
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNiobium- and zirconium-catalyzed reactions of substituted 2 alkynylamines with Et2Zn†
Rita N. Kadikova *,
Ilfir R. Ramazanov,
Azat M. Gabdullin,
Oleg S. Mozgovoj and
Usein M. Dzhemilev
*,
Ilfir R. Ramazanov,
Azat M. Gabdullin,
Oleg S. Mozgovoj and
Usein M. Dzhemilev
Institute of Petrochemistry and Catalysis of Russian Academy of Sciences, 141 Prospekt Oktyabrya, Ufa 450075, Russian Federation. E-mail: kadikritan@gmail.com
First published on 22nd January 2021
Abstract
The NbCl5–EtMgBr-catalyzed reaction of N,N-disubstituted 2-alkynylamines with Et2Zn followed by hydrolysis or deuterolysis affords (2Z)-alkenylamines (reduction products of alkyne) in high yields. The reaction of N,N-disubstituted 2-alkynylamines with Et2Zn catalyzed by the Cp2ZrCl2–EtMgBr system occurs as 2-zincoethylzincation, resulting, after deuterolysis or iodinolysis, in the regio- and stereoselective formation of the corresponding dideuterated and diiodinated 2-alkenylamine derivatives with a trisubstituted double bond. This study demonstrates the difference between the catalytic effects of NbCl5 and Cp2ZrCl2 on the pathway of reaction of tertiary 2-alkynylamines with Et2Zn in the presence of catalytic amounts of EtMgBr.
Introduction
Metal alkyne complexes of Ti,1–10 Zr,11–15 Ta,16–22 and Nb23–30 have a high value for organic synthesis, as they are efficient precursors for the preparation of a broad range of organic compounds. A considerable contribution to the chemistry of zirconocene–alkyne derivatives was made by Buchwald31–33 and Negishi.34,35 It is known that halides of low-valent transition metals such as Mo36,37 and W36 also readily react with acetylene substrates to give low-valent metal complexes of the alkyne molecule. Considering low-valent niobium complexes with acetylene substrates, the first synthetic application of niobium complexes was demonstrated in relation to the reaction of a niobium cyclopropene complex with phthalaldehyde to give 2,3-disubstituted 1-naphthols.38 The chemistry of low-valent niobium complexes with unsaturated substrates is poorly developed; no methods for the preparation of niobium cyclopropene complexes based on functionally substituted acetylene compounds are available from the literature. Studies into the chemistry of low-valent niobium are often coupled with studies of analogous tantalum complexes, which is due to the similarity of their electronic and chemical properties. The first studies of niobium16 and tantalum23,24 complexes with acetylene derivatives were performed by Cotton and co-workers. They demonstrated that reactions of niobium(III) or tantalum(III) compounds with acetylene derivatives such as diphenylacetylene and tert-butylmethylacetylene are accompanied by generation of chloride-bridged bimetallic complexes of the pentavalent metal. From analysis of the published data, it follows that Zn,39 Mg, Al,40–43 and Na/Hg amalgam44–49 are the most popular reducing agents suitable for efficient generation of low-valent niobium and tantalum in the presence of alkyne molecules. Oshima and co-workers reported for the first time that the reaction of non-functionalized disubstituted acetylenes with low-valent niobium, generated by reduction of NbC15 with NaAlH4, results in the selective formation of monodeuterated Z-olefins.50 An interesting approach to generation of highly reactive low-valent niobium was demonstrated in the cyclotrimerization of isocyanates using Nb(OEt)5 and organomagnesium reagents such as i-PrMgCl and EtMgCl.51 However, there are no examples of preparation of low-valent niobium or tantalum by reduction of niobium or tantalum compounds with zinc or aluminum organic derivatives. Currently, we have demonstrated that 2-zincoethylzincation of nitrogen- and phosphorus-containing acetylene derivatives with Et2Zn is accompanied by generation of low-valent titanium diisopropoxide complex.52–54 Meanwhile, it follows from analysis of the literature that reduction of five-coordinate niobium compounds in the presence of acetylene substrates is accompanied by the formation of metal alkyne complexes. We were interested in studying the behavior of NbC15 in the Ti–Mg-catalyzed reaction of functionally substituted alkynes with Et2Zn, which we currently study. We were faced with the following questions: (1) can the replacement of titanium tetraisopropoxide by niobium(V) chloride promote the catalytic carbometallation of alkyne molecules with Et2Zn? (2) Are niobium cyclopropene intermediates, which are hydrolyzed to give 1,2-disubstituted olefins (reduction products of alkyne molecules), generated during the organozinc synthesis? The reduction of acetylene compound with NbC15 in the presence of diethylzinc would attest to generation of low-valent niobium under the action of dialkylzinc and to formation of niobium cyclopropene intermediates. Previously, 1-alkynylamines demonstrated high reactivity in Ti–Mg-catalyzed reaction with Et2Zn.54,55 Therefore, first of all, we studied the reaction of substituted 2-alkynylamines with Et2Zn in the presence of catalytic amounts of NbC15 and EtMgBr.Results and discussion
The reaction of 2-alkynylamines 1a–g with 4 equivalents of Et2Zn (1 M in hexane) in the presence of 30 mol% EtMgBr (1.4 M in diethyl ether) and 15 mol% NbCl5 in diethyl ether at 40 °C for 18 h resulted in the selective formation of reduction products 2a–g, 3b, e in 64–89% yields (Scheme 1). When N,N-dimethyl-3-phenylprop-2-yn-1-amine was used as an alkyne substrate, the reduction product yield after 48 h was 27%. In this case, the amount of the original unreacted propargylamine was 77% (GC/MS data). The reaction of N,N-dimethyl-5-phenylpent-2-yn-1-amine 1f with Et2Zn catalyzed by the NbCl5–EtMgBr system for 18 h resulted in the selective formation of reduction product 2f in 64% yield. The structural identification of the products was carried out by 1D and 2D NMR spectroscopy techniques. The stereochemistry of the resulting allylamine molecules was studied by analyzing coupling of HC-1 and HC-2 ethylene protons (δ ∼ 5.44–5.49 ppm and δ ∼ 5.53–5.58 ppm) and H2C-3 (δ ∼ 2.05–2.07 ppm) and H2C-5 (δ ∼ 2.05–2.07 ppm) methylene protons. The Overhauser effects detected in the NOESY spectra between the H2C-3 (δ ∼ 2.05–2.07 ppm) and H2C-5 (δ ∼ 2.94–2.96 ppm) methylene protons attest to the Z-configuration of the double bond of compound 2b (Scheme 1). The positions of deuterium atoms in the dideuterated alkenylamines were also established on the basis of NMR spectra. In the 13C NMR spectra of the compounds 3b, e, no signals were presented for the sp2-hybridized carbon atoms that is typical of deuterated alkenes. Also, a signal of double bond hydrogen atom is absent in the 1H NMR spectra of the compounds 3b, e. Thus, replacement of titanium tetraisopropoxide in the 2-zincoethylzincation of 2-alkynylamines, which we reported previously,54 by NbCl5 changes the reaction pathway and leads to generation of organometallic cyclopropene intermediates instead of metallacyclopentenes.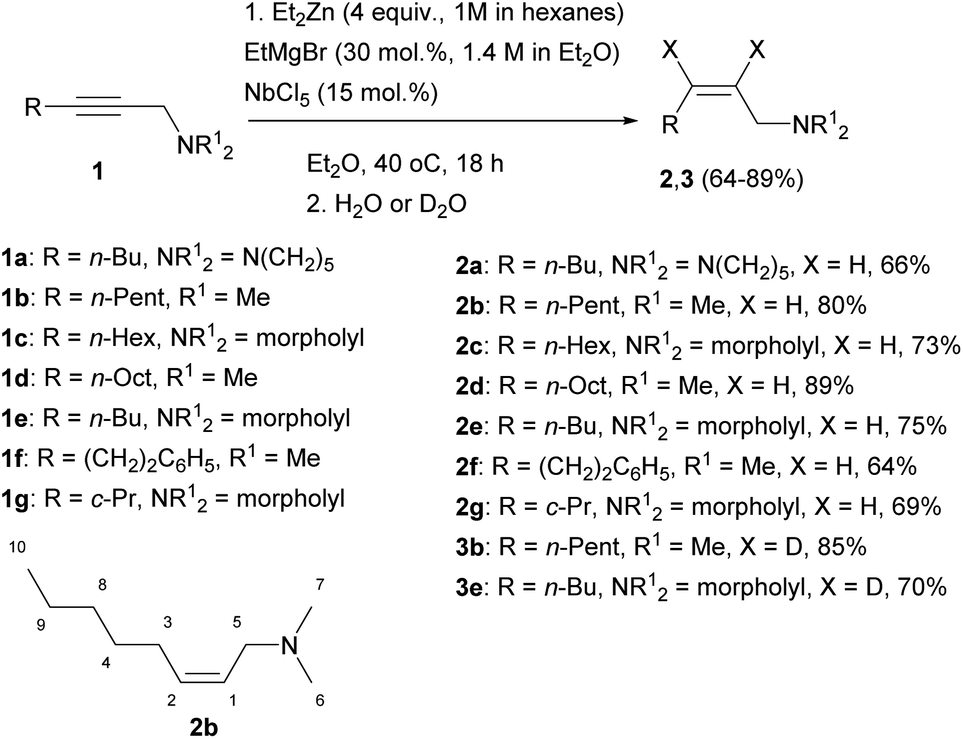 | ||
| Scheme 1 Nb–Mg-catalyzed reaction of N,N-disubstituted 2-alkynylamines with Et2Zn. | ||
Gas chromatography and gas chromatography/mass spectrometry analysis of the reaction mixture demonstrated that the reduction of the substituted propargylamines in the organozinc synthesis is accompanied by side formation of compounds that can be described as carbozincation products (9–15% yield) and products of 2-alkynylamine homocoupling catalyzed by low-valent niobium (2–5% yield).
In our opinion, allylamines 2 and 3 with Z-configuration of the double bond are formed in the following way. Previously, Negishi,55 who studied the Ti–Mg-catalyzed carbocyclization of non-functionalized and oxygenated enynes with Et2Zn, suggested that diisopropoxytitanium ethylene complex or diisopropoxytitanacyclopropane is the key intermediate of this reaction. The EtMgBr–Ti(OiPr)4-catalyzed 2-zincoethylzincation of 2-alkynylamines with Et2Zn is also presumably initiated by the generation of diisopropoxytitanacyclopropane intermediate.54 Urabe and Sato et al.10,56 previously reported that the use of Ti(OiPr)4 in conjunction with i-PrMgCl led to the formation of the corresponding low-valent alkoxytitanium species. Relying on these results, we assumed that the exchange reaction between NbCl5 and EtMgBr leads to generation of diethylniobium complex Cl3NbEt2 (Scheme 2). Upon disproportionation, this unstable complex is rapidly converted to niobium ethylene complex or niobacyclopropane intermediate A. According to literature,57–59 we assumed intermediate formation the structure A. For example, TaCl5-catalyzed carbomagnesiation of alkenes with n-alkyl Grignard reagents is initiated by the generation of alkene complex of tantalum(III) chloride. It was proposed that this complex is formed from the dialkyltantalum complex [TaCl3R2] (R is an alkyl group) as a result of beta-hydride transfer and elimination of alkane. Since niobium and tantalum are metals of close nature, we assumed a similar route for the formation of niobacyclopropane intermediate A. Subsequently, propargylamine, being a stronger nucleophile, displaces ethylene from the niobium coordination sphere to give niobacyclopropene complex B. The generated niobacyclopropene intermediate undergoes transmetallation under the action of Et2Zn to give zincacyclopropene intermediate C, the deuterolysis of which affords dideuterated allylamine D. The formation of low-valent niobium under the action of magnesium alkyl halides is additionally supported by the cyclotrimerization reaction of isocyanates induced by low-valent niobium generated upon the reaction of Nb(OEt)5 with Grignard reagents such as i-PrMgCl or EtMgCl.51
 | ||
| Scheme 2 Putative mechanism of Nb–Mg-catalyzed reaction of N,N-disubstituted 2-alkynylamines with Et2Zn. | ||
The use of EtMgBr in this reaction is important. We found that, without EtMgBr, the reaction of N,N-dimethyloct-2-yn-1-amine with 4 equivalents of Et2Zn carried out in the presence of 15 mol% NbCl5 in diethyl ether for 18 h at 40 °C affords a mixture of reduction, 2-zincoethylzincation, and dimerization products in 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:10 ratio. This indicates that low-valent niobium is also generated in the reaction of NbCl5 with Et2Zn. However, in the absence of EtMgBr, the selectivity of reduction of 2-alkynylamines decreases. Since it was ascertained that NbCl5 reacts with Et2Zn giving intermediates that show reactivity towards 2-alkynylamines, it was of interest to study this reaction using stoichiometric amounts of NbCl5 and Et2Zn with respect to functionally substituted alkyne. In the reaction of N,N-dimethyloct-2-yn-1-amine with 2 equivalents of NbCl5 and 3 equivalents of Et2Zn in the absence of EtMgBr, the proportion of the carbozincation product increased to 60%. We believe that the selectivity observed in the reduction of 2-alkynylamines in the presence of catalytic amounts of EtMgBr may be attributable to different compositions of low-valent niobium complexes formed upon reduction of NbCl5 with EtMgBr and Et2Zn. Meanwhile, the use of stoichiometric amount of EtMgBr without Et2Zn also produces unsatisfactory results. Indeed, the reaction of N,N-dimethylundec-2-yn-1-amine with 4 equivalents of EtMgBr in the presence of 15 mol% NbCl5 in diethyl ether at 40 °C is accompanied by complete conversion to give, after 2 days, a mixture of reduction and carbometallation products in 1:1 ratio. Thus, the optimal conditions for the reduction of 2-alkynylamines to Z-allylamines via organozinc synthesis imply the presence of catalytic amounts of NbCl5 and EtMgBr.
:40:10 ratio. This indicates that low-valent niobium is also generated in the reaction of NbCl5 with Et2Zn. However, in the absence of EtMgBr, the selectivity of reduction of 2-alkynylamines decreases. Since it was ascertained that NbCl5 reacts with Et2Zn giving intermediates that show reactivity towards 2-alkynylamines, it was of interest to study this reaction using stoichiometric amounts of NbCl5 and Et2Zn with respect to functionally substituted alkyne. In the reaction of N,N-dimethyloct-2-yn-1-amine with 2 equivalents of NbCl5 and 3 equivalents of Et2Zn in the absence of EtMgBr, the proportion of the carbozincation product increased to 60%. We believe that the selectivity observed in the reduction of 2-alkynylamines in the presence of catalytic amounts of EtMgBr may be attributable to different compositions of low-valent niobium complexes formed upon reduction of NbCl5 with EtMgBr and Et2Zn. Meanwhile, the use of stoichiometric amount of EtMgBr without Et2Zn also produces unsatisfactory results. Indeed, the reaction of N,N-dimethylundec-2-yn-1-amine with 4 equivalents of EtMgBr in the presence of 15 mol% NbCl5 in diethyl ether at 40 °C is accompanied by complete conversion to give, after 2 days, a mixture of reduction and carbometallation products in 1:1 ratio. Thus, the optimal conditions for the reduction of 2-alkynylamines to Z-allylamines via organozinc synthesis imply the presence of catalytic amounts of NbCl5 and EtMgBr.
In order to study the effect of various transition metals on the reduction of 2-alkynylamines via organozinc synthesis, we carried out a number of experiments with various metals. It was found that the replacement of NbCl5 in the Nb–Mg-catalyzed reaction of 2-alkynylamines with Et2Zn (entry 1, Table 1) by ZrCl4 (entry 2, Table 1) inhibited the conversion of N,N-dimethyloct-2-yn-1-amine (Table 1). According to GLC analysis of the reaction mixture, no reduction product was formed in this case. The low conversion of N,N-dimethyloct-2-yn-1-amine is associated with the formation of carbometallation product (according to GC/MS data) in minor amounts (2%) (entry 2). In the case of using TaCl5, apart from the trace amount of compound 2b (5%), the reaction gave a hard-to-separate mixture of macromolecular olefins (25%) (entry 3). When NbCl5 was replaced by TiCl4, the reaction was no longer chemoselective, and the conversion of the starting propargylamine after 24 h was 27%. The reduction product 2b was formed in 13% yield, while the contents of compounds that can be described as carbometallation and dimerization products were 8% and 4%, respectively (GC/MS data) (entry 4). To our surprise, when Cp2ZrCl2 was used as the catalyst, the reaction pathway crucially changed (entry 5).
We found that the reaction of 2-alkynylamines 1 with 2.5 equivalents of Et2Zn (1 M in hexane) in the presence of 10 mol% Cp2ZrCl2 and 20 mol% EtMgBr (2.5 M in Et2O) carried out in diethyl ether at room temperature for 18 hours and followed by deuterolysis, hydrolysis, or iodinolysis furnished substituted allylamines 5, 6, or 7 with Z-configuration of the double bond (Scheme 3). The reactions were regio- and stereoselective. The structures of the substituted 2-alkenylamines were established using 1D and 2D NMR spectroscopy of the products of their hydrolysis 5a–d, deuterolysis 6d, and iodinolysis 7b. We believe that in this case, the reaction follows the 2-zincoethylzincation pathway due to similar natures of zirconium and titanium atoms. As noted above, the use of titanium tetraisopropoxide as a catalyst in the reaction of substituted alkynes with Et2Zn also results in the formation of 2-zincoethylzincation products.54 Thus, the nature of the transition metal of the organometallic catalyst affects the conversion pathway of acetylene substrates in the reaction with Et2Zn.
 | ||
| Scheme 3 Zr–Mg-catalyzed reaction of N,N-disubstituted 2-alkynylamines with Et2Zn. | ||
It is noteworthy that no examples of Zr-catalyzed 2-zincoethylzincation of functionally substituted alkynes were reported in the literature. At the same time, it should be noted that addition of alkynylboronates to the reagent Cp2ZrCl2/2EtMgBr leads to selective formation of the zirconacyclopentenes which, upon hydrolysis, afforded (Z)-2-(2-ethylhex-1-enyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolanes in high yield.60 In this study, we demonstrated for the first time that the Zr-catalyzed reaction of 2-alkynylamines with Et2Zn leads to regio- and stereoselective formation of 2-zincoethylzincation products. Regarding carbozincation of non-functionalized alkynes during the organozinc synthesis, the only relevant example reported in the literature61 is Cp2ZrCl2-catalyzed 2-zincoethylzincation of dec-5-yne, which selectively gives the dideuterated product. Meanwhile, the conversion of dec-5-yne under conditions we developed in the presence of 4 equivalents of Et2Zn (1 M in hexane), 30 mol% EtMgBr (1.4 M in diethyl ether), and 15 mol% NbCl5 in diethyl ether is not selective and gives, together with the reduction product (10–15%), a hard-to-analyze product mixture, apparently, composed of oligo- and polymerization products of dialkyl-substituted acetylene induced by low-valent niobium complexes. Thus, we showed for the first time that the reaction of 2-alkynylamines with Et2Zn in the presence of catalytic amounts of EtMgBr and NbCl5 results in stereoselective conversion of 2-alkynylamines to (2Z)-alkenylamines. A regio- and stereoselective method was developed for the synthesis of 2-alkenylamines with a trisubstituted double bond by the reaction of 2-alkynylamines with Et2Zn catalyzed by the Cp2ZrCl2–EtMgBr system.
Conclusions
Thus, the NbCl5–EtMgBr-catalyzed reaction of N,N-disubstituted 2-alkynylamines with Et2Zn followed by hydrolysis or deuterolysis affords (2Z)-alkenylamines in high yields. On other hand, the reaction of N,N-disubstituted 2-alkynylamines with Et2Zn catalyzed by the Cp2ZrCl2–EtMgBr system occurs as 2-zincoethylzincation, resulting, after deuterolysis or iodinolysis, in the regio- and stereoselective formation of the corresponding dideuterated and diiodinated 2-alkenylamine derivatives with a trisubstituted double bond. The study demonstrates that the pathway of the reaction of tertiary 2-alkynylamines with Et2Zn depends on transition metal in the catalytic system.Experimental section
General information
The reagents were obtained from Sigma-Aldrich or Acros. Hexane were distilled over P2O5. Diethyl ether, benzene and 1,2-dimethoxyethane were dried over sodium. 2-Alkynylamines 1b, d, f were prepared by aminomethylation of terminal alkynes by bisamine.62 Alkynylamines 1a, e, c, g were prepared by aminomethylation of terminal alkynes with aqueous formaldehyde and secondary amines under CuI catalysis.63 Nuclear magnetic resonance spectroscopy was performed on a Brucker Avance 500. The 1H NMR spectra were recorded at 500 MHz and 13C-{1H} NMR spectra at 100 MHz in CDCl3. The chemical shifts are reported in ppm relative to tetramethylsilane (TMS) as the internal standard. The numbering of atoms in the 13C-{1H} and 1H NMR spectra of the compounds 2a–g, 3b, e, 5a–d, 6d, 7b is shown in Fig. 1 and 2. Elemental analysis was performed using a Carlo-Erba CHN 1106 elemental analyser. Mass spectra were obtained on a Finnigan 4021 instrument. The yields were calculated from the isolated amount of allylamines obtained from starting 2-alkynylamines. | ||
| Fig. 1 The numbering of atoms in the 13C- and 1H-NMR spectra of the compounds 2a–g, 3b, e. | ||
 | ||
| Fig. 2 The numbering of atoms in the 13C- and 1H-NMR spectra of the compounds 5a–d, 6d, 7b. | ||
Preparation of allylamines 2a–g, 3e, b via Nb–Mg-catalyzed reaction of substituted propargylamines with Et2Zn.
(Z)-N,N-Dimethyloct-2-en-1-amine; typical procedure
To a solution of N,N-dimethyloct-2-yn-1-amine (306 mg, 2 mmol) and Et2Zn (1 M in hexanes, 8 mL, 8 mmol) in Et2O (6 mL) was added NbCl5 (0.081 g, 0.30 mmol). Ethylmagnesiurn bromide (1.4 M in Et2O, 0.428 mL, 0.6 mmol) was then added and the reaction mixture rapidly turned black. After 18 h at 40 °C, the reaction mixture was diluted with Et2O (5 mL), and 25 wt% KOH solution (3 mL) was added dropwise while the reaction flask was cooled in an ice bath. The aqueous layer was extracted with diethyl ether (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous MgSO4. The reaction mixture was filtered through a filter paper and concentrated in vacuo to give crude product as a yellow oil. The residue was distilled through a micro column at 20 mmHg to give 2b (248 mg, 80%) as a colourless oil. b.p. 77–79 °C (20 mmHg).1H NMR (500 MHz, CDCl3): δ = 0.91 (s, 3H, C(10)H3), 1.31 (s, 4H, C(9,8)H2), 1.36–1.41 (m, 2H, C(4)H2), 2.05–2.09 (m, 2H, C(3)H2), 2.25 (s, 6H, C(6,7)H3), 2.95 (d, J = 6 Hz, 2H, C(5)H2), 5.44–5.49 (m, 1H, C(1)H), 5.53–5.61 (m, 1H, C(2)H).
13C NMR (500 MHz, CDCl3): δ = 14.04 (C(10)), 22.54 (C(9)), 27.43 (C(3)), 29.24 (C(4)), 31.48 (C(8)), 45.22 (C(6,7)), 56.13 (C(5)), 126.59 (C(1)), 132.97 (C(2)).
MS (EI): m/z, % = 155 (18) [M+], 98 (29), 84 (53), 58 (89), 45 (100).
Anal. calcd for C10H21N, (%): C, 77.35; H, 13.63; N, 9.02. Found, %: C, 77.58; H, 13.58; N, 8.71.
1H NMR (500 MHz, CDCl3): δ = 0.88 (t, J = 7 Hz, 3H, C(11)H3), 1.31 (s, 4H, C(4,10)H2), 1.41 (m, 2H, C(12)H2), 1.55–1.59 (m, 4H, C(8,9)H2), 2.03 (q, J = 6 Hz, 2H, C(3)H2), 2.36 (s, 4H, C(6,7)H2), 2.95 (d, J = 6 Hz, 2H, C(5)H2), 5.43–5.53 (m, 1H, C(1,2)H).
13C NMR (500 MHz, CDCl3): δ = 13.93 (C(11)), 22.68 (C(10)), 24.37 (C(12)), 25.99 (C(8,9)), 27.16 (C(3)), 31.73 (C(4)), 54.51 (C(6,7)), 55.88 (C(5)), 126.43 (C(1)), 132.71 (C(2)).
MS (EI): m/z, % = 181 (7) [M+], 138 (4), 124 (10), 98 (29), 84 (100), 55 (30), 41 (15).
Anal. calcd for C12H23N, (%): C, 79.49; H, 12.79; N, 7.72; found, %: C, 79.45; H, 12.92; N, 7.52.
1H NMR (500 MHz, CDCl3): δ = 0.83 (t, J = 6 Hz, 3H, C(13)H3), 1.22–1.25 (m, 6H, C(10–12)H2), 1.26–1.30 (m, 2H, C(4)H2), 1.98–2.02 (q, J = 7 Hz, 2H, C(3)H2), 2.39 (s, 4H, C(6,7)H2), 2.94 (d, J = 7 Hz, 2H, C(5)H2), 3.64–3.66 (m, 4H, C(8,9)H2), 5.36–5.40 (m, 1H, C(1)H), 5.49–5.53 (m, 1H, C(2)H).
13C NMR (500 MHz, CDCl3): δ = 13.99 (C(13)), 22.55 (C(12)), 27.45 (C(3)), 28.87 (C(10)), 29.43 (C(4)), 31.65 (C(11)), 55.43 (C(5)), 53.59 (C(6,7)), 66.94 (C(8,9)), 125.33 (C(1)), 133.67 (C(2)).
MS (EI): m/z, % = 211 (3) [M+], 126 (5), 87 (100), 86 (40), 57 (30), 40 (15).
Anal. calcd for C13H25NO, (%): C, 73.88; H, 11.92; N, 6.63; found, %: C, 74.03; H, 12.08; N, 6.77.
1H NMR (500 MHz, CDCl3): δ = 0.90 (t, J = 7 Hz, 3H, C(13)H3), 1.29 (s, 8H, C(9–12)H2), 1.35–1.38 (m, 4H, C(4,8)H2), 2.04–2.09 (m, 2H, C(3)H2), 2.25 (s, 6H, C(6,7)H3), 2.95 (d, J = 6 Hz, 2H, C(5)H2), 5.44–5.49 (m, 1H, C(1)H), 5.52–5.57 (m, 1H, C(2)H).
13C NMR (500 MHz, CDCl3): δ = 14.11 (C(13)), 22.68 (C(12)), 27.47 (C(3)), 29.29 (C(9,10)), 29.49 (C(8)), 29.57 (C(4)), 31.89 (C(11)), 45.26 (C(6,7)), 56.16 (C(5)), 126.66 (C(1)), 132.93 (C(2)).
MS (EI): m/z, % = 197 (9) [M+], 110 (4), 98 (24), 84 (52), 58 (89), 45 (100).
Anal. calcd for C13H27N, (%): C, 79.11; H, 13.79; N, 7.10; found, %: C, 79.16; H, 13.65; N, 6.95.
1H NMR (500 MHz, CDCl3): δ = 0.89 (s, 3H, C(11)H3), 1.21–1.25 (m, 4H, C(4,10)H2), 2.04–2.07 (m, 2H, C(3)H2), 2.45 (s, 4H, C(6,7)H2), 3.01 (d, J = 6 Hz, 2H, C(5)H2), 3.72 (s, 4H, C(8,9)H2), 5.42–5.46 (m, 1H, C(1)H), 5.55–5.59 (m, 1H, C(2)H).
13C NMR (500 MHz, CDCl3): δ = 13.94 (C(11)), 22.29 (C(10)), 27.20 (C(3)), 31.68 (C(4)), 53.46 (C(5)), 53.59 (C(6,7)), 66.98 (C(8,9)), 125.26 (C(1)), 133.79 (C(2)).
MS (EI): m/z, % = 183 (10) [M+], 140 (4), 110 (28), 87 (100), 57 (70), 41 (21).
Anal. calcd for C11H21NO, (%): C, 72.08; H, 11.55; N, 7.64; found, %: C, 72.22; H, 11.56; N, 7.37.
1H NMR (500 MHz, CDCl3): δ = 2.24 (s, 6H, C(6,7)H3), 2.43 (q, J = 7 Hz, 2H, C(3)H2), 2.71 (t, J = 7 Hz, 2H, C(4)), 2.94 (s, 2H, C(5)H2), 5.51–5.55 (m, 1H, C(1)H), 5.59–5.64 (m, 1H, C(2)H), 7.30 (t, J = 7 Hz, 2H, C(9)H), 7.21 (d, J = 7 Hz, 3H, C(10,11)H).
13C NMR (500 MHz, CDCl3): δ = 29.46 (C(3)), 35.80 (C(4)), 45.01 (C(6,7)), 55.93 (C(5)), 125.89 (C(11)), 127.38 (C(1)), 128.33 (C(9)), 128.49 (C(10)), 131.73 (C(2)), 141.74 (C(8)).
MS (EI): m/z, % = 189 (16) [M+], 144 (11), 143 (11), 129 (59), 98 (45), 91 (64), 58 (100), 45 (90).
Anal. calcd for C13H19N, (%): C, 82.48; H, 10.12; N, 7.40; found, %: C, 82.44; H, 9.97; N, 7.27.
1H NMR (500 MHz, CDCl3): δ = 0.26–0.28 (m, 2H(A), C(4,10)H2), 0.68–0.71 (m, 2H(B), C(4,10)H2), 1.26–1.30 (m, 2H, C(4)H2), 1.49–1.57 (m, 1H, C(3)H), 2.44 (s, 4H, C(6,7)H2), 3.08 (d, J = 7 Hz, 2H, C(5)H2), 3.66–3.67 (m, 4H, C(8,9)H2), 5.29–5.34 (m, 1H, C(1)H), 4.87 (t, J = 10 Hz, 1H, C(2)H).
13C NMR (500 MHz, CDCl3): δ = 6.46 (C(4,10)), 9.76 (C(3)), 53.61 (C(6,7)), 55.84 (C(5)), 66.98 (C(8,9)), 123.39 (C(1)), 137.76 (C(2)).
MS (EI): m/z, % = 167 (10) [M+], 138 (33), 87 (70), 79 (87), 56 (69), 40 (100).
Anal. calcd for C10H17NO, (%): C, 71.81; H, 10.25; N, 8.37; found, %: C, 71.98; H, 10.35; N, 8.35.
1H NMR (500 MHz, CDCl3): δ = 0.90 (s, 3H, C(10)H3), 1.27–1.30 (m, 4H, C(8,9)H2), 1.35–1.39 (m, 2H, C(4)H2), 2.03–2.07 (m, 2H, C(3)H2), 2.24 (s, 6H, C(6,7)H3), 2.94 (d, J = 6 Hz, 2H, C(5)H2).
13C NMR (500 MHz, CDCl3): δ = 14.04 (C(10)), 22.54 (C(9)), 27.28–27.42 (C(3)), 29.23 (C(4)), 31.48 (C(8)), 45.25 (C(6,7)), 56.09 (d, J = 11 Hz, C(5)), 126.57 (d, J = 17 Hz, C(1)), 132.85 (d, J = 15 Hz, C(2)).
MS (EI): m/z, % = 157 (26) [M+], 100 (21), 86 (36).
Anal. calcd for C10H19D2N, (%): C, 76.36; N, 8.90; found, %: C, 76.39; N, 9.02.
1H NMR (500 MHz, CDCl3): δ = 0.91 (t, J = 6 Hz, 3H, C(11)H3), 1.23–1.28 (m, 4H, C(4,10)H2), 2.07 (t, J = 6 Hz, 2H, C(3)H2), 2.47 (s, 4H, C(6,7)H2), 3.03 (s, 2H, C(5)H2), 3.73 (s, 4H, C(8,9)H2).
13C NMR (500 MHz, CDCl3): δ = 13.96 (C(11)), 22.32 (C(10)), 27.07 (C(3)), 31.68 (C(4)), 53.59 (C(6,7)), 55.34 (C(5)), 66.99 (C(8,9)).
MS (EI): m/z, % = 185 (7) [M+], 156 (1), 128 (6), 112 (19), 87 (100), 57 (70), 57 (70), 42 (13).
Anal. calcd for C11H19D2NO, (%): C, 71.30; N, 7.56; found, %: C, 71.46; N, 7.42.
Preparation of allylamines 5a–d, 6d, 7b via Zr–Mg-catalyzed reaction of substituted propargylamines with Et2Zn.
(Z)-3-Ethyl-N,N-dimethylundec-2-en-1-amine; typical procedure
To a solution of N,N-dimethylundec-2-yn-1-amine (390 mg, 2 mmol) and Et2Zn (1 M in hexanes, 5 mL, 5 mmol) in Et2O (6 mL) was added Cp2ZrCl2 (0.058 g, 0.20 mmol). Ethylmagnesiurn bromide (1.6 M in Et2O, 0.25 mL, 0.4 mmol) was then added and the reaction mixture rapidly turned black. After 18 h at r.t. °C, the reaction mixture was diluted with Et2O (5 mL), and 25 wt% KOH solution (3 mL) was added dropwise while the reaction flask was cooled in an ice bath. The aqueous layer was extracted with diethyl ether (3 × 10 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous MgSO4. The reaction mixture was filtered through a filter paper and concentrated in vacuo to give crude product as a yellow oil. The residue was distilled through a micro column at 1 mmHg to give 5d (401 mg, 89%) as a colourless oil. b.p. 104–107 °C (1 mmHg).1H NMR (500 MHz, CDCl3): δ = 0.89 (t, J = 6 Hz, 3H, C(15)H3), 1.01 (t, J = 7 Hz, 3H, C(4)H3), 1.29 (s, 8H, C(10–13)H2), 1.32–1.38 (m, 4H, C(9,14)H2), 2.03–2.06 (m, 4H, C(3,8)H2), 2.23 (s, 6H, C(6,7)H3), 2.91 (d, J = 6 Hz, 2H, C(5)H2), 5.22 (t, J = 6 Hz, 1H, C(1)H).
13C NMR (500 MHz, CDCl3): δ = 12.74 (C(4)), 14.10 (C(15)), 22.07 (C(14)), 28.49 (C(9)), 29.29 (C(10)), 29.52 (C(12)), 29.57 (C(11)), 29.79 (C(3)), 30.58 (C(8)), 31.89 (C(13)), 45.26 (C(6,7)), 56.86 (C(5)), 120.48 (C(1)), 144.41 (C(2)).
MS (EI): m/z, % = 225 (32) [M+], 210 (15), 196 (17), 180 (14), 151 (19), 112 (47), 95 (100), 82 (81), 67 (74), 58 (79), 46 (96).
Anal. calcd for C15H31N, (%): C, 79.92; H, 13.86; N, 6.21. Found, %: C, 79.80; H, 13.82; N, 6.01.
1H NMR (500 MHz, CDCl3): δ = 0.91 (t, J = 6 Hz, 3H, C(13)H3), 1.03 (t, J = 8 Hz, 3H, C(4)H3), 1.28–1.31 (m, 6H, C(10–12)H2), 1.33–1.39 (m, 2H, C(9)H2), 2.03–2.07 (m, 4H, C(3,8)H2), 2.25 (s, 6H, C(6,7)H3), 2.93 (d, J = 6 Hz, 2H, C(5)H2), 5.23 (t, J = 7 Hz, 1H, C(1)H).
13C NMR (500 MHz, CDCl3): δ = 12.75 (C(4)), 14.09 (C(13)), 22.65 (C(12)), 28.47 (C(9)), 29.59 (C(10)), 29.70 (C(3)), 31.79 (C(11)), 45.24 (C(6,7)), 56.84 (C(5)), 120.41 (C(1)), 144.51 (C(2)).
MS (EI): m/z, % = 197 (32) [M+], 182 (17), 168 (20), 152 (22), 123 (55), 112 (49), 95 (82), 82 (93), 67 (74), 58 (88), 46 (100).
Anal. calcd for C13H27N, (%): C, 79.11; H, 13.79; N, 7.10. Found, %: C, 79.10; H, 13.74; N, 6.89.
1H NMR (500 MHz, CDCl3): δ = 0.90 (t, J = 6 Hz, 3H, C(15)H3), 1.02 (qv, J = 7 Hz, 2H, C(4)H2D), 1.29 (s, 8H, C(10–13)H2), 1.31–1.35 (m, 4H, C(9,14)H2), 2.06–2.11 (m, 4H, C(3, 8)H2), 2.19 (s, 6H, C(6,7)H3), 2.87 (s, 2H, C(5)H2).
13C NMR (500 MHz, CDCl3): δ = 12.57 (t, J = 19 Hz, C(4)), 14.12 (C(15)), 22.69 (C(14)), 27.45 (C(8)), 28.28 (C(3)), 29.30 (C(10)), 29.37 (C(12)), 29.54 (C(11)), 30.03 (C(9)), 31.91 (C(13)), 45.47 (C(6,7)), 58.31 (C(5)).
MS (EI): m/z, % = 227 (12) [M+], 212 (20), 210 (11), 198 (23).
Anal. calcd for C15H29D2N, (%): C, 79.22; N, 6.16. Found, %: C, 79.36; N, 6.12.
:1) gave a yellow oil; yield: 487 mg, (56%); Rf = 0.68 (hexane/ethyl acetate, 5:1). The spectral properties (1H NMR, 13C NMR, MS) were in good agreement with those that were reported in the literature.54 Anal. calcd for C13H25I2N, (%): C, 33.12; H, 5.33; N, 3.22. Found, %: C, 32.91; H, 5.30; N, 3.21.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Russian Science Foundation (grant No. 19-73-10113).References
- R. D. Broene and S. L. Buchwald, Science, 1993, 261, 1696–1701 CrossRef CAS.
- T. Kawaji, N. Shohji, K. Miyashita and S. Okamoto, Chem. Commun., 2011, 47, 7857–7859 RSC.
- U. Rosenthal, V. V. Burlaakov, M. A. Bach and T. Beweries, Chem. Soc. Rev., 2007, 36, 719–728 RSC.
- K. Fukuhara, S. Okamoto and F. Sato, Org. Lett., 2003, 5, 2145–2148 CrossRef CAS.
- I. L. Lysenko, K. Kim, H. G. Lee and J. K. Cha, J. Am. Chem. Soc., 2008, 130, 15997–16002 CrossRef CAS.
- M. A. Tarselli and G. C. Micalizio, Org. Lett., 2009, 11, 4596–4599 CrossRef CAS.
- T. Hanamoto and K. Yamada, J. Organomet. Chem., 2009, 74, 7559–7561 CAS.
- G. J. Balaich and I. P. Rothwell, J. Am. Chem. Soc., 1993, 115, 1581–1583 CrossRef CAS.
- E. S. Johnson, G. J. Balaich and I. P. Rothwell, J. Am. Chem. Soc., 1997, 7685–7693 CrossRef CAS.
- K. Harada, H. Urabe and F. Sato, Tetrahedron Lett., 1995, 36, 3203–3206 CrossRef CAS.
- T. Takahashi, F.-Y. Tsai, Y. Li, K. Nakajima and M. Kotora, J. Am. Chem. Soc., 1999, 121, 11093–11100 CrossRef CAS.
- K.-i. Kanno, E. Igarashi, L. Zhou, K. Nakajima and T. Takahashi, J. Am. Chem. Soc., 2008, 130, 5624–5625 CrossRef CAS.
- T. D. Anthopoulos, F. B. Kooistra, H. J. Wondergem, D. Kronholm, J. C. Hummelen and D. M. de Leeuw, Eur. J. Org. Chem., 2009, 1679–1684 Search PubMed.
- Y. Nishihara, M. Miyasaka, M. Okamoto, H. Takahashi, E. Inoue, K. Tanemura and K. Takagi, J. Am. Chem. Soc., 2007, 129, 12634–12635 CrossRef CAS.
- A. Ohff, S. Pulst, N. Peulecke, P. Arndt, V. V. Burlakov and U. Rosenthal, Synlett, 1996, 111–118 CrossRef CAS.
- F. A. Cotton and W. J. Roth, Inorg. Chim. Acta, 1984, 85, 17–21 CrossRef.
- H. Shimizu and S. Kobayashi, Tetrahedron Lett., 2005, 46, 7593–7595 CrossRef CAS.
- T. Oshiki, K. Tanaka, J. Yamada, T. Ishiyama, Y. Kataoka, K. Mashima, K. Tani and K. Takai, Organometallics, 2003, 22, 464–472 CrossRef CAS.
- K. Takai, M. Yamada and K. Utimoto, Chem. Lett., 1995, 851–852 CrossRef CAS.
- I. Shibata, T. Kano, N. Kanazawa, S. Fukuoka and A. Baba, Angew. Chem., Int. Ed., 2002, 41, 1389–1392 CrossRef CAS.
- W. W. Brennessel, J. E. Ellis, M. K. Pomije, V. J. Sussman, E. Urnezius and V. G. Young, J. Am. Chem. Soc., 2002, 124, 10258–10259 CrossRef CAS.
- T. Oshiki, H. Nomoto, K. Tanaka and K. Takai, Bull. Chem. Soc. Jpn., 2004, 77, 1009–1011 CrossRef CAS.
- F. A. Cotton and W. T. Hall, Inorg. Chem., 1981, 20, 1285–1287 CrossRef CAS.
- F. A. Cotton and W. T. Hall, Inorg. Chem., 1980, 19, 2352–2354 CrossRef CAS.
- E. J. Roskamp and S. F. Pedersen, J. Am. Chem. Soc., 1987, 109, 6551–6553 CrossRef CAS.
- E. J. Roskamp, P. S. Dragovich, J. B. Hartung and S. F. Pederson, J. Org. Chem., 1989, 54, 4736–4737 CrossRef CAS.
- Y. Kataoka, J. Miyai, K. Oshima, K. Takai and K. Utimoto, J. Org. Chem., 1992, 57, 1973–1981 CrossRef CAS.
- K. Fuchibe and T. Akiyama, J. Am. Chem. Soc., 2006, 128, 1434–1435 CrossRef CAS.
- S. Arai, S. Takita and A. Nishida, Eur. J. Org. Chem., 2005, 5262–5267 CrossRef CAS.
- Y. Kataoka, K. Takai, K. Oshima and K. Utimoto, Tetrahedron Lett., 1990, 31, 365–368 CrossRef CAS.
- S. L. Buchwald, R. T. Lum and J. C. Dewan, J. Am. Chem. Soc., 1986, 108, 7441–7442 CrossRef CAS.
- S. L. Buehwald, B. T. Watson and J. C. Huffman, J. Am. Chem. Soc., 1987, 109, 2544–2546 CrossRef.
- S. L. Buchwald and R. B. Nielsen, Chem. Rev., 1988, 88, 1047–1058 CrossRef CAS.
- T. Takahashi, D. Swanson and E. Negishi, Chem. Lett., 1987, 623–626 CrossRef CAS.
- E. Negishi, S. J. Holmes, J. M. Tour, J. A. Miller, F. E. Cederbaum, D. R. Swanson and T. Takahashi, J. Am. Chem. Soc., 1989, 111, 3336–3346 CrossRef CAS.
- E. C. Walborsky, D. E. Wigley, E. Roland, J. C. Dewan and R. R. Schrock, Inorg. Chem., 1987, 26, 1615–1621 CrossRef CAS.
- A. Greco, F. Pirinoli and G. Dall'asta, J. Organomet. Chem., 1973, 60, 115–124 CrossRef CAS.
- J. B. Hartung Jr and S. F. Pedersen, J. Am. Chem. Soc., 1989, 111, 5468–5469 CrossRef.
- M. Fieser and L. F. Fieser, Reagents for Organic Synthesis, Wiley, New York, 1967, vol. 1, p. 1276 Search PubMed.
- M. D. Curtis and R. Real, J. Organometallics, 1985, 4, 940–942 CrossRef CAS.
- M. D. Curtis, J. Real and D. Kwon, Organometallics, 1989, 8, 1644–1651 CrossRef CAS.
- D. Kwon and M. D. Curtis, Organometallics, 1990, 9, 1–5 CrossRef CAS.
- A. Galindo, M. Gomez, P. Gomez-Sal, A. Martin, R. D. Del and F. Sanchez, Organometallics, 2002, 21, 293–304 CrossRef CAS.
- M. L. H. Green and B. Jousseaume, J. Organomet. Chem., 1980, 193, 339–344 CrossRef CAS.
- F. Guerin, D. H. McConville and J. J. Vittal, Organometallics, 1995, 14, 3154–3156 CrossRef CAS.
- R. Serrano and P. Royo, J. Organomet. Chem., 1983, 247, 33–37 CrossRef CAS.
- A. Antinolo, M. Martinez-Ripoll, Y. Mugnier, A. Otero, S. Prashar and A. M. Rodriguez, Organometallics, 1996, 15, 3241–3243 CrossRef CAS.
- A. Antiñolo, S. Garcia-Liedó, J. M. d. Ilarduya and A. Otero, J. Organomet. Chem., 1987, 335, 85–90 CrossRef.
- C. Garcia-Yebra, F. Carrero, C. Lopez-Mardomingo, M. Fajardo, A. Rodriguez, A. Antinolo, A. Otero, D. Lucas and Y. Mugnier, Organometallics, 1999, 18, 1287–1298 CrossRef CAS.
- M. Sato and K. Oshima, Chem. Lett., 1982, 157–160 CrossRef CAS.
- M. Ozaki, Y. Obora, Y. Tada and Y. Ishii, J. Organomet. Chem., 2013, 741–742, 109–113 CrossRef CAS.
- R. N. Kadikova, I. R. Ramazanov, A. M. Gabdullin, O. S. Mozgovoi and U. M. Dzhemilev, RSC Adv., 2020, 10, 17881–17891 RSC.
- R. N. Kadikova, I. R. Ramazanov, A. M. Gabdullin, O. S. Mozgovoi and U. M. Dzhemilev, Catalysts, 2019, 9, 1022–1031 CrossRef CAS.
- R. N. Kadikova, I. R. Ramazanov, O. S. Mozgovoi, A. M. Gabdullin and U. M. Dzhemilev, Synlett, 2019, 30, 311–314 CrossRef CAS.
- J.-L. Montchamp and E. Negishi, J. Am. Chem. Soc., 1998, 120, 5345–5346 CrossRef CAS.
- F. Sato, H. Urabe and S. Okamoto, Chem. Rev., 2000, 100, 2835–2886 CrossRef.
- R. M. Sultanov, R. R. Ismagilov, N. R. Popod'ko, A. R. Tulyabaev and U. M. Dzhemilev, J. Organomet. Chem., 2013, 724, 51–56 CrossRef CAS.
- R. M. Sultanov, R. R. Ismagilov, N. R. Popod’ko, A. R. Tulyabaev, D. Sabirov and U. M. Dzhemilev, J. Organomet. Chem., 2013, 745–746, 120 CrossRef CAS.
- R. M. Sultanov, U. M. Dzhemilev, E. V. Samoilova, R. R. Ismagilov, L. M. Khalilov and N. R. Popod’ko, J. Organomet. Chem., 2012, 715, 5–8 CrossRef CAS.
- A. A. Quntar and M. Srebnik, Org. Lett., 2004, 6, 4243–4244 CrossRef CAS.
- E. I. Negishi and T. Takahashi, Product Class 11: Organometallic Complexes of Zirconium and Hafnium, Sci. Synth., 2003, 681–848 CAS.
- M. G. Shaibakova, I. G. Titova, A. G. Ibragimov and U. M. Dzhemilev, Russ. J. Org. Chem., 2008, 44, 1126–1129 CrossRef CAS.
- L. W. Bieber and M. F. da Silva, Tetrahedron Lett., 2004, 45, 8281–8428 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10132j |
| This journal is © The Royal Society of Chemistry 2021 |