mahato, S. Santra, G. V. Zyryanov and A. Majee, ChemistrySelect, 2018, 3, 4058–4066 CrossRef CAS.
- T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10142g |
| This journal is © The Royal Society of Chemistry 2021 |