DOI:
10.1039/D0RA09833G
(Paper)
RSC Adv., 2021,
11, 4138-4146
First cycloruthenation of 2-alkenylpyridines: synthesis, characterization and properties†
Received
19th November 2020
, Accepted 5th January 2021
First published on 20th January 2021
Abstract
Several cyclometalated ruthenium complexes 1–5 with 2-alkenylpyridines as C,N-chelating ligands were synthesized and then characterized by NMR, MS, IR and UV-Vis spectra. According to the single crystal of complex 2, it is evident that carbon from vinyl group is successfully bonded to Ru(II) center. Moreover, the Ru–N bond trans to the Ru–C bond is elongated (2.127(5) Å), which is consistent with the strong trans effect of the carbon atom compared to that of the nitrogen atom. With different electron-donating groups linked to vinyl, these complexes exhibited regular changes in MLCT absorption bands, which were identified by UV-Vis and CV spectra in combination with DFT and TD-DFT. Interestingly, protonated intermediate species of these complexes in acidic solutions were tracked by the absorption changes and MS spectra, which displayed a possible protonation process of these complexes with the cleavage of Ru–C σ bonds.
Introduction
Cyclometalated complexes have attracted considerable interest for a wide variety of applications,1 since the first cyclometalated platinum complex was reported in 1965.2 There are now many examples of reactions involving metalation of ligands by transition metals such as Ir(III), Rh(III), Pt(IV), Ru(II), and Os(II). It is noted that the metalated ligand should contain at least one common donor atom (N, P, O, S) and one carbon donor atom.3 The carbon donor atoms usually involve alkyl,4 aryl,5 or benzyl.4,6 In comparison with aryl carbons, the metalation of alkenyl carbons is much less common. Up to now, some transition metals such as rhodium,7 platinum8 and iridium9 have been reported by metalations at alkenyl carbons involving 2-vinylpyridines.
Cyclometalated ruthenium complexes have been investigated for their significantly improved absorptions in the visible region especially when compared with the classic Ru(bpy)32+.10,11 To date, many cycloruthenated complexes have been synthesized and used as energy acceptors,12 electron transfer sensitizers in dyads,13 redox mediators in oxidase chemistry,14 sensitizers for dye sensitized solar cells,15 chemosensors,16 and PDT sensitizers17 by personalized modifications of cyclometalated ligands such as expanding π-systems,13,15 and grafting photosensitive groups.16b,17 However, most of ligand frameworks used in these complexes have been based on 2-arylpyridine derivatives11–17 and N-heterocyclic carbenes18 with rare examples with 2-phenylimidazole,19 and N,N-dimethylbenzylamine.14b,20 To the best of our knowledge, few cycloruthenations of 2-vinylpyridine and derivatives have been reported.
Therefore, our interest in the design of new organic ligands for cyclometalated ruthenium complexes has led us to synthesize a series of 2-alkenylpyridines. Herein, we investigated in details a series of ruthenium(II) metalations at alkenyl carbons involving 2-alkenylpyridine and related ligands. The synthetic route is shown in Scheme 1. After being characterized by NMR, MS, FT-IR and elemental analysis, photophysical properties of these complexes were investigated by UV-Vis and FL spectra in combination with CV spectra and theoretical calculations. To the best of our knowledge, deprotonations of ligands are necessary for the formation of cyclometalated ruthenium complexes. However, there have been rare examples on the protonation of cycloruthenated complexes despite that. Therefore, these complexes incubated in acidic solutions were investigated in details. Then, possible acid–base equilibria for these complexes were depicted.
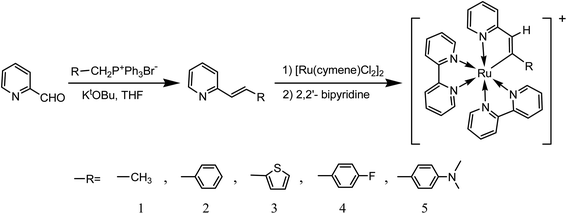 |
| | Scheme 1 The reactions of 2-alkenylpyridine and related ligands with ruthenium(II). | |
Experimental sections
Materials
Ethyltriphenylphosphonium bromide, benzyltriphenylphosphonium bromide, triphenyl-(4-fluorobenzyl)phosphonium bromide, triphenyl-(4-N,N-dimethylbenzyl)phosphonium bromide, and triphenyl-(thiophen-2-ylmethyl)phosphonium bromide were prepared by the methods described in literatures21 and used directly in the synthesis of 2-vinylpyridine derivatives. All solvents and reagents were commercially available and used without further purification unless specified.
Physical measurements
NMR spectra were recorded on a Bruker Avance-400 spectrometer. Mass spectra were obtained on a Bruker autoflex speed MALDI-TOF spectrometer by using CHCA (α-cyano-4-hydroxycinnamic acid) as matrix and elemental analyses were determined by a VarioEL III O-Element Analyzer system. Absorption spectra were recorded using a Shimadzu UV-2550 spectrophotometer. Fluorescence spectra were recorded on a Varian Cary Eclipse spectrophotometer in a 4.5 mL (1 cm in diameter) cuvette with 2 mL solution at 77 K. All pH measurements were made with a Model pHS-3B pH meter (Shanghai, China). Cyclic voltammogram spectra of 1–5 were collected in MeCN with [n-Bu4N]PF6 as the supporting electrolyte at 100 mV s−1, using the standard three-electrode setup with a platinum disk as working electrode, platinum wire as counter electrode, and AgNO3/Ag as reference electrode. Herein, potentials reported relative vs. ferrocenium/ferrocene (Fc+/Fc) used as an internal standard. The single-crystal X-ray data were collected on a Bruker P4 diffractometer with Mo Kα radiation (λ = 0.71073 Å) using the ω-scan mode. The crystal of 2 was kept at a steady T = 293(2) K during data collection. The structure of 2 was solved with the Olex2.solve22a solution program by using Olex2 1.3-dev22b as the graphical interface. The model was refined with ShelXL 2018/3 (ref. 22c) using full matrix least squares minimization on F2.
Preparation of solutions
Deionized water was used throughout all experiments. Stock solutions of 2-alkenylpyridine ligands (1.0 mM) and complexes 1–5 were prepared in acetonitrile and ethanol, respectively. Then, solutions of 2-alkenylpyridine ligands were diluted to 20 μM with acetonitrile, while complexes were diluted to 20 μM with ethanol. For complexes 1–5, their buffered solutions were prepared in ethanol and Briton–Robinson buffer at volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and then incubated for 4 h.
:2 and then incubated for 4 h.
Theoretical calculations
All the density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were performed with the Gaussian 09 package.23 The structures in the ground state (S0) were fully optimized by the Becke's three-parameter exchange functional along with the Lee–Yang–Parr correlation functional (B3LYP) and plus polarization function 6-31G* basis set. Calculations of electronic absorption spectra were computed with the time-dependent density functional theory (TD-DFT) method at the B3LYP/6-31G* level based on the optimized ground-state structures. For all the calculations, the associated basis set was applied to describe Ru with the “double-ζ” quality LANL2DZ basis set, whereas, for all other atoms, the 6-31G(d) basis set was employed. The contours of the HOMOs and LUMOs orbitals were plotted.
Synthesis of ligands
(E)-2-(Prop-1-en-1-yl)pyridine. In a three-necked flask, 2-pyridylaldehyde (45.6 mg, 0.43 mmol) and anhydrous tBuOK (98.7 mg, 0.88 mmol) were dried in vacuum for 30 min and followed by adding 20 mL of dry THF. Then, ethyltriphenylphosphonium bromide (160.2 mg, 0.43 mmol) dissolved in 5 mL of dry THF was added dropwise to the above solution with stirring under N2 atmosphere. The mixture was stirred at ambient temperature for another 3 h and then poured into ice-water (40 mL). The mixture was extracted with dichloromethane and concentrated in vacuum. After purified by silica gel column chromatography with CH2Cl2/petroleum ether (v/v = 2:1) as eluent, orange oil was obtained with yield of 61% (0.031 g). The 1HNMR was in good agreements with the ref. 9a.
(E)-2-Styrylpyridine. The procedure was the same as that of (E)-2-(prop-1-en-1-yl)pyridine. Yield: 0.052 g (67%). 1H NMR (CDCl3, 400 MHz), δ (ppm): 8.61 (d, J = 4.4 Hz, 1H), 7.48 (td, J = 8.0 Hz, J = 2.0 Hz 1H), 7.27–7.25 (m, 5H), 7.19 (d, J = 8.0 Hz, 1H), 7.12–7.09 (m, 1H), 6.87 (d, J = 12.4 Hz, 1H), 6.73 (d, J = 12.4 Hz, 1H).
(E)-2-(2-(Thiophen-2-yl)vinyl)pyridine. Yield: 0.046 g (58%). 1H NMR (CDCl3, 400 MHz), δ (ppm): 8.73 (d, J = 4.0 Hz, 1H), 7.68 (td, J = 1.6 Hz, J = 7.8 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.29 (d, J = 4.0 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H), 7.19 (dd, J = 7.8 Hz, J = 4.8 Hz, 1H), 7.00 (dd, J = 7.2 Hz, J = 3.6 Hz, 1H), 6.92 (d, J = 12.4 Hz, 1H), 6.51 (d, J = 12.4 Hz, 1H).
(E)-2-(4-Fluorostyryl)pyridine. Yield: 0.053 g (62%). 1H NMR (CD3OD, 400 MHz), δ (ppm): 8.51 (d, J = 5.2 Hz, 1H), 7.66–7.63 (m, 1H), 7.28–7.21 (m, 4H), 7.01–6.96 (m, 2H), 6.89 (d, J = 12.1 Hz, 1H), 6.70 (d, J = 12.1 Hz, 1H).
(E)-N,N-Dimethyl-4-(2-(pyridin-2-yl)vinyl)aniline. Yield: 0.044 g (46%). 1H NMR (CDCl3, 400 MHz), δ (ppm): 8.59 (s, 1H), 7.64–7.49 (m, 4H), 7.37 (d, J = 7.6 Hz, 1H), 7.09 (s, 1H), 7.03 (d, J = 16.0 Hz, 1H), 6.75 (d, J = 8.4 Hz, 2H), 3.02 (s, 6H).
Preparation of complexes
A suspension of [Ru(cymene)Cl2]2 (0.16 g, 0.26 mmol), triethylamine (0.05 mL), 2-vinylpyridine derivative (0.50 mmol) and KPF6 (0.182 g, 0.99 mmol) in 10 mL of acetonitrile was stirred at 50 °C for 24 h under argon. Then, the solvent was removed under vacuum. The orange red solid was obtained and added into the methanol solution (10 mL) containing 2,2′-bipyridine (0.169 g, 1.08 mmol). After the solution was refluxed for 2 h under argon, the solvent was removed by evaporation. The crude compound was purified directly by column chromatography on silica gel using CH3CN/CH2Cl2 as eluent to afford the product as black solid.
1. Yield: 0.079 g (45%). 1HNMR (CD3CN, 400 MHz), δ (ppm): 8.48 (dd, J = 5.6 Hz, J = 0.8 Hz, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.39 (d, J = 8.0 Hz, 1H), 8.35 (d, J = 8.0 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 7.99 (td, J = 8.0 Hz, J = 1.6 Hz, 1H), 7.91–7.85 (m, 2H), 7.82–7.77 (m, 1H), 7.74 (dd, J = 5.6 Hz, 2H), 7.68 (d, J = 5.6 Hz, 1H), 7.47 (td, J = 8.0 Hz, J = 1.6 Hz, 1H), 7.42–7.37 (m, 2H), 7.34 (m, 1H), 7.28–7.26 (m, 2H), 7.18–7.14 (m, 1H), 7.04 (d, J = 1.2 Hz, 1H), 6.60 (td, J = 7.2 Hz, J = 1.2 Hz, 1H), 1.84 (d, J = 1.2 Hz, 3H). 13CNMR (CD3CN, 100 MHz), δ (ppm): 215.54, 169.35, 157.54, 156.99, 156.79, 154.96, 154.71, 150.63, 149.99, 149.35, 148.68, 136.34, 134.94, 134.78, 133.67, 133.54, 126.87, 126.77, 126.37, 126.18, 125.86, 123.28, 122.95, 122.74, 122.60, 118.56, 117.30, 27.66. MS (Maldi-TOF, CHCA): m/z calculated for 532.11 (M-PF6−), found: 532.06. C28H24F6N5PRu calc. C, 49.71; H, 3.58; N, 10.35; found C, 50.06; H, 3.35; N, 10.43.
2. Yield: 0.058 g (30%). 1H NMR (CD3CN, 400 MHz), δ (ppm): 8.89 (d, J = 8.0 Hz, 1H), 8.47 (dd, J = 8.0 Hz, 2H), 8.19 (d, J = 8.0 Hz, 1H), 7.99–7.86 (m, 4H), 7.76 (d, J = 5.4 Hz, 1H), 7.71 (d, J = 5.4 Hz, 1H), 7.57–7.44 (m, 5H),7.39–7.35 (m, 3H), 7.13 (s, 1H), 6.98 (td, J = 8.0 Hz, J = 1.6 Hz, 1H), 6.85–6.83 (m, 3H), 6.69 (td, J = 8.0 Hz, J = 1.6 Hz, 1H), 6.43 (dd, J = 1.6 Hz, 2H). 13CNMR (CD3CN, 100 MHz), δ (ppm): 200.14, 169.49, 157.50, 156.94, 156.55, 155.53, 155.05, 151.59, 150.85, 150.07, 149.62, 148.91, 136.54, 135.24, 135.13, 133.95, 133.55, 129.74, 128.42, 128.17, 127.15, 126.91, 126.41, 126.08, 126.01, 125.67, 124.33, 123.46, 123.09, 122.35, 122.31, 120.07, 119.65, 118.34. MS (Maldi-TOF, CHCA): m/z calculated for 594.12 (M-PF6−), found: 594.09. C33H26F6N5PRu calc. C, 53.66; H, 3.55; N, 9.48; found C, 52.96; H, 3.35; N, 10.02.
3. Yield: 0.052 g (27%). 1H NMR (CD3CN, 400 MHz): 8.69 (d, J = 5.6 Hz, 1H), 8.44 (dd, J = 8.0 Hz, 2H), 8.28 (dd, J = 8.0 Hz, 2H), 7.99 (t, J = 8.0 Hz, 1H), 7.89 (dd, J = 8.0 Hz, 2H), 7.77 (t, J = 7.2 Hz, 1H), 7.67–7.65 (m, 3H), 7.54–7.28 (m, 7H), 7.13 (t, J = 6.8 Hz, 1H), 7.02 (d, J = 5.2 Hz, 1H), 6.65 (d, J = 5.6 Hz, 2H), 6.09 (d, J = 3.6 Hz, 1H); 13CNMR (CD3CN, 100 MHz), δ (ppm): 207.76, 168.92, 157.52, 156.96, 156.84, 155.56, 155.16, 154.20, 150.88, 150.27, 149.61, 148.87, 136.80, 135.32, 135.21, 134.18, 134.05, 129.38, 127.28, 127.12, 126.58, 126.15, 126.02, 123.97, 123.52, 123.20, 122.82, 122.63, 122.52, 120.30, 118.29. MS (Maldi-TOF, CHCA): m/z calculated for 600.03 (M-PF6−), found: 600.10. C31H24F6N5PRuS calc. C, 50.00; H, 3.25; N, 9.40. Found: C, 49.75; H, 3.64; N, 9.07.
4. Yield: 0.049 g (25%). 1HNMR (CD3CN, 400 MHz), δ (ppm): 8.84 (d, J = 5.2 Hz, 1H), 8.47–8.42 (dd, J = 8.0 Hz, 2H), 8.19 (d, J = 8.4 Hz, 1H), 8.02–7.87 (m, 4H), 7.74 (dd, J = 5.2 Hz, 2H), 7.62–7.51 (m, 3H), 7.47–7.35 (m, 5H), 7.14 (s, 1H), 7.01 (t, J = 7.2 Hz, 1H), 6.71–6.68 (m, 1H), 6.60 (t, J = 8.8 Hz, 2H), 6.43 (m, 2H); 13CNMR (d6-DMSO, 125 MHz), δ (ppm): 198.87, 169.18, 159.22, 157.39, 156.88, 156.56, 155.55, 155.03, 150.69, 149.92, 149.57, 148.91, 148.02, 137.31, 135.85, 135.72, 134.77, 134.42, 129.06, 127.83, 127.33, 126.95, 126.64, 125.31, 125.25, 124.32, 123.91, 123.30, 123.22, 120.66, 119.18, 114.31, 114.15. MS (Maldi-TOF, CHCA): m/z calculated for 612.09 (M-PF6−), found: 612.16. C33H25F7N5PRu calc. C, 52.38; H, 3.33; N, 9.26; found: C, 52.05; H, 3.84; N, 9.73.
5. Yield: 0.051 g (25%). 1H NMR (CD3CN, 400 MHz), δ (ppm): 8.79 (d, J = 5.6 Hz, 1H), 8.46 (dd, J = 8.0 Hz, 2H), 8.20 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 8.0 Hz, 1H), 7.97 (td, J = 7.2 Hz, J = 1.6 Hz, 1H), 7.89–7.83 (m, 2H), 7.73 (d, J = 5.6 Hz, 1H), 7.67–7.62 (m, 2H), 7.59 (d, J = 5.6 Hz, 1H), 7.49 (td, J = 8.0 Hz, J = 1.6 Hz, 1H), 7.40–7.29 (m, 5H), 7.11 (s, 1H), 7.03 (t, J = 6.4 Hz, 1H), 6.62 (t, J = 6.4 Hz, 1H), 6.46 (d, J = 8.0 Hz, 2H), 6.29 (d, J = 8.0 Hz, 2H), 2.76 (s, 6H); 13CNMR (d6-DMSO, 125 MHz), δ (ppm): 221.47, 167.33, 155.37, 154.75, 154.53, 153.51, 152.88, 148.51, 147.78, 147.20, 146.76, 146.48, 137.75, 135.11, 133.54, 133.30, 132.16, 132.10, 125.76, 125.41, 125.04, 124.58, 124.40, 123.68, 122.11, 121.71, 121.25, 121.11, 117.98, 116.03, 29.49, 28.16. MS (Maldi-TOF, CHCA): m/z calculated for 637.17 (M-PF6−), found: 637.12. C35H31F6N6PRu calc. C, 53.78; H, 4.00; N, 10.75; found: C, 53.55; H, 4.44; N, 10.93.
Results and discussion
Synthesis and characterization
The synthetic route of complexes 1–5 is presented in Scheme 1. Herein, 2-vinylpyridine derivatives involving different electron-withdrawing groups were firstly prepared by Wittig reactions between their corresponding phosphonium bromides and 2-pyridylaldehyde in the presence of t-BuOK with the moderate yields (46–67%), which were characterized by 1HNMR spectra (Fig. S1–S4†). Then, trimethylamine24 was used as a deprotonating agent to get their corresponding cyclometalated ruthenium complexes without purifying intermediates. And the yields were still satisfied in the range from 25 to 45%. All these complexes were characterized by NMR (Fig S5–S14†), MS (Fig. S5–S9† inset), IR (Fig. S15†) and elemental analysis spectra. It should be noted that the peaks attributed to Ru–C in 13CNMR spectra of these complexes were clearly observed in the range from 198 to 222 ppm, which are in accordance with those of [RuL(bpy)2]PF6 indicating successful cyloruthenations of these 2-vinylpyridine derivatives.11–17 Moreover, all the MS spectra of these complexes showed the parent peaks with the loss of one counter ion, which are in good agreements with the structures.
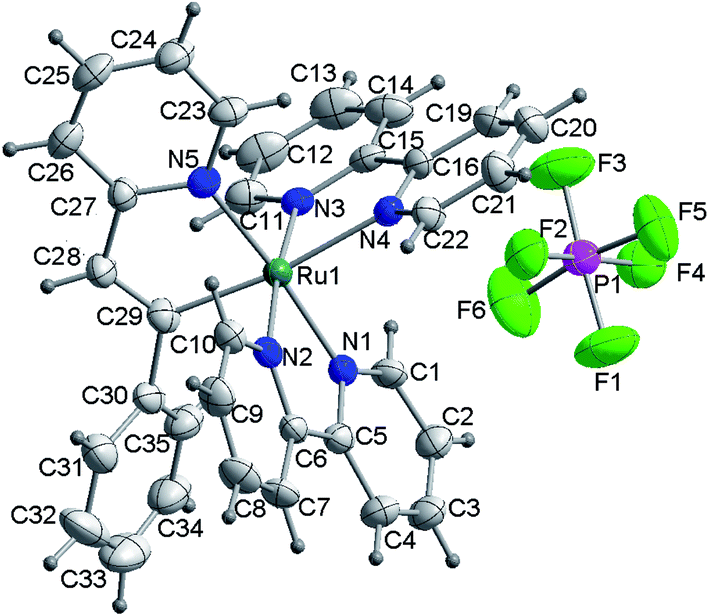
A single-crystal of 2 suitable for X-ray analysis was obtained by slow diffusion of petroleum ether into a solution of 2 in methanol. Then, complex 2 was further identified by X-ray crystallographic analysis. The structure of 2 is shown in Fig. 1. Further details of the crystal data, data collection, structure solution, and refinement parameters were summarized in Table S1.† Selected bond distances and angles were collected in Table 1. Herein, 2 adopts a triclinic, distorted octahedral coordination around the Ru(II) center with two chelating bipyridines and one (N,C)-bidentate ligand. The C(29)–Ru(1)–N(4) deviates slightly from linearity with the corresponding angle of 172.0(3)°. Along this line, the Ru(1)–C(29) bond was measured to be 2.037(7) Å, in the range of Ru–C distances observed for similar complexes.25 The Ru(1)–N(4) bond trans to the Ru(1)–C(29) bond is too elongated (2.127(5) Å) with respect to the Ru–N distances observed for bipyridine N(1) and N(2) atoms (2.047(5) and 2.056(6) Å, respectively). The result is consistent with the strong trans effect of the carbon atom compared with that of the nitrogen atom. However, it should be noted that the Ru(1)–N(5) bond is also somewhat elongated (2.067(5) Å).
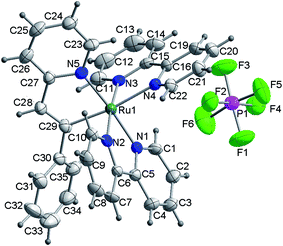 |
| | Fig. 1 ORTEP representation of the X-ray structure of complex 2. Herein, CH3OH is omitted for clarity. | |
Table 1 Selected bond lengths [Å] and angles [°] for 2
| Ru(1)–N(1) |
2.047(5) |
Ru(1)–N(2) |
2.056(6) |
Ru(1)–N(3) |
2.056(6) |
| Ru(1)–N(4) |
2.127(5) |
Ru(1)–N(5) |
2.067(5) |
Ru(1)–C(29) |
2.037(7) |
| C(28)–C(29) |
1.345(10) |
C(29)–C(30) |
1.490(10) |
C(29)–Ru(1)–N(4) |
172.0(3) |
| C(5)–N(1)–C(1) |
117.6(6) |
C(1)–N(1)–Ru(1) |
126.4(5) |
C(5)–N(1)–Ru(1) |
116.0(5) |
| C(6)–N(2)–Ru(1) |
115.4(5) |
C(10)–N(2)–Ru(1) |
126.6(5) |
C(11)–N(3)–Ru(1) |
125.7(5) |
| C(15)–N(3)–Ru(1) |
116.5(5) |
C(16)–N(4)–Ru(1) |
115.2(5) |
C(27)–N(5)–Ru(1) |
115.5(5) |
| C(28)–C(29)–Ru(1) |
114.4(6) |
C(30)–C(29)–Ru(1) |
124.6(5) |
C(28)–C(29)–C(30) |
120.4(7) |
Photophysical properties
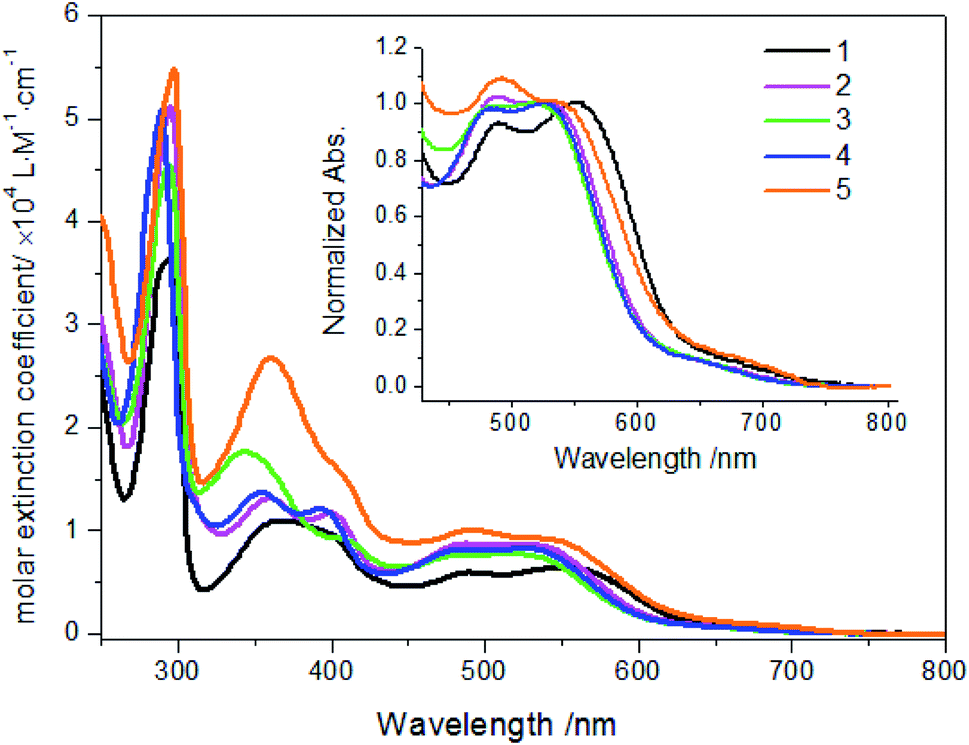
The spectroscopic properties of complexes 1–5 were investigated in details. As shown in Fig. 2 and Table 2, all these complexes display several absorption bands in UV-Vis spectral regions. According to the absorption spectra of these 2-alkenylpyridines in Fig. S16,† the bands between 300 and 450 nm can be assigned as LC transitions of 2-vinylpyridine derivatives mixed with LMCT transitions. The bands occurring between 450 and 800 nm are assigned as MLCT transitions. The first intense absorption band was observed at about 490 nm for these complexes, which is assigned as Ru(II) → cyclometalated ligands CT. The second intense absorption band was observed around 530 nm, which is assigned as Ru(II) → bpy CT transitions and noticeably red-shifted relative to Ru(bpy)32+ derivatives. Compared with absorptions of 2-alkenylpyridines, this red-shift can be attributed to the formation of Ru–C bond,11,25 which clearly indicated that ruthenations at alkenyl carbons involving 2-vinylpyridine derivatives were successful. In comparison with complex 1, the MLCT absorptions attributed to Ru(II) → bpy CT transitions are blue-shifted when methyl linked to vinyl was substituted by aryl such as 2-thienyl and phenyl derivatives. However, it should be noted that molar extinction coefficients of these complexes increased when aryl groups were linked to vinyl, as shown in Fig. 2 and Table 2. In addition, when electron donating or withdrawing groups were linked to phenyl in vinylphenyl parts, the absorption properties were also altered. As shown in Fig. 2 inset, Ru(II) → bpy CT transitions of complex 2 are centered at 527 nm while those of complex 4 with electron withdrawing group are centered at 524 nm. However, for complex 5 with N,N-dimethyl, the maximum absorption peak is centered at 538 nm. The results may be due to the different electron distributions on complexes induced by these groups. Nevertheless, there were no strong emissive spectra observed upon excited at ambient temperature. Then, the emission spectra at 77 K were investigated. As shown in Fig. S17,† the maximum emission peaks of complexes 1–5 are centered at 774, 710, 707, 708 and 726 nm, respectively. The results are in agreements with the regular changes of absorptions.
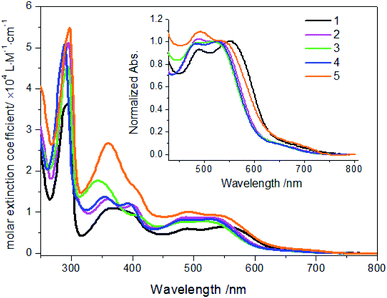 |
| | Fig. 2 Absorption spectra of complexes 1–5 (20 μM) in ethanol solution. Inset: normalized absorption spectra of complexes. | |
Table 2 Photophysical and electrochemical properties of complexes 1–5
| Complex |
λabsa nm, ε/104 M−1 cm−1 |
λemb/nm |
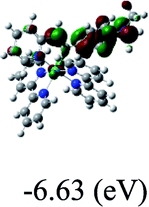
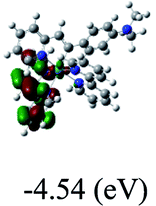
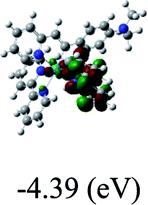
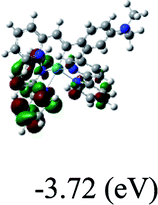
HOMO/LUMOc [eV] |
Band gapc/eV |
Eoxd/V for Ru2+/3+ (ΔEp/mV) |
| Absorptions measured in ethanol. Emissions measured in 2-methyltetrafuran glasses at 77 K. DFT/B3LPY calculated values. Recorded in MeCN with ferrocenium/ferrocene (Fc+/Fc) as an internal reference and converted to NHE by addition of 490 mV. |
| 1 |
245 (2.65), 287 (3.54), 295 (3.65), 368 (1.10), 401 (sh, 0.96), 490 (0.60), 552 (0.65) |
702, 774 |
−7.32/−4.69 |
2.63 |
0.64 (68) |
| 2 |
246 (3.16), 288 (4.64), 295 (5.12), 360 (1.32), 399 (1.18), 490 (0.88), 527 (0.87) |
654, 710 |
−7.30/−4.67 |
2.63 |
0.70 (63) |
| 3 |
244 (2.92), 286 (4.11), 294 (4.56), 344 (1.77), 408 (sh, 0.93), 495 (0.77), 519 (0.78) |
707 |
−7.22/−4.70 |
2.52 |
0.74 (99) |
| 4 |
241 (3.34), 282 (4.58), 292 (4.72), 356 (1.37), 395 (1.22), 484 (0.82), 524 (0.83) |
653, 708 |
−7.35/−4.72 |
2.63 |
0.72 (69) |
| 5 |
246 (0.75), 288 (4.96), 297 (5.47), 360 (2.70), 406 (sh, 1.55), 494 (1.01), 538 (0.93), 685 (sh, 0.085) |
726 |
−6.63/−4.54 |
2.09 |
0.67 (25) |
Electrochemical behaviors
The electrochemical behaviors of complexes 1–5 have been studied via cyclic voltammetry (CV) (Fig. S18†) and the summarized data are listed in Table 3. In the positive charge region, oxidation peaks attributed to Ru2+/3+ and anionic C,N-chelating ligands were observed. For complexes 1–5, the oxidation for anionic C,N-chelating ligands occurred at 0.99, 0.94, 0.89, 0.97 and 0.92 V, respectively. As expected, the peaks attributed to Ru2+/3+ redox couples appeared at 0.64, 0.70, 0.74, 0.72 and 0.67 V, respectively, which are substantially more negative than that of [Ru(bpy)3]2+.11,26 Moreover, Ru2+/3+ redox couples with ΔEp (25–99 mV) indicate that the processes are reversible (at least semi-reversible). It should be noticed that these cathodic shifts of Ru2+/3+ redox processes are in agreement with an increase in the donor strength of cyclometalated ligands induced by anion binding and a decrease in the overall charge of these complexes.16d,26 In these complexes, vinyl is an ideal electron delocalized channel. When it is used as a bridge between a pyridyl group and a substituted phenyl, the effect of electron donor strengths of the substituents is obvious. It can be seen in Table 2 that cyclometalated ligand in complex 5 renders the Ru(II) metal center easier to oxidize. The ligand involving a fluoride atom linked to phenyl shifts the oxidation potential to a bit more positive value. A similar trend for complex 1 and 2 was observed because methyl in complex 1 is a stronger electron donor than phenyl in complex 2. These results are consistent with those cyclometalated ruthenium complexes involving 2-arylpyridines.11,26b In particular, a new reversible oxidation peak at 0.56 V (versus NHE) (0.07 V versus Ag+/Ag) was observed for complex 5, which can be assigned as N,N-dimethylphenyl group.27 It indicated that there may be a possible intramolecular electron transfer from N,N-dimethylaminophenyl to Ru(III).27
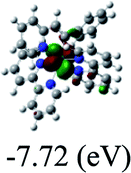
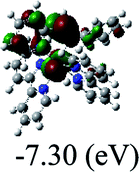
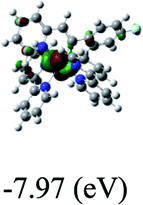
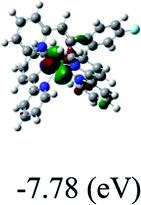
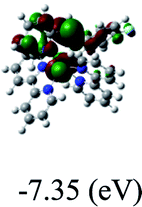
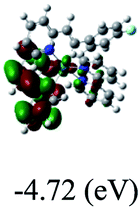
Table 3 Calculated molecular orbital distributions and energy levels of complexes 1–5
| |
HOMO−2 |
HOMO−1 |
HOMO |
LUMO |
LUMO+1 |
LUMO+2 |
| 1 |
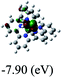 |
 |
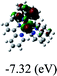 |
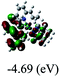 |
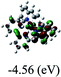 |
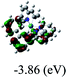 |
| 2 |
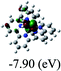 |
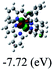 |
 |
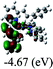 |
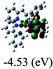 |
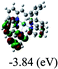 |
| 3 |
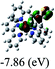 |
 |
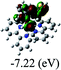 |
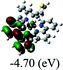 |
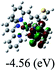 |
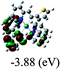 |
| 4 |
 |
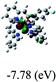 |
 |
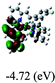 |
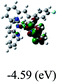 |
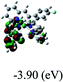 |
| 5 |
 |
 |
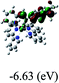 |
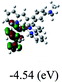 |
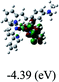 |
 |
Theoretical calculations
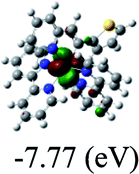
DFT calculations were carried out on complexes 1–5 to determine their electronic structures. Herein, calculated molecular orbital distributions and energy levels of complexes 1–5 are listed in Table 3. Obviously, the HOMOs of complexes 1–4 are primarily localized on ruthenium center and 2-vinylpyridine parts of the cyclometalated ligands. However, the HOMO of complex 5 is mainly delocalized on the double bond of vinyl and N,N-dimethylphenyl ring in the cyclometalated ligand with less distributions on Ru(II) and pyridine ring. It is clear that a set of π orbitals associated with 2-vinylpyridine parts of the cyclometalated ligands is located at slightly lower energy than vinylphenyl rings that comprise HOMO orbitals. In addition, all HOMO−1 and HOMO−2 orbitals primarily reside on Ru(II) center. And for complex 3, the thienyl ring also makes a partial contribution to comprise the HOMO−2. As anticipated, all LUMOs of these complexes are associated with one or two neutral bipyridyl ligands. Herein, the corresponding energy gaps between the HOMO and LUMO are calculated to be 2.63, 2.63, 2.52, 2.63 and 2.09 eV, respectively. For vinylphenyl based cyclometalated complexes 2, 4 and 5, the decrease of the HOMO–LUMO gap resulted in an obvious red shift in the absorption spectrum of complex 5.
TD-FDT calculations on optimized geometries in CH2Cl2 were then employed to model their corresponding absorption spectra. As suggested by TD-DFT calculations (see Table 4), the lowest singlet transitions responsible for the measured low-energy absorption bands of these complexes are assigned as S3 states, which are mainly composed of HOMO−1 → LUMO or HOMO−1 → LUMO+1 transitions. According to the orbital distributions, these absorption bands are mainly assigned as 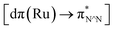 MLCT transitions and
MLCT transitions and 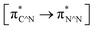 ligand-to-ligand charge-transfer (LLCT) transitions. For complexes 2, 4 and 5, TD-DFT shows that the absorption bands corresponding to S3 states are predicted at 523, 522 and 540 nm, respectively. It is consistent with the trend given by their experimental absorption spectra, in which their peaks occurred at 527, 524 and 538 nm, respectively.
ligand-to-ligand charge-transfer (LLCT) transitions. For complexes 2, 4 and 5, TD-DFT shows that the absorption bands corresponding to S3 states are predicted at 523, 522 and 540 nm, respectively. It is consistent with the trend given by their experimental absorption spectra, in which their peaks occurred at 527, 524 and 538 nm, respectively.
Table 4 Absorptions of complexes 1–5 in dichloromethane solutions from TDDFT calculations
| Complex |
States |
λ (nm) |
E (eV) |
Oscillator |
Main configurations |
CI coeff. |
Assignment |
| 1 |
S1 |
614 |
2.02 |
0.0061 |
HOMO → LUMO |
0.62 |
MLCT/LLCT |
| HOMO → LUMO+1 |
0.25 |
MLCT/LLCT |
| S2 |
605 |
2.0470 |
0.0002 |
HOMO → LUMO+1 |
0.62 |
MLCT/LLCT |
| S3 |
535 |
2.3135 |
0.0310 |
HOMO−1 → LUMO |
0.64 |
MLCT/LLCT |
| S4 |
506 |
2.4496 |
0.0022 |
HOMO−2 → LUMO |
0.50 |
MLCT/LLCT |
| S5 |
503 |
2.46 |
0.084 |
HOMO−1 → LUMO+1 |
0.54 |
MLCT/LLCT |
| S6 |
462 |
2.68 |
0.067 |
HOMO−2 → LUMO+1 |
0.46 |
MLCT/LLCT |
| 2 |
S1 |
600 |
2.06 |
0.0002 |
HOMO → LUMO+1 |
0.54 |
MLCT/LLCT |
| S2 |
596 |
2.0788 |
0.0045 |
HOMO → LUMO |
0.55 |
MLCT/LLCT |
| HOMO → LUMO+1 |
0.41 |
MLCT/LLCT |
| S3 |
523 |
2.3687 |
0.0150 |
HOMO−1 → LUMO |
0.65 |
MLCT/LLCT |
| S4 |
490 |
2.53 |
0.056 |
HOMO−3 → LUMO+1 |
0.50 |
MLCT/LLCT |
| S6 |
456 |
2.71 |
0.072 |
HOMO−2 → LUMO |
0.51 |
MLCT/LLCT |
| 3 |
S1 |
599 |
2.07 |
0.0004 |
HOMO → LUMO |
0.44 |
MLCT/LLCT |
| HOMO → LUMO+1 |
0.51 |
MLCT/LLCT |
| S2 |
594 |
2.0854 |
0.0036 |
HOMO → LUMO |
0.52 |
MLCT/LLCT |
| S3 |
519 |
2.3888 |
0.0094 |
HOMO−1 → LUMO |
0.66 |
MLCT/LLCT |
| S4 |
489 |
2.54 |
0.054 |
HOMO−1 → LUMO+1 |
0.46 |
MLCT/LLCT |
| HOMO−2 → LUMO+1 |
0.44 |
MLCT/LLCT |
| S6 |
456 |
2.72 |
0.059 |
HOMO−2 → LUMO |
0.45 |
MLCT/LLCT |
| HOMO → LUMO+2 |
0.39 |
MLCT/LLCT |
| 4 |
S1 |
598 |
2.07 |
0.0005 |
HOMO → LUMO |
0.49 |
MLCT/LLCT |
| S2 |
596 |
2.0790 |
0.0040 |
HOMO → LUMO |
0.49 |
MLCT/LLCT |
| S3 |
522 |
2.3729 |
0.0158 |
HOMO−1 → LUMO |
0.65 |
MLCT/LLCT |
| S4 |
490 |
2.5257 |
0.0285 |
HOMO−2 → LUMO+1 |
0.55 |
MLCT/LLCT |
| S5 |
489 |
2.53 |
0.071 |
HOMO−1 → LUMO+1 |
0.49 |
MLCT/LLCT |
| S6 |
456 |
2.72 |
0.072 |
HOMO−2 → LUMO |
0.49 |
MLCT/LLCT |
| 5 |
S1 |
637 |
1.94 |
0.0026 |
HOMO → LUMO |
0.59 |
MLCT/LLCT |
| S2 |
635 |
1.9511 |
0.0006 |
HOMO → LUMO+1 |
0.57 |
MLCT/LLCT |
| S3 |
540 |
2.2943 |
0.0050 |
HOMO−1 → LUMO+1 |
0.45 |
MLCT/LLCT |
| S4 |
535 |
2.31 |
0.011 |
HOMO−2 → LUMO |
0.58 |
MLCT/LLCT |
| S6 |
495 |
2.50 |
0.1002 |
HOMO−2 → LUMO+1 |
0.55 |
MLCT/LLCT |
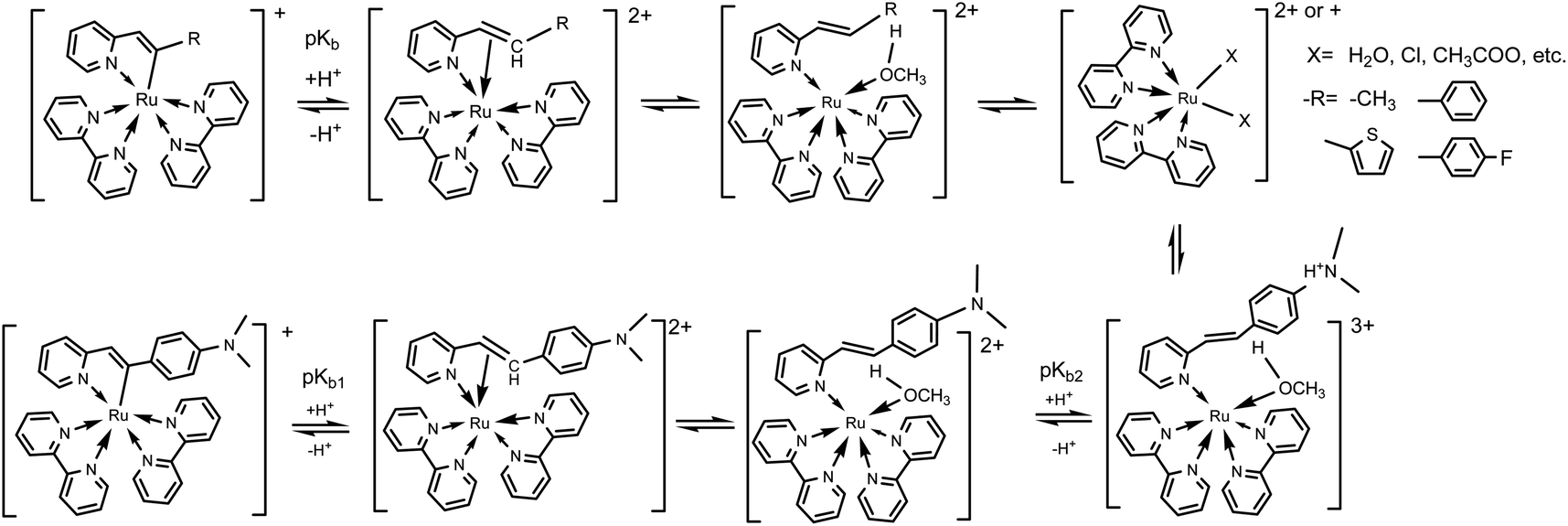
pH sensitivity
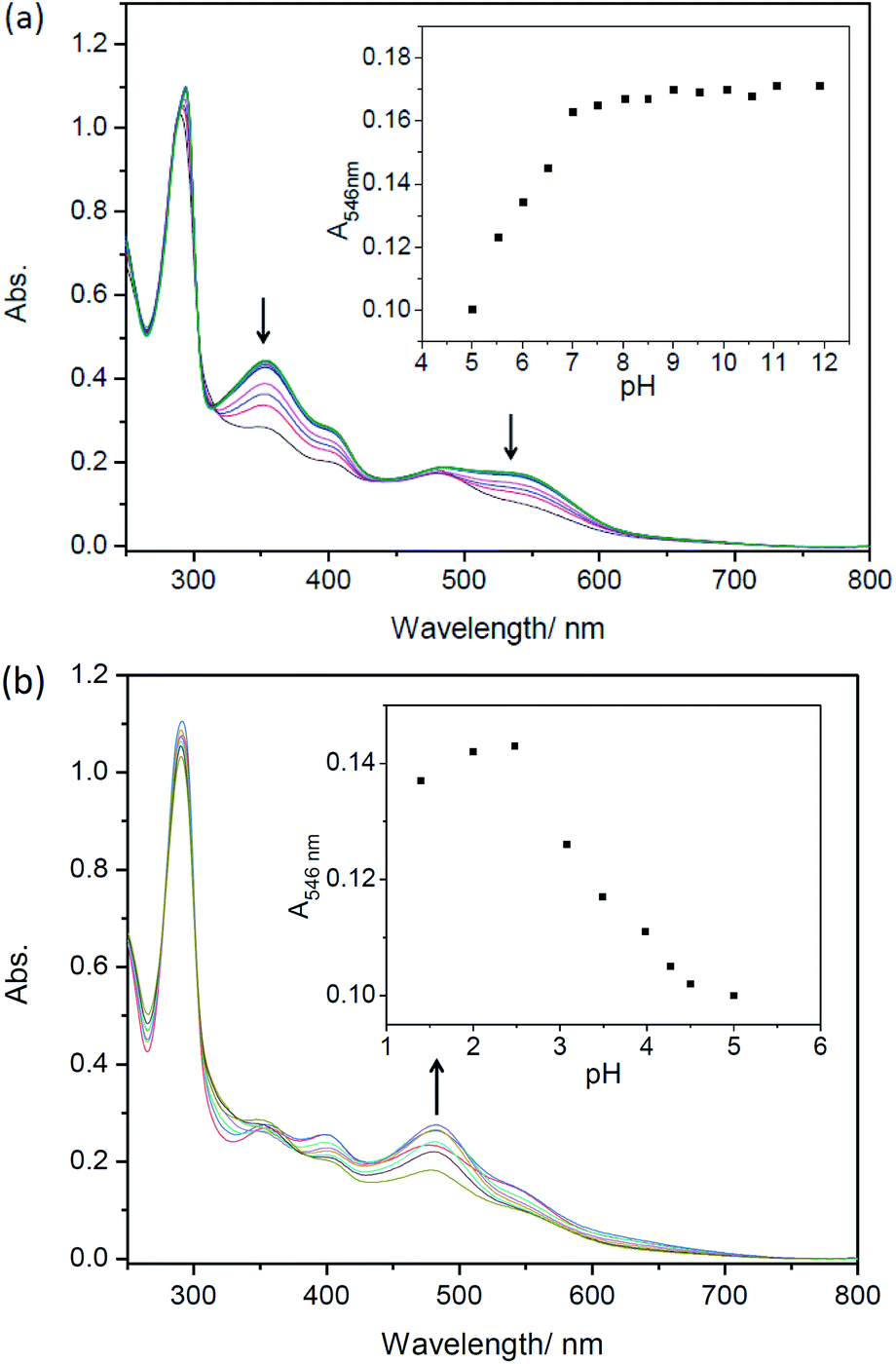
Considering the deprotonation of C–H in C,N-chelating ligands for synthesizing cyclometalated ruthenium complexes,10a,28,29a the presence of acids may lead to the intrusion of protons to carbanion, which could result in cleavages of Ru–C bonds and absorption spectral changes.25,29 However, there are only a few reports on the protonation of cyclometalated ruthenium complexes.25,29,30 Herein, UV-Vis absorption spectra of complexes 1–5 as a function of pH were then investigated. As shown in Fig. S19† and 3, in both alkaline and neutral environments, the intensities of absorption bands around 520 nm are almost unchanged for complexes 1–5, which indicates the presence of Ru(II) → bpy CT originated from cyclometaled ruthenium complexes. Upon decreasing pH, the intensities of absorption bands around 520 nm are gradually decreasing and new peaks are centered at around 485 nm for these five complexes, indicating the cleavages of Ru–C bonds. Obviously, the protonation process involved an attack of H+ to the anionic carbon atom, which weaken the Ru–C bond and resulted in great changes in MLCT absorptions. It is interesting that the newly formed band at 485 nm for complex 5 increased slightly in the intensities upon decreasing pH from 5.00 to 1.40 (Fig. 3b), which could be ascribed to the concurrent combination of one proton with N,N-dimethylamine.31
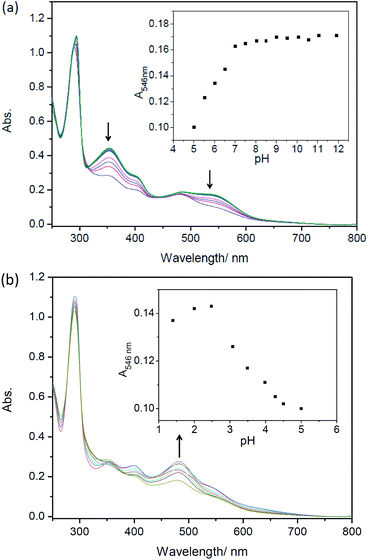 |
| | Fig. 3 Absorption spectral changes of complexes 5 (20 μM) after being incubated for 4 h in ethanol/Briton–Robinson buffer solutions (v/v = 1:2). (a) pH = 11.92–5.52; (b) pH = 5.00–1.40, respectively. Arrows show spectral changes upon decreasing pH. Inset: changes of absorption intensity at different pHs. | |
To better understanding the protonation process of these five cyclometalated complexes, absorption changes of Ru(bpy)2(ppy)+ (Hppy = 2-phenylpyrdine), Ru(bpy)2(dfppy)+ (Hdfppy = 2-(2,4-difluorophenyl)pyrdine) and Ru(bpy)2(thpy)+ (Hthpy = 2-(2-thienyl)pyridine) shared from our previous work29b are shown in Fig. S20.† It should be noted that the absorptions of complexes Ru(bpy)2(ppy)+, Ru(bpy)2(dfppy)+ and Ru(bpy)2(thpy)+ in acids are all centered at 450 nm and their corresponding products have been identified as [Ru(bpy)2(H2O)2]+ or [Ru(bpy)2X]+.25,29 However, absorptions of complexes 1–5 in acids are around at 485 nm, which demonstrated that there may be different species in given acidic solutions rather than [Ru(bpy)2(H2O)2]+ or [Ru(bpy)2X]+. Then, MS spectra of these complexes were tracked after being incubated in methanol/HAc solution for 4 h. As shown in Fig. S21,† there is an evident peak occurring at m/z 563.18 in the MS spectrum of complex 1, which could be attributed to the formation of [MH+–CH3OH–H+]+ (calc. m/z 563.63). For complexes 2–5 (Fig. S22–S25†), the peaks attributed to [MH+–CH3OH–H+]+ occurred at m/z 625.36 (calc. 625.71), 631.34 (calc. 631.64), 643.37 (calc. 643.69), 668.44 (calc. 668.77), respectively. These peaks demonstrated that the pronated species could have been formed as 2-alkenylpyridines attached to Ru(II) centers through N atoms when protons attacked carbonions and resulted in the cleavage of Ru–C bonds. Therefore, the different absorptions of complexes 1–5 in acidic solutions could be clearly explained by the presence of their protonated species. However, when these complexes were incubated in HAc/CH3OH for a long enough time, complexes 1–4 were decomposed by losing C,N-chelating ligands and then afforded [Ru(bpy)2CHCA–H+]+ (Fig. S26–S29†). However, for complex 5, the species [MH+–CH3OH–H+]+ was still observed (Fig. S30†). In addition, MS spectra of these complexes incubated in HCl/CH3OH were also investigated, [Ru(bpy)2Cl]+ and [Ru(bpy)2CHCA–H+]+ were found as the primary species for all complexes (Fig. S31–S35†). It should be noted that free 2-alkenylpyridines have been isolated and then characterized by 1HNMR spectra for all these complexes in these acidic solutions including HAc/CH3OH and HCl/CH3OH. It can be seen that these five cyclometalated complexes are thermally unstable in strong acidic conditions. These complexes finally decomposed and gave off free C,N-ligands. Furthermore, the C,N-ligand of complex 5 was chosen to mix with Ru(bpy)2Cl2 in CH3OH. As shown in Fig. S36,† some fragments that are the same as complex 5 in acid were observed after being treated by NaOH or Et3N. According to these results of MS and UV-Vis spectra, the possible acid–base equilibria of these complexes could be depicted as Scheme 2.
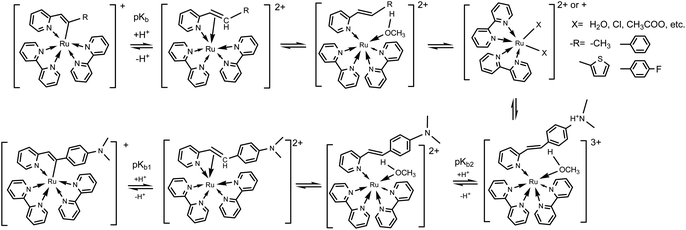 |
| | Scheme 2 The possible acid–base equilibria of complexes 1–5. | |
Then, the ground-state protonation constant values (pKb) of these five complexes were obtained by equation pH = pK − lg[(A − A0)/(Af − A)] via their specific titration curves, respectively.31 Herein, A0 and Af is the absorbance at the starting and ending pHs. As shown in Table 5, for complexes 1–4, there is only one protonation process occurring at the carbanions in the alkenyls that are bonded to Ru(II) centers. However, for complex 5, there are two protonation processes: the first is same to complexes 1–4, the second is protonation of N atom on N,N-dimethylaminophenyl.31 It can also be found that pK values depend on the electron properties of C,N-chelating ligands. For complexes involving vinylphenyl groups, a lower pKb value was obtained for complex 4 which involved a fluorine atom as the electron-withdrawing substituent. However, with an electron-donating group, complex 5 afforded a higher pKb value in its first protonation step. In comparison with phenyl attached to vinyl, methyl linked to vinyl is much stronger electron donator which resulted in a much higher pKb value than others. The pKb values of Ru(bpy)2(ppy)+, Ru(bpy)2(dfppy)+ and Ru(bpy)2(thpy)+ were also calculated and listed in Table 5, respectively. Obviously, an increase in the donor strength of the cyclometalated ligands could lead to easier decompositions of cyclometalated ruthenium complexes in acids.
Table 5 pKb values of complexes 1–5 and three classical cyclometaleted ruthenium complexes
| Complex |
pKb |
Complex |
pKb |
| 1 |
3.14 ± 0.02 |
|
|
| 2 |
2.28 ± 0.03 |
Ru(bpy)2(ppy)+ |
4.02 ± 0.03 |
| 3 |
2.53 ± 0.02 |
Ru(bpy)2(thpy)+ |
4.99 ± 0.03 |
| 4 |
2.16 ± 0.02 |
Ru(bpy)2(dfppy)+ |
2.44 ± 0.02 |
| 5 |
2.86 ± 0.02 |
|
|
| 5.53 ± 0.06 |
Conclusion
In summary, five cyclometalated ruthenium complexes with 2-vinylpyridine derivatives as C,N-ligands were successfully synthesized and characterized. Their structures were identified by NMR, MS, FT-IR and elemental analysis spectra. Then, their photophysical properties were primarily studied by CV, UV-Vis and FL spectra combined with theoretical calculations. Furthermore, the influence of acids on these new cyclometalated ruthenium complexes was investigated by UV-Vis spectra. The regular changes in absorptions and different absorption bands from cyclometalated 2-arylpyridine ruthenium complexes could be attributed to the cleavage of Ru–C bonds without release of C,N-chelating ligands, which were further identified and confirmed by the MS spectra. Then, possible acid–base equilibria for these complexes were proposed.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for National Science Foundation of China (21971258), and thanks to the help of Analytical & Measuring Center, School of Pharmaceutical Sciences, South-Central University for Nationalities.
References
-
(a) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2572 CrossRef CAS;
(b) M. Ghedini, I. Aiello, A. Crispini, A. Golemme, M. La Deda and D. Pucci, Coord. Chem. Rev., 2006, 250, 1373–1390 CrossRef CAS.
- A. C. Cope and R. W. Siekman, J. Am. Chem. Soc., 1965, 87, 3272–3273 CrossRef CAS.
- M. I. Bruce, Angew. Chem., Int. Ed., 1977, 16, 73–86 CrossRef.
- R. Romeo, M. R. Plutino, L. M. Scolaro, S. Stoccoro and G. Minghetti, Inorg. Chem., 2000, 39, 4749–4755 CrossRef CAS.
-
(a) J. B. Waern, C. Desmarets, L.-M. Chamoreau, H. Amouri, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Inorg. Chem., 2008, 47, 3340–3348 CrossRef CAS;
(b) P. A. Scattergood, A. Sinopoli and P. I. P. Elliott, Coord. Chem. Rev., 2017, 350, 136–154 CrossRef CAS;
(c) J. Yu, M. Li, C. Xu, F. Meng, J. Cao, H. Tan and W. Zhu, Dalton Trans., 2020, 49, 8785–8790 RSC.
-
(a) G. Minghetti, A. Zucca, S. Stoccoro and M. A. Cinellu, J. Organomet. Chem., 1994, 481, 195–204 CrossRef CAS;
(b) M. R. Churchil, H. J. Wasserman and G. J. Young, Inorg. Chem., 1980, 19, 762–770 CrossRef;
(c) K. Hiraki, Y. Fuchita and K. Takechi, Inorg. Chem., 1981, 20, 4316–4320 CrossRef CAS.
-
(a) R. J. Foot and B. T. Heaton, J. Chem. Soc., Dalton Trans., 1979, 295–298 RSC;
(b) R. J. Foot and B. T. Heaton, J. Chem. Soc., Chem. Commun., 1973, 838 RSC.
- X. Yang, X. Xu, J. Zhao, J.-S. Dang, Z. Huang, X. Yan, G. Zhou and D. Wang, Inorg. Chem., 2014, 53, 12986–13000 CrossRef CAS.
-
(a) B. M. Jennifer, S. Paulose, D. K. Rayabarapu, J.-P. Duan and C.-H. Cheng, Adv. Mater., 2004, 16, 2003–2007 CrossRef;
(b) D. Wang, Y. Wu, H. Dong, Z. Qin, D. Zhao, Y. Yu, G. Zhou, B. Jiao, Z. Wu, M. Gao and G. Wang, Org. Electron., 2013, 14, 3297–3305 CrossRef CAS;
(c) D. M. Kang, J.-W. Kang, J. W. Park, S. O. Jung, S.-H. Lee, H.-D. Park, Y.-H. Kim, S. C. Shin, J.-J. Kim and S.-K. Kwon, Adv. Mater., 2008, 20, 2003–2007 CrossRef CAS;
(d) S.-y. Takizawa, Y. Sasaki, M. Akhtaruzzaman, H. Echizen, J.-I. Nishida, T. Iwata, S. Tokito and Y. Yamashita, J. Mater. Chem., 2007, 17, 841–849 RSC.
-
(a) J.-P. Djukic, J.-B. Sortais, L. Barloy and M. Pfeffer, Eur. J. Inorg. Chem., 2009, 7, 817–853 CrossRef;
(b) L. Labat, J.-F. Lamère, I. Sasaki, P. G. Lacroix, L. Vendier, I. Asselberghs, J. Pérez-Moreno and K. Clays, Eur. J. Inorg. Chem., 2006, 15, 3105–3113 CrossRef.
-
(a) F. Barigelletti, B. Ventura, J.-P. Collin, R. Kayhanian, P. Gavina and J.-P. Sauvage, Eur. J. Inorg. Chem., 2000, 1, 113–119 CrossRef;
(b) M. L. Muro-Small, J. E. Yarnell, C. E. McCusker and F. N. Castellano, Eur. J. Inorg. Chem., 2012, 25, 4004–4011 CrossRef.
- S. Ott, M. Borgström, L. Hammarström and O. Johansson, Dalton Trans., 2006, 11, 1434–1443 RSC.
-
(a) M. Borgström, S. Ott, R. Lomoth, J. Bergquist, L. Hammarström and O. Johansson, Inorg. Chem., 2006, 45, 4820–4829 CrossRef;
(b) F. Barigelletti, L. Flamigni, M. Guardigli, A. Juris, M. Beley, S. Chodorowski-Kimmes, J.-P. Collin and J.-P. Sauvage, Inorg. Chem., 1996, 35, 136–142 CrossRef CAS.
-
(a) I. S. Alpeeva, V. S. Soukharev, L. Aexandrova, N. V. Shilova, N. V. Bovin, E. Csöregi, A. D. Ryabov and I. Y. Sakharov, JBIC, J. Biol. Inorg. Chem., 2003, 8, 683–688 CrossRef CAS;
(b) A. D. Ryabov, V. S. Sukharev, L. Alexandrova, R. L. Lagadec and M. Pfeffer, Inorg. Chem., 2001, 40, 6529–6532 CrossRef CAS.
-
(a) T. Bessho, E. Yoneda, J.-H. Yum, M. Guglielmi, I. Tavernelli, H. Imai, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2009, 131, 5930–5934 CrossRef CAS;
(b) P. G. Bomben, K. C. D. Robson, B. D. Koivisto and C. P. Berlinguette, Coord. Chem. Rev., 2012, 256, 1438–1450 CrossRef CAS;
(c) T.-D. Nguyen, Y.-P. Lan and C.-G. Wu, Inorg. Chem., 2018, 57, 1527–1534 CrossRef CAS.
-
(a) X. Li, Y. Wu, Y. Liu, X. Zou, L. Yao, F. Li and W. Feng, Nanoscale, 2014, 6, 1020–1028 RSC;
(b) X. Li, K. Du, C. Xie, Y. Wu, B. Zhang and D. Tang, Dalton Trans., 2020, 49, 2024–2032 RSC;
(c) X. Cheng, J. Li, X. Li, D. Zhang, H. Zhang, A. Zhang, H. Huang and J. Lian, J. Mater. Chem., 2012, 22, 24102–24108 RSC;
(d) C. R. Wade and F. P. Gabbaï, Inorg. Chem., 2010, 49, 714–720 CrossRef CAS;
(e) H.-S. Lo, K.-W. Lo, C.-F. Yeung and C.-Y. Wong, Anal. Chim. Acta, 2017, 990, 135–140 CrossRef CAS.
- Z. Lv, H. Wei, Q. Li, X. Su, S. Liu, K. Y. Zhang, W. Lv, Q. Zhao, X. Li and W. Huang, Chem. Sci., 2018, 9, 502–512 RSC.
-
(a) S. Aghazada, I. Zimmermann, V. Scutelnic and M. K. Nazeeruddin, Organometallics, 2017, 36, 2397–2403 CrossRef CAS;
(b) H.-J. Park, K. H. Kim, S. Y. Choi, H.-M. Kim, W. I. Lee, Y. K. Kang and Y. K. Chung, Inorg. Chem., 2010, 49, 7340–7352 CrossRef CAS;
(c) X. Xie and H. V. Huynh, Org. Chem. Front., 2015, 2, 1598–1603 RSC;
(d) J. Soellner, I. Císařová and T. Strassner, Organometallics, 2018, 37, 4619–4629 CrossRef CAS.
- V. S. Soukharev, A. D. Ryabov and E. Csöregi, J. Organomet. Chem., 2003, 668, 75–81 CrossRef CAS.
- S. Fernandez, M. Pfeffer, V. Ritleng and C. Sirlin, Organometallics, 1999, 18, 2390–2394 CrossRef CAS.
- X. Zhou, X. Li, Y. Liu, R. Li, K. Jiang and J. Xia, Org. Electron., 2015, 25, 245–253 CrossRef CAS.
-
(a) L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–71 CrossRef CAS;
(b) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS;
(c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- X. Su, R. Hu, X. Li, J. Zhu, F. Luo, X. Niu, M. Li and Q. Zhao, Inorg. Chem., 2016, 55, 745–754 CrossRef CAS.
- X. Su, X. Li, T. Ding, G. Zheng and Z. Liu, J. Organomet. Chem., 2015, 781, 59–64 CrossRef CAS.
- P. Reveco, R. H. Schmehl, W. R. Cherry, F. R. Fronczk and J. Selbin, Inorg. Chem., 1985, 24, 4078–4082 CrossRef CAS.
-
(a) P. Reveco, W. R. Cherry, J. Medley, A. Garber, R. J. Gale and J. Selbin, Inorg. Chem., 1986, 25, 1842–1845 CrossRef CAS;
(b) P. G. Bomben, B. D. Koivisto and C. P. Berlinguette, Inorg. Chem., 2010, 49, 4960–4971 CrossRef CAS.
-
(a) R. Argazzi and C. A. Bignozzi, J. Am. Chem. Soc., 1995, 117, 11815–11816 CrossRef CAS;
(b) X. Li, J. Gui, H. Yang, W. Wu, F. Li, H. Tian and C. Huang, Inorg. Chim. Acta, 2008, 361, 2835–2840 CrossRef CAS;
(c) N. Hirata, J.-J. Lagref, E. J. Palomares, J. R. Durrant, M. K. Nazeeruddin, M. Gräztel and D. D. Censo, Chem.–Eur. J., 2004, 10, 595–602 CrossRef CAS.
- B. Li, T. Roisnel, C. Darcel and P. H. Dixneuf, Dalton Trans., 2012, 41, 10934–10937 RSC.
-
(a) E. C. Constable, Polyhedron, 1984, 3, 1037–1057 CrossRef CAS;
(b) X. Su, R. Zeng, X. Li, W. Dang, K. Yao and D. Tang, Dalton Trans., 2016, 45, 7450–7459 RSC.
-
(a) E. C. Constable, S. J. Dunne, D. G. F. Rees and C. X. Schmitt, Chem. Commun., 1996, 10, 1169–1170 RSC;
(b) C. Moorlag, O. Clot, M. O. Wolf and B. O. Patrick, Chem. Commun., 2002, 3028–3029 RSC;
(c) C. Moorlag, M. O. Wolf, C. Bohne and B. O. Patrick, J. Am. Chem. Soc., 2005, 127, 6382–6393 CrossRef CAS.
-
(a) F. Liu, K. Wang, G. Bai, Y. Zhang and L. Gao, Inorg. Chem., 2004, 43, 1799–1806 CrossRef CAS;
(b) T. T. Meng, H. Wang, Z. B. Zheng and K. Z. Wang, Inorg. Chem., 2017, 56, 4775–4779 CrossRef CAS;
(c) X. L. Zhao, Z. S. Li, Z. B. Zheng, A. G. Zhang and K. Z. Wang, Dalton Trans., 2013, 42, 5764–5777 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectral data associated with this article can be found. CCDC 2032477 containing the supplementary crystallographic data for complex 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09833g |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Qiang Zhao
*a,
Qiang Zhao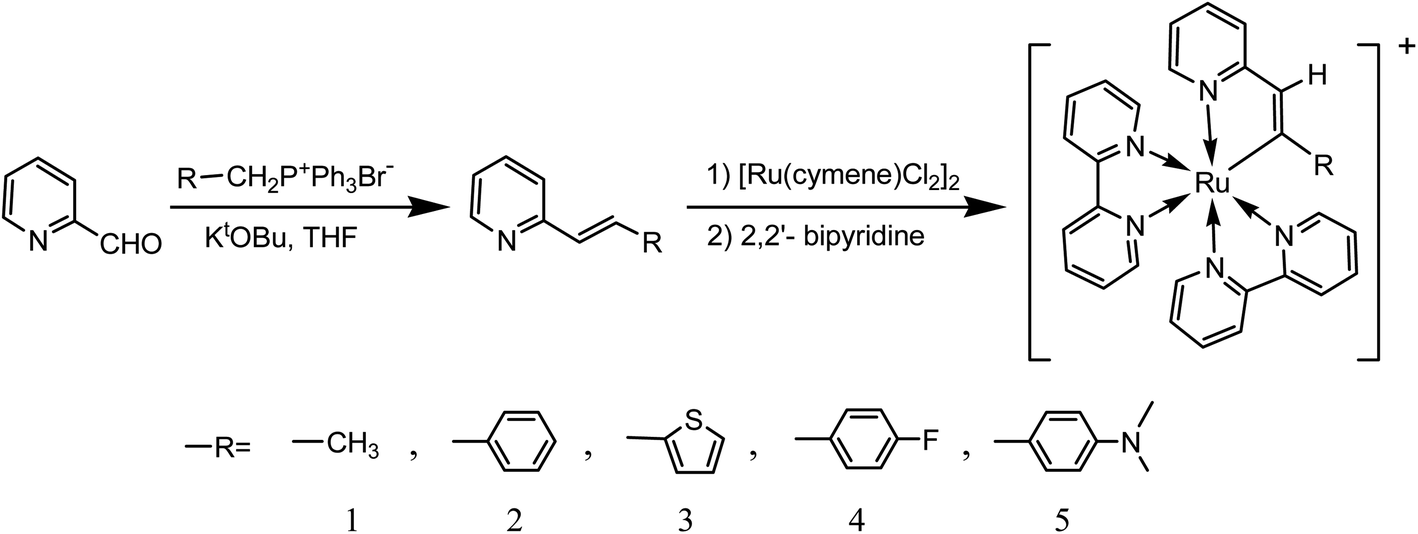
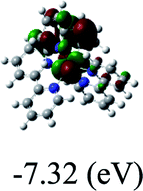
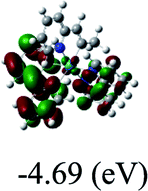
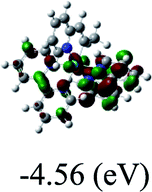
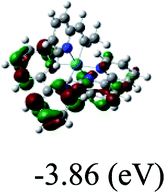
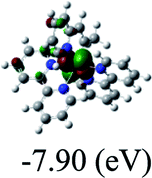
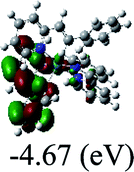
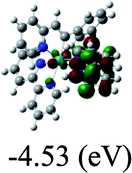
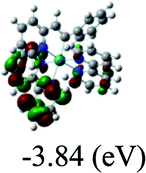
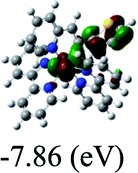
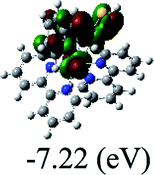
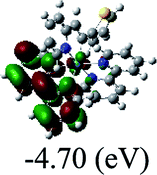
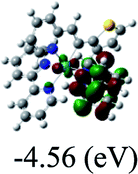
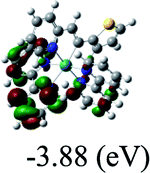
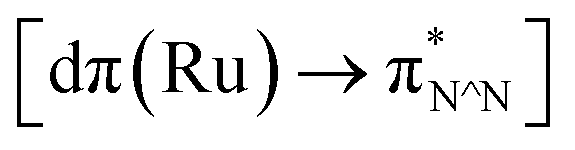 MLCT transitions and
MLCT transitions and 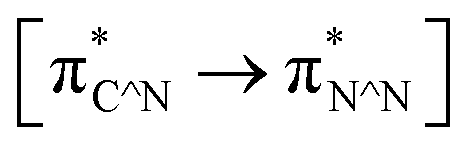 ligand-to-ligand charge-transfer (LLCT) transitions. For complexes 2, 4 and 5, TD-DFT shows that the absorption bands corresponding to S3 states are predicted at 523, 522 and 540 nm, respectively. It is consistent with the trend given by their experimental absorption spectra, in which their peaks occurred at 527, 524 and 538 nm, respectively.
ligand-to-ligand charge-transfer (LLCT) transitions. For complexes 2, 4 and 5, TD-DFT shows that the absorption bands corresponding to S3 states are predicted at 523, 522 and 540 nm, respectively. It is consistent with the trend given by their experimental absorption spectra, in which their peaks occurred at 527, 524 and 538 nm, respectively.