
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchical self-assembly of discrete bis-[2]pseudorotaxane metallacycle with bis-pillar[5]arene via host–guest interactions and their redox-responsive behaviors†
Gui-Yuan Wu *a,
Chao Lianga,
Yi-Xiong Hub,
Xu-Qing Wangb,
Guang-Qiang Yinb and
Zhou Lu*a
*a,
Chao Lianga,
Yi-Xiong Hub,
Xu-Qing Wangb,
Guang-Qiang Yinb and
Zhou Lu*a
aAnhui Province Key Laboratory of Optoelectronic Material Science and Technology, School of Physics and Electronic Information, Anhui Normal University, Wuhu, Anhui 241002, China. E-mail: zhoulu@ahnu.edu.cn; wgy@ahnu.edu.cn
bShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, 3663 N. Zhongshan Road, Shanghai, China
First published on 4th January 2021
Abstract
A discrete rhomboidal metallacycle R functionalized with bis-[2]pseudorotaxane of [Cu(phenanthroline)2]+ derivatives was successfully synthesized via coordination-driven self-assembly. Furthermore, the host–guest complexation of such a bis-[2]pseudorotaxane metallacycle with a bis-pillar[5]arene (bisP5) allowed for the formation of a new family of cross-linked supramolecular polymers R⊃(bisP5)2, which displayed interesting redox-responsive properties. By taking advantage of the substantial structural differences between the coordination geometries of [Cu(phenanthroline)2]+ and [Cu(phenanthroline)2]2+, the weight-average diffusion coefficients D of the supramolecular polymer were adjusted through changing the redox state of the Cu(I)/Cu(II) complexes.
Introduction
Hierarchical self-assembly (HSA) is defined as a multilevel spontaneous organizing process in which the precisely controlled self-assembly of components is realized by non-covalent interactions. It has become increasingly clear that HSA offers a powerful bottom-up strategy for constructing essential biomolecules such as DNA, RNA, and proteins, the structural integrities of which are shown to be superior to those of the nonhierarchical self-assembled ones. Inspired by nature, HAS has been widely applied to the fabrication of desirable functional supramolecular architectures with well-defined structures, intriguing properties, and multiple functions.1–3 Among these, coordination-driven self-assembly, as one of the most feasible and efficient methodologies, makes use of directional coordinative bonds between the building blocks and the metal ions to construct well-defined supramolecular coordination complexes (SCCs) such as two-dimensional (2-D) polygons and (3-D) three-dimensional polyhedron.4–9Based on coordination-driven self-assembly, successful strategies have been developed for designing supramolecular polymers cross-linked by discrete metallacycles or metallacages. The combination of relatively rigid, discrete metal–organic scaffolds with elastic polymer networks allows for the construction of a new class of hybrid soft materials with stimuli-responsive properties. The discrete metallacycles or metallacages were used either as metallacycle skeletons or stimulus response sites in these supramolecular polymers as reported by various research groups.10–19 Such metallosupramolecular architectures are of particular interest not only for their fascinating structures but also because of their stimuli-sensitivities to a wide variety of environmental conditions including solvent composition, pH, temperature, light irradiation, and so on.20,21 However, the metallacycle-based redox-responsive supramolecular polymers are still rare.
One way to access such redox-responsive supramolecular materials is to utilize host–guest repeating units which contain one or more redox-active species, for example disulphide, selenium, ferrocene, tetrathiafulvalene, transition metal ions, and viologen counterparts.22–24 In particular, coordination complexes based on transition metals and bidentate polypyridine ligands, notably Cu(I)–phenanthroline complexes, typically present desirable redox properties for diverse applications such as molecular machines, catalysis, ultrafast spectroscopy, solar energy conversion, optoelectronics, and luminescence.25,26 Moreover, the Cu(I)–phenanthroline complexes feature some promising characteristics including the extra structural rigidity afforded by the phenanthroline unit, and the Cu(I)–phenanthroline complexes being oxidized to Cu(II)–phenanthroline complexes that undergoes significant and rapid Jahn–Teller (J–T) distortion from D2d to D2 symmetry.27–33 With this strategy, Sauvage and co-workers have successfully constructed topologically spectacular architectures such as pseudorotaxanes, rotaxanes, catenates and knots with Cu(I)–phenanthroline complexes. It is therefore possible to further utilize the Cu(I)–phenanthroline complexes as the basic building blocks to develop redox-responsive supramolecular polymers with the topology of [2]pseudorotaxane.
Using the HSA method, we previously reported a novel supramolecular polymer which was based on hexagonal metallacycle skeletons containing the above-mentioned Cu(I)–phenanthroline moiety. It was displayed that the redox-responsive of the resulting supramolecular polymers are strongly dependent on the oxidation state of copper. In order to further examine the versatility of this synthesis strategy and investigate the effects of metallacycle skeleton on the redox-responsive behaviours of the supramolecular polymers, we herein report another type of redox supramolecular polymer constructed by the well-defined rhomboidal bis-[2]pseudorotaxanes metallacycle through the hierarchical orthogonal design strategy based on coordination-driven self-assembly and host–guest interactions. First, the rhomboidal metallacycle R containing two [2]pseudorotaxane moieties was constructed by the coordination between 1,10-phenanthroline(phen) derivatives containing [2]pseudorotaxane and 60° di-Pt(II) acceptors, resulting in a rhomboidal metallacycle R of which the skeleton size is less than the previously reported hexagonal metallacycle M.34 Subsequently, hyperbranched supramolecular polymer was successfully constructed in the high-concentration region through host–guest interactions of rhomboidal metallacycle with the bis-pillar[5]arene (bisP5). Notably, the host–guest interactions were similar to covalent bond end-capping process and the Cu(I)N4 [2]pseudorotaxanes could act as the active sites which respond to the redox stimuli in supramolecular polymers. Supramolecular polymerization was shown to help the otherwise unstable tetrahedral complex Cu(II)N4 maintain enough stability during redox reactions. In addition, the tunability of the diffusion constants upon redox stimuli was also compared between the supramolecular polymers made of the R and M, showing the influence caused by the skeleton size.
Results and discussion
Synthesis and characterization
With the 120° macrocycle 1 incorporating a phen moiety and ligand 2 possessing cyano sites in hand, the construction work of [2]pseudorotaxane with bis-pyridine(120°) donor D was carried out using an approach derived from the transition Cu(I) template strategy. The threading process leading to D, using Cu(I) as templating agent, was shown to be quantitative by 1H NMR.20 By simply mixing the donor building block D with the corresponding 60° acceptor A in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in acetone at 50 °C for five hours, a novel bis-[2]pseudorotaxanes rhomboidal metallacycle R was successfully obtained in near-quantitative yield via coordination-driven self-assembly (Scheme 1).29–33 It shall be noted that the skeleton size of such a rhomboidal metallacycle is less than that of the previously assembled hexagonal metallacycle M which used a 120° acceptor.34
:1 ratio in acetone at 50 °C for five hours, a novel bis-[2]pseudorotaxanes rhomboidal metallacycle R was successfully obtained in near-quantitative yield via coordination-driven self-assembly (Scheme 1).29–33 It shall be noted that the skeleton size of such a rhomboidal metallacycle is less than that of the previously assembled hexagonal metallacycle M which used a 120° acceptor.34
 | ||
| Scheme 1 Graphical representation of the self-assembled bis-[2]pseudorotaxanes rhomboidal metallacycle R. | ||
In the 1H NMR spectrum (Fig. 1 and S1†), the α- and β-pyridyl hydrogen signals both exhibited the obvious downfield shifts (Δδ ≈ 0.48 and 0.53 ppm for α-H; Δδ ≈ 0.47 and 0.54 ppm for β-H) because of the loss of electron density that occurred upon coordination of the pyridine-N atom with the Pt(II) metal center. Notably, two doublets were observed for both the α- and β-pyridine protons, which might be attributed to hindered rotation about the Pt–N(pyridyl) bond in a small cavity. The 31P NMR spectra of the metallacycle R displayed a sharp singlet (δ = 14.48 ppm), which shifted upfield from the signal of the starting platinum acceptor A by approximately 5.46 ppm (Fig. 1). This change, as well as the decrease in coupling of the flanking 195Pt satellites, is consistent with back-donation from the platinum atoms. Further characterization by 2-D 1H NMR spectroscopic techniques (1H–1H COSY and NOESY) were in agreement with the formation of the discrete bis-pseudorotaxanes metallacycles (Fig. S18†). For instance, the presence of cross-peaks between the signal of the pyridine protons (α-H and β-H) and the PEt3 protons (CH2 and CH3) were found in the 2-D NOESY spectrum, which supported the structure of metallacycle R as a result of the formation of N–Pt bonds (Fig. S18b†).
 | ||
| Fig. 1 (a) Partial 1H NMR spectra (500 MHz, 296 K) of 120° donor ligand D, and large metallacycle R in acetone-d6. (b) 31P NMR spectra (400 MHz, 296 K) of 60° acceptors A and metallacycle R in acetone-d6. | ||
The structure of metallacycle R was also confirmed by ESI-TOF-MS, which allowed the assembly to remain intact to the maximum extent during the ionization process while retaining the high resolution required for resolving the isotopic distribution. For instance, the ESI-TOF-MS spectrum of R revealed three signals that corresponded to different charge states resulting from the loss of PF6− counterions, [R–3PF6]3+, [R–4PF6]4+, and [R–5PF6]5+, respectively, in which R represents the intact assembly (Fig. 2). Further investigation revealed that each isotope pattern of these signals was in good agreement with the corresponding simulation result. All evidence above supports the successful formation of discrete bis-[2]pseudorotaxanes rhomboidal metallacycle R by the donor D and accepter A via coordination-driven self-assembly (Scheme 1).
 | ||
| Fig. 2 Theoretical (top) and experimental (bottom) ESI-TOF-MS spectra of bis-[2]pseudorotaxane rhomboidal metallacycles R. | ||
Cross-linked supramolecular polymers constructed through host–guest interactions
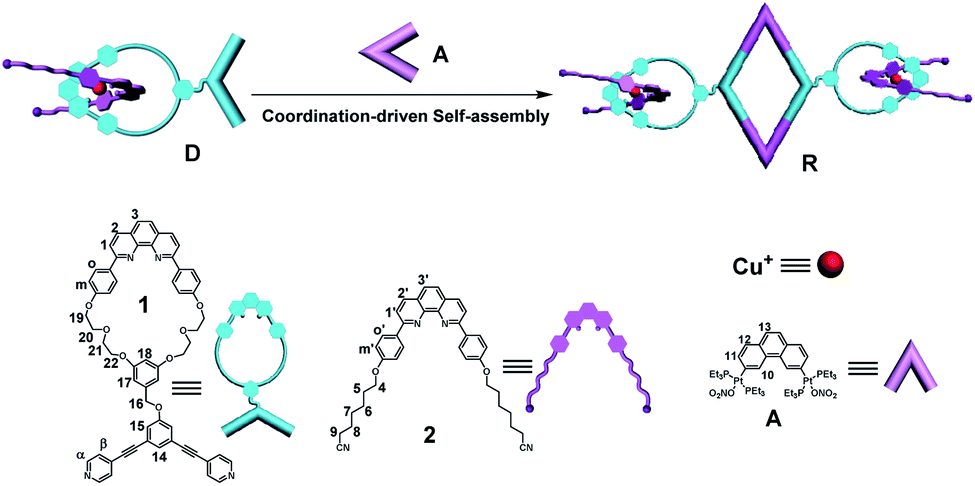
The supramolecular chemistry of pillar[n]arenes has developed immensely over the past ten years on account of their excellent inclusion properties and many intriguing applications in the constructions of molecular switches/machines, metal–organic frameworks, pH-responsive vesicles, as well as artificial transmembrane channels.35–53 Pillararenes are a new family of macrocyclic host molecules that possess symmetrical and columnar geometries with rigid and π-rich cavities. Compared with other macrocyclic hosts, the most peculiar host–guest properties of pillararenes are their strong affinities towards neutral guests by CH/π hydrogen bonds in organic media. For example, Yang's group previously reported a multiple stimuli-responsive supramolecular polymer through orthogonal coordination-driven self-assembly and host–guest complexation of multi-pillar[5]arene metallacycles and ditopic neutral guests.54 We have also previously reported the strong binding behavior between D and pillar[5]arene dimer bisP5 (Fig. S3†), showing that the linear long chain part of ditopic guest D was able to thread through the cavity of bisP5 to form poly[3]pseudorotaxanes in solution.34 By taking advantage of the strong affinities of pillar[5]arenes with neutral guests in organic solvents, we envisioned that cross-linked supramolecular polymers could be prepared from the bisP5 via host–guest interactions when the neutral tetratopic metallacycles R were employed as guests.By making use of the recognition motif between the ditopic guest D and the host bisP5 along with their good solubility in acetone, the mixtures of tetratopic metallacycles R with the host bisP5 (1:2 molar ratio of R and bisP5) at high concentration was expected to form a new family of cross-linked supramolecular polymers R⊃(bisP5)2. The concentration-dependent 1H NMR studies of complexes R⊃(bisP5)2 demonstrated the formation of such cross-linked supramolecular polymers. For instance, 1H NMR spectra (500 MHz, acetone-d6, 296 K) of the complex of R and bisP5 in a 1:2 molar ratio over a range of metallacycle R unit concentrations from 0.1 up to 3.0 mM were recorded with the purpose of probing the process of the supramolecular polymer formation (Fig. 3a). At low concentration (<0.5 mM), no obvious signal broadening was found, indicating that poly[3]pseudorotaxanes predominated in solution at low concentration. Upon increasing the concentrations, the signal of H5, H8 and H9 gradually became unobvious and finally disappeared. In the meanwhile, the Hm′ peak became broadened markedly at higher concentrations. These observations gave clean evidence for the formation of cross-linked supramolecular polymers through host–guest interactions (Fig. S4†) in the high concentration region.54
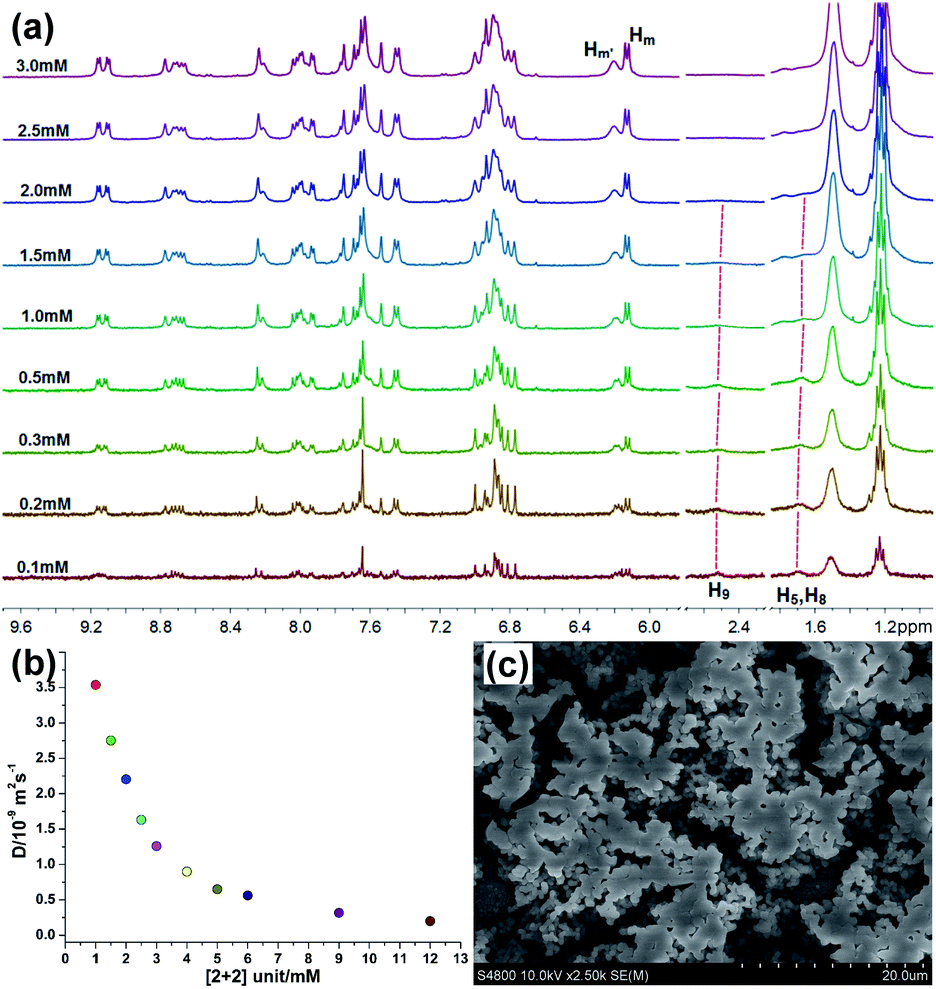 | ||
| Fig. 3 (a) Partial 1H NMR spectra (500 MHz, acetone-d6, 296 K) of R⊃(bisP5)2 at different concentrations. (b) 2-D DOSY (500 MHz, 296 K) plot of solutions in acetone-d6 of 1:2 molar ratio of R and bisP5 at different concentrations, respectively. (c) SEM image of cross-linked supramolecular polymers R⊃(bisP5)2 at concentration of 4.0 mM in CH3COCH3. | ||
Two-dimensional diffusion-ordered 1H NMR spectroscopy (DOSY) experiments were performed to provide further evidence for the formation of cross-linked supramolecular polymers. From Fig. 3b, the measured weight average diffusion coefficient (D) of R⊃(bisP5)2 decreased remarkably from 3.55 × 10−9 to 0.2 × 10−9 m2 s−1 (D 1.0 mM/D 12.0 mM = 17.8) as the concentration of R⊃(bisP5)2 increased from 1.0 to 12.0 mM. In general, a more than 10-fold decrease of the diffusion coefficient is believed to be the important evidence for the formation of a polymerization with high degree.41 Therefore, the results from Fig. 3b clearly indicates that the initially small poly[3]pseudorotaxanes grew into larger across-linked supramolecular polymers with the increase in R⊃(bisP5)2 concentration. These DOSY experimental results also further revealed that the generation of such supramolecular polymers was concentration dependent. Importantly, a higher diffusion coefficient (D) of the generated supramolecular aggregates were observed for R⊃(bisP5)2 when comparing with the previously reported M⊃(bisP5)3 at the same concentration of [Cu(phenanthroline)2]+ unit. For example, the D values of R⊃(bisP5)2 and M⊃(bisP5)3 were 1.26 × 10−9 m2 s−1 and 0.35 × 10−9 m2 s−1, respectively, when the concentration of [Cu(phenanthroline)2]+ unit was 6 mM in acetone (Fig. S5†). Such a difference in the diffusion constants indicates that the rhomboidal metallacycle R probably yielded in smaller supramolecular polymers than the hexagonal metallacycle M, therefore proving that the skeleton of the metallacycle has a significant effect on supramolecular polymers size.
In order to further demonstrate the role of bisP5 as the “cross-linking agent” in the formation of such supramolecular polymers. The scanning electron microscopy (SEM) studies were performed, which provided the direct evidence for the existence of supramolecular polymerization of R⊃(bisP5)2 and further insight into the morphological characteristics of cross-linked supramolecular polymers. As shown in Fig. S6,† the typical spherical morphologies for metallacycle R in acetone solvents were observed at low concentration (≤1.0 mM). With the increasing of concentrations, the formation of cube morphologies and the increase of spherical radius for metallacycle R in acetone solvents were observed, indicating the predominance of the oligomers when the concentration of metallacycle R is more than 1 mM. On the other hand, the more irregular and extended three-dimensional (3-D) networks for R⊃(bisP5)2 systems were observed with the presence of bisP5, which was consistent with the typical morphology of cross-linked supramolecular polymers (Fig. 3c and S7†).
Redox-responsive supramolecular polymer
As we know, the reversible redox state of the Cu(I)/Cu(II) complexes transition could be tuned by chemical oxidation and reduction processes. Thus, the redox-responsive properties of the resulting supramolecular polymers R⊃(bisP5)2 were further exploited by the manipulation of the redox reactions between copper(I) and copper(II) (Scheme 2). We have previously investigated the redox-responded behaviors of the polypseudorotaxanes D⊃bisP5 in acetone.34 The formation of poly[3]pseudorotaxanes was seen to be very stable between the bisP5 and cyanide chain through host–guest interactions, which was similar to a covalent bond end-capping process and prevented the structure 2 slipping off the macrocycle 1 in the unstable tetrahedral complex Cu(II)N4 when the Cu(I) was oxidized to Cu(II). | ||
| Scheme 2 Schematic representation of the formation of redox-responsive supramolecular polymer. | ||
In current work, the redox-responded behaviors of supramolecular polymers R⊃(bisP5)2 were also examined. Likewise, 1H NMR, 31P NMR, UV-vis, DOSY and SEM experiments showed that the supramolecular polymer R⊃(bisP5)2 were almost unbroken upon the redox stimulation. In the partial 1H NMR measurements, signals from the protons Hm and Hm′ on the phenanthroline moiety almost disappeared when Cu(I) was oxidized into Cu(II) by NOBF4. Interestingly, the proton peaks of R⊃(bisP5)2 were almost completely restored when the Cu(II) was reduced to Cu(I) by ascorbic acid(ASA) (Fig. 4a and S8†), showing the reversibility of redox manipulations on R⊃(bisP5)2. More importantly, the signal of the proton H9 remained absent during the redox process, indicating that the bisP5 exhibited very strong binding affinities towards the dinitriles guest which resulted in the stable cross-linked supramolecular polymers. 31P NMR and SEM of supramolecular polymers also showed negligible changes during the redox-responded processes (Fig. 4b and S9†). These results demonstrate that the formation of stable supramolecular polymers with rhomboidal metallacycle scaffolds in the extended network through host–guest interactions and degrees of such cross-linked polymerization remain unchanged during the redox reactions.
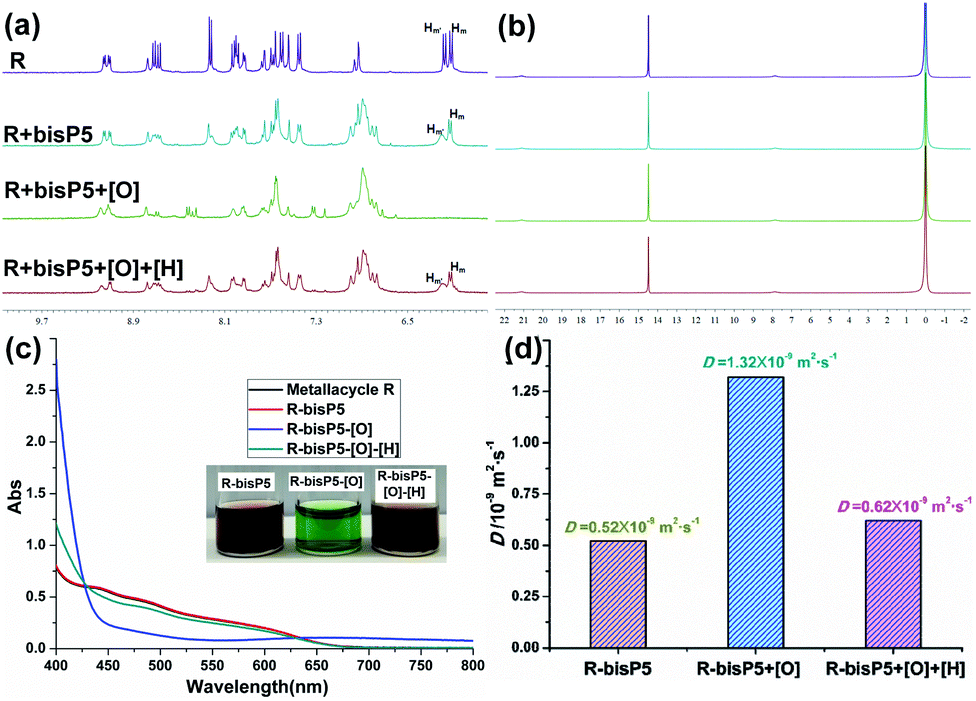 | ||
| Fig. 4 Partial 1H NMR spectra (a) and 31P NMR spectra (b) (500 MHz, acetone-d6, 296 K) of R (3.0 mM), R⊃(bisP5)2 (3 mM), R⊃(bisP5)2 + 4.0 equiv. NOBF4 and R⊃(bisP5)2 + 4.0 equiv. NOBF4 + 8.0 equiv. ASA. (c) Visible absorption spectra of a 0.1 mM solution of the R⊃(bisP5)2 in acetone, and of its oxidized form Cu(II)N4, and R⊃(bisP5)2 + 4.0 equiv. NOBF4 + 8.0 equiv. ASA. (d) 2-D DOSY (500 MHz, 296 K) plot of solutions in acetone-d6 of a 4.5 mM solution of the R⊃(bisP5)2 and of its oxidized form Cu(II)N4, and R⊃(bisP5)2 + 4.0 equiv. NOBF4 + 8.0 equiv. ASA. (NOBF4: [O], ASA: [H]). | ||
To verify the actual occurrence of the redox reactions, the UV-vis spectroscopic measurements on reduced and oxidized R⊃(bisP5)2 in acetone (0.1 mM) were carried out (Fig. 4c). A metal-to-ligand charge-transfer (MLCT) absorption band at 437 nm was observed in UV-vis absorption spectra for the original R⊃(bisP5)2 solution, indicated that the entwined arrangement of both phen units around Cu(I). When R⊃(bisP5)2 was oxidized by addition of NOBF4, the color of the solution changed from red-brown to emerald-green, while a new d–d absorption band appeared at 667 nm and the absorption peak at 550 nm became weaker. The addition of ascorbic acid triggered the reversed process: the color of solution turned back to red-brown and the resultant UV-vis absorption spectrum again became identical to that of the complex Cu(I)N4 (Fig. 4c).
The 2-D DOSY NMR experiment on R⊃(bisP5)2 was performed as well during the oxidation and reduction processes. As can be observed in Fig. 4d, the final weight-average diffusion coefficients D increased to 1.32 × 10−9 during the process of oxidation in R⊃(bisP5)2 ([Cu(phenanthroline)2]+ unit = 9 mM) (DCu(II)/DCu(I) = 2.54) (Fig. 4d and S10†). We have previously reported that the oxidation of M⊃(bisP5)3 ([Cu(phenanthroline)2]+ unit = 9 mM) resulted in a more dramatic increase of the measured weight-average diffusion coefficients D from 0.3 × 10−9 to 1.55 × 10−9 m2 s−1 (DCu(II)/DCu(I) = 4.70). These observations clearly demonstrated that the larger frame size of metallacycle has greater influence on the size changes of supramolecular polymer after oxidation (Table S1†). Moreover, the weight-average diffusion coefficient D of R⊃(bisP5)2 decreased to 0.62 × 10−9 when 8.0 equiv. ASA was added to the Cu(II)N4 on the R⊃(bisP5)2. Further analysis indicated that the above phenomena mainly resulted from the fact that the Cu(I)N4 complexes with the distorted tetrahedral geometry of approximate C2 symmetry were transformed into Cu(II)N4 with the D2 symmetry structures and the symmetry of the D2 structures was higher than that of the C2 symmetry structures. The change of metal complexes from the more asymmetric ones to highly symmetric structures drove a loose supramolecular polymer structure to form a more compact network, which might caused an increase in diffusion coefficients (Scheme 2).
Conclusions
In summary, we have constructed a discrete rhomboidal metallacycle R with bis[2]pseudorotaxanes via coordination-driven self-assembly. The structure of rhomboidal metallocycle R has been determined by 1H NMR, 31P NMR, CSI-TOF-MS, DOSY. The supramolecular polymers R⊃(bisP5)2, [3]pseudorotaxanes containing a Cu(I)–phenanthroline complex as a repeating units, was successfully constructed at the high-concentration region through host–guest interactions of bis[2]pseudorotaxanes metallacycles R with the bis-pillar[5]arene (bisP5). Subsequently, we have investigated interesting redox-responsive properties of the supramolecular polymers variation in R⊃(bisP5)2 system by switching the redox states of Cu(I)/Cu(II) ions. The R⊃(bisP5)2 system exhibit reversible chromatic at low concentration and the weight-average diffusion coefficients D at high-concentration controlled by changing the redox state of the Cu(I)/Cu(II) complexes. It's interesting that the smaller frame size of metallacycle has smaller influence on the size changes of redox-responsive supramolecular polymer. The redox-responsive supramolecular polymers are also promising as a unique material that can encapsulate and localize bioactive molecules and cells within the material matrix.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NSFC (Grant No. 21773256), Ministry of Education of Anhui Province and PhD research startup foundation of University Anhui Normal University.Notes and references
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC
.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. H. Mao, Nature, 2008, 452, 198–201 CrossRef CAS
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS
- G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233–2243 CrossRef CAS
- Y. Takezawa, J. Müller and M. Shionoya, Chem. Lett., 2017, 46, 622–633 CrossRef CAS
- L.-J. Chen and H.-B. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS
- W. Zheng, G. Yang, N. Shao, L.-J. Chen, B. Ou, S.-T. Jiang, G. Chen and H.-B. Yang, J. Am. Chem. Soc., 2017, 139, 13811–13820 CrossRef CAS
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC
- W. Zheng, W. Wang, S.-T. Jiang, G. Yang, Z. Li, X.-Q. Wang, G.-Q. Yin, Y. Zhang, H. Tan, X. Li, H. Ding, G. Chen and H.-B. Yang, J. Am. Chem. Soc., 2019, 141, 583–591 CrossRef CAS
- G.-Y. Wu, L.-J. Chen, L. Xu, X.-L. Zhao and H.-B. Yang, Coord. Chem. Rev., 2018, 369, 39–75 CrossRef CAS
- B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621–1636 RSC
- W. Tuo, Y. Sun, S. Lu, X. Li, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 16930–16934 CrossRef CAS
- Q. Zhang, D. Tang, J. Zhang, R. Ni, L. Xu, T. He, X. Lin, X. Li, H. Qiu, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 17909–17917 CrossRef CAS
- L. Xu, X. Shen, Z. Zhou, T. He, J. Zhang, H. Qiu, M. L. Saha, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 16920–16924 CrossRef CAS
- X. Yan, T. R. Cook, J. B. Pollock, P. Wei, Y. Zhang, Y. Yu, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 4460–4463 CrossRef CAS
- Z. Chen, M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2020, 142, 16471–16478 CrossRef CAS
- P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC
- J.-F. Chen, B.-B. Han, J.-F. Ma, X. Liu, Q.-Y. Yang, Q. Lin, H. Yao, Y.-M. Zhang and T.-B. Wei, RSC Adv., 2017, 7, 47709–47714 RSC
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS
- H. Jiang, Y. Jiang, J. Han, L. Zhang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 785–790 CrossRef CAS
- C.-B. Huang, L.-J. Chen, J. Huang and L. Xu, RSC Adv., 2014, 4, 19538–19549 RSC
- F. Michael, D.-R. Gilad, B. Yonatan, Y. Ravit, C. Rémi, A. A.-G. Miguel and W. Itamar, Polym. Chem., 2018, 9, 2905–2912 RSC
- C.-L. Sun, J.-F. Xu, Y.-Z. Chen, L.-Y. Niu, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Polym. Chem., 2016, 7, 2057–2061 RSC
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS
- M. C. Jimenez-Molero, C. Dietrich-Buchecker and J.-P. Sauvage, Chem.–Eur. J., 2002, 8, 1456–1466 CrossRef CAS
- A. Livoreil, J.-P. Sauvage, N. Armaroli, V. Balzani, L. Flamigni and B. Ventura, J. Am. Chem. Soc., 1997, 119, 12114–12124 CrossRef CAS
- J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS
- G. Chelucci and R. P. Thummel, Chem. Rev., 2002, 102, 3129–3170 CrossRef CAS
- W. W. Brandt, F. P. Dwyer and E. D. Gyarfas, Chem. Rev., 1954, 54, 959–1017 CrossRef CAS
- J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611–619 CrossRef CAS
- G.-Y. Wu, X.-Q. Wang, L.-J. Chen, Y.-X. Hu, G.-Q. Yin, L. Xu, B. Jiang and H.-B. Yang, Inorg. Chem., 2018, 57, 15414–15420 CrossRef CAS
- I. A. Bhat, D. Samanta and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 9497–9502 CrossRef CAS
- B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS
- P. S. Mukherjee, T. K. Maji, G. Mostafa, W. Hibbs and N. R. Chaudhuri, New J. Chem., 2001, 25, 760–763 RSC
- G.-Y. Wu, X. Shi, H. Phan, H. Qu, Y.-X. Hu, G.-Q. Yin, X.-L. Zhao, X. Li, L. Xu, Q. Yu and H.-B. Yang, Nat. Commun., 2020, 11, 3178–3189 CrossRef CAS
- G.-Y. Wu, B.-B. Shi, Q. Lin, H. Li, Y.-M. Zhang, H. Yao and T.-B. Wei, RSC Adv., 2015, 5, 4958–4963 RSC
- J. Murray, K. Kim, T. Ogoshi, W. Yao and B. C. Gibb, Chem. Soc. Rev., 2017, 46, 2479–2496 RSC
- K. Jie, Y. Zhou, E. Li and F. Huang, Acc. Chem. Res., 2018, 51, 2064–2072 CrossRef CAS
- T. Ogoshi, T.-a. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS
- T. Ogoshi, K. Maruyama, Y. Sakatsume, T. Kakuta, T.-a. Yamagishi, T. Ichikawa and M. Mizuno, J. Am. Chem. Soc., 2019, 141, 785–789 CrossRef CAS
- T. Kakuta, T.-a. Yamagishi and T. Ogoshi, Acc. Chem. Res., 2018, 51, 1656–1666 CrossRef CAS
- K. Jie, M. Liu, Y. Zhou, M. A. Little, A. Pulido, S. Y. Chong, A. Stephenson, A. R. Hughes, F. Sakakibara, T. Ogoshi, F. Blanc, G. M. Day, F. Huang and A. I. Cooper, J. Am. Chem. Soc., 2018, 140, 6921–6930 CrossRef CAS
- T. Ogoshi, S. Takashima and T.-a. Yamagishi, J. Am. Chem. Soc., 2018, 140, 1544–1548 CrossRef CAS
- S. Fa, M. Yamamoto, H. Nishihara, R. Sakamoto, K. Kamiya, Y. Nishina and T. Ogoshi, Chem. Sci., 2020, 11, 5866–5873 RSC
- K. Wada, T. Kakuta, T.-A. Yamagishi and T. Ogoshi, Chem. Commun., 2020, 56, 4344–4347 RSC
- T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS
- T. Kakuta, R. Nakanishi, T. Ogoshi and T.-A. Yamagishi, RSC Adv., 2020, 10, 12695–12698 RSC
- J. Fan, Y. Chen, D. Cao, Y.-W. Yang, X. Jia and C. Li, RSC Adv., 2014, 4, 4330–4333 RSC
- Q. Lin, L. Liu, F. Zheng, P.-P. Mao, J. Liu, Y.-M. Zhang, H. Yao and T.-B. Wei, RSC Adv., 2017, 7, 34411–34414 RSC
- T.-B. Wei, X.-B. Cheng, H. Li, F. Zheng, Q. Lin, H. Yao and Y.-M. Zhang, RSC Adv., 2016, 6, 20987–20993 RSC
- Z.-Y. Li, Y. Zhang, C.-W. Zhang, L.-J. Chen, C. Wang, H. Tan, Y. Yu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 8577–8589 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials, measurements, synthesis and characterization data, UV-vis spectra. See DOI: 10.1039/d0ra09920a |
| This journal is © The Royal Society of Chemistry 2021 |