
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTraceless solid-phase synthesis and β-turn propensity of 1,3-thiazole-based peptidomimetics†
Aizhan Abdildinova and
Young-Dae Gong*
and
Young-Dae Gong*
Innovative Drug Library Research Center, Department of Chemistry, College of Science, Dongguk University, 30, Pildong-ro 1-gil, Jung-gu, Seoul 04620, Korea. E-mail: ydgong@dongguk.edu
First published on 4th January 2021
Abstract
The design and solid-phase synthesis of 1,3-thiazole-based peptidomimetic molecules is described. The solid-phase synthesis was based on the utilization of a traceless linker strategy. The synthesis starts from the conversion of chloromethyl polystyrene resin to the resin with a sulfur linker unit. The key intermediate 4-amino-thiazole-5-carboxylic acid resin is prepared in three steps from Merrifield resin. The amide coupling proceeded at the C4 and C5 positions via an Fmoc solid-phase peptide synthesis strategy. After cleavage, the final compounds were obtained in moderate yields (average 9%, 11-step overall yields) with high purities (≥87%). Geometric measurements of Cα distances and dihedral angles along with an rmsd of 0.5434 for attachment with Cα of the β-turn template suggest type IV β-turn structural motifs. Additionally, the physicochemical properties of the molecules have been evaluated.
Introduction
Protein–protein interactions (PPIs) are involved in numerous biological processes at the cellular level, such as signal transduction, cell adhesion, cellular proliferation, growth, and programmed cell death.1,2 Thus, PPI regulators represent an attractive target for the development of new-generation therapeutics.3 Proteins bind to each other through a combination of hydrophobic bonding, van der Waals forces, and salt bridges at specific binding domains on each protein.4 The binding sites are often called hot-spot regions and represent key amino acid residues involved in the binding. The development of PPI modulators is based on structural features of hot-spot regions that are generally defined by secondary structural motifs such as turns, helices, and sheets. Efficient structure-based mimicking of hot-spot regions for the PPI modulation is a challenging research goal. Since PPIs often involve protein surfaces of 800–2000 Å2, small-molecule libraries show low hit rates after screening.3 Therefore, the development of compounds with higher molecular weight and large surfaces like peptides or natural product derivatives with greater structural diversity is preferred. However, even with improved medicinal properties, peptides are still limited by rapid proteolysis, poor membrane permeability, and low bioavailability.5 To solve those complications, several approaches, including peptidomimetic molecules, have been developed.6–10 Previously, our research team was interested in the development of peptidomimetic hybrid molecules containing both small-molecule and peptide moieties.1,11In continuation of our studies, we present a modified synthesis strategy for 1,3-thiazole-based peptidomimetics. The thiazole ring system is present in the core of natural product molecules, such as oriamide, cyclotheonaellazole A, and dendroamide A, and commercial drugs, such as thiamine (vitamin B1), niridazole (anthelmintic), abafungin (anesthetic), and azereonam (antibiotic).12–15 With C2, C4, and C5 atoms available for functionalization, thiazole is a perfect template for the construction of peptidomimetic molecules.
Peptidomimetic compounds were prepared via a solid-phase synthetic approach. Solid-phase synthesis is an advantageous tool for the construction of compounds from small molecules to peptides.16–18 In the previous studies, for the synthesis of heterocycle-based peptidomimetic molecules, we used backbone amide linker (BAL) utility. The introduction of BAL onto Merrifield resin 1 required up to three additional steps and to obtain heterocycle intermediate resins A and C we performed up to nine steps in total (Scheme 1a).1,11 In current work, we utilized a sulfur-based traceless linker. Thereby, the overall number of the reaction steps was reduced and 1,3-thiazole intermediate resin 4 was obtained in three steps (Scheme 1b). Traceless linkers release compounds without a trace of the linker that was used to tether the intermediates during the synthesis.19 However, by modifying cleavage conditions, those linkers can serve as a multifunctional unit. In our work, traceless cleavage was achieved by the nucleophilic attack of benzylamine derivatives allowing diversification of the thiazole scaffold at the C2 position. Dipeptide chain elongation proceeded on C4 and C5 positions of thiazole thus reducing distances and enhancing the probability of intramolecular hydrogen bonding between two strands. Herein, we present the design, synthesis, and computational validation of 1,3-thiazole-based peptidomimetics showing β-turn structural characteristics and discuss their potential as PPI modulators.
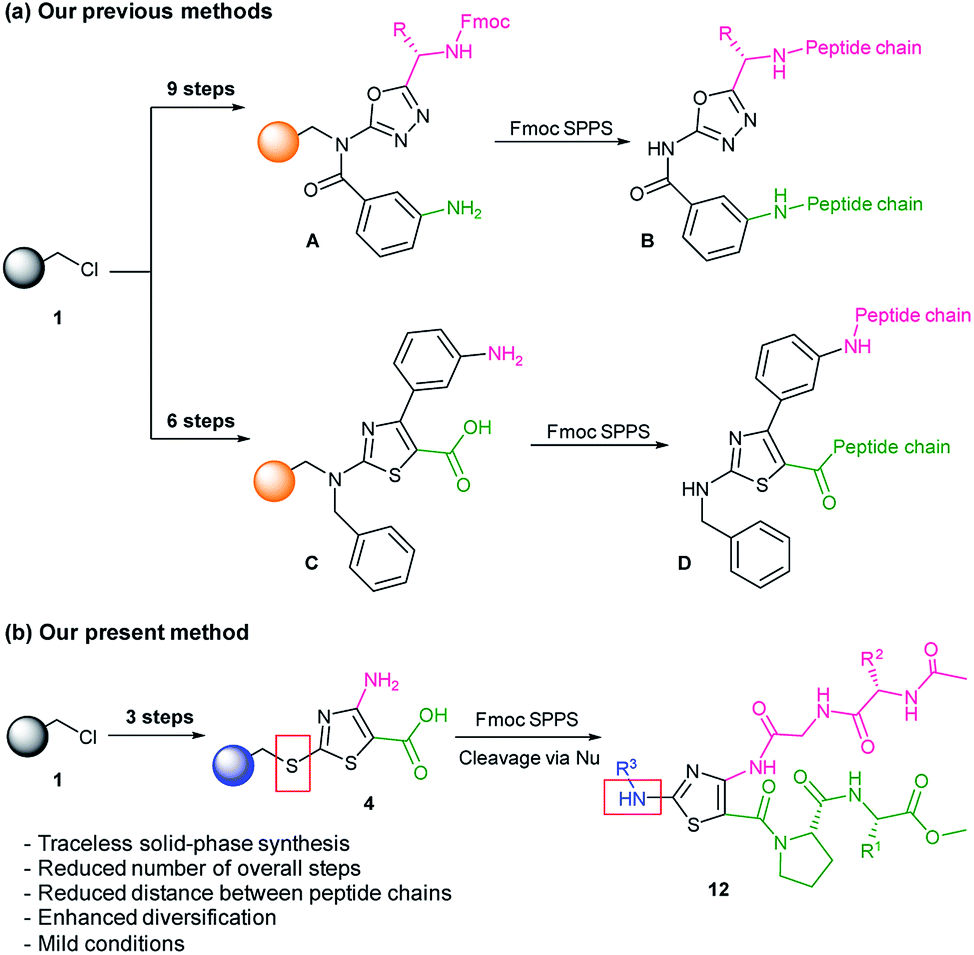 | ||
| Scheme 1 Solid-phase synthesis strategies for heterocycle-based peptidomimetics. | ||
Results and discussion
Design and synthesis of peptidomimetics
Thiazole (or 1,3-thiazole) is a five-membered heterocyclic aromatic compound that is broadly used in drug discovery. The thiazole was chosen as a template since it fits our major requirements for library construction: a promising pharmacophore capacity and functionalization (diversification) potential. Our design was based on the incorporation of amino acid chains in the β-turn mimetic template with a thiazole-L-Pro moiety as a turn inducing element. The β-turn is the most common secondary structural motif in the proteins. Generally, a β-turn consists of four amino acid residues, and the distance between Cαi and Cαi+3 carbons is ≤7 Å; the standard deviations of the Cα distances were suggested by Whitby et al. (Fig. 1a).20 β-Turns can be classified into nine types according to the φ and ψ dihedral angles of the loop region (Fig. 1b).20–22 Proline, which does not fit any other secondary structures, and glycine, which occurs in all secondary structural motifs, are often detected in the i + 1 or i + 2 positions of β-turns.23 On the opposite side of proline, glycine was introduced as a fixed member to facilitate intramolecular hydrogen bonding for additional molecular stabilization. Several amino acid residues were incorporated onto the thiazole-L-Pro scaffold at the R1, R2, and/or R3 positions in the molecular design. | ||
| Fig. 1 (a) Mean and standard deviations of the β-turn Cα distances; (b) schematic diagram of a β-turn. Reprinted with permission from Whitby et al., J. Am. Chem. Soc. 2011, 133, 10184–10194, DOI: 10.1021/ja201878v. Copyright (2011), American Chemical Society. | ||
Solid-phase synthesis on Merrifield resin with a traceless linker strategy has been developed for rapid access to the compounds. The overall synthetic scheme is shown in Scheme 2. The synthesis starts from the conversion of Merrifield resin 1 to cyanocarbonimidodithioate resin 2 using potassium cyanocarbonimidodithioate in DMF at room temperature (rt) with ATR-FTIR showing a nitrile band at 2160 cm−1 (Fig. S1a, ESI†).24 The treatment of resin 2 with ethyl bromoacetate and TEA in DMF at 60 °C for 6 h resulted in thiazole resin 3. The formation of resin 3 was confirmed by ATR-FTIR, showing the absence of the nitrile peak and formation of broad amine peaks at 3478 and 3365 cm−1 as well as the presence of the ester (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) peak at 1664 cm−1 (Fig. S1b, ESI†). The efficient hydrolysis of the ester group was achieved by treating the thiazole resin 3 with 5 M NaOH in THF:EtOH at 60 °C for 24 h with the broad OH peak detected at 3332 cm−1 (Fig. S1c, ESI†).
O) peak at 1664 cm−1 (Fig. S1b, ESI†). The efficient hydrolysis of the ester group was achieved by treating the thiazole resin 3 with 5 M NaOH in THF:EtOH at 60 °C for 24 h with the broad OH peak detected at 3332 cm−1 (Fig. S1c, ESI†).
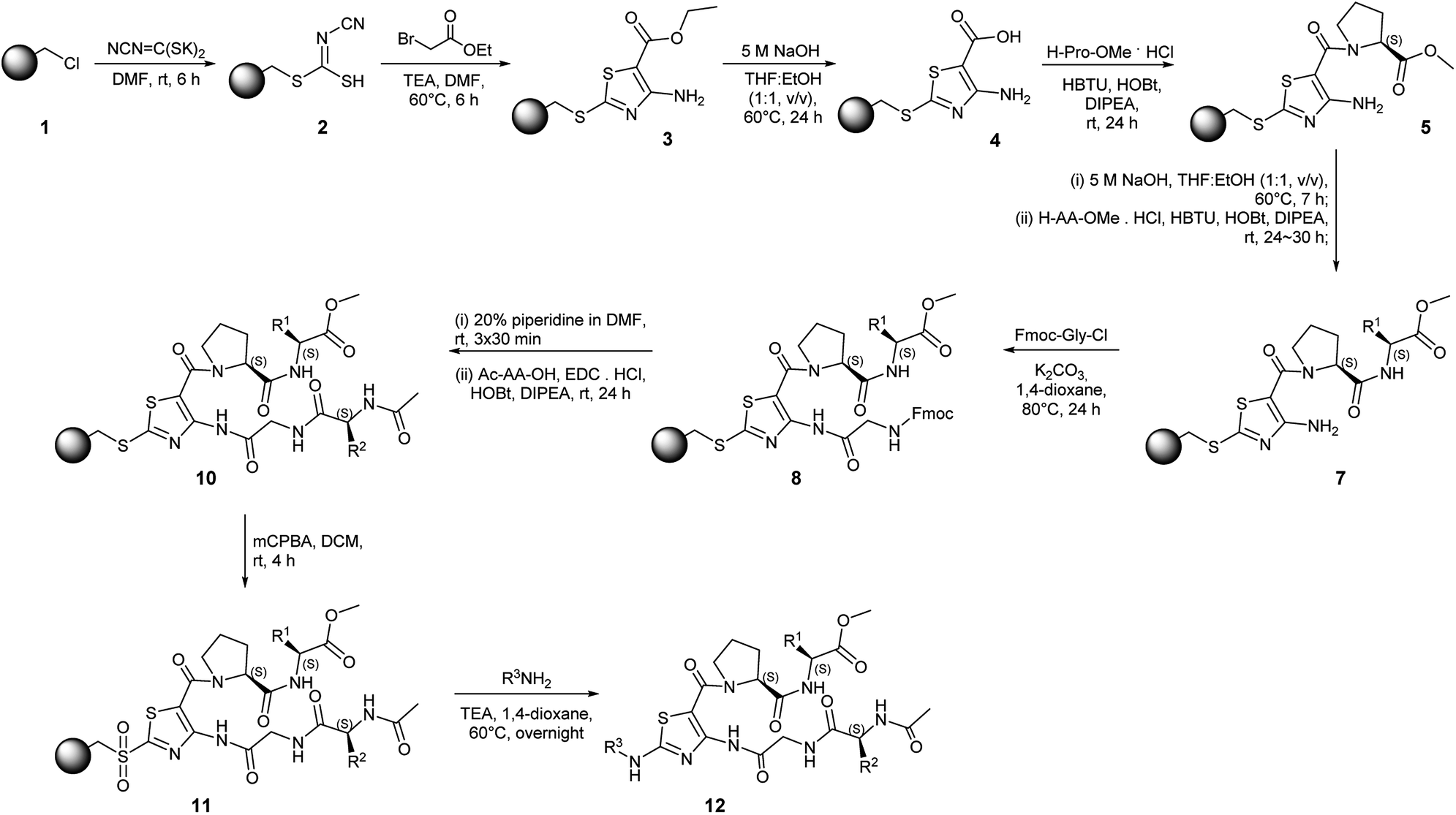 | ||
| Scheme 2 Solid-phase synthesis of thiazole-based peptidomimetics. | ||
The C-terminal amide coupling of resin 4 with amino acid methyl ester hydrochlorides was achieved in the presence of N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole (HOBt) with N,N-diisopropylethylamine (DIPEA). The amide bond formation was confirmed by ATR-FTIR showing the ester CO peak at 1738 cm−1 and CO–O stretching absorptions at 1172 cm−1 and 1197 cm−1 (Fig. S1d, ESI†). Subsequent hydrolysis of the methyl ester group and C-terminal amide coupling resulted in the formation of an intermediate resin 7 (Fig. S1e–f, ESI†).
Because of the low nucleophilicity of the primary amine group at the C4 position of thiazole, amide coupling did not yield the desired product 8. This limitation was solved by acylation reaction with activation of Fmoc-Gly-OH using SOCl2. Acylation reaction conditions initially were optimized in the solution phase (Table S1, ESI†). Accordingly, NaH, K2CO3, t-BuOK, Et3N, DIPEA, and pyridine were tested under different conditions. Interestingly, only treatment with K2CO3 in 1,4-dioxane afforded the desired product, yielding acylated compound in 36% yield. Increasing the reaction temperature up to 80 °C afforded product with a 78% yield while increasing temperatures up to 100 °C led to decomposition. The full conversion of the starting compound on the solid phase was achieved by extending the reaction time to 24 h with a typical amide bond peak occurring at 1703 cm−1 (Fig. S1g, ESI†). The removal of Fmoc and N-terminal amide coupling with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), HOBt, and DIPEA yielded resin 10, which is a resin-bound thiazole adorned with two dipeptide chains. The resin-bound intermediate 10 was oxidized with 3-chloroperbenzoic acid (mCPBA) in DCM for 4 h. The cleavage from the resin was achieved using benzylamine derivatives. The crude mixture was purified via flash column chromatography providing a peptidomimetic library in moderate yields (3.2–16.6% 11-step overall yield) with high purities (≥87%, Table 1). Peptidomimetics containing Trp residue showed a mass pattern of oxidized Trp reporting three +4, +16, +32 peaks corresponding to the formation of kynurenine (WKYN), hydroxy-Trp (Wox1), and N-formyl kynurenine/dihydroxy-Trp (WNFK/Wox2), respectively.25 Oxidation of Trp occurred during treatment of resin 10 with mCPBA; therefore, this synthetic route is unsuitable for the synthesis with unprotected amino acids.
| No. | R1 | R2 | R3 | Yielda (%) | Purityb (%) |
|---|---|---|---|---|---|
| a 11-step overall yield from Merrifield resin 1 (with a loading capacity of 2.28 mmol g−1).b All the purified products were analyzed by LC/MS. | |||||
| 12a | Leu | Leu | 4OHBn | 12.2 | ≥99 |
| 12b | Leu | Trp | 4OHBn | 7.7 | ≥99 |
| 12c | Tyr | Phe | 4OHBn | 16.6 | ≥99 |
| 12d | Tyr | Phe | Bn | 5.7 | 87 |
| 12e | Tyr | Trp | Bn | 3.2 | ≥99 |
Peptidomimetic 12a was analysed by 2D NMR spectroscopy in DMSO-d6 (Fig. S2†). TOCSY experiments showed cross-peaks for the 4-OH-Bn-NH coupling network. ROESY experiments showed several NOE cross-peaks at the Gly–Pro region, as well as NOE cross-peaks for the 4-OH-Bn-NH coupling network. These observations indicate that part of 12a exhibits a turn-type conformation that is stabilized by the intramolecular hydrogen bonding at the Gly–Pro region.
Computational analysis
To analyze potential β-turn conformations, we calculated the geometric features of the representative molecule 12a via Discovery Studio 2017. First, the distances between substituent Cα centers (i, i + 1, i + 2, i + 3) were calculated and compared with standard deviations proposed by Whitby et al.20 As shown in Table 2, the measured distances fall within the proposed ranges including the distance between Cαi and Cαi+3 of 5.83 Å. The calculated root-mean-square deviation (rmsd) value of 0.5434 Å (recommended ≤ 2 Å) for attachment of three substituent Cα centers of the molecule 12a with three α-carbons of β-turn template demonstrates the high potential of 12a in mimicking β-turn structural motifs. The type of β-turn was predicted by calculating the φ and ψ dihedral angles of central residues (Table S2, ESI†). The results suggest that synthesized molecules can be classified as a type IV β-turn mimetic. |
Measured Cα distances, Å | |
| i, i + 1 | 3.82 | |
| i, i + 2 | 7.03 | |
| i, i + 3 | 5.83 | |
| i + 1, i + 2 | 4.94 | |
| i + 1, i + 3 | 6.06 | |
| i + 2, i + 3 | 3.76 | |
Additionally, compounds were subjected to the computational calculation of the physicochemical properties such as calculated A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP, calculated pKa, molecular weight, number of hydrogen bond donors (HBD) and acceptors (HBA), number of rotatable bonds, molecular surface area, and molecular polar surface area (Fig. 2). Lipinski specified that molecular parameters of the compounds are crucial for their bioavailability as a drug. Accordingly, lipophilicity, the parameter responsible for the solubility, expressed in calculated logP should be no greater than 5, and good absorption or permeation is more likely to occur for compounds with five HBD and ten HBA.26 Those parameters fall within the recommended ranges for the compounds. The number of rotatable bonds is higher than ten which can affect the oral bioavailability.27 Calculated pKa varied from 1.65 to 9.83 and falls in a typical range of pKa values.28 Molecular weight is another important aspect. Although Lipinski's rule suggests molecular weight ≤ 500 Da, the PPI studies of Arkin et al. propose the tendency of clinical-stage PPI inhibitors to have a molecular weight ≥ 500 Da, which is suitable for peptidomimetic molecules.29 Molecular surface area and molecular polar surface area are expectedly higher than for small-molecules. Considering larger contact surfaces of PPI hot spots, the broader interaction surface is beneficial.30
logP, calculated pKa, molecular weight, number of hydrogen bond donors (HBD) and acceptors (HBA), number of rotatable bonds, molecular surface area, and molecular polar surface area (Fig. 2). Lipinski specified that molecular parameters of the compounds are crucial for their bioavailability as a drug. Accordingly, lipophilicity, the parameter responsible for the solubility, expressed in calculated logP should be no greater than 5, and good absorption or permeation is more likely to occur for compounds with five HBD and ten HBA.26 Those parameters fall within the recommended ranges for the compounds. The number of rotatable bonds is higher than ten which can affect the oral bioavailability.27 Calculated pKa varied from 1.65 to 9.83 and falls in a typical range of pKa values.28 Molecular weight is another important aspect. Although Lipinski's rule suggests molecular weight ≤ 500 Da, the PPI studies of Arkin et al. propose the tendency of clinical-stage PPI inhibitors to have a molecular weight ≥ 500 Da, which is suitable for peptidomimetic molecules.29 Molecular surface area and molecular polar surface area are expectedly higher than for small-molecules. Considering larger contact surfaces of PPI hot spots, the broader interaction surface is beneficial.30
 | ||
| Fig. 2 Physicochemical properties in bar charts: calculated AlogP, molecular weight (Da), number of HBD and HBA, number of rotatable bonds, molecular surface area (Å2), and molecular polar surface area (Å2). | ||
Next, for a biological docking evaluation, we conducted virtual screening with human MdmX (HdmX) protein. Mdm2 and MdmX (Hdm2 and HdmX in humans) belong to the Mdm family proteins and are known to negatively regulate the tumor suppressor, p53 protein.31–34 In contrast to Mdm2, MdmX has attracted less research interest but the number of studies has increased in recent years. The overexpression of MdmX has been reported in 40% tumor cell lines.35 Although some Mdm2 inhibitors can bind to MdmX, the structural difference in the p53-binding pockets of the oncoprotein decreases the efficiency of Mdm2 inhibitors against MdmX. Studies reveal a dual role of MdmX including direct inhibition of p53 and enhanced Mdm2-mediated degradation of p53.31,35 Therefore, the development of MdmX inhibitors is of therapeutic interest as studies suggest independent MdmX drug development apart from Mdm2 inhibitors.
Studies showed that crucial interaction of the p53 peptide to human MdmX are made by its hydrophobic Phe19, Trp23 and Leu26 residues that form an interface that fills up a hydrophobic pocket of human MdmX with key residues identified as Tyr99 and Met53 (PDB ID 3DAB).36 Additionally, it was reported that β-hairpin can mimic structural features of the helical structure of p53 enabling structural diversity for the p53 peptidomimetics.37 The crystal structure of human MdmX in complex with p53 analogues was reported previously (PDB ID 3FE7). Accordingly, 8-mer peptidomimetic of p53 (Ac–Phe–Met–Aib–Pmp–Trp–Clu–Ac3c–Leu–NH2) makes interactions with HdmX in a similar way as p53 at the hydrophobic binding pocket.38 However, structural side-chain reorientations were observed for Leu56, Val52, and Tyr99. Considering those reports, we run molecular docking of 12a with human MdmX. Compound 12a was docked at the binding site of HdmX at the resolution 1.35 Å using Discovery Studio software. In the docked mode, molecule 12a is bound to the hydrophobic pocket of HdmX resulting in major interactions with Tyr99, Met53, and Leu 98, which is in agreement with previous studies (Fig. 3). Accordingly, Pro of 12a fills the pocket and enters into hydrophobic bonding with Met53, Leu98, and Val92 of HdmX; while Leu binds via pi–alkyl and alkyl interactions with Tyr66, Ile60, and Met61 on the other side. N-terminal Leu of 12a binds to the target protein through hydrophobic interactions with Val92 and Lys93 and conventional hydrogen bonding with Gln71. Additionally, the secondary amine at the R3 position makes carbon hydrogen bonding with Tyr99.
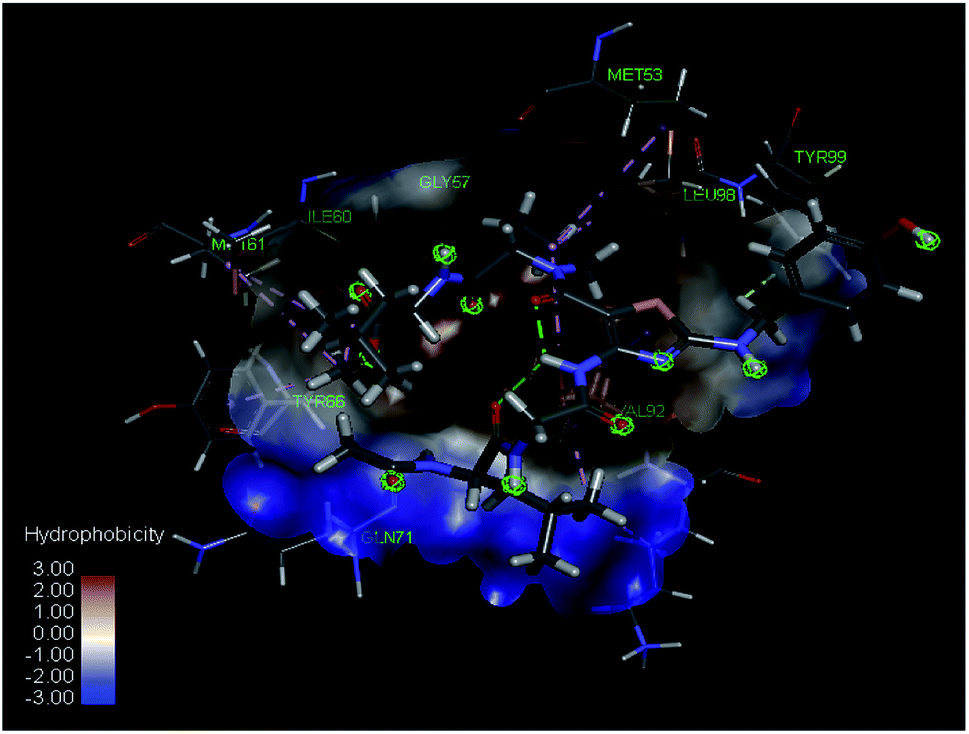 | ||
| Fig. 3 Superimposed view of molecule 12a binding to the active site of HdmX (PDB ID: 3FE7) at a resolution 1.35 Å (conventional hydrogen bond, carbon hydrogen bond, pi–alkyl and alkyl interactions in green, light green, and pink respectively). | ||
Conclusions
In conclusion, we established an accessible traceless solid-phase synthesis methodology for the thiazole-based peptidomimetics. The thiazole core incorporated with L-Pro was used as a peptidomimetic template. The key intermediate, 1,3-thiazole with amine and ester functional groups at positions C4 and C5 respectively, was prepared by cyclization of cyanocarbonimidodithioate intermediate resin with an α-bromo ketone. Peptide chain elongations proceeded on both C-terminal and N-terminal sites of the ethyl 4-aminothiazole-5-carboxylate core. Oxidation with further cleavage afforded final compounds in moderate yields and high purities. 2D NMR studies of 12a showed a part of 12a exhibits a turn-type conformation that is stabilized by the intramolecular hydrogen bonding at the Gly–Pro region. The geometric characterization of the molecules including Cα carbon distances, angles, and rmsd value suggests a type IV β-turn mimetic structure. Calculations of physicochemical properties showed molecular diversity and suitability of the compounds as PPIs inhibitors. Finally, the representative molecule 12a was subjected to computational molecular docking to check its potential as a regulator of HdmX/p53 PPIs. The predicted binding poses suggest that the peptidomimetic molecule interacts with the HdmX binding site similar to p53 via hydrophobic and hydrogen bond interactions between key residues such as Leu, Phe, and Trp. We believe that the tools and strategies developed in this work will facilitate the development of new peptidomimetic molecules and plan to conduct further studies in the future.Experimental section
General synthetic methods
All chemicals were of reagent grade and used as purchased. The Merrifield resin was used in 100–200 mesh beads with a loading capacity of 2.28 mmol g−1. The solution phase reactions were monitored by TLC (thin-layer chromatography) analysis using Merck silica gel 60 F-254 thin-layer plates. The solid-phase reactions were monitored using an ATR-FTIR spectrometer (Smiths Detection Group Ltd). The crude product mixtures were purified with a flash chromatography system using Isolera One (Biotage). Flash column chromatography was carried out on Merck silica gel 60 (230–400 mesh). NMR analysis was recorded in δ units relative to deuterated solvent as an internal reference using a 500 MHz NMR spectrometer (Bruker). Liquid chromatography-tandem mass spectrometry was performed using electrospray ionization (ESI) with a photodiode array detector (PDA) via 6460 Triple Quad LC/MS (Agilent). High-resolution mass spectrometry was performed using a Q6550 iFunnel Q-TOF LC/MS system (Agilent).![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1653 (CN), 1369, 1096, 1059, 943, 758, 697, 661.:1, v/v, 46 mL) was reacted with 5 M NaOH (45.6 mL, 228 mmol). The reaction mixture was shaken for 24 h at 60 °C. The resin was cooled to RT, filtered and washed successively with THF, H2O, MeOH, and DCM several times, and dried under high vacuum. The resulting yield of the resin 4 was 4.74 g; single-bead ATR-FTIR νmax/cm−1: 3332 (OH), 2918, 2102, 1559, 1491, 1375, 1339, 1240, 1064, 794, 757, 696.:EtOH (1:1, v/v, 20 mL) was reacted with 5 M NaOH (20 mL, 100 mmol). The reaction mixture was shaken for 7 h at 60 °C. The resin was cooled to RT, filtered and washed successively with THF, H2O, MeOH, and DCM several times, and dried under high vacuum. The resulting yield of the resin 6 was 2.09 g; single-bead ATR-FTIR νmax/cm−1: 3342 (OH), 2920, 2105, 1698 (CO), 1573, 1507, 1491, 752, 696.
N), 1653 (CN), 1369, 1096, 1059, 943, 758, 697, 661.:1, v/v, 46 mL) was reacted with 5 M NaOH (45.6 mL, 228 mmol). The reaction mixture was shaken for 24 h at 60 °C. The resin was cooled to RT, filtered and washed successively with THF, H2O, MeOH, and DCM several times, and dried under high vacuum. The resulting yield of the resin 4 was 4.74 g; single-bead ATR-FTIR νmax/cm−1: 3332 (OH), 2918, 2102, 1559, 1491, 1375, 1339, 1240, 1064, 794, 757, 696.:EtOH (1:1, v/v, 20 mL) was reacted with 5 M NaOH (20 mL, 100 mmol). The reaction mixture was shaken for 7 h at 60 °C. The resin was cooled to RT, filtered and washed successively with THF, H2O, MeOH, and DCM several times, and dried under high vacuum. The resulting yield of the resin 6 was 2.09 g; single-bead ATR-FTIR νmax/cm−1: 3342 (OH), 2920, 2105, 1698 (CO), 1573, 1507, 1491, 752, 696.Representative procedure for the acylation reaction
:1, v/v) affording desired product ethyl 4-(2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)acetamido)-2-(benzylthio)thiazole-5-carboxylate C (0.045 g, 78% yield). LC-MS (ESI): m/z = 574.2 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C30H27N3O5S2 574.1465; found 574.1456.:1, v/v) and dried under high vacuum to obtain a beige-colored solid (43 mg, 12.2%, 11-step overall yield): LC-MS (ESI): m/z = 702.4 [M + H]+. HRMS (ESI) m/z: [M + H]+ calcd for C33H47N7O8S 702.3280; found 702.3273.Computational studies
The molecular docking study of the library was performed via Discovery Studio 2017 (BIOVIA) at the binding site of HdmX (PDB code 3FE7) at a resolution of 1.35 Å. Flexible docking was performed via CDOCKER docking protocol with the CHARMm force field. Ligands were subjected to full energy minimalization and docked with tautomerization and isomerization features off. The most energetically favorable conformation for the top hit was set to 1 for each molecule. Docking results were analyzed according to -CDOCKER energy: the molecule with the highest -CDOCKER energy is considered as the most suitable ligand for the target molecule.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
All authors were supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2016R1D1A1B04932654).References
- A. Abdildinova, S.-J. Yang and Y.-D. Gong, Tetrahedron, 2018, 74, 684–691 CrossRef CAS.
- L. G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695–4748 CrossRef CAS.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed. Engl., 2015, 54, 8896–8927 CrossRef CAS.
- L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 RSC.
- N. Qvit, S. J. S. Rubin, T. J. Urban, D. Mochly-Rosen and E. R. Gross, Drug Discovery Today, 2017, 22, 454–462 CrossRef CAS.
- J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS.
- L. Mabonga and A. P. Kappo, Int. J. Pept. Res. Ther., 2019, 26, 225–241 CrossRef.
- M. S. Kumar, Front. Nutr., 2019, 6, 11 CrossRef.
- G. R. Marshall and F. Ballante, Drug Dev. Res., 2017, 78, 245–267 CrossRef CAS.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- M.-J. Cha, A. Abdildinova and Y.-D. Gong, Tetrahedron, 2020, 131702, DOI:10.1016/j.tet.2020.131702.
- M. Gümüş, M. Yakan and İ. Koca, Future Med. Chem., 2019, 11, 1979–1998 CrossRef.
- S. J. Kashyap, V. K. Garg, P. K. Sharma, N. Kumar, R. Dudhe and J. K. Gupta, Med. Chem. Res., 2011, 21, 2123–2132 CrossRef.
- M. V. N. de Souza, J. Sulfur Chem., 2005, 26, 429–449 CrossRef CAS.
- S. Pola, in Scope of Selective Heterocycles from Organic and Pharmaceutical Perspective, 2016, ch. 1, DOI:10.5772/62077.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- J.-A. F. Muriel Amblard, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef.
- V. Made, S. Els-Heindl and A. G. Beck-Sickinger, Beilstein J. Org. Chem., 2014, 10, 1197–1212 CrossRef.
- N. Cankarova, E. Schutznerova and V. Krchnak, Chem. Rev., 2019, 119, 12089–12207 CrossRef CAS.
- L. R. Whitby, Y. Ando, V. Setola, P. K. Vogt, B. L. Roth and D. L. Boger, J. Am. Chem. Soc., 2011, 133, 10184–10194 CrossRef CAS.
- A. J. Metrano, N. C. Abascal, B. Q. Mercado, E. K. Paulson, A. E. Hurtley and S. J. Miller, J. Am. Chem. Soc., 2017, 139, 492–516 CrossRef CAS.
- P. Kountouris and J. D. Hirst, BMC Bioinf., 2010, 11, 407 CrossRef.
- B. Eckhardt, W. Grosse, L. O. Essen and A. Geyer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18336–18341 CrossRef CAS.
- T. Lee, J.-H. Park, M.-K. Jeon and Y.-D. Gong, J. Comb. Chem., 2009, 11, 288–293 CrossRef CAS.
- I. Perdivara, L. J. Deterding, M. Przybylski and K. B. Tomer, J. Am. Soc. Mass Spectrom., 2010, 21, 1114–1117 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- D. T. Manallack, Perspect. Med. Chem., 2007, 1, 25–38 Search PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS.
- M. C. Smith and J. E. Gestwicki, Expert Rev. Mol. Med., 2012, 14, e16 CrossRef.
- G. Sanz, M. Singh, S. Peuget and G. Selivanova, J. Mol. Cell Biol., 2019, 11, 586–599 CrossRef CAS.
- A. Czarna, G. M. Popowicz, A. Pecak, S. Wolf, G. Dubin and T. A. Holak, Cell Cycle, 2009, 8, 1176–1184 CrossRef CAS.
- M. Wade, Y. C. Li and G. M. Wahl, Nat. Rev. Cancer, 2013, 13, 83–96 CrossRef CAS.
- K. Lenos and A. G. Jochemsen, J. Biomed. Biotechnol., 2011, 2011, 876173 Search PubMed.
- A. Macchiarulo, N. Giacchè, A. Carotti, F. Moretti and R. Pellicciari, RSC Med. Chem., 2011, 2, 455–465 RSC.
- G. M. Popowicz, A. Czarna and T. A. Holak, Cell Cycle, 2008, 7, 2441–2443 CrossRef CAS.
- R. Fasan, R. L. Dias, K. Moehle, O. Zerbe, D. Obrecht, P. R. Mittl, M. G. Grutter and J. A. Robinson, Chembiochem, 2006, 7, 515–526 CrossRef CAS.
- J. Kallen, A. Goepfert, A. Blechschmidt, A. Izaac, M. Geiser, G. Tavares, P. Ramage, P. Furet, K. Masuya and J. Lisztwan, J. Biol. Chem., 2009, 284, 8812–8821 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR and 2D NMR data, HRMS and LC/MS spectral data, and computational data. See DOI: 10.1039/d0ra10127c |
| This journal is © The Royal Society of Chemistry 2021 |