
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional investigation of ultra-small SnO2 nanoparticle decorated rGO for ozone sensing and supercapacitor applications
J. Jayachandirana,
J. Yesurajb,
M. Arivanandhan c,
B. Muthuraamanb,
R. Jayavelc and
D. Nedumaran*a
c,
B. Muthuraamanb,
R. Jayavelc and
D. Nedumaran*a
aCentral Instrumentation and Service Laboratory, University of Madras, Guindy Campus, Chennai 600 025, TN, India. E-mail: dnmaran@gmail.com; dnmaran@unom.ac.in
bDepartment of Energy, University of Madras, Guindy Campus, Chennai 600 025, TN, India
cCentre for Nanoscience and Technology, Anna University, Guindy, Chennai 600 025, TN, India
First published on 4th January 2021
Abstract
Ultrasmall SnO2 nanoparticles with an average size of 7 nm were synthesized by a hydrothermal method and composited with reduced graphene oxide (rGO) through an ultrasonic assisted solution process. The structural, functional, morphological and compositional properties of synthesised SnO2 and rGO/SnO2 were studied by XRD, FTIR, HRSEM, HRTEM, XPS and Raman analyses. The prepared materials were developed as a film over a PVA/KOH conductive layer coated substrate with varying thickness of 3, 5 and 7 μm to study their ozone sensing characteristics at room temperature. The physico-chemical properties reveal that the fabricated SnO2 and rGO/SnO2 nanocomposite films have a strong interaction with the ozone gas. Among the fabricated composite films rGO/SnO2-S1 film exhibits high ozone sensing response (38%) at room temperature. Additionally, the electrochemical performance of SnO2 and rGO/SnO2 nanocomposites was analysed and the rGO/SnO2 nanocomposite exhibited higher specific capacitance (545 F g−1) than that of pure SnO2 (236 F g−1) at a current density of 1 A g−1 with higher cyclic stability (96%) than that of pure SnO2 (86%) at the current density of 20 A g−1 for a continuous 5000 charge–discharge cycles. Thus, the rGO/SnO2 nanocomposite showed an excellent ozone sensing and energy storage performance.
Introduction
Globally, the energy demand and environmental pollution are two major challenges for the present and future generations. Rapidly increasing usage of automobiles and chemical industries produces toxic gases and hydrocarbons, which react with the atmospheric chemical species thus creating harmful diseases for humans and other living things. Ozone is one of the toxic and strong oxidizing gases, with wide usage in various industrial and medical fields.1–5 It can be produced naturally in the upper level of the atmosphere, called outdoor ozone, and the ground level ozone can be produced by electrical discharge, household materials, etc. This could create acute health effects like pulmonary, cardiovascular and chronic diseases6 when exceeding the limited threshold value of (0.08 ppm).7 Moreover, ozone gas can be used in water purification, food processing, textile and ozone therapy for cancer treatment. Therefore, due to its widespread applications and toxicity, ozone sensing devices are highly essential for monitoring the ozone concentration.An electric power-based device can be effectively used to overcome the addressed environmental issues like air pollution and generation of toxic molecules. In this process the performance of a device depends on the charge storage capacity of the energy storage devices like supercapacitors. But these devices may suffer from low energy density due to their limited storage capacity hence, it holds great demand to achieve long-life span with superior charge storage performance. The electrode material plays a vital role for the improvement of specific capacitance of the supercapacitor. These electrode materials store the energy based on different charge storage mechanism such as electric double layer capacitance (EDLC) and pseudo-capacitance. The charge balance between the electrode and electrolyte produces the EDLC behaviour and the faradic charge transfer between the electrode and electrolyte through oxidation–reduction reaction, which is responsible for the pseudo-capacitance behaviour. The electrochemical property of the electrode material can be enhanced by controlling the morphology,8 particle size9 and hybridisation of different types.10–12 Hence, development of hybrid nanocomposite material with high power density and life cycle stability is essential to provide a favourable solution for sustainable usage. Meanwhile, numerous efforts have been exerted on the synthesis of novel metal oxide nanocomposites with required shape and structures for improving the sensing and energy storage performance of the device.
Transition metal oxides possess broad structural and morphological variations owing to their capability to form phases of different metal to oxygen ratios showing multiple stable oxidation states of the metal ions. Among them, SnO2 has attracted cognizable interest owing to its strong chemical stability and out-standing optical and electrical features. It has been extensively used for gas sensing13–16 and energy storage applications.9 The chemical and physical features of tin oxide nano-crystalline material can be tuned by preparation methods that leads to the controlled surface morphology as well as grain size. Therefore, the preparation of SnO2 nanoparticles with desired structure and morphology provides attractive physical and chemical features, which enables enhanced performance in gas sensing17,18 and energy storage applications.9 Though SnO2 nanoparticles have a good interaction with ozone gas, its semiconducting nature and operating temperature restrict the sensing response. The problem associated with ozone sensing was resolved by coating Au or Ag conductive layer, before the deposition of the sensing material on the substrate.19,20 This type of fabrication process increases the material cost as well as the complicated instrumentation for thin film deposition. Therefore, it is very essential to develop ways for controlling the structure, dimensions, interface and surface characteristics. In addition, hybridisation of graphene-based materials into SnO2 nanoparticles is a wise-approach to enhance the surface area and conductivity of the sensing SnO2 film with more active sites.21–23 Further, the synergistic effect between graphene and metal oxides favours high transportation of electrolyte ions in the electrode material that leads to superior electrochemical performance than the pure metal oxides.24–29
In the present work, new approach has been adopted for the synthesis of ultra-small SnO2 nanoparticles and the average size of the synthesised SnO2 nanoparticles was achieved below 10 nm. The size reduced fine particles enables large surface area with more active sites, which in-turn enhanced the ozone sensing performance. The synthesised SnO2 nanoparticles were composited with rGO nanosheets, which provide high sensitivity and high electrical conductivity during the exposure of ozone gas on the surface of the sensing film. The synthesised rGO/SnO2 nanocomposite material showed improved ozone sensing response by morphological, optical and electrical variations than pure SnO2 nanoparticles in the room temperature. Further, the composite materials exhibited higher specific capacitance value (545 F g−1) at a current density of 1 A g−1 with higher cyclic stability of 96% at the current density of 20 A g−1 for continuous 5000 charge–discharge cycles.
Experimental
Chemicals
Pre-oxidized graphite flakes, Tin(II) chloride dihydrate (SnCl2·2H2O), diethylamine ((CH3CH2)2NH), sodium nitrate (NaNO3), potassium permanganate (KMnO4), hydrochloric acid (HCl), sulfuric acid (H2SO4) and hydrogen peroxide (H2O2) were purchased from Sigma-Aldrich and used for the preparation of the nanocomposites. Ultra-pure water with resistivity of 18.2 MΩ cm−1 (Milli-Q Water, Millipore System) was used for all the preparation process.Synthesis of tin oxide (SnO2) nanoparticles
Tin(II) chloride dihydrate (SnCl2·2H2O) precursor material (1.327 g) was dissolved in ethanol solvent and stirred for 15 min. After that poly-vinyl alcohol (PVA) surfactant was added slowly and stirred for 30 min. Then, diethylamine ((CH3CH2)2NH) was added as a precipitating agent, stirred for 15 min and transferred into the Teflon lined autoclave and maintained at 200 °C for 24 h. After cooling the autoclave to room temperature, the precipitate material was washed, filtered thrice and dried in a hot-air oven. Finally, the resultant material was grained in a mortar and stored in a vacuum desiccator for further studies.Synthesis of rGO
Initially, the graphene oxide (GO) was synthesized from pre-oxidized graphite flakes by modified Hummer's method.30,31 In an ice-bath, 1.5 g of NaNO3, 115 mL of H2SO4 and 3 g of pre-oxidized graphite were mixed and stirred for 20 min. Then, 9 g of KMnO4 was slowly added to the suspension under vigorous stirring condition and the temperature was maintained below 20 °C. After that, the temperature of the mixture was increased to 35 °C and continuously stirred for about 12 h. Then, 300 mL of DI water was added and heated to 98 °C. To minimize the residual manganese dioxide and permanganate, the suspension was diluted to 1 L using DI water and treated with 30 wt% of colorless H2O2. To neutralize the pH of the mixture, it was filtered and washed with DI water and 5 wt% of HCl aqueous solution. Subsequently, the product was dried at 40 °C under vacuum for about 24 h and the final form of GO was collected.32 The prepared GO was reduced to rGO by adding thiourea as a reducing agent at 100 °C followed by continuous stirring for 24 h.Preparation of rGO/SnO2 nanocomposite
The rGO/SnO2 nanocomposite material was prepared by the ultrasonic assisted solution process. Initially, 150 mL of rGO suspension was prepared by adding 0.125 g of rGO into the DI water and sonicated for 30 min. Then, 0.03125 g of SnO2 nanoparticles were added to 50 mL of DI water and sonicated for 30 min. Both the solutions were mixed and sonicated for 45 min. After that the sample was collected by drying in a hot-air oven. Finally, the rGO/SnO2 nanocomposite powder was obtained.Fabrication of rGO/SnO2 nanocomposite film
As reported in the previous work, the PVA/KOH conductive polymer coated sample exhibited very good response towards ozone gas sensing.33 Therefore, the PVA/KOH conductive polymer was coated on the cleaned quartz substrate by dip-coating method. Then, the variable thickness of rGO/SnO2 nanocomposite material was drop-casted layer by layer over the conductive layer and dried in a hot-air oven at 180 °C. The fabricated composite's sample thicknesses were measured and named as rGO/SnO2-S1 (3 μm), rGO/SnO2-S2 (5 μm) and rGO/SnO2-S3 (7 μm), respectively.Electrode preparation for electrochemical analysis
The electrochemical properties of prepared pure SnO2 and rGO/SnO2 nanocomposite were analysed by cyclic voltammetry (CV) with three electrode configurations using 1 M H2SO4 electrolyte at room temperature. The working electrode was fabricated by addition of 80 wt% of active material, 10 wt% acetylene black, and 10 wt% polyvinylidene fluoride (PVDF) binder in N-methyl-2-pyrrolidone (NMP) solvent. The resultant mixture was finely grained to prepare the slurry. The slurry was coated on the nickel foil using doctor blade method and dried in hot air oven for 24 h at 80 °C. The weight of the active materials was approximately 4 mg. The platinum foil, saturated calomel electrode (SCE) and active material coated nickel foil were used as counter, reference and working electrodes, respectively.Characterization
The structural, morphological, elemental composition and the corresponding valance states of the prepared rGO/SnO2 nanocomposites were characterized by the Bruker X-ray diffractometer (Model no. D8 Advance), HRSEM (Model no. Hitachi S4800), High-Resolution TEM (JEOL JEM-2000) and the Multi-technique X-ray Photoelectron Spectroscopy system Kratos AXIS ULTRA, respectively. The particle size of the SnO2 nanoparticles was measured by dynamic light scattering technique using nano Partica nano particle analyzer (SZ-100), HORIBA scientific. The sensing response of the prepared pure and composite films were investigated before and after ozone exposure as follows; the functional groups interaction of the material with the ozone molecules were examined by the Versatile FT-IR Laboratory Spectrometer (Model no. ABB MB3000) in the region of 400–4000 cm−1. The Raman spectra were recorded by the Horiba Jobin Yvon LabRAM HR micro Raman system with an excitation wavelength of 632 nm. The topographical variations of the sensing films were examined by Atomic force microscopy (Model no. MFP-3D Infinity AFM) for analyzing the surface properties of the films before and after ozone exposure. The ozone generator working on the basis of corona discharge method was used as an ozone source and the ppm of the ozone output was controlled by special dialing knob attached with the ozone generator. The electrochemical properties of the prepared materials were analyzed through cyclic voltammetry (CV), galvanostatic charge/discharge (GCD) and electrochemical impedance spectroscopic (EIS) analysis using Biologic Instrument (Model SP-150).Results and discussion
Structural, morphological and compositional studies of SnO2 and rGO/SnO2 nanocomposite
Fig. 1 shows the X-Ray Diffraction (XRD) patterns of rGO, SnO2 and rGO/SnO2 nanocomposites. The XRD pattern of rGO exhibits two prominent peaks at the diffraction angles of 24.42° and 42.21° corresponding to (002) and (100) planes, respectively59 and it is well-matched with the JCPDS no. 75-1621. Broad peaks are observed in the XRD pattern of SnO2 at 26.57°, 33.73°, 51.81°and 64.73° corresponding to (110), (101), (211) and (112) planes, respectively,34 which is matched with the JCPDS no. 01-072-1147. The broad diffraction peaks represent SnO2 nanocrystalline particles. Further, the XRD pattern of rGO/SnO2 nanocomposite shows rGO related peaks at 24.39° (002), 42.74° (110) and peaks related to SnO2 at 26.53° (110), 33.69° (101), 51.84° (211) and 64.70° (112). These results confirm the formation of rGO/SnO2 nanocomposite by the ultrasonic assisted solution process.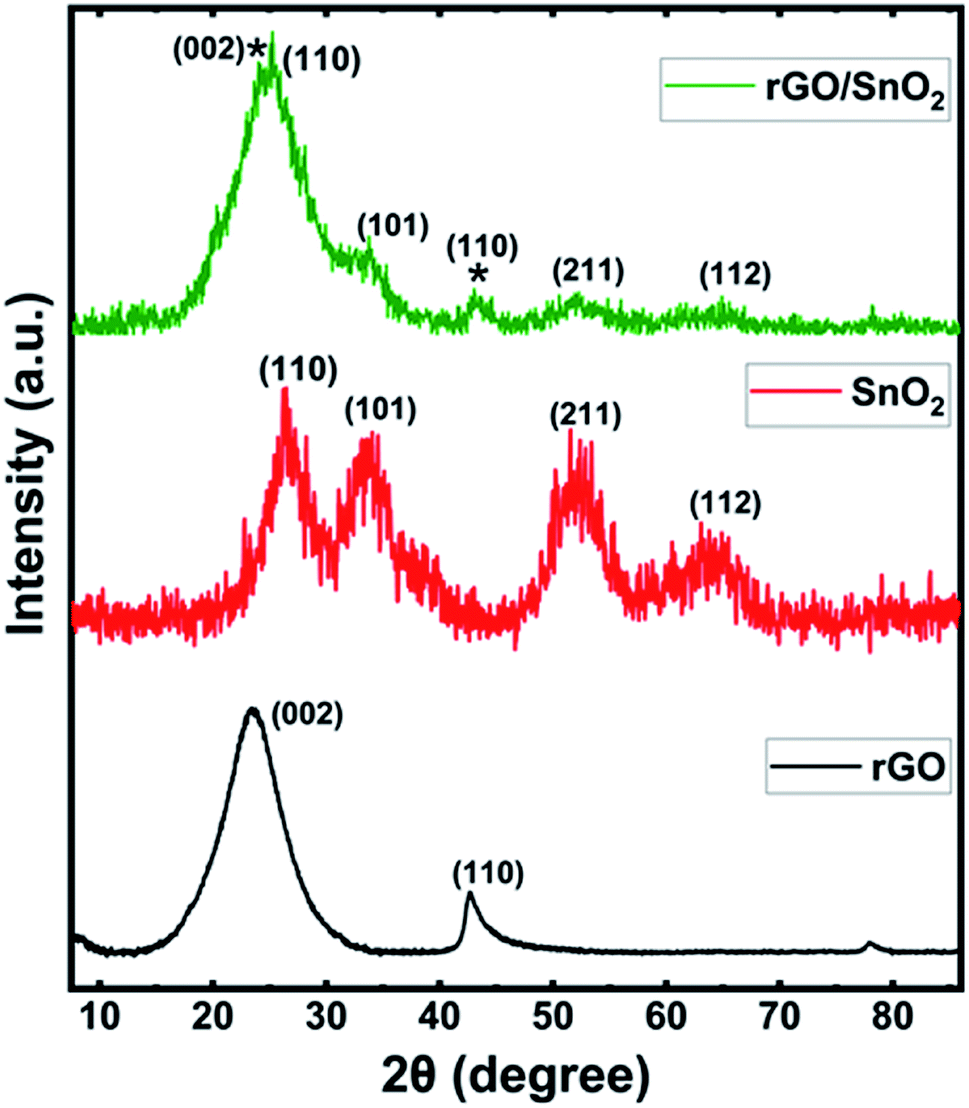 | ||
| Fig. 1 X-ray diffraction patterns of rGO, SnO2 and rGO/SnO2 nanocomposite. | ||
The mean particle size of the pure SnO2 was measured as 7 nm by dynamic light scattering (DLS) as shown in Fig. 2a. The distribution of particles is more uniform with a narrow range of size variation from 6 to 9 nm. The morphological image of the SnO2 is shown in Fig. 2b. It is significant to note that the ultra-small SnO2 nanoparticles are formed in the present synthesis method. It may be explained as follows; during the synthesis process, the polyvinyl alcohol act as template, which can inhibit the nanoparticle overgrowth and agglomeration. Conversely, the capping agent, diethyl amine effectively passivated the surface and thereby controlled the particle size, which resulted in the formation of ultra-small nanoparticles.
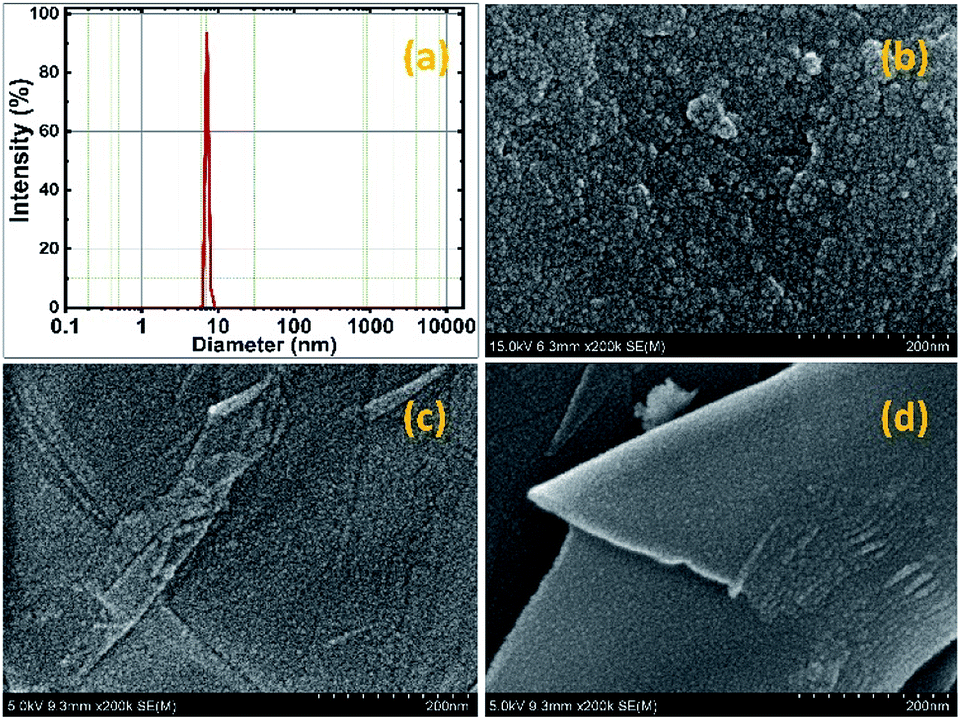 | ||
| Fig. 2 DLS particle size analysis curve of SnO2 nanoparticles (a), HR-SEM images of SnO2 (b) and rGO/SnO2 nanocomposite (c and d) in different area. | ||
Further, the morphological observations of rGO/SnO2 nanocomposite are shown in Fig. 2c and d. It is obvious that the ultra-small SnO2 nanoparticles are uniformly distributed on the rGO nanosheets. The SnO2 nanoparticles act as an inter-sheet spacer, which inhibits restacking and collapsing between the rGO nanosheets. Besides, it is expected that the ultra-small SnO2 nanoparticles provide high specific surface area and more active sites on the surface of the rGO/SnO2 nanocomposites. Further, investigation was carried out through the HR-TEM analysis and the obtained images of pure SnO2 and rGO/SnO2 nanocomposite are shown in Fig. 3.
 | ||
| Fig. 3 HR-TEM images of SnO2 (a), rGO/SnO2 nanocomposite (b and c) and SAED pattern of rGO/SnO2 nanocomposite (d). | ||
The uniformly grown ultra-small SnO2 particles could be seen from the magnified image shown in Fig. 3a. In the case of rGO/SnO2 nanocomposite (Fig. 3b and c), the ultra-small nanoparticles are found to be evenly embedded on the surface as well as at the interlayer spacing of rGO nanosheets as it is highlighted in dotted circle in Fig. 3b. The lattice fringes of rGO/SnO2 nanocomposites are clearly visible in Fig. 3c. The measured d-spacing values of 0.23 nm (111), 0.17 nm (211) correspond to inter planar spacing of SnO2 and 0.21 nm (100) is for rGO, which are well-matched with the observed XRD pattern of rGO/SnO2 nanocomposite. Additionally, the SAED pattern of the rGO/SnO2 nanocomposite, as shown in Fig. 3d, exhibits the ring pattern due to the presence of rGO and bright spots corresponding to SnO2 nanoparticles. These studies confirm the formation SnO2/rGO composite structure.
The energy dispersive X-ray (EDX) analysis of SnO2 and rGO/SnO2 nanocomposites confirm the material formation as shown in Fig. 4. The observed EDX spectrum of SnO2 (Fig. 4a) demonstrates the presence of Sn, O and Cu. Conversely, in the EDX spectrum of rGO/SnO2 (Fig. 4b), carbon (C) peak additionally appears owing to the hybridisation of rGO into the SnO2. The Cu peak is observed due to the copper substrate, which is used to hold the samples.
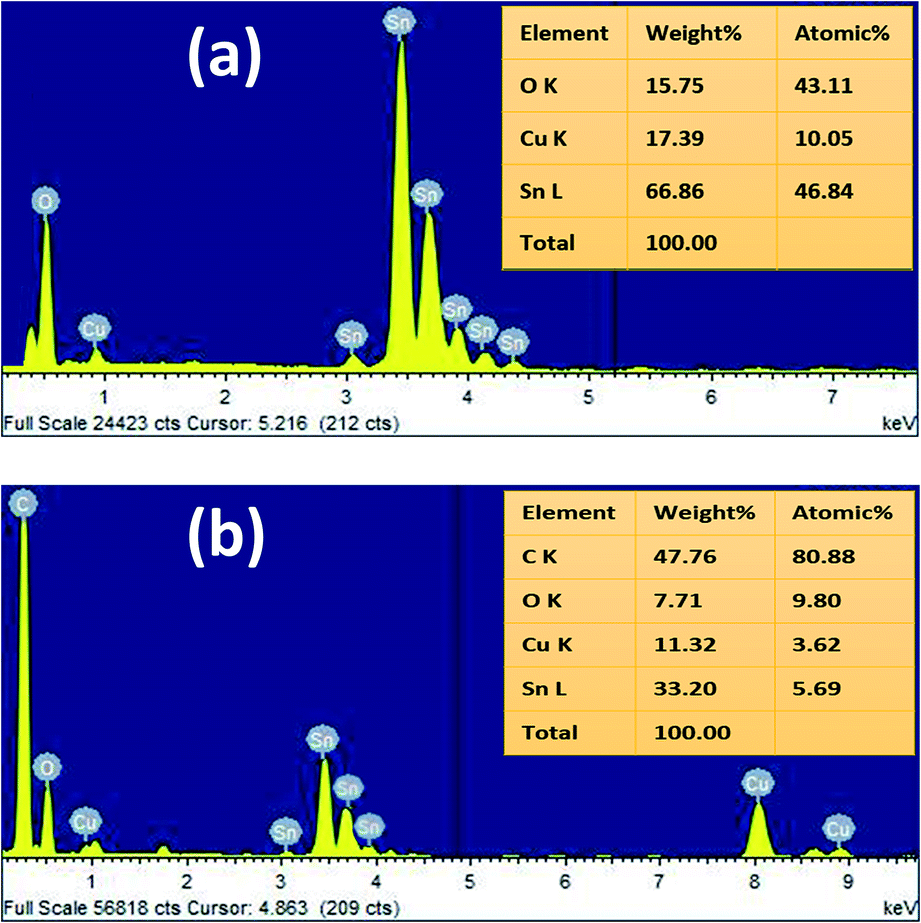 | ||
| Fig. 4 EDX spectra of SnO2 (a) and rGO/SnO2 nanocomposite (b). | ||
Fig. 5a shows the survey spectrum of rGO/SnO2 nanocomposite, which confirms the nanocomposite formation. In Fig. 5b–d, all the core level high resolution spectra of elements are seen with fitting and deconvolution using Gaussian distribution. The core level spectrum of Sn 3d region of SnO2 shows two characteristic peaks at 486.3 eV and 494.6 eV corresponding to Sn 3d5/2 and Sn 3d3/2, respectively as shown in Fig. 5b.35 Fig. 5c demonstrates that the C 1s spectrum of rGO with high intense peak at 284.5 eV assigned to the graphite (sp2) C–C bond and relatively very low intense shoulder peaks are observed at 285.5 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and 286.4 eV (C–OH), respectively.36–38
C) and 286.4 eV (C–OH), respectively.36–38
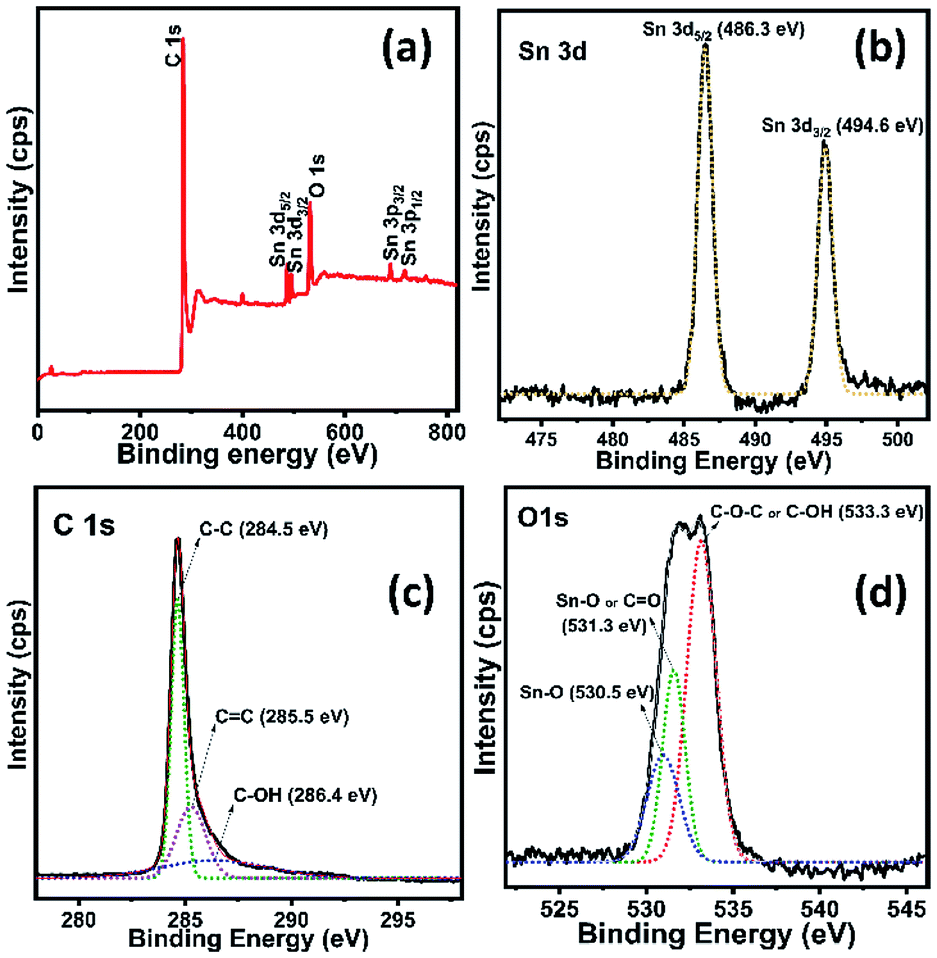 | ||
| Fig. 5 XPS survey spectra of rGO/SnO2 nanocomposite (a) and high-resolution spectra of Sn 3d (b), C 1s (c) and O 1s (d). | ||
Further, the deconvoluted spectrum of O 1s (Fig. 5d) shows a most intensive peak at 533.3 eV assigned to the C–OH and/or C–O–C39 and relatively two low intense peaks are noticed, owing to the binding energies of Sn–O (530.5 eV)40 and CO (531.3 eV).39,40 Consequently, these peaks appear due to the interaction between the functional groups of rGO and SnO2, that confirms the formation of rGO/SnO2 nanocomposite. After the successful confirmation of the composite formation the prepared rGO/SnO2 nanocomposite material was further investigated for the feasibility of ozone gas sensing and energy storage performances.
Investigations of the fabricated SnO2 and rGO/SnO2 films under ozone atmosphere
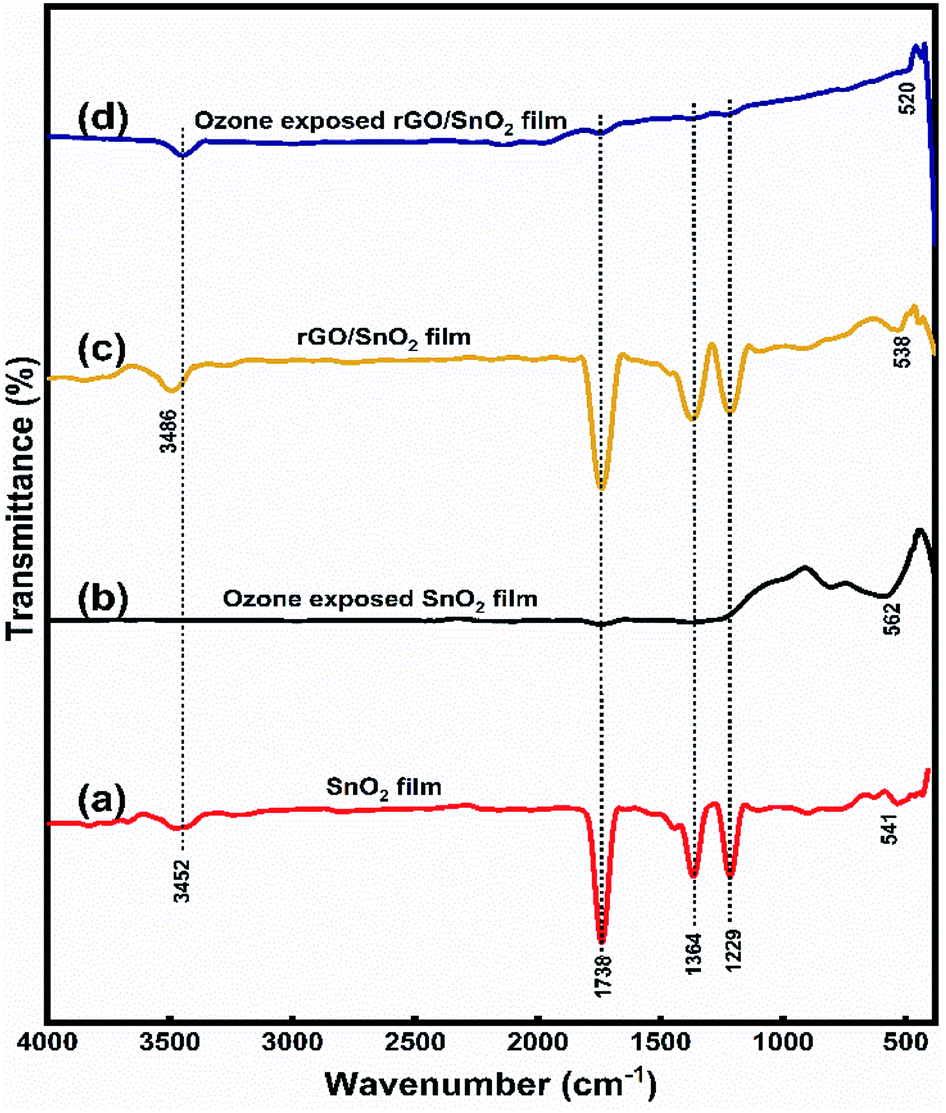 | ||
| Fig. 6 FTIR spectra of SnO2 film (a), ozone exposed SnO2 (b), rGO/SnO2 film (c) and ozone exposed rGO/SnO2 film (d). | ||
The Raman spectra of SnO2 film before and after ozone exposure are shown in Fig. 7a. As can be seen from the Fig. 7a, three characteristic vibrational modes of SnO2 at 473 cm−1 (Eg), 643 cm−1 (A1g) and 786 cm−1 (B2g)46 are observed. The PVA peaks observed around 880 cm−1, 982 cm−1 and 1173 cm−1 correspond to the fan or twist modes in cyndio or isotactic sequences.47 After exposing the ozone gas on the surface of the film, SnO2 related peaks are clearly visible, in particular, the intensity of the peak at 786 cm−1 (B2g) is found to increase, which contributes that the ozone interacts on the SnO2 film surface. Conversely, in the rGO/SnO2 nanocomposite film, the SnO2 related peaks appear at 490 cm−1, 647 cm−1 and 786 cm−1 (B2g), due to the introduction of PVA and rGO (Fig. 7b). Further, SnO2 related peaks are shifted to higher frequency region, which supports the rGO/SnO2 nanocomposite formation. The PVA peaks at 818 cm−1 and 914 cm−1 correspond to the fan or twist modes in cyndio or isotactic sequences.47 The peaks at 1098 cm−1 and 1230 cm−1 are attributed to the O–H deformation and C–O valance bond vibrational modes of secondary alcohols in the PVA matrix.47
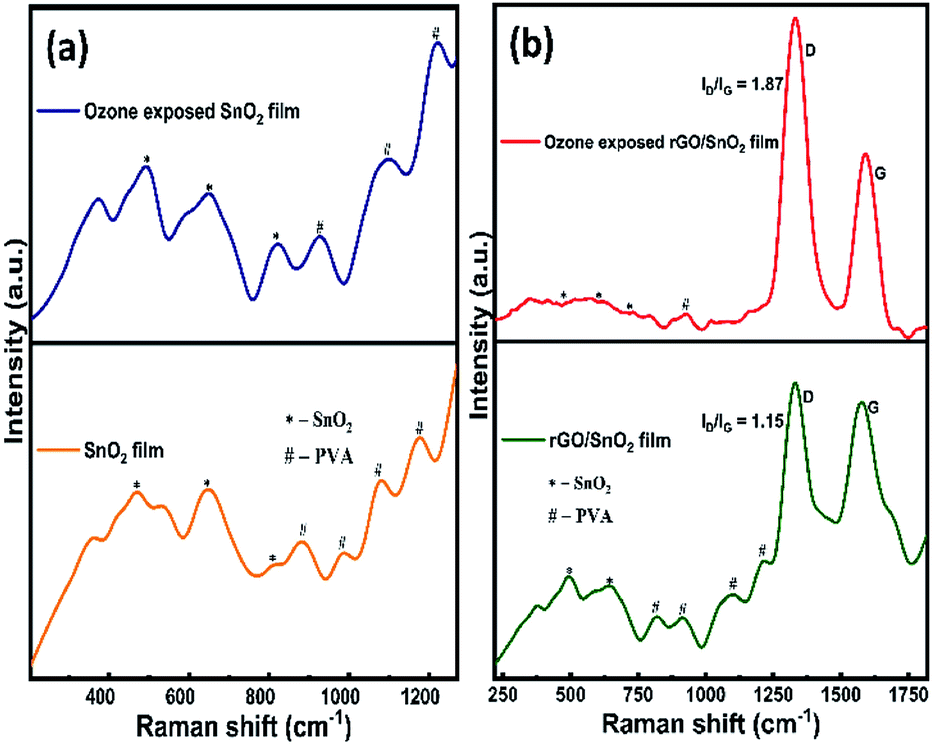 | ||
| Fig. 7 Raman spectra of SnO2 & ozone exposed SnO2 film (a) and rGO/SnO2 & ozone exposed rGO/SnO2 film (b). | ||
In Fig. 7b, the D and G bands are observed at 1330 cm−1 and 1574 cm−1, respectively. The G band corresponds to E2g phonon vibrations of sp2 hybridized carbon atoms in the isolated double bonds of graphene lattice and the D band analogous to the K-point phonons' one-phonon defect-assisted process of vibrations with A1g symmetry.48 Therefore, the ID/IG ratio provides the structural imperfections in the rGO, owing to the attachment of functional groups on the plane of rGO.48 The ID/IG ratio of the rGO/SnO2 nanocomposite film is calculated as 1.15, which strongly supports the attachment of the SnO2 nanoparticles on the surface and interlayers of rGO as seen in the morphological studies (HRSEM and HRTEM).
After passing the ozone gas, the intensity of the D and G bands is found to increase and the bands are noticed at 1331 cm−1 and 1590 cm−1, respectively (Fig. 7b). Most of the PVA peaks disappear and the SnO2 related peak intensities decreased owing to the interaction of the ozone molecules. The ID/IG ratio of the ozone exposed rGO/SnO2 film is calculated as 1.87, which explains the enrichment of graphene content after the ozone interaction.
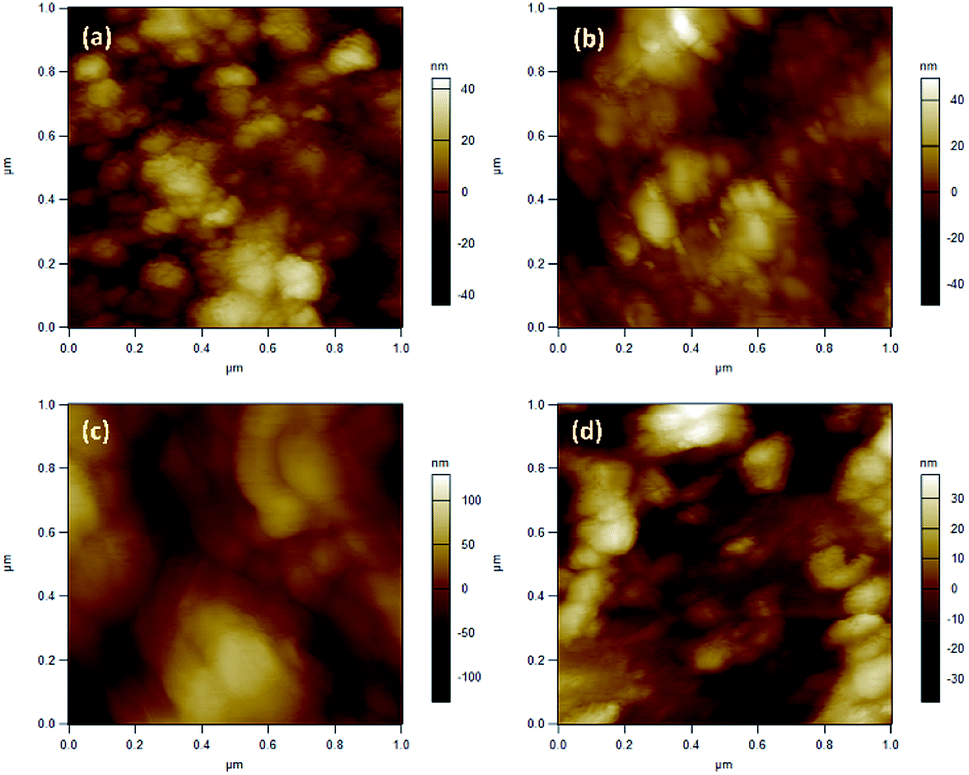 | ||
| Fig. 8 AFM images of SnO2 (a) ozone exposed SnO2 film (b), rGO/SnO2 nanocomposite (c) and ozone exposed rGO/SnO2 nanocomposite film (d). | ||
The surface roughness profiles of SnO2, ozone exposed SnO2 film, rGO/SnO2 film and ozone exposed rGO/SnO2 film have been analysed for studying the morphological variations. Fig. 9a shows the variation of surface roughness of SnO2 film and it varied from −2 to 2 nm, which is found to be decreased to −1.3 to 1 nm, after exposing the ozone gas on the surface of the SnO2 film (Fig. 9b). The variations of surface roughness of rGO/SnO2 nanocomposite are in the range of −4 to 4 nm as shown in Fig. 9c and it is found to decrease after exposure of ozone, which may be due to the removal of rGO, PVA functional groups from the surface of the rGO/SnO2 film (Fig. 9d). Therefore, the ozone exposure induces morphological variations based on the ozone environment that leads to the electrical variation in the sensing film.
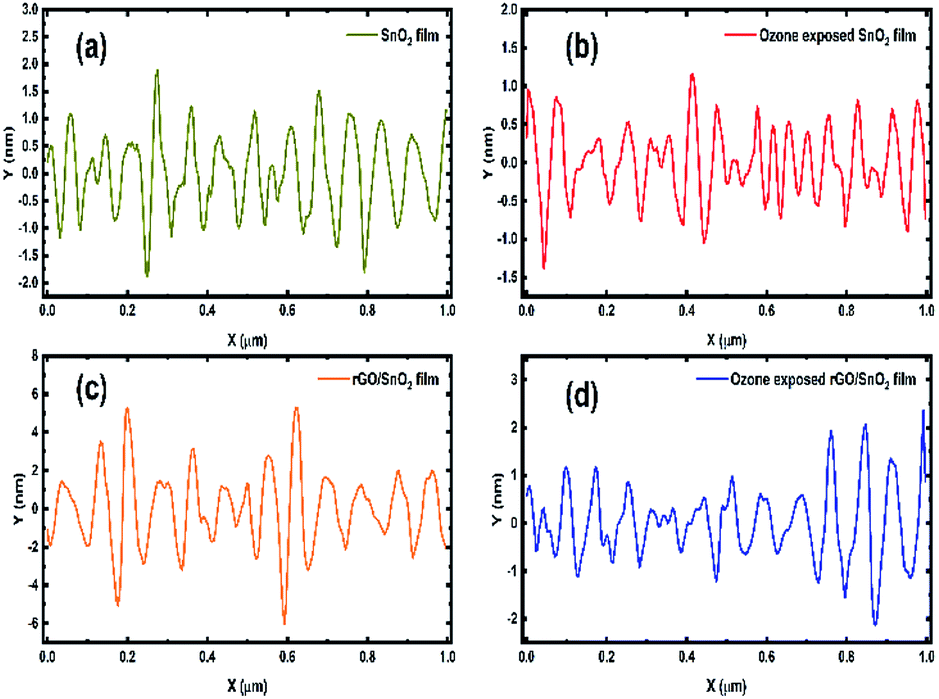 | ||
| Fig. 9 Surface roughness profiles of SnO2 (a) ozone exposed SnO2 film (b), rGO/SnO2 nanocomposite (c) and ozone exposed rGO/SnO2 nanocomposite film (d). | ||
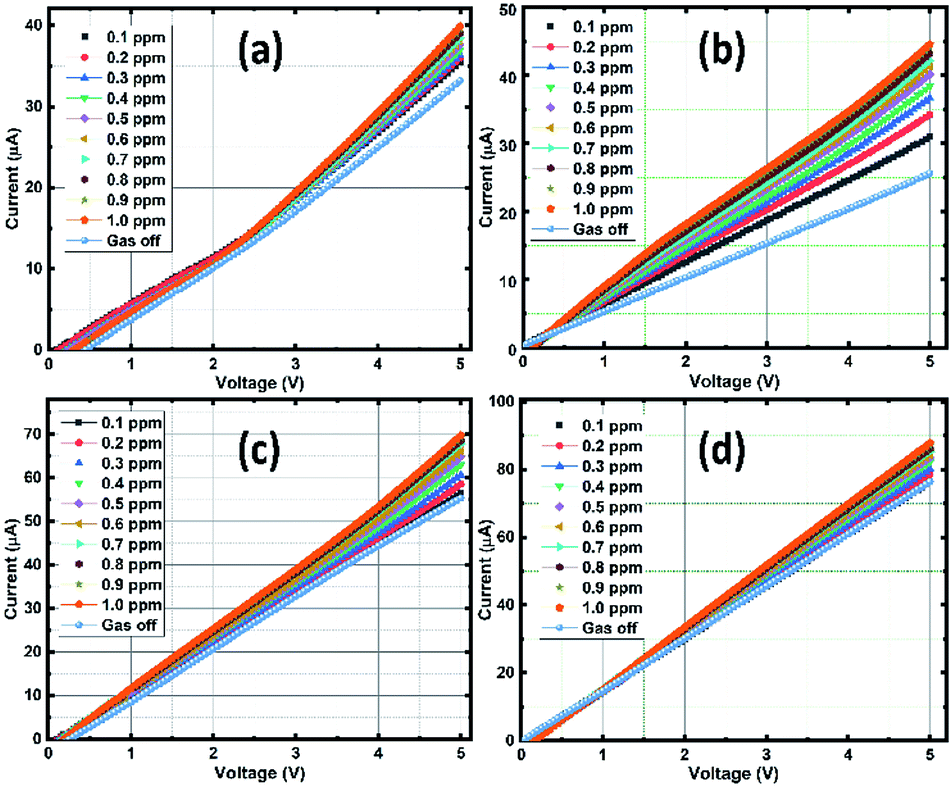 | ||
| Fig. 10 V–I measurements of the sensing films at different ozone concentration: SnO2 (a), rGO/SnO2-S1 (b), rGO/SnO2-S2 (c) and rGO/SnO2-S3 (d). | ||
In Fig. 10a, the pure SnO2 film exhibits a gradual current increment, while increasing the ozone concentration from 0.1 to 1 ppm. It is observed that the V–I characteristic lines start to bunch at a threshold voltage that ranges approximately between 2 to 3 V and then increases with respect to the higher applied potential and ozone concentrations. The observed increase in conductivity beyond the threshold voltage regime (2–3 V) promises the optimum sensing performance of the SnO2 film. Whereas, in the case of composite films of rGO/SnO2-S1, rGO/SnO2-S2 & rGO/SnO2-S3, the current rate is varied with the concentration of ozone in the voltage range of approximately 0.5–1.5 V as shown in the Fig. 10b and c.
The higher electrical response is achieved for the rGO/SnO2-S2 and rGO/SnO2-S3 films, due to the presence of rGO. However, the rGO/SnO2-S1 show a quantifiable current variation with respect to the ozone concentration at 1 V that suggest the best sensing performance and lower operating voltage of the rGO/SnO2-S1 film. Hence, based on the V–I data, the electrical resistance of the ozone exposed sensing films was calculated, and the results were plotted as a function of ozone concentration (Fig. 11a). Further investigations revealed that the rGO/SnO2 nanocomposite films exhibit relatively high response towards the ozone concentrations compared to pure SnO2. As can be seen from the Fig. 11b, the composite films show the maximum ozone response (20–38%) at 1.0 ppm compared to the SnO2 film (18%). However, it must be noted that the rGO/SnO2-S1 with 3 μm thickness shows the proportional response to the entire range (0.1–1 ppm) of exposed ozone concentrations.
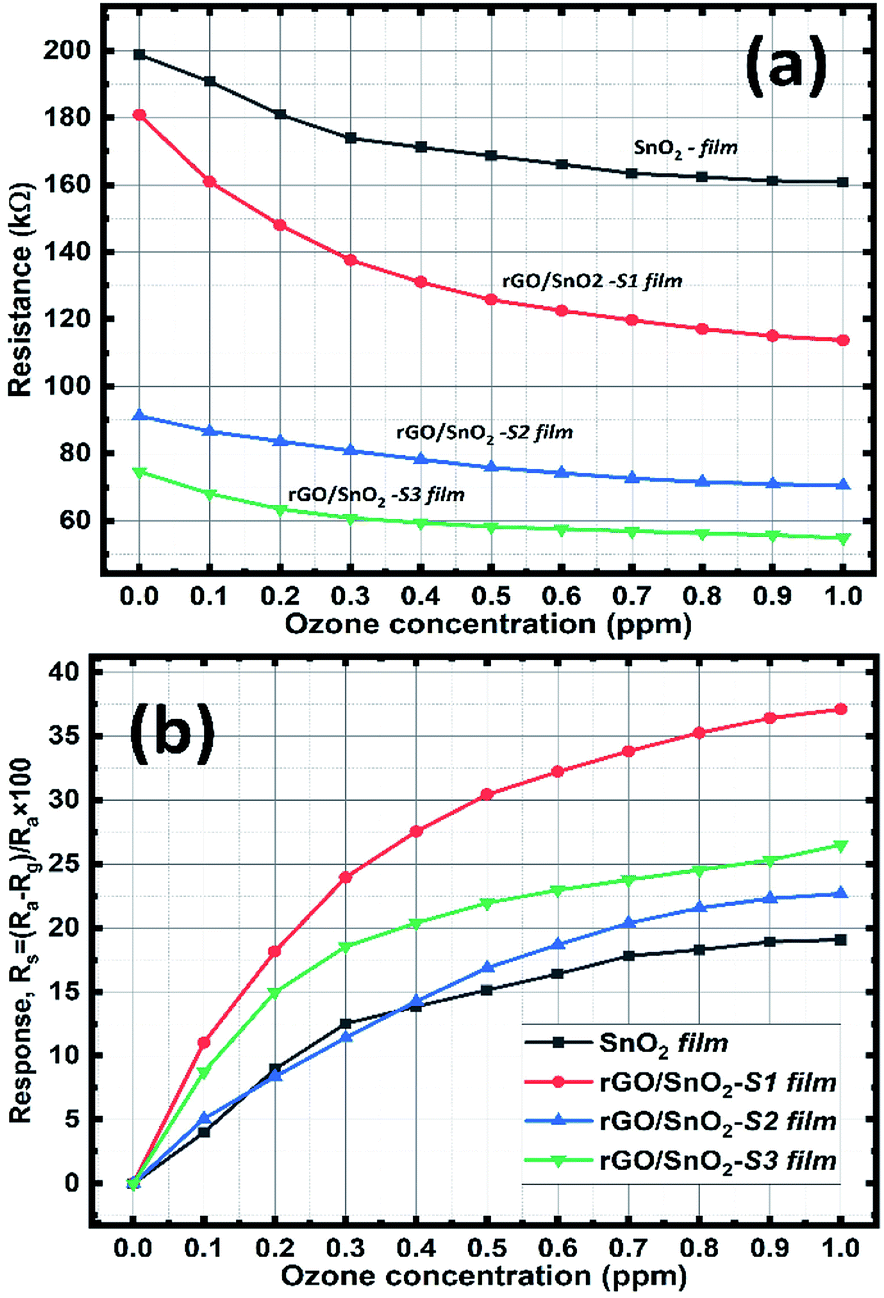 | ||
| Fig. 11 Resistance variation (a) and the ozone sensing response curves of the SnO2 & rGO/SnO2 nanocomposite films as a function of ozone concentration (b). | ||
Electrochemical performance of the rGO/SnO2 nanocomposite
The CV analysis were carried out by varying the scan rates from 5 to 100 mV s−1 within the potential range from −1.0 to 0.1 V in 1 M of H2SO4 electrolyte solution.49 The recorded CV curves are shown in Fig. 12a and b, the shape of the curves explains the capacitive behaviour of the electrode materials and the CV curves doesn't show any redox peaks but all curves exhibited roughly in rectangular shape, which signifying the formation of electric double layer capacitor (EDLC) charge storage mechanism.24 The CV curves remain stable and no distortion occurred when increasing scan rate up to 100 mV s−1 for both SnO2 and rGO/SnO2 electrodes representing high stability and quick interfacial charge transfer of the electrode materials.50 The current peak increased gradually with an increase of scan rate from 5 to 100 mV s−1, which is attributed to the superior electrochemical features of SnO2 and rGO/SnO2 electrodes.25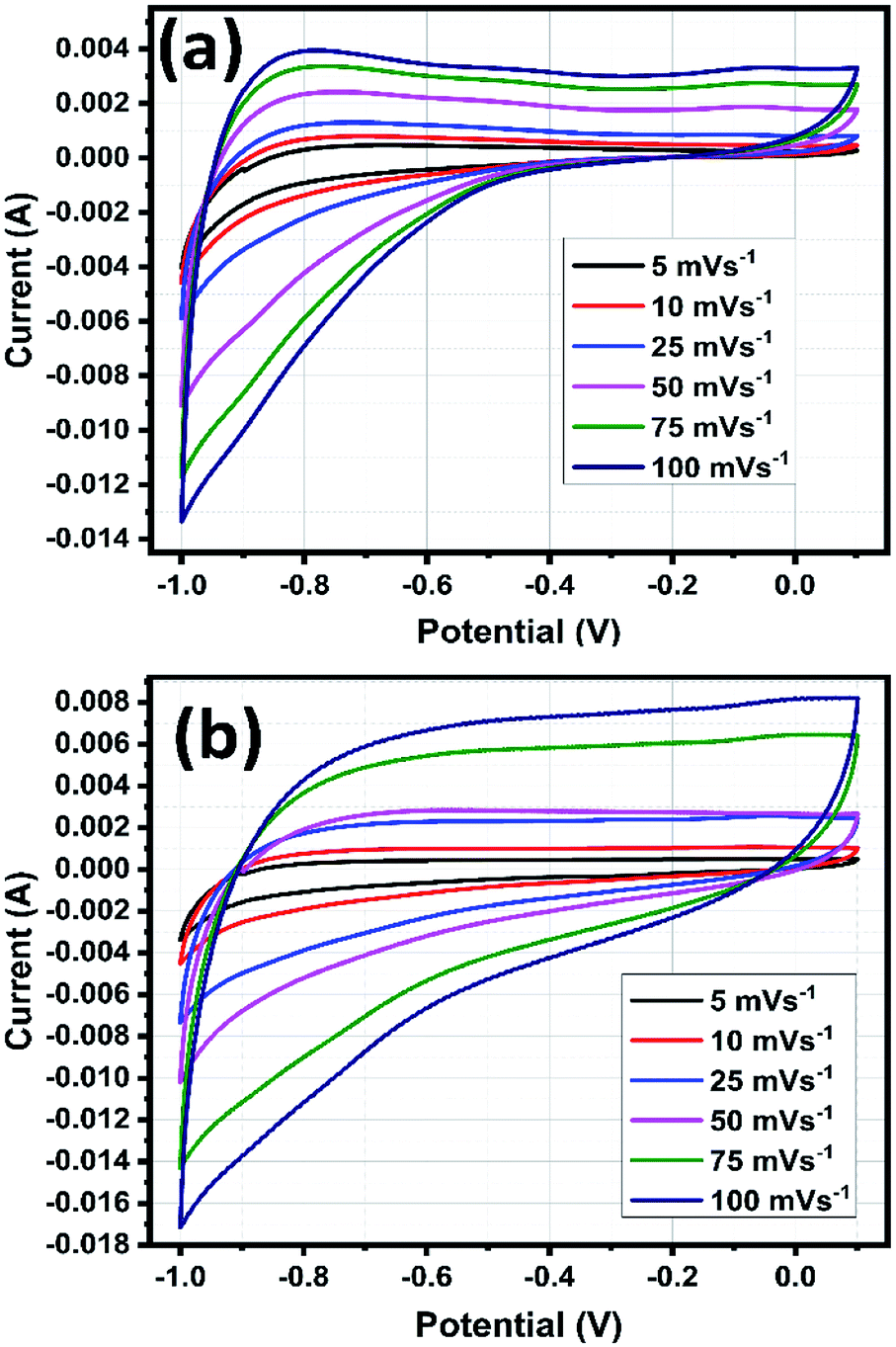 | ||
| Fig. 12 Cyclic voltammetric curves of SnO2 (a) and rGO/SnO2 nanocomposite (b). | ||
Also, the larger voltage window 1.1 V indicates the high energy density of SnO2 and rGO/SnO2 electrode materials. It is well known that an integral area of the CV curves is directly proportional to the specific capacitive behaviour of the electrodes. In the present study, rGO/SnO2 electrode material exhibited higher integral area than that of pure SnO2, which implies the superior specific capacitance feature of the rGO/SnO2 electrode material.
The electrochemical properties of the electrode materials were further evaluated by galvanostatic charge/discharge (GCD) analyses as shown in Fig. 13a and b. The linear profile of GCD curves confirms the EDLC behavior which is more consistent with the CV results.51 When compared with the SnO2 electrode, the rGO/SnO2 provides long discharge time confirming the superior specific capacitive nature. At lower current densities, the charge/discharge process took longer time which is ascribed to the adequate interaction of electrolyte ions during charging discharging process as shown in Fig. 13c. The specific capacitance (Csp) of the electrode can be calculated using eq.,52
| Csp = IΔt/mΔV |
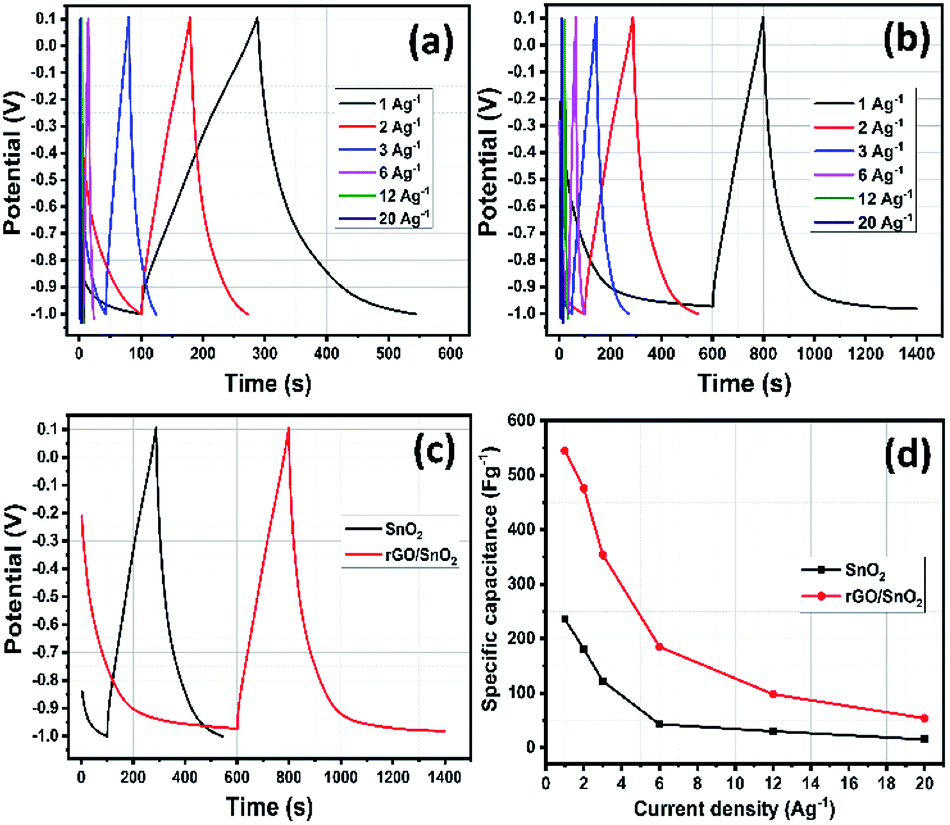 | ||
| Fig. 13 GCD studies of SnO2 (a) and rGO/SnO2 nanocomposite (b) at different current densities, GCD study of SnO2 & rGO/SnO2 nanocomposite at the current density of 1 A g−1 (c) and specific capacitance versus current density (d). | ||
| Material | Electrolyte | Specific capacitance (F g−1) | Current density (A g−1) | Ref. |
|---|---|---|---|---|
| rGO/SnO2 | 1 M KOH | 35 | 0.1 | 53 |
| Graphene–SnO2 | 1 M Na2SO4 | 158 | 0.1 | 54 |
| SnO2/GNS | 1 M Na2SO4 | 184.6 | 0.1 | 55 |
| SnO2/rGO@NF | PVA/KOH | 229.1 | 1 | 56 |
| SnO2QDs/rGO | 1 M Na2SO4 | 253.3 | 1 | 57 |
| SnO2/rGO | 1 M Na2SO4 | 262.2 | 0.1 | 28 |
| SnO2/rGO | 2 M KOH | 310 | 2 | 29 |
| SnO2/rGO | 1 M H2SO4 | 337.52 | 0.5 | 49 |
| SnO2/graphene | 1 M Na2SO4 | 364.3 | 1 | 24 |
| SnO2@NGO | 6 M KOH | 378 | 4 | 26 |
| rGO/SnO2 | 1 M H2SO4 | 545 | 1 | This work |
The higher specific capacitance of rGO/SnO2 composite electrode is due to the following reason: (i) the homogeneous distribution of ultrasmall SnO2 materials on rGO sheets in the course of influence of synergistic effect, which is more encouraging for electrochemical process and thus, ensuing high specific capacitance (ii) the ultra-small SnO2 materials provide more active sites for electrochemical progression and reduced the electrolyte ion transport pathways. (iii) Thin rGO nanosheets exhibit attractive features such as high electrical conductivity, high surface area, and high flexibility which may help to enhance the specific capacitance of the composite materials.
Rate capability is one of the important parameters for supercapacitor electrode materials. For evaluation of rate capability of SnO2 and rGO/SnO2 electrodes, the GCD studies were carried out with various current densities as shown in Fig. 13d. It is perfectly visible that the specific capacitance slightly decreases with increasing of current density due to time limitation process. At low current density, the electrolyte ions experience larger time to meet the electrode materials, which enhance the electrode materials utilization and thus increases the specific capacitance. On the other hand, the outer surface electrode materials only utilized by electrolyte ions due to shorter time at higher current densities which decreases the specific capacitance.27
Long term cycling stability is a significant factor for evaluating the electrode materials for supercapacitor applications. Cycling stability analyses were carried out for both SnO2 and rGO/SnO2 electrodes at 20 A g−1 current density for continuous 5000 charge discharge cycles as shown in Fig. 14. Fascinatingly, the specific capacitance slightly increases to 102% at 1400th cycles in rGO/SnO2 electrode, which signifies the activation process of electrode materials.58
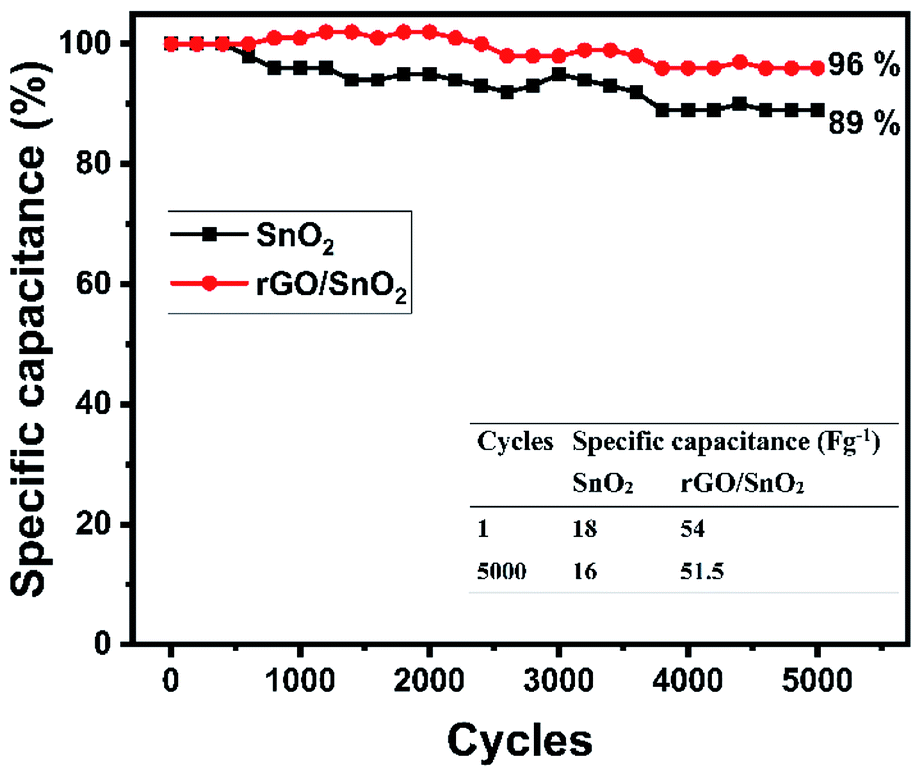 | ||
| Fig. 14 Cyclic stability of SnO2 and rGO/SnO2 nanocomposite electrode at 20 A g−1 for 5000 charge/discharge cycles. | ||
In the endeavour, the accumulation of electrolyte ions contributes to the activation of electrode materials which is advantageous to stimulate the internal active region and enhancing the specific capacitance. After that, the specific capacitance gradually decreases and attains 89 and 96% for SnO2 and rGO/SnO2 electrodes, respectively. When compared with SnO2, the rGO/SnO2 electrode exhibiting good cyclic stability features. This phenomenon is due to the fact that the SnO2 material may act as a spacer material between the two-neighbourhood reduced graphene oxide nanosheets and thus it helps to reduce the restacking of rGO nanosheets which always maintain the high surface area during cyclic stability analysis.59 Furthermore, this factor enhances the surface area utilization feature of electrolyte ions and thus it keeps specific capacitance maximum for 5000 cycles. In addition, the aforementioned key advantage of rGO/SnO2 electrode is attributed to excellent contact between SnO2 and rGO nanosheets. Hence, rGO/SnO2 is a significant electrode material for future supercapacitor device application.
The electrochemical impedance spectra were recorded for the prepared electrode materials of SnO2 and rGO/SnO2 nanocomposite as shown in Fig. 15. Both the spectra exhibited a depressed semicircle in the high frequency region followed by a linear region appeared in the low frequency range. The depressed semicircle explains the interfacial charge transfer process at the electrode/electrolyte interface and the linear region describes about the Warburg impedance owing to the diffusion dominant charge transport in the electrolyte and surface of the electrode materials.60
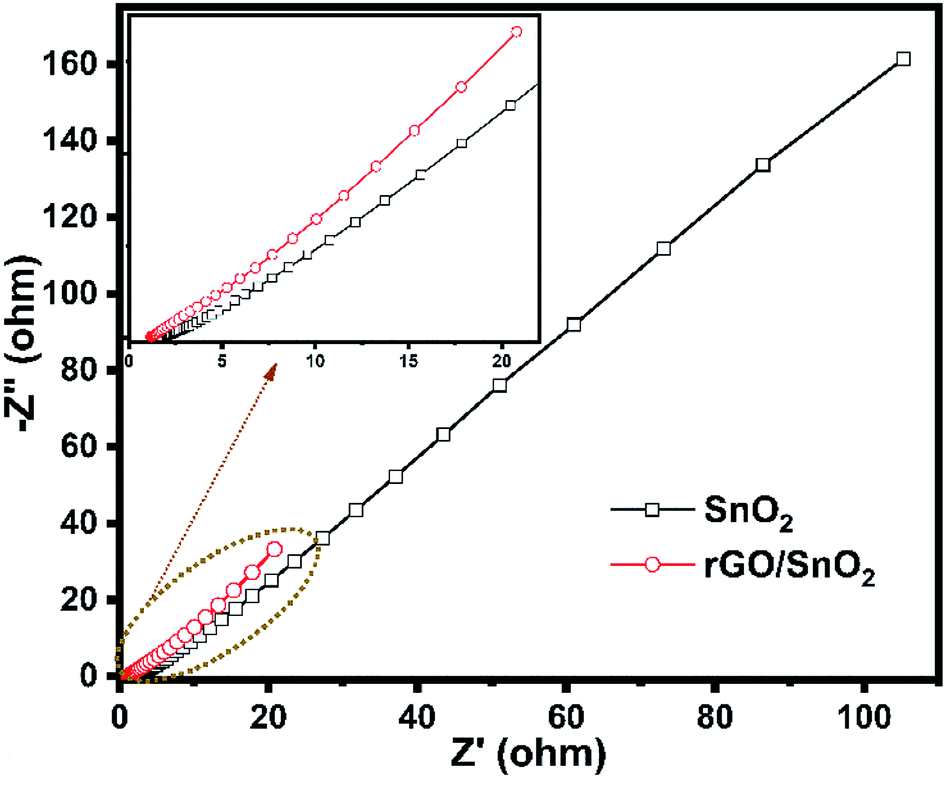 | ||
| Fig. 15 Electrochemical impedance spectra of SnO2 and rGO/SnO2 nanocomposite. | ||
During the charging process the charges carried by the ions may diffuse towards the electrode material and thereby produces the electric double-layer capacitance (EDLC) as shown schematically in Fig. 16. Further, the intercalation of ultra-small SnO2 between the stacks of rGO nanosheets favors a highly porous nature (Fig. 16) and fast charge transfer process thus leads to the high charge storage capacity of the rGO/SnO2 electrode material than that of pure SnO2 nanoparticles.
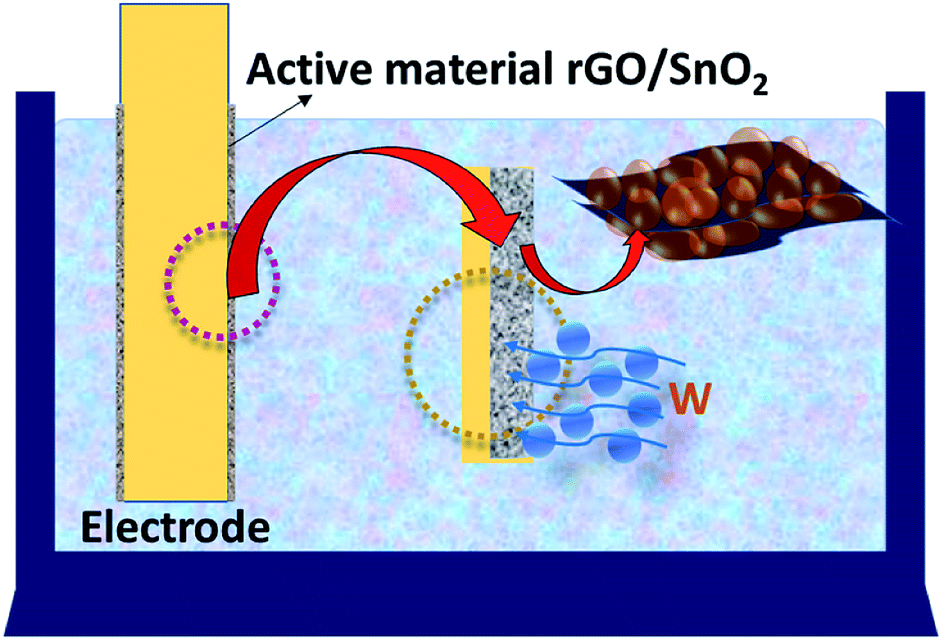 | ||
| Fig. 16 Schematic representation of the charge storage mechanism of rGO/SnO2 nanocomposite. | ||
Conclusions
Ultra-small SnO2 nanoparticles synthesised by hydrothermal route were composited with chemically synthesised rGO nanosheets. The morphological analyses show a uniform distribution of SnO2 nanoparticles on the surface and at the interlayer spacing of the rGO nanosheets. The XRD, FTIR and Raman analyses confirmed the formation of pure ultra-small SnO2 and rGO/SnO2 nanocomposite. XPS studies strongly support the rGO/SnO2 nanocomposite formation during ultrasonication process. Structural, functional, morphological and electrical conductivity studies reveal that the fabricated SnO2 and rGO/SnO2 nanocomposite films have a good interaction with the ozone gas. Among the fabricated composite films. The rGO/SnO2-S1 film exhibits high ozone sensing response (38%) at room temperature. Additionally, the electrochemical performance of SnO2 and rGO/SnO2 nanocomposites exhibited higher specific capacitance value (545 F g−1) at 1 A g−1 and higher cyclic stability of 96% at 20 A g−1 for continuous 5000 charge–discharge cycles than the pure SnO2. The synthesised rGO/SnO2 nanocomposite material showed best ozone sensing and energy storage performance than the pure SnO2 nanoparticles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by University Grants Commission (UGC) sponsored Rajiv Gandhi National Fellowship for Students with Disabilities (RGNFD), New Delhi. We acknowledge the facilities extended by the University of Madras DST PURSE research facility for DLS and AFM analysis.Notes and references
- A. Barone, M. Otero-Losada, A. M. Grangeat, G. Cao, F. Azzato, A. Rodriguez and J. Milei, Int. J. Cardiol., 2016, 223, 258–261 CrossRef CAS
.
- J. Xue, Y. Zhang, Y. Liu and M. Gamal El-Din, Water Res., 2016, 105, 444–455 CrossRef CAS
- L. Bilińska, M. Gmurek and S. Ledakowicz, Process Saf. Environ. Prot., 2017, 109, 420–428 CrossRef
- R. G. Rice, Commercial applications of ozone in food processing, Woodhead Publishing Ltd, 2010, vol. 3 Search PubMed
- M. F. Kamaroddin, J. Hanotu, D. J. Gilmour and W. B. Zimmerman, Algal Res., 2016, 17, 217–226 CrossRef
- D. Nuvolone, D. Petri and F. Voller, Environ. Sci. Pollut. Res., 2018, 25, 8074–8088 CrossRef CAS
- J. E. Goodman, R. L. Prueitt, J. Chandalia and S. N. Sax, J. Appl. Toxicol., 2014, 34, 516–524 CrossRef CAS
- X. He, J. E. Yoo, M. H. Lee and J. Bae, Nanotechnology, 2017, 28, 245402 CrossRef
- S. N. Pusawale, P. R. Deshmukh and C. D. Lokhande, Appl. Surf. Sci., 2011, 257, 9498–9502 CrossRef CAS
- D. Ganguly, D. Pahari, N. S. Das, P. Howli, B. Das, D. Banerjee and K. K. Chattopadhyay, J. Electroanal. Chem., 2016, 778, 12–22 CrossRef CAS
- S. Liu, J. Wu, J. Zhou, G. Fang and S. Liang, Electrochim. Acta, 2015, 176, 1–9 CrossRef CAS
- M. Saranya, R. Ramachandran and F. Wang, J. Sci. Adv. Mater. Devices, 2016, 1, 454–460 CrossRef
- M. Tonezzer, J. H. Kim, J. H. Lee, S. Iannotta and S. S. Kim, Sens. Actuators, B, 2019, 281, 670–678 CrossRef CAS
- M. Hijazi, M. Rieu, V. Stambouli, G. Tournier, J. P. Viricelle and C. Pijolat, Sens. Actuators, B, 2018, 256, 440–447 CrossRef CAS
- G. Korotcenkov, V. Brinzari and B. K. Cho, Sens. Actuators, B, 2017, 243, 507–515 CrossRef CAS
- S. Mills, M. Lim, B. Lee and V. Misra, ECS J. Solid State Sci. Technol., 2015, 4, S3059–S3061 CrossRef CAS
- M. Belaqziz, M. Amjoud, A. Gaddari, B. Rhouta and D. Mezzane, Superlattices Microstruct., 2014, 71, 185–189 CrossRef CAS
- J. Du, S. He, R. Zhao, S. Chen, T. Guo and H. Wang, Mater. Lett., 2017, 186, 318–321 CrossRef CAS
- S. Mohammad-Yousefi, S. Rahbarpour and H. Ghafoorifard, Mater. Chem. Phys., 2019, 227, 148–156 CrossRef CAS
- G. Korotcenkov, V. Brinzari, L. B. Gulina and B. K. Cho, Appl. Surf. Sci., 2015, 353, 793–803 CrossRef CAS
- Y. Fukai, Y. Kondo, S. Mori and E. Suzuki, Electrochem. Commun., 2007, 9, 1439–1443 CrossRef CAS
- F. Chen and M. Liu, Chem. Commun., 1999, 1829–1830 RSC
- N. Hu, Z. Yang, Y. Wang, Z. Liling, Y. Wang, X. Huang, H. Wei, L. Wei and Y. Zhang, Nanotechnology, 2013, 25, 025502 CrossRef
- S. P. Lim, N. M. Huang and H. N. Lim, Ceram. Int., 2013, 39, 6647–6655 CrossRef CAS
- H. Zhang, A. Xie, C. Wang, H. Wang, Y. Shen and X. Tian, ChemPhysChem, 2014, 15, 366–373 CrossRef CAS
- S. Ramesh, H. M. Yadav, Y. J. Lee, G. W. Hong, A. Kathalingam, A. Sivasamy, H. S. Kim, H. S. Kim and J. H. Kim, Sci. Rep., 2019, 9, 1–10 CrossRef
- T. Liu, H. Chai, D. Jia, Y. Su, T. Wang and W. Zhou, Electrochim. Acta, 2015, 180, 998–1006 CrossRef CAS
- Y. Zhang, M. Liu, S. Sun and L. Yang, Adv. Compos. Lett., 2020, 29, 2633366X2090983 CrossRef
- M. Chen, H. Wang, L. Li, Z. Zhang, C. Wang, Y. Liu, W. Wang and J. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 14327–14337 CrossRef CAS
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS
- M. Shanmugam and R. Jayavel, J. Nanosci. Nanotechnol., 2015, 15, 7195–7201 CrossRef CAS
- H. C. Hsu, I. Shown, H. Y. Wei, Y. C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L. C. Chen, Y.-C. Lin and K. H. Chen, Nanoscale, 2012, 262–268 Search PubMed
- J. Jayachandiran, M. Arivanandhan, O. Padmaraj, R. Jayavel and D. Nedumaran, Adv. Compos. Hybrid Mater., 2020, 3, 16–30 CrossRef CAS
- J. Du, R. Zhao, Y. Xie and J. Li, Appl. Surf. Sci., 2015, 346, 256–262 CrossRef CAS
- A. Kar, S. Sain, D. Rossouw, B. R. Knappett, S. K. Pradhan and A. E. H. Wheatley, Nanoscale, 2016, 8, 2727–2739 RSC
- C.-H. Chuang, Y.-F. Wang, Y.-C. Shao, Y.-C. Yeh, D.-Y. Wang, C.-W. Chen, J. W. Chiou, S. C. Ray, W. F. Pong, L. Zhang, J. F. Zhu and J. H. Guo, Sci. Rep., 2014, 4, 4525 CrossRef
- A. Ariharan, B. Viswanathan and V. Nandhakumar, Graphene, 2017, 06, 41–60 CrossRef CAS
- N. A. Kumar, H. Nolan, N. McEvoy, E. Rezvani, R. L. Doyle, M. E. G. Lyons and G. S. Duesberg, J. Mater. Chem. A, 2013, 1, 4431–4435 RSC
- R. Tian, Y. Zhang, Z. Chen, H. Duan, B. Xu, Y. Guo, H. Kang, H. Li and H. Liu, Sci. Rep., 2016, 6, 19195 CrossRef CAS
- J. Liang, C. Yuan, H. Li, K. Fan, Z. Wei, H. Sun and J. Ma, Nano-Micro Lett., 2018, 10, 21 CrossRef
- A. S. Roy, S. Gupta, S. Sindhu, A. Parveen and P. C. Ramamurthy, Composites, Part B, 2013, 47, 314–319 CrossRef CAS
- M. M. Rashad, A. A. Ismail, I. Osama, I. A. Ibrahim and A. H. T. Kandil, Arabian J. Chem., 2014, 7, 71–77 CrossRef CAS
- S. Sagadevan and J. Podder, Mater. Res., 2016, 19, 420–425 CrossRef CAS
- T. A. Dontsova, S. V. Nagirnyak, V. V. Zhorov and Y. V. Yasiievych, Nanoscale Res. Lett., 2017, 12, 332 CrossRef
- M. Shanmugam, A. Alsalme, A. Alghamdi and R. Jayavel, Mater. Express, 2015, 5, 319–326 CrossRef CAS
- V. Agrahari, M. C. Mathpal, M. Kumar and A. Agarwal, J. Alloys Compd., 2015, 622, 48–53 CrossRef CAS
- I. Y. Prosanov and A. A. Matvienko, Phys. Solid State, 2010, 52, 2203–2206 CrossRef CAS
- S. Navazani, A. Shokuhfar, M. Hassanisadi, A. Di Carlo and N. Shahcheraghi, Mater. Sci. Semicond. Process., 2018, 88, 139–147 CrossRef CAS
- P. J. Sephra, P. Baraneedharan, M. Sivakumar, T. D. Thangadurai and K. Nehru, Mater. Res. Bull., 2018, 106, 103–112 CrossRef CAS
- K. H. Lee, Y. W. Lee, S. W. Lee, J. S. Ha, S. S. Lee and J. G. Son, Sci. Rep., 2015, 5, 1–10 Search PubMed
- T. Y. Kim, H. W. Lee, M. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2011, 5, 436–442 CrossRef CAS
- X. Li, X. Li, G. Wang, X. Wang and J. Ji, J. Mater. Chem. A, 2013, 1, 10103–10106 RSC
- A. Choudhari, B. A. Bhanvase, V. K. Saharan, P. H. Salame and Y. Hunge, Ceram. Int., 2020, 46, 11290–11296 CrossRef CAS
- S. R. Eedulakanti, A. K. Gampala, K. Venkateswara Rao, C. Shilpa Chakra, V. Gedela and R. Boddula, Mater. Sci. Energy Technol., 2019, 2, 372–376 Search PubMed
- M. Liu, S. Sun, L. Yang and S. Yin, Int. J. Mater. Res., 2018, 109, 743–750 CrossRef CAS
- R. Hu, J. Zhao and J. Zheng, Mater. Lett., 2017, 197, 59–62 CrossRef CAS
- R. Hu, J. Zhao and J. Zheng, Int. J. Electrochem. Sci., 2020, 15, 6257–6268 Search PubMed
- J. Xiao and S. Yang, J. Mater. Chem., 2012, 22, 12253–12262 RSC
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS
- H. T. Rahal, A. M. Abdel-Gaber and R. Awad, Int. J. Electrochem. Sci., 2017, 12, 10115–10128 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2021 |