
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA Brønsted acidic ionic liquid anchored to magnetite nanoparticles as a novel recoverable heterogeneous catalyst for the Biginelli reaction†
Hourieh Sadat Oboudatian ,
Hossein Naeimi and
Mohsen Moradian*
,
Hossein Naeimi and
Mohsen Moradian*
Department of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan, 87317, I. R. Iran. E-mail: m.moradian@kashanu.ac.ir; Fax: +98-03155912397; Tel: +98-03155913055
First published on 11th February 2021
Abstract
In this study, simple and effective methods were used for the preparation of an ionic liquid that immobilized magnetite nanoparticles. Fe3O4 nanoparticles were prepared via a chemical co-precipitation method. Then, a SiO2 shell was coated on the magnetic core via the Stober method. Finally, CPTES (chloropropyltriethoxysilane) and morpholine were coated on the SiO2 shell. Morpholine sulfate, an acidic ionic liquid, was successfully bound to magnetite nanoparticles (Mag@Morph-AIL) and this was used as an efficient catalyst for the preparation of 3,4-dihydropyrimidinones. Compared to previous works, the easy separation of the nanocatalyst using an external magnet and the recyclability, non-toxicity, versatility, and high stability of the catalyst, combined with low reaction times and excellent yields, make the present protocol very useful for the synthesis of the title products. The synthesized products and catalyst were confirmed via 1H-NMR, 13C-NMR, FT-IR, scanning electron microscope, X-ray diffraction, and elemental analysis.
Introduction
Dihydropyrimidinone and its derivatives have attracted significant attention because of their potential activities as calcium channel modulators, antihypertensive agents, and antagonists agents for neuropeptide Y (NPY) and α-1a-adrenergic receptors.1–3 Moreover, these compounds are bio-isosteres of dihydropyridines, which have attracted widespread attention because of their vital pharmaceutical and therapeutic properties, for example, anticancer,4 antifungal,5 anti-inflammatory,6 antibacterial,7 antiviral,8 antidiabetic,9 antithyroid,10 antimuscarinic,11 and hypolipidemic activities.8 Besides, various alkaloids including the dihydropyrimidine moiety have been isolated from natural and marine sources, and they also display interesting biological properties. Hence, the synthesis of a variety of these molecules is desirable and of much current importance.12The synthesis of 3,4-dihydropyrimidinone compounds can be performed under different conditions, including under strongly acidic conditions in a protic solvent.13–16 It has traditionally been catalysed using strong Brønsted acids; Lewis acids;17–21 homogeneous acid catalysts, such as polyphosphate esters,22 ZnCl2, or CuCl2;23 heterogeneous acid catalysts, like Al-MCM-41 or FeCl3 embedded in Al-MCM-41;24 nanocomposites;25,26 nanoparticles;27 magnetic nanoparticles;28 metal oxides; and enzymes. Also, this reaction can be modified using microwave irradiation,29,30 ultrasound irradiation,31 ionic liquids,32–34 and organocatalysis,35 or using a combination of these.
However, despite their potential utility, many of these reported one-pot protocols suffer from shortcomings, such as the use of expensive reagents, long reaction times, low yields, and volatile strongly acidic conditions. Therefore, the introduction of milder and more efficient methods with higher yields is needed. We have investigated the synthesis of 3,4-dihydropyrimidinone derivatives utilizing Fe3O4@SiO2 nanoparticles as recyclable catalysts and eco-friendly materials, based on the principles of green chemistry and with the aim of developing an ideal synthesis approach. Magnetic iron oxide nanoparticles have attracted much research interest in recent years because of their unique physicochemical properties and great potential for various biomedical applications.
The reactivity of catalytic nanoparticles is largely determined by the energy of surface atoms, which can be easily gauged based on the number of neighboring atoms, the bonding modes, and the accompanying energies of the small molecules to be transformed on the surfaces of nanoparticles.36,37 Applications of magnetic nanoparticles (MNPs) in different industrial and biological fields, such as in magnetic resonance imaging, drug delivery, bioseparation, hyperthermia treatment, and catalytic processes, have been demonstrated.36–39 Among the various magnetic nanoparticles under investigation, Fe3O4 nanoparticles (Fe3O4 NPs) are arguably the most extensively studied NPs.
The main characteristics of these nanoparticles are their simplicity and their easy separation from reaction media using a magnetic field. Furthermore, the surfaces of magnetic metal oxide NPs, such as magnetic Fe3O4 nanoparticles, can be functionalized and modified with various organic and inorganic materials.40,41 In this study, a morpholine sulfate acidic ionic liquid was bonded to magnetite nanoparticles (Mag@Morph-AIL) as an efficient catalyst, and it was used for the preparation of 3,4-dihydropyrimidinones. Also, we managed to synthesize two new derivatives of 3,4-dihydropyrimidinones (4k and 4o).
Experimental
Materials and method
The chemicals used in this work were purchased from Fluka and Merck Chemical Companies and were used without purification. Infrared (IR) spectra were obtained in KBr pellet form using a PerkinElmer 781 spectrophotometer and an Impact 400 Nicolet FT-IR spectrophotometer. Proton and carbon nuclear magnetic resonance (1H-NMR and 13C-NMR) spectra were recorded in CDCl3 or DMSO solvent using a Bruker DRX-400 spectrometer with tetramethylsilane as an internal reference. The X-ray diffraction analysis (XRD) patterns of samples were obtained using a Philips Xpert X-ray powder diffractometer (CuK radiation, λ = 0.154056 nm). Energy-dispersive X-ray spectroscopy (EDX) spectra of the nanoparticles were obtained using a Sigma ZEISS, Oxford Instruments field-emission scanning electron microscope. The melting points of products were determined using Electrothermal 9200 apparatus and values were uncorrected. Determination of the purity of substrates and reaction monitoring was accomplished via thin-layer chromatography (TLC) on silica gel Polygram SILG/UV 254 plates (from Merck Company). Thermogravimetric and differential thermal analysis (TGA-DTA) data were obtained using a Bahr STA-503 instrument in air at a heating rate of 10 °C min−1. Transmission electron microscopy (TEM) images were recorded using a Zeiss-EM10C microscope with an acceleration voltage of 100 kV.General procedure for the synthesis of the functionalized magnetic nanocatalyst
General procedure for the synthesis of 3,4-dihydropyrimidinones
Ethyl acetoacetate (1 mmol), aromatic aldehyde (1 mmol), urea (1.5 mmol) and 0.06 g of Mag@Morph-AIL as catalyst were added to 30 mL of absolute ethanol in a 50 mL round-bottomed flask, and the mixture was mechanically stirred at 80 °C for an appropriate amount of time. After the completion of the reaction as monitored via TLC, 10 mL of ethanol was added to the reaction mixture and stirring was continued at ambient temperature for 10 min. The catalyst was easily isolated using a bar magnet, and the product was obtained after the removal of the solvent under reduced pressure. Then, the product was treated with water followed by crystallization from EtOH. The products were characterized based on 1H-NMR, FT-IR, and melting point analysis, and the spectral data of the synthesized compounds were compared with authentic samples. The data is given below.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1647, 1464 (CC), 1223, 1092 (C–O); 1H-NMR (CDCl3, 400 MHz) δ (ppm): 1.18 (t, J = 14 Hz, 3H, CH3), 2.36 (s, 3H, CH3), 4.07 (q, 2H, CH2), 5.40 (s, 1H, CH), 5.64 (s, 1H, NH), 7.28–7.33 (m, 5H), 7.72 (s, 1H, NH).O), 1643, 1444 (CCarom), 1221, 1080 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 0.97 (t, J = 14 Hz, 3H, CH3), 2.28 (s, 3H, CH3), 3.88 (q, 2H, CH2O), 5.61 (s, 1H, CH), 7.21–7.37 (m, 3H), 7.39 (d, J = 6.8 Hz, 1H), 7.71 (s, 1H, NH), 9.26 (s, 1H, NH).O), 1647, 1461 (CC), 1222, 1089 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.07 (t, J = 12.4 Hz, 3H, CH3), 2.23 (s, 3H, CH3), 3.95 (q, 2H, CH2O), 5.12 (s, 1H, CH), 7.23 (d, J = 7.2 Hz, 2H), 7.38 (d, J = 6 Hz, 2H), 7.77 (s, 1H, NH), 9.25 (s, 1H, NH).O), 1639, 1455 (CC), 1222, 1092 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.10 (t, J = 14 Hz, 3H, CH3), 2.22 (s, 3H, CH3), 3.98 (q, 2H, CH2O), 5.08 (s, 1H, CH), 6.58–6.70 (m, 3H), 7.08 (dd, J = 15.2 Hz, 1H), 7.68 (s, 1H, NH), 9.15 (s, 1H, NH), 9.36 (1H, OH).O), 1643 (CC), 1456 (CC), 1225, 1096 (C–O); 1H-NMR (CDCl3, 400 MHz) δ (ppm): 1.00 (t, J = 12 Hz, 3H, CH3), 2.27 (s, 3H, CH3), 3.90 (q, 2H, CH2O), 5.58 (s, 1H, CH), 7.41 (d, J = 8 Hz, 1H), 7.31 (d, J = 6 Hz, 1H), 7.55 (s, 1H, NH), 7.75 (s, 1H), 9.31 (s, 1H, NH).O), 1639, 1450 (CC), 1227, 1096 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.06 (t, J = 12 Hz, 3H, CH3), 2.44 (s, 3H, CH3), 4.01 (q, 2H, CH2O), 5.85 (s, 1H, CH), 7.16 (dd, J = 14.8 Hz, 1H), 7.23–7.28 (m, 2H), 7.27 (s, 1H, NH), 7.55 (d, J = 8 Hz, 1H), 8.82 (s, 1H, NH).O), 1642, 1459 (CC), 1228, 1100 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.12 (t, J = 12 Hz, 3H, CH3), 2.41 (s, 3H, CH3), 4.04 (q, 2H, CH2O), 5.65 (s, 1H, CH), 5.7 (d, J = 14.4 Hz, 1H), 7.03 (d, J = 8 Hz, 1H), 7.08 (dd, J = 12 Hz, 1H), 7.14 (s, 1H, NH), 7.23 (dd, J = 16 Hz, 1H), 8.16 (s, 1H, NH).O), 1628, 1455 (CC), 1526, 1374 (N–O), 1223, 1088 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.08 (t, J = 13.6 Hz, 3H, CH3), 2.25 (s, 3H, CH3), 3.99 (q, 2H, CH2O), 5.28 (s, 1H, CH), 7.63 (d, J = 7.6 Hz, 1H), 7.68 (dd, J = 20 Hz, 1H), 7.91 (s, 1H, NH), 8.07 (s, 1H), 8.13 (d, J = 7.6 Hz, 1H), 9.38 (s, 1H, NH).O), 1644, 1463 (CC), 1521, 1394 (N–O), 1217, 1092 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.09 (t, J = 13.2 Hz, 3H, CH3), 2.25 (s, 3H, CH3), 3.97 (q, 2H, CH2O), 5.26 (s, 1H, CH), 7.49 (d, J = 7.2 Hz, 2H), 7.89 (s, 1H, NH), 8.21 (d, J = 7.2 Hz, 2H), 9.36 (s, 1H, NH).O), 1635, 1451 (CC), 1230, 1099 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 0.17 (t, J = 14 Hz, 3H, CH3), 2.22 (s, 3H, CH3), 3.34 (q, 2H, CH2O), 3.39 (s, 1H, CH), 7.00 (d, J = 10 Hz, 1H), 7.49 (t, J = 20 Hz, 4H), 7.69 (s, 1H, NH), 8.07 (d, J = 8 Hz, 2H), 8.41 (s, 1H), 8.55 (d, J = 13.6 Hz, 1H), 9.26 (s, 1H, NH).O), 1673 (CO), 1639, 1447 (CC), 1223, 1087 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.00 (t, J = 8 Hz, 3H, CH3), 2.30 (s, 3H, CH3), 3.90 (q, 2H, CH2O), 5.60 (s, 1H, CH), 7.64 (s, 1H, NH), 7.67 (d, J = 8 Hz, 1H), 7.87 (s, 1H), 7.92 (d, J = 8 Hz, 1H), 7.99 (d, J = 8 Hz, 2H), 8.15 (dd, J = 24 Hz, 2H), 9.30 (s, 1H, NH).O), 1647, 1460 (CC), 1222, 1089 (C–O), 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.11 (t, J = 8 Hz, 3H, CH3), 2.22 (s, 3H, CH3), 2.24 (s, 3H, CH3), 3.95 (q, 2H, CH2O), 5.09 (s, 1H, CH), 7.06 (d, J = 9.6 Hz, 2H), 7.10 (d, J = 9.6 Hz, 2H), 7.66 (s, 1H, NH), 9.15 (s, 1H, NH).O), 1652, 1459 (CC), 1232, 1092 (C–O); 1H-NMR (CDCl3, 400 MHz) δ (ppm): 1.20 (t, J = 14 Hz, 3H, CH3), 2.35 (s, 3H, CH3), 3.84 (s, 3H, CH3), 3.87 (s, 3H, CH3), 4.10 (q, 2H, CH2O), 5.61 (s, 1H, CH), 5.66 (s, 1H), 6.75 (s, 1H, NH), 6.80 (d, J = 6.4 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 7.89 (s, 1H, NH).O), 1646, 1453 (CC), 1225, 1090 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.09 (t, J = 12 Hz, 3H, CH3), 2.21 (s, 3H, CH3), 2.83 (s, 6H, CH3), 3.94 (q, 2H, CH2O), 5.01 (s, 1H, CH), 6.64 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 7.57 (s, 1H, NH), 9.26 (s, 1H, NH).O), 1651, 1466 (CC), 1219, 1085 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 0.84 (d, J = 6 Hz, 6H, CH3), 1.10 (q, 2H, CH2), 1.18 (t, J = 14 Hz, 3H, CH3), 1.35 (q, 2H, CH2), 1.69 (m, 1H, CH), 2.14 (s, 3H, CH3), 3.32 (q, 2H, CH2O), 4.04 (t, J = 12.8 Hz, 1H, CH), 7.43 (s, 1H, NH), 8.95 (s, 1H, NH).
O), 1647, 1464 (CC), 1223, 1092 (C–O); 1H-NMR (CDCl3, 400 MHz) δ (ppm): 1.18 (t, J = 14 Hz, 3H, CH3), 2.36 (s, 3H, CH3), 4.07 (q, 2H, CH2), 5.40 (s, 1H, CH), 5.64 (s, 1H, NH), 7.28–7.33 (m, 5H), 7.72 (s, 1H, NH).O), 1643, 1444 (CCarom), 1221, 1080 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 0.97 (t, J = 14 Hz, 3H, CH3), 2.28 (s, 3H, CH3), 3.88 (q, 2H, CH2O), 5.61 (s, 1H, CH), 7.21–7.37 (m, 3H), 7.39 (d, J = 6.8 Hz, 1H), 7.71 (s, 1H, NH), 9.26 (s, 1H, NH).O), 1647, 1461 (CC), 1222, 1089 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.07 (t, J = 12.4 Hz, 3H, CH3), 2.23 (s, 3H, CH3), 3.95 (q, 2H, CH2O), 5.12 (s, 1H, CH), 7.23 (d, J = 7.2 Hz, 2H), 7.38 (d, J = 6 Hz, 2H), 7.77 (s, 1H, NH), 9.25 (s, 1H, NH).O), 1639, 1455 (CC), 1222, 1092 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.10 (t, J = 14 Hz, 3H, CH3), 2.22 (s, 3H, CH3), 3.98 (q, 2H, CH2O), 5.08 (s, 1H, CH), 6.58–6.70 (m, 3H), 7.08 (dd, J = 15.2 Hz, 1H), 7.68 (s, 1H, NH), 9.15 (s, 1H, NH), 9.36 (1H, OH).O), 1643 (CC), 1456 (CC), 1225, 1096 (C–O); 1H-NMR (CDCl3, 400 MHz) δ (ppm): 1.00 (t, J = 12 Hz, 3H, CH3), 2.27 (s, 3H, CH3), 3.90 (q, 2H, CH2O), 5.58 (s, 1H, CH), 7.41 (d, J = 8 Hz, 1H), 7.31 (d, J = 6 Hz, 1H), 7.55 (s, 1H, NH), 7.75 (s, 1H), 9.31 (s, 1H, NH).O), 1639, 1450 (CC), 1227, 1096 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.06 (t, J = 12 Hz, 3H, CH3), 2.44 (s, 3H, CH3), 4.01 (q, 2H, CH2O), 5.85 (s, 1H, CH), 7.16 (dd, J = 14.8 Hz, 1H), 7.23–7.28 (m, 2H), 7.27 (s, 1H, NH), 7.55 (d, J = 8 Hz, 1H), 8.82 (s, 1H, NH).O), 1642, 1459 (CC), 1228, 1100 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.12 (t, J = 12 Hz, 3H, CH3), 2.41 (s, 3H, CH3), 4.04 (q, 2H, CH2O), 5.65 (s, 1H, CH), 5.7 (d, J = 14.4 Hz, 1H), 7.03 (d, J = 8 Hz, 1H), 7.08 (dd, J = 12 Hz, 1H), 7.14 (s, 1H, NH), 7.23 (dd, J = 16 Hz, 1H), 8.16 (s, 1H, NH).O), 1628, 1455 (CC), 1526, 1374 (N–O), 1223, 1088 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.08 (t, J = 13.6 Hz, 3H, CH3), 2.25 (s, 3H, CH3), 3.99 (q, 2H, CH2O), 5.28 (s, 1H, CH), 7.63 (d, J = 7.6 Hz, 1H), 7.68 (dd, J = 20 Hz, 1H), 7.91 (s, 1H, NH), 8.07 (s, 1H), 8.13 (d, J = 7.6 Hz, 1H), 9.38 (s, 1H, NH).O), 1644, 1463 (CC), 1521, 1394 (N–O), 1217, 1092 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.09 (t, J = 13.2 Hz, 3H, CH3), 2.25 (s, 3H, CH3), 3.97 (q, 2H, CH2O), 5.26 (s, 1H, CH), 7.49 (d, J = 7.2 Hz, 2H), 7.89 (s, 1H, NH), 8.21 (d, J = 7.2 Hz, 2H), 9.36 (s, 1H, NH).O), 1635, 1451 (CC), 1230, 1099 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 0.17 (t, J = 14 Hz, 3H, CH3), 2.22 (s, 3H, CH3), 3.34 (q, 2H, CH2O), 3.39 (s, 1H, CH), 7.00 (d, J = 10 Hz, 1H), 7.49 (t, J = 20 Hz, 4H), 7.69 (s, 1H, NH), 8.07 (d, J = 8 Hz, 2H), 8.41 (s, 1H), 8.55 (d, J = 13.6 Hz, 1H), 9.26 (s, 1H, NH).O), 1673 (CO), 1639, 1447 (CC), 1223, 1087 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.00 (t, J = 8 Hz, 3H, CH3), 2.30 (s, 3H, CH3), 3.90 (q, 2H, CH2O), 5.60 (s, 1H, CH), 7.64 (s, 1H, NH), 7.67 (d, J = 8 Hz, 1H), 7.87 (s, 1H), 7.92 (d, J = 8 Hz, 1H), 7.99 (d, J = 8 Hz, 2H), 8.15 (dd, J = 24 Hz, 2H), 9.30 (s, 1H, NH).O), 1647, 1460 (CC), 1222, 1089 (C–O), 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.11 (t, J = 8 Hz, 3H, CH3), 2.22 (s, 3H, CH3), 2.24 (s, 3H, CH3), 3.95 (q, 2H, CH2O), 5.09 (s, 1H, CH), 7.06 (d, J = 9.6 Hz, 2H), 7.10 (d, J = 9.6 Hz, 2H), 7.66 (s, 1H, NH), 9.15 (s, 1H, NH).O), 1652, 1459 (CC), 1232, 1092 (C–O); 1H-NMR (CDCl3, 400 MHz) δ (ppm): 1.20 (t, J = 14 Hz, 3H, CH3), 2.35 (s, 3H, CH3), 3.84 (s, 3H, CH3), 3.87 (s, 3H, CH3), 4.10 (q, 2H, CH2O), 5.61 (s, 1H, CH), 5.66 (s, 1H), 6.75 (s, 1H, NH), 6.80 (d, J = 6.4 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 7.89 (s, 1H, NH).O), 1646, 1453 (CC), 1225, 1090 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 1.09 (t, J = 12 Hz, 3H, CH3), 2.21 (s, 3H, CH3), 2.83 (s, 6H, CH3), 3.94 (q, 2H, CH2O), 5.01 (s, 1H, CH), 6.64 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 7.57 (s, 1H, NH), 9.26 (s, 1H, NH).O), 1651, 1466 (CC), 1219, 1085 (C–O); 1H-NMR (DMSO-d6, 400 MHz) δ (ppm): 0.84 (d, J = 6 Hz, 6H, CH3), 1.10 (q, 2H, CH2), 1.18 (t, J = 14 Hz, 3H, CH3), 1.35 (q, 2H, CH2), 1.69 (m, 1H, CH), 2.14 (s, 3H, CH3), 3.32 (q, 2H, CH2O), 4.04 (t, J = 12.8 Hz, 1H, CH), 7.43 (s, 1H, NH), 8.95 (s, 1H, NH).Results and discussion
Structural analysis of the Mag@Morph-AIL nanocatalyst
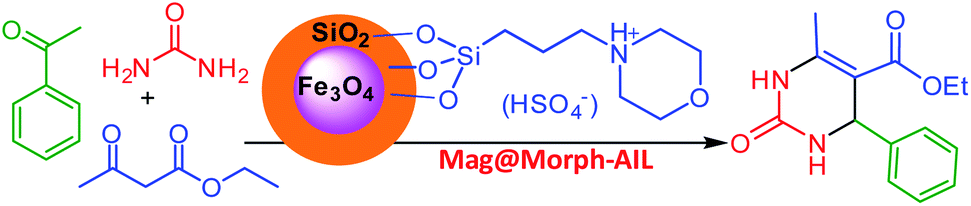
In this work, green and effective methods were used to produce Fe3O4@SiO2/TES-Mo+HSO4− nanoparticles. Fe3O4 nanoparticles were prepared via a chemical co-precipitation method. Then, a SiO2 shell was coated on the magnetite core via the Stober method. Finally, CPTES (chloropropyltriethoxysilane) and morpholine were coated on the SiO2 shell. The Mag@Morph-AIL nanoparticles were an efficient catalyst that were used for the preparation of 3,4-dihydropyrimidinones (Scheme 1).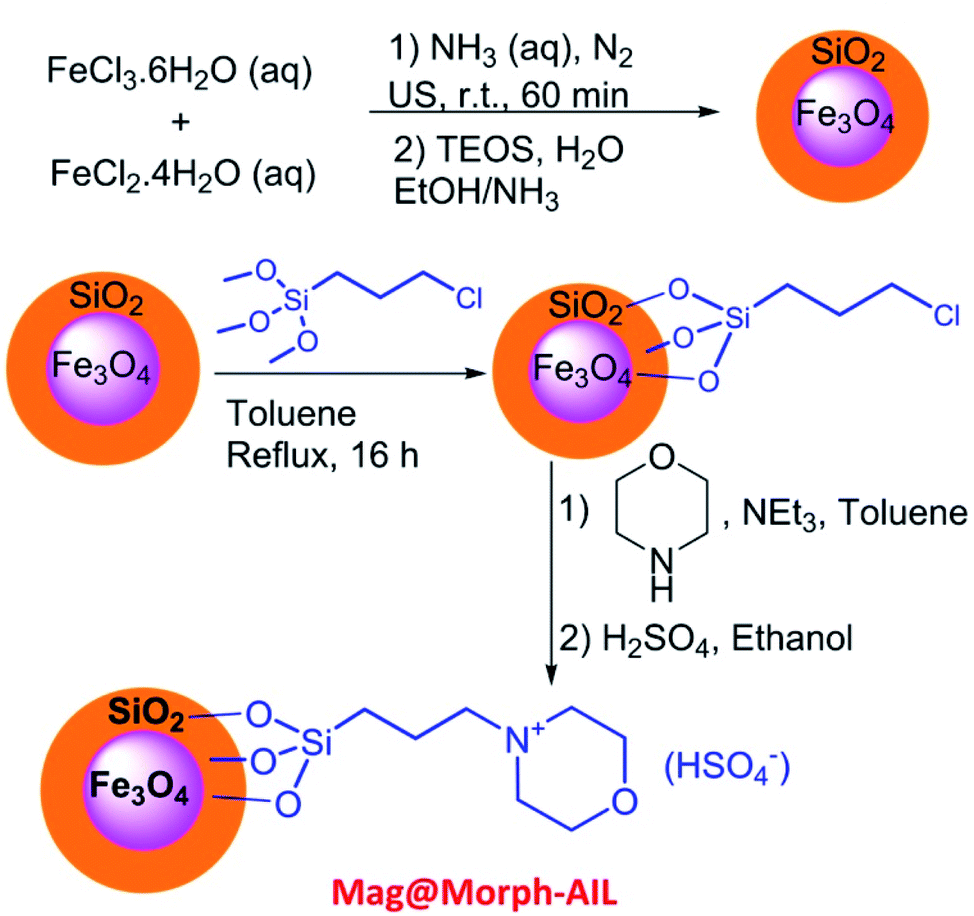 | ||
| Scheme 1 The preparation of the catalyst Mag@Morph-AIL. | ||
FT-IR spectra of Fe3O4, Fe3O4@SiO2, and Mag@Morph-AIL are shown in Fig. 1. In the FT-IR spectrum of Fe3O4, the peaks at 3410 and 1625 cm−1 correspond to O–H stretching and bending vibrations, respectively, and the peak at 583 cm−1 corresponds to Fe–O stretching vibrations. In the FT-IR spectrum of Fe3O4@SiO2 (Fig. 1b), the peak at 1092 cm−1 corresponds to Si–O stretching vibrations.
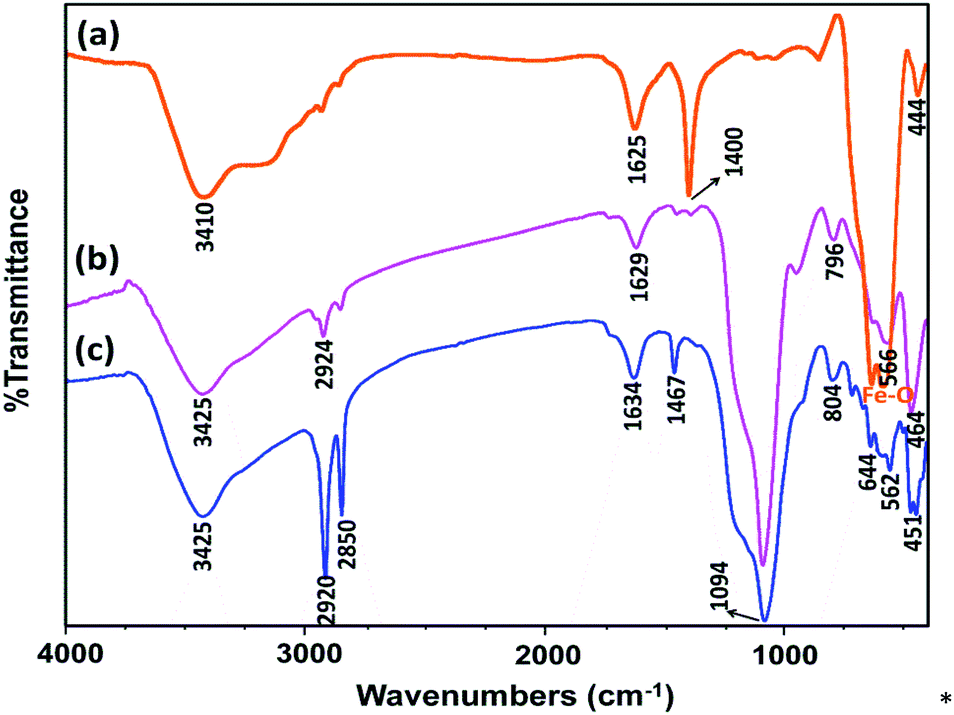 | ||
| Fig. 1 FT-IR spectra of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Mag@Morph-AIL. | ||
In the FT-IR spectrum of Mag@Morph-AIL (Fig. 1c), the SO stretching vibrations of the –HSO4 group appeared at 1100–1250 cm−1 with a high relative intensity. The peaks at 1467 and 1634 cm−1 correspond to N–H and O–H bending vibrations, respectively, and the peak at 3425 cm−1 corresponds to N–H and O–H stretching vibrations.
As shown in Fig. 2, the XRD patterns of Fe3O4, Fe3O4@SiO2, and Mag@Morph-AIL nanoparticles are provided. The patterns agree well with the reported pattern for Fe3O4 nanoparticles. The XRD pattern of Mag@Morph-AIL includes peaks from Fe3O4 and the silica layer. According to the Scherrer equation utilizing the FWHM, the average crystalline size of the Mag@Morph-AIL nanoparticles that were obtained was calculated to be 10–15 nm.
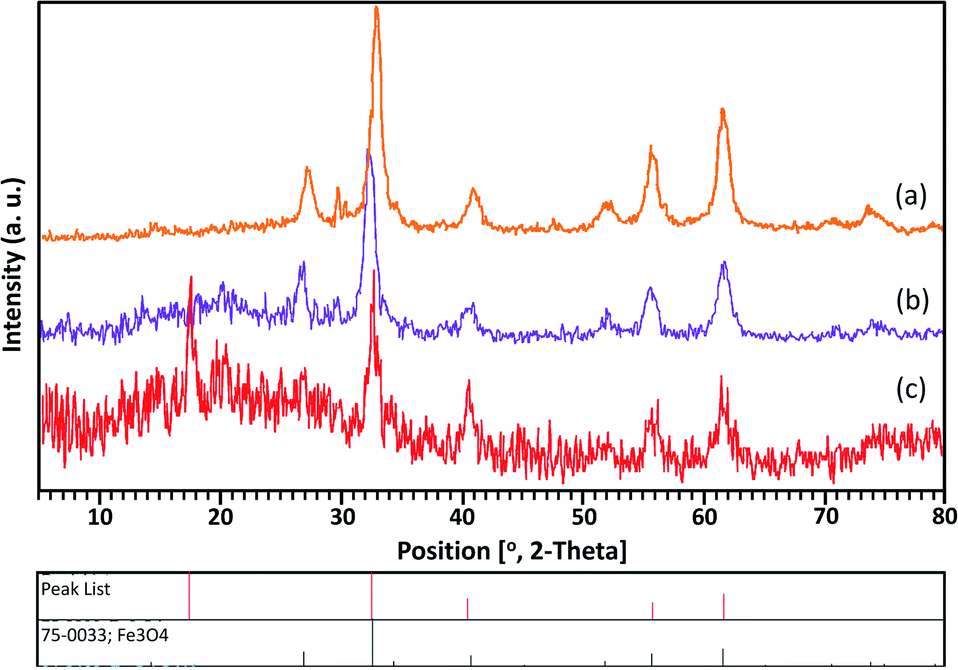 | ||
| Fig. 2 The XRD patterns of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Mag@Morph-AIL. | ||
As shown in Fig. 3, the hysteresis loops of the samples are completely reversible, confirming their superparamagnetic nature. The catalyst shows high magnetic permeability and high hysteresis loop reversibility. The decrease in the level of catalyst magnetism relative to the initial state indicates the presence of organic groups on the primary surface.
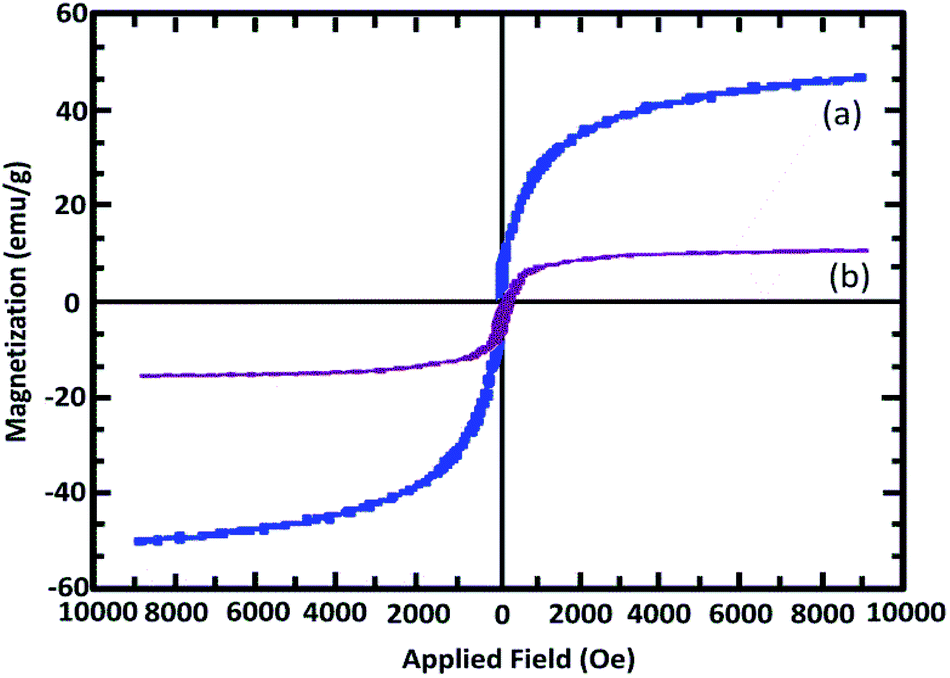 | ||
| Fig. 3 Magnetization versus the applied field for (a) Fe3O4@SiO2 and (b) Mag@Morph-AIL. | ||
As indicated in Fig. 3, the levels of saturation magnetization for Fe3O4@SiO2 and Mag@Morph-AIL are 50.86 emu g−1 and 13.84 emu g−1, respectively. These results determine that the magnetization properties decrease upon coating and functionalization. According to these results, the catalyst can be easily separated and recovered using an external magnetic field.
As shown in Fig. 4, scanning electron microscopy (SEM) imaging indicates that the prepared Mag@Morph-AIL NPs possess an average diameter of about 65 nm, and the nanoparticles show good dispersity with spherical morphology. A TEM image of the Mag@Morph-AIL nanoparticles is shown in Fig. 4d. Here, the TEM image gives more accurate information about the particle size and morphology of the nanomaterial. The TEM image reveals the spherical shape of the magnetic nanomaterial, with an average size of 69 nm, which shows close agreement with the value determined via SEM, while being larger than the value obtained via XRD analysis. Also, as seen in Fig. 5, the EDX spectrum of the synthesized Mag@Morph-AIL NP catalyst was obtained, in which the expected elemental composition (C, N, O, Si, S, and Fe) is shown clearly.
 | ||
| Fig. 4 SEM images of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Mag@Morph-AIL, and (d) a TEM image of Mag@Morph-AIL. | ||
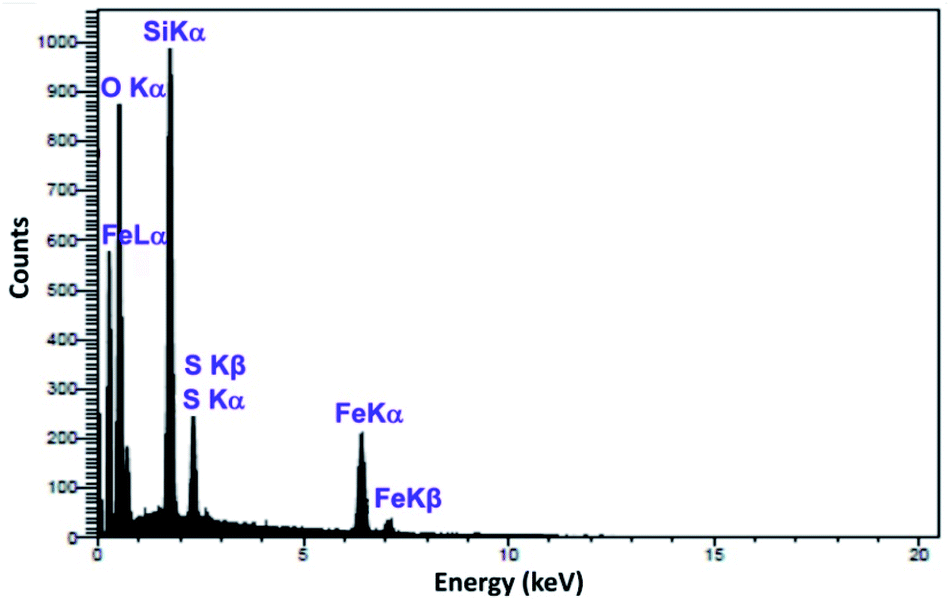 | ||
| Fig. 5 The EDX spectrum of Mag@Morph-AIL. | ||
As shown in Fig. 6, the thermal behavior of the Mag@Morph-AIL nanocatalyst was studied. The TG profile exhibited two weight loss steps. An initial mass loss of 4% with an endothermic peak in the DTA curve is revealed in the temperature range of 100–150 °C. This can be related to the release of physically absorbed water on the surface of the nanomaterial. The second mass loss of 30% occurred in a wide temperature range of 420–500 °C, which overlapped with a broad endothermic DTA peak. This corresponds mainly to the thermal decomposition of the organic complex. The results of thermal analysis revealed that the thermal decomposition of the organic moieties was completed at a temperature of 790 °C.
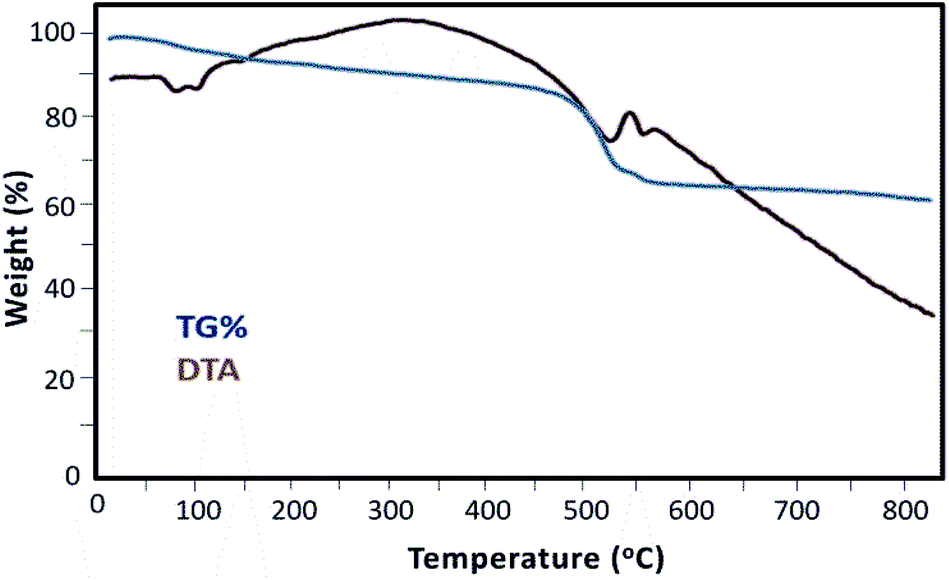 | ||
| Fig. 6 TGA and DTA curves of Mag@Morph-AIL. | ||
Thermogravimetric analysis can be used to estimate the amount of catalytically active species on the solid nanocomposite material. To calculate the amount of organic moieties (mmol g−1), the solid catalyst weight-loss value of 26% w/w that can be attributed to organic functional groups was used in conjunction with the molecular weight of the organic moieties. According to these data, the amount of organic active species anchored on the surface of the solid catalyst is 1.070 mmol g−1.
The amount of acid in the catalyst was quantitatively estimated via ion-exchange pH analysis.47 For this purpose, 75 mg of solid catalyst was added to an aqueous solution of NaCl (1 M, 50 mL), and the resulting mixture was stirred for 3 days. The final mixture was titrated using 0.05 M NaOH solution. The amount of acid in the Mag@Morph-AIL catalyst was determined to be 1.862 mmol g−1, which appears to be a good approximation because it is twice the amount of organic species estimated via TGA and each organic moiety has two acidic hydrogens.
Catalytic performance of the solid acidic ionic liquid for the Biginelli reaction
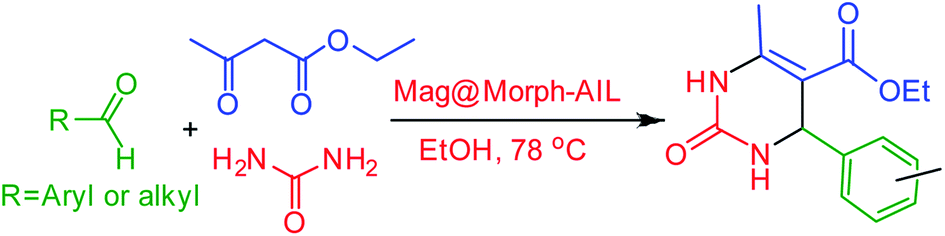
Mag@Morph-AIL, as an organic–inorganic hybrid nanomaterial, when used as a catalyst in the Biginelli reaction, showed significant advantages, such as biocompatibility, magnetic separability and recoverability, thermal stability, a high surface area, and a higher loading of active sites. Moreover, the introduction of sulfonic acid and organic groups onto the surface of Fe3O4@SiO2 is interesting since the combination of both acidic and hydrophobic functionalities allows for the creation of a less-polar organic environment with relatively strong acidity, leading to a “quasi-homogeneous” catalyst for acid-catalyzed reactions. However, in many acid-catalyzed reactions using silica-based solid acids, the water produced as a by-product can be adsorbed on the surface of silanol, thus poisoning the surface, reducing the performance of the catalyst, and deactivating the active acidic sites of the catalyst.To begin this study, the reaction conditions, such as the solvent, temperature, and the amount of catalyst, were optimized (Scheme 2). For this purpose, the reaction between benzaldehyde (1.0 mmol), ethyl acetoacetate (1.0 mmol), and urea (1.5 mmol) as substrates with the prepared nanocomposite material as a catalyst under reflux conditions was selected as the model reaction. At first, the optimum amount of catalyst was investigated in ethanol solvent (Table 1). It was found that the best results were obtained when the reaction was carried out in the presence of 0.06 g of catalyst (Table 1, entry 4).
 | ||
| Scheme 2 The use of Mag@Morph-AIL in the Biginelli reaction. | ||
| Entry | Catalyst amount (g) | Time (min) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: p-methyl benzaldehyde (1 mmol), ethyl acetoacetate (1 mmol), urea compound (1.5 mmol), and Mag@Morph-AIL catalyst were added to EtOH as the solvent (5 mL) at 80 °C.b Isolated yield. | |||
| 1 | 0.01 | 150 | 70 |
| 2 | 0.02 | 100 | 75 |
| 3 | 0.04 | 80 | 92 |
| 4 | 0.06 | 60 | 92 |
| 5 | 0.08 | 60 | 92 |
In order to optimize the temperature of the reaction, the reaction was performed at different temperatures in the presence of 0.06 g of catalyst and with ethanol as the solvent. With reference to the results shown in Table 2, it was found that the best results were obtained when the reaction was carried out at 80 °C (Table 2, entry 3).
In an effort to obtain better yields and the most effective solvent, various solvents were used for the synthesis of 3,4-dihydropyrimidinones with a catalytic amount of Mag@Morph-AIL NPs in a simple reaction. With reference to the results in Table 3, polar and protic solvents increased the rate of reaction, and EtOH provided excellent yields and proved to be the solvent of choice.
| Entry | Catalyst amount (g) | Time (min) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: p-methyl benzaldehyde (1 mmol), ethyl acetoacetate (1 mmol), urea compound (1.5 mmol), and Mag@Morph-AIL catalyst (0.06 g) were refluxed in the selected solvent (5 mL) at 80 °C.b Isolated yields. | |||
| 1 | THF | 150 | 50 |
| 2 | CH3CN | 130 | 44 |
| 3 | EtOH | 60 | 92 |
| 4 | DMSO | 120 | 35 |
In continuation of this research, as shown in Table 4, the corresponding products were obtained in excellent yields and with short reaction times under the optimum conditions. To ascertain the scope and limitations of this reaction, some aliphatic and aromatic aldehydes were refluxed with urea and ethyl acetoacetate in the presence of 0.06 g of Mag@Morph-AIL NPs with ethanol as the solvent at 80 °C. A best isolated yield of product of 93% was obtained within 58 min (Table 4). The desired 3,4-dihydropyrimidinone derivatives were synthesized and then characterized via spectroscopic methods.
|
|||||
|---|---|---|---|---|---|
| Entry | Name | Productb | Time (min) | Yieldc (%) | MPd (°C) |
| a Reaction conditions: benzaldehyde (1 mmol), ethyl acetoacetate (1 mmol), urea compound (1.5 mmol) and Mag@Morph-AIL catalyst (0.06 g) were refluxed in EtOH as the solvent (5 mL).b All products were characterized based on their spectroscopic data (FT-IR and 1H-NMR).c Isolated yield.d Literature reference. | |||||
| 1 | 4a | 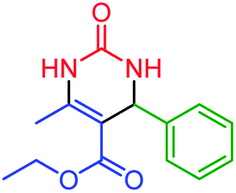 |
64 | 86 | 204–206 (ref. 47) |
| 2 | 4b | 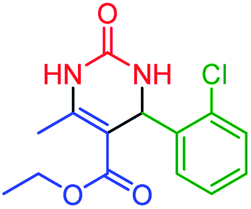 |
65 | 88 | 252–253 (ref. 48) |
| 3 | 4c | 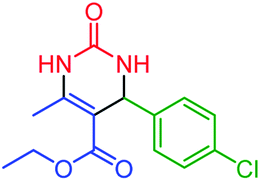 |
60 | 87 | 206–208 (ref. 49) |
| 4 | 4d | 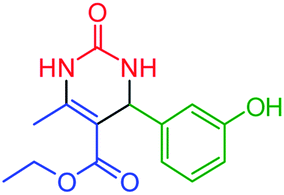 |
70 | 85 | 167–170 (ref. 49) |
| 5 | 4e | 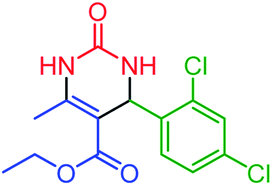 |
60 | 89 | 251–254 (ref. 49) |
| 6 | 4f | 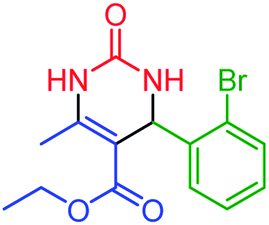 |
65 | 86 | 185–188 (ref. 44) |
| 7 | 4g | 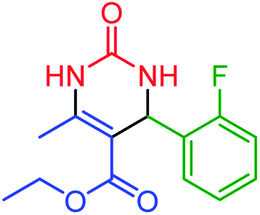 |
70 | 88 | 224–225 (ref. 52) |
| 8 | 4h | 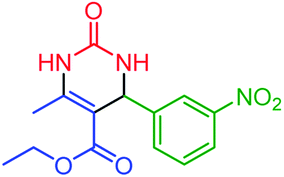 |
60 | 90 | 123–126 (ref. 47) |
| 9 | 4i | 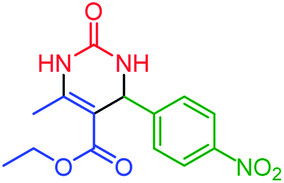 |
58 | 93 | 205–208 (ref. 51) |
| 10 | 4j | 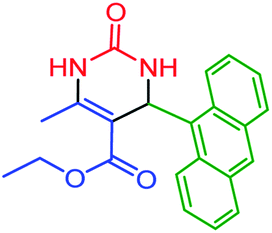 |
70 | 89 | 283–285 (ref. 53) |
| 11 | 4k | 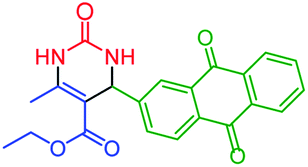 |
66 | 91 | 272–275 (ref. 52) |
| 12 | 4l | 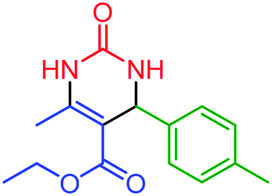 |
60 | 92 | 206–207 (ref. 49) |
| 13 | 4m | 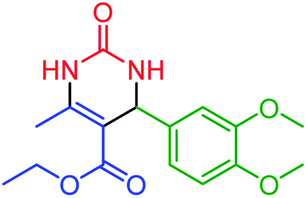 |
75 | 85 | 176–179 (ref. 49) |
| 14 | 4n | 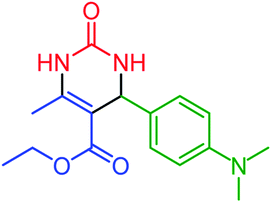 |
75 | 87 | 258–261 (ref. 50) |
| 15 | 4o | 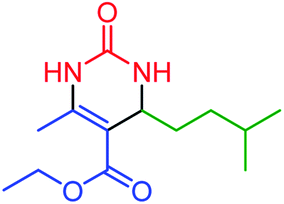 |
80 | 80 | 169–173 (ref. 44) |
Reusability of the Mag@Morph-AIL nanocatalyst
The Mag@Morph-AIL nanocatalyst is recoverable without a considerable loss of catalytic activity. After the completion of the reaction, 5 mL of ethanol was added to the reaction mixture and the nanocatalyst was recycled using an external magnetic field and washed with acetone to remove residual product or other organic material. The nanocatalyst was reused for a new Biginelli reaction between p-methyl benzaldehyde, ethyl acetoacetate, and urea under similar reaction conditions. As shown in Fig. 7, the NPs could be reused for up to eight cycles with an insignificant loss of catalytic activity, providing the product in high yields.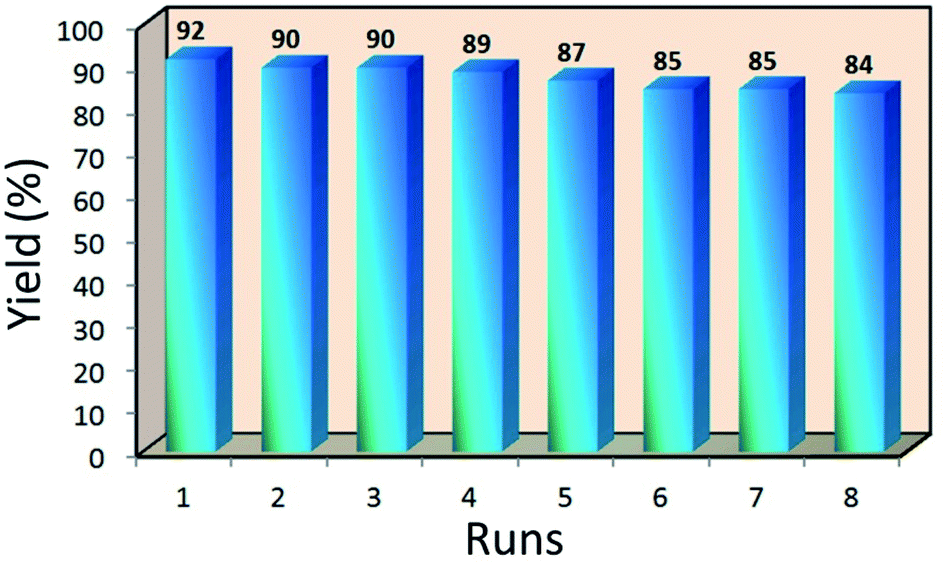 | ||
| Fig. 7 Recyclability study of Mag@Morph-AIL. | ||
Conclusions
In this report, we have developed a straightforward and efficient method for the synthesis of 3,4-dihydropyrimidinones using a Mag@Morph-AIL nanocomposite material as a new metal-free catalyst. The novel magnetic recoverable nanocatalyst was characterized using FT-IR, SEM, EDX, XRD, VSM, and BET techniques. The catalyst showed excellent efficiency and could convert >92% of the substrates to the target molecules within 60 min at 78 °C. The catalyst was recovered using an external magnet from the reaction mixture and could be recycled for at least 8 runs without a major decrease in catalytic activity. The method offers several advantages, including clean reaction profiles, good availability, high yields, short reaction times, simple experimental procedures, catalyst reusability, and low catalyst loading.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the University of Kashan for supporting this work (grant no. 159148).Notes and references
- T. Peters, H. Lindenmaier, W. Haefeli, J. Weiss and N. S. Arch, Pharmacology, 2006, 372, 291–299 CAS
.
- J. C. Cochran, J. E. Gatial, T. M. Kapoor and S. P. Gilbert, J. Biol. Chem., 2005, 280, 12658–12667 CrossRef CAS
- S. Tcherniuk, R. van Lis, F. Kozielski and D. A. Skoufias, Biochem. Pharmacol., 2010, 79, 864–872 CrossRef CAS
- K. De, S. Chandra, B. Sarkar, S. Ganguly and M. Misra, J. Radioanal. Nucl. Chem., 2010, 283, 621–628 CrossRef CAS
- I. M. T. C. Crevel, M. C. Alonso and R. A. Cross, Curr. Biol., 2004, 14, R411–R412 CrossRef CAS
- S. H. Choi and D. McCollum, Curr. Biol., 2012, 22, 225–230 CrossRef CAS
- J. C. Cochran and S. P. Gilbert, Biochem, 2005, 44, 16633–16648 CrossRef CAS
- L. Duan, T.-Q. Wang, W. Bian, W. Liu, Y. Sun and B.-S. Yang, Spectrochim. Acta, Part A, 2015, 137, 1086–1091 CrossRef CAS
- S. DeBonis, J. P. Simorre, I. Crevel, L. Lebeau, D. A. Skoufias, A. Blangy, C. Ebel, P. Gans, R. Cross, D. D. Hackney, R. H. Wade and F. Kozielski, Biochemistry, 2003, 42, 338–349 CrossRef CAS
- K. Drosopoulos, C. Tang, W. C. H. Chao and S. Linardopoulos, Nat. Commun., 2014, 5, 3686 CrossRef CAS
- C. J. Funk, A. S. Davis, J. A. Hopkins and K. M. Middleton, Anal. Biochem., 2004, 329, 68–76 CrossRef CAS
- R. V. Chikhale, R. P. Bhole, P. B. Khedekar and K. P. Bhusari, Eur. J. Med. Chem., 2009, 44, 3645–3653 CrossRef CAS
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS
- L. H. S. Matos, F. T. Masson, L. A. Simeoni and M. Homem-de-Mello, Eur. J. Med. Chem., 2018, 143, 1779–1789 CrossRef CAS
- P. Biginelli, Gazz. Chim. Ital., 1883, 23, 360–413 Search PubMed
-
(a) S. S. Panda, P. Khanna and L. Khanna, Curr. Org. Chem., 2012, 16, 507–520 CrossRef CAS
- A. S. Paraskar, G. K. Dewkar and A. Sudalai, Tetrahedron Lett., 2003, 44, 3305–3308 CrossRef CAS
- K. A. Kumar, M. Kasthuraiah, C. S. Reddy and C. D. Reddy, Tetrahedron Lett., 2001, 42, 7873–7875 CrossRef
- M. Gohain, D. Prajapati and J. S. Sandhu, Synlett, 2004, 2, 0235–0238 Search PubMed
- C. Simon, T. Constantieux and J. Rodriguez, Eur. J. Org. Chem., 2004, 4957–4980 CrossRef CAS
- J. S. Yadav, B. V. S. Reddy, P. Sridhar, J. S. S. Reddy, K. Nagaiah, N. Lingaiah and P. S. Saiprasad, Eur. J. Org. Chem., 2004, 552–557 CrossRef CAS
- C. O. Kappe and S. F. Falsone, Synlett, 1998, 718–720 CrossRef CAS
-
(a) S. Xue, Y.-C. Shen, Y.-L. Li, X.-M. Shen and Q.-X. Guo, Chin. J. Chem., 2002, 20, 385–389 CrossRef CAS
-
(a) V. R. Choudhary, V. H. Tillu, V. S. Narkhede, H. B. Borate and R. D. Wakharkar, Catal. Commun., 2003, 4, 449–453 CrossRef CAS
- J. Safari and S. G. Ravandi, J. Mol. Catal. A: Chem., 2013, 373, 72–77 CrossRef CAS
- J. Safaei-Ghomi, R. Teymuri and A. Ziarati, Monatsh. Chem., 2013, 144, 1865–1870 CrossRef CAS
- H. R. Memarain and M. Ranjbar, J. Mol. Catal. A: Chem., 2012, 356, 46 CrossRef CAS
- J. Javidi, M. Esmaeilpour and F. N. Dodeji, RSC Adv., 2015, 5, 308–315 RSC
- B. K. Banik, A. T. Reddy, A. Datta and C. Mukhopadhyay, Tetrahedron Lett., 2007, 48, 7392–7394 CrossRef CAS
- J. Safari and S. Gandomi-Ravandi, New J. Chem., 2014, 38, 3514–3521 RSC
- J. T. Li, J. F. Han, J. H. Yang and T. S. Li, Ultrason. Sonochem., 2003, 10, 119–122 CrossRef CAS
- J. Peng and Y. Deng, Tetrahedron Lett., 2001, 42, 5917–5919 CrossRef CAS
- A. Zhu, Q. Li, L. Li and J. Wang, Catal. Lett., 2013, 143, 463–468 CrossRef CAS
- M. Rahman, A. Sarkar, M. Ghosh, A. Majee and A. Hajra, Tetrahedron Lett., 2014, 55, 235–239 CrossRef CAS
- S. Verma, S. L. Jain and B. Sain, Tetrahedron Lett., 2010, 51, 6897–6900 CrossRef CAS
- M. Faraji, Y. Yamini and M. Rezaee, J. Iran. Chem. Soc., 2010, 7, 1–37 CrossRef CAS
- L. M. Rossi, N. J. S. Costa, F. P. Silva and R. V. Goncalves, Nanotechnol. Rev., 2013, 2, 597–614 CAS
- S. J. Wang, Z. Y. Wang and Z. G. Zha, Dalton Trans., 2009, 43, 9363–9373 RSC
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC
- Q. He, W. Wu and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397–415 CrossRef
- R. Y. Hong, B. Feng, G. Liu, S. Wang, H. Z. Li, J. M. Ding, Y. Zheng and D. G. Wei, J. Alloys Compd., 2009, 476, 612–618 CrossRef CAS
- M. H. Valkenberg, C. de Castro and W. F. Holderich, Green Chem., 2002, 4, 88–93 RSC
- S. A. Galema, Chem. Soc. Rev., 1907, 26, 233–238 RSC
- M. Bagherzadeh, M. Haghdoost, F. Matlobi-Moghaddam, B. Koushki-Foroushani, S. Sarazdi and E. Payab, J. Coord. Chem., 2013, 66, 3025–3036 CrossRef CAS
- A. Shaabani, A. Bazgir and F. Teimouri, Tetrahedron Lett., 2003, 44, 857–859 CrossRef CAS
- D. Azarifar, O. Badalkhani and Y. Abbasi, J. Sulfur Chem., 2016, 37, 1–18 CrossRef CAS
-
(a) Q. Zhang, H. Su, J. Luo and Y. Wei, Green Chem., 2012, 14, 201–208 RSC
- C. O. Kappe, Tetrahedron, 1993, 49, 6937–6963 CrossRef CAS
- A. Mobinikhaledi and M. A. Bodaghi-Fard, Acta Chim. Slov., 2010, 57, 931–935 CAS
- Y. Ma, C. Qian, L. Wang and M. Yang, J. Org. Chem., 2000, 65, 3864–3868 CrossRef CAS
- J. Lu and Y. Bai, Synthesis, 2002, 0466–0470 CrossRef CAS
- G. Aridoss and Y. T. Jeong, Bull. Korean Chem. Soc., 2010, 31, 863–868 CrossRef CAS
- K. Konkala, N. M. Sabbavarapu, R. Katla, N. Y. V. Durga, V. K. Reddy, B. L. A. Prabhavathi-Devi and R. B. N. Prasad, Tetrahedron Lett., 2012, 53, 1968–1973 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR and H-NMR spectroscopy data. See DOI: 10.1039/d0ra09929e |
| This journal is © The Royal Society of Chemistry 2021 |