
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of magnetic field to CO hydrogenation using a confined-space catalyst: effect on reactant gas diffusivity and reactivity
Waleeporn Donphaiab,
Naphaphan Kunthakudeea,
Sirapat Munpollasriab,
Pariyawalee Sangteantongab,
Surangrat Tonlublaoc,
Wanwisa Limphiratc,
Yingyot Poo-arpornc,
Sirapassorn Kiatphuengpornd and
Metta Chareonpanich *ab
*ab
aKU-Green Catalysts Group, Department of Chemical Engineering, Faculty of Engineering, Kasetsart University, Bangkok 10900, Thailand. E-mail: fengmtc@ku.ac.th
bNanocatalysts and Nanomaterials for Sustainable Energy and Environment Research Network of NANOTEC, Kasetsart University, Bangkok 10900, Thailand
cSynchrotron Light Research Institute, Nakhon Ratchasima 30000, Thailand
dNational Nanotechnology Center, National Science and Technology Development Agency, Pathumthani 12120, Thailand
First published on 21st January 2021
Abstract
An external magnetic field has recently been applied in reaction processes to promote movement and avoid agglomeration of magnetic particles, and also reduce the activation energy through improving the gas–solid contact. In this work, the effect of an external magnetic field on reactant gas diffusivity and reactivity in CO hydrogenation within a confined-space catalyst was investigated for the first time using a conventional reactor packed with a bimetallic 5Fe–5Co/ZSM-5 molecular sieve catalyst. The synergistic effect between magnetic field and limited mass transfer within zeolite cavities improved the mass transfer ability and reaction phenomena of the reactant molecules, leading to enhancement of catalytic activity with tailored reaction pathways. As a result, CO conversion and CH4 selectivity were increased by factors of 1.9 and 1.3 compared to those without a magnetic field. These synergistic interactions are able to provide an innovative challenge for green and sustainable chemical processes and separation processes by means of selective reactant and product mass transfer designed for selective catalytic conversion in the future.
Introduction
Carbon monoxide hydrogenation reactions have been explored and investigated by many researchers. Among these reactions, Fischer–Tropsch synthesis (FTS) is known as one important route for converting synthesis gas (CO and H2) to high-quality and environmentally friendly hydrocarbon fuels.1–3 To improve the conversion and selectivity for CO hydrogenation, suitable catalysts such as Fe and Co-based catalysts are generally applied. Fe-based catalysts show high activities for both the FTS reaction and water–gas shift (WGS) reaction, allowing for efficient utilization of CO,4–6 while Co-based catalysts are superior at promoting long-chain hydrocarbons with low WGS activity at low operating temperatures.7–9 In addition, catalyst supports also play crucial roles as regards various factors including cluster size of active metal particles,5 mass transfer of reactants and products,10 and chemisorption ratio of H2 and CO on the active surfaces.11 Among the catalyst supports, ZSM-5 zeolite exhibits superior performance in the CO hydrogenation reaction due to the confined spaces in the zeolite microstructure which help promote the re-adsorption ability and secondary reactions (i.e. cracking, isomerization, and aromatization) of the primary products, and possesses shape-selective features, leading to product selectivity towards gasoline-range hydrocarbons.12–15In addition to the catalyst and support, various key parameters, such as reactant gas pressure, reaction temperature, and space velocity, also strongly influence the CO hydrogenation reaction. Recently, an external magnetic field has been applied for the first time in CO2 reforming,16 photocatalytic reaction,17 Fischer–Tropsch synthesis,18,19 hydrogen production,20 in order to control and improve the movement of magnetic particles, avoid agglomeration of magnetic particles, and reduce the activation energy through improving gas–solid contact, respectively.21–23 In previous work,24–26 we fitted a catalytic packed bed reactor with an external magnetic field and observed significant improvement in CO2 hydrogenation over Fe–Cu based catalysts by means of increased adsorption ability and selectivity to longer hydrocarbon products.
Accordingly, it is of a great interest to apply an external magnetic field to the CO hydrogenation reaction especially that in a conventional reactor packed with a confined-space bimetallic 5Fe–5Co/ZSM-5 catalyst. The mass transfer ability and reaction phenomena of reactant molecules are expected to be improved under magnetic field and magnified within a confined-space ZSM-5 molecular sieve, leading to enhancement of catalytic activity with tailored reaction pathways. These synergistic interactions are able to provide an innovation challenge for simultaneous separation–reaction processes in the future.
Experimental
ZSM-5 zeolite and 5Fe–5Co/ZSM-5 catalyst preparation
ZSM-5 zeolite support was prepared following the synthesis process reported by Chareonpanich et al. (2018) using sodium silicate solution (Na2Si3O7: 4 wt% NaOH; 27 wt% SiO2) as a silica source, aluminium nitrate (Al(NO3)3·9H2O: 98% purity, UNILAB) as an alumina source and tetrapropyl ammonium bromide (C12H28NBr or TPABr: 98% purity, Fluka Chemicals) as a structure-directing agent. Regarding the synthesis process, sodium silicate solution was primarily added into the mixture of TPABr and aluminium nitrate solution, while pH of the mixture was maintained at 10.5 throughout the mixing process by adding 1 M HCl solution. The mixture was kept stirring for 0.5 h and the obtained gel was transferred to a Teflon-lined autoclave and hydrothermally heated at 240 °C for 24 h. The solid product was filtered and washed with deionized water, dried at 80 °C for 24 h, and calcined in air at 550 °C for 4 h using a heating rate of 2 °C min−1 to remove TPABr and other volatile contaminants.In this study, 5 wt% Fe and 5 wt% Co were loaded onto ZSM-5 zeolite support using iron nitrate (Fe(NO3)3·6H2O: 98%, Univar) and cobalt nitrate hexahydrate (Co(NO3)2·6H2O: 99.95%, Univar) as metal precursors via incipient wetness impregnation method. The obtained mixture was dried using microwave irradiation at 560 W for 2 min, and consecutively calcined in air at 500 °C for 5 h using a heating rate of 5 °C min−1. The obtained catalyst was denoted as 5Fe–5Co/ZSM-5.
Characterization of textural, chemical, and magnetic properties of ZSM-5 zeolite and 5Fe–5Co/ZSM-5catalyst
The textural properties of ZSM-5 zeolite support and 5Fe–5Co/ZSM-5 zeolite catalysts were examined by N2 physisorption technique using a Quantachrome Autosorb-1C instrument at −196 °C. Prior to sorption measurement, the sample was degassed at 200 °C for 12 h. The specific surface area and pore size distribution were obtained by using Brunauer–Emmett–Teller (BET) analysis and t-plot method, and the pore volume was measured at a relative pressure of 0.995.The crystallographic structures of ZSM-5 zeolite support and 5Fe–5Co/ZSM-5 catalyst were analyzed by X-ray diffraction spectroscopy (XRD: TTRAX III) using Cu Kα radiation at high angle in the 2θ range of 5–60°.
The surface morphology of ZSM-5 zeolite support and 5Fe–5Co/ZSM-5 catalyst were observed by using field emission scanning electron microscopy (FE-SEM: JEOL, JSM-7600F) operated at 15 keV with Pt-coated on the samples. Fe and Co elements on the catalyst surface were mapped by using energy-dispersive X-ray spectroscopy (EDS: OXFORD, XMaxN). The catalyst structure was examined by using transmission electron microscopy (TEM: JEOL JEM-2010) with an acceleration voltage of 200 kV.
The reducing temperatures of Fe and Co catalysts and interaction with ZSM-5 zeolite support were examined by using H2 temperature programmed reduction (H2-TPR: Micromeritics AutoChem II chemisorption analyzer). In the TPR experiment, 0.2 g of catalyst was heated in a flow of H2/Ar mixture gas (10% H2) using a total flow rate of 30 ml min−1. The experiment was carried out in temperature range of 50–1000 °C with a heating rate of 5 °C min−1.
Local geometry changes of Fe and Co species in catalyst during reduction stage with H2 gas at a temperature range of 30–400 °C and holding stage of 400 °C for 6 h were investigated by time resolved X-ray absorption spectroscopy (TR-XAS) at Beamline 2.2 of the Synchrotron Light Research Institute (SLRI), Thailand. XANES spectra of Fe K-edge and Co K-edge were collected in energy ranges of 6975–7525 eV and 7690–7840 eV, respectively, in the transmission detection mode using ionization chamber.
Magnetic properties were measured using a vibrating sample magnetometer (VSM: Lake-Shore7404) with an external magnetic field of 5 kOe at room temperature.
Catalytic CO hydrogenation in external magnetic field
CO hydrogenation reaction over 5Fe–5Co/ZSM-5 zeolite catalyst was examined using a packed-bed reactor (SUS-316, O.D. 3/8′′) assembled with a magnetic system (a ring cast Alnico permanent magnet: Bunting Magnetics Co. Ltd.; O.D., 1.0 in; I.D., 0.75 in; thickness, 0.5 in; grade 5) with a curie temperature of 840 °C and a maximum operating temperature of 538 °C. Magnetic field orientations (north to south (N–S) and south to north (S–N) alignments) and magnetic flux density at the catalyst bed were controlled by varying numbers of magnet pairs.In this study, magnetic flux intensities at the catalyst bed were −20.8, −25.1 and −27.4 mT for two pairs, three pairs, and four pairs of magnets, respectively, with magnetic field orientation of N–S alignment. Prior to each experiment, 0.2 g catalyst packed at the center of magnetic pairs was reduced at 400 °C for 6 h using a H2 flow rate of 50 ml min−1 at atmospheric pressure. After catalyst activation, the reactor was cooled down to room temperature using He gas, then reactant gases (H2 and CO with molar ratio of 2) were introduced into the reactor. The reaction was performed at 320 °C at a total pressure of 10 bar.
Effluent gases including H2, CO, CO2, and CH4 were analyzed by using Shimadzu gas chromatograph (GC-2014) with a thermal conductivity detector (TCD) and a Unibead-C packed column, while hydrocarbon products (C1–C5+) were analyzed by Shimadzu gas chromatograph (GC-8A) with a flame ionization detector (FID). A Porapak-Q packed column and OV-1 Uniport HP packed column were used to analyze C1–C4 and C5+ hydrocarbons, respectively. Activity and selectivity of 5Fe–5Co/ZSM-5 catalyst without an external magnetic field were also examined in comparison.
Results and discussion
In this work, use of an external magnetic field and confined-space 5Fe–5Co/ZSM-5 catalyst reactant gas diffusivity and catalytic reactivity in CO hydrogenation reaction were systematically investigated. Accordingly, ZSM-5 zeolite was prepared by hydrothermal synthesis, followed by loading of the active metals via incipient wetness impregnation method. The crystalline structures of ZSM-5 support and 5Fe–5Co/ZSM-5 catalyst were examined by X-ray diffraction spectroscopy (Fig. 1). The diffraction peaks of both ZSM-5 and 5Fe–5Co/ZSM-5 catalyst corresponded well to the MFI-type framework,27,28 indicating good dispersion of Fe2O3 and CuO over ZSM-5 support. The specific surface areas of ZSM-5 support and 5Fe–5Co/ZSM-5 catalyst were found to be 271 and 227 m2 g−1, respectively, representing a type I isotherm with H4 hysteresis loop according to IUPAC classification (Fig. 2)—indicating the existence of a microporous structure with narrow slit pores. Pore size distribution of ZSM-5 zeolite was in the range of 0.3–0.7 nm, with a maximum of 0.6 nm, while a narrower pore was observed for 5Fe–5Co/ZSM-5 catalyst (0.4 nm). The fraction of micropore volume of 5Fe–5Co/ZSM-5 catalyst was reduced by approximately 13% after Fe and Co loading due to partial blockages of metal clusters, which led to this lower specific surface area. The morphology of 5Fe–5Co/ZSM-5 catalyst, examined by FE-SEM (Fig. 3), exhibited uniform, aggregated cubic particles with a mean diameter of approximately 1.1 μm. The EDS result evidently confirmed the good distribution of iron oxide and cobalt oxide nanoclusters over the surface of ZSM-5 zeolite (Fig. 3 (inset)).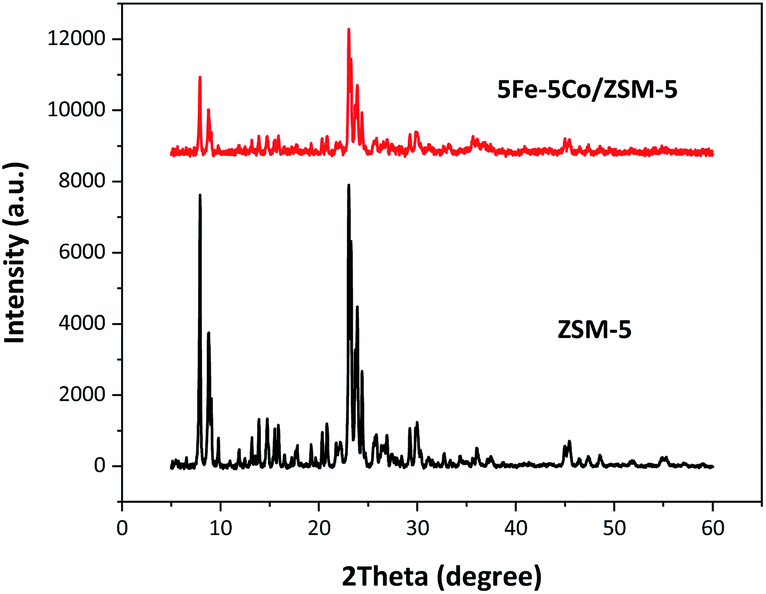 | ||
| Fig. 1 XRD patterns of ZSM-5 support and 5Fe–5Co/ZSM-5 catalyst. | ||
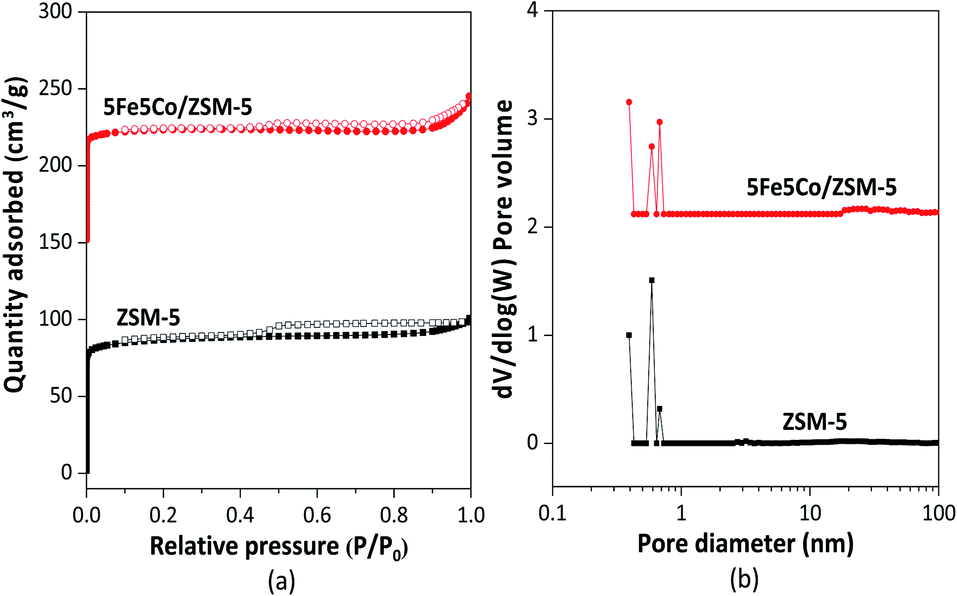 | ||
| Fig. 2 N2 adsorption–desorption isotherm (a) and pore size distribution (b) of ZSM-5 support and 5Fe–5Co/ZSM-5 catalyst. | ||
 | ||
| Fig. 3 SEM images of 5Fe–5Co/ZSM-5 catalyst (a) with EDS mapping of Fe (b), Co (c) dispersions and EDS spectrum (d). | ||
The reducibility of the catalysts was evaluated by H2-TPR, and the reduction profiles are presented in Fig. 4. Three deconvolution peaks of reduction in the temperature range of 200–800 °C are observed in 5Fe–5Co/ZSM-5 catalyst. The first peak at lower temperature (317 °C) can be assigned to transformations of Fe2O3 to Fe3O4 and of Co3O4 to CoO.5,6,12 The second peak (414 °C) is attributed to the reduction of Fe3O4 and FeO to metallic Fe and of CoO to metallic Co, while the last peak (452 °C) can be ascribed to the reduction of remaining FeO or of hardly reducible Fe species.7,29
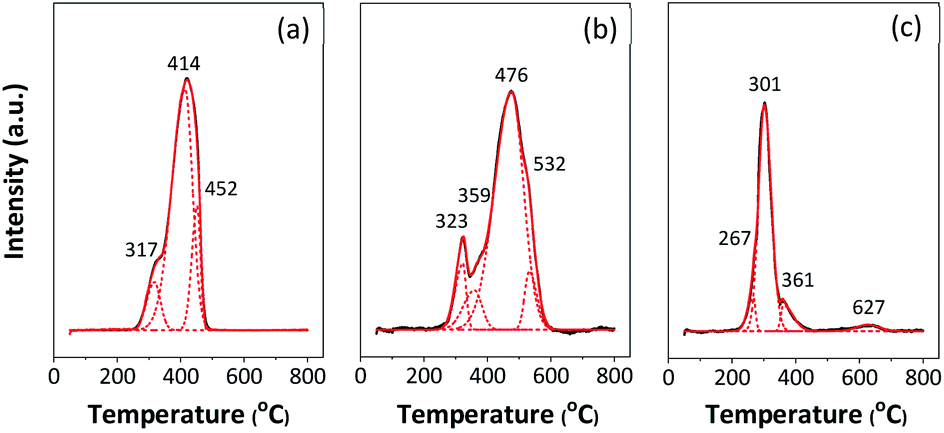 | ||
Fig. 4 H2-TPR profile of (a) 5Fe–5Co/ZSM-5, (b) 5Fe/ZSM-5 and (c) 5Co/ZSM-5 catalysts. ![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) TPR profile (experiment); TPR profile (experiment);  TPR profile (program); TPR profile (program);  deconvoluted profile of Fe and Cu oxide species. deconvoluted profile of Fe and Cu oxide species. | ||
In situ XANES technique was applied to confirm the transformation of Fe and Co species on 5Fe–5Co/ZSM-5 (Fig. 5). The XANES spectra in the range 50–340 °C exhibited absorption edge energy at 7127 eV, indicating the presence of Fe2O3 and Fe3O4. Increasing the reduction temperature to above 340 °C under a flow of H2, a gradual shift of the absorption edge energy towards lower energy was observed, indicating the reduction of oxidation state of Fe species. In the case of Co species, the reduction of oxidation state occurred when increasing the reduction temperature to those higher than 300 °C. In order to determine the evolution of the oxidation states of Fe and Co species on ZSM-5 support, linear combination fitting was performed on each XANES spectrum at the reduction temperature of 400 °C, and compared to the XANES spectra of the references consisting Fe2O3, Fe3O4, FeO and Fe for Fe species, as well as Co3O4, CoO and Co for Co species. As shown in Fig. 6, the metal oxide catalysts were transformed to approximately 20% FeO and 70% Fe, and approximately 20% CoO and 80% Co, after the reduction process.
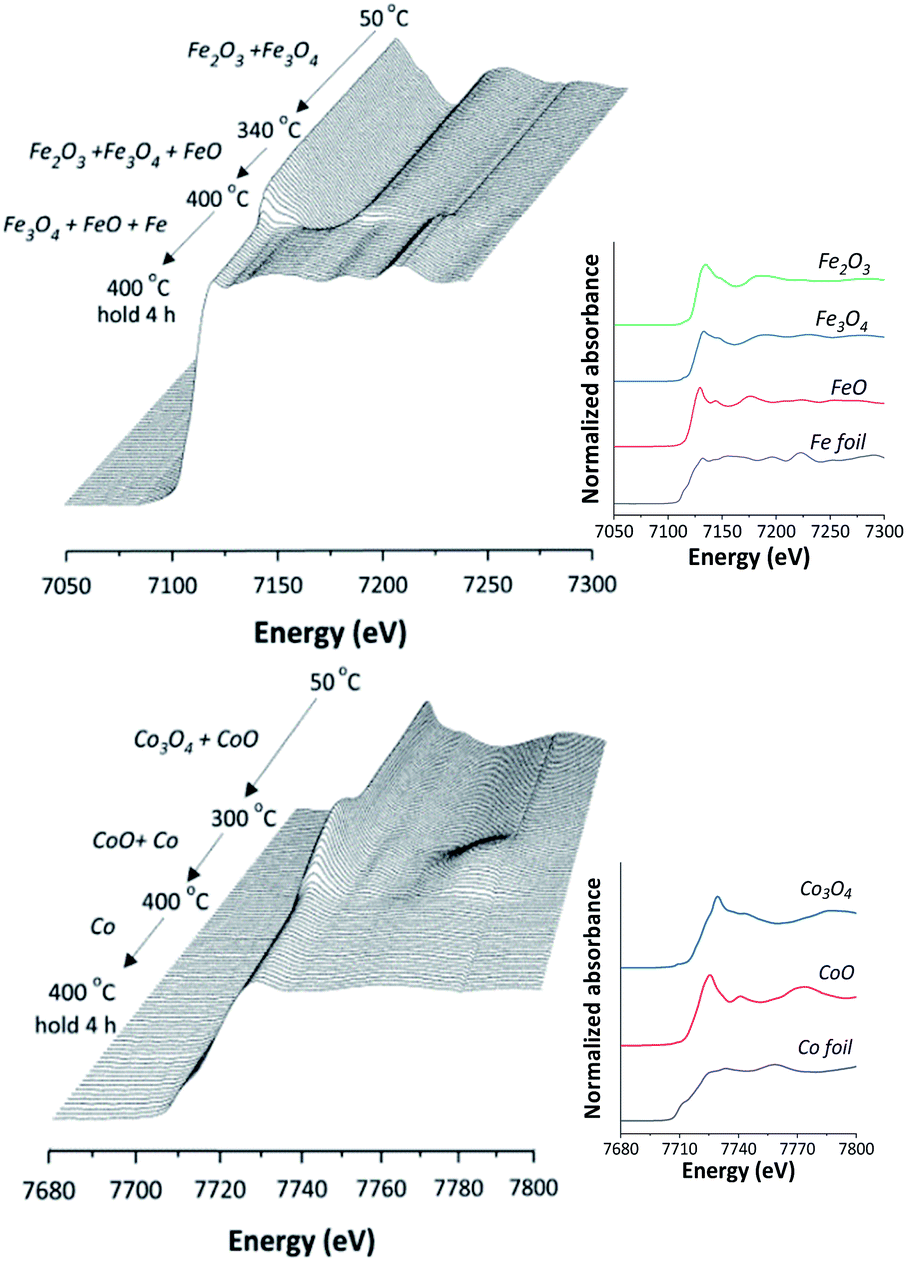 | ||
| Fig. 5 In situ Fe K-edge and Co K-edge XANES spectra of 5Fe–5Co/ZSM-5 catalyst reduction from 50–400 °C and for 4 h holding time. | ||
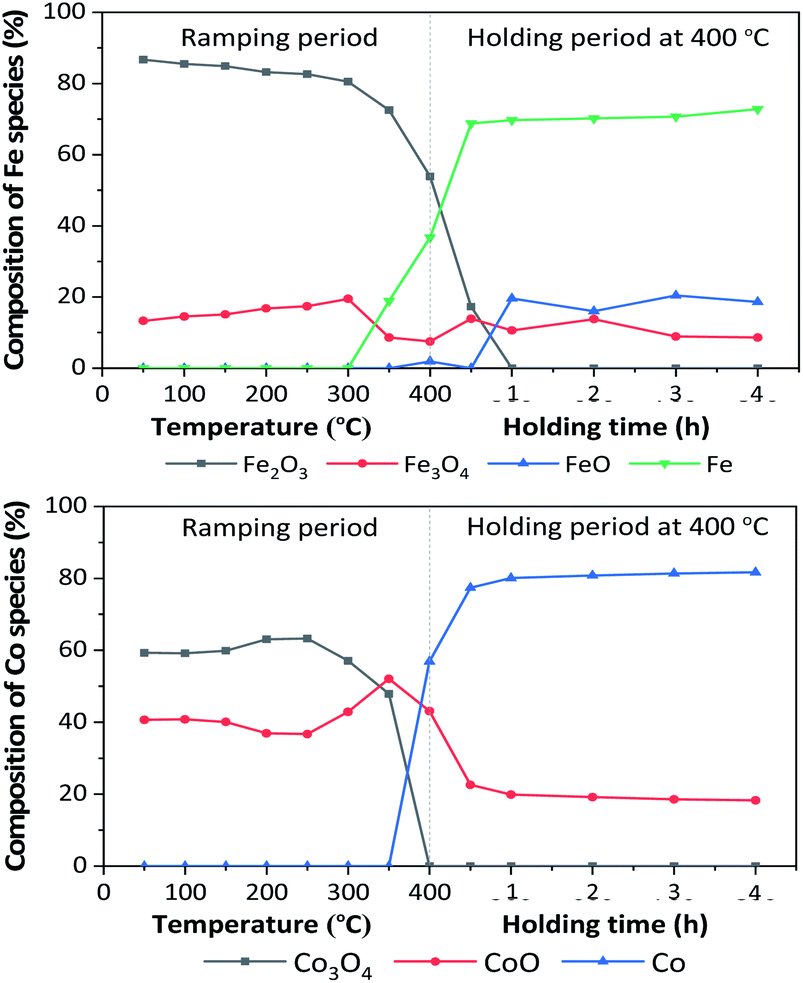 | ||
| Fig. 6 Evolution of Fe and Co species during the reduction of 5Fe–5Co/ZSM-5 catalyst. | ||
The magnetic properties of 5Fe–5Co/ZSM-5 catalyst before and after the reduction process at 400 °C were investigated using a vibrating sample magnetometer (VSM) (Fig. 7). Before the reduction process, 5Fe–5Co/ZSM-5 catalyst exhibited a small magnetic hysteresis loop, indicating paramagnetic nature of the catalyst.30,31 After the reduction process, a narrower hysteresis loop was clearly observed. The saturation (Ms) and remanence (Mr) magnetizations of the reduced 5Fe–5Co/ZSM-5 catalyst both significantly increased, indicating higher magnetic responses compared to the unreduced catalyst due to the existence of α-Fe phase with ferromagnetic property within the catalyst. In addition, a higher saturation magnetization of 19.0 emu g−1 was observed on the reduced catalyst, compared to 3.6 emu g−1 for the catalyst before reduction.
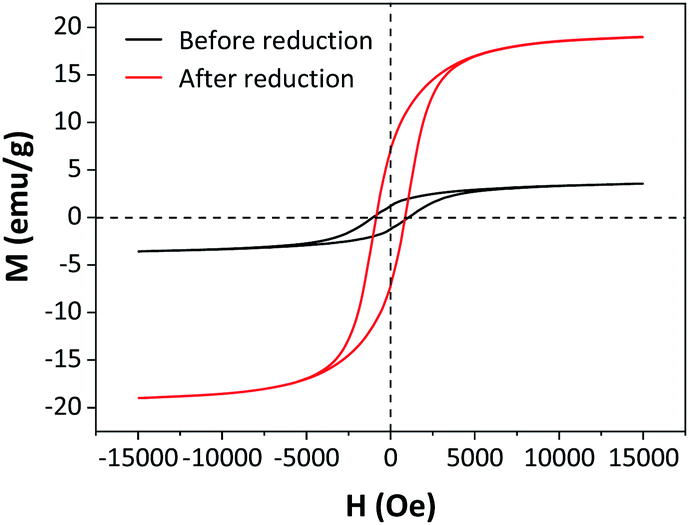 | ||
| Fig. 7 Magnetic hysteresis loops of 5Fe–5Co/ZSM-5 catalyst before and after reduction process at 400 °C. | ||
The performances of 5Fe–5Co/ZSM-5 catalyst in CO hydrogenation reaction were examined using a magnetic field-assisted packed bed reactor operated at 320 °C and 10 bar with a total flow rate of 30 ml min−1; the results are shown in Fig. 8 and 9. Magnetic flux intensities were varied in the range of 0–27.7 mT (the geomagnetic field thereby being approx. 0.03–0.06 mT). It was found that CO conversion under external magnetic field increased by a factor of 1.9 compared to that without magnetic field. Compared to the reaction over Co/Al2O3 catalyst, CO conversion at 15 mT increased by a factor of 1.1.19 However, the CO conversion showed no significant variation using magnetic flux intensities ranging from 20.8–27.4 mT.
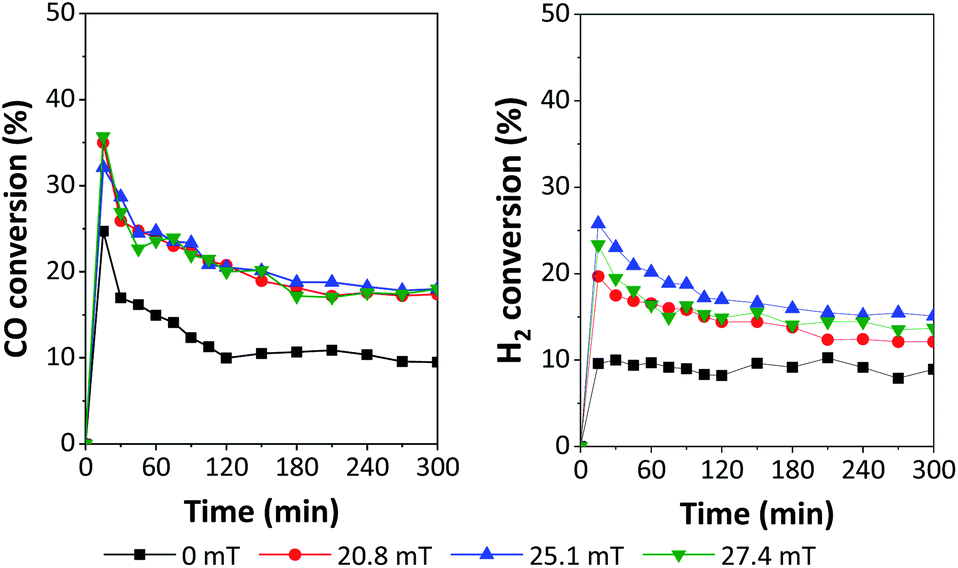 | ||
| Fig. 8 Catalytic performances in terms of H2 and CO conversion over 5Fe–5Co/ZSM-5 using different magnetic flux intensities. | ||
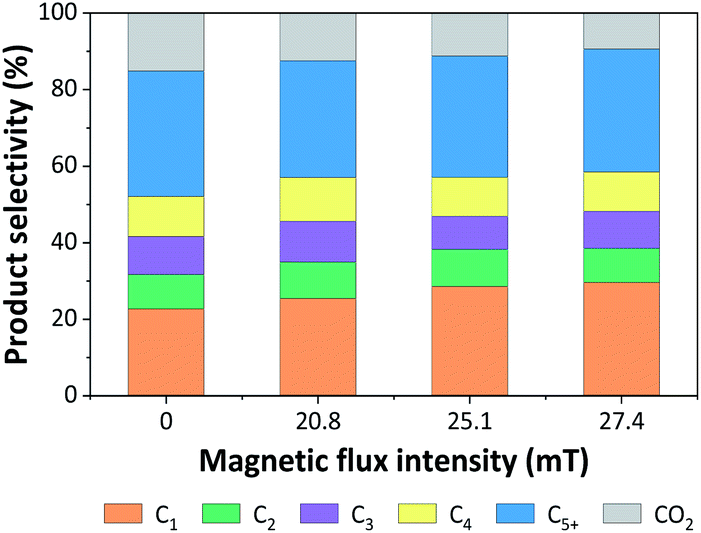 | ||
| Fig. 9 Product distributions from CO hydrogenation over 5Fe–5Co/ZSM-5 catalyst for different magnetic flux intensities. | ||
It was noted that methane yield significantly increased, while CO2 obviously decreased with increasing magnetic flux intensities in the range of 0–27.4 mT. These results indicated that the external magnetic field facilitated the activity of 5Fe–5Co/ZSM-5 catalyst through increasing the reverse water–gas shift reaction.32 This may attributed to the effective diffusion of reactant molecules and their interaction with the catalyst surface under magnetic field.
Based on the above results, it was suggested that the selective CO conversion and product selectivities under external magnetic field could possibly rely not only on the improvement of catalyst active surfaces, but also on the diffusion rate of the reactants into and the product gases from the confined-space ZSM-5 zeolite based catalyst. In order to confirm the synergistic effect of the external magnetic field and limited mass transfer within zeolite cavities, the diffusion of CO and H2 reactant molecules through 5Fe–5Co/ZSM-5 catalyst bed under external magnetic field were observed and compared to that without magnetic field, as shown in Fig. 10.
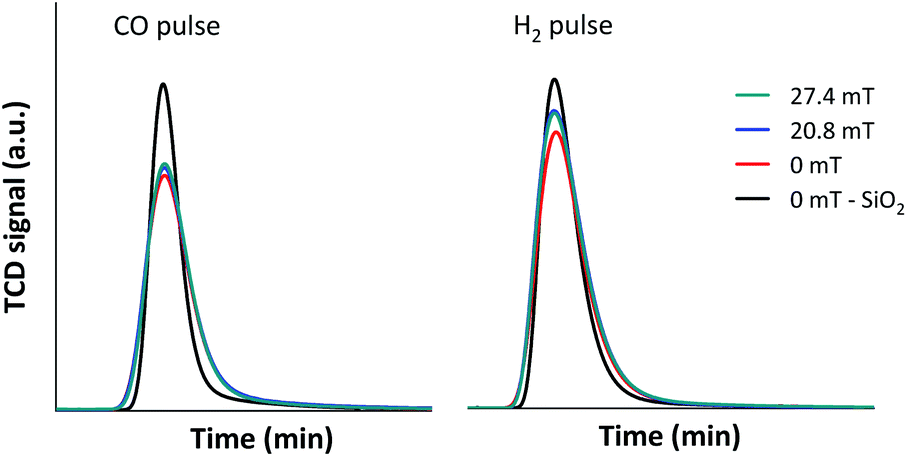 | ||
| Fig. 10 CO and H2 diffusion over 5Fe–5Co/ZSM-5 catalyst at 50 °C. | ||
As shown in Fig. 10, the diffusion rates of both CO and H2 reactant gases were significantly lowered principally due to the interaction with –OH functional group on ZSM-5 zeolite surfaces and limited mass transfer within zeolite cavities compared to inert silica. In addition, H2 diffusivity improved considerably, more than that of CO under external magnetic field as its kinetic diameter (289 pm) is 1.3 times smaller than that of CO (376 pm). The synergistic effect of the external magnetic field and limited mass transfer within zeolite cavities therefore led to selective CO conversion to methane and larger hydrocarbon products.
In order to understand the effect of external magnetic field on the reactant gas diffusion within the confined space catalyst, the effective diffusion coefficients (De) of CO and H2 were calculated using eqn (1)–(5);33 the results are shown in Table 1.
| De = Dporeε/τ | (1) |
 | (2) |
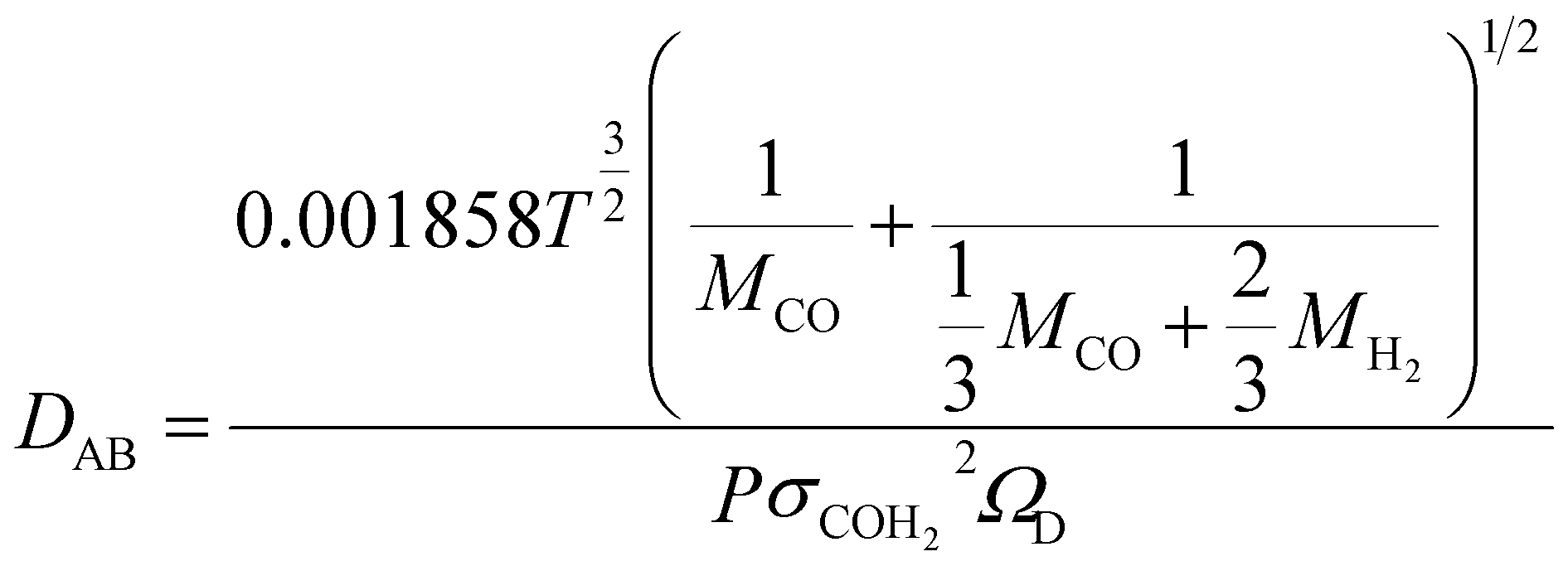 | (3) |
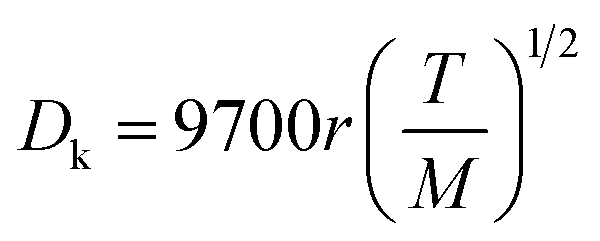 | (4) |
| Magnetic flux intensity (mT) | Diffusivities in pores × 10−3 (cm2 s−1) | Effective diffusion coefficient × 10−4 (cm2 s−1) | ||
|---|---|---|---|---|
| CO | H2 | CO | H2 | |
| 0 | 0.89 | 3.30 | 4.29 | 15.97 |
| 20.8 | 1.75 | 4.83 | 8.49 | 23.41 |
| 25.1 | 2.33 | 9.57 | 11.30 | 46.32 |
| 27.4 | 1.84 | 6.71 | 8.92 | 32.51 |
The tortuosity (τ) of 5Fe–5Co/ZSM-5 catalyst in eqn (1), can be calculated by using eqn (5).34
 | (5) |
Table 1 shows that effective diffusion coefficients of CO and H2 in pores of 5Fe–5Co/ZSM-5 catalyst under external magnetic field were improved by approximately 2.6 and 2.9 times, respectively, compared to those without magnetic field. This is in good agreement with the experimental results observed in Fig. 10. As a result, it can be concluded that the application of an external magnetic field to catalytic CO hydrogenation over a confined-space ZSM-5 zeolite based catalyst can facilitate the synergy between the external magnetic field and limited mass transfer within zeolite cavities. These phenomena consequently led to differences in effective diffusion abilities of CO and hydrogen during the reaction within the catalyst pores, leading to significant improvement in CO conversion.
As the reactor system consisting an external magnetic field in cooperation with a confined-space zeolite catalyst can facilitate different mass transfer abilities of reactant gases and products, the potential application for reactive separation processes by means of selective reactant and product mass transfers designed for separation—selective catalytic conversion with tailored reaction pathways are therefore able to provide a potential challenge for larger—scale green and sustainable chemical processes in the future.
Conclusions
In this work, the external magnetic field was applied for the first time for catalytic CO hydrogenation in a conventional reactor packed with a confined-space 5Fe–5Co/ZSM-5 catalyst. It was found that magnetic flux intensities at the catalyst bed in the range of −20.8 and −25.1 mT with magnetic field orientation of N–S alignment could successfully promote the CO conversion and CH4 selectivity by factors of 1.9 and 1.3 compared to those without magnetic field, respectively. In addition, the observation on CO and H2 diffusivities over the 5Fe–5Co/ZSM-5 catalyst evidently confirmed the synergistic effect between magnetic field and limited mass transfer within zeolite cavities as external magnetic field could promote mass transfer ability and enhance the catalytic activity with tailored reaction pathways. As a result, the concepts of magnetic field-assisted confined-space catalyst utilization are therefore able to provide an innovation challenge for green and sustainable chemical processes and separation processes by means of selective reactant and product mass transfers designed for selective catalytic conversion–separation in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Program Management Unit for Human Resources & Institutional Development, Research and Innovation [grant number B05F630097]; the Research Network of NANOTEC (RNN), the Ministry of Science and Technology, Thailand; the Center for Advanced Studies in Nanotechnology for Chemical, Food and Agricultural Industries, Kasetsart University; the Kasetsart University Research and Development Institute (KURDI); and the Faculty of Engineering (postdoc grant), Kasetsart University. The authors would like to thank the Synchrotron Light Research Institute (BL2.2: TR-XAS) for support in XAS measurements.References
- H. Li, J. Wang, C. Chen, L. Jia, B. Hou and D. Li, RSC Adv., 2017, 7, 9436–9445 RSC.
- C. I. Méndez and J. Ancheyta, Catal. Today, 2020, 353, 3–16 CrossRef.
- Z. Qi, L. Chen, S. Zhang, J. Sub and G. A. Somorjai, Appl. Catal. Gen., 2020, 602, 117607 CrossRef.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- H. Dong, M. Xie, J. Xu, M. Li, L. Peng, X. Guo and W. Ding, Chem. Commun., 2011, 47, 4019–4021 RSC.
- V. Benedetti, S. S. Ail, F. Patuzzi, D. Cristofori, R. Rauch and M. Baratieri, Renewable Energy, 2020, 147, 884–894 CrossRef CAS.
- T. Numpilai, N. Chanlek, Y. Poo-Arporn, S. Wannapaiboon, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, P. Kongkachuichay, M. Chareonpanich, G. Rupprechter, J. Limtrakulg and i Witoon, Appl. Surf. Sci., 2019, 483, 581–592 CrossRef CAS.
- H. Song, Q. Zhao, X. Zhou, Z. Cao and M. Luo, Fuel, 2018, 229, 144–150 CrossRef CAS.
- B. Gu, A. Y. Khodakov and V. V. Ordomsky, Chem. Commun., 2018, 54, 2345–2348 RSC.
- H. Becker, R. Güttel and T. Turek, Catal. Sci. Technol., 2016, 6, 275–287 RSC.
- J. Wang, H. Li, D. Li, J. P. den Breejen and B. Hou, RSC Adv., 2015, 5, 65358–65364 RSC.
- A. Carvalho, M. Marinova, N. Batalha, N. R. Marcilio, A. Y. Khodakov and V. V. Ordomsky, Catal. Sci. Technol., 2017, 7, 5019–5027 RSC.
- H. X. Vu, M. Schneider, U. Bentrup, T. T. Dang, B. M. Q. Phan, D. A. Nguyen, U. Armbruster and A. Martin, Ind. Eng. Chem. Res., 2015, 54(6), 1773–1782 CrossRef CAS.
- R. Liu, Z. Ma, J. D. Sears, M. Juneau, M. L. Neidig and M. D. Porosoff, J. CO2 Util., 2020, 41, 101290 CrossRef CAS.
- C. Liu, Y. Chen, Y. Zhao, S. Lyu, L. Wei, X. Li, Y. Zhang and Ji. Li, Fuel, 2020, 263, 116619 CrossRef CAS.
- G. H Yao, F. Wang, X. B Wang and K. T Gui, Energy, 2010, 35, 2295–2300 CrossRef.
- W. Zhang, X. Wang and X. Fu, Chem. Commu., 2003, 2196–2197 RSC.
- A. N. Pour, J. Karimi, S. Taghipoor, M. Gholizadeh and M. Hashemian, J. Iran Chem. Soc., 2017, 14, 1477–1488 CrossRef.
- P. Nikparsa, A. Nikparsa and A. Mirzaei, Phys. Chem. Res., 2020, 8, 645–656 CAS.
- J. Li, L. Zhou, Q. Zhu and H. Li, Ind. Eng. Chem. Res., 2013, 52, 6647–6654 CrossRef CAS.
- J. Li, J. Li, Q. Zhu and H. Li, Chem. Eng. J., 2018, 350, 496–506 CrossRef CAS.
- B. Stuyven, Q. Chen, W. V. de Moortel, H. Lipkens, B. Caerts, A. Aerts, L. Giebeler, B. Van Eerdenbrugh, P. Augustijns, G. Van den Mooter, J. Van Humbeeck, J. Vanacken, V. V. Moshchalkov, J. Vermant and J. A. Martens, Chem. Commun., 2009, 47, 47–49 Search PubMed.
- L. Chen, Q. Zhu, Z. Hao, T. Zhang and Z. Xie, Int. J. Hydrogen Energy, 2010, 35(6), 8494–8502 CrossRef CAS.
- S. Kiatphuengporn, P. Jantaratan, J. Limtrakul and M. Chareonpanich, Chem. Eng. J., 2016, 306, 866–875 CrossRef CAS.
- S. Kiatphuengporn, W. Donphai, P. Jantaratan, N. Yigit, K. Föttinger, G. Rupprechter and M. Chareonpanich, J. Clean. Prod., 2017, 142, 1222–1233 CrossRef CAS.
- W. Umchoo, C. Sriakkarin, W. Donphai, C. Warakulwit, Y. Poo-arporn, P. Jantaratana, T. Witoon and M. Chareonpanich, Energ. Convers. Manage., 2018, 159, 342–352 CrossRef CAS.
- B. Liu, Q. Duan, C. Li, Z. Zhu, H. Xi and Y. Qian, New J. Chem., 2014, 38, 4380–4387 RSC.
- W. Wannapakdee, C. Wattanakit, V. Paluka, T. Yutthalekha and J. Limtrakul, RSC Adv., 2016, 6, 2875–2881 RSC.
- L. Niu, X. Liu, X. Wen, Y. Yang, J. Xu and Y. Li, Catal. Today, 2020, 343, 101–111 CrossRef CAS.
- C. Sriakkarin, W. Umchoo, W. Donphai, Y. Poo-arporn and M. Chareonpanich, Catal. Today, 2018, 314, 114–121 CrossRef CAS.
- W. Donphai, N. Piriyawate, T. i Witoon, P. Jantaratana, V. Varabuntoonvit and M. Chareonpanich, J. CO2 Util, 2016, 16, 204–211 CrossRef CAS.
- K. Oshima, T. Shinagawa, Y. Nogami, R. Manabe, S. Ogo and Y. Sekine, Catal. Today, 2014, 232, 27–32 CrossRef CAS.
- W. L. McCabe, J. C. Smith and P. Harriott, Unit Operations of Chemical Engineering, McGraw-Hill, New York, 7th edn, 2005 Search PubMed.
- J. W. Beeckman, Chem. Eng. Sci., 1990, 45, 2603–2610 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |