
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of fragments inducing conformational effects in dynamic proteins using a second-harmonic generation biosensor†
Edward A. FitzGerald ab,
Margaret T. Butko‡
c,
Pierre Boronatd,
Daniela Cederfelta,
Mia Abramssona,
Hildur Ludviksdottira,
Jacqueline E. van Muijlwijk-Koezend,
Iwan J. P. de Eschd,
Doreen Dobritzscha,
Tracy Youngc and
U. Helena Danielson*ae
ab,
Margaret T. Butko‡
c,
Pierre Boronatd,
Daniela Cederfelta,
Mia Abramssona,
Hildur Ludviksdottira,
Jacqueline E. van Muijlwijk-Koezend,
Iwan J. P. de Eschd,
Doreen Dobritzscha,
Tracy Youngc and
U. Helena Danielson*ae
aDepartment of Chemistry – BMC, Uppsala University, Uppsala, 751 23, Sweden. E-mail: helena.danielson@kemi.uu.se
bBeactica Therapeutics, Virdings allé 2, Uppsala, 754 40, Sweden
cBiodesy, Inc., 170 Harbor Way, South San Francisco, 94080, CA, USA
dAmsterdam Institute of Molecular and Life Sciences (AIMMS), Division of Medicinal Chemistry, Faculty of Science, Vrije Universiteit Amsterdam, De Boelelaan 1108, 1081, HZ Amsterdam, The Netherlands
eScience for Life Laboratory, Uppsala University, Sweden
First published on 17th February 2021
Abstract
Biophysical screening of compound libraries for the identification of ligands that interact with a protein is efficient, but does typically not reveal if (or how) ligands may interfere with its functional properties. For this a biochemical/functional assay is required. But for proteins whose function is dependent on a conformational change, such assays are typically complex or have low throughput. Here we have explored a high-throughput second-harmonic generation (SHG) biosensor to detect fragments that induce conformational changes upon binding to a protein in real time and identify dynamic regions. Multiwell plate format SHG assays were developed for wild-type and six engineered single-cysteine mutants of acetyl choline binding protein (AChBP), a homologue to ligand gated ion channels (LGICs). They were conjugated with second harmonic-active labels via amine or maleimide coupling. To validate the assay, it was confirmed that the conformational changes induced in AChBP by nicotinic acetyl choline receptor (nAChR) agonists and antagonists were qualitatively different. A 1056 fragment library was subsequently screened against all variants and conformational modulators of AChBP were successfully identified, with hit rates from 9–22%, depending on the AChBP variant. A subset of four hits was selected for orthogonal validation and structural analysis. A time-resolved grating-coupled interferometry-based biosensor assay confirmed the interaction to be a reversible 1-step 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 interaction, and provided estimates of affinities and interaction kinetic rate constants (KD = 0.28–63 μM, ka = 0.1–6 μM−1 s−1, kd = 1 s−1). X-ray crystallography of two of the fragments confirmed their binding at a previously described conformationally dynamic site, corresponding to the regulatory site of LGICs. These results reveal that SHG has the sensitivity to identify fragments that induce conformational changes in a protein. A selection of fragment hits with a response profile different to known LGIC regulators was characterized and confirmed to bind to dynamic regions of the protein.
:1 interaction, and provided estimates of affinities and interaction kinetic rate constants (KD = 0.28–63 μM, ka = 0.1–6 μM−1 s−1, kd = 1 s−1). X-ray crystallography of two of the fragments confirmed their binding at a previously described conformationally dynamic site, corresponding to the regulatory site of LGICs. These results reveal that SHG has the sensitivity to identify fragments that induce conformational changes in a protein. A selection of fragment hits with a response profile different to known LGIC regulators was characterized and confirmed to bind to dynamic regions of the protein.
1 Introduction
Protein function is often regulated by ligand-induced structural changes, both via direct effects on their inherent characteristics and via indirect effects on their interactions with other biomolecules. Understanding the dynamics of such effects is critical for development of protein targeted therapeutics. However, there remains a need for screening methods that can monitor conformational changes in real time. Motivated by the lack of such methods, we have searched for a method which has the potential to be applied to a range of conformationally dynamic proteins and that could be used for identifying and characterizing conformational modulators in a (fragment) hit identification context.Ligand-gated ion channels (LGICs) represents a target class where conformational changes are necessary for function and for which the discovery of both agonists and antagonists would be of relevance for therapeutic development. The water-soluble Acetylcholine Binding Protein (AChBP), a homolog of the ligand-binding domain of nicotinic receptors and other LGICs, has been established as a model system for studies of fundamental mechanisms of ligand-binding, gating and ion transport in these ion channels, revealing important structural dynamic processes involved.1 Structural studies have revealed that all Nicotinic Acetylcholine Receptor (nAChR) ligands induce significant conformational changes in AChBP.2 Intriguingly, binding of agonists, partial agonists and antagonists result in different structural changes or functional outcomes. nAChR agonists co-crystallized with AChBP display an agonist-induced clockwise rotation of the inner sheets in the amino-terminal domains of two α subunits which is followed by an inward movement of loop C (also called loop C capping) which tightens the binding pocket.2,3 Conversely, antagonists push the loop in the opposite direction, thus opening the binding site.2,3 These structural insights are being exploited to further develop molecular probes to study a variety of LGICs.4–6
Our previous work on LGICs and the closely related AChBP has used surface plasmon resonance (SPR) biosensor technology to identify interactions and explore conformational changes. We have previously established strategies for immobilizing and studying complete ion channels,7 and have focused on understanding gating mechanisms, allosteric modulation, and screening strategies to identity agonists or antagonists of the receptor.8,9 However, the interpretation of the data is often elusive due to complex sensorgrams.
In this study we therefore explored Second Harmonic Generation (SHG) as a method that could provide direct evidence of conformational changes. It has been applied previously in a drug discovery context, against a series of difficult targets including Intrinsically Disordered Proteins (IDPs), KRAS and RNA,10–12 conducted in a well-plate format and with a detection method based on an optical readout whereby biomolecules of interest are made second-harmonic active (SH-active) through the incorporation of SH-active dye probes. Conformational changes are detected spectroscopically using SHG, a non-linear process where two photons from an incident laser are converted into a single photon of twice the energy,13 the efficiency of which is highly dependent on the angular orientation of the SH-active probes conjugated to the biomolecule of interest with respect to the surface normal where the biomolecules are tethered Fig. 1a.10 Any ligand-induced conformational change, which results in a net dye movement will be detected with a change in the SHG signal.14 The detection is independent of the size of the target and ligand, and can be applied (and is ideally suited) to large proteins such as the AChBP pentamer. Assays can be developed irrespective of the degree of structural knowledge about a given interaction. SHG technology does not require engineering of the target if it has suitable free amino groups (lysine residues) for conjugation, but was done here to provide an additional level of detail.
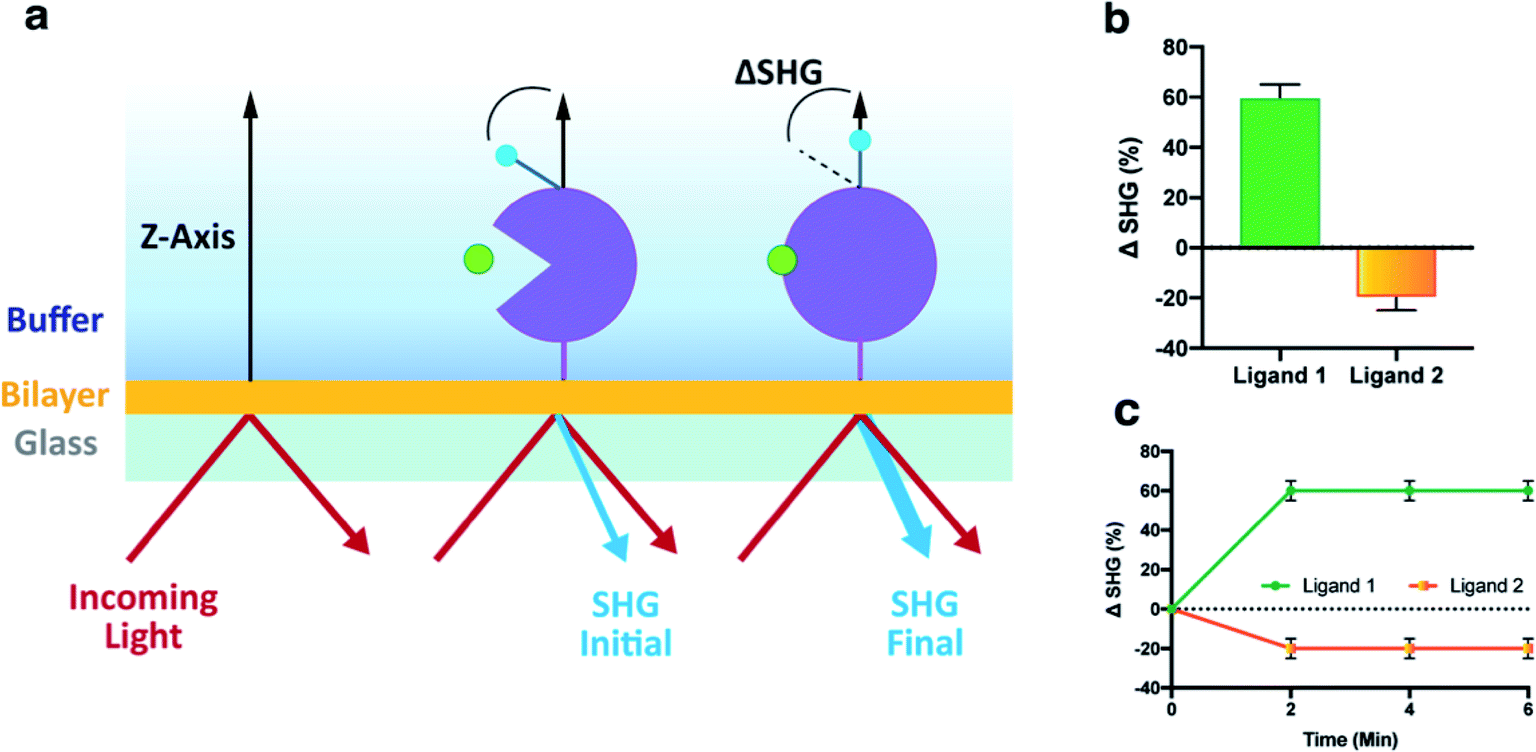 | ||
| Fig. 1 Principle for mass independent detection of structural changes in biomolecules by SHG. (a) Affinity-tagged biomolecules are conjugated with an SH-active dye (blue) and tethered onto a lipid bilayer (orange) through either His-tag:Ni/NTA or biotinylated Avi-tag:avidin interactions. Incoming light at 800 nm (red arrow) is directed at the dye, which transforms two photons of this light into one photon of light with twice the energy (400 nm), the second-harmonic light (blue light). The intensity of this second harmonic light is highly dependent on the orientation of the dye with respect to the surface normal (Z-axis). Ligand-induced structural changes in the biomolecule alter the net dye orientation changing the SHG intensity, which is detected by the instrument (b) SHG signal change upon movement as depicted in (a) is reported as ΔSHG (%). A ligand can cause an increase (Ligand 1) or a decrease in signal (Ligand 2). (c) The signal change is reported as either an end point reading (shown here, e.g. 6 minutes after ligand injection) or as a time course. | ||
The method is sensitive to both large and small (sub-Å) structural changes and therefore very suitable to study protein-fragment interactions. The magnitude of the change in SHG signal is not based upon potency but is instead a function of the overall structural change. It can differentiate between ligands with different binding modes. SHG has many advantages in a HTS setup but is also well suited to fragment-based drug discovery (FBDD). The well-based assay format can allow experiments to be performed in 1536 well-microtiter plate, which is an advantage for uncurated fragment libraries, where compounds may suffer from solubility or aggregation issues under the experimental conditions.
The first concepts and methods of FBDD emerged over 20 years ago, and its subsequent use continues to increase. Its core principles are accepted as viable means for finding hits in chemical biology or drug discovery projects.15–19 FBDD in its essence, is a reductionist alternative to high-throughput screening (HTS), built on the theory of probing a much broader chemical space by using structurally diverse compounds with molecular weight <300 Da, i.e. lower than one would conventionally find in a HTS or drug-like lead library. Since chemical space can be more efficiently explored using small compounds than large ones, fragment libraries contain usually hundreds to thousands, rather than hundreds of thousands of compounds. Accordingly, it is possible to explore novel binding sites and chemical moieties in the early stages of a discovery program.
However, fragments only have weak and transient interactions with their targets, due to their small size and therefore provide only few intermolecular contact points. Detection of functional effects from fragments is often difficult due to their usually fast and low affinity interactions.20 The binding of a fragment to a target is therefore commonly detected directly using very sensitive biophysical methods, enabling relatively high concentrations of fragments to be screened, without running into experimental artifacts. They include, but are not limited to, X-ray crystallography,21 Nuclear Magnetic Resonance (NMR), Surface Plasmon Resonance (SPR), thermal shift, and in silico methods, functional screening and Isothermal Calorimetry (ITC), in order of popularity.22–25 However, not all aforementioned methods are useful for screening purposes or for a certain target, and the method used for screening must be complemented by an orthogonal method for validation of hits. Here we have used SHG as a starting point for discovery of fragments affect the function of conformationally flexible targets.
2 Results and discussion
2.1 Development of an SHG assay for AChBP
An assay that can detect ligands inducing a conformational change upon binding to AChBP was developed as outlined in (Fig. 2a). The protein was labelled with an SH-active probe and subsequently tethered to the lipid bilayer of an analysis plate, generating a sensor surface. The functionality of the surface was validated by first recording the baseline intensity (SHGB), followed by the injection of control compounds and recording of the final intensity (SHGF). The difference between these two recordings is ΔSHG, which can be positive or negative.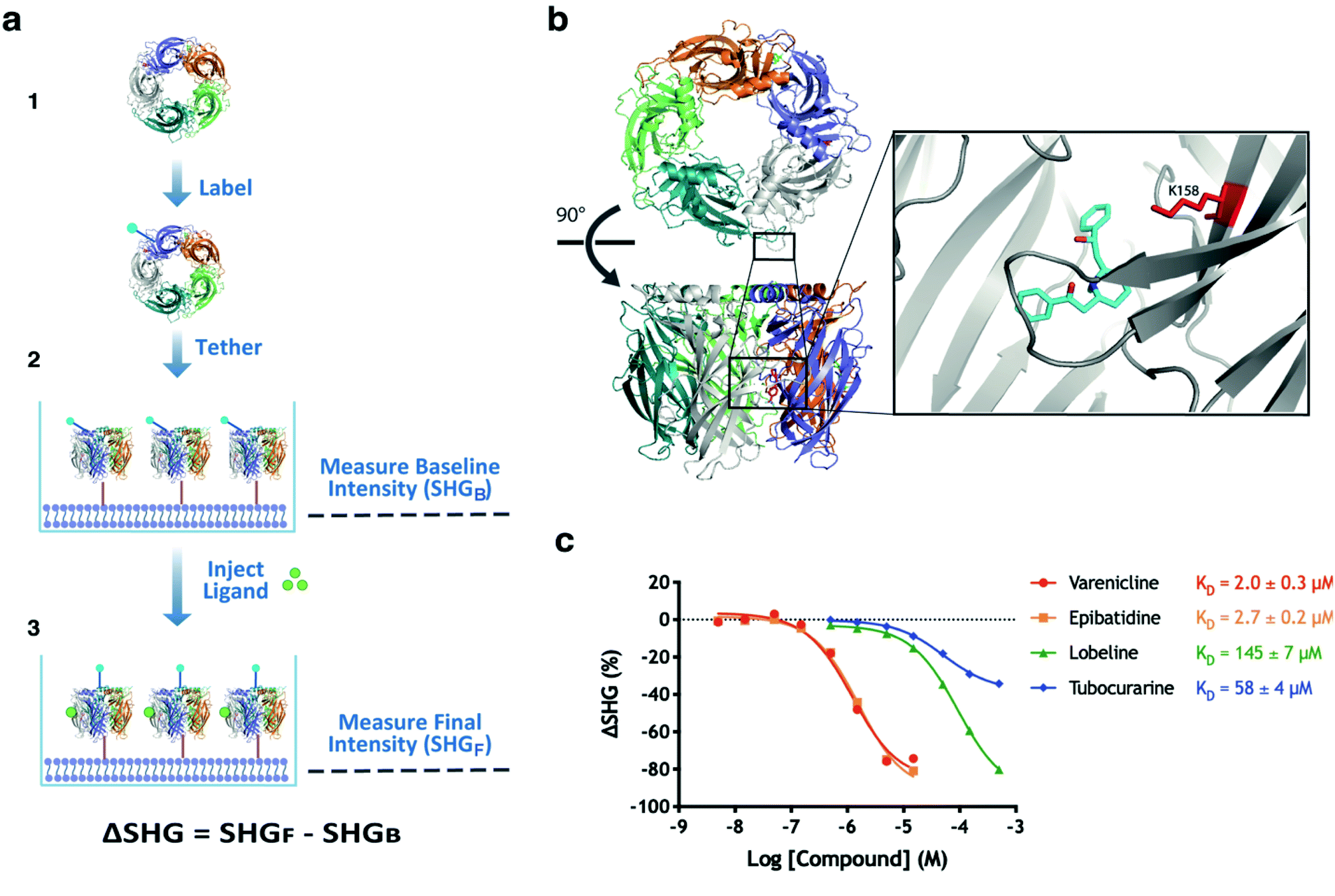 | ||
| Fig. 2 Development and validation of an SHG assay for AChBP. (a) Schematic overview of assay workflow; (1) AChBP was labelled with an SH-active probe, (2) the labelled AChBP was tethered to an analysis plate and the baseline intensity was recorded (SHGB), (3) the ligand was injected, and the final intensity measurements recorded (SHGF). (b) Structure of wildtype AChBP in complex with lobeline (cyan) and labelled with SHG1-SE probe on K158 (red). Lobeline from PDB 5AFH was inserted into AChBP from PDB 1UW6, after alignment of the binding sites. (c) concentration response curves for a set of agonists (varenicline, epibatidine), a partial agonist (lobeline) and an antagonist (tubocurarine) of nAChR injected in a concentration series over an AChBP conjugate labelled on K158, demonstrating differences in the induced conformational changes. KD values were determined from the 8-point concentration series data by non-linear regression analysis and an equation specific to SHG-derived CRCs.10 | ||
The sensitivity and stability of the AChBP assay was optimised by varying labelling conditions and incubation times for attachment to the bilayer, as well as parameters such as dye:protein ratios, pH, reaction time and the addition of glycerol to the reaction. The most successful coupling that routinely led to a homogenous population of labelled protein was achieved using SHG1-SE, a probe which labels accessible amine groups using N-hydroxy succinimide chemistry at pH 7.5. Various parameters were adjusted, resulting in a robust method which routinely led to an SH-active probe on K158 (Fig. 2b) as verified by peptide mapping mass spectrometry.
The functionality of the developed sensor surface and assay was validated by injecting nAChRs (partial) agonists (varenicline, epibatidine, lobeline) and nAChRs antagonist (tubocurarine) in a concentration series (Fig. 2c). Structural studies have confirmed that ligands with different efficacies on nAChRs also induce different conformations of AChBP.26–28 In the Concentration Response Curves (CRC's) tubocurarine (antagonist) shows a smaller ΔSHG than the other compounds, confirming that the SHG assay was able to distinguish conformational changes induced by these compounds in an AChBP conjugate labelled on K158. The experiment also indicated that the probe's location in the orthosteric site did not block the binding of these well-studied ligands. Having successfully developed an SHG assay for AChBP that can be used to understand the interactions of protein–ligand complex formation, and the subsequent conformational changes induced by known (partial) agonists and antagonists, we deemed this experimental setup suitable for screening of a fragment library to identify novel ligands interacting with AChBP.
2.2 Engineering single cysteine mutants for exploring the conformational landscape of AChBP
To explore the conformational landscape of AChBP, and to identify hits which modulate the protein by binding to different regions, six variants of AChBP were engineered. Each variant had a cysteine introduced at a single position in each subunit of the protein to which SH-active probes could specifically be conjugated (Fig. 3). They were designed to map regions of the protein subject to conformational changes upon ligand binding, and are able to differentiate between distinct ligand-induced conformational changes, thereby providing a more complete conformational landscape of AChBP.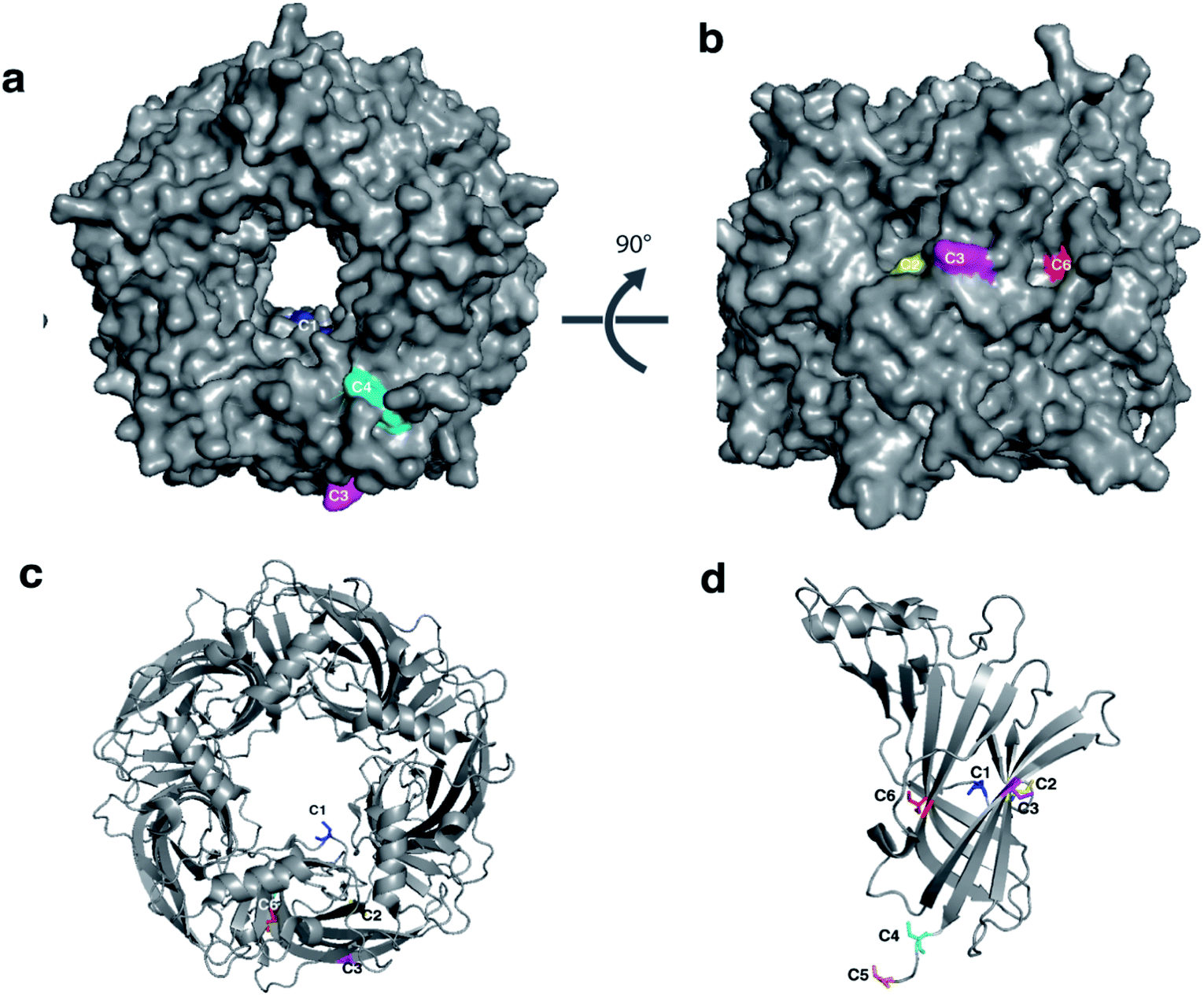 | ||
| Fig. 3 Engineered single cysteine mutants of AChBP. Visualisation of the mutated residues with (a) surface representation of complete pentamer shown in grey, (b) 90° degree rotation, (c) cartoon representation of (a), and (d) monomer of AChBP. The mutation sites are coloured as follows: C1 K98C (Blue), C2 K138C (yellow), C3 K178C (magenta), C4 K203C (cyan), C5 S206C (orange), C6 K33C (red). Note that each mutation gives rise to a total of five substitutions per pentamer. (AChBP structure from PDB 1UW6). | ||
Each of the AChBP variants were expressed and purified, and the pentameric structure was verified using native PAGE. NanoDSF was used to analyse protein quality and batch-to-batch variability (ESI Fig. S1a†), and to confirm that the mutants interacted with the control compounds in solution, thus verifying that the engineered variants were functional (ESI Fig. S1b, Table. S1†).
Of the six mutants that were brought forward to conjugation with SHG2-Mal, an alternative SH-active probe which specifically labels thiol groups of cysteine residues, two were omitted from further experiments (C4 & C6) due to the degree of labelling (DoL, i.e. the number of SH-active probes per protein molecule) varying from batch-to-batch even after multiple rounds of optimizing conjugation conditions. These inconsistencies could be indicative of an inherent stability issue upon conjugation. It could also be a potential artifact of manipulating the native protein structure by introducing point mutations. Spectroscopic analysis showed that cysteines in the wild-type protein were not conjugated by maleimide chemistry, indicating that only cysteines introduced in our protein engineering would be labelled by the maleimide dye.
2.3 Screening of fragment library using engineered single cysteine mutants of AChBP
A structurally diverse fragment library comprised of 1056 compounds including 3D fragments29 was screened against WT and engineered AChBP variants. The screening cascade was split into three distinct experiments (Fig. 4a). Initial hit calling of the fragment library screened at 250 μM (primary screen) (Fig. 4b), positive controls (varenicline, tubocurarine), and negative controls (running buffer) were dispersed throughout the screening plates across all screened constructs (Fig. 4b insert). Hits were picked by calculating the mean and standard deviation of all negative control responses across the screen. Fragments which induced SHG responses at a minimum of ±3 standard deviations from the mean negative control response were considered hits. These hit calling criteria can be adjusted to be more or less stringent depending on ambition and resources for subsequent follow-up.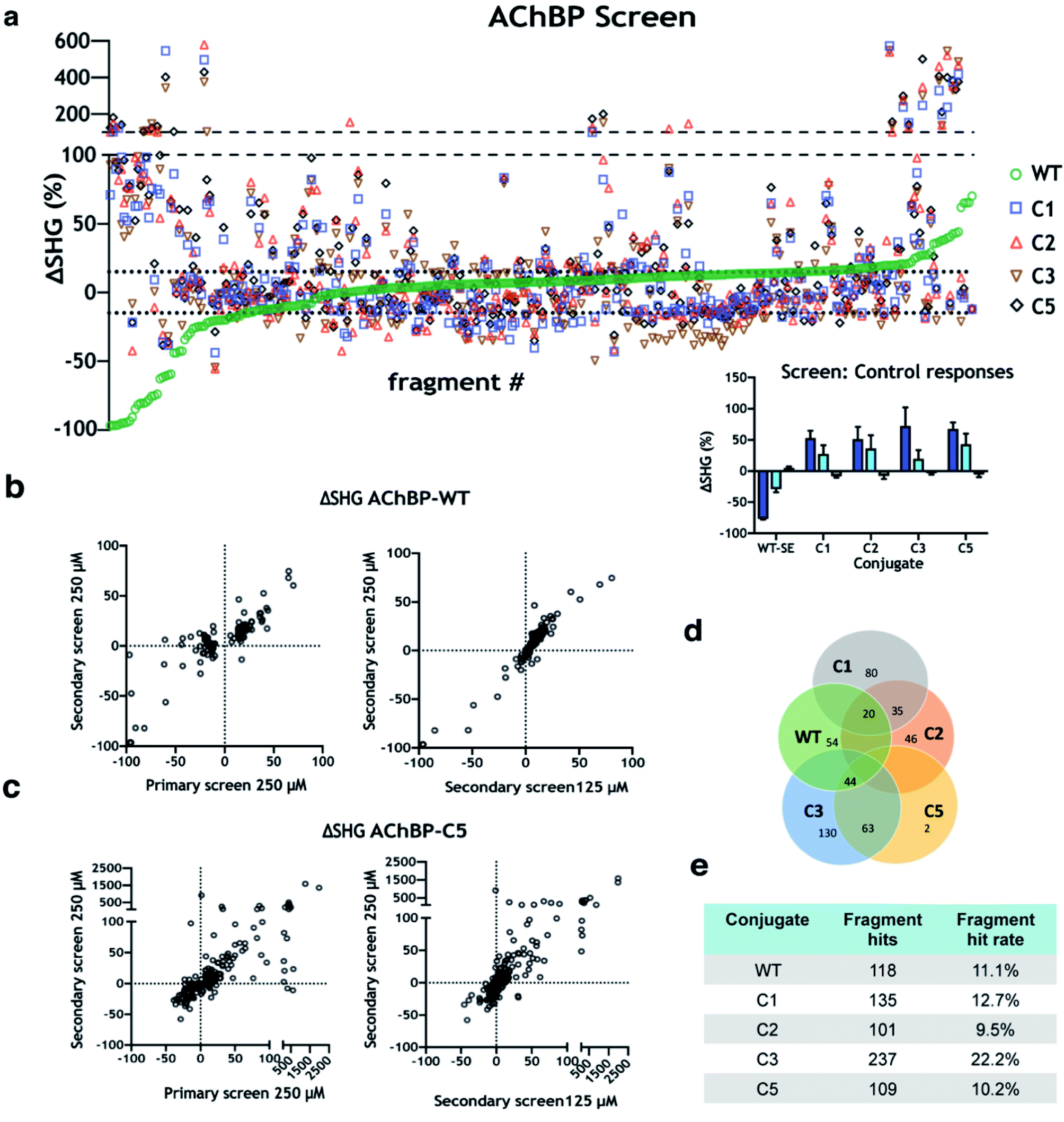 | ||
| Fig. 4 Screening of 1056-membered fragment library against AChBP variants. (a) Screening data for complete library and AChBP variants. The dotted lines around the x-axis represent the average ±3 SD for the WT negative control, compounds outside these lines are considered hits. Dashed lines show where there is a break in the y-axis, which uses two scales. Inset: screen control responses from varenicline, agonist (blue), tubocurarine, antagonist (cyan), and negative control (grey). (b) AChBP-WT screening data shown as ΔSHG for primary screen at 250 μM vs. secondary screen at 250 μM (left), and for secondary screen at 250 μM vs. 125 μM (right). (c) AChBP-C5 screening data shown as ΔSHG for primary screen at 250 μM vs. secondary screen at 250 μM (left), and for secondary screen at 250 μM vs. 125 μM (right). (d) Venn diagram illustrating hit rate and overlap across screened variants. (e) Summary of number of hits and hit rates (% of original library) for each AChBP variant. | ||
Following, fragment hits from tier one calling were brought forward and tested again at 125 & 250 μM concentrations (secondary screen). Many hits overlapped between assays, but also unique hits were observed. Fragments which induce a structural change upon the protein:fragment complex formation can be considered a responder. Depending on the conformational changes, these responders can have positive or negative ΔSHG shifts. Most positive responders in the primary screen gave the same response in the secondary screen, while many negative responders dropped out in the WT assay (Fig. 4c). Representative data from AChBP-WT and AChBP-C5 are shown in (Fig. 4c–d). Hit rates varied between 11–22% depending on screened construct (Fig. 4f), interestingly some hits were unique to individual constructs but many overlapped, as illustrated in a Venn diagram (Fig. 4e). Hits verified at this stage were brought forward to test dose dependency using concentration response curves (CRC) (CRC follow-up). This strategy led to the identification and subsequent validation of hits across all the screened constructs. To streamline the experimental workflow, AChBP-WT and AChBP-C5 were selected as representative constructs to illustrate this screening methodology and subsequent orthogonal validation. After inspecting overlap across each of the constructs, the largest overlap identified was between WT, C3, & C5 (44 fragments, Fig. 4e). For a fragment to be considered for final confirmation by CRC, it must have met the following criteria: response magnitude at 250 μM passed tier 1 hit calling thresholds in both screen and follow up plates, and response magnitude at 125 μM was at least 75% of the response magnitude at 250 μM. This will bias the follow up for higher potency hits where CRC will be more informative. A total of 24 fragments were brought to CRC for WT and 16 fragments for C5 (Fig. 5). Hits which showed a clear dose dependency and with time courses reaching a steady state at CRC were selected for validation via orthogonal methods.
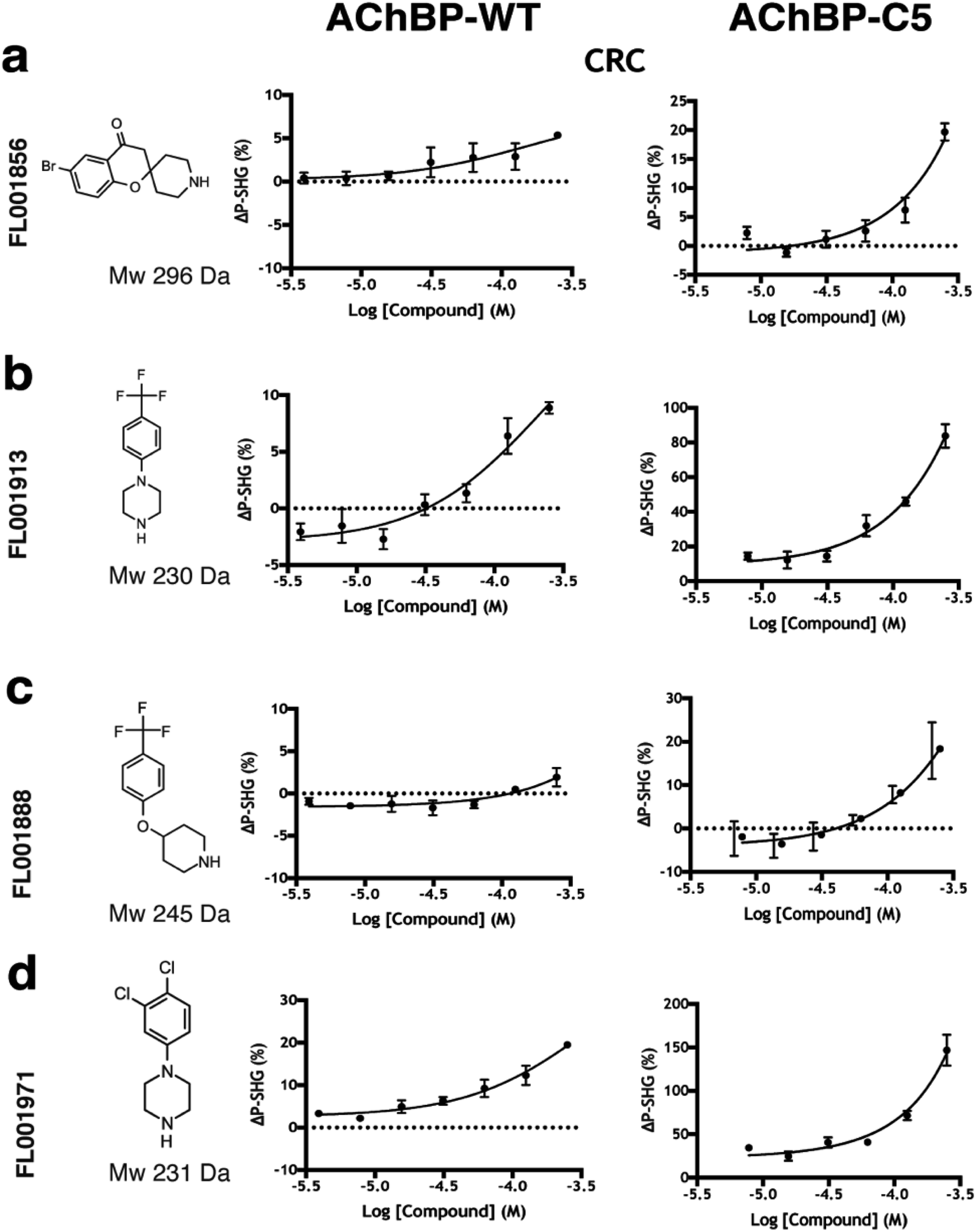 | ||
| Fig. 5 Hit confirmation by follow-up with concentration response curves (CRCs). Fragments which overlapped between WT and C5 assays showing a clear dose dependency with time courses reaching steady state (ESI Fig. S2†) were selected for orthogonal validation compounds shown at a highest concentration of 250 μM in a two-fold concentration series in rows; (a) FL001856 (b) FL001913 (c) FL001888 (d) FL001971. | ||
2.4 Orthogonal validation, kinetic characterization and structural elucidation of fragment hits
To illustrate the robustness of the devised screening strategy, four of the identified fragments which were identified as hits for WT, C3, & C5 AChBP were selected and carried through to orthogonal validation. These fragments passed all three stages in the SHG screening cascade, and showed a clear dose dependency in CRCs (data for non-selected hits ESI Fig. S3 & S4†). The binding of selected fragment hits to immobilized AChBP was evaluated using Grating Coupled Interferometry (GCI) biosensor analysis. All of the hits (Table in Fig. 6) were confirmed to bind (Fig. 6a–d) and their KD-values and kinetic rate constants were determined (Table in Fig. 6e). Two of the fragments had nanomolar affinities while the other two were in the low micromolar range. Note that KD values can more reliably be determined from the GCI than the SHG experiments since they monitor the equilibrium of fragment–target complex formation, while the CRC's from the SHG assay detects also the conformational change that this interaction may result in, i.e. an additional subsequent step. | ||
| Fig. 6 Validation of AChBP fragment hits from SHG assay using GCI biosensor-based interaction kinetic analysis. (a) FL001856, (b) FL001913, (c) FL001858, and (d) FL001971. (e) Kinetic parameters (ka, kd, and KD) were determined from the interaction kinetic curves for 10-point concentration series (up to125 μM) by global fitting using a 1:1 interaction kinetic model (a–d, blue lines). A steady state analysis was also performed and KD values estimated (see ESI, Fig. S5†). | ||
Fragments with highest affinities for AChBP were selected for validation by X-ray crystallography, the structures of two fragments with different scaffolds were determined (FL001856 and FL001888). Both fragments were found to bind to at least one subunit per pentamer, at an orthosteric site at the interface of each monomer previously recognized as a conformationally dynamic region of the protein. This binding site is formed by the five aromatic residues Y108, Y204, W162 (from one subunit), W72, and Y183 (from the neighbouring subunit) and capped by the C-loop. Based on fragment chemical structures and loop C capping we believe that these fragments may form the basis for the discovery of partial agonists or agonists (Fig. 7).
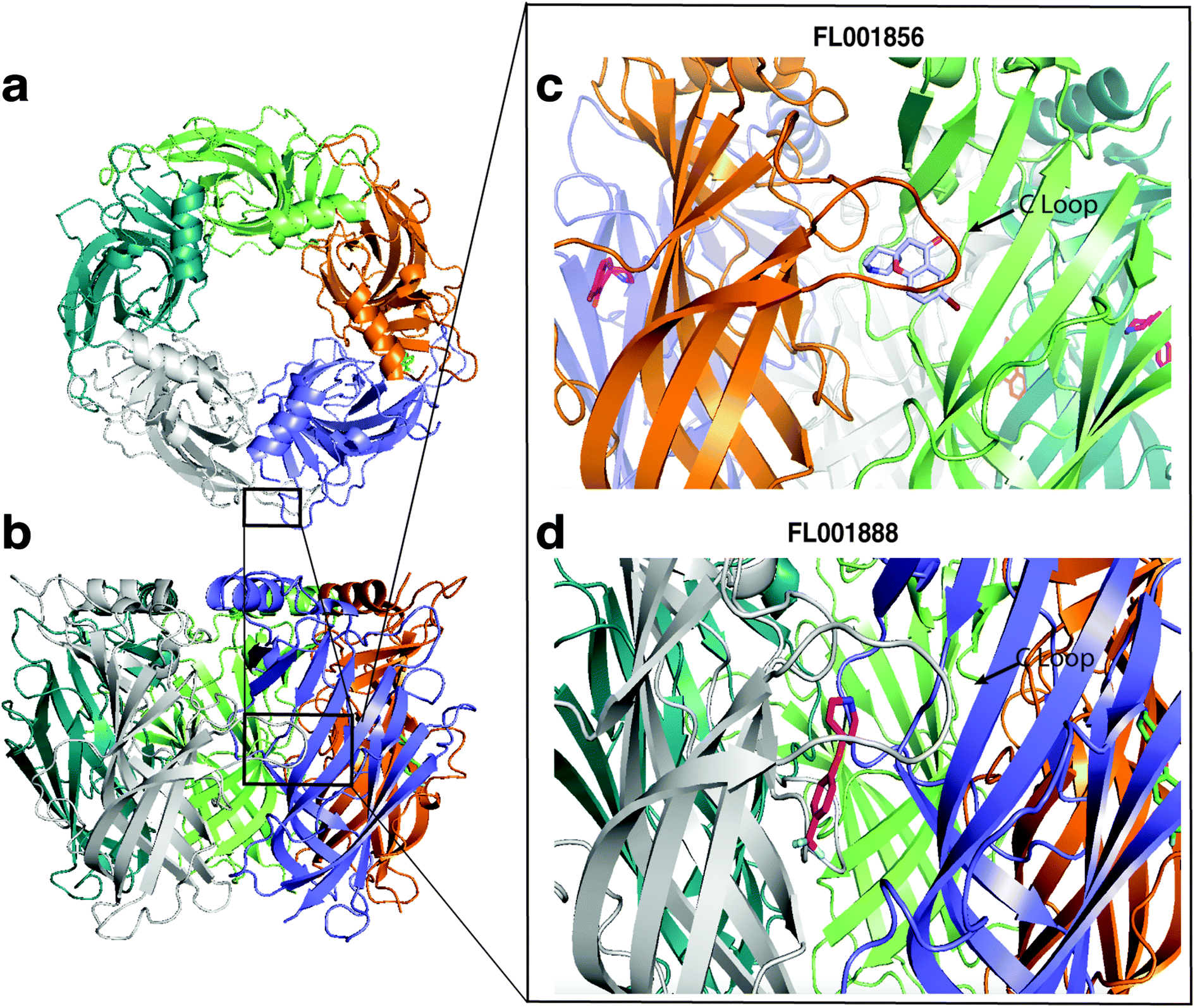 | ||
| Fig. 7 Structures of complexes between fragment hits and AChBP. (a and b) Structure of the AChBP homopentamer (top and side views), and close up view of fragment binding at the orthosteric C-loop site at the interface of each monomer: (c) FL001856 (PDB: 7NDV), (d) FL001888 (PDB: 7NDP). | ||
3 Experimental
Protein engineering, production and purification
A pFastBac1 plasmid containing Ls-AChBP cDNA was a gift from Chris Ulens (KU Belgium) and was used for protein expression and purification. Single cysteine mutants were engineered by choosing a surface accessible residue in various regions of the protein which surrounded regions of interest. Six mutations, C1 K98C, C2 K138C, C3 K178C, C4 K203C, C5 S206C, C6 K33C, were introduced into the wild type clone using the QuikChange Lightning kit (Agilent).The expression and purification were carried out as previously described.30 Spodoptera frugiperda insect cell line (Sf9) was utilized for expression of His-tagged Lymnaea stagnalis (Ls)-AChBP by infection with pre-isolated baculoviral stock (passage five, P5) with pFastBac1- Ls-AChBP gene fused in the viral genome. The cells were grown in supplemented Insect-XPRESS™ (Lonza) (penicillin and streptomycin; 100 u mL−1) at a cell density of 2 × 106 cells per mL. 1 mL per 100 mL cell culture of P5 viral stock was added to initiate protein expression. The cells were left to incubate for 72 hours at 27 °C at 90 revolutions per minutes (rpm) in a Minitron incubator Shaker (Infors HT).
Infected cells were centrifuged for 20 minutes at 4000 rpm in an Avanti J-26S XP (Beckman Coulter), supernatant was decanted into a separate flask. Ni-SepharoseTM excel beads (Cytiva) were prepared by rinsing the beads in a wash buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl). Approximate 1 mL of pre-rinsed beads were added to 1 L of supernatant and left with gentle stirring for two hours at 4 °C. Next, the beads were collected by filtering the medium with a filter funnel, beads were transferred to a PD 10 column. The column was rinsed with an imidazole containing washing buffer (20 mM Tris–HCl, 40 mM imidazole pH 8.0, 300 mM NaCl) for three column volumes. Protein was eluted with an elution buffer (20 mM Tris–HCl, 300 mM imidazole pH 8.0, 300 mM NaCl) and fractions collected. The protein concentration was estimated from the absorbance on ND-1000 spectrophotometer (NanoDrop®). The fractions containing protein were combined for protein concentrated with a 30 K Amicon® Ultra Centrifugal Filter spin column (Merck KGaA) to a storage buffer (20 mM HEPES pH 7.4, 137 mM NaCl and 2.7 mM KCl). Protein purity was assessed by SDS PAGE, to account for batch-to-batch variability the protein stability the protein was evaluated with nanoDSF on Tycho (Nanotemper).
Protein labeling with SH-active dye
His-tagged AChBP was lysine or cysteine labeled with SH-active dye using (SHG1-SE or SHG2-MAL; Biodesy, Inc.) via succinimidyl ester or maleimide chemistry. AChBP was buffer exchanged into PBS containing 10% (v/v) glycerol. AChBP was labeled at 50 μM with a 5:1 dye to protein molar ratio. The reaction was terminated by buffer exchange with ZebaSpin Desalting Columns, 7 K molecular weight cut-off (MWCO), 0.5 mL (Thermo Scientific) into PBS. After the conjugation, the average number of dye molecules per protein was determined by measuring absorbance at 280 and 410 nm with a Nanodrop. The Degree of Labeling (DoL = [Dye]/[Protein]) was calculated using the following equations:| [Protein] = (A280 − (A410 × 0.65))/Extinction coefficientprotein (M−1 cm−1) |
| [Dye] = A410/Extinction coefficientdye (M−1 cm−1) |
200 M−1 cm−1 and SHG2-Mal = 23000 M−1 cm−1.
SHG assay preparation and measurements
Supported lipid bilayers containing Ni-NTA were prepared according to the manufacturer's instructions (Biodesy, Inc.) and were formed by fusion to the well surface of 384-well Biodesy plates.10 AChBP-SHG1 was tethered to the lipid bilayer membrane at a concentration between 0.25–1 μM depending on the experiment, in AChBP assay buffer PBS-P + (20 mM phosphate buffer pH 7.4 2.7 mM KCl, 137 μM NaCl, 0.05% Surfactant P20) and incubated overnight at 4 °C. After it was tethered, wells were washed with assay buffer to remove unbound protein.Ligand injections and SHG detection were carried out on the Biodesy Delta as follows: after reading the baseline SHG signal, 20 μL of ligand at 2 times the desired concentration was injected onto 20 μL of solution volume. The SHG signal change was defined as the percentage change in SHG intensity, ΔSHG (%), and calculated as ((It − It0)/It0) × 100, where It is the SHG intensity at time t and It0 is the SHG baseline intensity before injection.
KD values for control compounds were determined using SHG data points from a concentration series. The data was fitted by non-linear regression using Prism (GraphPad Software, San Diego, CA, USA) and an equation specific to SHG-derived CRCs.10
Interaction kinetic analysis – GCI
All interaction kinetic experiments were conducted with a GCI – flow-based biosensor (WAVEdelta, Creoptix AG). The analysis temperature and running buffer composition, if not otherwise stated, were 25 °C with PBS-P+ buffer (Cytiva) supplemented with 1% DMSO (running buffer). The GCI data referencing and analysis were performed using WAVEcontrol software (Creoptix AG). AChBP was immobilized on a PCH WAVEchip (Creoptix AG) on the WAVEdelta. Sensor chips were conditioned using injections of borate buffer (10 mM sodium tetraborate pH 8.5, 1 M NaCl). Protein was diluted to the desired concentration in sodium acetate (10 mM pH 5.0) depending on the required immobilization of the target. The sensor chip was functionalized for 420 s with EDC and NHS (Cytiva) with a final protein immobilization level of 6000 surface mass (pg mm−2) with an injection time of 400 s and a flow rate of 10 μL min−1. After immobilization, the surface was deactivated with ethanolamine-HCl (1.0 M pH 8.5) for 420 s.Kinetic measurements for AChBP controls and fragments were performed with a two-fold serial dilution starting at 250 μM for each compound. Solvent correction was performed ranging from 0–2% DMSO. Blank samples of the running buffer, 1× PBS-P + buffer (20 mM phosphate buffer pH 7.4 2.7 mM KCl, 137 μM NaCl, 0.05% Surfactant P20) or HBS-P + buffer (0.01 M HEPES pH 7.4 0.15 M NaCl and 0.05% v/v Surfactant P20) both containing 1% DMSO, were injected during the measurements every fifth cycle. Samples were applied to the immobilized surface and reference channel. The sensorgrams were adjusted to account for solvent correction and blank subtraction. Kinetic fitting was performed with WAVEcontrol software (Creoptix AG) with a suitable fitting model.
Thermal unfolding assays
Label free DSF was conducted measuring Intrinsic protein fluorescence using a TychoNT.6 nanoDSF instrument (NanoTemper Technologies, Germany). Intrinsic fluorescence was recorded at 25 μM protein in a series of different buffers which were used in aforementioned assays. Protein stability of WT and engineered single cysteine mutants were assessed. Thermal shift of tool compounds and putative fragment hits were tested at a final concentration of 1 mM and a protein concentration of 1 μM.X-ray crystallography
AChBP at concentrations between 10 and 13 mg mL−1 in storage buffer (20 mM HEPES, 137 mM NaCl, 2.7 mM KCl, pH 7.4) was incubated with compound dissolved in DMSO, resulting in a final concentration of 2.5 mM compound and 5% DMSO. The drops of 2 μL contained a 1:1 ratio of protein–compound mix and reservoir solution (100 mM citric acid at pH 4.8–5.2 and 1.5–2 M ammonium sulphate). The crystallization experiments, performed in a hanging drop vapour diffusion setup at RT, resulted in crystals of various morphologies forming after 1–2 weeks. The crystals were cryo-protected in a reservoir solution supplemented with 20% glycerol before snap-freezing in liquid nitrogen. Diffraction data was collected at the Diamond Light Source (Oxford, UK) IO4 beamline and the MAXIV (Lund, Sweden) BioMAX beamline. Indexing, merging and scaling was done using XDS,31 XSCALE,32 and XDSCONVERT.32 Molecular replacement was done with PhaserMR33 with the structure deposited with PDB accession code 1UW6 as search model.30 The ligand dictionaries were created using AceDRG.34 Model building and structure refinement were done using Coot35 and REFMAC5,36 respectively. Figures were prepared with PyMol.37
4 Conclusions
SHG proved to have the sensitivity required to identify low molecular weight ligands that induce conformational changes in a protein. This is of relevance for initiating fragment-based drug discovery projects involving conformationally dynamic targets.5 Author contributions
E.A.F. & M.B. designed the experiments. E.A.F. collated and curated the fragment library. E.A.F., H.L. and M.A. produced the proteins. E.A.F. and M.B. performed SHG experiments. E.A.F. & M.A. performed biophysical experiments. EAF., P.B., D.C. and D.D. did crystallographic studies. U.H.D. supervised the project. E.A.F and U.H.D wrote the manuscript.Conflicts of interest
M. B. & T. Y. were employees of Biodesy, Inc.Acknowledgements
The authors wish to acknowledge support from Eldar Abdurakhmanov, SciLifeLab Drug Discovery and Development Platform, and library access from the Chemical Biology Consortium Sweden (CBCS). To members of the Danielson Lab for helpful discussions. The authors wish to acknowledge Hannah Klein and Peter O'Brien at the University of York and David J Hamilton and Maikel Wijtmans at the VU Amsterdam for additional compounds. To Prof. Chris Ulens, Laboratory of Structural Neurobiology, KU Leuven for AChBP expression plasmids. This project has received funding from the European Union's Framework Programme for Research and Innovation Horizon 2020 (2014–2020) under the Marie-Skoldowska-Curie grant agreement number ID 675899 Fragment based drug discovery Network (FRAGNET).References
- J. Monod, J. Wyman and J. Changeux, On the Nature of Allosteric Transitions, J. Mol. Biol., 1965, 12, 88–118 CrossRef CAS.
- S. B. Hansen, et al., Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations, EMBO J., 2005, 24, 3635–3646 CrossRef CAS.
- P. Purohit and A. Auerbach, Loop C and the mechanism of acetylcholine receptor-channel gating, J. Gen. Physiol., 2013, 141, 467–478 CrossRef CAS.
- M. H. P. Verheij, et al., Design, synthesis, and structure-activity relationships of highly potent 5-HT receptor ligands, J. Med. Chem., 2012, 55, 8603–8614 CrossRef CAS.
- C. Ulens, et al., Use of acetylcholine binding protein in the search for novel alpha7 nicotinic receptor ligands. In silico docking, pharmacological screening, and X-ray analysis, J. Med. Chem., 2009, 52, 2372–2383 CrossRef CAS.
- A. J. Thompson, et al., The binding characteristics and orientation of a novel radioligand with distinct properties at 5-HT3A and 5-HT3AB receptors, Neuropharmacology, 2014, 86, 378–388 CrossRef CAS.
- C. Seeger, et al., Histaminergic pharmacology of homo-oligomeric b 3 g -aminobutyric acid type A receptors characterized by surface plasmon resonance biosensor technology, Biochem. Pharmacol., 2012, 84, 341–351 CrossRef CAS.
- M. Geitmann, et al., Interaction Kinetic and Structural Dynamic Analysis of Ligand Binding to Acetylcholine-Binding Protein, Biochemistry, 2010, 49, 8143–8154 CrossRef CAS.
- R. Spurny, et al., in Molecular blueprint of allosteric binding sites in a homologue of the agonist-binding domain of the α 7 nicotinic acetylcholine receptor, 2015, DOI:10.1073/pnas.1418289112.
- T. A. Young, et al. Chapter Eight - Second-Harmonic Generation (SHG) for Conformational Measurements: Assay Development, Optimization, and Screening, in Modern Approaches in Drug Discovery, ed. C. A. Lesburg, Academic Press, 2018, vol. 610, pp. 167–190 Search PubMed.
- E. Donohue, et al., Second harmonic generation detection of Ras conformational changes and discovery of a small molecule binder, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17290–17297 CrossRef CAS.
- Y. Birman, S. Khorsand, E. Tu, R. B. Mortensen and M. T. Butko, Second-harmonic generation-based methods to detect and characterize ligand-induced RNA conformational changes, Methods, 2019, 167, 92–104 CrossRef CAS.
- R. J. Tran, K. L. Sly and J. C. Conboy, Applications of Surface Second Harmonic Generation in Biological Sensing, Annu. Rev. Anal. Chem., 2017, 10, 387–414 CrossRef CAS.
- B. Moree, et al., Protein Conformational Changes Are Detected and Resolved Site Specifically by Second-Harmonic Generation, Biophys. J., 2015, 109, 806–815 CrossRef CAS.
- P. J. Hajduk, et al., Discovery of Potent Nonpeptide Inhibitors of Stromelysin Using SAR by NMR, J. Am. Chem. Soc., 1997, 119, 5818–5827 CrossRef CAS.
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Discovering High-Affinity Ligands for Proteins: SAR by NMR, Science, 1996, 274, 1531–1534 CrossRef CAS.
- M. J. Harner, A. O. Frank and S. W. Fesik, Fragment-based drug discovery using NMR spectroscopy, J. Biomol. NMR, 2013, 56, 65–75 CrossRef CAS.
- M. J. Hartshorn, C. W. Murray, A. Cleasby, M. Frederickson and I. J. Tickle, Fragment-Based Lead Discovery Using X-ray Crystallography, J. Med. Chem., 2005, 48, 403–413 CrossRef CAS.
- B. Lamoree and R. E. Hubbard, Current perspectives in fragment-based lead discovery (FBLD), Essays Biochem., 2017, 453–464 Search PubMed.
- P. Brandt, M. Geitmann and U. H. Danielson, Deconstruction of Non-Nucleoside Reverse Transcriptase Inhibitors of Human Immunodeficiency Virus Type 1 for Exploration of the Optimization Landscape of Fragments, J. Med. Chem., 2011, 54, 709–718 CrossRef CAS.
- J.-P. Renaud, C. Chung, U. H. Danielson, U. Egner, M. Hennig, R. E. Hubbard and H. Nar, Biophysics in drug discovery: impact, challenges and opportunities, Nat. Rev. Drug Discov., 2016, 15, 679 CrossRef CAS.
- D. Erlanson, Poll results: affiliation and fragment-finding methods in 2019, available at: https://practicalfragments.blogspot.com/2019/12/poll-results-affiliation-and-fragment.html Search PubMed.
- M. Majewski, S. Ruiz-carmona and X. Barril, An investigation of structural stability in protein-ligand complexes reveals the balance between order and disorder, Commun. Chem., 2019, 2, 110 CrossRef.
- M. Rachman and A. Scarpino, DUckCov: a Dynamic Undocking-Based Virtual Screening Protocol for Covalent Binders, 2019, pp. 1011–1021, DOI:10.1002/cmdc.201900078.
- M. M. Rachman, X. Barril and R. E. Hubbard, ScienceDirect Predicting how drug molecules bind to their protein targets, Curr. Opin. Pharmacol., 2018, 42, 34–39 CrossRef CAS.
- N. Tabassum, Q. Ma, G. Wu, T. Jiang and R. Yu, Exploring the binding energy profiles of full agonists, partial agonists, and antagonists of the α7 nicotinic acetylcholine receptor, J. Mol. Model., 2017, 23, 251 CrossRef.
- B. Billen, et al., Molecular actions of smoking cessation drugs at α4β2 nicotinic receptors defined in crystal structures of a homologous binding protein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9173–9178 CrossRef CAS.
- M. Nys, D. Kesters and C. Ulens, Structural insights into Cys-loop receptor function and ligand recognition, Biochem. Pharmacol., 2013, 86, 1042–1053 CrossRef CAS.
- E. A. FitzGerald, et al., Multiplexed experimental strategies for fragment library screening using SPR biosensors, bioRxiv, 2020 DOI:10.1101/2020.12.23.424167.
- P. H. N. Celie, et al., Nicotine and Carbamylcholine Binding to Nicotinic Acetylcholine Receptors as Studied in AChBP Crystal Structures, Neuron, 2004, 41, 907–914 CrossRef CAS.
- W. X. D. S. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS.
- W. Kabsch, Integration, scaling, space-group assignment and post-refinement, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS.
- A. J. McCoy, Solving structures of protein complexes by molecular replacement with Phaser, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 32–41 CrossRef CAS.
- F. Long, et al., AceDRG: a stereochemical description generator for ligands, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 112–122 CrossRef CAS.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS.
- G. N. Murshudov, et al., REFMAC5 for the refinement of macromolecular crystal structures, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS.
- L. L. C. Schrödinger, The {PyMOL} Molecular Graphics System, Version∼1.8, 2015 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09844b |
| ‡ Current address: Adverum Biotechnologies, Inc., 800 Saginaw Drive, Redwood City, CA 94063, USA. |
| This journal is © The Royal Society of Chemistry 2021 |