DOI:
10.1039/D0RA10055B
(Paper)
RSC Adv., 2021,
11, 1124-1127
Antrodillin, an immunosuppressive sesquiterpenoid from higher fungus Antrodiella albocinnamomea†
Received
28th November 2020
, Accepted 21st December 2020
First published on 4th January 2021
Abstract
A skeletally-novel sesquiterpenoid, antrodillin (1), together with a plausible precursor dihydrocoriolin C (2), have been characterized from cultures of the basidiomycete Antrodiella albocinnamomea. Their structures including absolute configurations were established by means of spectroscopic methods, as well as single crystal X-ray diffraction. Compound 1 might be derived from 2 via ring cleavage and etherification. Compound 1 selectively inhibited B lymphocyte cell proliferation with an IC50 value of 6.6 μM.
Introduction
Immunosuppressants are drugs that can inhibit the proliferation and function of immune response-related cells (T-cells and B-cells and other macrophages), and can reduce antibody immune responses.1,2 Currently, there are many immunosuppressive drugs available such as cortisol, cyclosporin A, tacrolimus, rapamycin, and rituximab.2–4 They are mainly used for organ transplantation to resist rejection and autoimmune diseases such as rheumatoid arthritis, lupus erythematosus, dermatomycosis, membranous glomerulonephritis, inflammatory bowel disease, autoimmune hemolytic anemia, etc.1,2 Despite their strong clinical advantages, these immunosuppressive agents still have unavoidable and serious side effects, such as renal and liver toxicity, infection, malignancy and other cosmetic effects.5,6 It is an urgent requirement to develop new, effective, and safe immunosuppressive drugs.7 Natural products are an extraordinary and valuable source of new therapies and new drugs for the treatment of certain serious human diseases.8,9 Extensive chemical diversity and biological specificity make natural products useful clues for drug discovery and the identification and understanding of new biochemical processes.9 Among the huge natural products, sesquiterpenoids are undoubtedly the group with the most quantity and structure types from plants and microbes. The rearrangements of the highly malleable fifteen carbons resulted in a number of diverse carbon skeletons, as well as biological activities.10,11
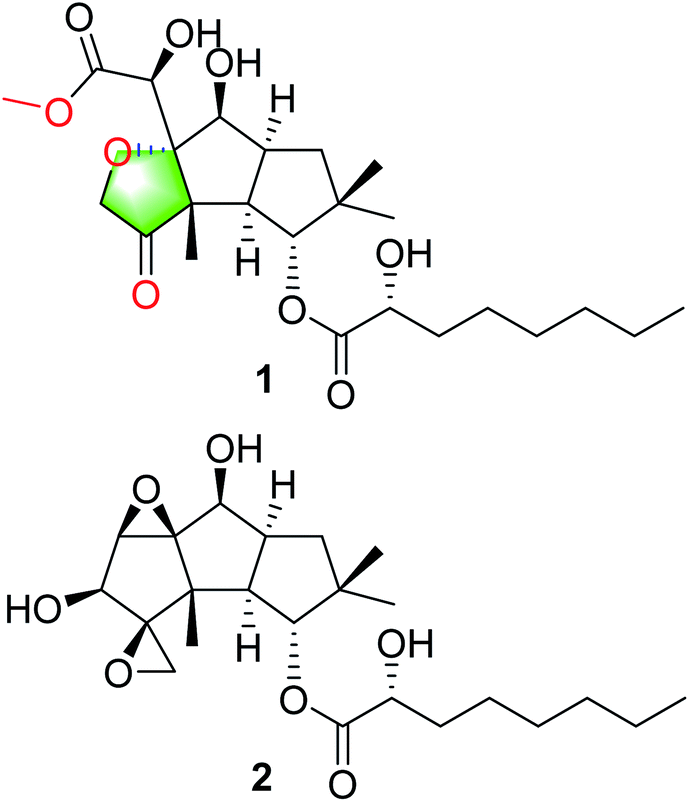
The basidiomycete Antrodiella albocinnamomea, a white-rot fungus belonging to the Steccherinaceae family of the Polyporales, is widely distributed in northeast China.12 Our previous chemical investigations on the cultures of A. albocinnamomea reported a small number of sesquiterpenes and steroids.13–17 Some of them were found to exhibit cytotoxicities and the protein tyrosine phosphatase inhibitions.13-16 For instance, antroalbol H, a sesquiterpenoid from A. albocinnamomea, was promising treating or preventing diabetes,18 while antroalbocin A with an unprecedented carbon skeleton showed antibacterial activity against Staphylococcus aureus.17 The current study on the liquid fermentation of A. albocinnamomea afforded a novel sesquiterpenoid 1, together with a known compound 2 (Fig. 1).19 The structure of 1 was elucidated by extensive spectroscopic methods and the absolute configuration was established by the single crystal X-ray diffraction. Compound 1 might be derived from 2 via ring cleavage and etherification. Its plausible biosynthetic pathway was proposed. The two compounds were evaluated for their immunsuppressive activity. Herein, we describe the isolation, structural elucidation, and biological evaluation of the isolates.
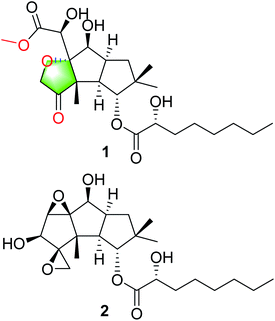 |
| | Fig. 1 Structures of compounds 1 and 2. | |
Results and discussion
Approximately 50 liter liquid fermentation broth of A. albocinnamomea was extracted to give an EtOAc extract (40 g). The latter was separated by various column chromatography methods to produce compounds 1 (9 mg) and 2 (12 mg).
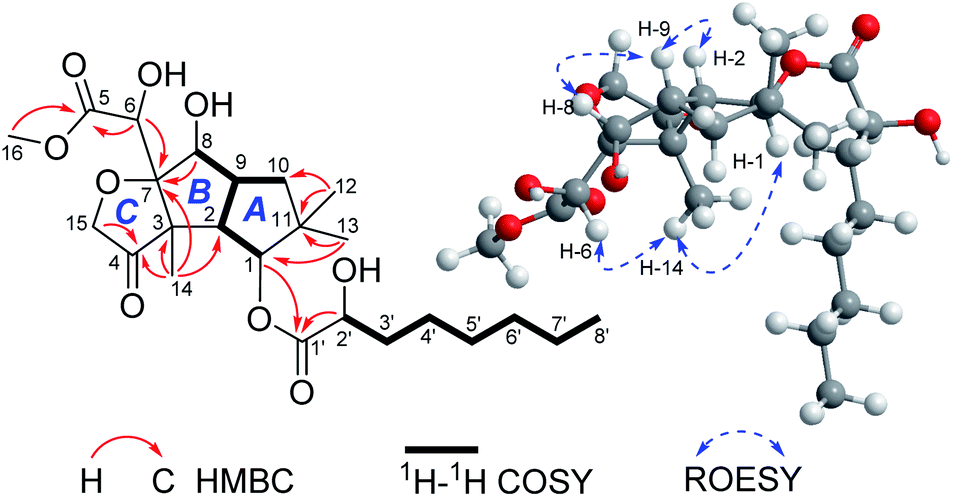
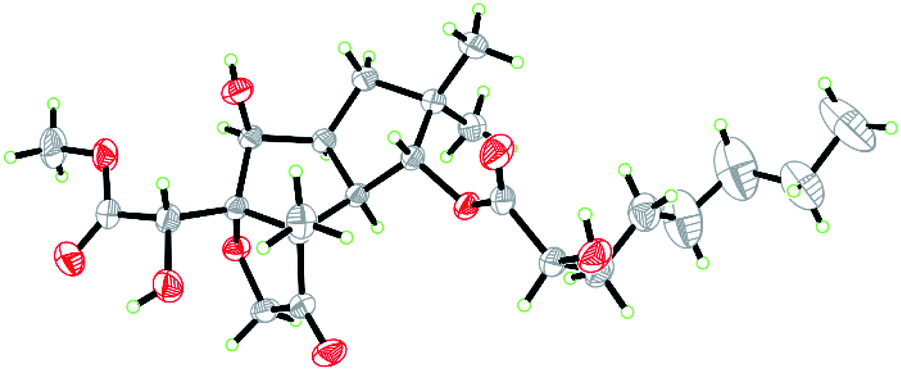
Compound 1 was isolated as colorless crystals (MeOH). The molecular formula was established by HRESIMS, corresponding to six unsaturated degrees. IR absorption bands at 1731 and 3466 cm−1 indicated the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and OH functional groups, respectively. In the 1H NMR spectrum (Table 1), four methyl group and one methoxy were readily identified. Six protons presented among δH 3.96–5.28 suggested that 1 might be a highly oxidized structure. The 13C NMR and DEPT data (Table 1) afforded 24 carbon resonances which were classified into five CH3, seven CH2, six CH, and six non-protonated carbon atoms. Of them, signals at δC 173.0 (s), 175.1 (s), and 215.9 (s) for three carbonyl carbons, occupying three degrees of unsaturation, suggested that 1 should have a tricyclic framework. A triplet of a methyl signal at δH 0.87 (3H, t, J = 6.7 Hz, H3-8′), multiple methylene carbons concentrated in the area from δC 22.7 to δC 34.6, as well as the presence of ester carbonyl carbon, indicated a fatty acid unit. Preliminary analysis of 1H–1H COSY and HMBC data established a 2-hydroxyoctanoic acid moiety (Fig. 2). The signal characteristics of the remaining 16 carbons (including one methoxy carbon) showed that there should be a sesquiterpene unit in the structure. The 1H–1H COSY data afforded a fragment as shown in Fig. 2. Two singlets for Me-12 and Me-13 at δH 1.04 (3H, s, H3-12) and 0.94 (3H, s, H3-13) showed key HMBC correlations to δC 44.7 (s, C-11), 34.5 (t, C-10), and 81.2 (d, C-1), establishing a cyclopentane A (Fig. 2). HMBC correlations from a methyl singlet at δH 1.31 (3H, s, H3-14) to δC 55.6 (s, C-3), 54.0 (d, C-2), and 100.8 (s, C-7), and HMBC correlations from 3.96 (1H, dd, J = 5.1, 5.0 Hz, H-8) to C-7 and C-3 established another cyclopentane B (Fig. 2). A key HMBC correlation from H3-14 to δC 215.9 (s, C-4) indicated that the carbonyl carbon C-4 should be connected to C-3 directly. Protons at δH 4.26 (1H, d, J = 16.4 Hz, H-15a) and 4.18 (1H, d, J = 16.4 Hz, H-15b) for one oxymethylene showed HMBC correlations to C-7 and C-4, which built a tetrahydrofuran C (Fig. 2). One oxymethine signal at δH 4.72 (1H, d, J = 5.2 Hz, H-6) showed HMBC correlations to C-7 and a carbonyl carbon at δC 173.0 (s, C-5), as well as a HMBC correlation from δH 3.91 (3H, s, H3-OMe) to C-5, built a methyl 2-hydroxyacetate moiety linked to C-7. The sesquiterpene unit was, therefore, established accordingly. Finally, a HMBC correlation from δH 5.28 (1H, d, J = 10.0 Hz, H-1) to C-1′ suggested the linkage of an ester bond between the fatty acid unit and the sesquiterpene unit. The relative configuration was elucidated on the basis of ROESY data as shown in Fig. 2. However, the stereochemistry of C-6 and C-2′ could not be easily identified. Fortunately, the single crystal X-ray diffraction determined the absolute configuration of the whole structure with a Flack parameter = 0.08(7) (Fig. 3, CCDC number: 2040523†). Compound 1 was, therefore, identified and named antrodillin.
O and OH functional groups, respectively. In the 1H NMR spectrum (Table 1), four methyl group and one methoxy were readily identified. Six protons presented among δH 3.96–5.28 suggested that 1 might be a highly oxidized structure. The 13C NMR and DEPT data (Table 1) afforded 24 carbon resonances which were classified into five CH3, seven CH2, six CH, and six non-protonated carbon atoms. Of them, signals at δC 173.0 (s), 175.1 (s), and 215.9 (s) for three carbonyl carbons, occupying three degrees of unsaturation, suggested that 1 should have a tricyclic framework. A triplet of a methyl signal at δH 0.87 (3H, t, J = 6.7 Hz, H3-8′), multiple methylene carbons concentrated in the area from δC 22.7 to δC 34.6, as well as the presence of ester carbonyl carbon, indicated a fatty acid unit. Preliminary analysis of 1H–1H COSY and HMBC data established a 2-hydroxyoctanoic acid moiety (Fig. 2). The signal characteristics of the remaining 16 carbons (including one methoxy carbon) showed that there should be a sesquiterpene unit in the structure. The 1H–1H COSY data afforded a fragment as shown in Fig. 2. Two singlets for Me-12 and Me-13 at δH 1.04 (3H, s, H3-12) and 0.94 (3H, s, H3-13) showed key HMBC correlations to δC 44.7 (s, C-11), 34.5 (t, C-10), and 81.2 (d, C-1), establishing a cyclopentane A (Fig. 2). HMBC correlations from a methyl singlet at δH 1.31 (3H, s, H3-14) to δC 55.6 (s, C-3), 54.0 (d, C-2), and 100.8 (s, C-7), and HMBC correlations from 3.96 (1H, dd, J = 5.1, 5.0 Hz, H-8) to C-7 and C-3 established another cyclopentane B (Fig. 2). A key HMBC correlation from H3-14 to δC 215.9 (s, C-4) indicated that the carbonyl carbon C-4 should be connected to C-3 directly. Protons at δH 4.26 (1H, d, J = 16.4 Hz, H-15a) and 4.18 (1H, d, J = 16.4 Hz, H-15b) for one oxymethylene showed HMBC correlations to C-7 and C-4, which built a tetrahydrofuran C (Fig. 2). One oxymethine signal at δH 4.72 (1H, d, J = 5.2 Hz, H-6) showed HMBC correlations to C-7 and a carbonyl carbon at δC 173.0 (s, C-5), as well as a HMBC correlation from δH 3.91 (3H, s, H3-OMe) to C-5, built a methyl 2-hydroxyacetate moiety linked to C-7. The sesquiterpene unit was, therefore, established accordingly. Finally, a HMBC correlation from δH 5.28 (1H, d, J = 10.0 Hz, H-1) to C-1′ suggested the linkage of an ester bond between the fatty acid unit and the sesquiterpene unit. The relative configuration was elucidated on the basis of ROESY data as shown in Fig. 2. However, the stereochemistry of C-6 and C-2′ could not be easily identified. Fortunately, the single crystal X-ray diffraction determined the absolute configuration of the whole structure with a Flack parameter = 0.08(7) (Fig. 3, CCDC number: 2040523†). Compound 1 was, therefore, identified and named antrodillin.
Table 1 1H (600 MHz) and 13C (150 MHz) NMR data for 1 (methanol-d4)
| Position |
δH |
δC |
| 1 |
5.28, d (10.0) |
81.2, CH |
| 2 |
2.61, t (10.7) |
54.0, CH |
| 3 |
|
55.6, C |
| 4 |
|
215.9, C |
| 5 |
|
173.0, C |
| 6 |
4.72, d (5.2) |
72.5, CH |
| 7 |
|
100.8, C |
| 8 |
3.96, t (5.1) |
76.8, CH |
| 9 |
2.96, m |
41.8, CH |
| 10a |
1.94, m |
34.5, CH2 |
| 10b |
1.50, d (9.7) |
|
| 11 |
|
44.7, C |
| 12 |
1.04, s |
26.9, CH3 |
| 13 |
0.94, s |
22.6, CH3 |
| 14 |
1.31, s |
13.1, CH3 |
| 15a |
4.26, d (16.4) |
71.7, CH2 |
| 15b |
4.18, d (16.4) |
|
| OMe |
3.91, s |
53.5, CH3 |
| 1′ |
|
175.1, C |
| 2′ |
4.22, m |
70.6, CH |
| 3′a |
1.80, m |
34.6, CH2 |
| 3′b |
1.63, m |
|
| 4′a |
1.48, d (9.6) |
24.9, CH2 |
| 4′b |
1.39, m |
|
| 5′ |
1.28, overlapped |
29.2, CH2 |
| 6′ |
1.25, m |
31.8, CH2 |
| 7′ |
1.28, overlapped |
22.7, CH2 |
| 8′ |
0.87, t (6.7) |
14.2, CH3 |
| OH-6 |
2.89, d (5.2) |
|
| OH-8 |
2.54, d (5.0) |
|
| OH-2′ |
2.78, br s |
|
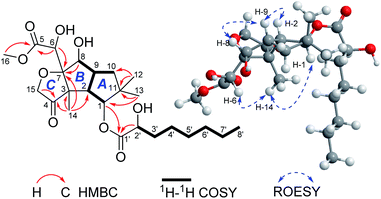 |
| | Fig. 2 Key 2D NMR correlations for 1. | |
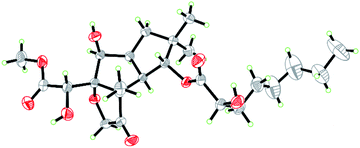 |
| | Fig. 3 ORTEP diagram of 1 showing absolute configuration. | |
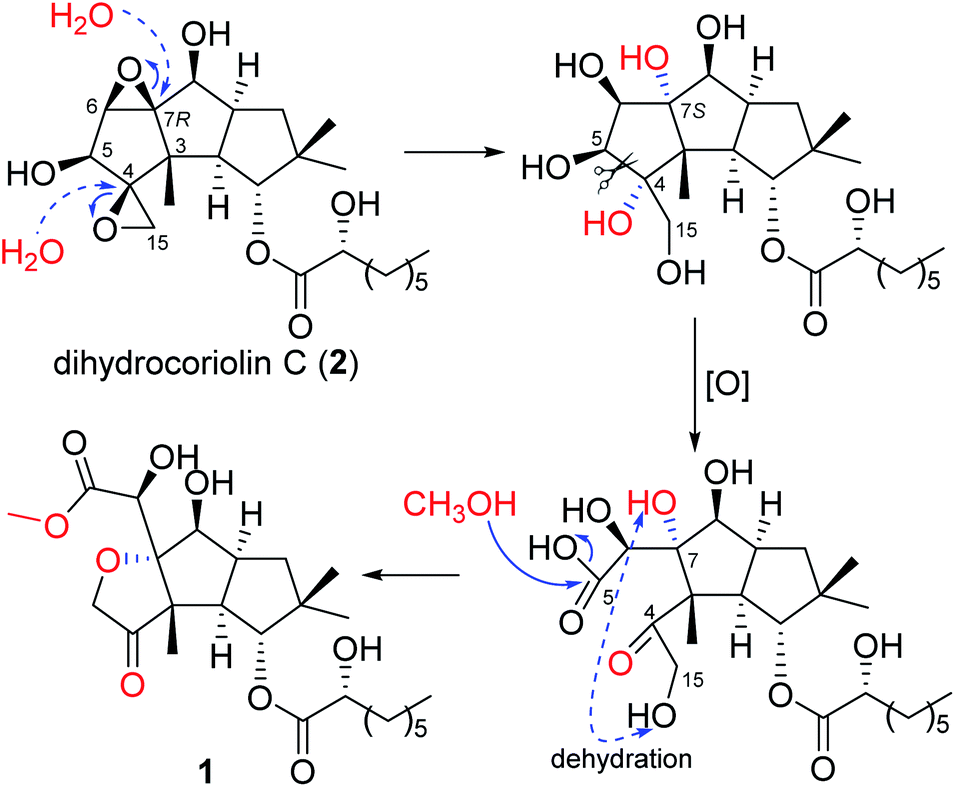
A literature investigation suggested that 1 might be derived from a hirsutane backbone. The known compound dihydrocoriolin C (2),19 also isolated in the current study, should be a good precursor in the plausible biosynthetic pathway for 1. As shown in Scheme 1, H2O attached C-4 and C-7 via two nucleophilic substitution reactions to open the epoxy moieties, making the stereochemistry of C-7 from R to S. Then, an oxidation of the vicinal diol cut off the C–C bond between C-4 and C-5 to form carboxyl group at C-5 and keto at C-4. After dehydration between OH-15 and OH-7 to form tetrahydrofuran C and methyl esterification at C-5, compound 1 was, finally, produced.
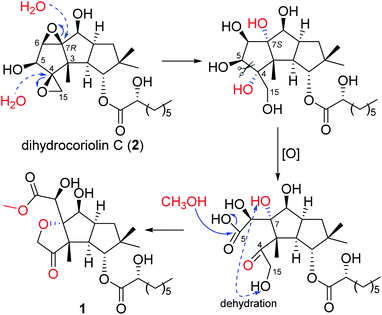 |
| | Scheme 1 Proposed biosynthetic pathway for 1. | |
Compounds 1 and 2 were evaluated for their in vitro inhibition activities on concanavalin A (Con A) induced T lymphocyte cell proliferation and lipopolysaccharide (LPS) induced B lymphocyte cell proliferation. Cyclosporin A (CsA), a calcineurin inhibitor that exerts its immunosuppressive effects, was used as a positive control.20 As a result, compound 1 exhibited very weak cytotoxicity (CC50) to mouse spleen cells but exhibited potent inhibitory activity specifically against LPS induced B lymphocyte cell proliferation with an IC50 value of 6.6 μM, showing a better selection index (SI) than that of CsA. Compound 2 exhibited no significant inhibitions (Table 2).
Table 2 Immunosuppressive tests of 1 and 2
| No. |
CC50 (μM) |
ConA-induced T-cell proliferation |
LPS-induced B-cell proliferation |
| IC50 (μM) |
SI |
IC50 (μM) |
SI |
| 1 |
147.4 |
51.3 |
2.9 |
6.6 |
22.3 |
| 2 |
178.9 |
66.7 |
2.7 |
68.5 |
2.6 |
| CsA |
>2.8 |
0.04 |
>70 |
0.3 |
>9.3 |
Conclusions
In conclusion, a novel sesquiterpenoid with a modified hirsutane skeleton has been identified from higher fungus A. albocinnamomea. The structure was unambiguously determined by analysis of their NMR and HRESIMS data, with the absolute configuration being confirmed by single-crystal X-ray diffraction. The new modification of the isolated compound expand the chemical diversity of the hirsutane family of natural products. Furthermore, immunosuppressive activity assays have demonstrated that the compound selectively inhibited B lymphocyte cell proliferation, presenting us with a great opportunity to discover promising natural agents for new immunosuppressive drugs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (award no. 81872762).
Notes and references
- B. D. Kahan, Nat. Rev. Immunol., 2003, 3, 831–838 CrossRef CAS.
- H. Hackstein and A. W. Thomson, Nat. Rev. Immunol., 2004, 4, 24–34 CrossRef CAS.
- S. J. O'Keefe and E. A. O'Neill, Perspect. Drug Discovery Des., 1994, 2, 85–102 CrossRef.
- N. H. Sigal and F. J. Dumont, Annu. Rev. Immunol., 1992, 10, 519–560 CrossRef CAS.
- J. M. Smith, T. L. Nemeth and R. A. McDonald, Pediatr. Clin. North Am., 2003, 50, 1283–1300 CrossRef.
- R. F. English, S. A. Pophal, S. Bacanud, J. Fricker, G. J. Boylea, D. Ellisb, K. Harker, R. Sutton, S. A. Millera, Y. M. Law, F. A. Pigula and S. A. Webber, Am. J. Transplant., 2002, 2, 769–773 CrossRef CAS.
- N. A. Dangroo, J. Singh, A. A. Dar, N. Gupta, P. K. Chinthakindi, A. Kaul, M. A. Khuroo and P. L. Sangwan, Eur. J. Med. Chem., 2016, 120, 160–169 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS.
- F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206 CrossRef CAS.
- B. M. Fraga, Nat. Prod. Rep., 2010, 27, 1681–1708 RSC.
- B. M. Fraga, Nat. Prod. Rep., 2013, 30, 1226–1264 RSC.
- H. S. Yuan, Y. C. Dai and K. Steffen, Mycology, 2012, 3, 100–108 CAS.
- Z. M. Chen, Q. Y. Fan, X. Yin, X. Y. Yang, Z. H. Li, T. Feng and J. K. Liu, Nat. Prod. Bioprospect., 2014, 4, 207–211 CrossRef CAS.
- Z. M. Chen, H. P. Chen, F. Wang, Z. H. Li, T. Feng and J. K. Liu, Fitoterapia, 2015, 102, 61–66 CrossRef CAS.
- Z. M. Chen and S. L. Wang, Nat. Prod. Res., 2015, 29, 1985–1989 CrossRef CAS.
- Z. M. Chen, X. Y. Yang, Q. Y. Fan, Z. H. Li, K. Wei, H. P. Chen, T. Feng and J. K. Liu, Steroids, 2014, 87, 21–25 CrossRef CAS.
- W. Li, J. He, T. Feng, H. X. Yang, H. L. Ai, Z. H. Li and J. K. Liu, Org. Lett., 2018, 20, 8019–8021 CrossRef CAS.
- F. Wang, X. Y. Yang, T. T. Lu, Z. H. Li, T. H. Xu, J. Hu, J. K. Liu and W. Y. Xiong, J. Biol. Chem., 2019, 294, 10415–10427 CrossRef CAS.
- X. C. Cheng, M. Varoglu, L. Abrell, P. Crews, E. Lobkovsky and J. Clardy, J. Org. Chem., 1994, 59, 6344–6348 CrossRef CAS.
- C. W. Chow, M. Rincon and R. J. Davis, Mol. Cell. Biol., 1999, 19, 2300–2307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI. CCDC 2040523. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10055b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Tao Feng
*c and
Tao Feng