
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the utility of hybrid siloxane-phosphocholine (SiPC) liposomes as drug delivery vehicles†
Mark B. Frampton‡
 a,
Andrea Blaisa,
Zachary Raczywolskia,
Alan Castleb and
Paul M. Zelisko*a
a,
Andrea Blaisa,
Zachary Raczywolskia,
Alan Castleb and
Paul M. Zelisko*a
aDepartment of Chemistry, Centre for Biotechnology, Brock University, 1812 Sir Isaac Brock Way, St. Catharines, Ontario, Canada. E-mail: pzelisko@brocku.ca
bDepartment of Biological Sciences, Centre for Biotechnology, Brock University, 1812 Sir Isaac Brock Way, St. Catharines, Ontario, Canada
First published on 6th April 2021
Abstract
Hybrid siloxane-phosphocholines (SiPCs) are a unique class of lipids that possess the capacity to spontaneously form unilamellar vesicles (ULVs) that are ∼100 nm in diameter upon exposure to aqueous media without the need for extrusion. These hybrid SiPCs do not negatively impact the growth of a number of bacterial strains and were subsequently explored as candidates for the delivery of a model compound, calcein. Liposomes that were formed spontaneously encapsulated larger quantities of calcein than liposomes that were formed via extrusion (both SiPCs and commercially available lipids), indicating that the extrusion process results in the loss of the material being entrapped and decreased entrapment efficiency. Although the percentage of calcein released was virtually identical across liposome samples (extruded and non-extruded), the greater entrapment efficiency of the spontaneously formed liposomes ultimately results in a greater payload delivery.
Introduction
One of the ongoing avenues of research within the pharmaceutical industry is the design and development of effective drug delivery systems.1 Many different modalities for the delivery of bioactive compounds have been developed including those based on polysaccharides,2 proteins,3,4 nanoparticles,5,6 and emulsions7–9 to name but a few. In principle, drug delivery systems offer the capacity to enhance the efficacy of therapeutic agents by increasing the physiological concentration of these agents, as well as allowing therapeutic agents to be targeted to specific subcellular locations. Ideally, a drug candidate would be delivered for a desired amount of time, while minimizing deleterious side effects to surrounding healthy tissues.10–12Liposomes were first approved as nanoscale vehicles for clinical use in 1995 and are of particular interest in the area of drug delivery technology.13–20 Liposomes have been shown to be excellent candidates for the controlled release of therapeutic agents, particularly in the fight against cancer, although they are not without their drawbacks.21,22 The success of liposomes as delivery systems can be traced to their amphipathic characteristics which allows for the selective encapsulation of drugs with varying lipophilicities; hydrophobic drugs can be entrapped within the fatty acid tails of the lipid bilayer while hydrophilic drugs can be solubilized for encapsulation within the hydrophilic inner core.17,23 Not only can therapeutics with differing solubilities be encapsulated, but specific cells or organs can be targeted to limit or reduce potential harmful side effects caused by the distribution of the drug to non-targeted tissues.24 Lipids with long chain (i.e., >10 carbon atoms in their tails) tend to form multilamellar vesicles (MLVs) rather than unilamellar vesicles (ULVs). Although MLVs can be used to entrap and deliver various types of molecules, they tend to have shorter circulation half-lives.25,26
There have a been number of reports in the literature examining the addition of silicon-based compounds, most notably silicones, to liposome structures.27–29 These silicon-based compounds were not part of the lipids themselves but were added to stabilize/protect the liposomal structures. In 1987 Thompson and co-workers reported the first chlorosilane-modified lipid followed nearly 25 years later by a trialkoxysilane-modified lipid synthesized by Yasuhara et al.30,31 These silane moieties were not added to the lipids to change the overall properties of the lipids but were respectively added to facilitate binding of the lipid to a solid support and incorporation of the lipid into a sol–gel reaction.
It was not until 2017 that siloxane moieties were first grafted onto lipid structures with the express purpose of modifying the lipids' interfacial behaviour.32–34 Siloxane-phospholipids (SiPCs) possessing identical lipid tail structures to biologically relevant lipid species self-assemble in aqueous media to form low dispersity, unilamellar vesicles (ULVs) that are on average 120 nm in diameter, unlike natural phosphocholines such as 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyl-sn-3-glycerophosphocholine (DOPC), or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), which typically form larger (>200 nm) multilamellar liposomes. Hybrid SiPCs, possessing a siloxane fatty acid tail in the sn-2 position and an aliphatic fatty acid tail in the sn-1 position, self-assembled into ULVs or multilamellar vesicles (MLVs) depending on the chain length of the sn-1 fatty acid. Fatty acids with a C8 to C14 chain length provided ULVs (Fig. 1).34 When palmitic acid (C16), or stearic acid (C18) were incorporated at the sn-1 position, SAXS analysis revealed the presence of MLVs as well as ULVs.34 Liposomes derived from hybrid SiPCs generally do not exhibit any detectable thermotropic phase transitions between 5 °C and 60 °C. The exception to this was the SiPC, which had stearic acid in the sn-1 position and displayed a phase transition temperature at 17 °C. The absence of phase transitions suggests that SiPC liposomes are comparable to liposomes composed of phosphocholines (PCs) such as POPC, DOPC with unsaturated fatty acids, or the archaebacterial PC 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhyPC).35–38
 | ||
| Fig. 1 SiPCs and their capacities to spontaneously form ULVs or MLVs.32,34 | ||
The suitability of artificial liposomes for drug delivery purposes is largely dependent on the specific release kinetics of the bilayer. Transport across the bilayer is controlled by the polarity of the encapsulant, as well as the permeability and fluidity of the liposomal bilayer, bilayer thickness, and the number of bilayers through which the drug must migrate.39 Drug delivery systems typically rely on diffusion-controlled processes that are dependent on the solubility of the drug and the diffusion coefficient within the release medium.39
Calcein, a commonly employed fluorophore used as a model drug in release studies involving liposomal structures, is a negatively charged, water soluble fluorescent probe that self-quenches at concentrations above 70 mM. Calcein has been used in examining release kinetics from phosphocholine-derived liposomes to characterize the effects of membrane fluidity, liposome size, the influence of membrane lipids, and peptide induced membrane destabilization.40,44
These SiPCs represent unique lipid structures that have the capacity to spontaneously self-assemble into unilamellar structures that are ∼100 nm in diameter by simple and economic agitation. These lipids have the potential to overcome some of the negative aspects of commercially available biologically relevant lipids when it comes to the delivery of bioactive agents, namely the need for extrusion processes and the associated loss of both lipid and bioactive material.
In this study, we examined the release of calcein from liposomes composed of hybrid siloxane-phosphocholines to ascertain the suitability of siloxane-phosphocholines as components in drug delivery systems; POPC, DPPC, and DPhPC were included as commercially available comparators. The release of calcein was carried out at pH 4.5, 7.5 and 10.5 to mimic the different physiological environments within the human body.
Experimental
Materials
Calcein, Novozyme-435 (lipase B acrylic resin from Candida antarctica (≥5000 U g−1)), palmitic acid (≥95% purity), 4-pentenoic acid (≥98% purity), Sepharose 4B, N,N′-dicyclohexylcarbodiimide (DCC, 99% purity), 4-dimethylaminopyridine (DMAP, 99% purity), platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution in xylenes (Karstedt's catalyst), activated carbon, hexanes, ethyl acetate, Celite® 545, and hydrochloric acid were acquired from Sigma-Aldrich (Oakville, Ontario, Canada). 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospcocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), and 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhyPC) were acquired from Avanti Polar Lipids Inc. (Alabaster, Alabama, USA). Glycerophosphocholine (GPC) was acquired from Toronto Research Chemicals (Toronto, Ontario, Canada). 1,1,3,3,3-Pentamethyldisiloxane was acquired from Gelest, Inc. (Morristown, PA, USA). Chloroform-d (99.8% deuterated) and methanol-d4 (MeOH-d4) were obtained from Cambridge Isotope Laboratories, Inc. (Landover, MD, USA). Triton® X-100 was obtained from Alfa Aesar (Ward Hill, MA, USA). Tris-(hydroxymethyl)aminomethane (Tris) was supplied by ACP Chemicals (Montréal, QC, Canada). Bacteria were maintained as stock cultures on Difco Nutrient Agar (NA; Becton-Dickson). Test samples were grown in Difco Nutrient Broth (NB). All materials were used as received unless otherwise stated.Synthesis of lipids
Hybrid SiPCs Cap-ValDS-PC 1, Cpc-ValDS-PC 2, Lau-ValDS-PC 3, Myr-ValDS-PC 4, Pal-ValDS-PC 6, and Ste-ValDS-PC 7 were synthesized as previously described.34 The general procedures for the synthesis of lyso-PCs and hybrid SiPCs are included below. More detailed syntheses and complete spectroscopic characterization can be found in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CHCl3:MeOH. The immobilized lipase beads were removed by filtering the mixture through a medium porosity, glass-fritted Büchner funnel. The solvents were removed in vacuo to give viscous oils. The crude product was purified by column chromatography using 400 mesh silica gel, applied as a silica gel-chloroform slurry. Each column was preconditioned with 10 mL of 9:1 CHCl3:MeOH before loading the crude mixture and eluting with an isocratic elution solvent of 65:25:4 CHCl3:MeOH:H2O.:1 CHCl3:MeOH and the eluted with 65:25:4 CHCl3:MeOH:H2O to give hybrid SiPCs as opaque gels. Detailed synthetic information can be found in the ESI.†
:1 CHCl3:MeOH. The immobilized lipase beads were removed by filtering the mixture through a medium porosity, glass-fritted Büchner funnel. The solvents were removed in vacuo to give viscous oils. The crude product was purified by column chromatography using 400 mesh silica gel, applied as a silica gel-chloroform slurry. Each column was preconditioned with 10 mL of 9:1 CHCl3:MeOH before loading the crude mixture and eluting with an isocratic elution solvent of 65:25:4 CHCl3:MeOH:H2O.:1 CHCl3:MeOH and the eluted with 65:25:4 CHCl3:MeOH:H2O to give hybrid SiPCs as opaque gels. Detailed synthetic information can be found in the ESI.†Ultraviolet/visible (UV/vis) and fluorescence spectroscopy
UV-vis spectra were recorded using a SpectroVis Plus spectrometer/fluorimeter. Wavelength (λmax) selection depended on the nature of the sample (i.e., the pH of the buffer solution). The wavelength of maximum absorbance (λmax) for calcein is typically reported as 490 nm, however, the emission intensity has been shown to vary with pH.39 Since we were interested in examining the release of calcein at various pH values, the emission wavelengths (λem) values were experimentally determined for calcein at pH 4.5, 7.5, and 10.5 (Table 1) and used as appropriate in the subsequent experiments – for all pH values the samples were excited at 405 nm. Absorption and emission spectra were analyzed using the Logger Pro v3.10.2 software platform.| pH | λem |
|---|---|
| 4.5 | 526 |
| 7.5 | 523 |
| 10.5 | 535 |
General method for liposome preparation
Commercially available phospholipids (100 mg) were dissolved into 1.0 mL of chloroform, aliquoted into 10 mg portions in glass ampules and dried under vacuum in a desiccator containing Drierite® for 24 h to produce lipid thin films. A similar methodology was used to prepare thin films of SiPCs – the volume of chloroform was adjusted based on the available mass of the particular SiPC. Ampules were heat-sealed and subsequently stored at −20 °C. Lipid films were rehydrated with 0.7 mL of a 100 mM calcein solution (5 mL of the pH 7.5 Tris buffer to which calcein was added) to make a 20 mM solution of the lipid. The sample was then vortexed at 1500 rpm for 10 min at room temperature to initiate liposome formation (non-extruded liposomes). Samples of liposomes were additionally extruded through a 100 nm Nanosizer MINI Sterile Extruder (T&T Scientific Corporation, Knoxville, TN, USA) (extruded liposomes). The extruder was conditioned with 10 mL of Tris buffer (pH 7.5, 50 mM Tris, 100 mM NaCl). Extrusion was performed with 31 passages through the membrane.General method for liposome purification
Liposomes were isolated from free calcein using size exclusion chromatography (SEC) on Sepaharose 4B. A slurry of Sepharose 4B was prepared applying 5 mL of Sepharose into a polypropylene column. The column was then conditioned with 10 mL of Tris buffer (pH 7.5, 50 mM Tris, 100 mM NaCl) prior to loading the liposome suspension. The calcein-containing liposomes were eluted with Tris buffer to remove unincorporated calcein from liposomes. Unincorporated calcein eluted from the column first, as evidenced by a bright green coloured solution, followed by calcein-containing liposomes, deep red in colour.Dynamic light scattering (DLS)
Siloxane phosphocholines were diluted to a concentration of approximately 30 mg mL−1 in an ultrapure H2O suspension and vortexed for 15 minutes. The DLS (Protein Solutions Dyna Pro Light Scattering System and Temperature Controlled Microsampler) was calibrated using silver nanoparticles in a 70% solution of ethanol from Cytodiagnostics that are 100 nm in diameter with a concentration of 0.02 mg mL−1 and have a refractive index of 0.135. All siloxane phosphocholine measurements were obtained using a quartz cuvette with a 1 cm pathlength. The refractive index was set to 0.16 and the radius was calculated using a globular protein model.Liposome release studies
The liposomes formulated with calcein were analyzed for calcein release immediately after SEC. Liposome suspensions (30 μL) were combined with 3 mL of Tris buffer (50 mM Tris, 100 mM NaCl) at three different pH values (pH 4.5, 7.5, and 10.5) in 5 mL polystyrene fluorescence cuvettes (Spectrecology, Dunedin, FL, USA) using a SpectroVis Plus Fluorimeter with an excitation wavelength of 405 nm with the appropriate maximum emission wavelength for the pH value (Table 2) being recorded. The release profile was then calculated using eqn (1):
 | (1) |
| Total carbon number | Trivial name | Three-letter abbreviation |
|---|---|---|
| C1 | Formic acid | For |
| C2 | Acetic acid | Ace |
| C3 | Propionic acid | Pro |
| C4 | Butyric acid | But |
| C5 | Valeric acid |
Val |
| C6 | Caproic acid | Cpr |
| C7 | Enanthic acid | Ena |
| C8 | Caprylic acid | Cap |
| C9 | Pelargonic acid | Pel |
| C10 | Capric acid | Cpc |
| C11 | Hendecanoic acid | Hen |
| C12 | Lauric acid | Lau |
| C13 | Tridecylic acid | Try |
| C14 | Myristic acid | Myr |
| C15 | Pentadecylic acid | Pen |
| C16 | Palmitic acid | Pal |
| C17 | Margaric acid | Mar |
| C18 | Stearic acid | Ste |
SiPC lipid stability
The lipid (POPC or Myr-ValDS-PC, 10 mg) was added to 1 mL of buffer (pH 4.5, 7.5, or 10.5). Once suspended the lipid was extruded 31 times and allowed to sit at room temperature for 24 h after which time the buffer was removed in vacuo. Following removal of the buffer the lipid was suspended in either CDCl3 (POPC) or MeOD-d4 (Myr-ValDS-PC) and the corresponding 1H NMR spectra were acquired and compared with the virgin lipids.General methods for microbial toxicity studies
Bacteria were grown overnight in NB in a shaking water bath at 28 °C. Hybrid siloxane-phosphocholine lipids (50 μg mL−1, 25 μg mL−1, 12.5 μg mL−1, 6.25 μg mL−1, 3.125 μg mL−1, and 0 μg mL−1) were added to microtitre plate wells containing sufficient bacteria (Erwinia amylovora, Pantoea agglomerans, Bacillus subtilus, Streptococcus saprophyticus) in NB to give an optical density (OD595) of approximately 0.1 (between 5 × 106 and 1 × 107 cells per mL depending upon the species). Each test was repeated eight times. Test plates were incubated at 28 °C with gentle agitation to prevent cell settling. The optical density of bacterial samples was measured at 595 nm in an iMark Microplate Absorbance Reader (Bio-Rad, Mississauga ON) at 0, 2, 4, 6, and 24 h.Results and discussion
Siloxane-containing lipid structures
We have been studying siloxane phosphocholines which feature at least one fatty acid tail that has been modified to contain a pentamethyldisiloxane moiety. We previously reported the synthesis of double tailed siloxane-phosphocholines which featured the enzymatic esterification of siloxane-containing carboxylic acids with glycerol, to generate 1,2- and 1,3-substituted diacylglycerides, which was followed by installation of the phosphocholine head group.31 Both 1,2-SiPC and 1,3-SiPC were found to self-assemble into ULVs with low size dispersity.30 Employing a lipase-mediated esterification to produce lyso-PCs (C8 to C18) substituted in the sn-1 position, we then installed the second fatty acid into the sn-2 position using a modification of the Steglich esterification to provide access to a library of SiPCs (Fig. 1).To aid in the identification of these unique species, a nomenclature system based on a general A–BC–D system (Fig. 2) was developed where “A” represents the trivial name of the carboxylic acid in the sn-1 position, “B” represents the trivial name for the carboxylic acid portion at the sn-2 position, “C” represents the silicon-containing portion at the sn-2 position, and “D” represents the head group at the sn-3 position.
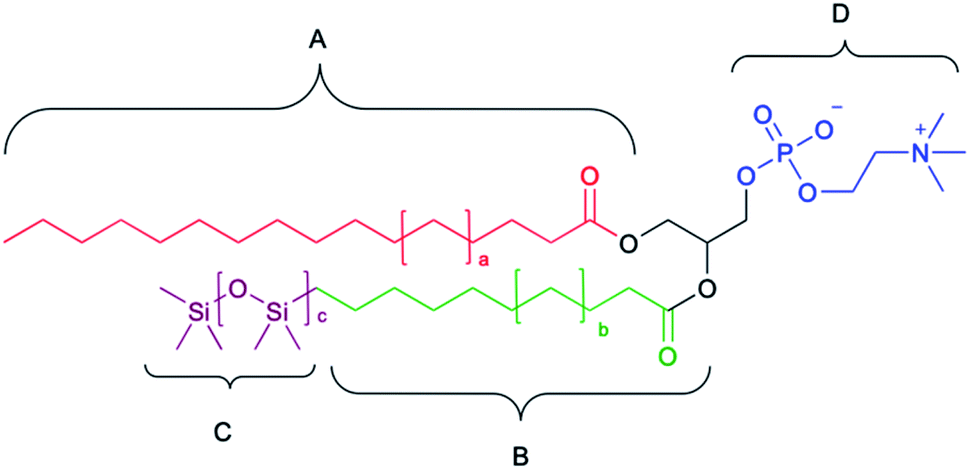 | ||
| Fig. 2 The foundational components for hybrid SiPC nomenclature. | ||
The conventional two-letter abbreviation (e.g., PC, PE, PG, etc.) for the phosphate head groups of biologically relevant phospholipids is employed for “D”. The silicon-containing portion of the lipid is abbreviated “XS”, where “X” represents the first letter of the quantifier (i.e., di-, tri-, etc.) indicating the extent of the siloxane unit, “S”. The trivial name for the carboxylic acid moieties is abbreviated as outlined in Table 2.
We synthesized six hybrid SiPCs (Fig. 1) to examine the release of calcein from 100 nm extruded vesicles and non-extruded liposomes at three different pH values (4.5, 7.5, and 10.5). POPC, DPPC, and DPhPC (Fig. 3), three commercially available phospholipids that have been utilized in release studies and to mimic biological membranes, were included for comparison.45–53 It was of particular interest to include DPhPC in this study given the similarities that had been observed between this lipid and siloxane-containing lipids.31
 | ||
| Fig. 3 Commercially available lipids used as comparators for the calcein entrapment and release experiments. | ||
Bacterial toxicity
Four bacterial species (Erwinia amylovora, Pantoea agglomerans, Bacillus subtilis, Streptococcus saprophyticus) were used to examine the microbial toxicity of the hybrid siloxane-phosphocholine molecules at concentrations of 50.00 μg mL−1, 25.00 μg mL−1, 12.50 μg mL−1, 6.250 μg mL−1, and 3.125 μg mL−1. Toxicity trials were replicated eight-fold and some of the average values are reported in Fig. 4 for example. All of the species tolerated all of the hybrid siloxane-phosphocholine compounds indicating that, for these bacteria at the very least, the lipid compounds were not antimicrobial. At this point it is not clear whether the lipids are being incorporated into the membrane structure or if the lipids are simply being used as a nutrient source by the bacteria. The tolerance of bacteria for the lipids suggested that liposomes based on the hybrid siloxane-phosphocholine molecules have the potential to behave as drug delivery vehicles. | ||
| Fig. 4 Growth of bacterial cultures on hybrid SiPC lipids. OD595 readings of time 0 and time 24 h cultures grown in 50 μg mL−1 lipid. | ||
Dynamic light scattering
Symmetrical, double-tailed SiPCs have been shown to spontaneously self-assemble into ULVs of approximately 120–200 nm diameters upon hydration with ultra-pure water.32 Furthermore, we have shown that these hybrid SiPCs also self-assemble into ULVs.34 The sizes of vesicles formed from hybrid SiPCs were analyzed by DLS and the results are summarized in Table 3. Hybrid SiPCs were found to self-assemble into vesicles of ∼100 nm in diameter, with the exception of Cpc-ValDSPC (2), which was determined to be 168 nm in diameter and Lau-ValDSPC (3), which had a diameter of 364 nm.| Lipid | Diameter (nm) | %Polydispersity |
|---|---|---|
| Cap-ValDS-PC | 97.1 | 0.1 |
| Cpc-ValDS-PC | 168.3 | 0.1 |
| Lau-ValDS-PC | 364.3 | 11.8 |
| Myr-ValDS-PC | 106.3 | 0.1 |
| Pal-ValDS-PC | 94.7 | 0.1 |
| Ste-ValDS-PC | 99.6 | 0.1 |
Calcein entrapment/release studies
The tendency of calcein to self-quench at concentrations over 70 mM makes it a convenient compound for probing the release capacity of liposomal systems.39–43 When entrapped within the liposomes the calcein should produce little to no fluorescence. However, upon exiting the liposomes the calcein will become more dilute and will fluoresce.38–41 The increase in fluorescence intensity serves as an indicator for the rate of release from the liposomes.We have previously reported that Cap-ValDS-PC 1, Cpc-ValDS-PC 2, Lau-ValDS-PC 3, and Myr-ValDS-PC 4 spontaneously form ULVs when dispersed in aqueous media and it was of interest to examine the release of calcein from both extruded and non-extruded liposomal systems. Since the benchmark lipids (POPC, DPPC, and DPhPC) are known to spontaneously form MLVs,32,54 these lipids were all extruded to form ULVs.
Calcein release profiles were very similar in overall appearance across the lipids that were studied (see ESI†), with the vast majority of the liposomes displaying a burst release period followed by a plateau or slower release rate; linear release profiles were typically observed over the first 60–90 min. In these liposomal systems, the release of calcein is best described by the migration of the molecule through the lipid bilayer into the surrounding medium. The movement of calcein across the bilayer does not require the input of energy and relies on the concentration gradient across the lipid membrane.39 Previous work has suggested that calcein migrates through the hydrocarbons portion of the lipid bilayer rather than through pores found in the lipid membrane.42 The observed differences in the rate of release, monitored as an increase in fluorescence can be attributed to the overall ionization state of calcein, the differences in the physical characteristics of the lipid bilayer, the area per lipid, and the electrochemical gradient across the lipid membrane.38 Unsaturated lipids generally possess a larger mean molecular area compared to saturated lipids of the same chain length, and tend to release more of their entrapped cargo.39 SiPCs more closely resemble unsaturated lipids, than saturated lipids, architecturally, and typically have larger molecular volumes than either saturated or unsaturated lipids of comparable chain length.32,34
Not only was it of interest to examine the release profiles of the various liposomal systems, but it was also of interest to correlate these release profiles with the amount of material that was actually released from the liposomes (Table 4). For all of the hybrid SiPC liposomal systems, save for those based on Myr-ValDS-PC, the non-extruded liposomes entrapped a greater amount of calcein than the extruded liposomes (Fig. 5 and Table 4).
| Liposome | pH | Avg. calcein entrapped (mg mL−1) | Avg. calcein released after 6.5 h (%) | Avg. calcein released after 6.5 h (mg mL−1) |
|---|---|---|---|---|
| Cap-ValDS-PC (non-extruded) | 4.5 | 0.029 | 58 | 0.017 |
| 7.5 | 48 | 0.014 | ||
| 10.5 | 66 | 0.019 | ||
| Cap-ValDS-PC (extruded) | 4.5 | 0.009 | 74 | 0.007 |
| 7.5 | 93 | 0.009 | ||
| 10.5 | 63 | 0.006 | ||
| Cpc-ValDS-PC (non-extruded) | 4.5 | 0.029 | 45 | 0.019 |
| 7.5 | 66 | 0.019 | ||
| 10.5 | 69 | 0.020 | ||
| Cpc-ValDS-PC (extruded) | 4.5 | 0.011 | 78 | 0.008 |
| 7.5 | 93 | 0.010 | ||
| 10.5 | 73 | 0.008 | ||
| Lau-ValDS-PC (non-extruded) | 4.5 | 0.026 | 62 | 0.016 |
| 7.5 | 64 | 0.016 | ||
| 10.5 | 71 | 0.018 | ||
| Lau-ValDS-PC (extruded) | 4.5 | 0.009 | 81 | 0.007 |
| 7.5 | 97 | 0.008 | ||
| 10.5 | 70 | 0.006 | ||
| Myr-ValDS-PC (non-extruded) | 4.5 | 0.032 | 68 | 0.022 |
| 7.5 | 64 | 0.20 | ||
| 10.5 | 68 | 0.022 | ||
| Myr-ValDS-PC (extruded) | 4.5 | 0.017 | 72 | 0.012 |
| 7.5 | 78 | 0.014 | ||
| 10.5 | 63 | 0.011 | ||
| Pal-ValDS-PC (non-extruded) | 4.5 | 0.037 | 62 | 0.023 |
| 7.5 | 59 | 0.022 | ||
| 10.5 | 60 | 0.022 | ||
| Pal-ValDS-PC (non-extruded) | 4.5 | 0.013 | 68 | 0.009 |
| 7.5 | 87 | 0.012 | ||
| 10.5 | 60 | 0.008 | ||
| Ste-ValDS-PC (non-extruded) | 4.5 | 0.022 | 56 | 0.012 |
| 7.5 | 56 | 0.012 | ||
| 10.5 | 58 | 0.013 | ||
| Ste-ValDS-PC (extruded) | 4.5 | 0.012 | 74 | 0.009 |
| 7.5 | 93 | 0.011 | ||
| 10.5 | 63 | 0.007 | ||
| POPC (extruded) | 4.5 | 0.013 | 70 | 0.009 |
| 7.5 | 77 | 0.010 | ||
| 10.5 | 51 | 0.006 | ||
| DPPC (extruded) | 4.5 | 0.016 | 64 | 0.010 |
| 7.5 | 75 | 0.012 | ||
| 10.5 | 49 | 0.008 | ||
| DPhPC (extruded) | 4.5 | 0.011 | 65 | 0.007 |
| 7.5 | 86 | 0.009 | ||
| 10.5 | 58 | 0.006 |
 | ||
| Fig. 5 Calcein entrapment efficiency in the liposomal systems using a pH 7.5 buffer. | ||
In all instances the SiPC liposomes that had been extruded released a greater percentage of their cargo than the non-extruded liposomes (Table 4). Extruded vesicles tend to be smaller than unextruded vesicles, leading to decreased packing between lipid molecules. Shimanouchi et al. suggested that liposome size contributes to the permeability of a lipid bilayer.42
In spite of the propensity for Pal-ValDS-PC 6 and Ste-ValDS-PC 7 to spontaneously form MLVs (i.e., larger vesicles) in addition to ULVs. Presumably the majority of the entrapped calcein in these liposomes is found in the central aqueous core of the MLVs and the overall release of calcein would be retarded by the increasing number of lipid bilayers standing between the calcein and the external aqueous environment. In some instances, the number of bilayers present is larger owing to smaller vesicles nested inside of larger vesicles, potentially even after extrusion.55
However, in spite of releasing a lower percentage of the entrapped calcein, the non-extruded liposomes, in all cases, both entrapped and subsequently released a greater overall amount of calcein than the liposomes that had not been subjected to extrusion (Table 4). This suggests that calcein is lost during the extrusion process and is therefore not incorporated into the liposomes. Extrusion of liposomes through a 100 nm filter reduces not only the size of the resulting liposomes, but as a result, the packing of the lipid molecules is reduced. Reduced lipid packing would account for the higher percentage of calcein that was released for the extruded liposomes.39
Degradation of the lipid membrane
The effect of pH on the calcein release was examined. Previous work had shown that in the simulated gastric and duodenal digestion of POPC, lowered pH values resulted in a rapid release of calcein.39 The physical integrity of the liposome remained the same over the two-hour experimental time frame. Upon the addition of bile salts and increasing the pH to 6.5 to mimic duodenal digestion, liposome disruption and the subsequent burst release of calcein was observed. Contrasting these results, we have found that extruded hybrid SiPC liposomes, as well as extruded liposomes of POPC, DPPC, and DPhyPC, released a lower percentage of their calcein at pH 4.5 and 10.5 than at 7.5, suggesting that there is no liposomal degradation at these pH values (see ESI†). Non-extruded liposomes did not generally indicate such differences across pH values.In an effort to gain insights into the release mechanism(s) from the SiPC liposomes, the structural stability of the lipids in acid media were determined; POPC and Myr-ValDS-PC were utilized as models for the commercially available lipids and for the hybrid SiPCs explored in this paper, respectively. The lipid was exposed to buffer (pH 4.5, 7.5, or 10.5) for a period of 24 h. After this time the buffer was removed via freeze drying to yield the isolated lipid species. 1H and 31P NMR analysis of the isolated lipid material (without any purification) produced virtually identical spectra to the pure, virgin, lipid compounds (Fig. 6–9); residual water can be observed in the 1H NMR spectra of both lipids. This data indicated that there was no significant degradation of the lipid structures upon exposure to the buffers used as the release media in these experiments. This lack of observable degradation supports the assertion that lipid degradation does not play a role in calcein being released from liposomes and that release of the entrapped material occurred via simple diffusion through the membrane.
 | ||
| Fig. 6 1H NMR spectra of virgin POPC (purple), POPC exposed to pH 4.5 buffer (green), pH 7.5 buffer (red), and pH 10.5 buffer (blue). Samples possess some entrained water. | ||
 | ||
| Fig. 7 31P NMR spectra of virgin POPC (purple), POPC exposed to pH 4.5 buffer (green), pH 7.5 buffer (red), and pH 10.5 buffer (blue). | ||
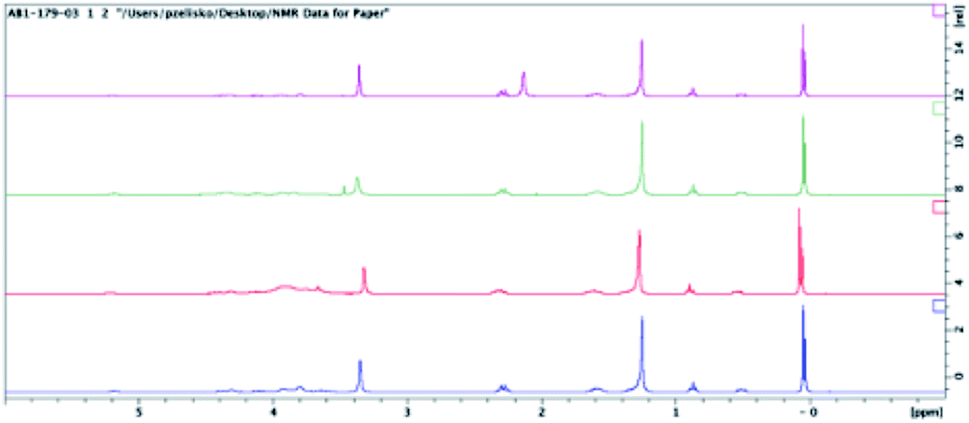 | ||
| Fig. 8 1H NMR spectra of virgin Myr-ValDSPC (purple), Myr-ValDSPC exposed to pH 4.5 buffer (green), pH 7.5 buffer (red), and pH 10.5 buffer (blue). Samples possess some entrained water. | ||
 | ||
| Fig. 9 31P NMR spectra for virgin Myr-ValDSPC (purple), Myr-ValDSPC exposed to pH 4.5 buffer (green), pH 7.5 buffer (red), and pH 10.5 buffer (blue). | ||
Conclusions
Hybrid siloxane-phosphocholine liposome structures did not inhibit the growth of Erwinia amylovora, Pantoea agglomerans, Bacilis subtilis, or Streptococcus saprophyticus. A model compound, calcein, was efficiently entrapped within both extruded and non-extruded liposomes based on hybrid SiPC systems, even in comparison with the control systems. Not surprisingly, the non-extruded liposomes ultimately possessed greater entrapment efficiency than their extruded counterparts, presumably because material was lost on the membrane and in the syringes during the extrusion process. Both the extruded and non-extruded hybrid SiPC liposomes demonstrated moderate to high (>60%) absolute releases of their entrapped cargo; for many of the hybrid SiPC systems this release rate was somewhat greater than the release observed for the model, extruded, biologically relevant, liposomes based on POPC, DPPC, and DPhyPC. Given the propensity for the majority of the hybrid SiPC compounds to spontaneously form ULVs that are ∼100 nm in diameter, these compounds represent a useful tool for not only delivering biologically active compounds, but also for the efficient entrapment of said compounds when compared to commercially available biologically relevant lipids currently being explored for the delivery of active compounds.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would thank the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Brock University Advanced Biomanufacturing Centre (ABC) for funding. ZR was the recipient of an NSERC Undergraduate Student Research Award (USRA). The authors would also like to thank Prof. T. Yan (Brock University) and Jesse Vanloon (Brock University) for their assistance with the DLS.Notes and references
- D. Jain, S. S. Mahammad, P. P. Sing and R. Kodipyaka, Drug Dev. Ind. Pharm., 2019, 45, 1403 CrossRef CAS PubMed.
- T. G. Barclay, C. M. Day, N. Petrovsky and S. Garg, Carbohydr. Polym., 2019, 221, 94 CrossRef CAS PubMed.
- P. H. L. Tran, W. Duan, B.-J. Lee and T. T. D. Tran, Int. J. Pharm., 2019, 566, 557 CrossRef CAS PubMed.
- G. Yang, Y. Lu, H. N. Bomba and Z. Gu, Curr. Med. Chem., 2019, 26, 1377 CrossRef CAS PubMed.
- D. Warther, Y. Xiao, F. Li, Y. Wang, K. Huffman, W. R. Freeman, M. Sailor and L. Cheng, Drug Delivery, 2018, 25, 1537 CrossRef CAS PubMed.
- W. Chen, C. A. Glackin, M. A. Horwitz and J. I. Zink, Acc. Chem. Res., 2019, 52, 1531 CrossRef CAS PubMed.
- P. M. Zelisko, K. K. Flora, J. D. Brennan and M. A. Brook, Biomacromolecules, 2008, 9, 2153 CrossRef CAS PubMed.
- P. M. Zelisko, A. Lopez Aguilar and M. A. Brook, Langmuir, 2007, 23, 3620 CrossRef CAS PubMed.
- P. M. Zelisko and M. A. Brook, Langmuir, 2002, 18, 8982 CrossRef CAS.
- M. Piffoux, A. Nicolás-Boluda, V. Mulens-Arias, S. Richard, G. Rahmi, F. Gazeau, C. Wilhelm and A. K. A. Silva, Adv. Drug Delivery Rev., 2019, 138, 247 CrossRef CAS PubMed.
- J. M. Gudbergsson, K. Jønsson, J. B. Simonsen and K. B. Johnsen, Adv. Drug Delivery Rev., 2019, 306, 108 CAS.
- J. C. Cuggino, E. R. O. Blanco, L. M. Gugliotta, C. I. A. Igarzabal and M. Calderón, Adv. Drug Delivery Rev., 2019, 307, 221 CAS.
- V. Nekkanti and S. Kalepu, Pharm. Nanotechnol., 2015, 3, 35 CrossRef CAS.
- S. S. Lokhande, Pharma Sci. Monit., 2018, 9, 188 CAS.
- M. Singh, S. Devi, V. S. Rana, B. B. Mishra, J. Kumar and V. Ahluwalia, J. Microencapsulation, 2019, 36, 215 CrossRef CAS PubMed.
- C. Zylberberg and S. Matosevic, Drug Delivery, 2016, 23, 3319 CrossRef CAS PubMed.
- J.-S. Kim, J. Pharm. Invest., 2016, 46, 387 CrossRef CAS.
- N. Oku, Biol. Pharm. Bull., 2017, 40, 119 CrossRef CAS PubMed.
- C. Y. Wong, H. Al-Salami and C. R. Dass, Int. J. Pharm., 2018, 549, 201 CrossRef CAS PubMed.
- A. S. Abu Lila and T. Ishida, Biol. Pharm. Bull., 2017, 40, 1 CrossRef CAS PubMed.
- V. Sreekanth and A. Bajaj, ACS Biomater. Sci. Eng., 2019, 5, 4148–4166 CrossRef CAS PubMed.
- H. Daraee, A. Etemadi, M. Kouhi, S. Alimizalu and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 381 CrossRef CAS PubMed.
- T. Imai, Y. Takahashi, M. Nishikawa, K. Kato, M. Morishita, T. Yamashita, A. Matsumoto, C. Charoenviriyakul and Y. Takakura, J. Extracell. Vesicles, 2015, 4 DOI:10.3402/jev.v4.26238.
- G. Bozzuto and A. Molinari, Int. J. Nanomed., 2015, 10, 975 CrossRef CAS PubMed.
- R. L. Juliano and D. Stamp, Biochem. Biophys. Res. Commun., 1973, 63, 651 CrossRef.
- D. B. Fenske and P. R. Cullis, Phys. Can., 2004, 60, 187 Search PubMed.
- J. Lewandowska, M. Kępczyński, J. Bednar, E. Rzad, V. Moravcikova, B. Jachimska and M. Novakowska, Colloid Polym. Sci., 2010, 288, 37 CrossRef CAS.
- V. J. Mohanraj, T. J. Barnes and C. A. Prestidge, Int. J. Pharm., 2010, 392, 285 CrossRef CAS.
- J. Lewandowska-Łańucka, K. Mystek, A. Gilarska, K. Kamiński, M. Romek, B. Sulikowski and M. Nowakowska, Colloids Surf., B, 2016, 143, 359 CrossRef PubMed.
- R. K. Kallury, U. J. Krull and M. Thompson, J. Org. Chem., 1987, 52, 5478 CrossRef CAS.
- K. Yasuhara, S. Miki, H. Nakazono, A. Ohta and J. Kikuchi, Chem. Commun., 2011, 47, 4691 RSC.
- M. B. Frampton, D. Marquardt, I. Letovsky-Papst, G. Papst and P. M. Zelisko, Langmuir, 2017, 33, 4948 CrossRef CAS PubMed.
- M. B. Frampton and P. M. Zelisko, Eur. J. Lipid Sci. Technol., 2017, 119 DOI:10.1002/ejlt.201600248.
- M. B. Frampton, M. H. Nguyen, M. DiPasquale, R. Dick, D. Marquardt and P. M. Zelisko, Chem. Phys. Lipids, 2018, 216, 1 CrossRef CAS PubMed.
- H. Binder and K. Gawrisch, Biophys. J., 2001, 81, 969 CrossRef CAS PubMed.
- R. Thakur, A. Das and A. Charaborty, RSC Adv., 2014, 4, 14335 RSC.
- A. Das, C. Adhikari and A. Chakraborty, Langmuir, 2016, 32, 8889 CrossRef CAS PubMed.
- A. Yasmann and S. Sukharev, Langmuir, 2015, 31, 350 CrossRef CAS PubMed.
- B. Maherani, E. Arab-Tehrany, A. Kheirolomoom, D. Geny and M. Linder, Biochimie, 2013, 95, 2018 CrossRef CAS PubMed.
- H. Heerklotz and J. Seelig, Eur. Biophys. J., 2007, 36, 305 CrossRef CAS PubMed.
- T. Wieprecht, M. Dathe, R. M. Epand, M. Beyermann, E. Krause, W. L. Maloy, D. L. MacDonald and M. Bienert, Biochemistry, 1997, 36, 12869 CrossRef CAS PubMed.
- T. Shimanouchi, H. Ishii, N. Yoshimoto, H. Umakoshi and R. Kuboi, Colloids Surf., B, 2009, 73, 156 CrossRef CAS PubMed.
- G. D. Sprott, C. J. Dicaire, L. P. Flemming and G. P. Patel, Cells Mater., 1996, 6, 143 CAS.
- J. Davidsen, K. Jørgensen, T. L. Andresen and O. G. Moritsen, Biochim. Biophys. Acta, 2003, 1609, 95 CrossRef CAS.
- S. Punnamaraju, H. You and A. J. Steckl, Langmuir, 2012, 28, 7657 CrossRef CAS PubMed.
- N. Essam Eldin, A. S. Abu Lila, K. Kawazoe, H. M. Elnahas, M. A. Mahdy and T. Ishida, Eur. J. Pharm. Sci., 2016, 81, 60 CrossRef PubMed.
- A. Rodríguez-Pulido, A. I. Kondrachuk, D. K. Prusty, J. Gao, M. A. Loi and A. Herrmann, Angew. Chem., Int. Ed., 2013, 52, 1008 CrossRef PubMed.
- C. F. de Freitas, I. R. Calori, A. L. Tessarob, W. Caetano and N. Hioka, Colloids Surf., B, 2019, 181, 837 CrossRef CAS PubMed.
- N. Pippa, A. Meristoudi, S. Pispas and C. Demetzos, Int. J. Pharm., 2015, 485, 374 CrossRef CAS PubMed.
- S. Damiati, An. Scheberl, S. Zayni, S. A. Damiati, B. Schuster and U. B. Kompella, Biophys. Chem., 2019, 251, 106178 CrossRef CAS PubMed.
- B. L. Spaller, J. M. Trieu and P. F. Almeida, J. Membr. Biol., 2013, 246, 257 CrossRef CAS PubMed.
- T. Lu and T. L. M. ten Hagen, Adv. Drug Delivery Rev., 2017, 247, 64 CAS.
- A. Svetlova, J. Ellieroth, F. Milos, V. Maybeck and A. Offenhäusser, Langmuir, 2019, 35, 8076 CrossRef CAS PubMed.
- M.-P. Nieh, J. Katsaras and X. Q. Biochim, Biochim. Biophys. Acta, 2008, 1778, 1467 CrossRef CAS PubMed.
- H. L. Scott, A. Skinkle, E. G. Kelley, M. N. Waxham, I. Levental and F. A. Heberle, Biophys. J., 2019, 117, 1381 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10052h |
| ‡ Current address: Loyalist College, Belleville, Ontario, Canada. |
| This journal is © The Royal Society of Chemistry 2021 |