
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and applications of sodium sulfinates (RSO2Na): a powerful building block for the synthesis of organosulfur compounds
Raju Jannapu Reddy
 * and
Arram Haritha Kumari
* and
Arram Haritha Kumari
Department of Chemistry, University College of Science, Osmania University, Hyderabad 500 007, India. E-mail: rajuchem77@osmania.ac.in; rajuchem08@yahoo.co.in
First published on 1st March 2021
Abstract
This review highlights the preparation of sodium sulfinates (RSO2Na) and their multifaceted synthetic applications. Substantial progress has been made over the last decade in the utilization of sodium sulfinates emerging as sulfonylating, sulfenylating or sulfinylating reagents, depending on reaction conditions. Sodium sulfinates act as versatile building blocks for preparing many valuable organosulfur compounds through S–S, N–S, and C–S bond-forming reactions. Remarkable advancement has been made in synthesizing thiosulfonates, sulfonamides, sulfides, and sulfones, including vinyl sulfones, allyl sulfones, and β-keto sulfones. The significant achievement of developing sulfonyl radical-triggered ring-closing sulfonylation and multicomponent reactions is also thoroughly discussed. Of note, the most promising site-selective C–H sulfonylation, photoredox catalytic transformations and electrochemical synthesis of sodium sulfinates are also demonstrated. Holistically, this review provides a unique and comprehensive overview of sodium sulfinates, which summarizes 355 core references up to March 2020. The chemistry of sodium sulfinate salts is divided into several sections based on the classes of sulfur-containing compounds with some critical mechanistic insights that are also disclosed.
 Raju Jannapu Reddy | Dr Raju Jannapu Reddy obtained his PhD (with Prof. Deevi Basavaaih) from the School of Chemistry, University of Hyderabad, India. He moved to National Taiwan Normal University, Taiwan as a postdoctoral fellow (with Prof. Kwunmin Chen) and then worked as a JSPS postdoctoral fellow (with Prof. Jun'ichi Uenishi) at Kyoto Pharmaceutical University, Japan. As a Marie Curie Research Fellow (IIF), he has carried out his research at the University of Birmingham (with Dr Paul W. Davies), UK. Presently, he is working as a UGC – Assistant Professor at the Department of Chemistry, University College of Science, Osmania University, Hyderabad, India. His research focuses on the areas of organosulfur chemistry including sulfonyl radical-triggered photoredox catalysis and electrochemical synthesis on developing divergent and sustainable methodologies. |
 Arram Haritha Kumari | Mrs Arram Haritha Kumari was born in 1989 in Warangal, Telangana, India. She graduated with a BSc degree from Kakatiya University, Warangal and obtained her MSc (Chemistry) in 2011 from the National Institute of Technology Warangal. She qualified for the CSIR-UGC-NET in 2011, and she worked as a lecturer for two years. Since 2018, she has been pursuing her PhD under the supervision of Dr Raju Jannapu Reddy in the Department of Chemistry, Osmania University, Hyderabad, India. She is the recipient of the Women Scientist Award (WOSA) under DST schemes. Her research interests include the photoredoxcatalysis for intramolecular vicinal difunctionalization of alkynes and alkenes using sodium sulfinates and thiosulfonates. |
1. Introduction
Among various sulfinate salts (RSO2Met; Met = Li, Na, Zn, Fe, etc.),1 sodium sulfinates (RSO2Na) have become most popular and have received significant attention due to their versatile reactivity. Compared to conventional sulfonylating agents, such as sulfonyl chlorides, in general, sulfinate salts are odorless, moisture-insensitive, easy-to-handle, and bench-stable colorless solids. Only a few sulfinate sodium salts are commercially available but they can be readily prepared from inexpensive sulfonyl chlorides. Typically, this classical approach limits the functional group tolerance, which is a significant drawback. Consequently, organic chemists are continually searching for alternative convergent and straightforward synthetic routes. Tremendous efforts have been revolutionized in preparing sodium sulfinates in recent years2,3 (see Section 2). Despite advancements, efficient and sustainable methodologies for the synthesis of sodium sulfinates are still highly desirable.Due to a great interest in organosulfur chemistry,4,5 numerous reviews have been published recently.6,7 In 2014, Messaoudi, Hamze and co-workers8 reported a brief survey of sulfinate derivatives and subsequently, desulfinative cross-couplings using arylsulfinates were also documented.9,10 During the preparation of this review article, Hamze and co-workers11 provided an update on sulfinate derivatives, which covered sulfinic acids, sulfinate salts, sulfonyl chlorides, sulfonyl hydrazides, etc. In contrast, the present review exclusively focuses on further exploration of sodium sulfinates to cover all aspects, from the established to emerging aspects. Despite many synthetic applications and a large number of articles published on sodium sulfinates, to the best of our knowledge, there has been no general and exclusive review article. As such, a comprehensive and systematic review is undoubtedly required for synthetic chemists and biologists. Herein, we present a unique and exhaustive overview of sodium sulfinates to cover all references, from the originating reports up to early 2020. We have summarized 355 core references on sodium sulfinates in several sections based on the classes of sulfur-containing organic compounds.
Sodium sulfinates play indispensable roles as sulfonylating (RSO2–), sulfenylating (RS–) and sulfinylating (RSO–) agents. Over the last decade, sodium sulfinates have demonstrated incredible flexible reactivity, such as being nucleophilic, electrophilic, and radical reagents by providing suitable reaction conditions. As a result, sodium sulfinates have emerged as powerful building blocks for synthesizing many valuable sulfur-containing organic compounds (Fig. 1). Consequently, sodium sulfinates are widely useful coupling partners for constructing mainly three types of bonds that are largely studied in organic synthesis: (i) the S–S bonds for the synthesis of thiosulfonates (R–SO2S–R1);12 (ii) the N–S bonds to generate sulfonamides (R–SO2N–R1R2);13 (iii) the S–C bond to form sulfides (R–S–R1)14,15 and sulfones (R–SO2–R1).16 Among the C–S bonds forming reactions of sodium sulfinates, vinyl sulfones,17,18 allylic sulfones and β-keto sulfones are formed.19,20 These types of organosulfur compounds are broadly used in different pharmaceutical applications, which may lead to forthcoming medicinal therapies.21
 | ||
| Fig. 1 Comprehensive overview of sodium sulfinates for the synthesis of organosulfur compounds. | ||
Several attractive features of sodium sulfinates have been recently envisioned: the S-centered sulfonyl radical-triggered ring-closing sulfonylation, multicomponent reactions of sodium sulfinates, and remote site-selective C–H sulfonylation.22,23 The most promising photocatalytic carbon–sulfur bond formation24 and electrochemical transformation25 of sulfinate salts have also been effectively summarized in this review article.
Fig. 2 represents the number of articles published each year (total 244) on sodium sulfinates (RSO2Na), from 2010 to 2020, through the SciFinder® search profile. Based on these citations, the data reveal that the synthesis and utility of sodium sulfinates have become exponentially growing fields over the last decade. The preparation of sodium sulfinates, including sodium triflinate (F3CSO2Na), and their extensive applications have attracted great interest from synthetic chemists on a large scale.
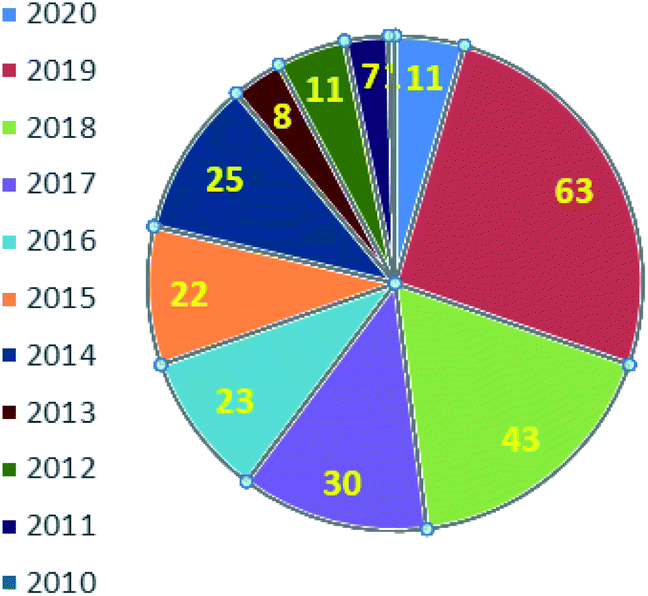 | ||
| Fig. 2 Representative articles on sodium sulfinates (total 244) published by year (2010 to 2020). | ||
2. Synthesis of sodium sulfinates
Only a few sodium sulfinates are commercially available, but they can be easily prepared from the inexpensive sulfonyl chlorides. Many other methods to synthesize structurally different sodium sulfinate salts have recently been achieved,2 some of which could be scalable on industrial and commercial levels.2.1. Sodium (hetero)aryl/alkylsulfinates (RSO2Na)
Among the variety of conventional synthetic methods, one of the methods involving the Michael addition of acrylonitrile with thiols resulted in sulfides, which were oxidized with hydrogen peroxide in glacial acetic acid to form the corresponding sulfones. The subsequent treatment of sulfone with an equivalent amount of a thiol sodium salt afforded the desired sodium sulfinates in good yields (Scheme 1).26 A substantially improved synthesis of aromatic and aliphatic sodium sulfinates was also achieved using ethyl propiolate in a similar manner.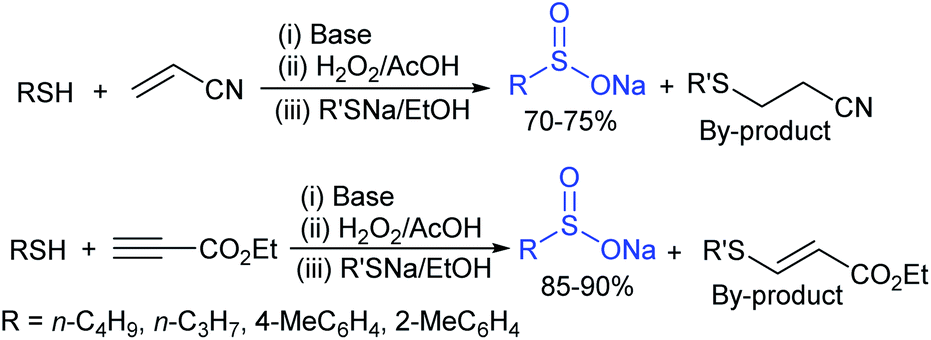 | ||
| Scheme 1 Conventional synthesis of sodium sulfinates. | ||
Alternatively, there is a straightforward method for the reduction of the corresponding p-toluenesulfonyl chloride with zinc/sodium carbonate in the water to afford sodium p-toluenesulfinate hydrate (Scheme 2).27 Until now, the most common method for the preparation was the reduction of the corresponding sulfonyl chloride by sodium sulfite (Na2SO3) in the presence of sodium bicarbonate in water at 70–80 °C (Scheme 2).28 Recrystallization from ethanol produced sodium benzenesulfinate and sodium p-toluenesulfinate in pure form in high yields.
 | ||
| Scheme 2 Conventional synthesis of sodium sulfinates. | ||
Field and co-workers29,30 prepared a wide range of di- and trisulfide-derived sodium sulfinates whose biological activities were successfully examined. In particular, the disulfide-derived sodium 4-(2-acetamidoethyldithio)butanesulfinate and the trisulfide disulfinate are promising antiradiation drugs at low doses with low toxicity. From a synthetic point of view, few interesting disulfides-derived sulfinates (1 and 2) were obtained from the corresponding cyclic thiosulfonates (Scheme 3).30
 | ||
| Scheme 3 Sulfinate salts as antiradiation drugs. | ||
In 1984, Uneo et al. demonstrated the synthesis of various 2-alkyl/aryl-benzo[d]thiazol-2-yl sulfones (3) that were cleanly cleaved with sodium borohydride for the formation of the corresponding sodium sulfinates (Scheme 4A).31 This method was extended to the chiral (enantiomerically enriched) sulfinate salt derived from (R)-(+)-α-methylbenzylalcohol. The authors stated that due to the hygroscopic nature of sulfinates, the yields were not determined. Later, the Uneo procedure was successfully modified by Hu and co-workers32 for the synthesis of fluorinated sodium sulfinates from readily available difluoromethylbenzo[d]thiazol-2-yl sulfones (3) in excellent yields (Scheme 4B). It is worth noting that these sodium sulfinates were obtained as white solids, stable and with high purity, confirmed by elemental analysis. The preparation and purification of fluorinated sulfinate salts are readily scalable under these straightforward procedures.
 | ||
| Scheme 4 Uneo and Hu's methods for the synthesis of sodium sulfinates. | ||
In 2018, Ratovelomanana-Vidal and co-workers33 described a three-step synthesis of functionalized aliphatic and benzyl sulfinates. A series of 2-alkylthiobenzothiazoles were prepared through the alkylation of 2-mercaptobenzothiazole with alkyl halides (R–X; the authors did not mention the specific halides used), followed by oxidation to afford sulfones in 50% to quantitative yields (Scheme 5). Subsequently, the sulfones were treated with sodium borohydride to produce the corresponding sulfinates with diverse functional groups, including alkenes, alkynes, ethers, acetals, etc.
 | ||
| Scheme 5 Preparation of benzyl and aliphatic sulfinates. | ||
The Prakash and Olah group realized that the pyridine moiety served as a good leaving group in 2-sulfonyl pyridines. The alkylation of Hu's sulfone (4) with different alkyl iodides (R–X; X = I, Br) in the presence of LiHMDS formed a series corresponding sulfones (5). Following cleavage of 5 with EtSNa and ethyl mercaptan (EtSH), alkyl α,α-difluorinated sulfinates were formed (one representative example PhCH2CF2SO2Na was confirmed), which were directly converted into the corresponding sodium sulfonate salts in the presence of H2O2 (Scheme 6A).34 Afterwards, Baran and co-workers35 successfully adapted the Olah–Prakash alkylation of Hu's sulfone (4) with representative alkyl iodides followed by the cleavage of 5 with sodium hydride and EtSH to afford three analogs of α,α-difluoro alkylsulfinates (Scheme 6B). Among them was the scalable preparation of sodium difluoroethylsulfinate (DFES-Na) as a stable white solid, which was made commercially available.
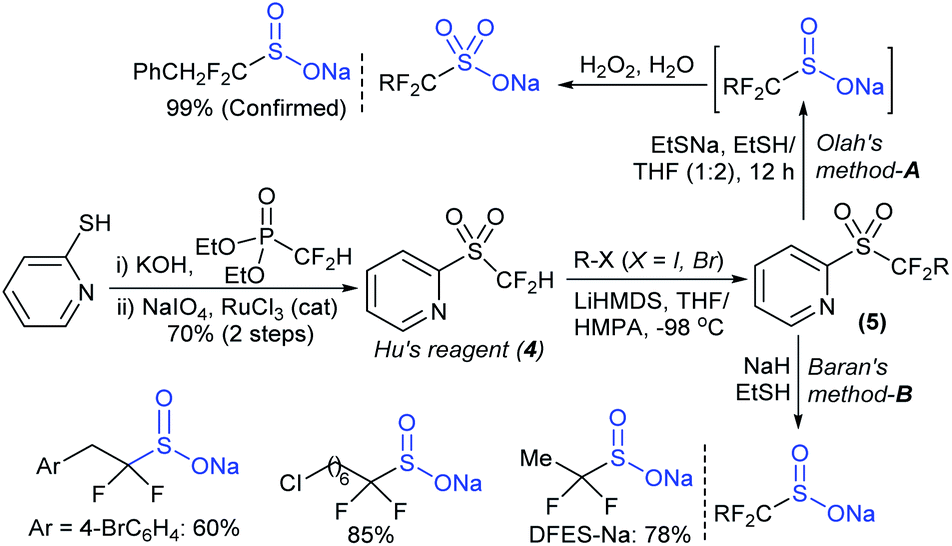 | ||
| Scheme 6 Preparation of alkyl α,α-difluoro sodium sulfinates. | ||
Subsequently, in 2014, the Baran group36 elegantly prepared structurally varied sodium sulfinates via a key Barton-type decarboxylation reaction, as shown in Scheme 7. The photolytic decarboxylation of the Barton ester (6) was followed by RuCl3-catalyzed oxidation conditions to form 2-pyridyl sulfones (7) in 30–92% yields. The removal of the pyridine moiety with alkoxides or thiolates provided various alkylsulfinates in moderate to high yields. Some of these sodium sulfinates were further validated through a commercialization partnership with MilliporeSigma.2
 | ||
| Scheme 7 Synthesis of new types of sodium sulfinates. | ||
In 2015, Harrity and co-workers37 reported the high-yield synthesis of azetidine and oxetane-based sulfinates in three simple steps, as presented in Scheme 8. The sulfenylation of 3-iodoheterocycle derivatives with 2-mercatopyridine and the oxidation of 8 with m-CPBA gave the corresponding 2-pyridyl sulfones, which undergo cleavage of the pyridine moiety with sodium thiolates. These sulfinate salts are readily accessible at the gram scale in overall good yields.
 | ||
| Scheme 8 Synthesis of azetidine and oxetane sulfinate salts. | ||
Paras and co-workers38 reported an improved and alternative two-step synthesis of optically pure sulfinate salts from 2-mercaptopyrimidines. The oxidation of a variety of chiral 2-mercaptopyrimidines (9) with m-CPBA provided pyrimidinyl sulfones (10) in high yields, which were cleaved smoothly with sodium methoxide in methanol to form the desired enantiomerically pure sulfinates in high yields (Scheme 9). It is worth mentioning that the pyrimidinyl sulfones (10) are sufficiently stable and quickly liberate the pyrimidinyl group under mild conditions.
 | ||
| Scheme 9 Synthesis of optically pure sodium sulfinate salts. | ||
Odell and co-workers39 developed a convenient method for the synthesis of sodium arylsulfinates from aryl bromides with 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABSO, 11) as a SO2 surrogate. A range of aryl/heteroaryl bromides were used for the in situ generation of aryl magnesium or aryl lithium reagents and trapped with DABSO (11), followed by treatment with aqueous Na2CO3 (Table 1). The purification was performed via liquid–liquid and solid–liquid extraction to avoid sulfonic acids and obtain the corresponding sodium arylsulfinates in good to high yields. The reaction of 2-bromo-thiophene with n-BuLi at −78 °C resulted in lithium–bromine exchange and nucleophilic attack by 2-lithiothiophene on BuBr; subsequent reaction with DABSO gave 5-butyl thiophenesulftnate in 68% yield.
|
||
|---|---|---|
| Sodium arylsulfinates | Method-A | Method-B |
 |
R = H: 72% | R = H: 85% |
| R = 4-Ph: 87% | R = 4-Ph: 42% | |
| R = 4-CF3: 91% | R = 4-CF3: 48% | |
| R = 2-CF3: 75% | R = 2-CF3: 99% | |
| R = 4-OMe: 48% | R = 4-OMe: 59% | |
| R = 3-OMe: 85% | R = 3-OMe: 91% | |
 |
48% | 82% |
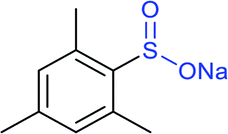 |
69% | 64% |

Later in 2018, Wang, Zhang and co-workers40 reported a direct and straightforward preparation of sodium arenesulfinate salts from arenes and DABSO (11) in the presence of excess AlCl3 (Scheme 10). The reaction proceeded smoothly and arenes bearing electron-donating and halide groups gave good to excellent yields. The nitrobenzene declined to provide the desired product. Mechanistic studies of Friedel–Crafts-type sulfination with DABSO showed an electrophilic aromatic substitution pathway.
 | ||
| Scheme 10 The preparation of sodium arylsulfinates through Friedel–Crafts-type sulfination with DABSO (11). | ||
Simultaneously, Maruoka and co-workers41 described a new two-step protocol for preparing sodium α-aminoalkane-sulfinate salts, as shown in Scheme 11. Various N-Boc-protected dialkylamines were treated with sec-BuLi to generate an α-lithiated alkyl-amine that was successfully trapped with DABSO, aqueous workup with sodium carbonate to provide sodium α-aminoalkanesulfinates in variable yields.
 | ||
| Scheme 11 Two-step synthesis of sodium α-aminoalkanesulfinate salts. | ||
2.2. Sodium perfluoroalkylsulfinates (RFSO2Na)
In the late 1980s, sodium trifluoromethanesulfinate (sodium triflinate)42 was prepared on a large scale by the Rhône-Poulenc Co., through the single-electron reduction of bromotrifluoro-methane (Scheme 12A).43 Unfortunately, this method was expelled because of its ozone-depleting effect in the process. In 2007, two independent methods were published for the preparation of sodium perfluoroalkanesulfinates. The Chen group demonstrated the sulfinatodehalogenation of chlorotrifluoro-methane with sodium dithionite (Na2S2O4) to give the desired sodium triflinate in quantitative yield based on the 19F NMR (Scheme 12B).44 Similarly, several perfluoroalkanesulfinates and bis-sulfinates were successfully achieved in high yields. Concurrently, Langlois and co-workers developed sustainable and mild reaction conditions to synthesize trifluoromethane-sulfinate (triflinate) salts via β-elimination of aliphatic triflones using NaOMe in MeOH (Scheme 12C).45 As a result, this reagent is well known as the Langlois reagent.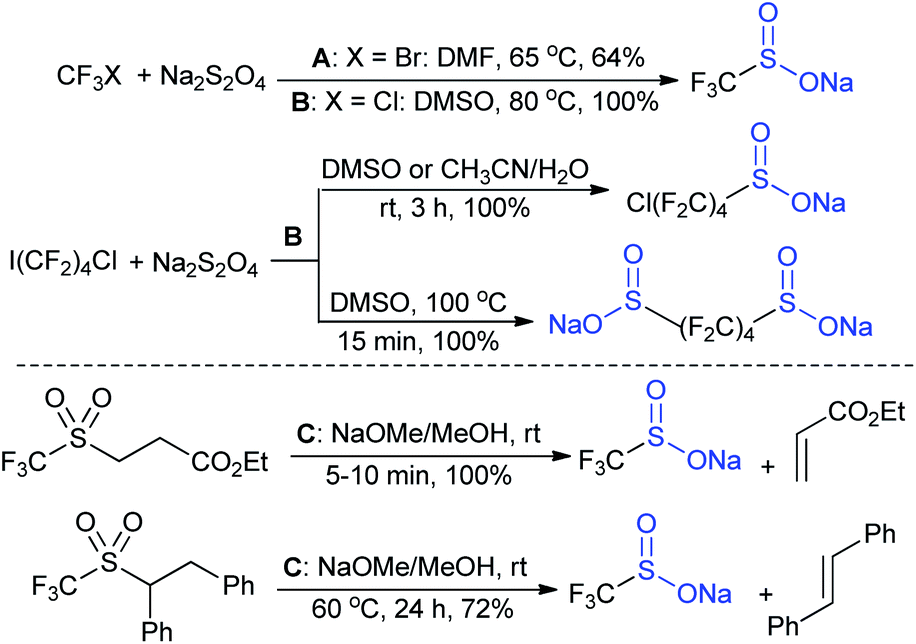 | ||
| Scheme 12 Synthesis of sodium perfluoromethanesulfinate (RFSO2Na). | ||
3. Applications of sodium sulfinates
As already highlighted in the introduction (see Fig. 1), sodium sulfinates possess vast synthetic applications with a significant number of different strategies. Accordingly, we have classified the applications of sodium sulfinates into several sections based on the types of organosulfur compounds with some exciting reaction mechanisms. The following Sections 3.1–3.5 are also further subdivided based on the transformations.3.1. Synthesis of thiosulfonates (R–SO2S–R1)
In 1972, Bentley et al. described the silver nitrate (in aqueous acetone)-assisted reaction between sodium methanesulfinate and alkyl di-sulfides for the formation of thiosulfonates (thiol esters are also known as thiolsulfonates), as described in Scheme 13.46 The nucleophilic sulfinate attack on the silver–disulfide complex led to the desired methyl thiosulfonates, and silver mercaptans were identified by gas chromatography.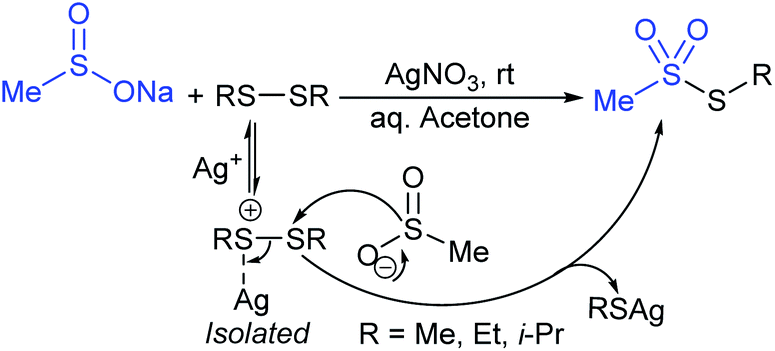 | ||
| Scheme 13 Synthesis of thiosulfonates from alkyl disulfides. | ||
Consequently, Abe and Tsurugi prepared various aryl thiosulfonates in good yields by the sulfenylation reaction of N-(arylthio)succinimides (12) and sulfinate salts were vigorously shaken for 5–10 min (Table 2A).47 In 2012, Chen and co-workers48 examined Sc(OTf)3-catalyzed sulfenylation using various N-(organothio)succinimides (12) and sodium sulfinates for the formation of symmetrical and unsymmetrical thiosulfonates in good to high yields (Table 2B). The scope and generality was explored with a range of substitutions, including aliphatic and aromatic succinimides, as well as arylsulfinates, which were adequately tolerated. The sulfenylation reaction was smoothly accelerated in ionic liquids (ILs), such as [BMIM]/PF6, and water played a critical role in the reaction; probably the solubility of sodium sulfinates may have been enhanced. Interestingly, Sc(OTf)3/ILs was recovered and reused for sulfonylation without any significant loss in the catalytic activity.
|
||
|---|---|---|
| Thiosulfonates | Method-A | Method-B |
 |
R = H: 92% | R = Cl: 78% |
| R = Cl: 92% | R = Me: 92% | |
| R = Me: 95% | ||
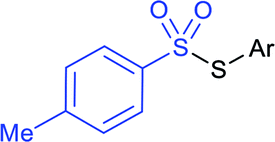 |
Ar = 4-ClC6H4: 80% | Ar = 4-FC6H4: 85% |
| Ar = 4-MeC6H4: 87% | Ar = 4-BrC6H4: 79% | |
| Ar = 4-CF3C6H4: 51% | Ar = 4-NO2C6H4: 77% | |
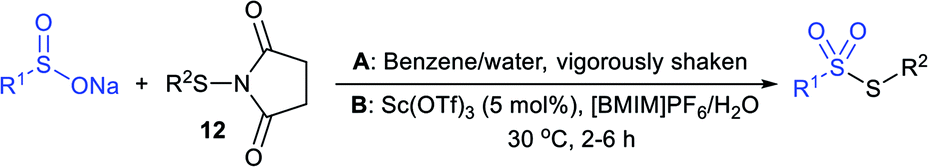
In 1996, Langlois and co-workers49 described the synthesis of a wide range of alkyl and aryl thiosulfonates in high yields. The sulfenylation reaction of disulfides with sodium benzenesulfinate and trifluoromethanesulfinate was conducted in the presence of bromine (Scheme 14). Various disulfides have been studied, and primary disulfides are generally more reactive than the secondary ones; the hindered tert-butyl disulfide disappointed in delivering the desired thiosulfonate.
 | ||
| Scheme 14 Synthesis of thiosulfonates from disulfides. | ||
Fujiki and co-workers50 demonstrated the efficient and straightforward synthesis of various unsymmetrical thiosulfonates in good to high yields. I2-catalyzed oxidative sulfenylation of various acyclic and cyclic disulfides with several sulfinates occurred in the presence or absence of solvent at room temperature (Scheme 15). An interesting outcome of this protocol is cyclic disulfides cleaving S–S bonds with arenesulfinates to synthesize valuable bis-thiosulfonates in high yields.
 | ||
| Scheme 15 Synthesis of bis-thiosulfonates using cyclic disulfides. | ||
NBS-promoted sulfenylation reaction between disulfides and sulfinates to access unsymmetrical and symmetrical thiosulfonates was described. Wu and co-workers51 successfully explored broad functional group tolerance and atom-economical and practical procedures to synthesize a series of thiosulfonates in good to excellent yields. A few representative examples are presented in Scheme 16.
 | ||
| Scheme 16 The synthesis of thiosulfonates using disulfides. | ||
The Taniguchi and Yadav groups independently reported S–S coupling between thiols and sodium sulfinates to synthesize various thiosulfonates under aerobic conditions. The CuI–Phen·H2O (Phen = 1,10-phenanthroline)-catalyzed sulfenylation reaction of thiols with sulfinates gave the desired thiosulfonates in 40–96% yields (Scheme 17A).52 Similarly, the FeCl3-catalyzed coupling of thiols with sulfinates provided a wide variety of symmetrical and unsymmetrical thiosulfonates in 83–96% yield (Scheme 17B).53 Both methods proceeded smoothly and are widely applicable to various arene- and alkanethiols with aromatic and aliphatic sulfinates.
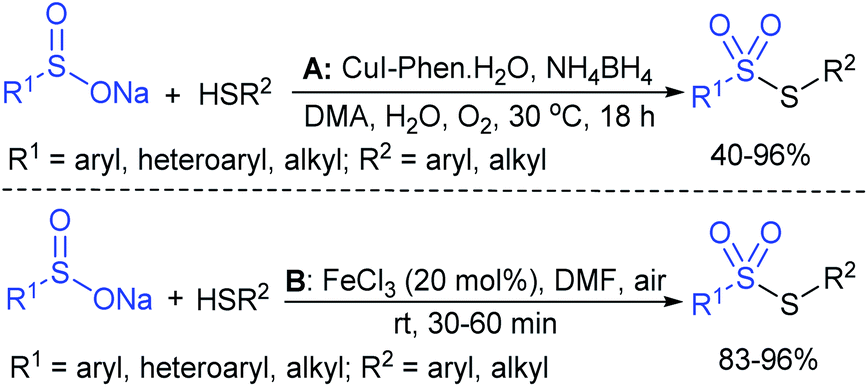 | ||
| Scheme 17 Sulfenylation of thiols with sodium sulfinates. | ||
Taniguchi also reported a copper-catalyzed sulfenylation of disulfides and diselenides with sodium sulfinates under air at ambient temperature (Scheme 18).54 The formation of S–S and Se–S bonds efficiently gave various thiosulfonates and selenosulfonates in good to high yields. The use of ditelluride with sodium 4-toluenesulfinate did not yield the desired product under the same reaction conditions. Notably, the same reaction was performed under a nitrogen atmosphere instead of air (oxygen) and the corresponding products were produced at lower yields. To understand the role of oxygen, the formation and the reactivity of PhXCu(I) was examined with sodium 4-toluenesulfinate under the same conditions and gave the corresponding thio(seleno)sulfonates in low yields.
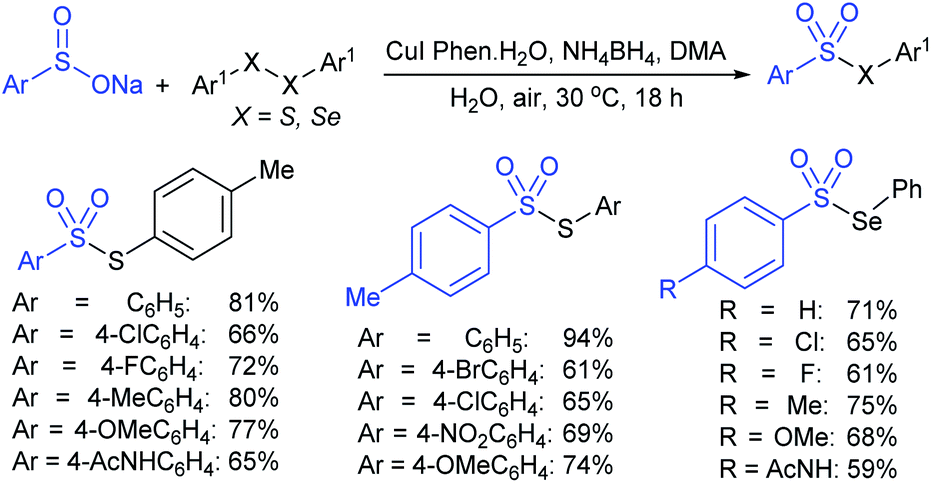 | ||
| Scheme 18 Copper-catalyzed sulfenylation of disulfides with sodium sulfinates. | ||
Lu, Shen and co-workers55 used trifluoromethanesulfinate (13) as an electrophilic thiolating reagent. The treatment of sodium aryl and heteroaryl sulfinates with 13 in acetic acid at room temperature for 12 h generated the corresponding trifluoromethyl thiosulfonates in high to excellent yields. A few representative examples are presented in Scheme 19.
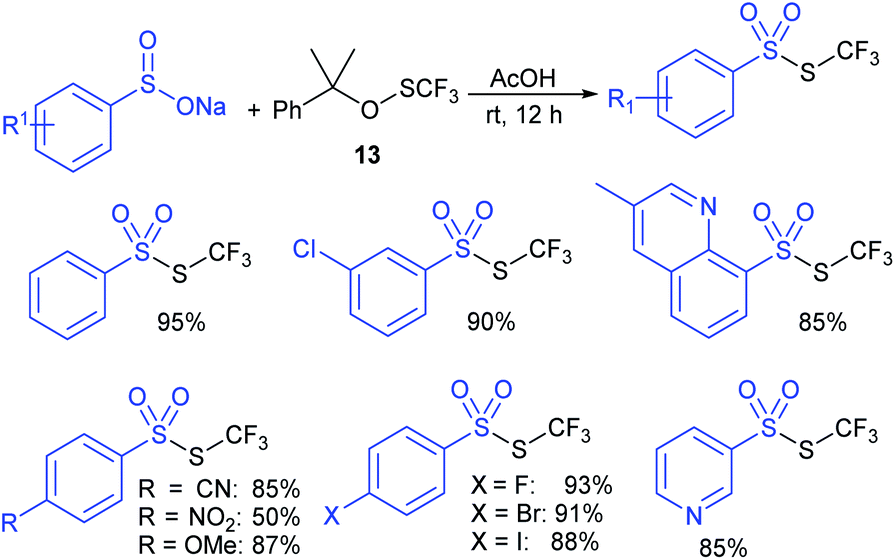 | ||
| Scheme 19 Synthesis of trifluoromethylthiosulfonates using 13. | ||
Recently, Wang et al. found a BF3·OEt2-mediated radical disproportionate coupling reaction of sodium sulfinates to synthesize thiosulfonates in good yields (Scheme 20).56 The simple and practical protocol had good functional group tolerance and can also be applied to prepare both symmetrical and unsymmetrical thiosulfonates under mild reaction conditions. Generally, the sodium arylsulfinates containing electron-donating groups perform better than those with electron-withdrawing groups for symmetrical thiosulfonates. More interestingly, the disproportionate coupling reaction of various sodium alkylsulfinates and sodium arylsulfinates gave unsymmetrical thiosulfonate products in moderate to good yields.
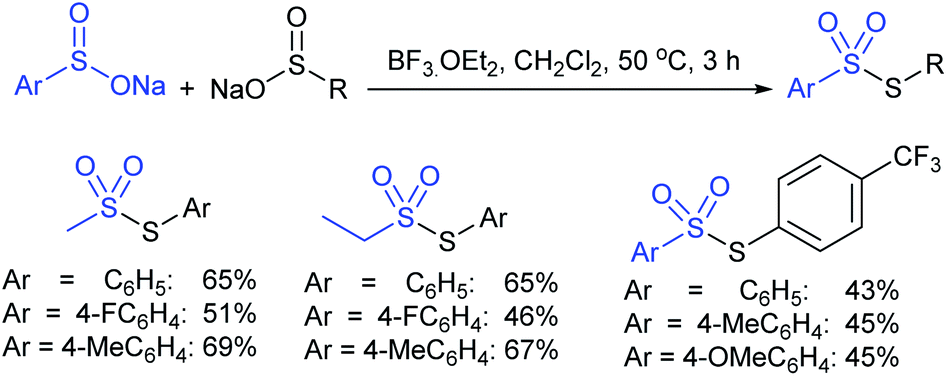 | ||
| Scheme 20 BF3·OEt2-mediated radical disproportionate coupling reaction of sodium sulfinates. | ||
Chen and co-workers57 revealed the hypervalent iodine-mediated synthesis of selenosulfonates from diaryl diselenides with sodium sulfinates, as presented in Scheme 21. The three different sodium arenesulfinates were well reacted with diaryl diselenides in the presence of [(bis(trifluoroacetoxy)iodo)]-benzene in methylene chloride and gave the corresponding aryl selenosulfonates in 68–83% yields.
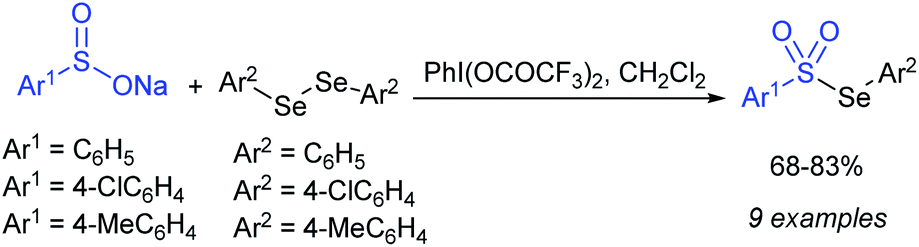 | ||
| Scheme 21 Synthesis of selenosulfonates from diaryl diselenides with sodium sulfinates. | ||
3.2. Synthesis of sulfonamides (R–SO2N–R1R2)
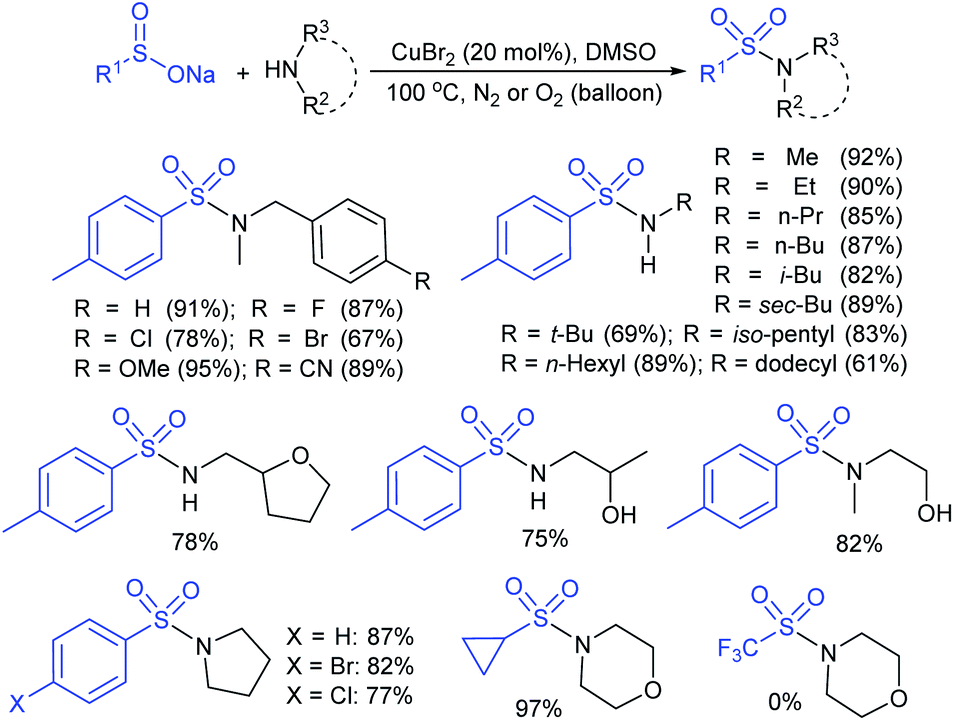 | ||
| Scheme 22 Copper-catalyzed construction of sulfonamides. | ||
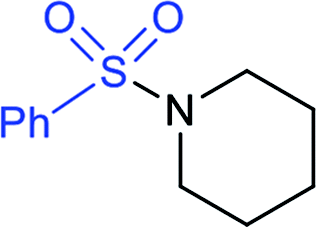
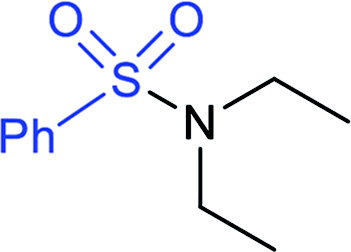
In 2015, the metal-free synthesis of sulfonamides was carried out using the molecular iodine-mediated coupling of various amines with sodium sulfinates at room temperature. The Wang59 and Song60 groups independently reported using a stoichiometric amount of iodine for the formation of series of sulfonamides in good to excellent yields (Table 3). Both methods established the sulfonylation using a wide range of primary and secondary amines, including substituted aromatic, aliphatic, acyclic, and cyclic amines. Further, the amination reaction was explored for various substituted aromatic, heteroaromatic, and aliphatic sulfinates as suitable substrates for this protocol.
|
|||||
|---|---|---|---|---|---|
| Sulfonamides | A | B | Sulfonamides | A | B |
 |
97% | 40% |  |
95% | 64% |
 |
98% | 95% |  |
85% | 80% |
 |
90% | 71% |  |
77% | 50% |
 |
78% | 82% |  |
96% | 46% |

Simultaneously, Yuan and co-workers61 demonstrated an efficient and eco-friendly way to synthesize sulfonamides using a sub-stoichiometric amount of iodine (50 mol%) in H2O at room temperature. Both aromatic and aliphatic amines conveniently reacted with various sodium sulfinates under the optimal reaction conditions, giving the desired products in moderate to excellent yields (Table 4A). In the same year, the iodine-catalyzed oxidative amination of sodium sulfinates with amines in the presence of sodium percarbonate as an oxidant was developed by Yotphan and co-workers.62 A wide range of aromatic, aliphatic, heteroaromatic amines and hydrochloride salts of amines with varied sulfinate substrates were successfully employed for this transformation (Table 4B).
|
|||||
|---|---|---|---|---|---|
| Sulfonamides | A | B | Sulfonamides | A | B |
 |
92% | 81% |  |
93% | 62% |
 |
85% | 42% |  |
93% | 62% |
 |
92% | 74% |  |
94% | 72% |

Very recently, the Peng and Dietrich group reported innovative work for the preparation of DNA-conjugated sulfonamides from DNA-conjugated amines and sodium sulfinates in the presence of iodine under mild conditions (Scheme 23).63 A wide range of highly functionalized sulfinates were synthesized, made to commercially available and successfully employed to generate desired sulfonamides in moderate to good yields. A vast number of unnatural amino acids bearing a DNA skeleton, including primary and secondary amines participated and gave corresponding sulfonamides in reasonable yields.
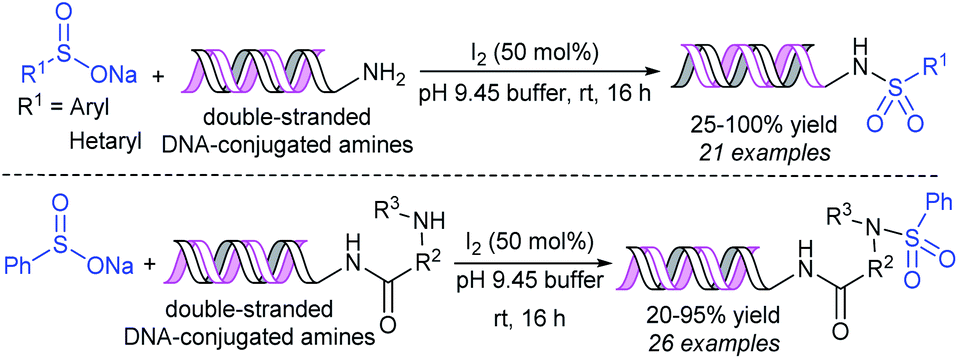 | ||
| Scheme 23 I2-mediated DNA-conjugated sulfonamides from DNA-conjugated amines with sodium sulfinates. | ||
Zhao et al. developed an alternative procedure for the preparation of sulfonamides via the TBAI-catalyzed coupling of amines with sodium sulfinates under mild conditions (Table 5A).64 An oxidative N-sulfonylation of various primary and secondary amines bearing different functional groups was conducted, which smoothly reacted with arylsulfinates and delivered a wide variety of sulfonamides in good to high yields. Challenging substrates like alkylsulfinate salts also served as coupling partners, but trifluoromethanesulfinate was unsuccessful in yielding the desired trifluoromethanesulfonimide. In 2018, Fu and co-workers reported sulfonamides prepared by the NaI-catalyzed oxidative amination of primary and secondary amines with sodium sulfinates using ethylene dibromide (EDB) under aerobic conditions (Table 5B).65 Various amines, such as aliphatic, aromatic, and benzylic amines reacted well with sodium p-toluenesulfinate to give the corresponding sulfonamides in moderate to good yields. Additionally, various aryl and heteroaryl sulfinates also afforded the corresponding sulfonamides in acceptable yields. The authors stated that the instability of the aliphatic sulfonyl radicals was presumably because aliphatic sulfinates did not participate.
|
|||
|---|---|---|---|
| Sulfonamides | A or B | Sulfonamides | A or B |
 |
R = Ph: A: 70% B: 78% | 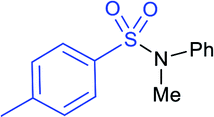 |
A: 74% B: 82% |
| R = Bn: A: 60% B: 75% | |||
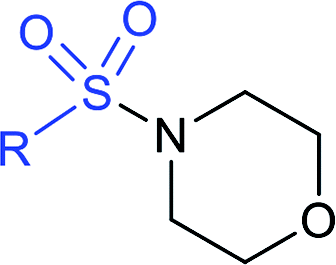
 |
X = O; A: 96% |  |
A: 80% B: 58% |
| X = CH2; B: 72% | |||
 |
R = Me; A: 98% | 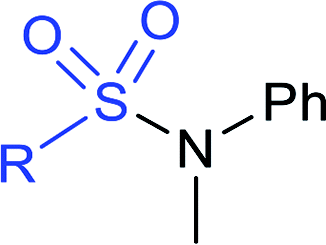 |
R = Me: B: 0% |
| R = CF3; A: 0% | R = CF3: B: 0% | ||

Yan and co-workers66,67 prepared N-sulfonyl benzotriazoles through the iodine-catalyzed sulfonylation of benzotriazoles with sodium sulfinates under air at room temperature. The catalytic radical sulfonylation proceeded efficiently using various benzotriazoles with aryl and alkyl sulfinates. The monosubstituted benzotriazoles (5-Me and 5-Cl) afforded a mixture of 5- and 6-substituted products and the ratios ranged from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 2:3 (Scheme 24A).66 Subsequently, the same group reported a simple procedure using NaBr/mCPBA for the preparation of N-sulfonylbenzotriazoles from benzotriazoles with aryl and methyl sulfinates at room temperature in 40–90% yields (Scheme 24B).68
:1 to 2:3 (Scheme 24A).66 Subsequently, the same group reported a simple procedure using NaBr/mCPBA for the preparation of N-sulfonylbenzotriazoles from benzotriazoles with aryl and methyl sulfinates at room temperature in 40–90% yields (Scheme 24B).68
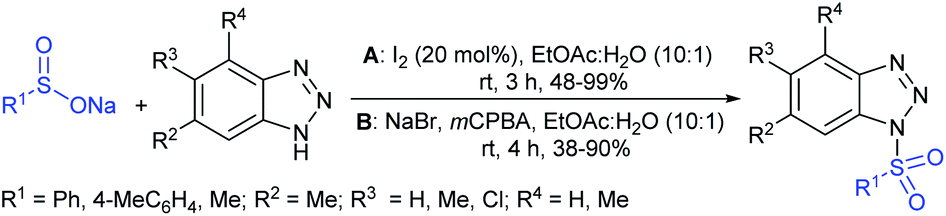 | ||
| Scheme 24 N-Sulfonylation of benzotriazoles with sodium sulfinates. | ||
Fu et al. developed the NXS (X = I, Br)-mediated direct N-sulfonylation of azoles with sodium sulfinates to synthesize sulfonamide derivatives (Scheme 25).68 Several azoles, such as substituted benzoimidazoles, benzotriazole and 1,2,4-triazole were coupled with a range of electron-rich and electron-deficient sodium arylsulfinates to afford the desired products in high yields. Remarkably, pyrazole substrates underwent unusual halogenation-sulfonylation in the presence of NXS with sodium sulfinates. A variety of substituted pyrazoles smoothly reacted with sodium arylsulfinates to give 4-halo-1-sulfonyl pyrazole derivatives in good to high yields.
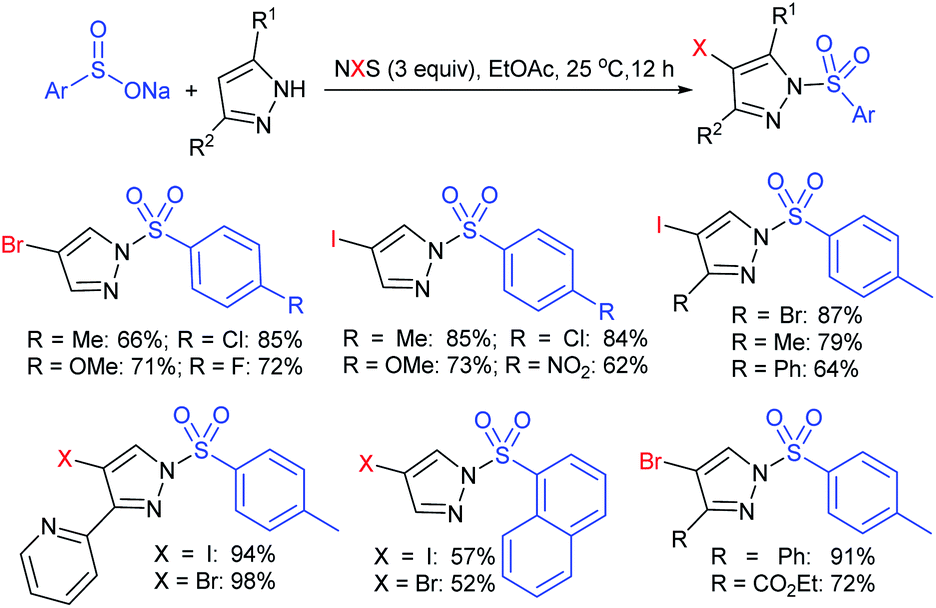 | ||
| Scheme 25 NXS-mediated halogenation-sulfonylation of substituted pyrazoles with sodium arylsulfinates. | ||
Zeng and co-workers69 developed a graphite-nickel-based electrochemical reaction for the synthesis of sulfonamides. The electrochemical oxidative amination of a range of 1°/2° aryl and alkyl amines and aqueous ammonia were smoothly reacted and afforded the desired sulfonamides in acceptable yields. Additionally, varied sodium arenesulfinates were explored in this protocol to synthesize sulfonamides in moderate to good yields, as shown in Scheme 26.
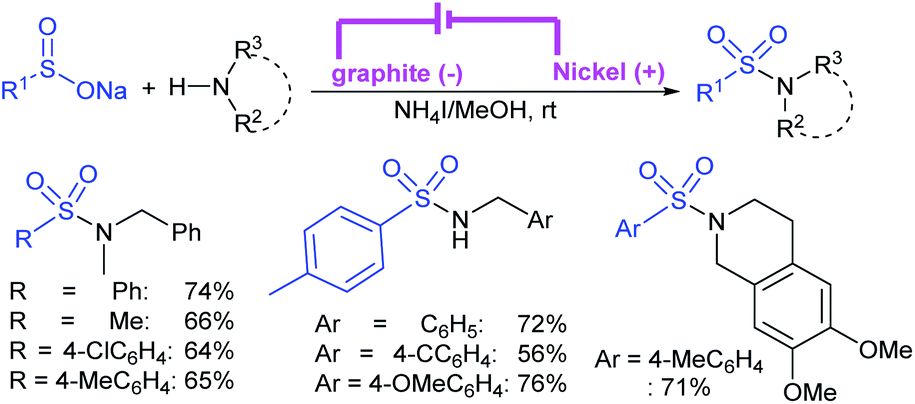 | ||
| Scheme 26 Electrochemical oxidative amination of sodium sulfinates with amines. | ||
In the same year, Yuan and co-workers70 developed an electrochemical method for the synthesis of sulfonamides in the presence of NaI, which served as a supporting electrolyte at room temperature. A variety of amines were readily sulfonylated with substituted aromatic sulfinates in water to give the corresponding aryl sulfonamides in good to high yields (Scheme 27). However, sodium trifluoromethanesulfinate (F3CSO2Na) was ineffective and did not provide the desired product.
 | ||
| Scheme 27 Electrochemical oxidative amination of amines with sulfinates. | ||
Yuan elegantly synthesized sulfonamides and β-arylsulfonyl enamines using sodium sulfinates via the cleavage of C–N and C–H bonds of tertiary amines, respectively.71 The combination of iodine and t-butyl hydroperoxide (TBHP) allowed the reaction between tertiary amines and sodium sulfinates. The oxidative-sulfonylation of tertiary amines occurred through cleavage of the C–N bond in water and provided desired sulfonamides in good to high yields (Scheme 28). The reactivity dramatically changed from H2O and DMSO, and the C–H bond cleavage of triethylamine in dimethyl sulfoxide with substituted aromatic sulfinates to corresponding β-arylsulfonyl enamines yielded in 69–81% range. A few control experiments were performed to gain mechanistic insights for this novel protocol. With the use of the radical scavenger 2,2,6,6-tetramethylpiper-idine-1-oxyl (TEMPO), no desired product was detected, indicating that the reaction presumably underwent a radical pathway.
 | ||
| Scheme 28 Synthesis of sulfonamides and β-arylsulfonyl enamines. | ||
Later, Yan and co-workers72 prepared sulfonamides from amines and sodium sulfinates using (n-C4H9)4NBr and m-chloroperbenzoic acid at room temperature (Scheme 29). A series of primary and secondary amines including cyclic amines, participated in coupling with sodium benzenesulfinate and sodium p-toluenesulfinate to furnish the corresponding sulfonamides in 39–89% yields.
 | ||
| Scheme 29 Synthesis of sulfonamides from amines. | ||
Recently, Zhou and He described a hypervalent iodine-catalyzed reaction between various primary and secondary amines with sodium arylsulfinates under mild reaction conditions to provide the corresponding sulfonamides in 30–81% yields (Scheme 30).73 The actual hypervalent iodine (iodosylpropane) catalyst was generated in situ from the catalytic amount of 1-iodopropane with a stoichiometric amount of m-CPBA as an oxidant for this protocol.
 | ||
| Scheme 30 Hypervalent iodine-mediated synthesis of sulfonamides. | ||
Our research group74 successfully demonstrated a convenient and metal-free protocol for the highly regioselective sulfonylation of NH-1,2,3-triazoles with sodium sulfinates. A range of disubstituted NH-1,2,3-triazoles were readily sulfonylated with various arylsulfinates in the presence of molecular iodine. A variety of synthetically viable N2-sulfonyl triazoles was obtained in moderate to high yields with excellent regioselectivities (Scheme 31). Disappointingly, the monosubstituted NH-1,2,3-triazoles afforded the corresponding sulfonamides in a mixture of regioisomers.
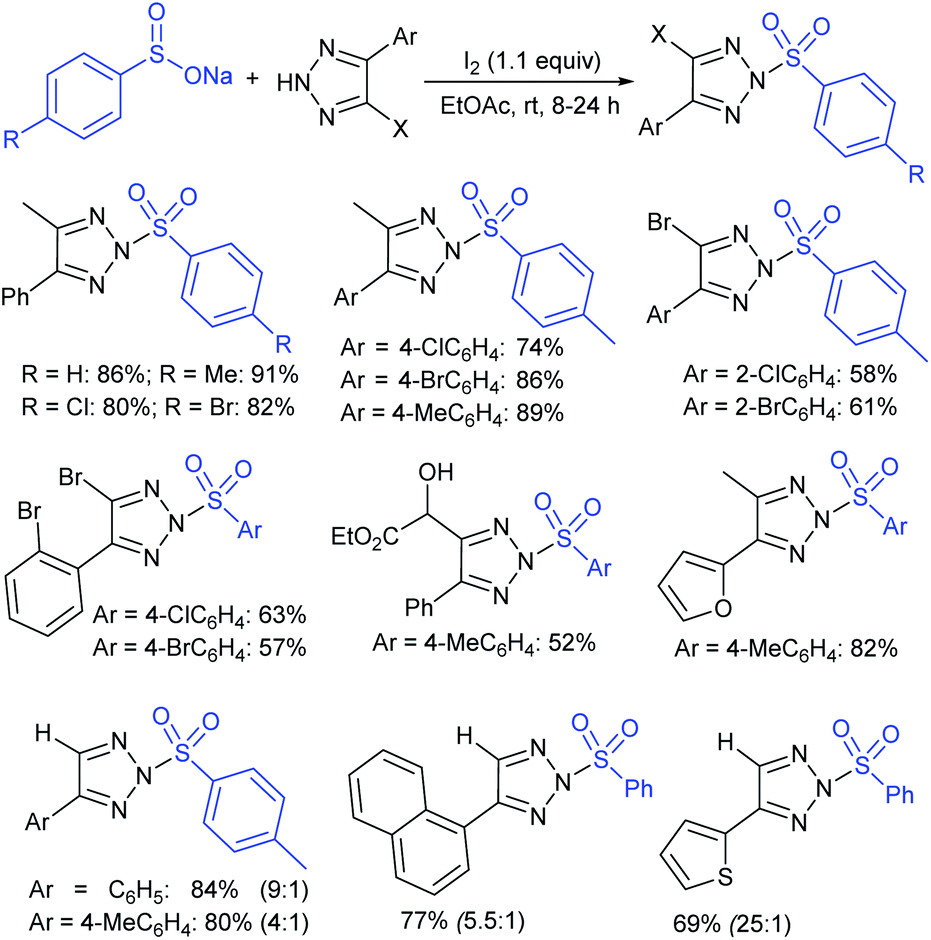 | ||
| Scheme 31 N2-regioselective sulfonylation of NH-1,2,3-triazoles with sulfinates. | ||
 | ||
| Scheme 32 Synthesis of sulfonamides. | ||
Tu and co-workers76 demonstrated an attractive and efficient copper-catalyzed electrophilic amination using O-benzoyl hydroxylamines with sodium sulfinates under ambient conditions. Only 2 mol% CuBr2 catalyst was sufficient for coupling several acyclic and cyclic O-benzoyl hydroxylamines and provided a broad range of sulfonamides in high yields (Scheme 33). Additionally, a series of aryl, heteroaryl, and alkyl sulfinates were readily coupled with O-benzoyl hydroxylamine and there was no reaction with sodium trifluoromethanesulfinate. Moreover, the protocol was readily scalable up to the gram level when the reaction was performed at a 5 mmol with O-benzoyl hydroxymorpholine and sodium benzenesulfinate under standard conditions. Some control experiments were performed, including radical scavenger TEMPO, and no sulfonamide was detected. A plausible catalytic radical pathway for the oxidative addition–reductive elimination was proposed as shown in Scheme 33. Initially, CuBr was readily generated from CuBr2 by the coordination of copper to sodium sulfonate to produce the CuI intermediate A via a free sulfonyl radical. Oxidative addition with O-benzoyl hydroxylamine formed the CuIII complex B, then the sulfonamide product. Subsequently, the resulting BzO–CuI reacted with NaBr to regenerate CuBr and complete the catalytic cycle.
 | ||
| Scheme 33 Cu-catalyzed electrophilic amination of O-benzoyl hydroxyl-amines with sodium sulfinates. | ||
Xia et al. developed the KOtBu mediated decarbonylation of N,N-disubstituted formamides as the amine source for direct S–N bond formation (Scheme 34).77 The NIS-promoted oxidative amination of N,N-disubstituted formamides with arene-sulfinates bearing electron-donating or electron-withdrawing groups was conducted for the preparation of a series sulfonamide in good to high yields. In addition to formamide, the N,N-diethyl-formamide and N-acetylmorpholine-compatible substrate gave the desired sulfonamides in this protocol.
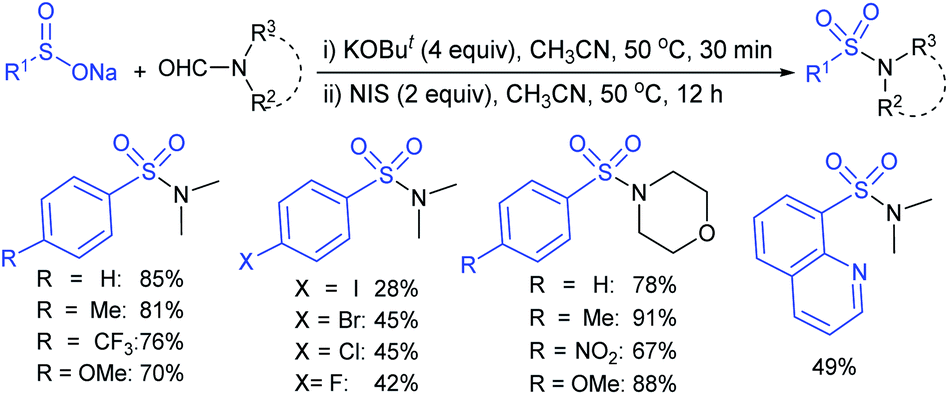 | ||
| Scheme 34 Cu-catalyzed electrophilic amination of O-benzoyl hydroxyl-amines with sodium sulfinates. | ||
The enzymatic oxidative-sulfonylation of 4-substituted urazoles with sodium arenesulfinates was performed in phosphate buffer in the air as an external oxidant (Scheme 35).78 The successive aerobic oxidation of urazoles followed by sulfonylation with only commercially available arylsulfinates using laccase-50U as an eco-friendly biocatalyst was carried out to furnish a range of 1-sulfonyl-1,2,4-triazolidine-3,5-dione derivatives in 85–97% yields.
 | ||
| Scheme 35 Laccase biocatalyst for the coupling of 4-substituted urazoles with arylsulfinates. | ||
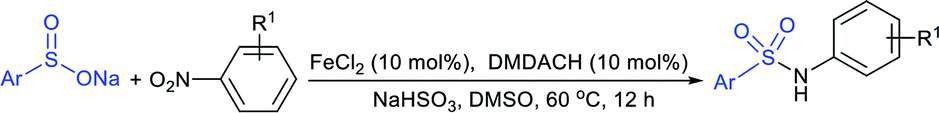
Luo and co-workers79 realized a convenient one-pot reduction-sulfonylation via the FeCl2-catalyzed direct coupling of nitroarenes with sodium sulfinates in the presence of NaHSO3 and N,N′-dimethyl-1,2-diaminocyclohexane (DMDACH) in DMSO. The reduction of nitroarenes by NaHSO3 followed N–S bond construction to generate many N-aryl sulfonamides in good to excellent yields (Table 6). In this protocol, both nitroarenes and sulfinate salt substrates having a broad range of functional groups were tolerated. No desired sulfonamides were detected when using nitromethane and sodium benzylsulfinate under the same reaction conditions.
|
|||
|---|---|---|---|
| Sulfonamides | Yield | Sulfonamides | Yield |
 |
R = H: 93% |  |
X = N: 81%, X = CH: 94% |
| R = F: 85% | |||
| R = Cl: 89% | |||
| R = Br: 91% | |||
| R = OMe: 92% | |||
 |
R = Me: 88% |  |
93% |
| R = Cl: 93% | |||
| R = CN: 97% | |||
| R = OMe: 85% | |||
| R = CO2H: 78% | |||
| R = CO2Me: 86% | |||
 |
R = Cl: 96% |  |
90% |
| R = Me: 85% | |||
| R = CF3: 98% | |||
| R = COMe: 84% | |||
| R = CO2Me: 95% | |||
Afterwards, the Andrioletti group described the reductive-sulfonylation of nitroarenes and sulfinate salts in the presence of sodium bisulfite (NaHSO3) alone as the reducing agent in water at 60 °C (Scheme 36A).80 A variety of water-soluble nitroarenes were successfully converted into the corresponding sulfonamides in 25–78% yields. This protocol was restricted to only electron-withdrawing nitroarenes. Consequently, the copper-catalyzed redox coupling of nitroarenes with sodium sulfinates in NMP at 120 °C was described by Zhang and co-workers (Scheme 36B).81 A series of aromatic sulfonamides were obtained in moderate to good yields using various nitroarenes coupled with sodium 4-methylbenzenesulfinate. Aryl and alkyl sulfinates were smoothly reacted with 4-methyl nitrobenzene to give the desired N-aryl sulfonamides in reasonable yields. The reductive-sulfonylation proceeded without any external reducing additive, assuming that sodium sulfinates acted as reducing reagent.
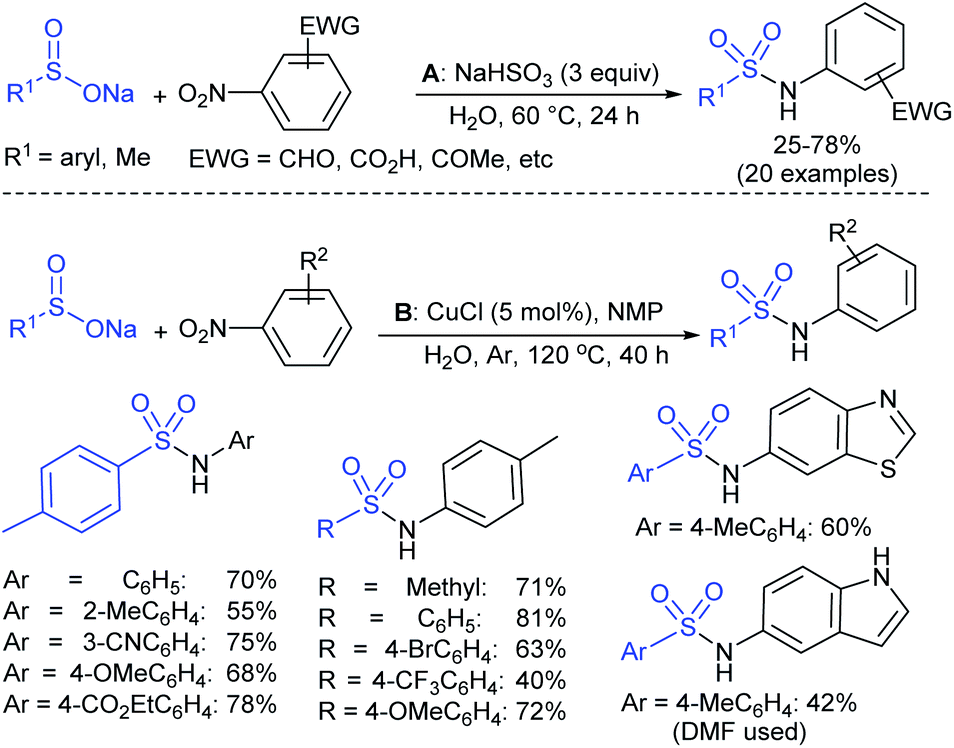 | ||
| Scheme 36 NaHSO3 or CuCl-mediated direct coupling of sodium arylsulfinates with nitroarenes. | ||
3.3. Synthesis of thioethers (R–S–R1)
|
|||
|---|---|---|---|
| Thioethers | A or B | Thioethers | A or B |
 |
A: 85% |  |
Ar = 4-MeC6H4, A: 90%; B: 85% |
| B: 75% | Ar = 4-ClC6H4, A: 93%; B: 97% | ||
 |
A: 65% | 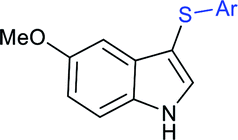 |
Ar = C6H5, A: 90% |
| B: 44% | Ar = 4-MeC6H4, B: 88% | ||
 |
Ar = C6H5, A: 82% |  |
Ar = C6H5, A: 86% |
| Ar = 4-MeC6H4, B: 88% | Ar = 4-MeC6H4, B: 83% | ||
 |
Ar = C6H5, A: 87% |  |
Ar = C6H5, A: 0% |
| Ar = 4-MeC6H4, B: 89% | Ar = 4-MeC6H4, B: 75% | ||

Rao et al. examined various persulfates, such as (NH4)2S2O8, Na2S2O8, K2S2O8 and oxone for thiolation of indole with sodium arenesulfinate salts and K2S2O8 was found to be the best choice to afford 3-arylthioindoles (Scheme 37).84 The scope of C3-sulfenylation was efficiently investigated with varied indoles as well as arenesulfinates to give a range of 3-arylthioindoles in good to excellent yields. Interestingly, 3-methylindole also participated in producing the C2-sulfenylated product in low yield. The transformation was suitable for the Gram-scale synthesis of indole-3-thioether without any significant variation in the outcome.
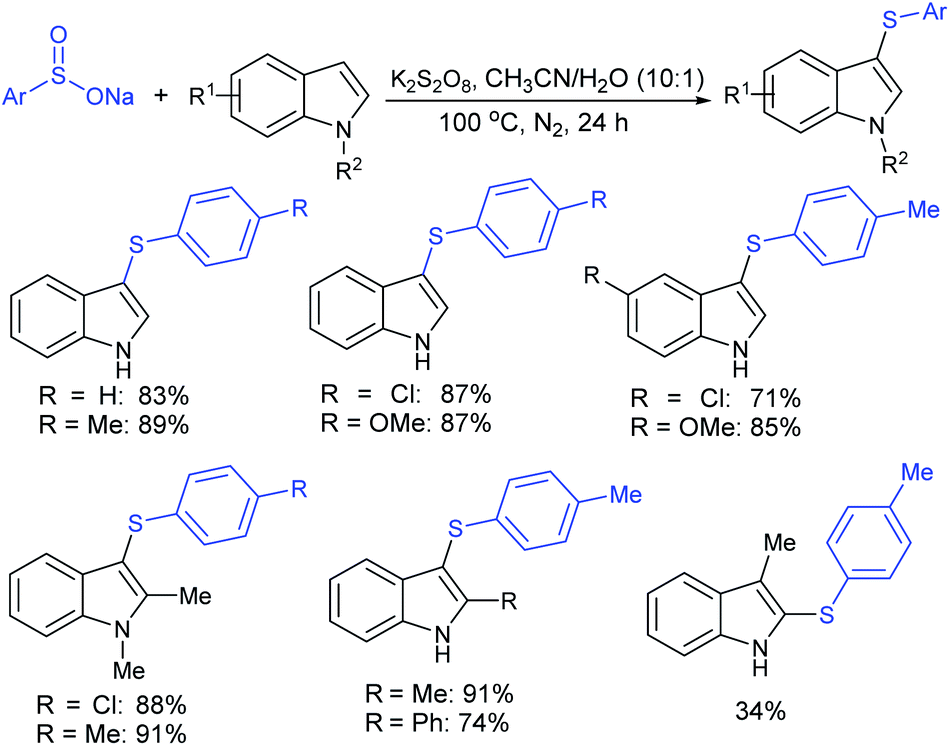 | ||
| Scheme 37 K2S2O8-mediated thiolation of indole with arenesulfinate salts. | ||
Yi and Zhang carried out interesting work on the copper-catalyzed direct trifluoromethylthiolation of indoles, pyrroles, and enamines. A series of substituted indoles were sulfenylated with sodium trifluoromethanesulfinate (Langlois reagent) and a wide variety C3-sulfenyalted products were obtained in good to excellent yields (Scheme 38A).85 Next, pyrroles and enamines were utilized in trifluoromethylthiolation to afford the desired thioethers in good yields. Further, the method was successfully extended to direct perfluoroalkyl-thiolation using different sodium perfluoroalkane sulfinates (RfSO2Na) having different Rf groups to give the desired products in high yields. The authors carefully examined the 19F NMR to understand the actual species for this protocol. The intermediates I–V were detected based on fluorine values where either the F3CSOH (III) or F3CS+ (V) precursors underwent an electrophilic attack on the indoles. Similarly, Cai and co-workers86 disclosed a metal-free trifluoromethylthiolation of indoles, pyrroles, and enamines with sodium trifluoromethane-sulfinate under mild conditions (Scheme 38B). A series of trifluoromethyl thioether products were obtained in comparable yields. It was assumed that a reactive F3CSCl was generated from the F3CSO2Na in the presence of the Ph3P and N-chlorophthalimide reagent system. The reaction was not inhibited by the addition of TEMPO and as a result, the radical pathway was ruled out.
 | ||
| Scheme 38 Thiolation of indoles, pyrroles and enamines with sulfinates. | ||
Subsequently, Liu and co-workers described the efficient and metal-free direct C-3 sulfenylation and sulfinylation of indoles with sodium trifluoromethanesulfinate under mild conditions. The versatile reactivity of sodium trifluoromethane-sulfinate was observed by using different phosphorus reagents, such as phosphorus trichloride (PCl3) and phosphorus oxychloride (POCl3) in DMF at room temperature (Table 8).87 By using PCl3 reagent the trifluoromethylthiolation of indole derivatives gave 3-trifluoromethylthiolated indoles in moderate to good yields, whereas the corresponding sulfoxides were delivered in good to high yields in the presence of phosphorus oxychloride. Moreover, this protocol was extended to other sodium perfluoroalkanesulfinates to obtain the corresponding products in high yields. Similar to Zhang's work, 19F NMR revealed that CF3SCl and CF3SOCl were key intermediates in trifluoro-methylthiolation and trifluoromethylsulfinylation, respectively.
|
|||
|---|---|---|---|
| Thioethers | With PCl3 | Sulfoxides | With POCl3 |
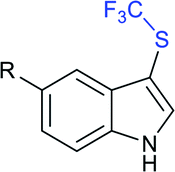 |
R = H: 79% |  |
R = H: 83% |
| R = Cl: 75% | R = Cl: 87% | ||
| R = Me: 82% | R = Me: 81% | ||
| R = NO2: 68% | R = OMe: 94% | ||
| R = OMe: 81% | R = CHO: 80% | ||
| R = CHO: 57% | |||
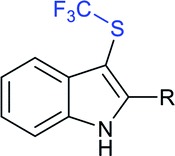 |
R = Me: 66% |  |
R = Me: 87% |
| R = Ph: 78% | R = Ph: 63% | ||
 |
69% |  |
RF = C4F9: 81% |
| RF = CF2CF2Cl: 77% | |||
 |
41% |  |
80% |

In 2017, Zhang and co-workers88 further extended the metal-free approach for the direct sulfenylation of various electron-rich arenes and heteroarenes with sodium fluoroalkylsulfinates (HCF2SO2Na, CF3SO2Na, or RFSO2Na). Significantly diverse substrates such as indoles, pyrroles, azaindoles and electron-rich arenes formed the desired fluoroalkyl thioethers in good to high yields (Scheme 39). The protocol was also further expanded to difluoromethylthiolation, trifluoromethylthiolation and perfluoroalkylthiolation using the corresponding sodium sulfinates. For the mechanism, the fluoroalkyl sulfinate salts are expected to reduce sulfinates under the influence of (EtO)2P(O)H and TMSCl and generate related actual species HCF2S+, CF3S+, or RFS+ for electrophilic thiolation reactions.
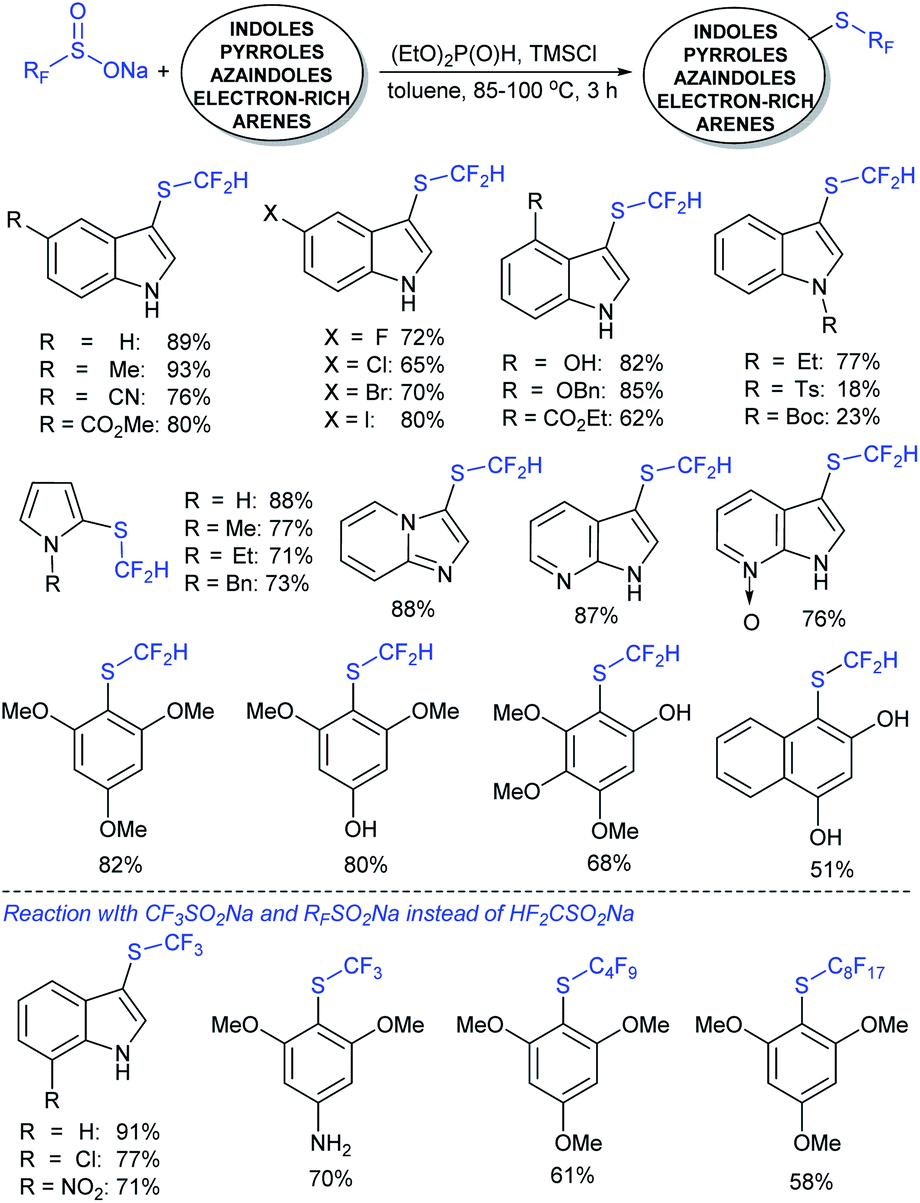 | ||
| Scheme 39 Trifluoromethylthiolation and trifluoromethylsulfinylation reactions using sodium fluoromethanesulfinates. | ||
A combined experimental/theoretical investigation on the glucosamine-promoted regioselective C3-sulfenylation of indoles under the catalytic influence of Cu(OAc)2 and NH4I as an additive (Scheme 40).89 Zhou and co-workers have utilized various indoles bearing methyl groups at the C4, C5, C6, and C7 positions with sodium benzenesulfinate to afford the desired 3-indole thioethers in 73–93% yields; however, the C2 sulfenylated product was not detected under the same conditions. Subsequently, the various para-substituted (4-Me/F/Cl) arylsulfinates were smoothly reacted with indole to generate the expected thioether products in high yields, whereas there was no reaction using sodium methanesulfinate under standard reaction conditions.
 | ||
| Scheme 40 Cu(OAc)2-glucosamine-promoted regioselective C3 sulfenylation of indoles with sodium arylsulfinates. | ||
In 2018, the Chen group reported the copper-catalyzed sulfenylation of indoles with sodium sulfinates in DMF to provide 3-sulfenyl-indoles in good to high yields (Scheme 41).90 A variety of substituted indoles (–OMe, –F, –Cl, –Br, –CO2Me and NO2), as well as N-methylindole and 2-methylindole were readily sulfenylated with different sodium arylsulfinates under the same reaction conditions.
 | ||
| Scheme 41 Cu(OAc)2-catalyzed sulfenylation of indoles with sodium sulfinates. | ||
The iodine-Ph3P-mediated mono- and bis-sulfenylation of indoles with sodium sulfinates in water was described by Xie and co-workers (Scheme 42).91 The reaction of various substituted indoles with sodium arylsulfinates afforded monosulfenylated indoles in moderate to excellent yields. The extensive investigations of the synthesis of 3-sulfenylindoles were realized, but the synthesis of 2,3-bis-sulfenylindoles was limited. Gratifyingly, the double C–H thiolation of indoles at the 2- and 3-positions readily proceeded using excess sodium sulfinates and provided the desired 2,3-disulfenylindoles in moderate to high yields. Overall, this protocol displays broad substrate scope and several functional groups, such as methoxyl, fluoro, ester, ketone, and amide groups on indole rings that are well tolerated under metal-free conditions.
 | ||
| Scheme 42 I2/PPh3-mediated mono- and bis-sulfenylation of indoles with sodium arylsulfinates. | ||
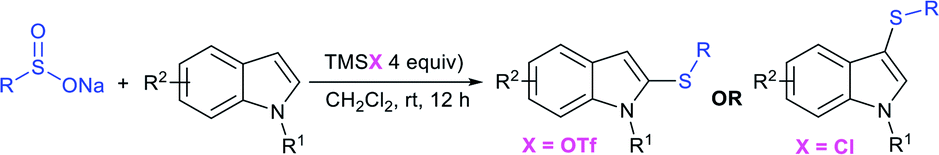
Very recently, the Wu group recognized that the switchable C–H thiolation of indoles with sodium arylsulfinates provided for the synthesis of both C2- and C3-sulfenylindoles (Table 9).92 TMSCl-mediated a highly regioselective approach and the exclusive C3-thiolation of indoles with sodium arylsulfinates was achieved. In contrast, instead of TMSCl, the use of TMSOTf realized the regiospecific C2–H thiolation of indoles with the same set of substrates. Both the reactivity and regioselectivity swapping of the counteranions of TMS from triflate to chloride led to a regioselective shift between the C2– and C3–H thiolation of indoles. To demonstrate the practicability of the C3–H thiolation reaction, 5-bromo-3-((3,4,5-trimethoxy-phenyl)thio)-1H-indole (anticancer agent) was used under the standard conditions.
|
|||
|---|---|---|---|
| C3-thioethers | TMSOTf yield | C2-thioethers | TMSCl yield |
 |
X = H: 92% |  |
R = H: 78% |
| X = F: 86% | R = Br: 67% | ||
| X = Br: 73% | R = OMe: 53% | ||
| X = Cl: 86% | |||
 |
32% (anticancer) |  |
R = F: 53% |
| R = tBu: 87% | |||
| R = OMe: 93% | |||
The Liu and Yang groups independently reported an efficient vicinal bifunctionalization of indoles with sodium sulfinates under metal-free conditions. The vicinal halo-trifluoromethyl-thiolation of indoles with sodium trifluoromethanesulfinate occurred under the influence of phosphorus oxyhalide (Table 10A).93 The protocol could readily be extended to substituted indoles, and other sodium perfluoroalkanesulfinates provided 2-halo-3-sulfenylindoles. Later, the triphosgene-mediated chloro-alkylthiolation of indoles with sodium methane-sulfinate and sodium cyclopropanesulfinate gave the corresponding 2-chloro-3-sulfenylindoles in good to high yields (Table 10B).94 Surprisingly, only mono-functionalization, that is trifluoro-methylsulfenylation of indoles, was observed, where the use of CF3SO2Na instead of CH3SO2Na afforded the corresponding 3-trifluoromethylsulfenyl-indoles in moderate to good yields. The phenomenon explained that the highly active electrophilic intermediates, CH3SCl and CF3SOCl, were responsible for the contrast reactivity.
|
|||
|---|---|---|---|
| Thioethers | Method: A yield | Thioethers | Method: B yield |
 |
R = H: 75% |  |
R = H: 83% |
| R = Me: 78% | R = Me: 85% | ||
| R = OMe: 81% | R = OMe: 87% | ||
 |
R = Me: 83% | 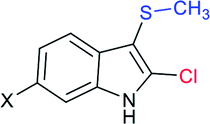 |
X = F: 63% |
| R = Et: 79% | X = Cl: 75% | ||
| R = Bn: 68% | X = Br: 62% | ||
 |
RF= C4F9: 60% | 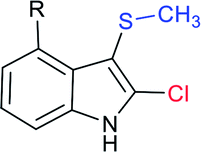 |
R = CN: 38% |
| RF = C6F13:53% |
R = Me: 51% | ||
| RF = (CF2)2Cl: 67% | R = OMe: 52% | ||
 |
R = Me: 41% |  |
R = Br: 68% |
| R = Et: 39% | R = OMe: 60% | ||
| R = Bn: 26% | |||
An efficient iodine-triphenylphosphine-mediated direct thiolation of imidazo[1,2-a]pyridines with sodium sulfinates in DMF at 80 °C was developed by Ge, Li and co-workers (Scheme 43A).95 A wide variety of 2-substituted imidazo[1,2-a]pyridines were explored with a number of sodium aryl and alkylsulfinates to provide a diverse range of 3-imidazo[1,2-a]pyridine-derived thioethers in moderate to high (43–99%) yields. Other classes of fused imidazoheterocycles, such as 6-phenylimidazo[2,1-b]thiazole, 2-phenylbenzo[d]imidazo[2,1-b]thiazole and 2-phenylimidazo[1,2-a]pyrimidine were also suitable substrates in this sulfenylation process to give the desired thioethers in high yields. It is worth noting that the unsubstituted imidazo[1,2-a]pyridine favors providing the only C-3 sulfenylated product, and no C-2 sulfenylated product was observed. In 2018, Guo et al. reported a similar iodine-triphenylphosphine-mediated direct sulfenylation of imidazo[1,2-a]pyridines with sodium sulfinates in DCM at 100 °C instead of DMF (Scheme 43B).96 A broad range of imidazo[1,2-a]pyridine-derived thioethers were obtained in good to high yields by using imidazo[1,2-a]pyridine bearing substituents on the C6, C7 and C8 positions with a variety of aromatic and aliphatic sulfinates. Although the electronic property tendency is less significant, pyridine-3-sulfinate provided the desired product in only 18% yield. Both methods involved generating sulfenyl iodide (RSI) species from sodium sulfinates, which were reduced using I2/Ph3P to attack the C-3 position of imidazo[1,2-a]pyridines.
 | ||
| Scheme 43 The I2-PPh3-mediated thiolation of fused imidazo-heterocycles. | ||
Zhou and co-workers97 utilized a cheap ammonium iodide for the regioselective sulfenylation of chromones (flavones), indole, and arylimidazo[1,2-a]pyridines with sodium arenesulfinates, as shown in Scheme 44. A wide variety of corresponding thioethers were generated via the transition metal-free direct C–H sulfenylation in good to high yields. Control experiments proved the regioselective sulfenylation using substituents at the α- and β-positions of flavones. The α-methyl flavone did not give the desired product; however, the methyl group at the β-position of flavone provided a significantly low yield of the corresponding thioether, possibly due to the steric effects from the adjacent methyl group. A plausible mechanism involves the generation of the electrophilic ArS–I, which reacted with flavone, indole, and aryl-imidazo[1,2-a]pyridine to obtain the desired thioethers.
 | ||
| Scheme 44 Sulfenylation of imidazo[1,2-a]pyridines and chromones using sulfinates. | ||
 | ||
| Scheme 45 I2-mediated thiolation of 2-naphthols and N,N-dimethyl aniline with sulfinates. | ||
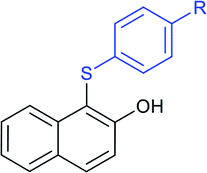
Later, the Deng group successfully employed a metal-free and environmentally friendly protocol to synthesize aryl sulfides and sulfones under aqueous conditions. Iodine promoted the coupling of various substituted 2-naphthols with a series of aromatic and aliphatic sodium sulfinates to provide the desired thioethers in good to high yields (Table 11).99 Additionally, 2-methoxy/2-ethoxy-naphthalenes, phenol derivatives, methoxy-benzenes, and N,N-dimethylaniline were well reacted with sodium benzenesulfinate to afford the desired aryl sulfides in good to high yields. The iodine and formic acid reagent system reduced RSO2Na to R-SI as an electrophilic species for the sulfenylation pathway. Alternatively, sodium sulfinates react with iodine by using K3PO4 to generate sulfonyl iodide (RSO2I), which readily promotes the sulfonylation process.
|
|||
|---|---|---|---|
| Thioethers | Method-A | Sulfones | Method-B |
 |
R = H: 80% |  |
R = H: 82% |
| R = F: 70% | R = F: 73% | ||
| R = Br: 75% | R = Br: 77% | ||
| R = Cl: 75% | R = Cl: 75% | ||
| R = Me: 89% | R = Me: 84% | ||
| R = CF3: 71% | R = CF3: 67% | ||
| R = OMe: 85% | R = OMe: 72% | ||
| R = OCF3: 66% | R = OCF3: 70% | ||
 |
R = Me: 60% | 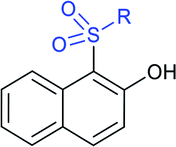 |
R = Me: 68% |
| R = n-Pr: 80% | R = n-Pr: 45% | ||

Similarly, Lu and coworkers100 described the reaction of electron-rich arenes with sodium arylsulfinates in the presence of HCl/[Hmim]Br (heaxylmethyl-imidazolium bromide) for the synthesis of a range of diaryl sulfides under acidic conditions (Scheme 46). The scope and generality of the reaction studied concerning sodium arylsulfinates with 2-naphthol derivatives. Various electron-rich arenes such as naphthalen-2-ol or 2-methoxynaphthalene, electron-donating aromatic amines, arenol (estrone) and heteroarenes also worked under similar conditions. Only a trace amount of disulfide from sodium p-toluenesulfinate was detected in the absence of HCl, suggesting that the acid plays a crucial role in the reduction process. The ionic liquid [Hmim]Br was reused and recycled ten times without any deviation in the outcome.
 | ||
| Scheme 46 HCl/[Hmim]Br-mediated thiolation of electron-rich arenes with arylsulfinates. | ||
Lu, Yi and co-workers employed the regioselective sulfenylation of aromatic amines, arenols and ketones with sodium sulfinates under the influence of I2 and PPh3 in aqueous media (Scheme 47).101 A series of anilines and phenols underwent sulfenylation with various arenesulfinates to furnish the diaryl thioethers in moderate to good yields. Subsequently, the C–H bond sulfenylation of ketones and α,β-unsaturated ketones was also explored with sodium arylsulfinates to afford the desired products in good to high yields. The complex molecules like estrone and progesterone were also coupled; interestingly, the progesterone undergoes di-sulfenylation under identical conditions. The authors also designed and investigated the radical trapping experiments and studies of 1H, 13C NMR and EPR, suggesting that the transformations might involve a radical process.
 | ||
| Scheme 47 I2-Ph3P-mediated thiolation of aromatic amines, arenols and ketones with sulfinates. | ||
:1 ratio) was observed in most of the cases. The reaction with an aliphatic alkyne, oct-4-yne, afforded the desired product in 20% yield with 1:1 regioisomers. The authors insisted that the exact reaction mechanism is not precise for the reaction process.
 | ||
| Scheme 48 Fe-catalyzed sulfenylation and arylation of alkynes with sodium arylsulfinates. | ||
Yi and co-workers103 developed a simple method for β-iodoalkenyl sulfides via the vicinal functionalization of alkynes with sodium arenesulfinates. Iodine-induced the regio- and stereoselective construction of C–S bonds in a one-pot operation with high functional group compatibility. A series of terminal alkynes and different kinds of sodium arenesulfinates were explored in the sulfenylation to afford numerous β-iodoalkenyl sulfides in good to high yields (Scheme 49). Disappointingly, the sodium alkylsulfinates were ineffective for this transformation. The protocol was also extended to internal alkynes to form the (E)-β-iodoalkenyl sulfides in satisfactory yields. The iodoalkenyl sulfides readily reacted with PhB(OH)2 and phenylacetylene via the Suzuki and Sonogashira coupling reactions, respectively. The reaction was completely inhibited under the influence of TEMPO, indicating that the reactions proceeded through a radical pathway. The reduction of ArSO2Na using I2–Ph3P leads to the arenesulfenyl iodide, and subsequent homolytic cleavage yields a sulfenyl radical, which is a crucial precursor for the mechanistic aspects.
 | ||
| Scheme 49 I2-induced synthesis of β-iodoalkenyl sulfides. | ||
The Lu and Yi group demonstrated the H2SO4-Ph3P-promoted hydrothiolation of alkynes, alkenes, and H-phosphine oxides with sodium arylsulfinates. A range of terminal aryl/alkyl-ethynes and -alkenes participated in hydrothiolation with various sodium arylsulfinates and gave the corresponding thioethers in moderate to excellent yields (Scheme 50).104 However, most electron-deficient alkenes were unsuccessful in the standard reaction conditions. Interestingly, the phosphonothioates were also easily generated in acceptable yields in the reaction between H-phosphine oxides and sodium aryl/alkylsulfinates. The authors performed radical-trap control experiments, and EPR results confirmed the in situ generated arylsulfenyl radical for the mechanistic pathway.
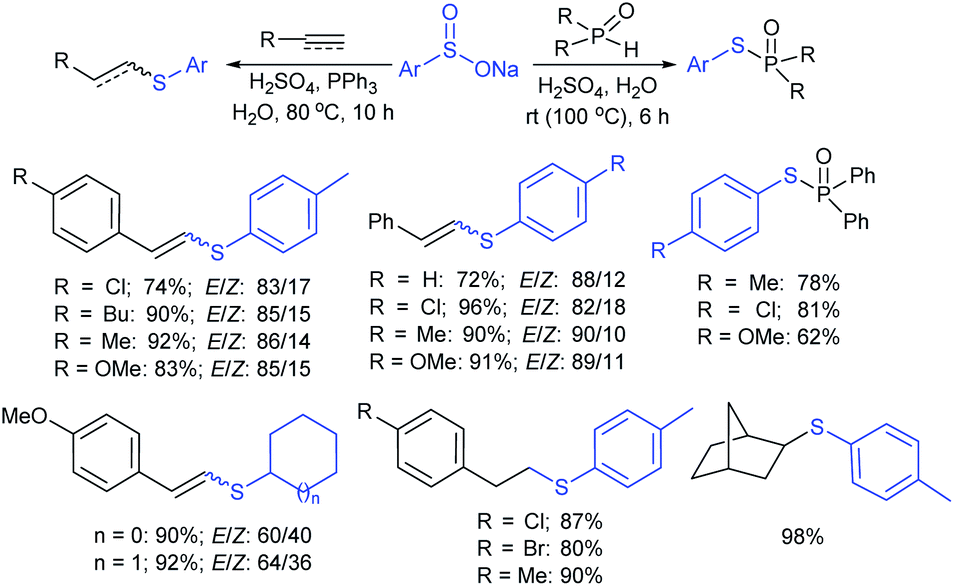 | ||
| Scheme 50 H2SO4-Ph3P-induced thiolation of alkynes, alkenes and H-phosphine oxides with sodium arylsulfinates. | ||
Wu and co-workers established a new and efficient copper-catalyzed ring-closing-oxysulfenylation of enolates with sodium sulfinates to synthesize sulfenylated cyclic ethers (Scheme 51).105 Various aromatic and aliphatic sodium sulfinate substrates were reacted with pent-4-en-1-ol to afford a broad range of arylsulfenylated tetrahydrofurans in good to high yields. Furthermore, this transformation extended to various enol/enolates for the construction of successive C–O and C–S bonds to provide four to seven-membered cyclic thioethers in good to high yields. Moreover, the oxysulfenylation of 2-allylphenol with different sodium sulfinates gives the corresponding 2,3-dihydrobenzofuran thioether products in 80–88% yields. The control experiments were performed using TEMPO radical scavenger and the reaction was fully inhibited. As a result, the reaction mechanism likely proceeded as a radical pathway for this oxysulfenylation protocol.
 | ||
| Scheme 51 Cu-catalyzed oxysulfenylation of enols with sodium sulfinates. | ||
Jiang and co-workers106 successfully recognized the iodine-catalyzed cascade cyclization/sulfenylation of alkynoates and alkynamides with sodium arylsulfinates for the assembly of 3-sulfenylcoumarin and 3-sulfenylquinolinone derivatives (Scheme 52). Various sodium arylsulfinates bearing electron-donating groups, as compared with weak electron-withdrawing groups, reacted with phenyl 3-phenylpropiolate to give the corresponding 3-sulfenylated coumarin derivatives in moderate to good yields. The strong electron-withdrawing substituted benzene-sulfinates, pyridine-4-sulfinate, and ethanesulfinate, were challenging substrates for this transformation. On the other hand, representative classes of alkynoate derivatives were explored, and various substituents on the phenyl ring of alkynoates as well as multi-substituted aryl and alkyl alkynoates were compatible, thus giving the expected products in variable yields. In contrast, the 4-methoxyphenyl-3-phenylpropiolate undergoes a distinctive dearomatization-sulfenylation under the same conditions, providing the spiro-compound in reasonable yield. Various N-alkyl arylpropiolamides were further utilized for cascade cyclization/sulfenylation using arylsulfinates to furnish 3-sulfenylquinolinone derivatives in 66–83% yields. The free N–H arylpropiolamide was also suitable, albeit the cyclization product was obtained in low yield. The control experiments revealed three key factors: (a) the desired product was detected only in trace amounts in the absence of DMSO, which acted as the oxidant; (b) the disulfide could be a possible intermediate; (c) there was no effect with the use of the radical scavenger TEMPO. From this observation, the protocol should not be a radical pathway.
 | ||
| Scheme 52 The I2-catalyzed cascade cyclization/sulfenylation of alkynoates and alkynamides with sodium arylsulfinates. | ||
Very recently, Xie and co-workers conveniently constructed 2-sulfenyl-indenones using the iodine–Ph3P-mediated one-pot cascade reaction of propargyl alcohols and sodium sulfinates (Scheme 53).107 The protocol proceeded through a metal-free cascade Meyer–Schuster rearrangement/radical addition/oxidative C–H cyclization reaction. The reaction scope is reasonably general, involving a wide range of propargyl alcohols that reacted smoothly with different sodium sulfinates and obtained a series of 2-sulfenylindenones in moderate to good yields. Moreover, sodium methanesulfinate also underwent this reaction to give the corresponding thioether in 50% yield.
 | ||
| Scheme 53 I2-catalyzed cascade cyclization/sulfenylation of propargyl alcohols with sodium sulfinates. | ||
A plausible mechanism was proposed for this new transformation, as shown in Scheme 54. Initially, the sodium sulfinate was reduced using I2/PPh3 to form the corresponding aryl disulfides, followed by homolytic cleavage to generate the ArS˙ free radical. The propargyl alcohol is expected to undergo the Meyer–Schuster rearrangement to generate the allenol-type intermediate A, and the subsequent selective attack of ArS˙ to afford the allylic radical intermediate B. The intramolecular radical cyclization followed by keto–enol tautomerism led to the formation of C, then the single electron transfer under the influence of K2S2O8. Finally, the deprotonation of C delivered the dihydroindenone D and the oxidation of D produced the corresponding 2-sulfenylindenone derivatives.
 | ||
| Scheme 54 Mechanism for the formation of 2-sulfenylindenone derivatives. | ||
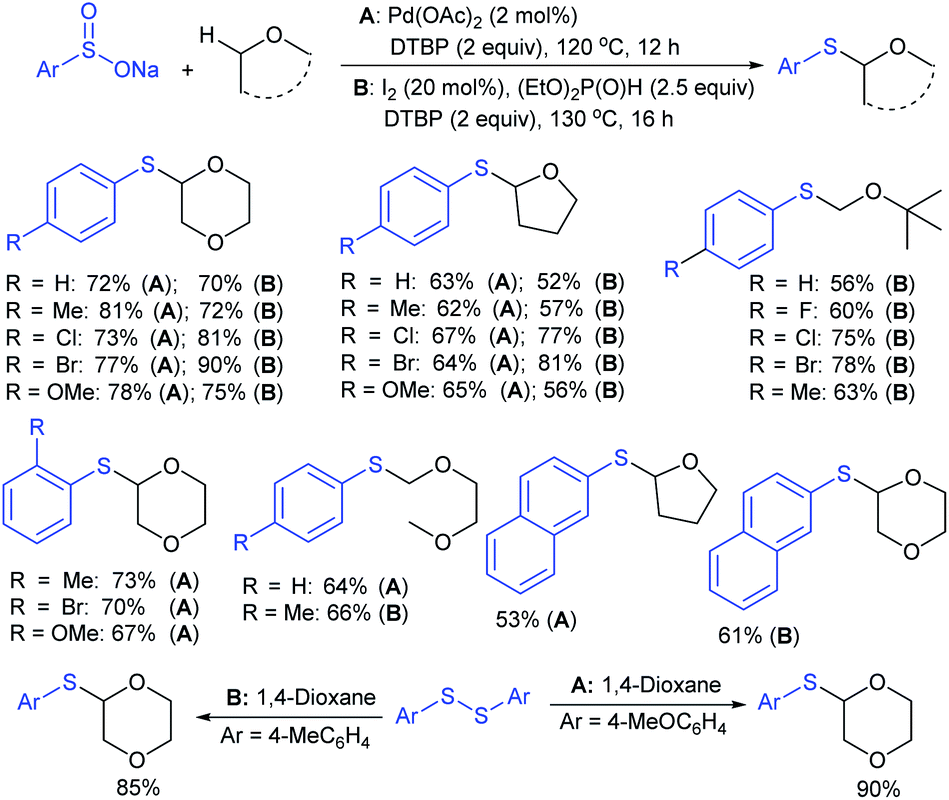 | ||
| Scheme 55 Oxidative arylthiolation of ethers with sodium sulfinates. | ||
Deng and co-workers reported the I2-catalyzed sulfenylation-dehydrogenation of cyclohexanones (as a reliable phenol source) with sodium sulfinates in the presence of diethyl phosphite (Scheme 56).110 A series of 2-sulfenylphenols were obtained in good to high yields using different classes of cyclohexanones bearing 4-alkyl or aryl substituents smoothly coupled with sodium p-toluenesulfinate. Moreover, various substituted arylsulfinates reacted with cyclohexanone to afford the corresponding 2-arylsulfonylphenols in satisfactory yields. Disappointingly, aliphatic sulfinates, for instance, methanesulfinate and ethanesulfinate were not suitable for this transformation. The roles of diethyl phosphite and I2 were explained under mechanistic investigation, which generated an electrophilic species R-SI from the corresponding sodium sulfinates.
 | ||
| Scheme 56 I2-catalyzed sulfenylation-dehydrogenation of cyclohexanones with sodium sulfinates. | ||
3.4. Synthesis of sulfones (R–SO2–R1)
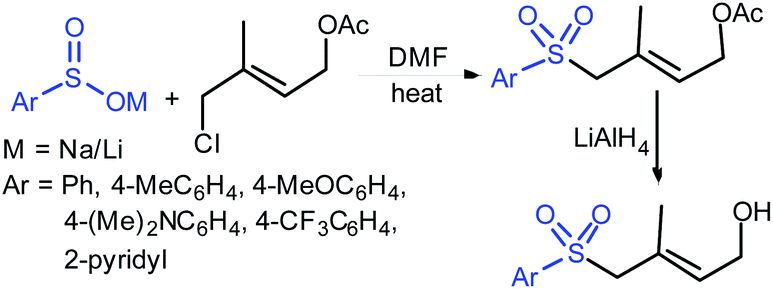 | ||
| Scheme 57 Synthesis of trans-acetoxy sulfones. | ||
In 1986, Hiroi and Makino112 reported the Pd-catalyzed asymmetric allylic sulfonylation of allylic acetate with sodium p-toluenesulfinate using the chiral bisphosphine ligand to form the corresponding allylic sulfone in 73% yield and 88% ee. Later, in 1995, Gais and co-workers113 also similarly reported that the palladium-catalyzed asymmetric sulfonylation of allylic acetate or allyl chloride in the chiral phosphino-oxazoline ligand (16) gave the allylic sulfones. The highly regioselective sulfonylation of racemic diphenyl-substituted allylic substrates under the catalytic influence of the oxazoline-based ligand (16) and ent-16, gave enantiomerically pure allylic sulfones, as shown by representative examples in Scheme 58.
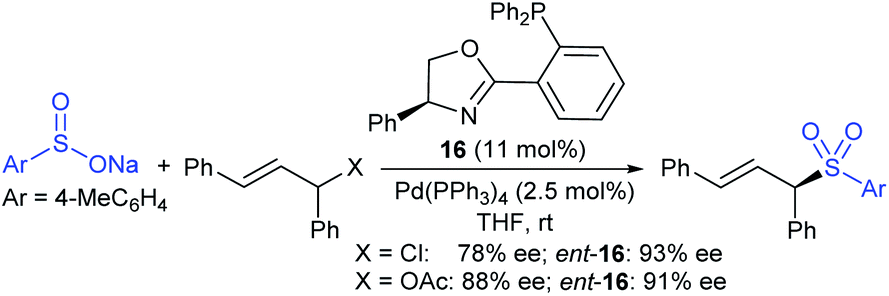 | ||
| Scheme 58 Pd-catalyzed asymmetric sulfonylation of allylic substrates. | ||
Later, Gais and co-workers investigated the palladium-catalyzed kinetic resolution of racemic cyclic and allylic carbonates with sulfinate salts in the presence of N,N-(1R,2R)-1,2-cyclo-hexanediyl-bis[2-(diphenylphosphino)benzamide] (BPA-17) as a chiral ligand (Scheme 59).114 For instance, the racemic cyclohexenyl carbonate was resolved with sodium phenylsulfinate and sodium p-tolylsulfinate as nucleophiles in Hex4NBr (THAB). Interestingly, the corresponding allyl sulfones and unreacted carbonates were obtained in excellent enantioselectivities with moderate to good yields.
 | ||
| Scheme 59 Pd-catalyzed kinetic resolution of cyclohexenyl carbonates with aromatic sulfinates. | ||
Chandrasekar et al. described the synthesis of allyl phenyl sulfones from the Pd(OAc)2/Ph3P-catalyzed sulfenylation of allylic alcohols with sodium benzenesulfinate in the presence of Et3B. Varied allylic alcohols, including aryl, alkyl and conjugated dienols are equally useful in this transformation to obtain the desired sulfones with good to high yields (Scheme 60A).115 At the same time, Felpin and Landais reported using inexpensive Pd/C-catalyst for the sulfonylation of allylic acetates with sodium p-toluenesulfinate in water (Scheme 60B).116 The linear allylic acetate reacted smoothly with only 1 mol% of Pd/C; however, the branched allylic acetates required 5 mol% of Pd/C in the presence of Ph3P. More interestingly, the chiral allylic acetate (18) was successfully sulfonylated with sodium p-toluenesulfinate and provided the desired linear allyl sulfone (19) along with branched sulfone (20) as shown in Scheme 60.
 | ||
| Scheme 60 Synthesis of allyl phenyl sulfones from the allylic alcohols. | ||
An amphiphilic polystyrene-poly(ethyleneglycol) (PS-PEG) resin-supported phosphine–palladium complex (21)-catalyst served efficiently for the π-allylic substitution of allyl carbonates with sodium sulfinates in water (Scheme 61).117 A range of primary, secondary, and tertiary acyclic allylic carbonates and cyclic allylic carbonates were smoothly sulfonylated with sodium benzenesulfinate under the catalytic influence of Pd-PS-PEG resin in a heterogeneous reaction medium to obtain the desired allyl sulfones in good to high yields.
 | ||
| Scheme 61 The PS-PEG resin-supported Pd-catalyzed π-allylic sulfonylation of allyl carbonates with sodium benzenesulfinate. | ||
Chang et al. presented the ZnI2-promoted allylic sulfonylation of α-methylstyrenes with sodium sulfinates in MeCN to form α-sulfonylmethyl-styrenes (Scheme 62).118 A range of substituted α-methylstyrenes, including the electron-donating/neutral/withdrawing groups on benzene, did not affect the distributed yields. Further, the α-sulfonylation of methylstyrene using various aryl and alkyl sulfinates readily produced various α-sulfonyl methylstyrenes in good to high yields. Although there was no apparent electronic effect using varied sulfinates, Langlois' reagent (CF3SO2Na) was not compatible with this transformation. The oxygenated aryl α-methylstyrenes undergo the self-dimerization rather the than sulfonylation route due to a strong electron-rich group that readily stabilizes the intermediate with the tertiary carbocation under the influence of ZnI2 reaction conditions.
 | ||
| Scheme 62 ZnI2-mediated allylic sulfonylation of α-methyl styrenes with sodium sulfinates. | ||
Jegelka and Plietker have described the iron-catalyzed highly regioselective allylic sulfonation of allylic carbonates under the influence of the phosphane ligand (Scheme 63).119 A range of arenesulfinates were successfully allylated to give the corresponding allylic sulfones in good to excellent yields with a high level of regioselectivites. The yields and regioselectivities were significantly dropped for the ipso-sulfonylation of 2-substituted arylsulfinates. The multi-functionalized allylic carbonates were also successfully transformed into their corresponding sulfones in good to excellent regioselectivities and isolated yields. More interestingly, the enantiomerically pure allylic carbonates, for example (R)-linalool-derived carbonate (97:3 er), were also examined under the same sulfonylation conditions and yielded the desired sulfone with 96:4 enantiomeric ratio, retaining the same configuration. The tertiary carbonates are prominently limited in this protocol.
 | ||
| Scheme 63 Iron-catalyzed ipso-sulfonation of allyl carbonates with aromatic sulfinates. | ||
Tian and co-workers120 elegantly employed for the first time an efficient direct α-selective substitution of primary allylic amines with sodium sulfinate salts with excellent regio- and stereoselectivities. Here, 0.1 mol% [Pd(allyl)Cl]2, 0.4 mol% dppb (1,4-bis(diphenyl-phosphino)butane) and excess boric acid were used for the deamino-sulfonylation of various α-unbranched primary allylic amines (substituents aryl, heteroaryl, alkenyl, or alkyl groups at β- and γ-positions) with diverse sodium sulfinates to provide structurally varied allylic sulfones in good to excellent yields with exclusive E-selectivity (Scheme 64). Subsequently, the unsymmetric α-chiral primary allylic amines were successfully transformed into the corresponding allylic sulfones, in good to excellent yields with high level of configuration retention, by replacing dppb with chiral BINOL. In particular, varied unsymmetric α-chiral primary allylic amines were used to provide α-chiral allylic sulfones with high optical purity. In contrast, symmetric α-chiral primary allylic amines resulted in the product as a racemic mixture with high yield. The observed racemic mixture was explained through the putative π-allylpalladium intermediate for symmetrical substrates.
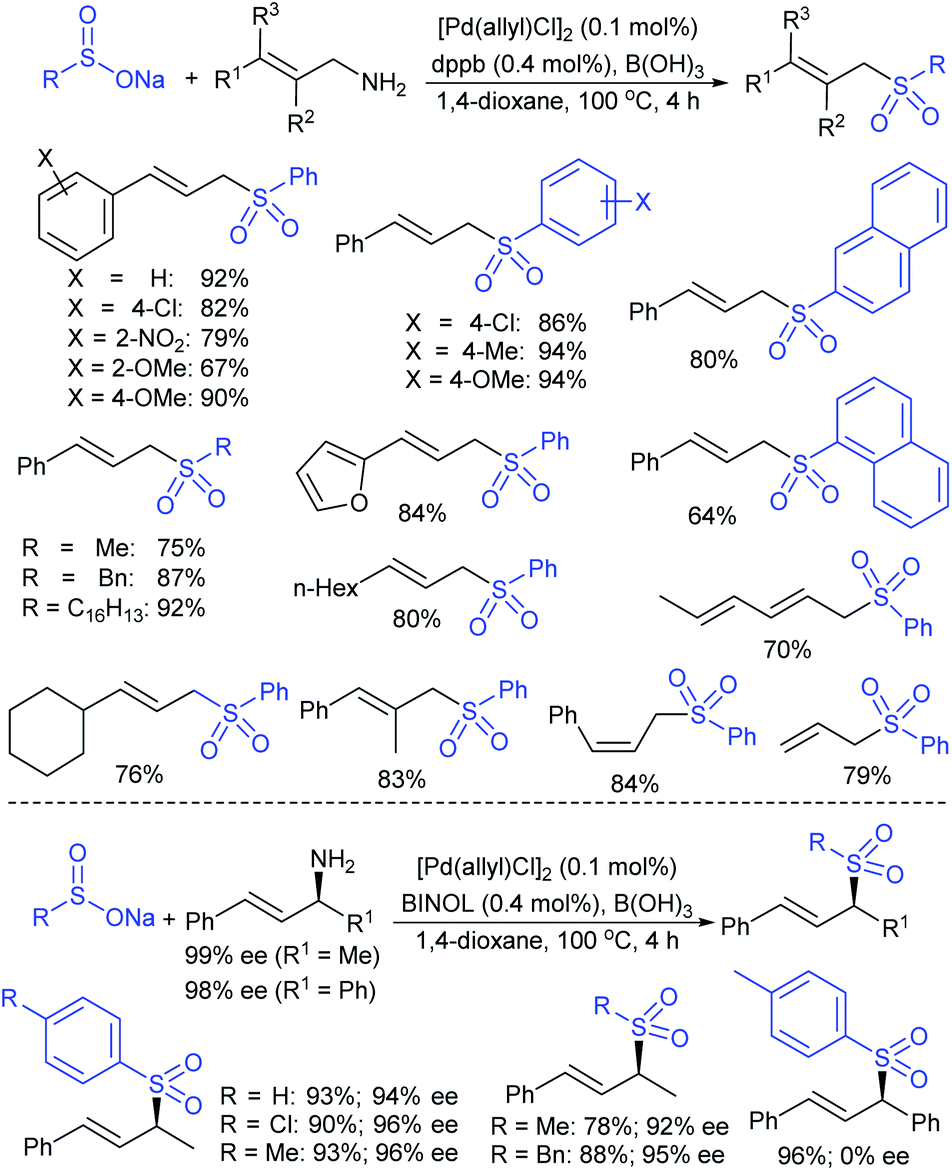 | ||
| Scheme 64 Direct substitution of α-unbranched primary allylic amines with sodium sulfinates. | ||
Gu, Tian and co-workers121 devised the direct stereospecific substitution reaction of enantioenriched allylic alcohols with sodium sulfinates in palladium diacetate/racemic 2,2′-BINAP (5 mol%; 1:1) as a catalytic system (Scheme 65). A series of aryl/heteroaryl/alkyl sulfinates were reacted with unsymmetrical enantioenriched allylic alcohol to provide the corresponding α-chiral allylic sulfones in moderate to excellent yields. Interestingly, these sulfone products were obtained with the same level of retention of configuration and alkene geometry. A variety of substitutions at the α- and γ-positions of enantioenriched allylic alcohols bearing aryl-, heteroaryl-, and alkyl smoothly underwent a selective reaction with sodium benzenesulfinate to give structurally diverse α-chiral allylic sulfones in satisfactory yields. Typically, this was an allylic substitution reaction and the regioselectivity was determined by the steric and electronic properties of the α- and γ-substituents in the allylic alcohols. Based on the control experiments, the stereospecific substitution of a regioisomer of allylic alcohol (90% ee) with sodium benzenesulfinate proceeded in a γ-selective fashion to afford the corresponding allyl sulfone with identical retention of configuration and exclusive E-selectivity. Moreover, the symmetrical enantioenriched allylic alcohol employed under the standard reaction conditions led to a racemic allylic sulfone. Of note, the chirality transfer was not valid due to the symmetry of the resulting π-allylpalladium intermediate.
 | ||
| Scheme 65 Stereospecific substitution of enantioenriched allylic alcohols with sodium sulfinates. | ||
Ueda and Hartwig described the highly regio- and enantioselective iridium-complex (22)-catalyzed allylation of allyl carbonates with sodium sulfinates to obtain branched sulfonylated products in high yields (Scheme 66).122 Various aromatic and aliphatic allylic carbonates were successfully explored to afford allyl sulfones in nearly complete regioselectivities and high enantioselectivities. Besides, the allyl sulfonylation using aromatic sulfinates having more electron-rich groups occurred in higher yield than those of the electron-neutral/poor arylsulfinates with somewhat lower regio- and enantioselectivity. Additionally, less nucleophilic aliphatic sulfinates were required in excess amounts and were compatible with p-methoxy-substituted cinnamyl carbonate to furnish the desired allyl sulfones with exceptionally high regioselectivity, high yield, and high enantioselectivity.
 | ||
| Scheme 66 Ir-catalyzed enantioselective sulfonation of allyl carbonates with sulfinates. | ||
Recently, Cai and Kleij elegantly revealed a general palladium-catalyzed regio- and enantioselective allylic substitution of racemic tertiary allylic carbonates with sodium sulfinates under phosphoramidite ligand (23) providing chiral α,α-allylic-disubstituted allylic sulfones (Scheme 67).123 Various ortho-, meta-, and para-substituted benzylsulfinates and other functionalized alkylsulfinates showed significant reactivity in the allylic sulfonylation and provided branched allylic sulfone products in good to high yields with high er values. Next, a series of aryl and heteroaryl sulfinates were successfully employed in the slightly modified conditions providing the desired chiral sulfone scaffolds in good yields and considerable regio- and enantio-selectivities. A wide range of allylic carbonates were treated with sodium benzylsulfinate to access α,α-disubstituted allylic sulfones in moderate to good yields with high levels of regio- and enantioselectivities. The asymmetric allylic sulfonylation successfully extended to the synthesis of the valuable acetoxy allylic sulfone precursor in 62% yield (82:12 er), which was transformed into antifungal and antimicrobial active (+)-agelasidine A in a single operation.
 | ||
| Scheme 67 Pd-catalyzed regio- and enantioselective allylic substitution of racemic tertiary allylic carbonates with sodium sulfinates. | ||
The Kleij group further elaborated on the Cu(OTf)2/chiral bisoxazolines (BOX-24)-catalyzed asymmetric propargylic sulfonylation of propargylic cyclic carbonates with sodium sulfinates to generate propargylic sulfones containing elusive quaternary stereocenters (Scheme 68).124 A wide range of propargylic cyclic carbonates with different steric and electronic effects on the aryl substituent were used and they afforded the desired propargylic sulfones with good to high yields and excellent enantiomeric purities. Bulky 2-naphthyl and heteroaryl-derived substrates also readily participated; however, aliphatic cyclic carbonates were unsuccessful even at higher temperatures and/or catalyst loadings. A variety of ortho-, meta-, and para-substitutions-derived arylsulfinates, including heteroaryl and alkyl sulfinates, are also suitable coupling partners with propargylic cyclic carbonates to give high levels of asymmetric induction in the corresponding sulfones.
 | ||
| Scheme 68 The Cu-catalyzed asymmetric sulfonylation of propargylic cyclic carbonates with sodium sulfinates. | ||
The mechanism is depicted in Scheme 69. The in situ formation of the chiral-[Cu] complex and base-mediated deprotonation would form a copper-acetylide species A. Subsequently, decarboxylation furnished a copper-acetylide intermediate B, which is in resonance with the copper-allenylidene intermediate C. The nucleophilic sulfinate attack occurred on Cu(allenylidene) preferentially at the Re-face of the copper-acetylide species C. Finally, protodemetalation allowed the formation of the desired propargylic sulfones in the presence of HFIP, whereas the active copper catalyst was regenerated for further catalytic cycle processes.
 | ||
| Scheme 69 Mechanism for the Cu-catalyzed asymmetric sulfonylation of propargylic cyclic carbonates with sodium sulfinates. | ||
Very recently, the Khan group established another efficient palladium-catalyzed asymmetric allylic substitution (AAS) of vinyl cyclic carbonates with sodium sulfinates (Scheme 70).125 Both Pd2(dba)3·CHCl3 and a chiral diphosphine DACH-naphthyl ligand (25) were essential for promoting sulfone-bearing quaternary carbon stereocenters in high yield, excellent enantiomeric excess, and branch selectivity. A wide array of aryl and heteroaryl sulfinate salts were effectively employed with vinyl cyclic carbonate to provide the desired branched allylic sulfones in high yields with excellent enantioselectivities. Further extended primary and secondary alkyl sulfinate salts were also readily converted into tertiary allylic sulfones in good to high yields with excellent regio- and enantioselectivities. Next, a variety of vinyl cyclic carbonates bearing diverse substitution patterns, particularly functionalized longer alkyl chains were tolerated and afforded the desired sulfones in moderate to good yields with high regio- and enantioselectivities. The aromatic-substituted allylcarbonate substrates were also suitable coupling patterns in allylic sulfonylation to provide their corresponding products in moderate yields and excellent enantioselectivities. Of particular note, asymmetric allylic substitution (AAS) was also well demonstrated for the synthesis of (+)-agelasidine-A from the corresponding allylic sulfones, which involved in three simple steps.
 | ||
| Scheme 70 Pd-catalyzed regio- and enantioselective allylic substitution of vinyl cyclic carbonates with sodium sulfinates. | ||
Very recently, the Chen group achieved a palladium-catalyzed stereoselective sulfonylation of vinylethylene carbonates with sodium sulfinates for the synthesis of (Z)-allylic sulfones (Scheme 71).126 Sodium aromatic sulfinates bearing various ortho-, meta-, and para-substituted groups on the benzene ring were well tolerated and gave the corresponding allylic sulfones in good to high yields and excellent stereoselectivties. Also, heteroaromatic and aliphatic-derived sulfinates were compatible for this protocol and provided allylic alcohols in acceptable yields and stereoselectivities. The different aryl and heteroaryl-derived vinylethylene carbonates were readily assembled with sodium p-toluenesulfinate, affording desired products with good yields and excellent Z-selectivity. Even sterically hindered vinyl-ethylene carbonates were also transformed into Z-tetrasubstituted allylic sulfones and other alkyl vinylethylene carbonates gave the related products exclusively as cis-isomers in satisfactory yields.
 | ||
| Scheme 71 The Pd-catalyzed stereoselective sulfonylation of vinylethylene carbonates with sodium sulfinates. | ||
Yuan and co-workers127 generated a series of densely functionalized tri-substituted allylic sulfones in good to excellent yields (71–98%) with good to high selectivity (Z/E from 79:21 to >99:1). A range of methyl acrylate-derived Morita–Baylis–Hillman (MBH) carbonates smoothly reacted with sodium benzenesulfinate and sodium p-toluenesulfinate under catalyst-free reaction conditions (Scheme 72). One representative example, the acrylonitrile-derived MBH carbonate, was also successfully transformed into the corresponding allyl sulfone with excellent yield (99%) and selectivity (E/Z: >99:1).
 | ||
| Scheme 72 Allylic sulfonylation of Morita-Baylis–Hillman (MBH) carbonates. | ||
The Jiang group developed a catalyst-free γ-substitution reaction of the MBH carbonates of isatins with sodium sulfinates under mild reaction conditions (Scheme 73).128 This protocol features easily accessible starting materials that are a variety of MBH carbonates of isatins and sodium arylsulfinates, which allowed the rapid synthesis of 3-alkenyloxindoles in high yields (up to 96%) and Z/E-selectivities (up to >20:1). Unfortunately, the MBH carbonates of isatins possessing a 5-nitro group or derived from acrylonitrile were unsuccessful because of unwanted side reactions that became dominant under the same reaction conditions.
 | ||
| Scheme 73 Sulfonylation of MBH carbonates of isatins with sodium sulfinates. | ||
Our research group successfully prepared various nitroallylic sulfones from the corresponding nitroallylic acetates with sodium benzenesulfinate and sodium p-toluenesulfinate in high yields with retained (E)-stereoselectivity (Scheme 74).129 Subsequently, these newly prepared allyl sulfones were treated with sodium azide to form the anticipated triazolylsulfones in good to high yields.
 | ||
| Scheme 74 Synthesis of nitroallylic sulfones with sodium sulfinates. | ||
Wu, Jiang and co-workers130 described the trifluoromethane-sulfonylation (triflation) of allylic alcohols/cinnamyl type esters with F3CSO2Na under transition metal-free conditions (Scheme 75). Various primary, secondary, and tertiary allyl alcohols were successfully investigated. A range of functional groups, including electron-donating and electron-withdrawing groups on the benzene ring, were tolerated in this transformation. Additionally, the steric impact of substituents did not influence the reaction, and the corresponding allylic triflones were generated in good to excellent yields. A wide range of cinnamyl alkyl esters, cinnamyl-2-aminobenzoate, and cinnamyl cinnamate were converted into the desired allylic triflones in moderate to good yields.
 | ||
| Scheme 75 Triflylation of primary and secondary allylic alcohols with NaSO2CF3. | ||
Recently, the Pd-catalyzed ring-opening sulfonylation of gem-difluorocyclopropanes with sodium arylsulfinates was reported by Zhang and co-workers.131 The reaction involved C–C bond cleavage, β-F elimination, and allylic sulfonylation to form the desired 2-fluoroallylic sulfones with Z-selectivity (Scheme 76). Several arylsulfinates bearing electron-donating or electron-withdrawing groups were found to be compatible and offered the corresponding 2-fluoroallylic sulfones in moderate to good yields. No desired product was obtained when CF3SO2Na was a coupling partner. Various substituted gem-difluorocyclopropanes with diverse electronic or steric substituents were examined and found to give the expected sulfones in acceptable yields. Further, the pharmaceutically important esterone, clofibrate, fenofibrate, and diacetone-D-glucose-derived substrates were illustrated to provide 2-fluoroallylic sulfone scaffolds in satisfactory yields.
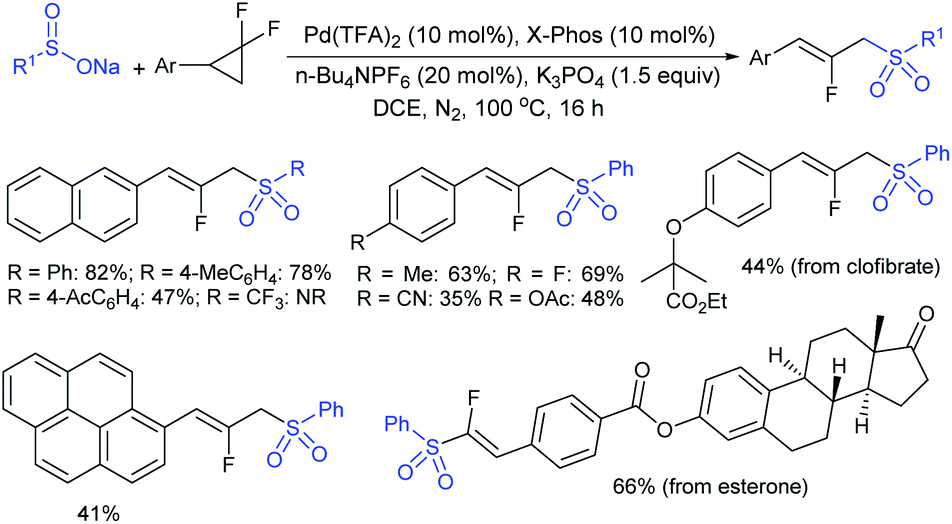 | ||
| Scheme 76 Pd-catalyzed ring-opening sulfonylation of gem-difluorocyclopropanes with sodium arylsulfinates. | ||
3.4.2.1. Nucleophilic sulfonylation. In 1997, Ochiai et al. demonstrated the double nucleophilic vinylic substitution of (Z)-(β-haloalkenyl)phenyliodonium tetrafluoroborates with sodium benzenesulfinate. The reaction involves Michael addition and the nucleophilic vinylic sulfonylation of (Z)-(β-chloro-1-alkenyl)iodonium tetrafluoroborate afforded (Z)-1,2-bis(benzenesulfonyl)alkenes in high yields with retention of the configuration (Scheme 77).132 the substitution of bromo and fluoro-derived iodonium salts, such as (Z)-(β-bromoalkenyl)iodonium tetrafluoroborates and (Z)-(2-fluoro-1-decenyl)phenyl-iodonium tetrafluoroborate with sodium benzenesulfinate afforded the (Z)- and (E)-isomers as the major product, respectively.
 | ||
| Scheme 77 Sulfonylation of (Z)-(β-haloalkenyl)iodonium salts with sodium benzenesulfinate. | ||
Yadav and co-workers133 reported a LiBr-catalyzed regio-selective synthesis of vinyl sulfones from terminal epoxides and sodium arenesulfinates using water as a reaction medium (Scheme 78). The generality of the protocol was demonstrated across a range of terminal epoxides bearing an electron-donating substituent, which afforded better yields of vinyl sulfones than that with an electron-withdrawing group. Additionally, the p-sodium toluenesulfinate gave slightly better results than the benzenesulfinate salt and reacted with 2-alkyl epoxides to produce internal (E)-vinyl sulfones with complete selectivity.
 | ||
| Scheme 78 Synthesis of vinyl sulfones via the opening of epoxides with sodium sulfinates. | ||
In 2007, an anion-functionalized ionic liquid, 1-ethyl-3-methylimidazolium-(S)-2-amino-3-methyl-butyric acid salt, [emim][Val] promoted the CuI-catalyzed coupling of either (Z)- or (E)-β-bromostyrenes with sodium sulfinates (Scheme 79A).134 At 100 °C in DMSO, different (E)-vinyl sulfones were obtained in 61–73% yields using sodium benzenesulfinate and sodium methanesulfinate salts. The transition-metal-free procedure for the synthesis of (E)-vinyl sulfones via the reaction between vinyl halides and sodium sulfinates in water was reported by Yu and co-workers (Scheme 79B).135 The sulfonylation was accelerated in the presence of n-Bu4NBr and HCl. Various arylvinyl bromides bearing either an electron-donating or -withdrawing substituents on the benzene ring afforded vinyl sulfones in good to high yields. As expected, the vinyl chlorides are less reactive than vinyl bromides; however, bromo- and chloro-acrylic acid also afforded the desired products in moderate yields.
 | ||
| Scheme 79 Synthesis of vinyl sulfones from vinyl halides. | ||
Consequently, in 2016, a simple and metal-free nucleophilic substitution reaction between 1-chloro-2-nitroethene and sodium arylsulfinates was described by Stoeva and co-workers (Scheme 80).136 Various p-substituted aromatic sulfinates bearing both electron-rich and electron-poor substituents were effectively explored in the vinylic sulfonylation to furnish a range of vinyl sulfones in 72–94% with only E-regioselectivity.
 | ||
| Scheme 80 Synthesis of vinyl sulfones from 1-chloro-2-nitroethene. | ||
A mild, efficient and metal-free synthesis of vinyl sulfones was developed by Liang and coworkers using 1,2-dibromides with sodium sulfinates (Scheme 81).137 At 80 °C in DMF, a wide variety of 1,2-dibromide derivatives were quickly coupled with sodium benzenesulfinate and sodium methanesulfinate without any catalyst to form phenyl and methyl (E)-vinyl sulfones in 65–88% yields.
 | ||
| Scheme 81 Synthesis of vinyl sulfones via 1,2-dibromides with sodium sulfinates. | ||
Chang and co-workers138 briefly reported the Ph3P-mediated disulfonylation of 1,3-dihalostyrenes with sodium sulfinates, as described in Scheme 82. Double nucleophilic substitution of 1,3-dihalostyrenes with different arene-sulfinates and methanesulfinate provided various 1,3-disulfonylstyrene derivatives in 80–86% yields.
 | ||
| Scheme 82 Disulfonylation of 1,3-dihalostyrenes with sodium sulfinates. | ||
Later, the same group reported a solvent-free sequential one-pot for the synthesis of β-sulfonyl styrenes through a polyphosphoric acid (PPA)-catalyzed 1,1-diacetoxylation of arylacetaldehydes with Ac2O, followed by deacetoxylative sulfonylation with sodium sulfinates under solvent-free conditions (Scheme 83).139 A series of vinyl sulfones was obtained on the gram-scale (5 mmol), and the yields ranged from good to high using different arylacetaldehydes and several aryl and alkyl sulfinates. Aryl vinyl sulfones, benzyl vinyl sulfones, and divinyl sulfones were obtained in acceptable yields. The aliphatic crotonaldehyde is also a suitable substrate to give the desired vinyl sulfone in good yield.
 | ||
| Scheme 83 Disulfonylation of 1,3-dihalostyrenes with sodium sulfinates. | ||
Reeves et al. utilized vinyl tosylates for palladium-catalyzed cross-coupling with sulfinate salts to synthesize vinyl sulfones. Several cyclic and acyclic vinyl tosylates were smoothly coupled with sodium p-toluenesulfinate under the catalytic influence of 2.5 mol% Pd2(dba)3 and 5.0 mol% XantPhos ligand and gave a range of desired alkenyl sulfones in moderate to good yields (Scheme 84).140 Acyclic vinyl tosylates formed a mixture of trans and cis isomers of vinyl sulfones. Unfortunately, the sulfonylation of vinyl tosylate with sodium methanesulfinate was unsuccessful; however, cyclopropyl-sulfinates provided the desired vinyl sulfone in 74% yield.
 | ||
| Scheme 84 Pd-catalyzed coupling of vinyl tosylates with sulfinate salts. | ||
Chang et al., reported the palladium-catalyzed bis-sulfonylation of propargylic bromides with sodium sulfinates in the presence of n-Bu4NF under the refluxing aqueous 1,4-dioxane (Scheme 85).141 The inexpensive Pd/C-catalyst enabled the coupling of propargylic bromides with various arylsulfinates to produce the diverse 2,3-bis-sulfonylpropene derivatives in 65–90% yields. Based on the outcomes, the electron-withdrawing substituents were slightly lower than the electron-donating groups on the benzene ring of arylsulfinates. Disappointingly, sodium methanesulfinate provided the similar bis-sulfone in traces.
 | ||
| Scheme 85 Pd/C-catalyzed reaction of propargylic bromides with sodium arenesulfinates. | ||
An eco-friendly method was developed by Wang and co-workers142 for the synthesis of sulfonylated-2(5H)-furanone derivatives (26). The TBAB-catalyzed sulfonylation of C(sp2)–X of 3,4-dihalo-5-alkoxy-2(5H)-furanones with sodium sulfinates occurred via a radical sulfonylation. A range of sodium arylsulfinates bearing electron-donating or electron-withdrawing groups readily participated with 3,4-dibromo-5-methoxy-2(5H)-furanone to produce the corresponding vinyl sulfones in moderate to high yields (Scheme 86). Several 5-substituted 3,4-dibromo-2(5H)-furanones were also employed with sodium p-toluenesulfinate and provided the desired products in satisfactory yields. Similarly, a series of 5-substituted 3,4-dichloro-2(5H)-furanones were also compatible and reacted well with different arylsulfinates to give the corresponding sulfones with slightly diminished yields as compared with the dibromo substrates. The large-scale experiments were successfully accomplished to prepare sulfonylated 2(5H)-furanones with acceptable yields. Notably, the radical trapping experiments were performed by adding TEMPO or BHT into the standard conditions. As expected, product formation was inhibited, thus indicating that a free radical pathway was involved in this process.
 | ||
| Scheme 86 TBAB-catalyzed sulfonylation of 3,4-dihalo-5-alkoxy-2(5H)-furanones with sodium sulfinates. | ||
3.4.2.2. Sulfonylation of alkenes and alkynes. In 2001, Nair et al., firstly reported the cerium(IV) ammonium nitrate (CAN)-mediated sulfonylation of various styrenes and alkenes with sodium p-toluenesulfinate and benzenesulfinate in the presence of sodium iodide to afford a series of vinyl sulfones in high yields (Scheme 87A).143 The oxidative addition of sulfinate and iodine to varied terminal alkynes led to the synthesis of β-iodovinyl sulfones in good to high yields. Subsequently, the authors treated the in situ generated β-iodovinyl sulfones with K2CO3 at reflux in the one-pot operation to synthesize acetylenic sulfones in good yields (Scheme 87B).144 Cyclohexene and phenylcyclohexene showed different reactivities under the same reaction conditions.
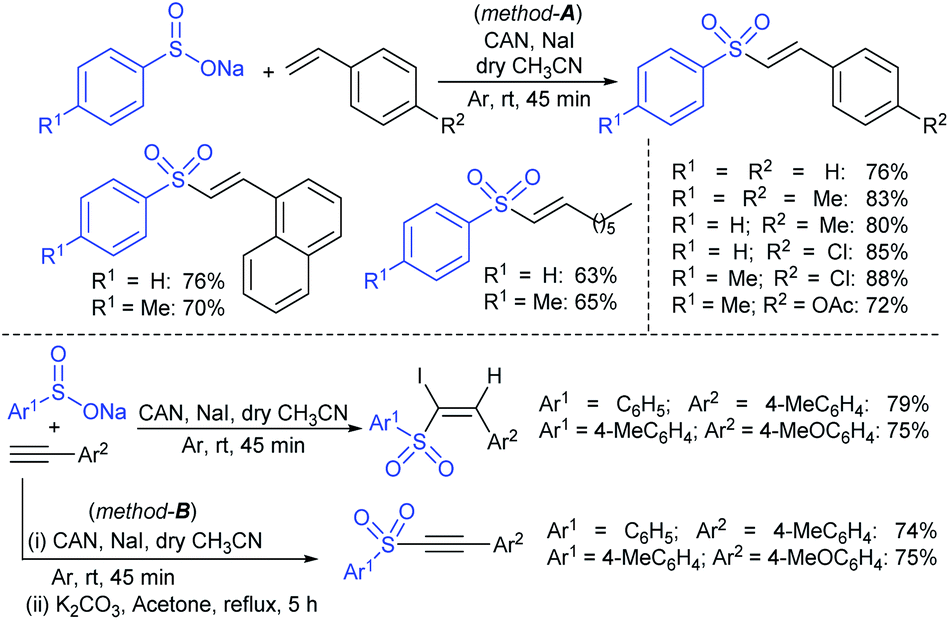 | ||
| Scheme 87 CAN-mediated sulfonylation of alkenes and alkynes with sodium sulfinate salts. | ||
In 2010, Kuhakarn and co-workers145 demonstrated the PhI(OAc)2 [(diacetoxyiodo)benzene, DIB]/KI-mediated sulfonylation of alkenes and alkynes with sodium arenesulfinates. Both the sodium p-toluenesulfinate and sodium benzenesulfinate reacted smoothly with a variety of styrene derivatives and afforded the corresponding vinyl sulfones in moderate to high yields (Scheme 88). The DBU treatment is required for functionalized aliphatic alkenes and cyclic alkenes to yield the corresponding vinyl sulfones exclusively, instead of the formation of β-iodosulfones. A range of α,β-unsaturated carbonyl derivatives were also examined in the oxidative sulfonylation and provided the desired vinyl sulfones in good yields. This method worked well with substituted arylacetylenes and 1-octyne to afford β-iodovinyl sulfones in high yields with a single regioisomer.
 | ||
| Scheme 88 PhI(OAc)2-mediated sulfonylation of alkenes and alkynes with sulfinates. | ||
Following Nair's and Kuhakarn's work, in 2011, Das and co-workers146 also synthesized a wide range of vinyl sulfones from the corresponding alkenes (Scheme 89). The combination of sodium periodate and potassium iodide catalyzed the oxidative sulfonylation of aromatic and aliphatic olefins, including sensitive functional groups (–OH, NHCbz, –OPMB, etc.)-derived alkenes, also smoothly coupled with sodium p-toluenesulfinate and sodium benzenesulfinate to produce the desired alkenyl sulfones in 87–95% yields.
 | ||
| Scheme 89 NaIO4/KI-catalyzed oxidative sulfonylation of alkenes with sulfinate salts. | ||
The Kuhakarn group further reported an improved method for preparing vinyl sulfones from alkenes with sodium sulfinates under the influence of molecular iodine and sodium acetate (Scheme 90).147 A wide variety of aromatic, aliphatic (acyclic and cyclic) alkenes as well as activated alkenes were successfully coupled with sodium benzene/p-toluenesulfinates under standard conditions to deliver various vinyl sulfones in moderate to high yields. The excess use of sulfinate salts and iodine would be a significant drawback for this protocol.
 | ||
| Scheme 90 I2-mediated sulfonylation of alkenes with sulfinate salts. | ||
Taniguchi established a copper-catalyzed oxidative sulfonylation of terminal or internal alkenes or alkynes using sodium sulfinates in DMSO at 100 °C. A range of terminal alkenes and alkynes were successfully employed for the synthesis of different types, (E)-alkenyl sulfones and (E)-haloalkenyl sulfones, respectively, in moderate to high yields (Scheme 91).148 The scope of internal alkenes or alkynes was significantly limited to obtain the desired products. Next, the same author reported the copper-catalyzed (5 mol%; 1:1 CuCl:L-27) oxidative hydrosulfonylation of a wide range of terminal or internal alkynes using sodium sulfinates in the air (Scheme 92).149,150 A variety of (E)-alkenyl sulfones were obtained in good to high yields via the reaction, which proceeded syn-selectively. The alkylacetylenes reacted slightly slower than the arylalkynes; unfortunately, 1-octyne resulted in a complex mixture. Notably, instead of an oxygen atmosphere, the reaction performed under nitrogen using parent substrates led to the desired sulfonylated product in 12% only.
 | ||
| Scheme 91 Cu-catalyzed oxidative sulfonylation of alkenes or alkynes using sodium sulfinates. | ||
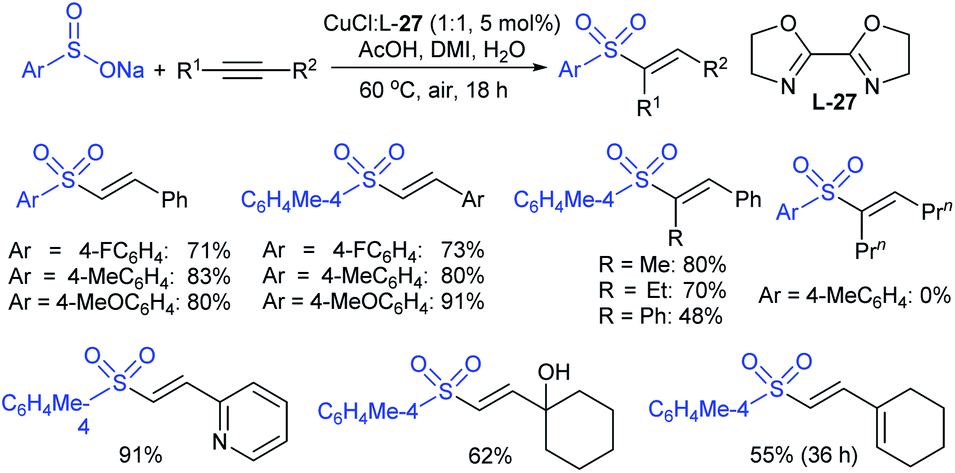 | ||
| Scheme 92 Cu-catalyzed hydrosulfonylation of alkynes using sodium sulfinates. | ||
Copper triflate catalyzed the hydrosulfonylation of alkynes with sodium sulfinates for the synthesis of vinyl sulfones under microwave irradiation, as shown in Scheme 93. Kumar and co-workers151 explored various terminal- and internal-alkynes with sodium p-toluenesulfinate and sodium benzene sulfinate and furnished diverse vinyl sulfones in good to excellent (71–89%) yields with high regio- and stereoselectivity. Additionally, the heteroaromatic alkynes, 2-ethynylpyridine conveniently reacted with both arenesulfinates to provide the corresponding vinyl sulfones in acceptable yields.
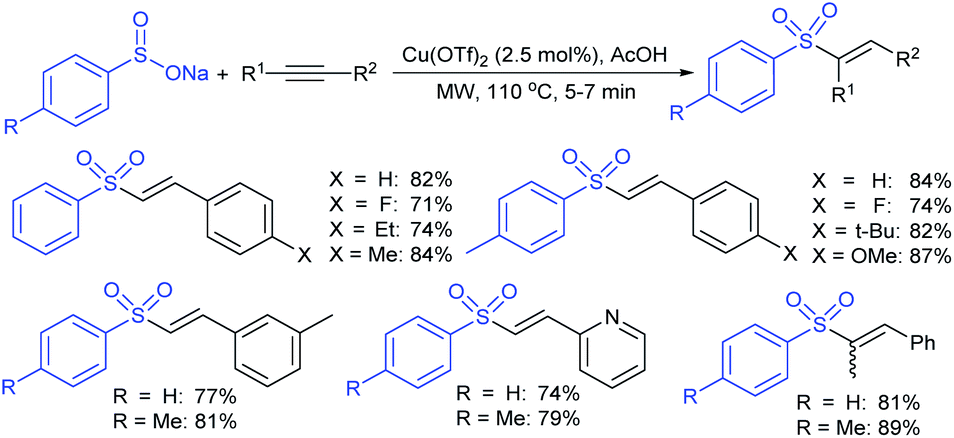 | ||
| Scheme 93 Cu(OTf)2-catalyzed hydrosulfonylation of alkynes with sulfinates. | ||
Bi and co-workers described the mechanistically interesting Ag2CO3-mediated sulfonylation of various allyl/propargyl alcohols with sodium sulfinates to synthesize γ-keto sulfones (Scheme 94).152 A variety of electron-donating or electron-withdrawing groups on the benzene of propargyl alcohols readily reacted with sulfinate salts to afford the desired vinyl sulfones in high to excellent yields. Similarly, a series of arylallyl alcohols resulted in a smooth conversion with various sulfinates to form the corresponding γ-keto sulfones in high yields. Although many arylsulfinates provided the desired sulfones, the reaction with MeSO2Na allowed the trimerization of propargyl alcohols. Simultaneously, the aliphatic alkynyl carbinols were ineffective in forming the desired γ-keto sulfones.
 | ||
| Scheme 94 Ag2CO3-mediated sulfonylation of allyl/propargyl alcohols using sodium sulfinates. | ||
König and co-workers153 developed an innovative and efficient method for the synthesis of vinyl sulfones using organic dye Eosin Y (EY)-catalyst for the visible-light photooxidation of sulfinate salts with alkenes. The striking feature of this work was the reaction of alkenes through the radical-initiated S-center of the sulfinate under optimized reaction conditions (Scheme 95). The substrate scope was explored using a wide range of alkyl and heteroaryl sulfinates with various cyclic and acyclic olefins to generate the corresponding sulfones in moderate to excellent (51–99%) yields. In particular, the bulky 10-camphor sulfinate is also a suitable substrate for this protocol. The possible mechanism was systematically investigated using various spectroscopic methods to understand the role of eosin Y (EY) and nitrobenzene or air as the terminal oxidant. The photooxidation of sulfinate salts proceeds through the EY radical cation as a key intermediate; nitrobenzene is a strong quencher, preferentially oxidizing the excited catalyst and regenerating the photocatalyst for the further catalytic processes.
 | ||
| Scheme 95 Eosin Y (EY)-mediated synthesis of vinyl sulfones. | ||
Silver catalyzed the C–S bond coupling of aromatic alkenes with sodium arylsulfinates for the synthesis of vinyl sulfones under the influence of K2S2O8 and TEMPO (cat) with a high E-selectivity (Scheme 96).154 Various substituted arylstyrenes, including electron-donating and electron-withdrawing groups, unambiguously proceeded to afford the desired vinyl sulfones in an efficient manner. Numerous arylsulfinate salts with the ortho-, meta- or para-substitutions on the benzene ring and heteroarylsulfinates were converted smoothly into the corresponding vinyl sulfones in moderate to high yields. The cyclic alkenes also proceeded efficiently in DMSO instead of toluene with sodium benzenesulfinate to yield the desired products. The major disadvantage of this protocol to aliphatic alkenes is that, 1-hexene was employed; however, no reaction occurred. A scale-up experiment (7 mmol) was also performed between styrene and sodium benzenesulfinate under the standard conditions to afford the desired product in 72% (1.23 g) yield.
 | ||
| Scheme 96 Ag-catalyzed sulfonylation of alkenes with sulfinate salts. | ||
Khalili reported the hydrosulfonylation-type reaction between sodium arylsulfinates and dialkyl acetylenedicarboxylates or alkyl propiolates in DMF at room temperature (Scheme 97).155
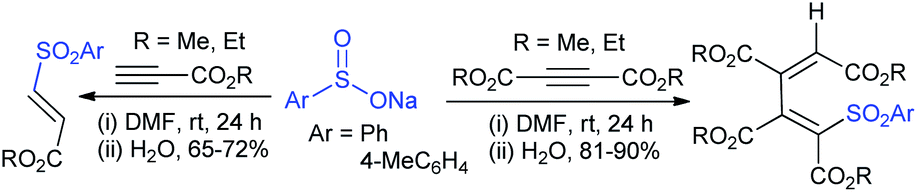 | ||
| Scheme 97 Hydrosulfonylation-type reaction sodium arylsulfinates and acetylenedicarboxylates or propiolates. | ||
Sodium benzenesulfinate and p-toluenesulfinate were treated with two equivalent dialkyl (Me or Et) acetylene dicarboxylates to furnish different sulfonylated 1,3-butadienes in 81–90% yields. The regioselective hydrosulfonylation of alkyl (Me or Et) propiolates with sodium arylsulfinates gave the desired vinyl sulfones in 65–72% yields.
Yuan and co-workers156 established an electrochemical sulfonylation between styrenes and sodium sulfinates with NaI as a supporting electrolyte in DMSO. A range of electron-poor, -rich and -neutral substituents on the benzene ring of styrenes were readily coupled with various sodium aryl and alkyl sulfinates to afford the corresponding vinyl sulfones in good to high yields (Scheme 98). However, the vinyloxybenzene was not a suitable substrate for this process to provide the desired product. Some control experiments were performed, and the authors assumed that the in situ electrogenerated I2 from NaI played a pivotal role in this electrochemical process.
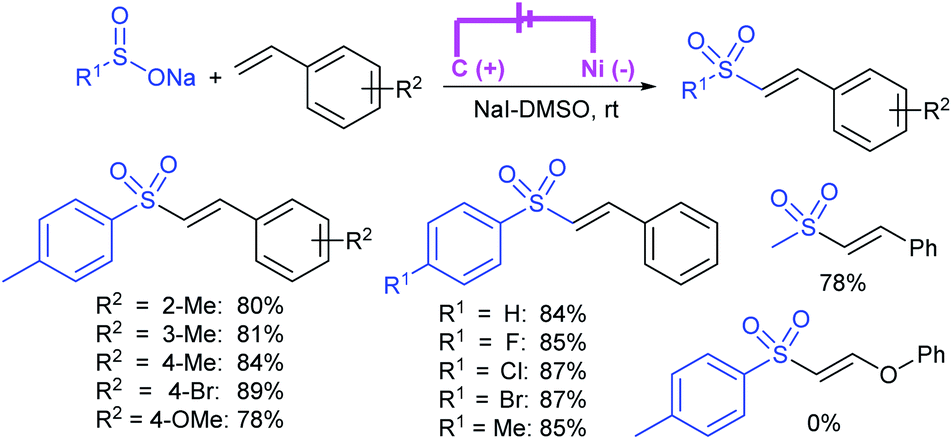 | ||
| Scheme 98 Electrochemical sulfonylation of styrenes with sodium sulfinates. | ||
Shi and co-workers157 developed a gold-catalyzed unusual Markovnikov addition of terminal alkynes and sodium sulfinates to synthesize α-substituted vinyl sulfones (Scheme 99). The efficient C–S bond was constructed with exclusive α-regioselectivity in the presence of cationic gold species and gallium triflate in dry 1,2-dichloroethane (DCE). Various substituted benzenesulfinates were reacted with phenylacetylene to provide various α-substituted aryl vinyl sulfones in promising yields. Heteroaryl-derived sulfinate was also a useful substrate, whereas aliphatic sulfinate salts gave the corresponding vinyl sulfones with somewhat low results in this transformation.
 | ||
| Scheme 99 Gold-catalyzed Markovnikov addition of phenyl acetylene and sulfinates. | ||
Similar analogs such as vinyl triflones were readily obtained via the iodine-mediated triflylation of styrenes with CF3SO2Na at room temperature (Scheme 100).158 A range of electron-donating and electron-withdrawing substituted styrenes were reacted regioselectively with CF3SO2Na to afford the desired vinyl triflones in modest yields. The benzofuran or thiophene-containing styrenes were also suitable, affording the corresponding vinyl triflones in 52% and 50% yields, respectively. However, the exact role of CuCN remained unclear for this protocol. The predicted mechanistic pathway involved the electrophilic addition of iodine or CF3SO2I (in situ generated by the reaction of CF3SO2Na and I2) to alkenes to form the three-membered cyclic iodonium ion (I). Next, the selective nucleophilic attack on cyclic iodonium ion (I) by CF3SO2N led to the iodotriflylated product (II). Finally, spontaneous HI elimination of II in the presence of a base gave vinyl triflones.
 | ||
| Scheme 100 I2-mediated C–H triflylation of styrenes with CF3SO2Na. | ||
In 2016, water-induced hydrosulfonylation was achieved by He and co-workers159 for the synthesis of Z-β-sulfonyl-α,β-unsaturated carbonyl products. The direct C–S coupling reaction of sodium aryl and alkyl sulfinates with a range of propargylic carboxylates led to the synthesis of the desired β-sulfonyl enoates with (Z)-stereoselectivity in good to high yields (Scheme 101). Moreover, several complex bioactive esters, including indanol, perillyl, benzoin, and farnesol-derived alkynes were well reacted to generate the corresponding tosylacrylates in 56–82% yields with a mixture of Z/E isomers. Additionally, other electron-withdrawing groups, such as amide, acetyl, and azine acetylenes were sulfonated to give similar products in moderate to good yields. The standard conditions investigated with radical inhibitors (TEMPO and BHT) did not prevent the reaction, representing that the sulfonylation might not proceed through a radical pathway.
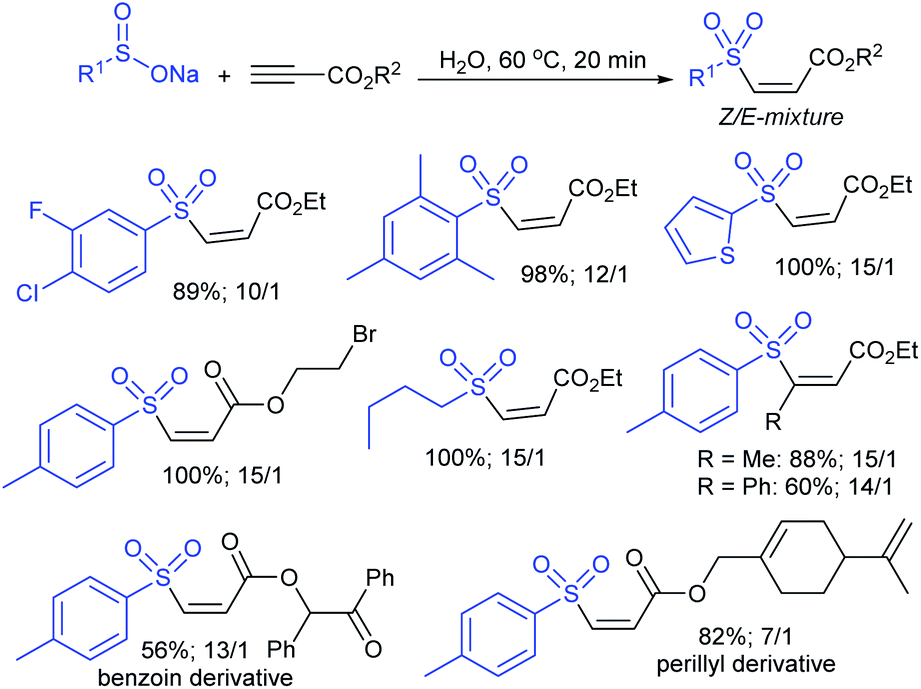 | ||
| Scheme 101 Sulfonylation of propargylic carboxylates with sodium sulfinates. | ||
At the same time, similar work was reported by Xiang and co-workers,160 namely, the rapid regioselective sulfonylation of propargyl esters with sodium sulfinates in acidic water to access highly stereoselective (Z)-β-sulfonyl enoates. A broad range of aromatic and heteroaromatic sodium sulfinates and propargyl esters-derived sensitive functional groups were tolerated in the hydrosulfonylation reaction and provided different classes of vinyl sulfones in 75–100% yields (Scheme 102). The deuterium experiment was performed with D2O instead of H2O, which indicated that the α-hydrogen atom of (Z)-β-sulfonyl enoates resulted from water. The study of the TEMPO radical scavenger revealed that the sulfonylation did not proceed via a free-radical pathway.
 | ||
| Scheme 102 Sulfonylation of propargylic carboxylates with sulfinates. | ||
Recently, Guan, He, and co-workers developed a simple and efficient hydrosulfonylation of electron-deficient alkynes with sodium sulfinates in acidic buffer solutions to access (E)-β-sulfonyl-α,β-unsaturated carbonyl compounds (Scheme 103).161 Various ynone derivatives with diverse substitution patterns at both ends reacted smoothly with sodium benzenesulfinate to afford the desired vinyl sulfones in moderate to good yields with high stereoselectivity. Alkynyl esters were easily converted into the desired product with diminished yield and stereoselectivity. Besides, sodium sulfinates including aryl and alkyl sulfinates also served as suitable reaction partners and gave the corresponding products with good yields and excellent E-selectivity. The 3-phenyl-propiolic acid, chalcone, and sodium trifluoromethanesulfinate were also subjected to the reaction but, unfortunately, they were sluggish to participate in this protocol.
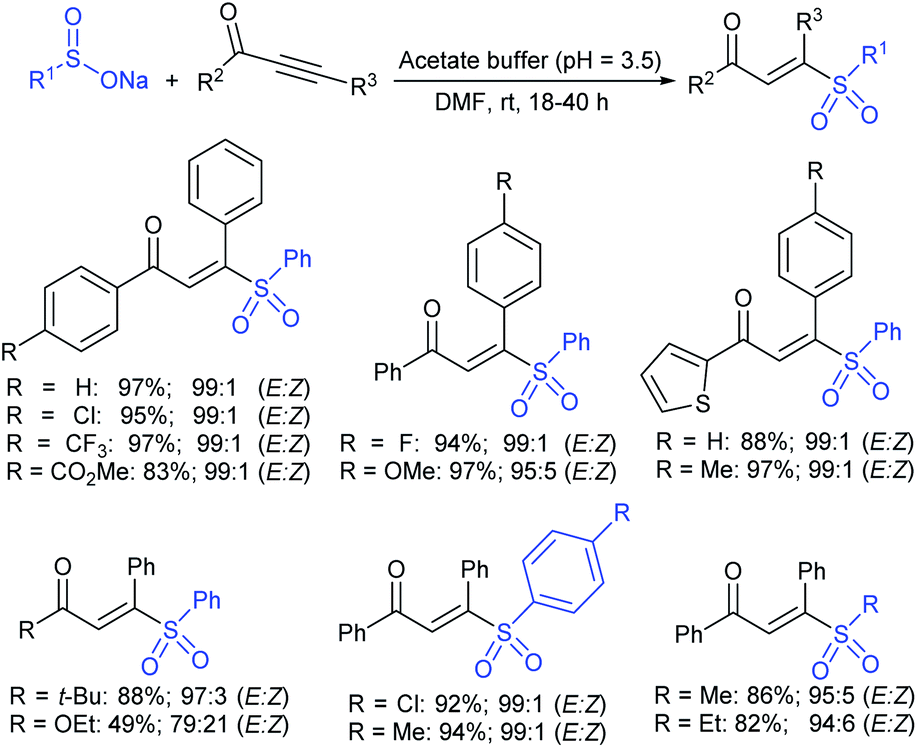 | ||
| Scheme 103 Sulfonylation of propargylic carboxylates with sulfinates. | ||
Yan and co-workers162 employed an inspirational method using quinine-thiourea-derived organocatalyst 28 and L-proline as a co-catalyst for the enantioselective synthesis of axially chiral vinyl sulfones at room temperature (Scheme 104). The vinylidene o-quinone methide (VQM) key intermediates were produced from o-alkynylnaphthols, which are readily sulfonylated with sodium sulfinates. Various p-substituted aromatic sulfinates bearing electron-donating and electron-withdrawing groups, as well as alkylsulfinates, were smoothly reacted with (phenylethynyl)-naphthalen-2-ol to form different sulfone-containing enantioenriched styrenes in good to high yields. They further explored a series of o-alkynylnaphthols with sodium benzenesulfinate to give the corresponding vinyl sulfones in acceptable yields with excellent enantioselectivities.
 | ||
| Scheme 104 Sulfonylation of o-alkynylnaphthols with sodium sulfinates. | ||
The mechanism of this methodology, particularly the key VQM intermediate was involved. Initially, the VQM intermediate was formed from o-alkynylnaphthols via prototropic rearrangement, which was synergistically promoted by the quinuclidine base and hydrogen bonding of the thiourea catalyst (Scheme 105). Next, the allene moiety was assumed as an absolute (R)-configuration. Meanwhile, the reaction between proline and sulfinate salt would generate a quaternary ammonium salt, enhancing the reactivity and solubility of the sulfinate salt. Subsequent nucleophilic addition of the highly active VQM intermediate with sulfinate anion furnished the sulfone-containing chiral styrenes. Boric acid played the role of reactivating proline due to the proton buffering properties.
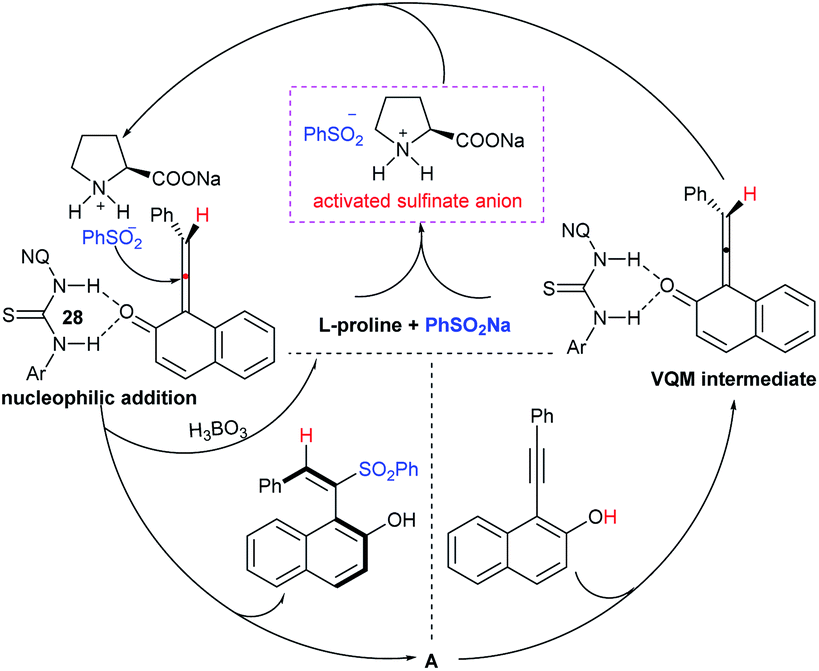 | ||
| Scheme 105 A plausible mechanism for the sulfonylation of o-alkynylnaphthols with sodium sulfinates. | ||
The vicinal disulfonylation of 1-bromoalkynes with sodium arylsulfinates in the presence of 12 M HCl in DMSO was developed by the Tang group (Scheme 106A).163 The control experiment indicated that weak acid (12 M HCl) was essential for this transformation. A broad range of substituted (E)-1,2-bis(arylsulfonyl)ethylenes were obtained in moderate to high yields when the reaction was conducted between various substituted arylalkynyl bromides with aromatic sulfinates under mild conditions. Although various alkynyl bromides responded, the strong electron-donating (methoxyl)-derived 1-bromoalkyne showed slightly decreased yield. Regrettably, the sterically hindered mesitylenesulfinate, and strongly electron-deficient 4-nitrobenzenesulfinate were unsuccessful in this transformation. In 2020, Gao, Tang, and co-workers demonstrated the copper-catalyzed vicinal disulfonylation of less reactive terminal alkynes with sodium sulfinates under the influence of bromodifluoroacetate for the synthesis of (E)-1,2-disulfonylethenes (Scheme 106B).164 Various aryl and heteroaryl alkynes were disulfonylated with sodium p-toluenesulfinate to obtain a wide range of (E)-1,2-disulfonylethenes in moderate to good yields, irrespective of their electronic and steric properties. Interestingly, eneynes were substantiated as a suitable coupling partner for the desired products in appreciable yields, whereas, there was no reaction with alkyl-derived alkynes. Further, a few substituted aryl and heteroaryl sulfinates were favourably coupled with phenylacetylene to give the corresponding vinyl sulfones in 70–82% yields. The ethanesulfinate salt did not afford the desired product.
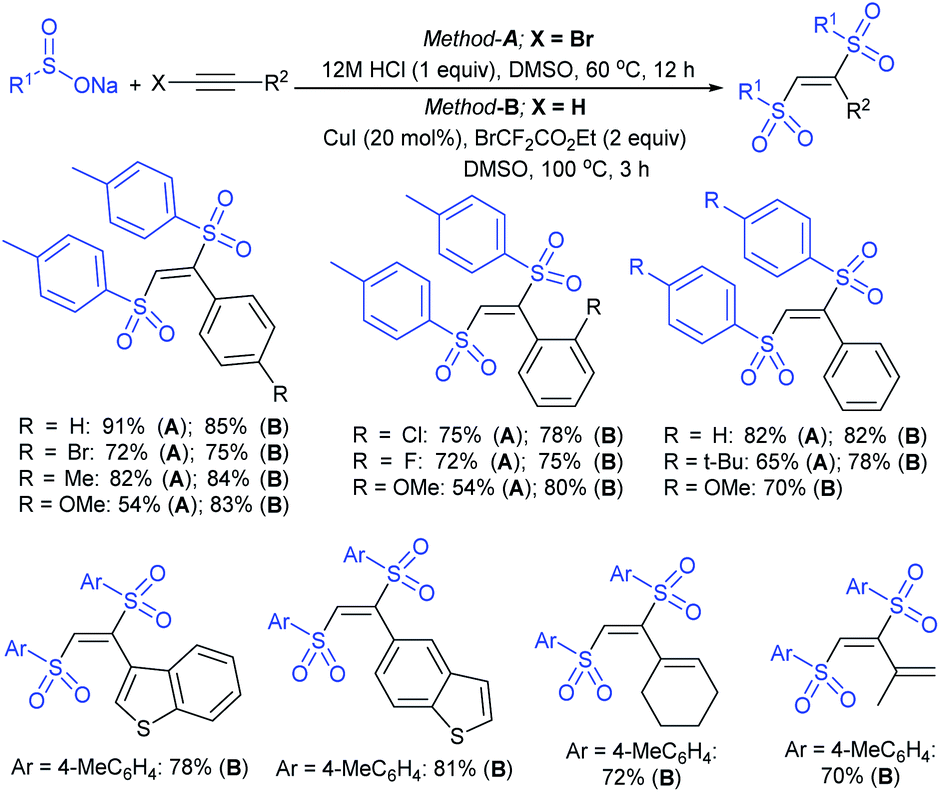 | ||
| Scheme 106 Disulfonylation of alknyl derivatives with sodium sulfinates. | ||
3.4.2.3. Sulfonylation of allenes. The Ren group reported an efficient solvent-dependent hydrosulfonylation of 3-cyclopropylidene-prop-2-en-1-ones with sodium sulfinates. The nucleophilic addition was controlled to β-sulfonylation in MeOH and the γ-sulfonylation occurred in DMSO as tunable selectivity (Scheme 107).165 A series of aryl and heteroaryl sulfinates reacted well with 3-cyclopropylideneprop-2-en-1-ones to afford β-adducts in good to high yields. The alkylsulfinates, cyclopropylsulfinate and (S)-10-camphorsulfinate were also suitable substrates in this protocol. Additionally, various substituted 3-cycloprop-ylideneprop-2-en-1-ones successfully reacted and gave the desired vinyl sulfones in slightly lower yields. Next, the authors gracefully extended γ-selective addition to a wide variety of sodium aryl, heteroaryl and alkyl-derived sulfinates that coupled with different 3-cyclopropylideneprop-2-en-1-ones to produce the expected γ-adducts in variable yields with good to high E-selectivity. Moreover, the effects of different substituents on 3-cyclopropylideneprop-2-en-1-one were carefully investigated and related products were obtained in moderate to high yields with a satisfactory selectivity.
 | ||
| Scheme 107 Solvent-dependent hydrosulfonylation of 3-cyclopropylidene-prop-2-en-1-ones with sodium sulfinates. | ||
The AcOH-mediated convenient sulfonylation of allenylphosphine oxides with sodium sulfinates for the synthesis of sulfonyl and phosphinyl bifunctionalized 1,3-butadienes or allenes was reported by Wu and co-workers (Scheme 108).166 A series of arylsulfinates bearing both electron-donating and electron-withdrawing substituents as well as aliphatic sulfinates were smoothly coupled with (3-cyclohexylidene-3-(phenylsulfonyl)-prop-1-en-2-yl)diphenyl-phosphine oxide to provide the desired vinyl sulfones in good to high yields. Besides, the cyclic and acyclic allenylphosphine oxides were compatible to react with sodium benzenesulfinate and generated sulfonyl-1,3-butadienes in 45–92% yields. The variation of acyclic allenylphosphine oxides produced stereodiversity in the products. The nucleophilic substitution between allenyl-phosphine oxides and sodium benzenesulfinate gave complementary sulfonylated phosphinyl allenes in acceptable (31–78%) yields. This regiodivergent phenomenon has also been explained by the effect of steric hindrance and stabilization of the substituents.
 | ||
| Scheme 108 AcOH-mediated sulfonylation of allenylphosphine oxides with sodium sulfinates. | ||
Following the inspired works on hydrosulfonylation, in 2020, the Voskressensky group also employed the hydrosulfonylation of inactivated allenes with sodium sulfinates under the influence of Eosin Y as the photocatalyst with green LED irradiation using one 30 W LED or two 15 W LEDs (Scheme 109).167 Various aryl- and heteroaryl allenes, such as indol-1-yl, pyrrol-1-yl, phenyl and naphthylallenes responded well in hydrosulfonylation with sulfinates and obtained Markovnikov-type vinyl sulfones in moderate to high yields. Besides, many sodium sulfinates, including aryl, heteroaryl and aliphatic (MeSO2Na) sulfinate sodium salts were successfully used to produce the corresponding vinyl sulfone in acceptable yields. The use of aliphatic allenes and trifluoromethane-sulfinate (Langlois reagent) were incompetent substrates for this transformation. The reaction was significantly inhibited either in the presence of TEMPO or under an oxygen atmosphere, proving a radical pathway.
 | ||
| Scheme 109 Eosin-Y-catalyzed hydrosulfonylation of allenes with sulfinates. | ||
3.4.2.4. Sulfonylation of enamides. The Mn(OAc)3-promoted oxidative C(sp2)–H sulfonylation of enamides and encarbamates with aryl, heteroaryl and aliphatic sulfinic acid salts was described by Manolikakes and co-workers.168 The method allowed access to synthetically valuable acyclic β-amidovinyl sulfones in good to high yields with excellent (E)-stereoselectivities (Scheme 110). This process proceeded at room temperature under mild conditions and tolerated various functional groups as well as common carbamate protecting groups on the nitrogen. The reaction of E or Z or E/Z-configured enamides were allowed to exclusively form the (E)-configured amidovinyl sulfone. Disappointingly, sodium 4-nitrobenzenesulfinate and trifluoro-methanesulfinate were unsuccessful in this transformation.
 | ||
| Scheme 110 Mn(OAc)3-promoted oxidative C(sp2)–H sulfonylation of enamides with sodium sulfinates. | ||
Visible-light induced the synthesis of β-acetylamino acrylosulfones from cyclic enamides and sodium sulfinates using the organic dye Rose Bengal (RB) as the photocatalyst, nitrobenzene and air as the oxidants (Scheme 111).169 The different substituents on cyclic enamides at variable positions were conveniently sulfonylated with sodium benzenesulfinate affording the cyclic vinyl sulfones in moderate to good yields. No desired product was observed in the case of tertiary enamides (in the absence of N–H), whereas acyclic enamides gave the desired products with an E/Z-mixture. Moreover, various aryl, heteroaryl and alkyl sulfinates readily reacted with enamide to give a range of sulfone products in moderate to good (68–93%) yields. The pragmatism was demonstrated at a gram-scale experiment under standard conditions without any significant impact on the outcome. Only a trace amount of the desired product was detected when the radical scavenger TEMPO was added, indicating that the reaction might involve a radical pathway.
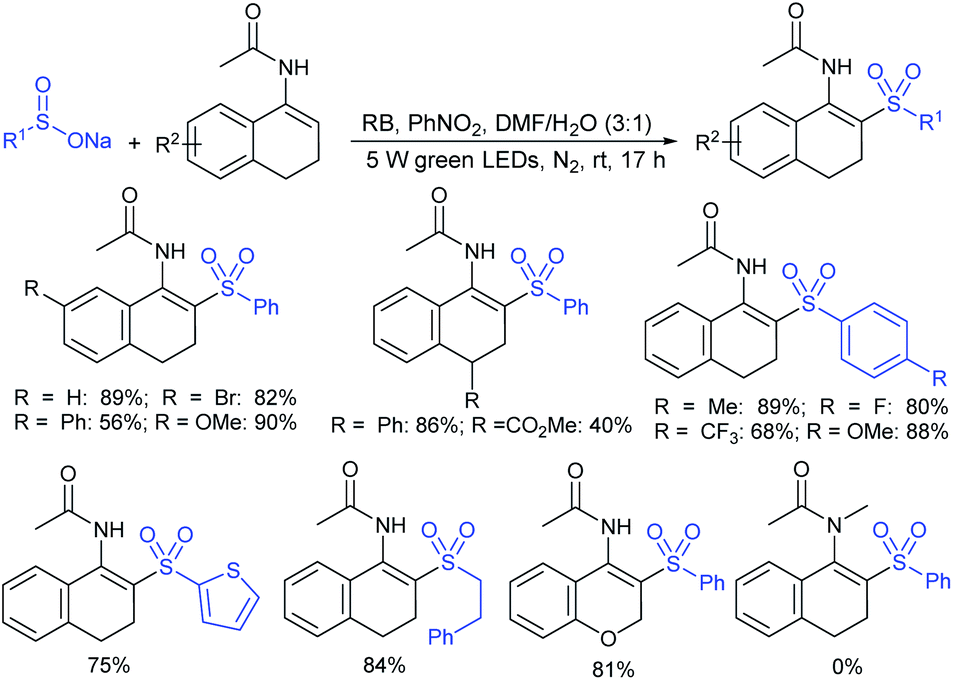 | ||
| Scheme 111 RB-catalyzed sulfonylation of enamides with sodium sulfinates. | ||
In 2019, Gui and co-workers disclosed the TBAI (tetrabutylammonium iodide)-catalyzed oxidative sulfonylation of tertiary amines with sodium sulfinates in the presence of TBHP (tert-butyl hydroperoxide) for the synthesis of β-arylsulfonyl enamines (Scheme 112).170 Various sodium arylsulfinates with electron-donating and electron-withdrawing groups on the benzene ring were coupled with triethylamine to afford the corresponding β-arylsulfonyl enamines in moderate to good yields. Also, other tertiary amines were efficiently converted into the desired β-arylsulfonyl enamines.
 | ||
| Scheme 112 TBAI-catalyzed oxidative sulfonylation of tertiary amines with sodium sulfinates. | ||
 | ||
| Scheme 113 Cu-catalyzed decarboxylative coupling of alkenyl carboxylic acids with sodium sulfinates. | ||
The copper-catalyzed decarboxylative radical sulfonylation for the C–S bond formation of α,β-unsaturated carboxylic acids with sodium arylsulfinates was described by Prabhu and co-workers (Scheme 114).172 The magnitude of the decarboxylative coupling was explored with a variety of electron-rich aromatic cinnamic acids and 2-thiophenyl-derived alkenic acids were reacted with various arenesulfinates to afford the expected vinyl sulfones in moderate to good yields of exclusively the (E)-isomer. One flaw was that the 2-nitrobenzenesulfinate, 4-methoxy-3-nitrobenzenesulfinate, and sodium methanesulfinate salts were ineffective under the optimal reaction conditions. Remarkably, the cis-4-methoxycinnamic acid was sulfonylated with sodium p-toluenesulfinate and furnished the E-isomer in 77% yield of the desired product exclusively. This protocol appears to be limited to only electron-rich aromatic cinnamic acids and other acrylic acids were not fruitful. The scaling-up experiment was efficient at the gram-scale of model substrates under the same reaction conditions. The reaction was inhibited under radical scavengers implying that the reaction probably proceeded via a free radical pathway.
 | ||
| Scheme 114 Cu-catalyzed decarboxylative coupling of alkenyl carboxylic acids with sodium sulfinates. | ||
At the same time, Guo et al., developed a Pd-catalyzed decarboxylative C–S coupling reaction between cinnamic acids with sodium arenesulfinates (Scheme 115).173 The catalyst was a combination of Pd(OAc)2 and dppb in the presence of Ag2CO3 as the base and oxidant in DMF for sulfonylation reaction. A wide variety of substituted aromatic cinnamic acids with a range of sodium sulfinates afforded the corresponding vinyl sulfones with retained stereochemistry as the E-isomer in yields ranging from 45–94%. The sodium methanesulfinate was an unsuccessful coupling partner under the same reaction conditions.
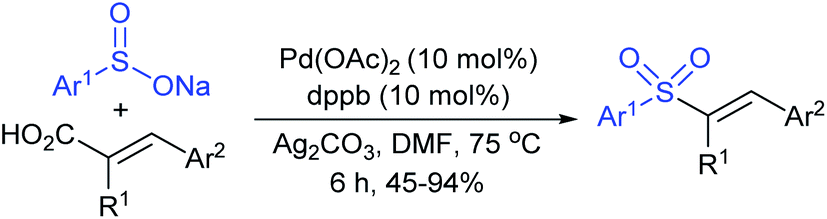 | ||
| Scheme 115 Pd-catalyzed decarboxylative coupling of cinnamic acids with sulfinates. | ||
Simultaneously, the Jiang and Kuhakarn research groups independently described the tandem cross-decarboxylative coupling reaction to synthesize vinyl sulfones (Table 12).174,175 Both these methods are widely applicable for a series of aryl, heteroaryl, and alkyl sulfinates, as well as a wide range of substituted aryl and heteroaryl cinnamic acids to readily construct the C–S bond under mild conditions. Jiang et al. developed a simple protocol using K2CO3 in DMSO at 100 °C to couple cinnamic acids with sodium sulfinates to form a wide range of alkenyl sulfones 66–94% yields (Table 12A).174 Later, the Kuhakarn group employed PhI(OAc)2 in DMF at 100 °C for the C–S bond coupling of cinnamic acids with sodium sulfinates to furnish vinyl sulfones in moderate to high yields (Table 12B).175
|
||
|---|---|---|
| Vinyl sulfones | Method-A | Method-B |
 |
R = H: 92% | R = H: 73% |
| R = Br: 79% | R = Br: 69% | |
| R = NO2: 94% | R = NO2: 43% | |
 |
R = Me: 87% | R = Me: 79% |
| R = Cl: 85% | R = Cl: 71% | |
| R = OMe: 77% | R = OMe: 80% | |
 |
73% | 61% |
 |
79% | 24% |

In 2015, the groups of Shi and Yuan independently reported a metal-free decarboxylative-sulfonylation of cinnamic acids with sodium sulfinates to furnish alkenyl sulfones (Table 13).176,177 The Shi group developed the iodine (2 equiv.)/TBHP-mediated C–S coupling between various aryl cinnamic acids, including 1-naphthyl and 2-thiophenyl-derived alkenyl acids, which were coupled with different arylsulfinates to form the anticipated vinyl sulfones in good to high yields (Table 13A).176 Sodium cinnamate was further examined with sodium benzenesulfinate under the same conditions to obtain the desired product in traces. Similarly, Yuan and co-workers developed an eco-friendly method by using I2 (1 equiv.)/K2CO3 in H2O for the aerobic cross-coupling of substituted-aryl and heteroaryl cinnamic acids with various sodium arylsulfinates to afford the corresponding vinyl sulfones in good to high yields (Table 13B).177 Encouragingly, the sodium methanesulfinate was also a suitable substrate to give the methyl vinyl sulfone in 45% yield. It is noteworthy that the model reaction was performed at the gram-scale, thus indicating that the protocol was practical and scalable. Both the oxidative sulfonylation approaches were probably occurring in a free radical pathway.
|
||
|---|---|---|
| Vinyl Sulfones | Method-A | Method-B |
 |
X = H: 92% | X = H: 78% |
| X = Br: 83% | X = Br: 81% | |
| X = Cl: 86% | X = Cl: 83% | |
 |
Ar = Ph: 91% | Ar = 4-MeC6H4: 79% |
 |
Ar = Ph: 84% | Ar = 4-MeC6H4: 57% |

In 2016, the electrochemical decarboxylative-sulfonylation of cinnamic acids with sodium sulfinates was successfully demonstrated under mild conditions (Scheme 116). Zha, Wang and co-workers178 have shown a broad substrate scope with respect to cinnamic acids and sodium arylsulfinates to provide a variety of (E)-vinyl sulfones in 26–84% yields. The β-substituted cinnamic acid afforded the corresponding products in moderate yields, whereas α-methyl-substituted cinnamic acid gave a trace amount. The 2-cyclohexylideneacetic acid was also examined, and no desired product was detected. The major drawback of the protocol is its incompatibility with the more electron-rich (4-MeO), electron-poor (4-NO2), sterically bulky (mesitylene)-derived sulfinates, as well as MeSO2Na and F3CSO2Na under the standard conditions. EPR and CV experiments detected the radical intermediate, and a radical-based pathway was proposed as a plausible mechanism.
 | ||
| Scheme 116 Electrochemical decarboxylative sulfonylation of cinnamic acids with sulfinates. | ||
Mao, Zhang and co-workers179 described a new phosphoric acid-mediated decarboylative coupling of arylpropiolic acids with sodium sulfinates in DMSO at 80 °C. A range of electron-rich/neutral/-poor substituted derived arylpropiolic acids were explored with various aryl- and alkyl-sulfinates to produce vinyl sulfones in moderate to excellent yields (Scheme 117). Although the decarboxylative-sulfonylation reactions worked well, very strong electron-withdrawing sodium arylsulfinates were less effective substrates to yield the desired products in somewhat lower yields. Almost the same level of yields were obtained using sodium phenylpropioate and benzenesulfinic acid under the prevailing conditions. Cross-experimental studies were conducted using TEMPO (a radical scavenger); the reaction was inhibited, and this suggests that a radical process was likely to be involved.
 | ||
| Scheme 117 H3PO4-mediated decarboxylative coupling of arylpropiolic acids with sodium sulfinates. | ||
The Xiang group disclosed the Mn(OAc)2-catalyzed aerobic decarboxylative coupling of cinnamic acids with sodium arenesulfinates in DMSO at 110 °C without any base or additive for the synthesis of vinyl sulfones (Scheme 118).180 A variety of cinnamic acids with ortho-, meta- or para-substituents reacted well with sodium benzenesulfinate to form the corresponding vinyl sulfones in good yields. Further, the 4-methyl and 4-bromo benzenesulfinates were converted into the desired aryl vinyl sulfones in satisfactory yields; however, sodium methanesulfinate barely reacted to provide the desired product.
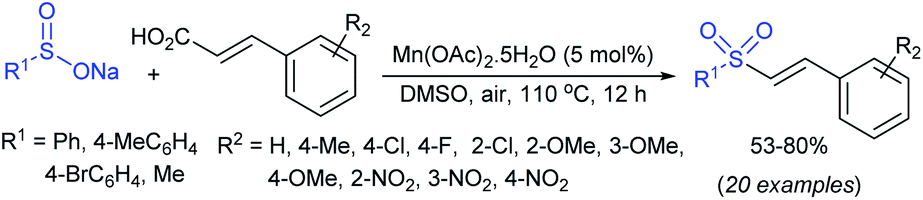 | ||
| Scheme 118 Decarboxylative coupling of cinnamic acids with sulfinate salts. | ||
Kuhakarn and co-workers181 developed the iodine-mediated sulfonylation of arylacetylenic acids and arylacetylenes with sodium sulfinates in the presence of TBHP for the synthesis of arylacetylenic sulfones. Only particular sodium arenesulfinates participated in the oxidative decarboxylative sulfonylation of 3-phenylpropiolic acid, giving the corresponding products in low to good yields (Scheme 119). Fortunately, sodium methane-sulfinate gave the corresponding product in low yield (20%) only. Various types of electronically different substituted arylacetylenic acid derivatives, including 3-(naphthalen-2-yl)propiolic acid and 3-(thiophen-2-yl)propiolic acid were sulfonylated with sodium p-toluenesulfinate to furnish the corresponding alkynyl sulfones in good yields. Efforts were unsuccessful use of β-alkyl- and β-silyl-substituted propiolic acids under developed conditions. In one representative example, a scale-up experiment (5 mmol) was performed and the desired product was obtained with similar efficiency. The protocol was further extended to the sulfonylation of arylacetylenes with different sodium arenesulfinates under the same conditions, and arylacetylenic sulfones were obtained in moderate yields. Next, the alkylacetylenic sulfone was not obtained by using the terminal aliphatic alkyne (1-octyne); instead, the known (E)-β-iodovinyl sulfone was observed.
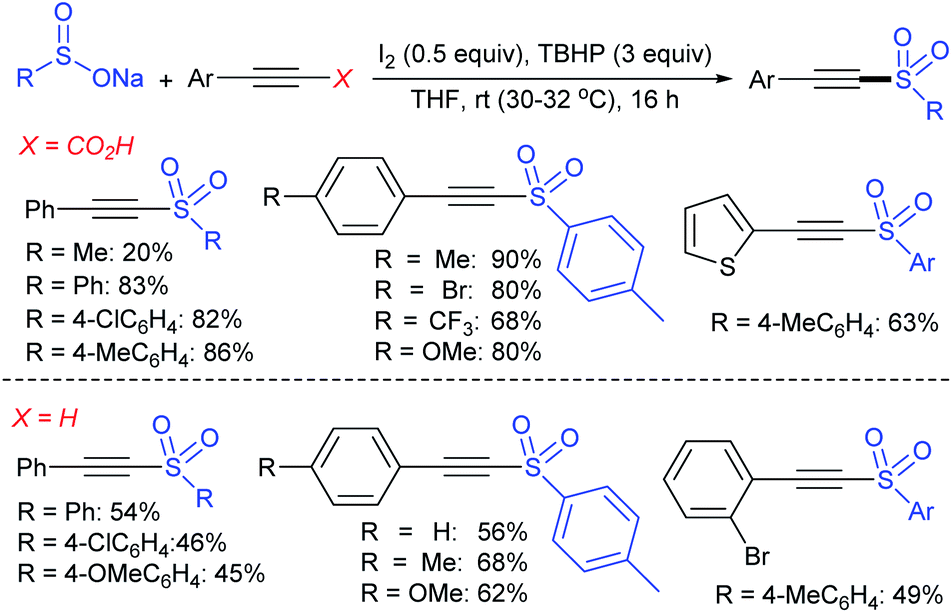 | ||
| Scheme 119 Iodine-mediated sulfonylation of arylacetylenic acids and arylacetylenes with sodium sulfinates. | ||
Visible-light promoted the decarboxylative aerobic coupling between cinnamic acids and sodium sulfinates under the catalytic influence of Merrifield resin (MS)-supported Rose Bengal (RB) ammonium salt under green LED (530–535 nm) irradiation for the synthesis of alkenyl sulfones in high yields (Scheme 120).182 A variety of aryl and heteroaryl cinnamic acids, together with sodium arylsulfinates were proved to be amenable substrates under mild conditions like tert-butyl hydroperoxide (TBHP, 70% in water) in aqueous DMSO at room temperature. The protocol did not proceed with the use of sodium methanesulfinate. Significantly, the MS supported RB catalyst was recycled six times without any loss of activity for generating the desired products in good yields.
 | ||
| Scheme 120 Decarboxylative coupling of cinnamic acids with sulfinate salts. | ||
 | ||
| Scheme 121 Denitrative sulfonylation of β-nitrostyrenes with sodium sulfinates. | ||
Lei et al. reported a metal-free direct denitro-sulfonylation reaction between β,β-disubstituted nitroalkenes and sodium arylsulfinates to give allyl sulfones (Scheme 122).186 The iodine-catalyzed direct condensation of both reactants of β,β-disubstituted nitroalkenes and sodium arylsulfinates bearing electron-donating and electron-withdrawing groups was successfully explored in this protocol. An array of allyl sulfones was obtained in good to high yields with high selectivity. Additionally, α-ethyl nitroolefins were also examined under standard reaction conditions and the expected allyl sulfones were obtained in high yields with a mixture of Z/E isomers. Further, the substrate scope was extended to a more diverse substrate, such as (nitromethylene)cyclohexane reacted with various sodium arenesulfinates to provide cyclohexenyl sulfone derivatives in 34–45% yields. These results showed the different reactivity between common nitroalkenes and β,β-alkyl-nitroalkenes under these radical addition conditions.
 | ||
| Scheme 122 Direct denitro-sulfonylation of β,β-disubstituted nitroalkenes with sodium arylsulfinates for allyl sulfones. | ||
 | ||
| Scheme 123 NBS/NIS-mediated halosulfonylation of alkynes using sodium sulfinates. | ||
In 2017, Sun, Liu, and co-workers188 reported an efficient and environmentally benign method for synthesizing (E)-β-iodovinyl sulfones through I2-mediated vicinal iodosulfonylation of alkynes with sodium sulfinates in water. Various aryl-, heteroaryl- and alkyl-substituted terminal alkynes responded well to form the corresponding β-iodovinylsulfones in moderate to excellent yields (Scheme 124). Also, the internal alkyne was successfully converted into the iodosulfonylated product in 64% yields. Additionally, various aromatic and aliphatic sodium sulfinates were favourably employed to offer sulfones varied products in satisfactory yields. However, the stability of the corresponding sulfone radicals plays a vital role in the transformation. As a result, sodium nitrobenzenesulfinate, sodium trifluoromethane-sulfinate, sodium thiophene-2-sulfinate, and sodium pyridine-3-sulfinate substrates were unsuccessful. The advantage of molecular iodine is that it plays a dual role that triggers the reaction and iodine source in this radical process.
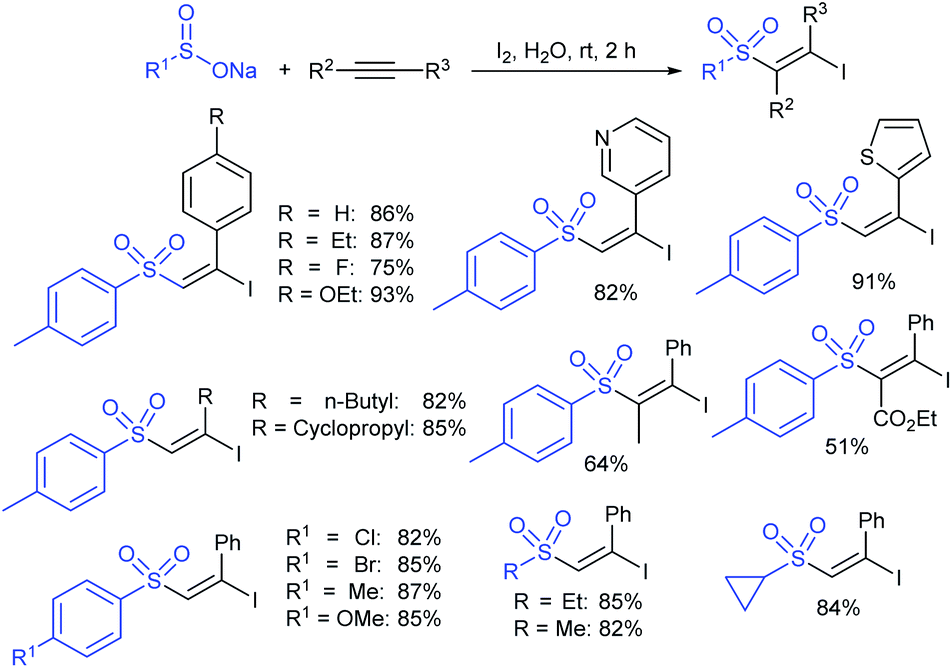 | ||
| Scheme 124 I2-mediated iodosulfonylation of alkynes using sodium sulfinates. | ||
Simultaneously, Bi et al. reported a simple protocol to synthesize (E)-β-iodovinyl sulfones via molecular iodine and di-tert-butyl peroxide (DTBP)-promoted iodosulfonylation of alkynes with sodium sulfinates under mild conditions (Scheme 125).189 Various aryl- and alkyl-acetylenes were successfully explored with sodium arylsulfinates or methanesulfinate to provide a series of functionalized (E)-β-iodovinyl sulfones in moderate to high yields. The electron-rich groups on phenylacetylene gave high yields as compared with electron-poor groups on phenylacetylene, which significantly decreased the yields. The present reaction conditions were not suitable for sodium trifluoromethanesulfinate to provide the desired product.
 | ||
| Scheme 125 I2-mediated iodosulfonylation of alkynes using sodium sulfinates. | ||
The stereo-selective vicinal iodosulfonylation of activated internal alkynes, sodium sulfinates and iodine for synthesizing tetra-substituted olefins was achieved by Reddy and co-workers. Particularly, various aryl, heteroaryl and alkyl ynones were accommodated by a series of sodium aromatic sulfinates to afford the corresponding β-iodosulfonyl enones in moderate to high yields (Scheme 126).190 Interestingly, an amino acid-derived ynone was also successfully converted to the desired product in 64% yield. The sodium methanesulfinate was also furnished to give the desired adduct in moderate yield, whereas the electron-deficient nitrophenyl sulfinate did not participate in the reaction. Furthermore, the alkyne bearing a variety of electron-withdrawing groups, such as esters, aldehyde, amide, cyano and sulfone groups were compatible substrates for coupling with sodium phenylsulfinate to yield β-iodovinyl sulfones. The haloalkynes also efficiently underwent the trans-iodosulfonylation to afford the vicinal dihalovinyl sulfones in good yields. Gratifyingly, aryl/alkyl-containing internal alkynes showed moderate reactivity and offered the corresponding iodovinyl sulfone in low to moderate yields.
 | ||
| Scheme 126 Iodine-mediated iodosulfonylation between activated internal alkynes and sodium sulfinates. | ||
In 2018, Zhang, Xie and group reported a convenient and straightforward protocol for synthesizing highly functionalized tetrasubstituted β-iodovinyl sulfones from different alkyne systems (acetylenic ketones, propargyl alcohols, or 1,3-diynes), sodium sulfinates, and iodine in aqueous medium at reflux (Scheme 127).191 Different substituted acetylenic ketones were employed with sodium sulfinates (phenylsulfinate, p-tolylsulfinate and methanesulfinate) to obtain the corresponding β-iodo-α-sulfonyl vinyl ketones in the range of 76–91% yields. Subsequently, the functionalized tetrasubstituted β-iodo-α-sulfonyl allylic alcohols were obtained in moderate to high yields when propargyl alcohols were reacted with sodium sulfinates and iodine in DCE/H2O at reflux. Further, the same reaction conditions were extended to the iodosulfonylation of 1,3-diynes with sulfinate salts, and iodine proceeded smoothly to provide polysubstituted conjugated enynes in 48–74% yields. The reactivity tendency of carbon–carbon triple bond noted as the α-alkynyl vinyl radical intermediate is more stable than the β-alkynyl vinyl radical intermediate due to the conjugation effects.
 | ||
| Scheme 127 Iodine-promoted iodosulfonylation between internal alkynes and sodium sulfinates. | ||
Cobalt catalyzed the aerobic anti-iodosulfonylation of alkynes using sodium sulfinates and KI to produce (E)-β-iodoalkenyl sulfones (Scheme 128).192 Numerous terminal and unsymmetrical internal alkynes were explored with aromatic and aliphatic sulfinates to obtain the desired β-iodovinyl sulfones in good to high yields. Unfortunately, the present procedure was not applicable to accommodate the diphenyl acetylene or terminal aliphatic alkyne (viz. 1-hexyne). The iodosulfonylation was entirely inhibited in the absence of oxygen atmosphere and the addition of TEMPO or TBC (4-t-butyl catechol) as a radical scavenger. These results suggest that the procedure requires oxygen and the reaction might proceed via radical intermediates.
 | ||
| Scheme 128 Cobalt-catalyzed iodosulfonylation between alkynes and sodium sulfinates. | ||
Chen, Yin, and co-workers reported iron(III) chloride hexahydrate-mediated regio- and stereoselective chloro-sulfonylation of alkynes and alkenes with sodium sulfinates under mild conditions. The anti-chlorosulfonylation reaction worked well among various aryl and heteroaryl alkynes bearing substituents of electron-withdrawing, electron-donating groups and steric groups to afford a range of β-chloroalkenyl sulfones with good to excellent yields (Scheme 129).193 A range of structurally varied alkenes were reaction partners and afforded the desired β-chloroalkyl sulfones in good to high yields. Surprisingly, strong electron-donating (OMe) group-derived alkynes and alkenes were not compatible, providing the desired products. Though several aryl and alkyl sodium sulfinates responded efficiently, sodium trifluoromethanesulfinate was not suitable for this process.
 | ||
| Scheme 129 FeCl3-mediated chlorosulfonylation of alkynes and alkenes using sodium sulfinates. | ||
 | ||
| Scheme 130 TPGS-750-M (29, 2% weight percent) used for aerobic oxidation of arylalkynes with sodium sulfinates. | ||
Consequently, Yadav and co-workers195 described the FeCl3/K2S2O8-catalyzed aerobic oxysulfonylation between alkynes and sodium arenesulfinates to form β-keto sulfones via a radical pathway. Various terminal aryl and heteroayl alkynes bearing electron-rich and electron-poor groups were smoothly oxysulfonylated with different sodium arenesulfinates and afforded a range of representative β-keto sulfones in 65–94% yields (Scheme 131). The internal alkyne prop-1-ynyl-benzene was also used and the desired product was obtained in moderate (56%) yield. As shown in the plausible mechanism, the catalyst system triggered the formation of the sulfonyl radical from the sulfinate salt. Then, the sulfonyl radical selectively attacked the triple bond of the alkynes to provide the alkenyl radical A, which was eventually trapped by dioxygen to form the dioxy radical B, which was also confirmed by oxygen labeling experiments. The radical B was expelled into enol (C) and tautomerization to give desired β-keto sulfones.
 | ||
| Scheme 131 FeCl3/K2S2O8-catalyzed aerobic oxysulfonylation of arylalkynes with sodium arenesulfinates. | ||
Histidine catalyzed the vicinal oxysulfonylation of terminal alkynes with arylsulfinic acid sodium salts in aqueous media at room temperature to synthesize β-keto sulfones (Scheme 132).196 A range of aryl acetylenes reacted well with sodium sulfinates to provide the desired β-keto sulfones in good to excellent yields; however, the electron-donating arylacetylenes have a slightly superior reactivity over electron-donating groups. Moreover, a variety sulfinate salts were smoothly coupled with phenylacetylene to afford the corresponding oxysulfonylated products in good yields. Further, a large-scale experiment (25 mmol) was also successfully performed with the same level outcome.
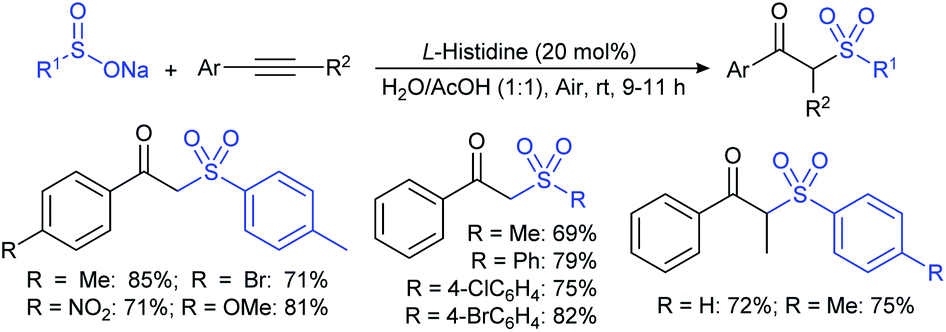 | ||
| Scheme 132 Histidine-catalyzed vicinal oxysulfonylation of terminal alkynes with sodium sulfinates. | ||
The Yadav group employed two independent methods for the direct radical sulfonylation of olefins with sodium sulfinates under aerobic conditions. K2S2O8 mediated the oxysulfonylation between aryl alkenes and arylsulfinates; these substrates bearing various electron-donating or electron-withdrawing groups were well tolerated to provide a broad range of β-keto sulfones in moderate to high yields (Table 14A).197 Next, the same group carried out the AgNO3-catalyzed aerobic oxysulfonylation of various aryl alkenes bearing electron-donating/withdrawing groups on the benzene ring with different arenesulfinate salts, leading to β-keto sulfones in good to high yields (Table 14B).198 Both approaches were applied to internal alkenes and the desired β-keto sulfones obtained in decent yields. The major drawback of these protocols involved not covering aliphatic or alicyclic alkenes due to unstable alkyl radicals compared with benzyl radicals in aryl alkenes.
|
||
|---|---|---|
| β-Keto sulfones | Method-A | Method-B |
 |
Ar = C6H5: 94% | Ar = C6H5: 96% |
| Ar = 4-BrC6H4: 88% | Ar = 4-BrC6H4: 88% | |
 |
R = H: 93% | R = H: 93% |
| R = Me: 90% | R = Me: 87% | |
| R = OMe: 85% | R = OMe: 72% | |
 |
R = F: 79% | R = F: 75% |
| R = Br: 81% | R = Br: 80% | |
| R = OMe: 82% | R = OMe: 86% | |
 |
R = Me: 87% | R = 85% |
| R = OMe: 84% | R = OMe: 71% | |
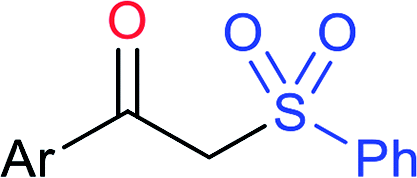 |
Ar = 2-naphthyl: 81% | Ar = 2-naphthyl: 82% |
 |
Ar = 2-naphthyl:76% | Ar = 2-naphthyl:83% |
 |
82% | 79% |

The Ponnapalli group demonstrated the Cu2+-doped zeolitic imidazolate framework-8 (Cu25%/ZIF-8)-catalyzed oxysulfonylation and hydrosulfonylation of terminal alkynes with sulfinate salts in MeOH and MeCN, respectively (Scheme 133).199 A range of substituted arylalkynes reacted with sodium benzenesulfinate and sodium p-toluenesulfinate to furnish β-keto sulfones in moderate to good yields. In contrast, the reaction performed in MeCN led to the formation of the corresponding vinyl sulfones with excellent E-selectivity in good to high yields. Notably, the catalyst was easily separated by centrifugation, washed with methanol, dried overnight at 100 °C and reused for five cycles without any significant effect on its catalytic activity.
 | ||
| Scheme 133 Cu25%/ZIF-8-catalyzed oxysulfonylation and hydrosulfonylation of terminal alkynes with sodium sulfinates. | ||
The Jiang group elegantly reported the copper-catalyzed oxidative sulfonylation of oxime acetates with sodium sulfinates to synthesize β-keto sulfone derivatives. In this process, oxime acetates served not only as a substrate but also as an oxidant. Various aromatic oxime acetates reacted smoothly with sodium p-tolylsulfinate to form the sulfonyl-vinylamine intermediate. Subsequent hydrolysis with HCl or AcOH of sulfonyl vinylamines produced β-keto sulfones in good to high yields (Scheme 134).200 Various para-substituted (F, Cl, Br, CF3 or OMe), 2-chloro group on the benzene, 2-naphthyl-derived sulfinates, and aliphatic sulfinate salts were well participated and afforded the desired products in moderate to good yields. Unfortunately, alkyl oxime acetates were not suitable and did not undergo the hydrolysis process.
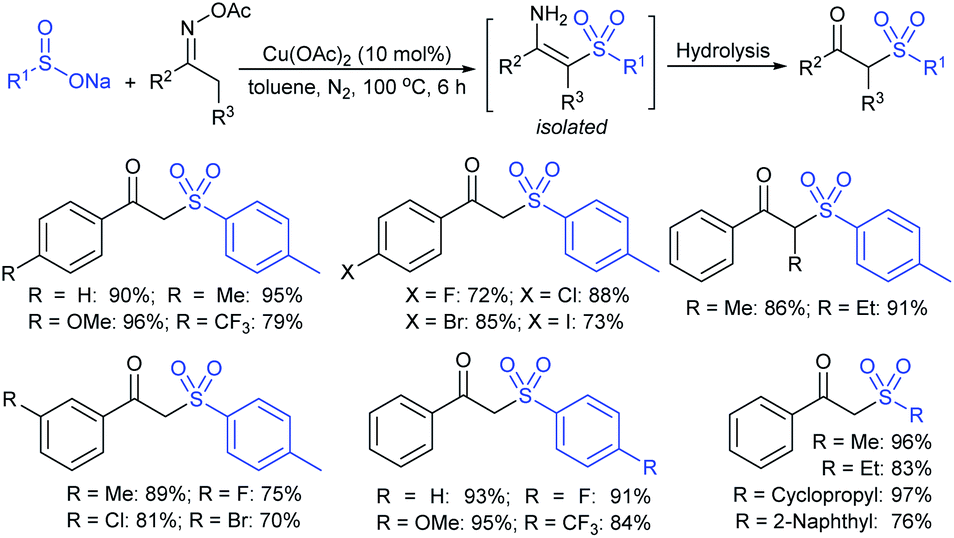 | ||
| Scheme 134 Copper-catalyzed oxidative coupling of aryl oxime acetates and sodium sulfinates. | ||
Jiang and co-workers proposed interesting mechanistic pathways to rationalize the observed results, as presented in Scheme 135. Path-A involving the acetophenone oxime acetate was transformed into a copper enamide intermediate (I) by the copper catalyst. The sulfonyl free radical could be generated from the CuII species and sodium sulfinates. Further, the sulfonyl free-radical reacted with copper enamide (I) to give a free radical intermediate (II). By releasing CuI through the single-electron-transfer (SET) process it forms intermediate (III), which undergoes the tautomeric form of the reasonably stable enamine intermediate (IV). Alternatively, the path-B mechanism might involve organo-copper(III) species (I′–III′), firstly the oxidative addition allowed the generation of intermediate (I′). Subsequently, there was coordination with sodium sulfinates to give (II′) and the simultaneous release of NaOAc. The expected vinyl CuIII species (III′) was obtained through tautomerization of the enamine intermediate (IV) generated from the reductive-elimination of III′ Finally, the hydrolysis of intermediate (IV) led to the corresponding β-keto sulfones.
 | ||
| Scheme 135 Plausible mechanistic pathways for the copper-catalyzed oxidative coupling of aryl oxime acetates and sodium sulfinates. | ||
In 2018, similar work from the Phan group was reported using metal–organic framework based Cu2(OBA)2(BPY), a recyclable heterogeneous catalyst for the oxidative-sulfonylation of a wide range of keto oxime acetates with sodium aryl and alkyl sulfinates to form β-sulfonyl vinylamines. The consequent hydrolysis of β-sulfonyl vinylamines with aqueous HCl produced a series of corresponding β-keto sulfones in 65–91% yields (Scheme 136).201 The steric and electronic properties of the substituents on the benzene ring of both substrates showed limited effect. Noticeably, the Cu-MOF catalyst was reused ten times without a considerable effect in the catalytic efficiency.
 | ||
| Scheme 136 Cu2(OBA)2(BPY)-catalyzed oxidative coupling of aryl oxime acetates and sodium sulfinates. | ||
Very recently, a similar oxidative coupling of oxime acetates with sodium sulfinates under the catalytic influence of MCM-41-supported Schiff base-pyridine bidentate copper(II) complex [MCM-41-Sb,Py-Cu(OAc)2] as the heterogeneous catalyst (Scheme 137) was reported.202 A range of oxime acetates also acted as an internal oxidant and effectively coupled with sodium p-tolylsulfinate with subsequent hydrolysis to afford the corresponding β-keto sulfones in good to excellent yields. A variety of aryl and alkyl sulfinates provided the desired β-keto sulfones in good to high yields. The electronic and steric nature of the substituents on the aromatic ring of the sodium benzenesulfinate substrate had limited influence. The heterogeneous copper catalyst can easily be prepared by a simple procedure and can be recovered by filtration of the reaction solution and recycled up to eight times with almost consistent activity.
 | ||
| Scheme 137 MCM-41-Sb,Py-Cu(OAc)2-catalyzed oxidative coupling of aryl oxime acetates and sodium sulfinates. | ||
In 2001, Xie and Chen developed a simple procedure for the synthesis of β-keto sulfones through α-tosyoxylation of ketones with [hydroxy(tosyloxy)iodo]benzene (HTIB) and the successive treatment with sodium sulfinates (Scheme 138).203 The reaction scope was widely applicable to aliphatic ketones, and several para-substituted aromatic ketones were successfully reacted with three different arylsulfinates. Since there was a regioselectivity issue, it was not suitable for unsymmetrical aliphatic ketones.
 | ||
| Scheme 138 α-Tosyoxylation of ketones with HTIB and sulfonylation using sodium sulfinates. | ||
In 2018, there was a similar report from the Tang group on the oxidative-sulfonylation of ketones with sodium sulfinates using the DMSO/HBr system to synthesize β-keto sulfones (Scheme 139).204 A variety of ortho-, meta- and para-substituted acetophenones were reacted with sodium p-toluenesulfinate, affording the expected β-keto sulfones in good to high yields. Additionally, propiophenone, 2-phenylaceto-phenone and 3,4-dihydro-naphthalen-1(2H)-one were used to provide the desired products in satisfactory yields. Heteroaromatic and alkyl ketones were also suitable and resulted in the formation of the related products in good yields. Subsequently, different substituted-arylsulfinates and sodium methanesulfinate were treated with acetophenone to target β-keto sulfones in good to high yields.
 | ||
| Scheme 139 DMSO/HBr-mediated oxidative-sulfonylation of ketones with sodium sulfinates. | ||
A mild and external oxidant-free protocol was disclosed by the Sekar group to synthesize β-keto sulfones from readily available secondary benzyl alcohols with sodium sulfinates. The successive oxidation and bromination of secondary benzyl alcohols with NBS to form α-bromoketones and subsequent nucleophilic displacement by sodium arenesulfinates led to β-keto sulfones (Scheme 140).205 A wide range of substituted secondary benzyl alcohols were explored with different para-substituted aromatic sulfinates to furnish the corresponding β-keto sulfones in good to high yields. The 2-Br and 4-NO2 aryl-substituted secondary benzyl alcohols did not afford the desired products. The transformation was also efficient at a gram-scale reaction without any influence on the outcome.
 | ||
| Scheme 140 NBS-mediated oxidative-sulfonylation of secondary benzyl alcohols with sodium sulfinates. | ||
A convenient CuBr2-catalyzed construction of α-alkyl-β-keto sulfones via C–H bond direct sulfonylation of aryl ketones and sodium sulfinates in the presence of 1,8-diazabicyclo-[5.4.1]undec-7-ene (DBU) was developed (Scheme 141).206 Various aromatic and aliphatic sulfinates were conveniently reacted with propiophenone to give the corresponding α-methyl-β-keto sulfones in moderate to good yields. Subsequently, aryl ketones with electron-withdrawing and electron-donating substituents on benzene ring were well tolerated and afforded a wide range of β-keto sulfones in varied yields. Typically, ortho, meta- and para-substituted propiophenones indicated that the transformation was not affected by the steric and electronic factors. It is worth noting that the use of 1,10-phenanthroline (20 mol%) was more suitable to obtain some of the analogs in high yields. Additionally, heteroaromatic ketones, n-butyrophenone and acetophenone also successfully led to the desired products. The low yield was obtained using α-tetralone; unfortunately, ethyl benzoyl acetate did not provide the product. Notably, a scale-up experiment also performed at 10 mmol proceeded smoothly without effect on the outcome.
 | ||
| Scheme 141 Tandem debromination/sulfonylation of α,α-dibromoketones with sodium sulfinates. | ||
A catalyst-free tandem debromination/nucleophilic sulfonylation sequence of α,α-dibromo ketones with sodium sulfinates in methanol afforded β-keto sulfones. A variety of aromatic and aliphatic α,α-dibromo ketones were found to be suitable substrates to react with sodium p-tolylsulfinate to furnish widespread β-keto sulfones in good to high yields (Scheme 142).207 Acyclic dibromo ketones and cyclic substrates also led to the tosylated products. Besides sodium p-tolylsulfinate, other aryl sulfinates bearing fluoro- and chloro-substituents, as well as sodium methanesulfinate were successfully employed to produce the desired products in good yields.
 | ||
| Scheme 142 Tandem debromination/sulfonylation of α,α-dibromoketones with sodium sulfinates. | ||
In 2016, a metal-free protocol was reported for the direct sulfonylation of enol acetates with sodium sulfinates using molecular iodine as an oxidizing reagent. Yadav and co-workers208 prepared different para-substituted β-keto sulfones in satisfactory (79–93%) yields from aryl enol acetates, and arylsulfinates bearing functional groups (methyl, methoxy, fluoro, chloro and bromo groups) were well tolerated in the present protocol (Scheme 143). The aliphatic enol acetates did not work to produce the desired β-keto sulfones.
 | ||
| Scheme 143 I2-mediated oxidative coupling of enol acetates and sodium sulfinates. | ||
Zou and co-workers employed NBS (N-bromosuccinimide)-promoted decarboxylative-sulfonylation of β-keto acids with sodium sulfinates under mild conditions.209 A broad substrate scope of aromatic β-keto acids as well as arylsulfinates were effectively coupled and furnished a wide range of β-keto sulfones in moderate to high yields (Scheme 144). Furthermore, aliphatic-substituted β-keto acids showed relatively low reactivity and the less reactive sodium methanesulfinate was also a suitable substrate. All these β-keto sulfones were probed for the inhibitory effect against CES1 and some of them showed promising results.
 | ||
| Scheme 144 NBS-promoted decarboxylative sulfonylation of β-keto acids with sodium sulfinates. | ||
K2S2O8 mediated the decarboxylative sulfonylation of cinnamic acids and sodium sulfinates for synthesizing β-keto sulfones under atmospheric oxygen (Scheme 145).210 The aerobic oxidative decarboxylative coupling of various phenyl-substituted cinnamic acids were readily reacted with sodium benzenesulfinate to produce the desired sulfones in good to high yields. The 3-(pyridin-2-yl)acrylic acid and hex-2-enoic acid were not suitable substrates for this protocol. Moreover, different substitutions on the benzene ring-derived arylsulfinates and methanesulfinate were suitable for producing the desired β-keto sulfones in good yields. The product was formed in traces in the presence of TEMPO (2 equiv.) under standard conditions, suggested a radical pathway. The reaction was performed under a N2 atmosphere instead of oxygen and no product was detected. The oxygen atoms in the obtained products originated from atmospheric oxygen, which was further confirmed by isotopic labeling experiments.
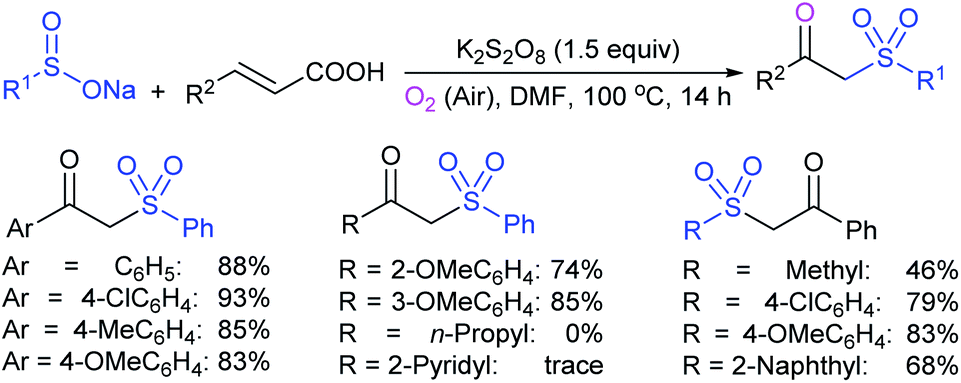 | ||
| Scheme 145 K2S2O8-mediated decarboxylative sulfonylation of cinnamic acids and sodium sulfinates. | ||
Li et al. described a metal-free protocol of unstrained Csp3–Csp3 bond cleavage of chlorohydrins under the catalytic influence of KI and NaHCO3 in THF. A wide range of α-chloro-β-hydroxy ketones underwent bond breaking and sulfonylation with various 4-substituted benzenesulfinates and MeSO2Na to afford β-keto sulfones in satisfactory yields (Scheme 146).211 Unfortunately, trifluoromethane sulfonylation failed on using CF3SO2Na, probably due to its lower nucleophilicity. The practicality of the methodology was also promptly established on the gram-scale without an apparent effect on the yield. Essentially, the sealed tube reaction was sluggish, indicating that the reaction could be an aerobic phenomenon. Next, the TEMPO control experiment suggested that a radical pathway might not be involved.
 | ||
| Scheme 146 KI-mediated oxidative cleavage-sulfonylation of chlorohydrins and sodium sulfinates. | ||
The Gao and Chang group disclosed that I2 catalyzed the direct C–H sulfonylation of β-dicarbonyl compounds with sodium sulfinates under TBHP oxidative conditions (Scheme 147).212 A series of β-dicarbonyl compounds readily coupled with sodium benzenesulfinate to give the corresponding β-dicarbonyl sulfones in good to high yields. Moreover, the aliphatic β-keto esters and β-diesters were also oxysulfonylated to give the desired sulfone products in moderate yields. Besides, the sulfonylation of β-diketones with different kinds of aromatic and methyl sulfinates was also suited to provide the β-diketo sulfones in high yields. In the absence of sodium sulfinates, the α-iodinated ester was quickly detected, which was treated with PhSO2Na and gave the desired product in high yield.
 | ||
| Scheme 147 Iodine-catalyzed oxidative sulfonylation reaction of 1,3-dicarbonyl compounds with sodium sulfinates. | ||
Gao et al. developed the I2-mediated sulfonylation of 1,3-diketones and β-keto esters with various sodium arenesulfinates and methanesulfinate to furnish the desired β-dicarbonyl sulfones in 57–95% yields (Scheme 148).213 The subsequent extension to the synthesis of a series of β-keto sulfones in 56–93% yield through the I2/Na2SO3-mediated deacylative-sulfonylation of 1,3-diketones and β-keto esters with aromatic and aliphatic sulfinates. The deacylative C–C bond cleavage process was also separately verified as being from β-dicarbonyl sulfone by treatment with Na2SO3 to obtain corresponding β-keto sulfone in quantitative yield.
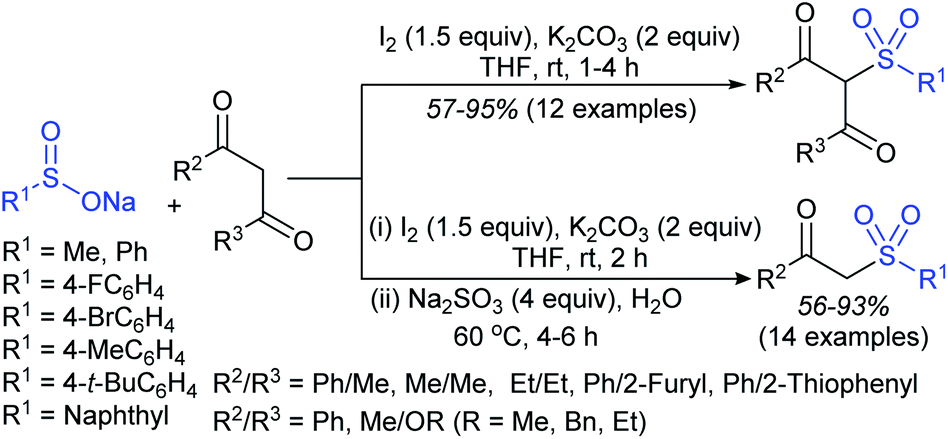 | ||
| Scheme 148 I2-mediated sulfonylation of 1,3-diketones and β-keto esters with sodium sulfinates. | ||
Subsequently, a combination of o-iodoxybenzoic acid (IBX) and a catalytic amount of iodine promoted the deacylative sulfonylation reaction of 1,3-dicarbonyl compounds with sodium sulfinates to yield β-keto sulfones. A series of 1,3-dicarbonyl compounds, including 1,3-diketones, β-keto esters, and β-keto amides efficiently reacted with sodium p-toluenesulfinate and gave the corresponding β-keto sulfones in good to high yields (Scheme 149).214 Furthermore, the deacylative sulfonylation was evaluated utilizing benzoyl acetone with varied aryl and methyl sulfinates to furnish the corresponding products in satisfactory yields. Sodium trifluoromethanesulfinate was found to not be a suitable substrate for this transformation.
 | ||
| Scheme 149 Iodine-catalyzed deacylative sulfonylation reaction of 1,3-dicarbonyl compounds with sodium sulfinates. | ||
The Terent'ev group examined the dramatic influence of the solvent and temperature for the regulated selective sulfonylation of β-ketoesters with sodium sulfinates in the presence of iron(III) salts (Scheme 150).215 Direct oxidative sulfonylation of different β-ketoesters with various aryl-substituted sulfinates in THF:H2O (2:1) at 40 °C led to a series of α-sulfonyl β-ketoesters in good to high yields. The successive deacylative-sulfonylation of aliphatic and aromatic dicarbonyl compounds with a range of sodium sulfinates in i-PrOH:H2O (2:1) under reflux (78 °C) resulted in the formation of different β-keto sulfones in satisfactory yields. Also, sodium methanesulfinate was successfully coupled to obtain the desired methyl sulfonylated product in moderate yield. The developed process involved sulfonyl radical generation via the single-electron oxidation of sodium sulfinate due to the dramatic influence of the Fe(III)-catalyst, the solvent, and the reaction temperature on the product formation.
 | ||
| Scheme 150 Fe(ClO4)3-mediated oxidative direct sulfonylation of β-ketoesters with sodium sulfinates. | ||
Yuan and workers described an efficient electrochemical synthesis of β-keto sulfones from 1,3-dicarbonyl compounds through deacylative sulfonylation under ammonium iodide as the supporting electrolyte in DMSO (Scheme 151).216 The present electrochemical sulfonylation is widely applicable to 3-oxobutanoates, β-keto amides and 1,3-butanediones and gave the corresponding β-keto sulfones in good to high yields. Further, various aryl and alkyl sulfinates reacted with ethyl 3-oxobutanoate and phenylbutane-1,3-dione to give β-oxo sulfones in good yields. Unfortunately, the sodium trifluoromethanesulfinate (F3CSO2Na) substrate was not well-matched in this transformation. According to control experiments, it was inferred that the in situ electrogenerated I2 played a vital role in this transformation.
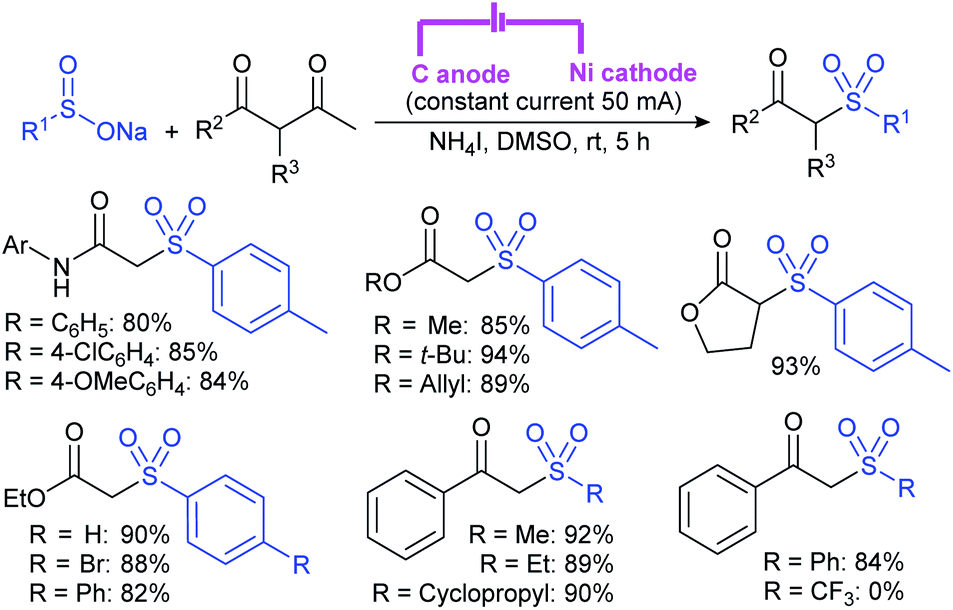 | ||
| Scheme 151 Electrochemical synthesis of β-keto sulfones from 1,3-dicarbonyl compounds with sodium sulfinates. | ||
Very recently, Yavari and Shaabanzadeh reported an electrochemical synthesis of β-keto sulfones via sulfonylation of aryl methyl ketones or aryl acetylenes with sodium sulfinates (Scheme 152).217 It is worth noting that, potassium iodide played the dual role of an oxidant and supporting electrolyte in the reaction. A variety of acetophenone derivatives bearing ortho and para-substituents were smoothly coupled with sodium p-toluenesulfinate and sodium benezenesulfinate provided a series of β-keto sulfones in moderate to good yields. The 2-acetylpyridine, 4′-nitroacetophenone, and sodium methanesulfinate were found to be ineffective under this electrochemical process. Next, aryl alkynes, such as phenyl-acetylene, 4-bromophenylacetylene, and 4-ethynyltoluene were also utilized to produce β-keto sulfones in 51–62% yields. The electrochemical synthesis of β-keto sulfones at a gram-scale (10 mmol) showed practical application for the process.
 | ||
| Scheme 152 Electrochemical sulfonylation of aryl methyl ketones or aryl acetylenes with sodium sulfinates. | ||
A combination of AgNO3/1,10-phen-catalyzed radical-carbene coupling of α-diazo carbonyl compounds with sodium sulfinates in the presence of K2S2O8 as an oxidant to yield β-keto arylsulfones was described by Wan and co-workers (Scheme 153).218 A variety of α-diazo carbonyl compounds were well tolerated by sodium benzenesulfinate, leading to the corresponding β-keto phenylsulfones in moderate to high yields. Next, an epiandrosterone-derived diazo ester was utilized as the substrate to furnish the target product in satisfactory yield. Both electron-withdrawing and electron-donating groups bearing arylsulfinates were well compatible to access the corresponding β-carbonyl arylsulfones in moderate to good yields. The 2-thiophenylsulfinate showed low efficiency, and unfortunately, 2-naphthyl and cyclopropyl-derived sulfinates were challenging substrates for this transformation. Further, the scalable-reaction was performed on 10 mmol and the parent product obtained in the same level yield. The authors also proposed an acceptable mechanism as shown in Scheme 153. K2S2O8 promoted the single electron transfer oxidation of sodium sulfinate and produced the corresponding sulfonyl radical A. The silver carbene intermediate B was obtained from α-diazo carbonyl compounds with AgNO3. The radical-carbene coupling between A and B facilitated the formation of intermediate C. Finally, the protonolysis of C gave the desired β-carbonyl arylsulfones.
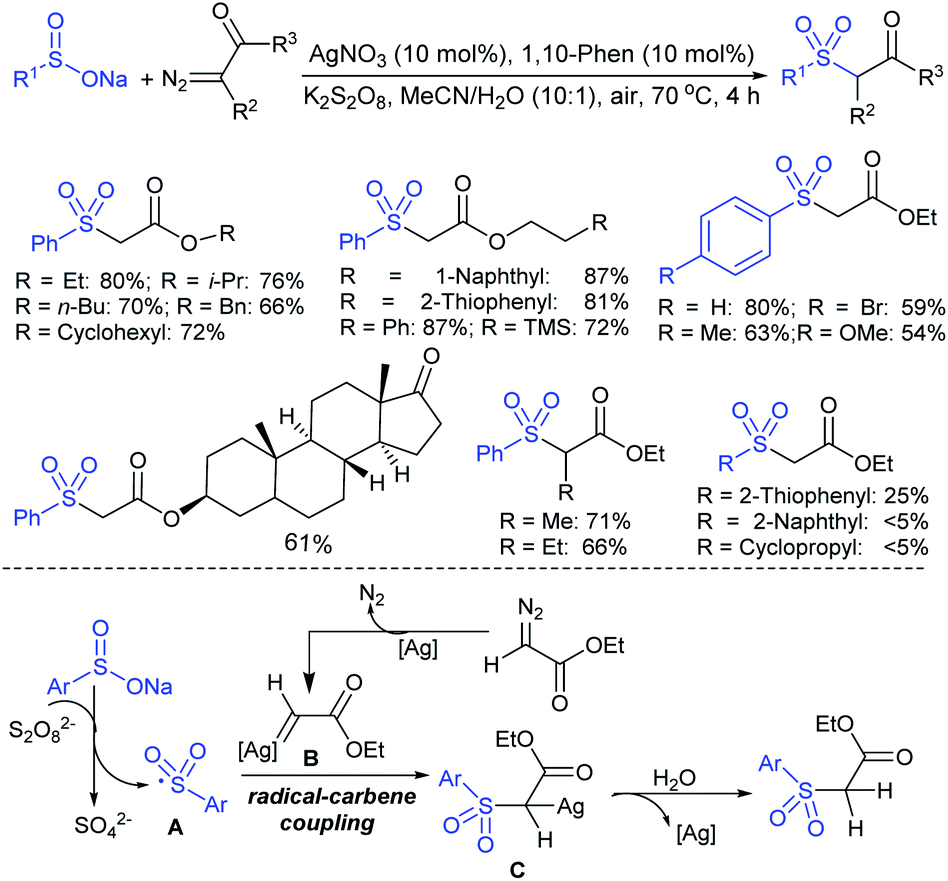 | ||
| Scheme 153 AgNO3-catalyzed radical-carbene coupling of α-diazo carbonyl compounds with sodium sulfinates and the proposed reaction mechanism. | ||
 | ||
| Scheme 154 Electrochemical oxysulfonylation of alkenes using sodium sulfinate salts. | ||
FeCl3-catalyzed the aerobic oxysulfonylation of different alkenes using sodium salts under ultra-sonication irradiation in a short reaction time to provide a mixture of β-keto sulfones and β-hydroxy sulfones in good yields (Scheme 155).220 Mostly, the 4-substituted aromatic alkenes worked well with sodium aryl/alkyl sulfinates and the use of trans-stilbene led to a complex mixture. In the case of α-methylstyrene, β-hydroxysulfone was formed exclusively in a 90% yield.
 | ||
| Scheme 155 FeCl3-catalyzed oxysulfonylation of alkenes using sodium sulfinates. | ||
A facile and straightforward protocol was developed to synthesize hydroxy sulfones and aryl methyl sulfones in aqueous conditions without any reagents, additives, and catalysts (Scheme 156). The Yuan group221 employed the radical sulfonylation of di-tert-butyl peroxide (DTBP) with aryl and heteroaryl sulfinates at 70 °C and obtained a variety of β-hydroxy sulfones in 63–82% yields. A series of aryl methyl sulfones were easily furnished in 61–93% isolated yields from the corresponding aryl, heteroaryl and alkylsulfinates at 110 °C under similar aerobic conditions.
 | ||
| Scheme 156 Radical sulfonylation of di-tert-butyl peroxide (DTBP) with sodium sulfinates. | ||
A convenient ring-opening of epoxides occurred with sodium sulfinates using ionic liquid, tetrapropyl ammonium hydroxide and L-proline [TPA][Pro] in aqueous medium at 80 °C for the synthesis of β-hydroxy sulfones (Scheme 157).222 Venkateswarlu and co-workers showed the reasonable generality for the regioselective sulfonylation of various mono- and di-substituted epoxides with different sodium aryl and alkyl sulfinates to give the corresponding β-hydroxy sulfones in good to high yields.
 | ||
| Scheme 157 Nucleophilic sulfonylation and Ru-catalyzed asymmetric reduction of terminal alkynes with sodium sulfinates. | ||
A vicinal electrochemical sulfonylation was established by Chen, Chang, and co-workers to synthesize β-hydroxy sulfones from α-methylstyrenes with sodium sulfinates using KI as a supporting electrolyte (Scheme 158).223 A variety of functionalized α-methylstyrenes reacted satisfactorily with different aromatic sulfinates bearing electron-withdrawing and electron-donating groups to give a broad range of desired β-hydroxy sulfones in good to high yields. Additionally, the aerobic electrochemical oxysulfonylation of cyclic alkenes occurred conveniently with sodium p-toluenesulfinate to form desired β-hydroxy sulfones in high yields. The authors explained that the role of the glassy carbon electrode (GCE) was to oxidize iodine ions to form I2, which produced the corresponding sulfonyl iodide. This intermediate decomposed to form a sulfonyl radical and iodine radical, which regenerated iodine to complete the electrochemical process.
 | ||
| Scheme 158 Electrochemical oxysulfonylation of alkenes using sodium sulfinate salts. | ||
In 2014, the Cheng and Liu group competently described a one-pot nucleophilic sulfonylation of α-bromo ketones and sodium sulfinates, followed by (S,S)-Ru-catalyzed (30) asymmetric transfer hydrogenation using HCO2Na as a hydrogen source, providing enantiomerically enriched β-hydroxy sulfones. Sodium benzenesulfinate was quickly sulfonylated with a range of α-bromoacetophenones bearing electron-donating or electron-withdrawing substituents and provided a variety of optically active β-hydroxy sulfones in good to high yields and excellent enantioselectivities (Scheme 159).224 Encouraged by these results, the authors utilized aliphatic α-bromoketones that were also able to furnish the desired products in good yields with slightly reduced enantioselectivities. Additionally, sodium p-toluenesulfinate and sodium methane-sulfinate successfully produced related products with high yields and enantioselectivities. Almost the same level of results was observed, even lowering the catalyst loading (2 mol%) in the one-pot reductive-sulfonylation.
 | ||
| Scheme 159 Nucleophilic sulfonylation and Ru-catalyzed asymmetric reduction of terminal alkynes with sodium sulfinates. | ||
In 2019, the Zhou group successfully investigated the one-pot synthesis of chiral β-hydroxy sulfones via a FeCl3-catalyzed aerobic oxysulfonylation of terminal alkynes with sodium sulfinates in MeOH/H2O (3:1) at 50 °C to generate β-keto sulfones. Subsequently, reduction was carried out using (S,S)-Ru-catalyzed (30) asymmetric transfer hydrogenation with HCO2Na as a hydrogen source in a one-pot operation (Scheme 160).225 A variety of enantiomerically enriched β-hydroxy sulfones were obtained in low to good yields and up to 99.9% ee values using a wide range of aryl alkynes and different sodium arylsulfinates. However, the sodium trifluoromethanesulfinate afforded the β-hydroxy sulfone in only 17% yield and 71.2% ee. This successive oxysulfonylation-reductive operation was demonstrated on the gram-scale (2.04 g with 98.7% ee) to synthesize the corresponding β-hydroxy sulfone with only 2 mol% catalyst loadings.
 | ||
| Scheme 160 Ru-catalyzed asymmetric reduction of terminal alkynes with sodium sulfinates. | ||
| Entry | Representative illustration of the synthesis of aryl sulfones | Range of yield | Ref. |
|---|---|---|---|
| 1 | 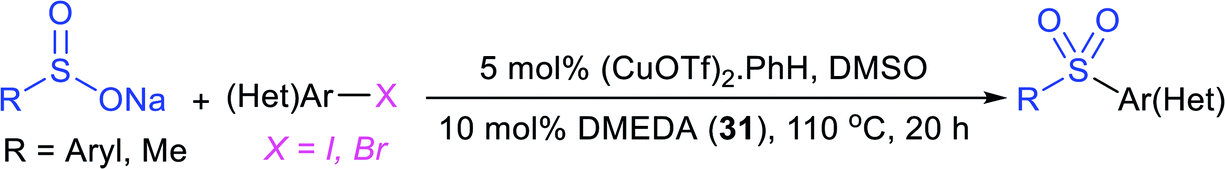 |
27–96% (X = I) | 228 |
| 24% (X = Br) | |||
| 2 |  |
46–96% | 229 |
| 3 | 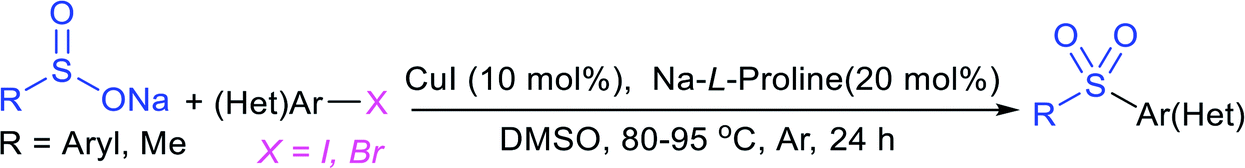 |
55–93% (X = I) | 230 |
| 46–89% (X = Br) | |||
| 4 |  |
81–98% (X = Cl) | 231 |
| 82% (X = Br) | |||
| 91% (X = I) | |||
| 75% (X = OTf) | |||
| 5 |  |
60–92% (X = Cl) | 232 |
| 75–85% (X = Br) | |||
| 50% (X = I) | |||
| 6 |  |
40–96% (X = Cl) | 233 |
| 38–98% (X = Br) | |||
| 55% (X = NO2) | |||
| 7 |  |
75–97% (X = I) | 234 |
| 72% (X = Br) | |||
| 8 |  |
65–95% (X = I) | 235 |
| 60–72% (X = Br) | |||
| 9 |  |
50–94% | 236 |
| 10 |  |
64–96% (X = I) | 237 |
| 47% (X = Br) | |||
| 11 | 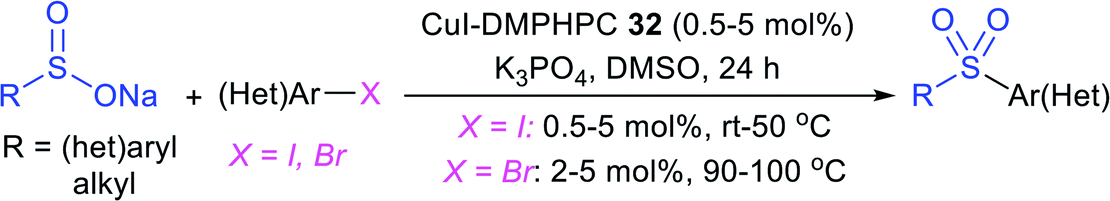 |
42–98% (X = I) | 238 |
| 29–98% (X = Br) | |||
| 12 |  |
27–89% (X = I) | 239 |
| 26–79% (X = Br) | |||
| 66% (X = Cl) | |||
| 12% (X = F) | |||
| 13 | 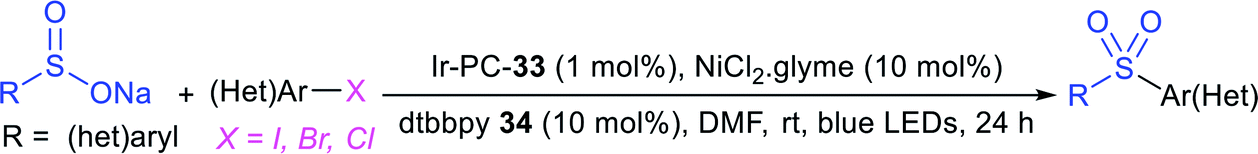 |
50–90% (X = I) | 240 |
| 32–94% (X = Br) | |||
| 35–43% (X = Cl) | |||
| 14 |  |
14–89% | 241 |
| 15 |  |
44–86% (X = I) | 242 |
| 33–83% (X = Br) | |||
| 16 |  |
32–95% | 243 |
| 17 |  |
21–86% (X = I) | 244 |
| 41–93% (X = Br) | |||
| 46% (X = Cl) | |||
| 18 |  |
52–99% (X = I) | 245 |
| 71–97% (X = Br) | |||
| 62–99% (X = Cl) | |||
| 89% (X = F) | |||
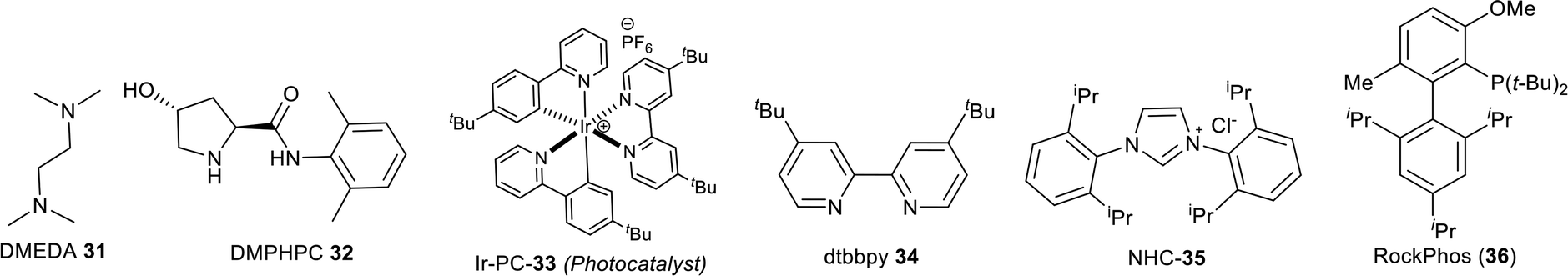 |
|||
Zhu and Ma described the CuI/L-proline sodium salt-catalyzed reaction of aryl and heteroaryl iodides that were readily coupled with MeSO2Na and PhSO2Na at 80–95 °C in DMSO to give the corresponding diaryl sulfones in good to high yields (Table 15; entry-3).230 The aryl iodides bearing a wide range of functional groups, including hydroxyl, amino, acetanilide, ketone, ester, nitrile groups, were compatible. Notably, various aryl bromides were also accommodated; however, those with electron-rich groups showed better reactivity than those with electron-deficient groups. Maloney et al. reported that a series of electron-deficient chloropyridines were successfully coupled with sodium sulfinates in the presence of a catalytic amount TBACl in DMAc (N,N-dimethylacetamide) at 100 °C, affording an array of sulfonylated pyridines in good to high yields (Table 15; entry-4).231 This metal-free sulfonylation was extended to iodo-, bromo- and triflate-derived pyridines and worked equally well, and the desired products were obtained in good to high yields. The authors stated that the role of TBACl in the in situ formation of n-Bu4NSO2Tol was to impart a significant rate acceleration. Similarly, Yuan and Guo revealed the CuCl-catalyzed coupling of electron-deficient aryl chlorides and chloropyridines with aryl and alkyl sulfinates under microwave irradiation within 3–30 min and delivered a broad range of unsymmetrical diaryl sulfones and aryl–alkyl sulfones in moderate to high yields (Table 15; entry-5).232 Moreover, 2-halothiophene derivatives were also well-tolerated in this coupling process.
Chen, Yu and co-workers disclosed transition metal-free sulfonylation via an SNAr reaction of five-membered haloheterocycles in DMSO at 110 °C. Widespread halo/nitro-thiophene derivatives were smoothly sulfonylated with different aryl/alkyl sulfinates to furnish aryl-thiophenyl sulfones in low to good yields (Table 15; entry-6).233 It is noted that chlorothiophene delivered the product with the same high yield as compared with bromothiophene. Additionally, 2-bromofuran, 2/3-bromoindoles, 2-bromothiazole, 2-halobenzothiazoles, 2-bromo-N-methyl-imidazole and 8-bromocaffein were efficiently converted into the corresponding sulfones. However, 2-bromo/2-nitro-thiophene, 5-chloro-N-methylimidazole and 2-chlorobenzoxazole remained as challenging substrates.
Zhang and co-workers also employed CuI-catalyzed coupling between various aryl iodides and aryl/alkyl sulfinates under the influence of D-glucosamine as a green ligand, and provided a variety of unsymmetrical aryl sulfones in good to high yields (Table 15; entry-7).234 Bromobenzene also participated at elevated temperature (120 °C) and longer reaction time than aryl iodides. Along the same lines, a chitosan-supported copper catalyst for the synthesis of aryl and alkyl sulfones from aryl halides with both aryl and alkyl sulfinates was presented by the same group (Table 15; entry-8).235 Less reactive aryl bromides worked well at higher reaction temperature (140 °C) and prolonged time. In particular, the chitosan@copper catalyst was recycled and reused five times without significant loss of catalytic activity. A concise route for the synthesis of the marketed drug zolimidine (antiulcer) was also successfully achieved.
The Handa group described the proline-based surfactant FI-750-M for the selective nucleophilic aromatic substitution of polyfluoro-(hetero)arenes by sodium sulfinate salts in water under nanomicelles with different binding sites (Table 15; entry-9).236 A variety of tetrafluoroarenes and tetrafluoropyridines were smoothly sulfonylated with a range of functionalized sulfinates bearing electronically different systems, which led to polyfluoroaryl(heteroaryl)sulfones in good to high yields. CuI/DMPHPC (4-hydroxy-L-proline-derived 2,6-dimethyl-aniline amide; 32) is an efficient catalytic system for the coupling of (hetero)aryl halides with sodium sulfinates (Table 15; entry-10).237 Remarkably, low catalyst loadings (0.5 to 5 mol%) and moderate reaction temperatures were used to synthesize a wide range of (hetero)aryl sulfones in good to high yields. As a result, a large number of substituted aryl(hetero) bromides were coupled smoothly with MeSO2Na and PhSO2Na at 90–100 °C. Additionally, several aryl and heteroaryl iodides are well compatible under mild conditions. Ge et al., developed an efficient catalytic system using Cu(OAc)2 (1 mol%)/DMEDA (31, 2 mol%) for the coupling of various substituted aryl iodides with a few aryl and alkyl sulfinate salts at 110 °C and obtained moderate to high yields of aryl sulfones (Table 15; entry-11).238 Although the catalytic amount was substantially reduced, unfortunately, the aryl bromide (bromobenzene) reacted poorly with benzenesulfinate as compared with iodobenzene. Iron-catalyzed sulfonylation occurred via the SNAr reaction of halopyridines with sodium sulfinates to provide pyridyl aryl sulfones. A variety of 2-halopyridines (F, Cl, Br, and I) were compatible with sodium p-toluenesulfinate; however, 2-bromo- or 2-iodopyridines gave better yields (Table 15; entry-12).239 The study of methyl-substitution at various positions of 2-halopyridines had no significant influence on the outcome. Although both 2-bromo- and 2-iodopyridines were sulfonylated with a wide range of arenesulfinates, 2-iodopyridines gave better yields.
In early 2018, the Rueping, Manolikakes, and Molander groups independently reported Ni/photoredox dual catalysis for constructing aryl- and heteroaryl sulfones under blue LED irradiation at room temperature without any base. Rueping and co-workers elegantly employed a powerful sulfonylation of a series of aryl, heteroaryl, and vinyl bromides and iodides, as well as more challenging aryl chlorides with a range of aryl and heteroaryl sulfinates under the catalytic influence of NiCl.glyme, Ir-PC (33) and dtbbpy (34) to give a broad spectrum of the corresponding sulfones in moderate to good yields (Table 15; entry-13).240 Similarly, the Manolikakes group used the NiCl2·6H2O/Ru(bpy)3Cl2·6H2O dual catalyst system for a cross-coupling of various aryl and heteroaryl iodides with different aryl and alkyl sulfinates to afford a wide range of aryl sulfones in moderate to good yields (Table 15; entry-14).241 This protocol was restricted to only (hetero)aryl iodides, however, the method was explored for the synthesis of sildenafil, which is a drug-like scaffold. Molander and co-workers successfully demonstrated dual catalysis, using Ni(phen)(H2O)4]Cl2/Ru(bpy)3(PF6)2, for the construction of aryl- and heteroaryl sulfones under blue LEDs irradiation at room temperature in the absence of base (Table 15; entry-15).242 A broad range of electrophilic partners, such as aryl iodides and aryl bromides bearing different functional groups, nitrogen-based heterocycles, amides, lactones, phenols were all easily coupled with sulfinates to give diaryl(heteroaryl) sulfones in moderate to good yields. Of note, both electron-rich/-poor arylsulfinates as well as heteroarylsulfinates proceeded smoothly, giving the corresponding sulfones in acceptable yields. In this study, the formation of the undesired byproduct thioether was observed in trace amounts.
Manolikakes and co-workers further extended the nickel/N-heterocyclic carbene (NHC-35) salt-catalyzed cross-coupling of aryl bromides with sodium sulfinates for the synthesis of diarylsulfones (Table 15; entry-16).243 A variety of electron-rich and electron-poor aryl bromides coupled with benzenesulfinate in moderate to good yields. The ortho-substituted aryl bromide substrates were ineffective under the same conditions. Additionally, different heterocyclic aryl bromides were employed as successful coupling partners, affording the heteroaryl–aryl sulfones in 50–95% although the various aryl sulfinates proceeded efficiently, heterocyclic and aliphatic sulfinates could not afford the desired sulfones. In 2019, Yan, Zhang, and co-workers presented the UV-light promoted coupling of aryl halides with sodium sulfinates for the synthesis of diaryl and aryl–heteroaryl sulfones via the single-electron transfer (SET) of the electron donor–acceptor (EDA) complex process (Table 15; entry-17).244 Various aryl iodides, bromides, and chlorides were coupled with sodium p-toluenesulfinate, however, aryl chloride exhibited low reactivity. A variety of aryl and heteroaryl iodides showed slighty better reactivity than the corresponding bromides. Different substituted aryl, pyridinyl- and alkyl sulfinates were also smoothly coupled with 4-iodobenzonitrile and the desired sulfones were obtained in moderate to good yields. The Larionov group employed an eco-friendly and efficient method of synthesizing N-heterocyclic sulfones in the presence of Na2S2O8 at room temperature conditions (Table 15; entry-18).245 Several kinds of nitrogen heterocycles, substituted 2/4-haloquinolines, 4-halopyridines and 1-haloisoquinolines including quinoxalines and quinazoline were easily sulfonylated and produced the heteroaryl sulfones in good and excellent yields. A variety of aryl-, thiophenyl- and methyl-derived sulfinates as well as dienyl sulfinates were also successfully reacted to provide the desired sulfones in satisfactory yields. Unusually, sodium hydroxymethylsulfinate served as a sulfonyl agent to prepare symmetrical bis-4-quinolinyl sulfones in acceptable yields.
In 2004, the cupric acetate-mediated cross-coupling of boronic acids with sodium sulfinates was introduced by the Beaulieu group. A series of aryl sulfones was easily accessed in good to high yields from a wide variety of functionalized aryl and heteroaryl boronic acids with MeSO2Na, PhSO2Na and 4-ClC6H4SO2Na. Generally, the electron-rich/poor groups and different substitution patterns on the benzene ring did not affect the sulfonylation coupling (Table 16; entry-1).246 Subsequently, Huang and Batey employed the cross-coupling protocol under a catalytic amount of the Cu(OAc)2/1,10-phenanthroline system under anhydrous conditions (Table 16; entry-2).247 A broad range of aryl- and alkenyl-boronic acids were well sulfonylated with the p-substituted arylsulfinates or methylsulfinate to form arylsulfones and vinyl sulfones in moderate to high yields. The use of potassium trifluorophenyl borate as the reaction partner instead phenylboronic acid led to a low yield of the corresponding sulfone. Concurrently, the Tse group utilized 1-benzyl-imidazole as a ligand for the Cu(OAc)2-catalyzed cross-coupling of aryl-, heteroaryl-, and alkenyl-boronic acids with MeSO2Na, PhSO2Na and p-tolylSO2Na to afford the corresponding sulfone analogs in satisfactory yields (Table 16; entry-3).248 Later, the [bmim][OTf] ionic liquid was found to be a suitable medium for the Cu(OAc)2-catalyzed sulfonylation of aryl boronic acids with sulfinic acid salts (p-tolylSO2Na and MeSO2Na) to furnish the desired aryl sulfones in good yields (Table 16; entry-4).249 In 2014, copper ferrite (CuFe2O4) nanoparticles were introduced by Sreedhar and co-workers to synthesize aryl sulfones (Table 16; entry-5).250 Various alkyl/aryl halides and aryl boronic acids were effectively sulfonylated with sodium benezesulfinate and sodium p-toluenesulfinate with the CuFe2O4/1,10-phen catalytic system to generate diaryl and aryl–alkyl sulfones in good to high yields. Noteworthily, the magnetically separable CuFe2O4 was recovered and reused for five consecutive cycles, which was entirely consistent with the catalytic efficiency. Furthermore, Cu(OTf)2 catalyzed the cross-coupling of aryl- and vinylboronic acids with PhSO2Na and 4-MeC6H4SO2Na under microwave irradiation for the synthesis of diaryl and vinyl sulfones in 41–81% yields (Table 16; entry-6).251





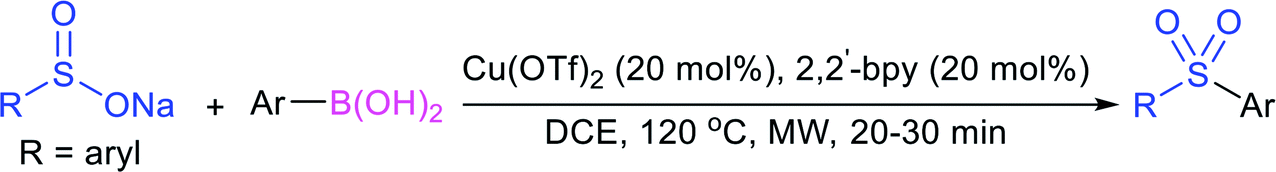


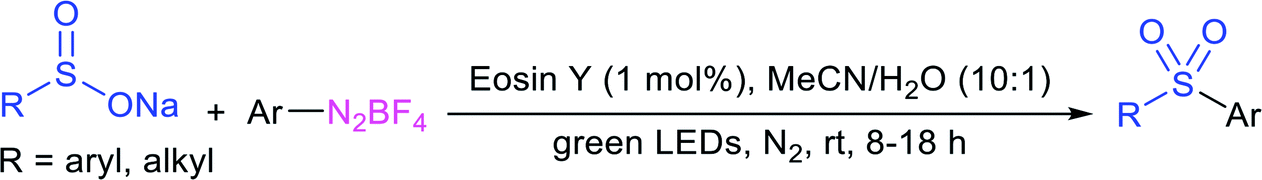



In 2014, Yang and co-workers firstly introduced arylsilanes for the copper-catalyzed cross-coupling with sodium arenesulfinates for the synthesis of diaryl sulfones (Table 16; entry-7).252 The reaction scope was proved with respect to various arylsilanes and different arylsulfinates to afford the diaryl sulfones in good to high yields. In 2016, the Shekhar group examined aryltriflates for cross-coupling with sodium trifluoromethanesulfinate. The combination of Pd2(dba)3, RockPhos (36), and TDA [tris(3,6-dioxaheptyl)amine] catalytic systems were compelling for triflination using NaSO2CF3 to afford a broad spectrum of corresponding triflones in 15–86% yields (Table 16; entry-8).253 Unfortunately, pyridin-2-yl-triflate could not be obtained under the same conditions. Further, the authors explained the order of the trifilation reactivity as PhOTf ≫ PhCl ≫ PhBr, which is reliable with transmetalation occurring. Aryldiazonium salts were also employed as coupling partners with sodium sulfinates under visible-light irradiation. The Yadav group demonstrated the eosin-Y catalyzed tosylation of aryldiazonium tetrafluoroborate bearing electron-withdrawing and electron-donating substituents on the benzene ring to deliver the corresponding diaryl sulfones in 48–95% yields (Table 16; entry-9).254 A few sodium arenesulfinates and alkyl sulfinates participated in obtaining representative diaryl sulfones.
Very recently, Ag2CO3-promoted the direct decarboxylative sulfonylation between aromatic carboxylic acids and sodium sulfinate for the synthesis of aryl sulfones (Table 16; entry-10).255 A variety of aryl and heteroaryl carboxylic acids were satisfactorily coupled with different aryl-substituted sulfinates to give the corresponding diaryl sulfones in moderate to good yields. Although cyclopropanesulfinate participated to deliver the desired alkyl aryl sulfone, nevertheless, CH3SO2Na and CF3SO2Na were unsuccessful for this protocol. The Manolikakes group successfully utilized diaryliodonium salts to synthesize diaryl sulfones in the absence of a reagent/catalyst. A wide variety of symmetrical and unsymmetrical diaryliodonium triflates were easily coupled with a range of aryl and heteroaryl sulfinates to furnish the desired sulfones in satisfactory yields (Table 16; entry-11).256 In the case of unsymmetrical salts, only the bulky aryl moiety was transferred to deliver sulfones. Notably, MeSO2Na was the inevitable substrate under these conditions. Simultaneously, the Cu2O-catalyzed cross-coupling of diaryl-iodonium salts with sodium trifluoromethanesulfinate was demonstrated by Shekhar and co-workers (Table 16; entry-12).257 Various symmetrical and unsymmetrical diaryl- and diheteroaryl-iodonium salts bearing different counter-ions (PF6, OTs, BF4, OTf) were systematically examined and coupled with F3CSO2Na to form the anticipated aryl trifluoromethylsulfones in moderate to high (41–88%) yields.
In 2014, Pandya and Mhaske established the TBAF-promoted sulfonylation of o-silyl aryl triflates, which generated in situ aryne precursors with sodium sulfinates for the synthesis of aryl sulfones (Scheme 161).258 An array of sulfones including diaryl-, aryl–heteroaryl-, and aryl–alkyl sulfones were easily obtained in good to high yields from different o-silyl aryl triflates with a range of aryl/heteroaryl/alkyl sulfinates.
 | ||
| Scheme 161 The TBAF-promoted sulfonylation of o-silyl aryl triflates with sodium sulfinates. | ||
3.4.9.1. Direct C–H sulfonylation. Tan and co-workers259 employed the copper-catalyzed direct ortho-sulfonylation via the C–H functionalization of 8-aminoquinoline (acts as a bidentate directing group)-derived benzamides with sodium sulfinates. Various functional groups on the benzene ring of benzamides are well tolerated in this transformation. Moreover, pyridine and thiophene derivatives were smoothly sulfonylated in 47% and 42% yields. Next, the scope of the sodium sulfinates was also successfully investigated, and substituted aromatic sulfinates were sulfonylated to generate the desired 2-arylsulfonyl benzamide derivatives in good to high yields (Scheme 162). The aliphatic sodium methanesulfinate showed low reactivity to provide the corresponding benzamide to produce the desired sulfone in only 46% yield. The standard condition was not inhibited when the reaction was conducted with TEMPO, indicating that the reaction may not involve a radical pathway. Next, the rate-determining step of the sulfenylation was confirmed by the intramolecular H/D competition experiment (kH/kD = 3.5), suggesting that the ortho C–H bond cleavage of benzamide was involved.
 | ||
| Scheme 162 Cu-mediated ortho-sulfonylation of 8-aminoquinoline benzamides. | ||
At the same time, Rao and Shi demonstrated a copper-catalyzed direct sulfonylation of C(sp2)–H bond of 2-pyridinyl-derived benzamides with sodium sulfinates using a removable pyridinyl directing group (Table 17A).260 This C–H sulfonylation allowed a wide range of functional groups, including electron-rich and electron-deficient benzamides, which were generally reacted with sodium benzenesulfinate to afford the desired sulfones in moderate to high yields. Subsequently, a series of electron-donating and electron-withdrawing groups-derived arylsulfinates were also found to be compatible with providing the corresponding diaryl sulfones in moderate to high yields. Similarly, in 2017, Gong, Song, and co-workers developed a Ni-catalyzed ortho-sulfonylation of C(sp2)–H bonds with sodium sulfinates directed by (pyridin-2-yl)isopropylamine (Table 17B).261 Various diaryl and alkyl aryl sulfones were prepared in 20–90% yields under the catalytic influence of NiBr2 (10 mol%) with Ag2CO3 (2.0 equiv.) as an oxidant in CHCl3 at 120 °C. In addition to arene and heteroarene substrates, the method can also be applied to alkene substrates to provide the desired vinyl sulfones. Moreover, a plausible Ni(I)/Ni(III) mechanism was rationalized by the authors based on control experiments and related precedents. A mixture of mono- and disulfonylated products was observed in some cases, which reflected a considerable drawback of this protocol.
|
||
|---|---|---|
| Sulfones | Method-A | Method-B |
 |
R = Cl: 76% | R = Cl: 76% |
| R = Me: 70% | R = Me: 52% | |
 |
R = Cl: 62% | R = Cl: 75% |
| R = CF3: 74% | R = CF3: 79% | |
| R = CO2Me: 76% | R = CO2Me: 85% | |
 |
R = Me: 74% | R = Me: 61% |
| R = CF3: 56% | R = CF3: 82% | |
| R = OMe: 62% | R = OMe: 50% | |
 |
Ar = 2-naphthyl: 80% | Ar = 2-naphthyl: 42% |
 |
— | 61% |

The Shi group performed elegant work on the Pd(OAc)2-catalyzed ortho-sulfonylation of the C(sp3)–H bond of the 8-aminoquinoline auxiliary-derived carboxamide with sodium sulfinates. This protocol particularly represented the first example of the transition-metal-catalyzed sulfonylation of unactivated C(sp3)–H bonds. This transformation featured excellent functional group tolerance of both the coupling partners to afford a broad range of aryl alkyl sulfones in moderate to good yields (Scheme 163).262 Further, the viability of C(sp3)–H of complex carboxamide-derived substrates, such as β-citronellol, (−)-santonin, and cholic acid, was investigated when they were successfully converted into their corresponding sulfones in 34%, 44%, and 43% yields, respectively.
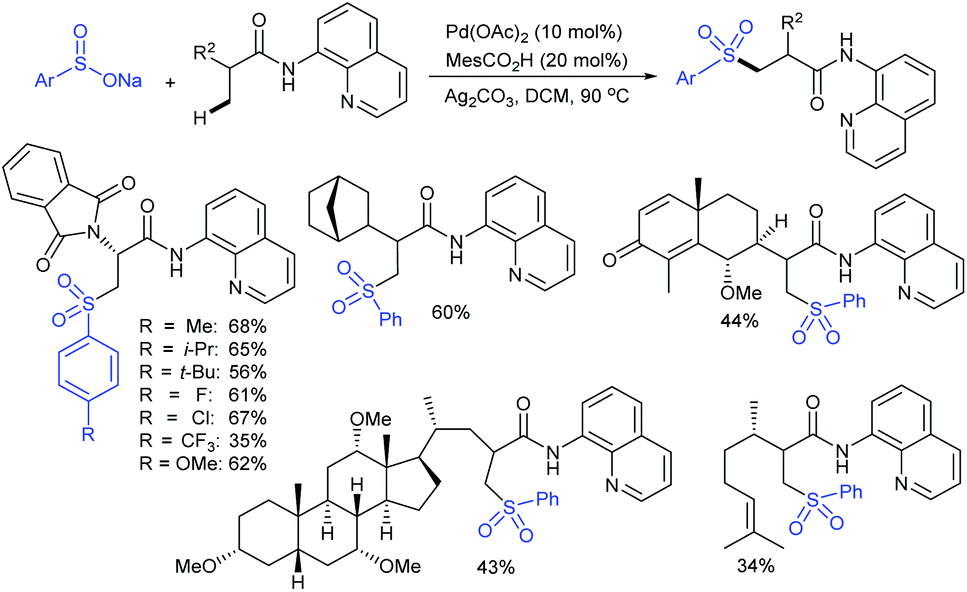 | ||
| Scheme 163 Pd(II)-catalyzed sulfonylation of C(sp3)–H functionalization. | ||
Xia et.al., reported an interesting copper acetate-catalyzed remote sulfonylation at the C5 position of N-(quinolin-8-yl)benzamide derivatives by utilizing sodium sulfinates (Scheme 164).263 An unusual regioselective C–H sulfonylation of N-(quinolin-8-yl)benzamide derivatives with a range of sodium sulfinates was carried out for the synthesis of diverse aminoquinolines-sulfones that were successfully obtained in moderate to high yields. Notably, sodium sulfinates containing electron-rich groups proved to have superior reactivity as compared to electron-poor groups, thus affording lower yields. Disappointingly, N-(naphthalen-1-yl)benzamide, N-methyl-N-(quinolin-8-yl)benzamide and quinolin-8-yl benzoate were unsuccessful at providing the sulfonylated products under the same reaction conditions. With the addition of TEMPO or BHT, this reaction was inhibited, or a trace of the product was detected. As a result, the sulfonylation may undergo a free radical mechanism via a single-electron-transfer (SET) process.
 | ||
| Scheme 164 Cu-catalyzed sulfonylation at the C5 position of N-(quinolin-8-yl)benzamide derivatives. | ||
Simultaneously, the Wu group demonstrated complementary catalysis strategies, Ru/Cu photocatalysis, or a traditional Cu/Ag cocatalysis for the remote C–H sulfonylation of 1-naphthylamides with sulfinate salts under mild reaction conditions (Scheme 165).264 The direct C4–H sulfonylation of various 1-naphthylamine derivatives with a series of sodium aryl/heteroaryl/alkyl sulfinates proceeded smoothly at room temperature and tolerated various functional groups. One representative example was performed in a gram-scale reaction using N-(naphthalen-1-yl)picolinamide to demonstrate the synthetic applications. The reaction was conducted under a nitrogen or an oxygen atmosphere instead of air and afforded the product in similar yields and did not affect the sulfenylation. The radical scavenger (TEMPO and BHT) experiments revealed that the sulfonylation might be via a radical process.
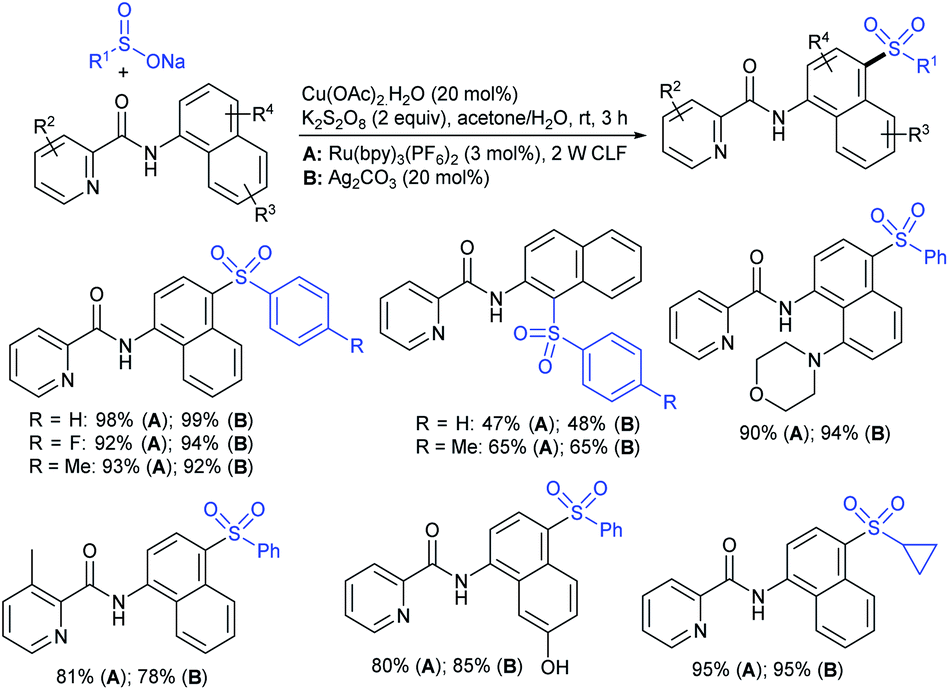 | ||
| Scheme 165 Ru/Cu or a Cu/Ag cocatalysis for remote C–H sulfonylation of 1-naphthylamides with sulfinate salts. | ||
The Cu(OAc)2-assisted direct three-component coupling of 8-aminoquinoline, acyl chlorides, and arylsulfinates was developed by Liu and co-workers toward synthesizing 5-sulfonyl aminoquinolines in a one-pot operation (Scheme 166).265 A step economical procedure was carried out for C5–H sulfenylation, without any further operation in preparing the 8-aminoquinoline-derived directing group substrates. Various linear and branched acyl chlorides as well as aroyl chlorides showed good functional group tolerance for the synthesis of the N-acyl/N-aroyl quinolinyl sulfones in moderate to good yields. Only three different para-substituted sodium arylsulfinates were effective in this transformation; unexpectedly, the 4-chlorophenylsulfinate or methylsulfinate declined to provide the desired products.
 | ||
| Scheme 166 Three-component coupling of 8-aminoquinoline, acyl chlorides and arylsulfinates. | ||
Manolikakes and co-workers266 utilized a new class of oxazolidine-based directing group amides for the direct C(sp2)–H sulfonylation with sodium sulfinates under the influence of copper acetate in trifluoroethanol (TFE) at 80 °C under air. The reaction scope was successfully assessed regarding the substituted benzamide as well as aryl/alkyl sulfinate components. A series of diaryl sulfones were generated in moderate to high yields with good functional group compatibility (Scheme 167). The heteroaromatic pyridine and furan-derived amide substrates were also sulfonylated in lower yields; however, heteroaryl sulfinates did not yield the desired sulfonylated products.
 | ||
| Scheme 167 Sulfonylation of oxazoline-based directing group amides with sulfinates. | ||
The same authors further envisioned a copper-catalyzed para-selective C–H functionalization via the cross-dehydrogenative coupling of anilines with sulfinate salts (Scheme 168).267 After careful investigation of various directing groups, the isoquinoline-1-carboxamide group was found to be a potential directing group involving a chelation complex with the copper catalyst for ortho-sulfonylation through an intriguing single-electron-transfer (SET) process. The para-sulfonylation scope was explored using different sodium sulfinates as well as isoquinoline-1-carboxamide-derived anilines, which afforded a range of diaryl sulfones in good to high yields. The major drawback is that the substrates bearing either electron-withdrawing or electron-donating substituents in the ortho-position did not provide the desired sulfonylated products. Unexpectedly, the sodium 2-pyridinesulfinate was not sustained in this transformation.
 | ||
| Scheme 168 The para-sulfonylation of isoquinoline-1-carboxamide-based directing group amides with sulfinates. | ||
Xiao, Deng, and co-workers illustrated the KI-mediated direct C(sp3)–H sulfonylation of 2-methylquinolines to synthesize 2-sulfoylmethyl quinoline in the presence TBHP as an oxidant. The oxidative sulfonylation of 2-methylquinoline with various aromatic substituted sulfinates and aliphatic sulfinates produced the desired sulfones in good to high yields (Scheme 169).268 Further, a series of substituted 2-methylquinolines, 2,3-dimethylquinoxaline and 2-methyl-3-phenylquinoline favorably employed the sulfonylation process with sodium benzenesulfinate to give the corresponding aryl sulfones in good yields. The stoichiometric amount of TEMPO was added in standard conditions, and no desired product was observed, thus indicating that the reaction may proceed through a radical pathway.
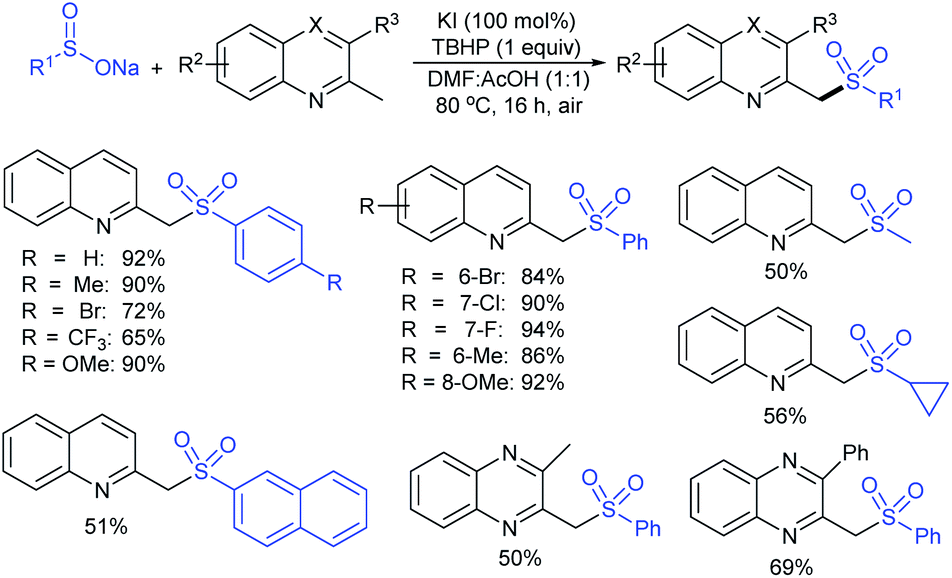 | ||
| Scheme 169 Sulfonylation of 2-methylquinolines and 2-methylquinoxalines. | ||
Karmakar and Samanta developed a direct Pd(II)-catalyzed site-selective sulfonylation of removable bidentate picolinamide-derived benzylamines using sodium arylsulfinates (Scheme 170).269 The selective ortho-sulfonylation was explored with various functional groups like alkyl and methoxy groups and notably, halogens also survived under Pd(II)-catalyzed conditions. Both coupling partners, benzylamines and sulfinate salts, have different electronic and steric properties that were systematically examined and well-tolerated. A series of desired sulfonylated products were obtained in moderate to high yields. No desired product was obtained when the reaction used a meta-CF3 benzenesulfinate or methanesulfinate.
 | ||
| Scheme 170 The Pd(II)-catalyzed direct sulfonylation of benzylamines with sodium sulfinates. | ||
Copper-catalyzed ortho-C–H disulfonylation of ferroceneamide with sodium sulfinate salts in the presence of Ag2CO3 under mild conditions was achieved by Kumar and co-workers. The 8-aminoquinoline directing group-derived ferrocene was smoothly sulfonylated with a variety of substituted arylsulfinates to give the desired ferroceneamide aryl sulfones in 42–71% yields (Scheme 171).270 Although monosulfonylation was carefully attempted by reducing sulfinates, the protocol was feasible only for disulfonylation. It is worth mentioning that the disulfonylation of ferroceneamide was successfully explored at a one-gram scale with the same level outcome as compared to small-scale experimentation.
 | ||
| Scheme 171 The Cu-catalyzed C–H disulfonylation of ferrocene amide with sulfinate salts. | ||
The disulfonylation reaction was initiated by deprotonation of 8-aminoquinoline-derived ferroceneamide with K2CO3 and the subsequent substitution with Cu(OAc)2 gave copper-amidate A (Scheme 172). The strong complexation between the quinoline-moiety and copper metal generated the cuprate intermediate B through a concerted metalation-deprotonation (CMD) process. Then, the addition of sodium sulfinic salt to the ferrocene copper complex gave the copper(III) species C via a single-electron transfer. A reductive-elimination followed by the ligand exchange of copper afforded C–SO2 to form intermediate D. Due to the strong bi-coordination of the aminoquinoline moiety, it was still bound with copper afterward, and the second C–H sulfonation took place in a similar path to afford disulfonated ferrocene. As a result, the authors observed only bisulfone ferrocene rather than the mono sulfonylated product and even decreased the sulfinate salt.
 | ||
| Scheme 172 A plausible mechanism for the Cu-catalyzed C–H disulfonylation of ferroceneamide with sulfinate salts. | ||
The Wu group disclosed the iodine/tert-butyl hydroperoxide (I2/TBHP)-mediated benzylic C–H sulfonylation of phenol derivatives with sodium arylsulfinates under metal-free conditions at room temperature (Scheme 173).271 The radical sulfonylation of 2,4,6-trimethylphenol with a series of sodium arylsulfinates (including electron-donating or electron-withdrawing substituents on the benzene ring) reacted smoothly to give the corresponding products in moderate to good yields. Similarly, various phenol derivatives were also explored with sulfinate salts to provide the corresponding products in good yields. The method was only suitable for arylsulfinates, where sodium methanesulfinate and sodium trifluoromethanesulfinate resulted in complications. Additionally, the tandem sulfonylation-peroxidation between 2,6-di-tert-butyl-4-methylphenol and sodium 4-methyl-benzenesulfinate under TBHP (4 equiv.) allowed unexpected peroxide-derived sulfone products in modest yields.
 | ||
| Scheme 173 I2/TBHP-mediated benzylic C–H sulfonylation of phenol derivatives with sodium arylsulfinates. | ||
A metal-free regioselective radical sulfonylation of ethers with sodium sulfinates was conducted using K2S2O8 as the oxidant in aqueous medium at room temperature. A variety of aromatic and aliphatic sulfinates reacted with tetrahydrofuran (THF) to produce the desired 2-alkyl/-aryl sulfonyl tetrahydrofurans in good to high yields (Scheme 174).272 Notably, arylsulfinates bearing an electron-donating group appeared to react faster than the electron-withdrawing group. The pyran and diethyl ether were also sulfonylated with sodium benzenesulfinate to provide the corresponding sulfones in high yields.
 | ||
| Scheme 174 K2S2O8-mediated regioselective sulfonylation of ethers with sodium sulfinates. | ||
A transition metal-free strategy was envisaged by Willis, Talbot and co-workers for the oxidative β-sulfonylation of tertiary amines with sodium sulfinates in the presence of NIS to provide enaminyl sulfones (Scheme 175).273 A straightforward dehydrogenative β-C–H functionalization of N-benzyl piperidine was smoothly sulfonylated using a wide range of substituted aryl, heteroaryl and alkyl sulfinates to provide a series of enaminyl sulfones in good to high yields. Variations of the amine involving different N-benzyl and N-alkyl substituents were useful substrates, whereas N-aryl substituents performed moderately well. Disappointingly, the five- and seven-membered N-benzyl amines were not tolerated well and provided the corresponding enaminyl sulfones in low yields. Further, non-cyclic amines and melperone (a marketed atypical antipsychotic) were also successfully sulfonylated under the same conditions. The larger-scale sulfonylation of amines delivered the desired products with the same level of yields to demonstrate the preparative utility.
 | ||
| Scheme 175 Oxidative β-sulfonylation of tertiary amines with sodium sulfinates. | ||
The Bull group recently established the oxidative coupling of secondary aldehydes and sulfinate salts using copper catalysis to form α-sulfonyl aldehydes. With the use of AcOH, the redox potential of MnO2 was enhanced as an effective oxidant in the sulfonylation. The cyclohexylcarboxaldehyde was successfully sulfonylated with a range of arylsulfinates bearing electron-rich/-neutral/-poor substituents to produce the desired β-formyl sulfones in moderate to good yields (Scheme 176).274 Additionally, methyl and cyclopropane sulfinate salts reacted effectively to access dialkyl sulfones in modest yields. Next, cyclic α,α-disubstituted aldehydes with ring sizes from 4 to 8 were coupled with sodium p-toluenesulfinate; however, higher yields were observed for the less strained ring systems. A variety of 4-substituted cyclohexanecarboxaldehydes, as well as tetrahydropyran and Cbz-protected piperidine carboxaldehydes, were compatible to give the quaternary sulfonylated products in satisfactory yields. Acyclic α,α-disubstituted aldehydes were sulfonylated with sodium p-toluenesulfinate and the desired sulfones were obtained in good yields. Although the mechanism is not entirely clear, two mechanistic pathways were predicted, which involved ionic and radical steps (Scheme 176). Initially, both pathways involve the coordination of the aldehyde to Cu(II)–complex A to generate the cationic complex B. Under acidic conditions, deprotonation of B at the α-position gave the copper enol/enolate C. The enol/enolate intermediate C could be oxidised to the Cu(III)–complex D through the ionic mechanism, then attacked by the sulfinate ion to give the desired sulfonylated aldehyde and Cu(I) intermediate E. Alternatively, a direct radical attack of C by a sulfonyl radical formed by the oxidation of the sulfinate salt could produce the sulfonylated aldehyde.
 | ||
| Scheme 176 Oxidative coupling of secondary aldehydes and sodium sulfinates and a possible mechanism. | ||
3.4.9.2. Oxidative sulfonylation of heteroarenes. Pan and co-workers275 developed the CuBr2-catalyzed deoxygenative C2-sulfonylation of quinoline N-oxides in the presence of K2S2O8 in 1,2-dichloroethane under mild conditions. A wide range of sodium aryl and alkyl sulfinates were evaluated with several substituted quinoline N-oxides to afford the corresponding 2-sulfonyl quinolines (Table 18A). Similarly, Sirilata and co-workers276 developed the iodine/TBHP-mediated regioselective 2-sulfonylation of quinoline N-oxides with sodium sulfinate salts (Table 18B). This metal-free deoxygenative sulfonylation of quinoline N-oxides was carried out and other heteroaromatic N-oxide substrates were successfully coupled with a variety of sodium aryl and alkyl(methyl) sulfinates to give the corresponding sulfonylated quinolines in good to excellent yields. Remarkably, gram scale (10 mmol) experiments were safely conducted to synthesize 2-sulfonylquinolines, which showed efficacy similar to small-scale reactions. In contrast, sodium triflinate did not participate, which was assumed to be due to a strong inductive effect. Later, the Zhao group described an improved protocol using iodine-catalyzed oxidative sulfonylation of various quinoline N-oxides with different aryl and methyl sodium sulfinates at room temperature (Table 18C).277 A series of 2-sulfonylquinolines were obtained in good yields and had some advantages, including short reaction time, absence of metals, and sub-stoichiometric amounts of iodine. The CuBr2-catalyzed coupling proceeded via the Minisci-type radical for deoxygenated sulfonylation, whereas I2-mediated sulfenylation ruled out the radical pathway.
|
|||
|---|---|---|---|
| Sulfones | Method-A | Method-B | Method-C |
 |
64% | 78% | 81% |
 |
75% | 82% | 73% |
 |
72% | 95% | 75% |
 |
77% | 76% | 69% |
 |
56% | 79%; | 66% |

The K2S2O8-mediated deoxygenative dual radical coupling of quinoline N-oxides with sodium sulfinates was described by the He group (Table 19A).278 Various functional groups, including both electron-rich and electron-poor substituents on quinoline N-oxides and sodium arenesulfinates were well tolerated and provided a series of 2-quinolinyl-aryl sulfones in moderate to good yields. The isoquinoline N-oxide was tosylated at the C1 and C3 positions in a 1:1 ratio but pyridine N-oxide or quinoxaline N-oxide led to only a trace amount of tosylated product. Surprisingly, no sulfonylation occurred using aliphatic sodium sulfinate. This sulfonylation protocol is also viable at the gram-scale (10 mmol) in delivering 2-tosylated quinoline. Simultaneously, Li et al. exemplified the FeCl3-catalyzed oxidative sulfonylation of quinoline N-oxides with sodium sulfinates to generate 2-sulfonylquinolines (Table 19B).279 A wide range of aromatic and aliphatic sulfinates were smoothly coupled with different substituted quinoline N-oxides and gave widespread 2-sulfonylatedquinolines in satisfactory yields. Later, the He group extended the sustainable TsCl-mediated sulfonylation of quinoline N-oxides with sodium sulfinates at ambient temperature in water (Table 19C).280 Several aryl, heteroaryl and alkyl sulfinates readily sulfonylated quinoline N-oxides to synthesize various 2-sulfonylquinolines in variable yields. The pyridine N-oxide and isoquinoline N-oxide furnished the corresponding products; hitherto, the quinoxaline N-oxide was not sulfonylated. A large-scale sulfonylation (5 mmol) experiment was also successfully demonstrated.
|
|||
|---|---|---|---|
| Sulfones | Method-A | Method-B | Method-C |
 |
74% | 82% | 84% |
 |
88% | 83% | 86% |
 |
75% | 72% | 87% |
 |
68% (R = Me) | 82% (R = H) | 78% (R = Me) |

In 2019, an exogenous oxidant and catalyst-free electro-chemical deoxygenative C2 sulfonylation of quinoline N-oxides with sodium sulfinates was employed by Lei and co-workers.281 The C2-sulfonylation of quinoline N-oxide with various substituted-aryl, 3-pyridyl, and ethyl-derived sulfinate salts furnished the desired quinoline-2-sulfones in 44–80% yields (Scheme 177). Next, various quinoline N-oxides bearing diverse substituents were easily sulfonylated with PhSO2Na to provide the respective 2-phenylsulfonylquinolines in 40–82% yields. Only the electron-rich methoxy group had some influence on the reaction efficiency. The pyridine N-oxide offered no desired 2-sulfonylated products under this electrochemical synthesis.
 | ||
| Scheme 177 Electrochemical deoxygenative C2-sulfonylation of quinoline N-oxides with sodium sulfinates. | ||
Recently, Harrity and co-workers37 utilized newly prepared azetidine and oxetane sulfinate salts (see Scheme 8) in the C-2 sulfonylation of indoles bearing electron-donating and electron-deficient groups in the presence of iodine. Electron-donating indoles were efficiently sulfonylated with azetidine and oxetane sulfinates to provide a range of corresponding sulfone derivatives in high yields (Scheme 178). Likewise, N-methyl indole reacted well to provide the corresponding C-2 sulfonylated products in high yields. In contrast, C-3 sulfenylation of 2-substituted indoles occurred under the influence of the I2/Ph3P reagent to afford the desired indolyl sulfides in high yields.
 | ||
| Scheme 178 C-2 sulfonylation and C-3 sulfenylation of indoles with azetidine and oxetane-derived sulfinates. | ||
Deng and co-workers282 disclosed the metal-free direct sulfonylation of indoles at the C-2 position with sodium sulfinates. The para-substituted arylsulfinates, 2-naphthyl-sulfinate and sodium alkylsulfinates reacted smoothly with indole in the presence of a catalytic amount of iodine. On the other hand, various substituents on the indole ring were also sulfonylated with sodium benzenesulfinate to produce 2-sulfonyl indoles. A series of substituted 2-sulfonyl indoles were obtained in moderate to high yields (Scheme 179). Further, 1-methylindole also coupled with sodium benzenesulfinate and gave the desired sulfone in 68% yield; however, the 2-methylindole afforded only trace amounts of the C-3 sulfonylated product. It is worth noting that direct C-2 sulfonylation exclusively occurred for the indole ring in all cases.
 | ||
| Scheme 179 I2-catalyzed C-2 sulfonylation of indoles with sodium sulfinates. | ||
At the same time, the Kuhakarn group283 also developed an iodine-mediated direct C-2 sulfonylation of indoles with sodium sulfinates for 2-sulfonyl indoles. A variety of substituted indoles were coupled with sodium benzenesulfinate, sodium 4-methylbenzenesulfinate, sodium 4-chlorobenzenesulfinate and sodium methanesulfinate to form 2-sulfonyl indoles in low to high yields. The N-alkyl (methyl, ethyl and benzyl) indoles gave moderate to high yields (54–93%) of the corresponding sulfones (Scheme 180). As expected, the N-EWG (Cbz, Bz, and Ms)-derived indoles were not sulfonylated under the same reaction conditions. The sulfenylation was ceased using excess TEMPO (5 equiv.), which implied that the reaction might proceed with a radical intermediate.
 | ||
| Scheme 180 I2-mediated C-2 sulfonylation of indoles with sodium sulfinates. | ||
The direct C-2 and C-3 sulfonylation of indoles with sodium sulfinates iodide and copper-salt catalysts occurred under mild conditions. An improved version of Deng's work was described by Zhang and co-workers284 using NH4I (10 mol%) at 60 °C instead of iodine. The selective C-2 sulfonylation by the coupling of substituted indoles with sodium arylsulfinates provided 2-sulfonylindole derivatives in 66–99% yield (Scheme 181A). The interesting part of Zhang's work was the Cu-catalyzed C3-sulfonylation of both electron-donating and electron-drawing groups-derived indoles that smoothly coupled with sodium arylsulfinates to produce 3-sulfonyl indoles in good to high yields (Scheme 181B). Notably, the C2 position is occupied by a substituent of the indole, and the iodide, and copper-salt catalysts both promote the installation of the sulfonyl group at the indole's C3-position. In contrast, no C-2 sulfonylation occurred using 3-methylindole under the influence of copper catalysis.
 | ||
| Scheme 181 C-2 and C-3 sulfonylation of indoles with sodium sulfinates. | ||
KI-catalyzed a convenient sulfonylation for the preparation of 2-sulfonyl indoles in moderate to good yields from indoles and sodium sulfinates in the presence of oxone in water (Scheme 182).285 Both electron-donating (Me and OMe) and electron-withdrawing (F and Br) substituents on the indole ring usually had no significant influence on the outcome. Unfortunately, 2-methylindole and 5-nitro indole proved to not be suitable substrates for this transformation.
 | ||
| Scheme 182 KI-catalyzed C-2 sulfonylation of indoles with sodium sulfinates. | ||
Chen, Yu and co-workers developed the electrochemical C-2 sulfonylation of 1H-indole with sodium sulfinates for the synthesis of 2-sulfonyl indoles at room temperature under mild reaction conditions by (Scheme 183).286 A variety of indoles bearing both electron-donating and electron-withdrawing groups underwent sulfonylation with sodium benzenesulfinate to give the indolyl phenyl sulfones in moderate to high yields. The 2-substituted indole sluggishly formed the 3-sulfonyl indole in trace amounts only. Several sodium arenesulfinates and alkylsulfinates afforded the corresponding indolyl phenyl sulfones in good to high yields. Subsequently, the electrochemical sulfonylation was used to prepare the Boc protected 2-sulfonyl indole, a biologically active 5-HT6 receptor modulator.
 | ||
| Scheme 183 Electrochemical α-sulfonylation of 1H-indole with sodium sulfinates. | ||
Silver nitrate-mediated site-selective 1,7-disulfonylation of diaryl(1H-indol-2-yl)methanols with sodium sulfinates under mild conditions was disclosed by Ji and co-workers (Scheme 184).287 A series of diaryl(1H-indol-2-yl)methanol derivatives bearing different substituents on the indole and benzene ring participated efficiently in the disulfonylation. The electron density on the indole ring had no influence, though the benzene ring had a critical impact on the reactivity. Sodium arylsulfinates having electron-donating or electron-withdrawing substituents on the benzene ring equally responded to form the desired 2-(diarylmethyl)-indole diarylsulfones in moderate to good yields. Additionally, sodium methanesulfinate was also well-tolerated and gave the target product in 67% yield; however, sodium trifluoromethanesulfinate did not detect the target product under the same conditions.
 | ||
| Scheme 184 Ag-mediated disulfonylation of diaryl(1H-indol-2-yl)methanols with sodium sulfinates. | ||
In 2018, Zhu, Shao and co-workers demonstrated the iodine-induced direct C–H sulfonylation of imidazo[1,2-a]pyridines with sodium sulfinates in Et2O at 100 °C (Scheme 185).96 A broad range of imidazo[1,2-a]pyridine-derived sulfones were generated in good to high yields by using imidazo[1,2-a]pyridine bearing substituents at C6, C7, and C8 positions with a series of aryl-substituted sulfinate salts. In most cases, both electron-donating and electron-withdrawing substituents on the para- and meta-positions of the phenyl ring were well-tolerated. The use of sodium pyridine-3-sulfinate was not suitable for providing the desired sulfonylated product. Unfortunately, NO2-, CN- and ortho-substituted imidazo-pyridine substrates gave low yields. No sulfonylation of 2-methylimidazo[1,2-a]pyridine and imidazo[1,2-a]pyridine was employed. Additionally, fused imidazoheterocycles, 6-phenyl-imidazo[2,1-b]thiazole, and 2-phenylbenzo[d]imidazo-[2,1-b]-thiazole were also sulfonylated to give the desired products in moderate yields.
 | ||
| Scheme 185 I2-PPh3-mediated sulfonylation of fused imidazoheterocycles. | ||
Oxindole-derived sulfones were obtained from sodium sulfinates using potassium iodide, tert-butyl hydroperoxide (TBHP) in DMSO and acetic acid. The oxidative sulfonylation of various oxindoles with various sodium sulfinates, including p-toluenesulfinate, benzenesulfinate, and methanesulfinate, was carried out to give the 3-sulfonyloxindoles (Scheme 186).288 Irrespective of the electronic influence of the substituents on the benzene ring, the corresponding sulfonylated products were obtained in moderate to good yields. The 3-benzyl-substituted oxindole was not a suitable substrate in the sulfonylation to give the quaternary-centered 3-sulfonyloxindole.
 | ||
| Scheme 186 KI-mediated sulfonylation of oxindoles with sodium sulfinates. | ||
A metal-free direct oxidative sulfonylation of pyrazolones was carried out with sodium sulfinates for the synthesis of sulfonated pyrazoles in the presence of TBHP as an oxidant. The Wang group successfully utilized the iodine-catalyst for various pyrazolones with aryl, heteroaryl and aliphatic sulfinates to generate a series of structurally diverse pyrazole-derived sulfones (Scheme 187A).289 The electron-withdrawing CF3 group at the 3 position of the pyrazolone to afford the desired product in moderate yield. Later, Li et al. used TBAI to catalyze the direct sulfonylation of pyrazolones with sodium sulfinates in the presence of TBHP in water as the solvent. A variety of sulfonyl pyrazolone derivatives were obtained from substituted pyrazolones with a broad range of sodium sulfinates (Scheme 187B).290 Sodium trifluoromethanesulfinate and 1,3-dimethyl-1-pyrazol-5(4)-one were also subjected under the same reaction conditions, and no desired products were detected.
 | ||
| Scheme 187 I2-catalyzed direct sulfonylation of pyrazolones with sodium sulfinates. | ||
3.4.9.3. Oxidative sulfonylation of electron-rich arenes. Wills and co-workers291 developed an attractive iridium-catalyzed sulfonylation of aniline derivatives with sodium sulfinates under blue LED (strips) irradiation. A new method without the need for the pre-functionalization of anilines was successfully employed under mild conditions for installing the sulfone moiety (Scheme 188). In a general survey of aniline derivatives, alkyl and aryl substituents on the aromatic ring were smoothly sulfonylated to afford the corresponding sulfones in good to high yields. Additionally, electron-donating aniline substrates bearing methoxy groups at the ortho-, meta- and para-positions all readily proceeded to give the anticipated sulfones in high yields. Next, the different substituents at the nitrogen atom revealed that aromatic groups participated well in the direct sulfonylation. A broad range of sulfinate salts, such as cycloalkyl, linear and branched alkyl, and aryl sulfinates were also suitable substrates in this transformation. A series of sulfonylated anilines having different functionalized groups were obtained in good to high yields. The broad applicability of this process was extended for the sulfonylation of promethazine and linezolid drugs. The promethazine (neuroleptic medication) underwent sulfonylation and yielded 92% with high regioselectivity; however, linezolid (antibiotic) was sulfonylated to provide two regioisomeric sulfones in 91% yield. Of note, in most cases, sulfonylation occurred predictably at the ortho- and para-positions concerning the amino substituent.
 | ||
| Scheme 188 Ir-catalyzed sulfonylation of anilines with sodium sulfinates. | ||
The Li group established an interesting electrochemical protocol to couple N,N-disubstituted anilines with sodium sulfinates in the absence of transition metal catalyst using undivided cells at room temperature (Scheme 189).292 The electrooxidative C–H sulfonylation of N,N-dimethyl aniline was coupled smoothly with a series of aryl, heteroaryl, and alkyl sulfinates and furnished the desired aniline-derived sulfones in moderate to excellent yields. The different substituents were at the nitrogen atom of aniline derivatives with sulfinate, and the site selectivity controlled by the steric hindrance effect of amino substituents led to ortho- and para-sulfonylated products or a mixture of both isomers.
 | ||
| Scheme 189 Electrochemical protocol to couple N,N-disubstituted anilines with sodium sulfinates. | ||
At the same time, Lei and co-workers reported a similar oxidative electrochemical ortho-sulfonylation of N,N-dialkyl anilines with sodium sulfinates by using undivided electrochemical cells at room temperature (Scheme 190).293 A broad range of aryl and heteroaryl sulfinates were smoothly reacted with 4-methyl N,N-dimethylanilines for generating the desired sulfones in moderate to high yields. Also, sodium alkyl sulfinates, such as methanesulfinate, ethanesulfinate, and octanesulfinate were suitable for electrooxidative sulfonylation. Moreover, different 4-substituted N,N-dimethyl-anilines and N,N-diethyl-4-methylaniline with electron-donating and electron-withdrawing groups afforded the corresponding diaryl sulfones in good yields. Moreover, the scalable electrochemical sulfonylation was successfully performed at a 5 mmol scale reaction in a beaker in open-air conditions.
 | ||
| Scheme 190 Electrochemical sulfonylation of arenes and aniline derivatives with sodium sulfinates. | ||
In 2019, Waldvogel and co-workers294 demonstrated the direct electrochemical sulfonylation of phenols with sodium sulfinates under boron-doped diamond electrodes (BDD) in an HFIP–water mixture at ambient temperature. A series of both diaryl and aryl sulfones was accessed in acceptable yields by using several substituted phenols and different aryl and alkylsulfinates substrates (Scheme 191A). Variations on the phenol moiety showed a significant influence on the yields. Also, a scalable electrolytic experiment was performed at almost the same level of yield. Subsequently, the same group successfully conducted oxidant-free electrochemical sulfonylation of anisole and aniline derivatives with sodium sulfinates (Scheme 191B).295 Various substituted anisoles with functionalized aryl and alkylsulfinates resulted in a wide range of aryl sulfones in moderate yields. Moreover, anilides and N,N-dimethylanilines were successfully sulfonylated to provide the desired products in low to moderate yields.
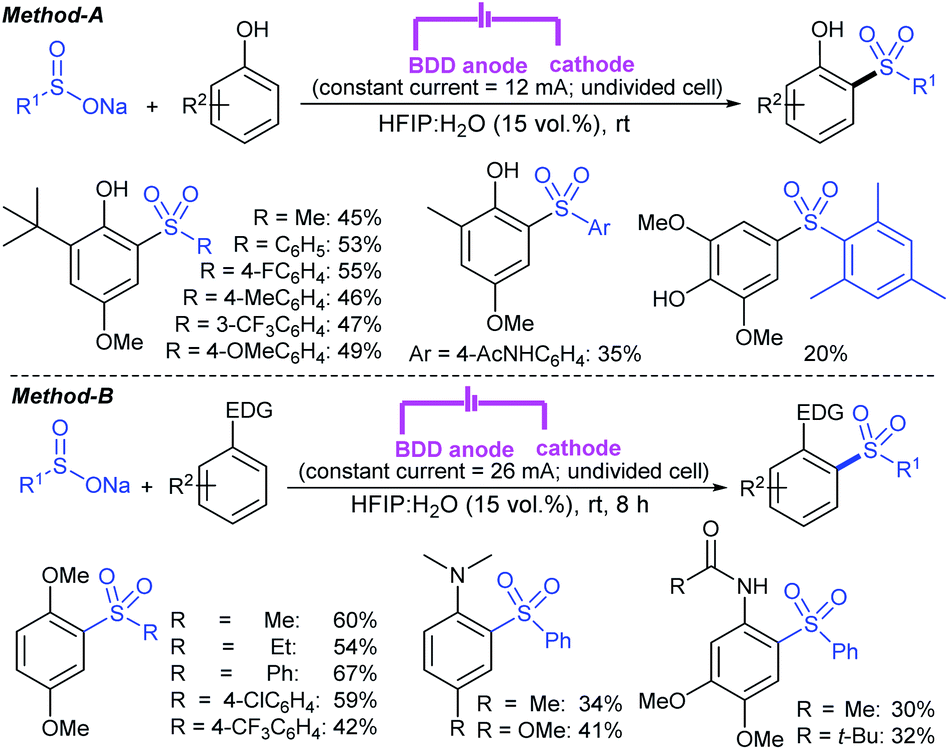 | ||
| Scheme 191 Electrochemical sulfonylation of arenes and aniline derivatives with sodium sulfinates. | ||
A highly dispersed and acid-resistant subnanometer cobalt catalyst (<1 nm) was designed using a MOF-template and prepared for selective C–H oxidative sulfonylation of tetrahydroquinoxalines (THQX) with sodium sulfinates (Scheme 192).296 Zhang and co-workers found that the variation of both coupling partners under the catalytic sulfonylation enabled the generation of a series of sulfonylquinoxalines in moderate to high yields. The merits of this protocol were good functional tolerence, broad substrate scope, and excellent regio- and chemoselectivity. Additionally, unsymmetrical THQX were reacted with sulfinates to produce two regioisomers in almost 1:1 ratios with no specific regioselectivity. The Co–N–C catalyst was reused to run the model reaction five consecutive times, where the catalytic activity and reaction selectivity well retained.
 | ||
| Scheme 192 Co–N–C catalysed selective C–H oxidative sulfonylation of tetrahydro-quinoxalines with sodium sulfinates. | ||
The Mn(OAc)3-promoted oxidative C–H sulfonylation of 1,4-dimethoxybenzene with sodium sulfinates was developed by Manolikakes and co-workers297 under mild reaction conditions. Various arenesulfinates bearing electron-withdrawing or electron-donating groups smoothly reacted with 1,4-dimethoxybenzenes and provided diaryl sulfones in moderate to high yields (Scheme 193). Additionally, naphthalene-sulfinate, 2-pyridinesulfinate and sodium methanesulfinate were also suitable substrates to furnish the corresponding sulfones in good yields. Moreover, the reaction of the unsymmetrical 1,4-dimethoxy-2-methylbenzene coupled with sodium 4-chlorobenzenesulfinate, led to the formation a regioisomeric mixture of sulfones. The oxidative coupling of other electron-rich arenes, such as anisole, 4-methyl anisole, 1,2/1,3-dimethoxybenzene, did not afford the desired products and was limited to 1,4-dimethoxybenzene derivatives only.
 | ||
| Scheme 193 Mn(OAc)3-promoted oxidative C–H sulfonylation of 1,4-dimethoxybenzene with sodium sulfinates. | ||
The Habibi group employed laccase-40 U (from Trametes versicolor)-promoted aerobic oxidative sulfonylation of catechols or hydroquinone with sodium benzenesulfinates in the presence of O2 as an oxidant and a phosphate buffer solution as a solvent at room temperature (Scheme 194).298 A series of diaryl sulfone analogs were obtained in moderate to high yields using a variety of catechols with substituted arylsulfinates under this green and eco-friendly transformation.
 | ||
| Scheme 194 Laccase-40 U-promoted oxidative sulfonylationof hydroquinone or catechols with sodium sulfinates. | ||
The Mhaske group employed an efficient oxidative sulfonylation of p-anisidine substrates via a reactive quinone imine ketal intermediate with sodium sulfinates to synthesize aryl sulfones involving two steps in a one-pot operation (Scheme 195).299 A variety of aryl, heteroaryl and alkyl sulfinate salts were smoothly coupled with N-tosyl p-anisidine to furnish different sulfonylated products in good to high yields. Instead of the N-tosyl group, other kinds of N-protected p-anisidines were also compatible, whereas N-pivaloyl p-anisidine furnished sulfone in a low yield. Various substituents on the aryl ring of p-anisidines required high temperature (60 °C) to participate under the developed protocol. The electron-withdrawing group on the p-anisidine moiety was well-tolerated rather than electron-donating substituent on p-anisidine. Similarly, Konovalova and co-workers300–302 employed the nucleophilic addition of the quinone imine ketal intermediate with sodium arenesulfinates to furnish 1,4-, 1,6-, and 6,1-addition products of N-arylsulfonyl, N-aroyl, and N-[arylsulfonylimino(phenyl)methyl] derivatives.
 | ||
| Scheme 195 Oxidative sulfonylation of p-anisidine substrates with sodium sulfinates. | ||
3.4.10.1. Construction of oxa-heterocycles. An efficient construction of various sulfonylated lactones via Ag-catalyzed radical cascade ring-closing sulfonylation of 1,6-enynes with sodium sulfinates was described by Jiang and co-workers (Scheme 196).303 The ring-closing sulfonylation reaction proceeded through a tandem of C–C and C–S bond formation under mild conditions. The radical sulfonylation of various substituted 1,6-enynes occurred on reaction with aromatic and aliphatic sulfinates to form the anticipated sulfonylated lactones in good to high yields. Surprisingly, the cascade cyclization-sulfonylation of 2-thiophenyl and 2-naphthyl-derived sulfinates failed to provide the desired products.
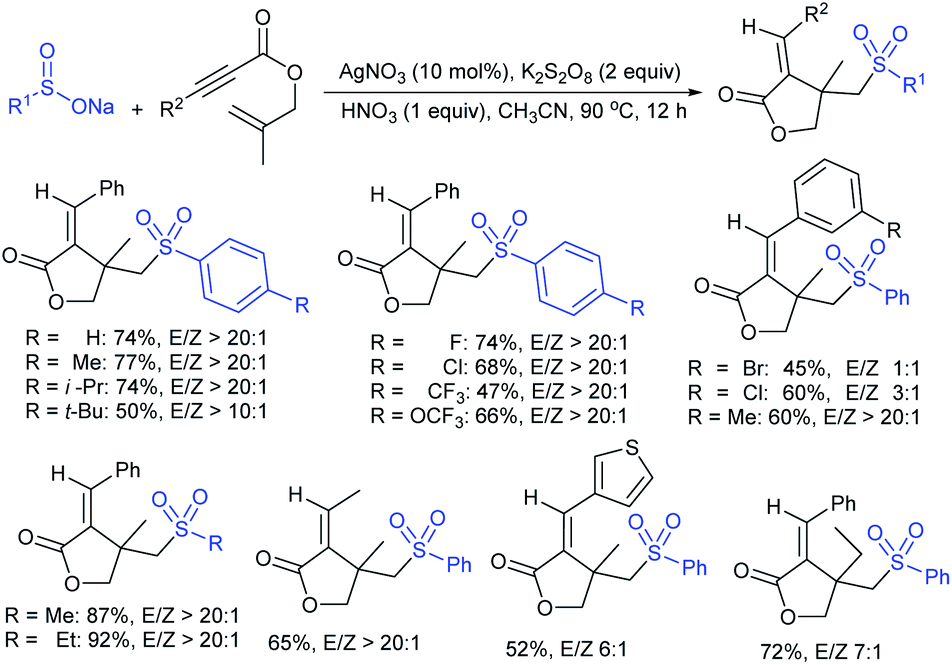 | ||
| Scheme 196 Ag-catalyzed radical cascade ring-closing sulfonylation of 1,6-enynes with sodium sulfinates. | ||
Afterwards, the same group efficiently developed the AgNO3-catalyzed oxidative cyclization reaction of 1,6-enynes and sodium sulfinates for the synthesis of various sulfonylated benzofurans. Both aryl and alkyl sodium sulfinate substrates were explored for sulfonylation with a wide range of 1,6-enynes to afford different 2,3-disubstituted benzofurans in moderate to good yields under mild conditions (Scheme 197).304 However, the trifluoromethanesulfinate and heteroaryl-derived sulfinates were not suitable substrates for this transformation. Generally, the relative reaction rate steadily decreased with the conversion from aryl- to alkyl-substituted 1,6-enynes, indicating that the conjugative effect has an influence. A variety of substituted aryl-linked 1,6-enynes showed excellent tolerance in the oxidative sulfonylation.
 | ||
| Scheme 197 Ag-catalyzed oxidative cyclization reaction of 1,6-enynes and sodium sulfinates. | ||
The efficient construction of 2-sulfonylbenzo[b]furans was induced by the CuCl2·2H2O/AgTFA system using trans-2-hydroxy-cinnamic acids and sodium sulfinates. The cascade decarboxylation reaction proceeded via tandem C–S and C–O bonds formation under mild conditions (Scheme 198).305 A variety of aromatic and aliphatic sulfinates easily reacted with trans-2-hydroxycinnamic acids, whereas the scope of trans-2-hydroxycinnamic acids was well-tolerated to give a series of 2-sulfonylbenzo[b]furan derivatives in moderate to good yields. Meanwhile, benzofuran-2-carboxylic acid treated with sodium sulfinates under the same conditions did not produce the expected 2-sulfonylbenzo[b]furans.
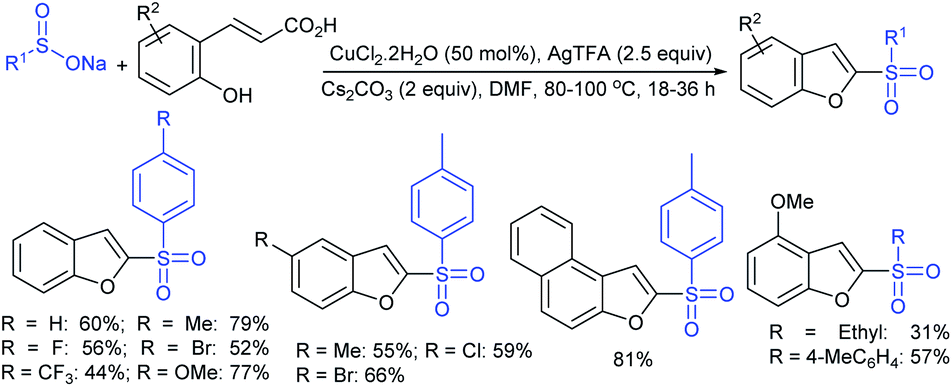 | ||
| Scheme 198 Co(OAc)2·4H2O/KI catalyzed ring-closing sulfonylation of N-alkyl-N-methacryloyl benzamides with different sodium sulfinates. | ||
A competent and straightforward approach to the synthesis of 3-sulfonylbenzofurans was achieved via o-hydroxyphenyl propargyl alcohols (o-HPPAs) and sodium sulfinates with no assistance from any reagent or catalyst (Scheme 199).306 The reaction proceeded through sulfa-Michael addition and oxy-cyclization of the in situ generated o-QM as a bifunctional transient intermediate. The effect of substitution on the alkyne terminus of o-HPPA was studied with electronically neutral and electron-rich substitutions that gave 2,3-disubstituted benzofurans in good to high yields. Further, the scope of the reaction with respect of aryl and methyl sulfinates was also explored to generate a few corresponding analogs in good yields. The efforts made toward the synthesis of 3-sulfonylindole variant was unsuccessful. A possible mechanism involves the o-QM key intermediate A from o-HPPA by expelling the water (Scheme 216). Next, the 1,4-addition of o-QM by sulfinate salt gives the sodium phenoxide intermediate B and regenerates the phenolic species C with liberating NaOH. Finally, deprotonation at a benzylic position with NaOH through allenyl isomerization gives the allene-type intermediate D, which undergoes 1,3-proton migration, leading to 2,3-disubstituted benzofuran derivatives.
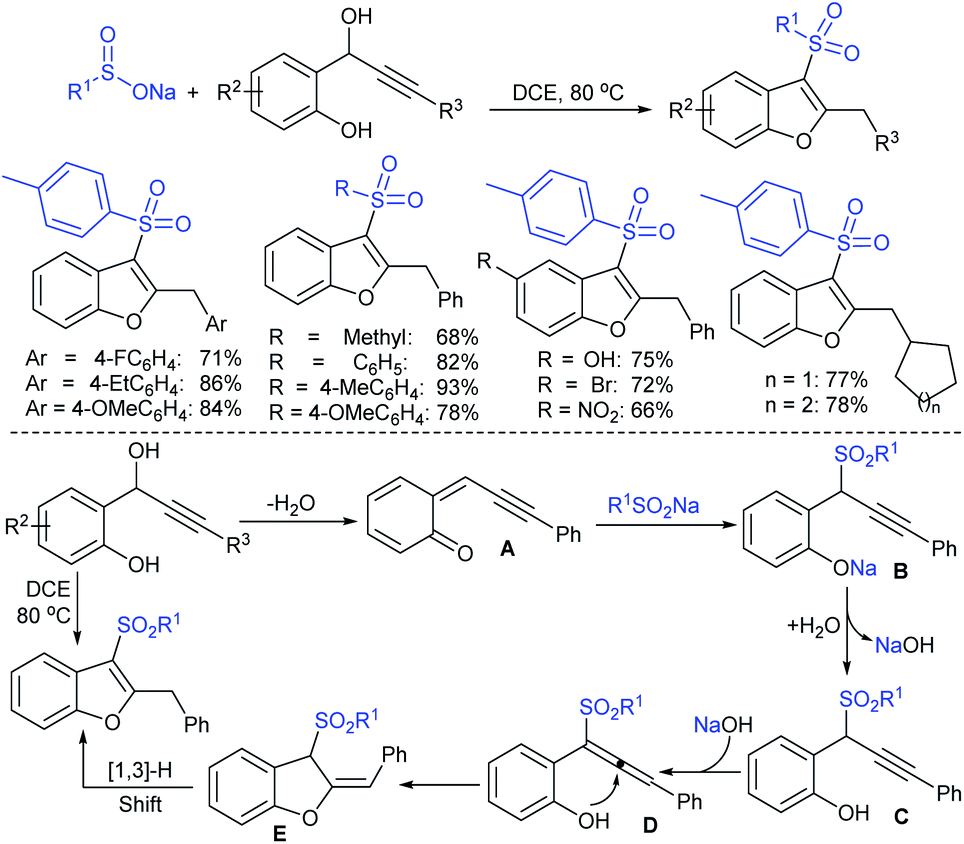 | ||
| Scheme 199 Ring-closing sulfonylation of o-hydroxyphenyl propargyl alcohols (o-HPPAs) and sodium sulfonylates with its mechanistic processes. | ||
The Zhu group accessed tetrasubstituted furans through the palladium-catalyzed ring-closing sulfonylation of homoallenyl amides with sodium sulfinates in the presence of PhI(O2CCF3)2 as the oxidant (Scheme 200).307 Gratifyingly, a wide variety of homoallenyl amides smoothly coupled with various aromatic and aliphatic sulfinates to afford the desired structurally diverse 2-amino-5-sulfonylmethylfurans in good to high yields. The reaction with sodium methanesulfinate or sodium trifluoromethanesulfinate generated the desired products in 67% or 35% yield, respectively. The cyclization-sulfonylation was fairly general, however, substitution at another end of the homoallenyl amides yielded the desired products in only trace amounts.
 | ||
| Scheme 200 Pd-catalyzed ring-closing sulfonylation of homoallenyl amides with sodium sulfinates. | ||
Highly functionalized 3-sulfonylfurans were furnished in a one-pot operation described by the Ren group308 via conjugate addition and 5-endo-trig cyclization of 3-cyclopropylideneprop-2-en-1-ones with sodium sulfinates under the influence of iodine (Scheme 201). A wide array of aromatic and aliphatic sulfinates were successfully surveyed without any special electronic effect and afforded a series of 3-sulfonylfurans in moderate to good yields. Furthermore, several representative 3-cyclopropylid-eneprop-2-en-1-ones also furnished the desired sulfonylated furan derivatives in acceptable yields. The one-pot method was also performed at 5.0 mmol scale reaction, thus indicating the transformation was easily to scalable without the loss of efficiency.
 | ||
| Scheme 201 One-pot method conjugate addition and 5-endo-trig cyclization of 3-cyclopropylideneprop- 2-en-1-ones with sodium sulfinates. | ||
Copper-catalyzed ring-closing sulfonylation of 2-vinylbenzoic acids with sodium sulfinates for the synthesis of sulfonyl phthalide derivatives. Weng, Lu and co-workers realized di-tert-butyl peroxide or molecular oxygen as the terminal oxidant for this protocol (Scheme 202).309 Various electron-donating and electron-withdrawing groups on the phenyl ring of 2-vinylbenzoic acids reacted smoothly with sodium benzenesulfinate to afford the corresponding products in good to high yields. Besides, a wide range of aromatic and aliphatic sulfinate salts efficiently underwent the construction of lactone-derived sulfones in moderate to high yields. The sodium trifluoromethanesulfinate failed to provide the desired product. A gram-scale reaction was performed successfully for the preparation of sulfonylated phthalide in high (90%) yield. The mechanism was believed to proceed through two possible pathways, either ionic or radical steps. Initially, the vinylbenzoic acid reacted with Cu(II) to form Cu-carboxylate A and sodium sulfinate was oxidized by DTBP or O2 to generate the sulfonyl radical. Subsquently, radical sulfonylation of the double bond of A afforded the rationally stable benzyl radical intermediate B. The path-A involves the C–O bond construction of B to form the Cu(III)-cyclic complex C, or the oxidation of B leads to the benzyl carbocation intermediate D via path-B. Finally, the reductive elimination of C or benzyl carbocation D undergoes cyclization to obtain the expected lactone. The Cu(II) species was regenerated to continue further catalytic cycle in both pathways.
 | ||
| Scheme 202 Cu-catalyzed ring-closing sulfonylation of 2-vinylbenzoic acids with sodium sulfinates and mechanistic pathways. | ||
The TFA-promoted ring-closing sulfonylation of 2-propynolphenols with sodium sulfinates was developed by Liang and co-workers for the synthesis of 4-sulfonyl 2H-chromenes under mild conditions (Scheme 203).310 The reaction occurred by the nucleophilic addition of sulfinates to the allylene carbocation intermediate followed by a 6-endo-trig pathway. Various symmetrical and unsymmetrical tertiary propargylic alcohols could be transformed into the desired products in moderate to high yields. However, substrates bearing alkyl groups failed to give the corresponding products. The limited scope of arylsulfinates and methanesulfinate was also investigated to generate the corresponding products in good yields. Further, secondary propargylic alcohols bearing electron-donating groups generated 2-aryl-4-sulfonyl 2H-chromenes in moderate to good yields. However, the electron-withdrawing substrates successfully afforded the desired products.
 | ||
| Scheme 203 Ag-catalyzed ring-closing sulfonylation of isocyanobiphenyls with sodium sulfinates. | ||
A combination of the potassium iodide and tert-butyl hydroperoxide reagent system for the ring-closing sulfonylation of phenyl propiolates with sodium sulfinates was presented by Zhang and co-workers (Scheme 204).311 A variety of functional groups were tolerated on phenyl 3-phenylpropiolates with a few aryl-substituted sulfinates to give the corresponding 3-phenyl-sulfonylcoumarins in good yields. Although no obvious electronic effect was observed, sodium cyclohexane-sulfinate did not give the desired product.
 | ||
| Scheme 204 KI/t-BuOOH for the ring-closing sulfonylation of phenyl propiolates with sodium sulfinates. | ||
In 2020, Han et al. developed the silver-catalyzed cascade radical cyclization of o-(allyloxy)arylaldehydes and sodium sulfinates using K2S2O8 as an oxidant for the synthesis of chroman-4-one derivatives (Scheme 205).312 By evaluating a variety of arylsulfinates and substituted o-(allyloxy)arylaldehydes, they were all well tolerated and afforded the corresponding analogs in satisfactory yields. Diverse electron-withdrawing and electron-donating groups did not show an appreciable effect on the outcome.
 | ||
| Scheme 205 Ag-catalyzed cascade radical cyclization of o-(allyloxy)arylaldehydes and sodium sulfinates. | ||
3.4.10.2. Construction of aza-heterocycles. An eosin Y-catalyzed net redox neutral process was conducted for the synthesis of 3-sulfonylindoles via the anionic oxidation of sodium sulfinates under the influence of visible light irradiation. A radical cascade ring-closing sulfonylation of 2-alkynyl-azido-arenes was developed by Kshirsagar and co-workers (Scheme 206).313 The substrate scope of 2-alkynyl-azido-arenes treated with sodium benzenesulfinate and proceeded well to the 3-sulfonylindoles in good to high yields. Moreover, a variety of aromatic and aliphatic sulfinate salts were subjected to 1-azido-2-(phenylethynyl)benzene and proceeded smoothly to deliver the corresponding products in 43–73% yields. The reaction offered metal and oxidant/reductant-free visible light-mediated vicinal sulfonamination of alkynes and led to 2-aryl/alkyl-3-sulfonylindoles.
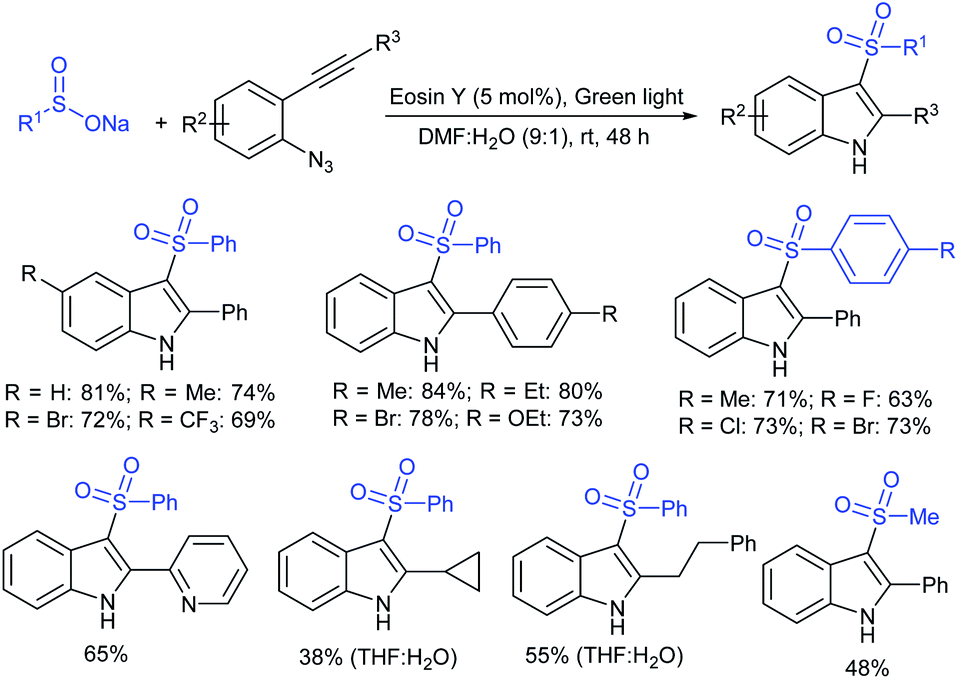 | ||
| Scheme 206 Eosin Y photoredox catalysis of 2-alkynyl-azido-arenes with sodium sulfinates. | ||
Electrochemical cyclization-sulfonylation was developed by Sun and co-workers314 for the synthesis of sulfone-derived oxindoles. A wide range of acrylamide and sodium sulfinate substrates were proved to be compatible in the presence undivided cells with NH4Br (10 mol%) as a redox catalyst (Scheme 207). Generally, a variety of N-substituted acrylamides reacted smoothly with sodium 4-methylbenzene sulfinate to provide a wide range of sulfonyl oxidoles in good to high yields, the N-unsubstituted and N-acetyl acrylamides were found to be ineffective under the same conditions. Next, the scope of the reaction was further explored with various aryl and alkyl sodium sulfinates to yield the desired products. Moreover, the practical and scalable experiment at the 10 mmol scale was also demonstrated for this protocol with comparable efficiency to the small scale reaction.
 | ||
| Scheme 207 Electrochemical ring-closing sulfonylation of N-substituted acrylamides with different sodium sulfinates. | ||
Copper mediated the alkene vicinal aminosulfonylation reaction between 2-vinylbenzamide derivatives and sodium sulfinates to access a variety of sulfonated lactams in moderate to good yields (Scheme 208).315 The ring-closing sulfonylation reaction proceeded through a tandem C–N and C–S bond formation under mild conditions. Various 2-vinylbenzamide derivatives with aromatic and aliphatic sulfinates in the presence of Cu(NO3)2·3H2O produced sulfone-derived lactams in moderate to good yields. The broad functional compatibility of the substitutions on the aryl group including electron-donating and electron-withdrawing group substrates participated well in the reaction.
 | ||
| Scheme 208 Copper-mediated alkene vicinal aminosulfonylation reaction between 2-vinylbenzamide derivatives and sodium sulfinates. | ||
Rao et al., reported a copper-catalyzed direct aminosulfonylation of unactivated alkenes with sodium sulfinates for the efficient synthesis of sulfonylated pyrrolidones via a 5-exo cyclization process (Scheme 209).316 Generally, the unsaturated quaternary amides reacted smoothly with sodium arylsulfinates and afforded the corresponding aminosulfonylation products in good to high yields. The reaction of α-substituted quaternary amides also generated the desired products in good yields with low diastereoselectivity ratios. Interestingly, α-spiro pyrrolidones were obtained in high yields by the aminosulfonylation of several α-cyclic substrates. A variety of arylsulfinates were suitable and provided the desired sulfonyl pyrrolidones in moderate to high yields, regardless of their electronic properties.
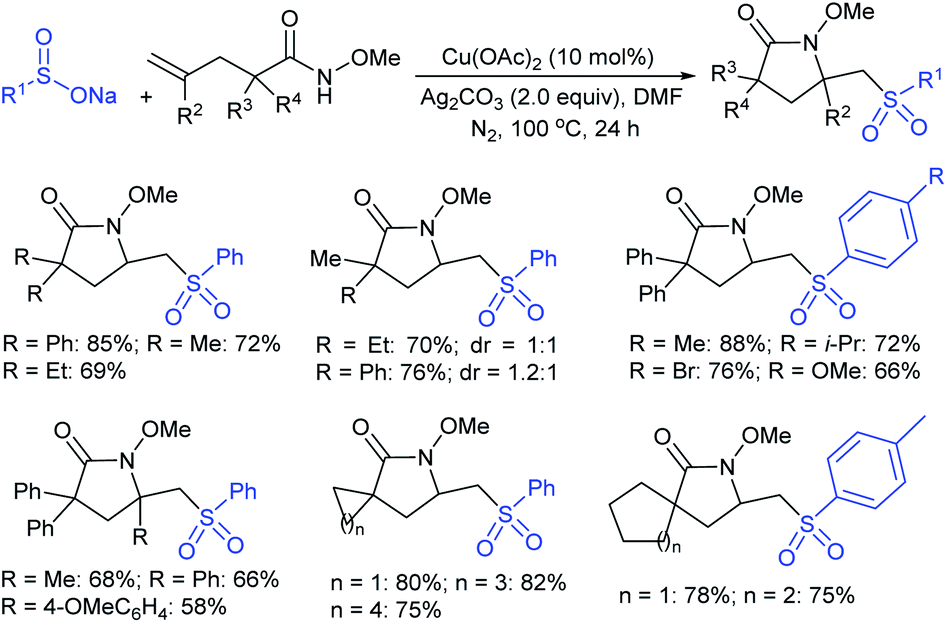 | ||
| Scheme 209 Copper-mediated alkene vicinal aminosulfonylation reaction between 2-vinylbenzamide derivatives and sodium sulfinates. | ||
An efficient radical imino-sulfonylation of γ,δ-unsaturated oxime esters with sodium sulfinates for functionalized pyrrolines was carried out by Chen, Zhu and co-workers (Scheme 210).317 The different aryl- and cyclohexyl-substituted oxime esters reacted smoothly with sodium p-toluenesulfinate to afford the desired imino sulfonylation products in moderate to good yields. Without a gem-dimethyl group on γ,β-unsaturated oxime ester was a poor substrate as explained by the lack of the Thorpe–Ingold effect and the alkyl oxime ester was found to not be a suitable substrate as well. A series of sulfinates with different electronic properties at the ortho, meta and para positions on the benzene ring were explored and were reacted with methanesulfinate to yield the corresponding pyrroline products.
 | ||
| Scheme 210 Copper-mediated imino sulfonylation of γ,δ-unsaturated oxime esters with sodium sulfinates. | ||
Li, Wang and co-workers described the construction of spiro[indole-3,3′-pyrrolidine]-derived sulfones via a silver promoted radical-induced dearomative cascade spiro-annulation of sodium sulfinates and N-[(1H-indol-3-yl)methyl]-methacrylamides (Scheme 211).318 A series of aliphatic and aromatic sulfinates were evaluated to give the desired spiro products in moderate to high yields. Additionally, a variety of N-[(1H-indol-3-yl)methyl]methacrylamides were employed, including the benzene substituents on the indole ring, 2-substituted indoles and N-substituted substrates all successfully participated in this transformation to provide sulfonylated spiro[indole-3,3′-pyrrolidines] in good to high yields. To prove the synthetic utility of the dearomative cascade cyclization, it was performed at a larger scale (4 mmol) and afforded the same level of outcome.
 | ||
| Scheme 211 Ag-promoted radical-induced dearomative cascade spiro-annulation of N-[(1H-indol-3-yl)methyl]methacrylamides with sodium sulfinates. | ||
A copper-mediated oxidative radical addition/cyclization cascade of o-cyanoarylacrylamides with sodium sulfinates was accomplished by Li and co-workers. The use of N-methyl- and benzyl-protected o-cyanoarylacrylamides with sodium benzenesulfinate afforded the quinoline-2,4-diones whereas the unprotected substrate was not compatible. Additionally, a variety of both electron-donating and-withdrawing groups on the aryl rings were well-tolerated and produced the corresponding quinoline-2,4-dione-derived sulfones in moderate to good yields (Scheme 212).319 The pyridineacrylamide substrate also reacted well with sodium benzenesulfinate and obtained the desired product in 91% yield. Other sodium sulfinates, such as 4-methyl- and 4-chloro-substituted benzenesulfinate salts as well as sodium methanesulfinate provided varied sulfonylated quinoline-2,4-diones in good yields.
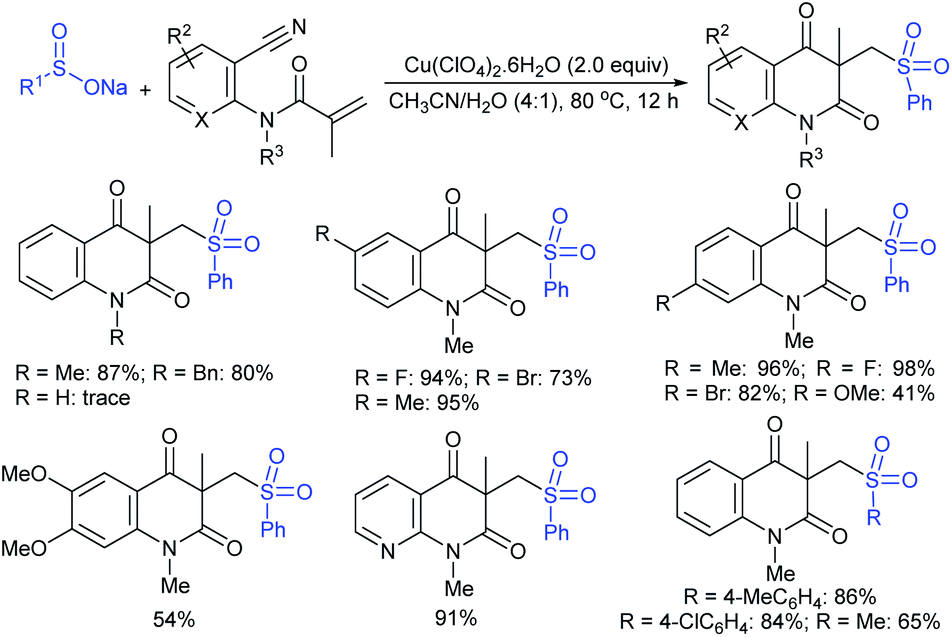 | ||
| Scheme 212 Cu-mediated oxidative radical addition/cyclization cascade of o-cyanoarylacrylamides with sodium sulfinates. | ||
A combination of Co(OAc)2·4H2O and KI-catalyzed ring-closing sulfonylation of N-alkyl-N-methacryloyl benzamides with different sodium sulfinates in CH3CN afforded isoquinoline-1,3(2H,4H)-dione derivatives. Zhou and co-workers employed both electron-donating and electron-withdrawing groups on the aryl rings of N-alkyl-N-methacryloyl benzamide, which did not affect the reaction yields with different aryl/alkyl sulfinate affording the corresponding products in 55–88% yields (Scheme 213A).320 Varying the N-alkyl (ethyl, methyl and isopropyl)groups had no obvious effect, however, N–H and N–OH-derived substrates did not afford the expected products. Subsequently, Zuo et al. reported that the eosin-Y catalyzed N-alkyl-N-methacryloyl benzamides with sodium sulfinates under visible light irradiation afforded isoquinoline-1,3(2H,4H)-diones (Scheme 213B).321 Several substituted N-methacryloylbenzamides at different positions of the benzene ring were smoothly reacted with a series of aryl/alkyl sulfinates to produce a wide range of corresponding products in good yields. Sodium trifluoromethanesulfinate was also used, however, the desired trifluoromethylsulfonylation product was not formed. As compared with the conventional method, photoredox catalysis showed inferior results.
 | ||
| Scheme 213 Co(OAc)2·4H2O/KI or Eosin-Y-catalyzed ring-closing sulfonylation of N-alkyl-N-methacryloyl benzamides with different sodium sulfinates. | ||
Mn(OAc)3-mediated the sulfonylation-cyclization reaction between 2-(2-alkynylphenyl)aminomaleates and arylsulfinic acid sodium salts to produce trisubstituted quinolines (Scheme 214).322 A variety of functional groups including the methoxy, halo, cyano, and carbonyl ester of aminomaleates are compatible with arylsulfonyl radical-triggered 6-endo-trig cyclization and aromatization. A sequential one-pot, two-step sulfonylation-cyclization was also developed via the three-component coupling of (2-ethynylphenyl)amine, dimethylacetylene dicarboxylate (DMAD) and sodium p-toluenesulfinate, which afforded 4-arylsulfonyl-methyl substituted quinolines in reasonable (48–80%) yields.
 | ||
| Scheme 214 Mn(OAc)3-mediated sulfonylation-cyclization reaction between 2-(2-alkynylphenyl)aminomaleates and sodium sulfinates. | ||
The Zhang group reported a facile method for the synthesis of 3-sulfonated quinolines via copper-catalyzed electrophilic cyclization of N-propargylamines using sodium sulfinates. A variety of N-propargylamines smoothly underwent the ring-closing sulfonylation with various aryl or alkyl sodium sulfinates and afforded a series of 3-sulfonylquinoline derivatives in moderate to high yields (Scheme 215A).323 More recently, Huang and co-workers also developed an alternative metal-free method for 3-sulfonated quinolines via visible-light irradiation of N-propargylanilines with sodium sulfinates. Eosin-Y catalyzed the wide-spread construction of 3-sulfonylquinolines in good to high yields by the ring-closing sulfonylation of various N-propargylanilines with sodium aryl and alkyl sulfinates (Scheme 215B).324 Both of these protocols represent mild and efficient methods for synthesizing 3-sulfonylquinolines for the formation of C–S and C–C bonds in a one step operation; particularly broad functional group tolerance is observed.
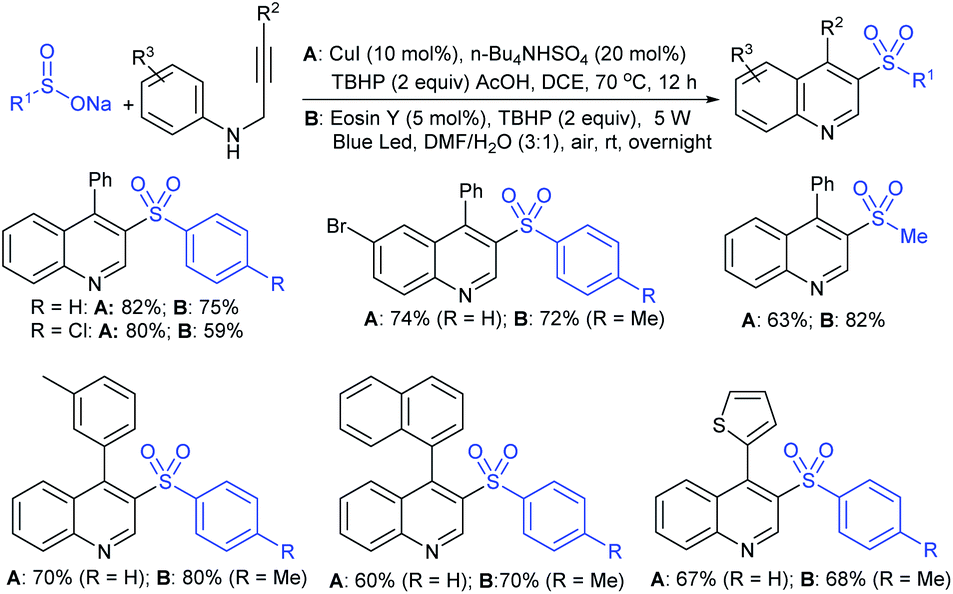 | ||
| Scheme 215 Electrophilic cyclization of N-propargylamines using sodium sulfinates. | ||
A silver-catalyzed tandem ring-closing azido-sulfonylation for the construction of sulfonated phenanthridine derivatives by the reaction of biphenyl acetylenes, TMSN3 and sodium sulfinates was reported. Bi and co-workers325 demonstrated that the silver catalyst played dual roles as the activator of the nitrogenation of biphenyl acetylene with TMSN3 as well as an oxidant for the generation of the reactive sulfonyl radical species from sodium sulfinates. The biphenyl acetylene substrates with either electron-donating or electron-withdrawing groups on both rings afforded the corresponding products in moderate to good yields (Scheme 216). Additionally, both aryl and alkyl sodium sulfinates were suitable substrates to provide the desired phenanthridine derivatives in 59–65% yields.
 | ||
| Scheme 216 Ag-catalyzed tandem ring-closing azido-sulfonylation of biphenyl acetylenes, TMSN3and sodium sulfinates. | ||
A direct and efficient silver-catalyzed synthesis of 6-sulfonylated phenanthridines from isocyanobiphenyls and sodium sulfinates using potassium persulfate as an oxidant at room temperature (Scheme 217) was reported.326 The sulfonylation was triggered by the insertion of sulfonyl radicals of 2-isocyanobiphenyls and successive cyclization and aromatization were successfully achieved. The generality of the protocol across a wide range of 2-isocyanobiphenyls and sodium sulfinates incorporating various functionalities, such as both electron-donating and electron-withdrawing groups produced the desired 6-sulfonyl phenanthridine derivatives in good to high yields, irrespective of any electronic and steric factors. The regioselectivity dispute was observed when the use of a 2-isocyanobiphenyl bearing an m-methoxy substituent was allowed to form a mixture of two regioisomers of the desired products in a ratio of 3:1.
 | ||
| Scheme 217 Ag-catalyzed ring-closing sulfonylation of isocyanobiphenyls with sodium sulfinates. | ||
The Kuhakarn group presented sulfonylation via the triggered 6-endo cyclization of o-alkynylisocyanobenzenes with sodium sulfinates to produce 2-sulfonylquinolines (Scheme 218).327 The scope of the reaction was successfully evaluated using various arenesulfinate salts bearing different groups and gave the corresponding products in variable yields. Additionally, sodium methanesulfinate also provided the mesylated quinoline in moderate yield. Unfortunately, sodium trifluoro-methanesulfinate was a challenging substrate for providing the desired product. Further, different types of o-alkynylisocyano-benzenes bearing various substituents around phenyl nucleus were well accommodated and generated a series of quinoline-derived sulfones in moderate to good yields. Various aliphatic isocyanobenzenes readily reacted with sodium 4-methylbenzenesulfinate to deliver the corresponding products in good yields.
 | ||
| Scheme 218 Sulfonylation via the triggered 6-endo cyclization of o-alkynyl isocyanobenzenes with sodium sulfinates. | ||
A copper-mediated oxysulfonylation reaction of alkenyl oximes with sodium sulfinates for the construction of sulfone-derived isoxazolines was established by Wang and co-workers. A wide variety of aromatic β,γ-unsaturated oximes with electron-donating and electron-withdrawing groups as well as a few aliphatic oximes reacted smoothly with sodium p-toluenesulfinate and provided the desired sulfones in moderate to good yields (Scheme 219).328 Oxysulfonylation was successfully applied to construct the desired product of a quaternary carbon-containing sulfone. A series of aromatic and aliphatic sulfinates participated in the cycloannulative-sulfonylation to provide varied sulfone derivatives in satisfactory yields. The 2-chlorophenyl sulfinate and trifluoro-methanesulfinate were poor substrates in this transformation.
 | ||
| Scheme 219 Copper-mediated oxysulfonylation reaction of alkenyl oximes with sodium sulfinates. | ||
A mild and convenient method was developed for the synthesis of various benzoxazines by using o-vinylanilides with sodium sulfinates via free radical ring-closing sulfonylation. Silver nitrate catalyzed the oxysulfonylation of various o-vinylanilides with aryl- or alkyl sodium sulfinates to produce a variety of valuable sulfonated benzoxazines in moderate to high yields (Scheme 220).329 A variety of substituents at the α-position of the styrenes and other substituted benzamide derivatives were successfully explored for this transformation.
 | ||
| Scheme 220 Ag-catalyzed ring-closing sulfonylation of isocyanobiphenyls with sodium sulfinates. | ||
3.4.10.3. Construction of carbocycles. Sodium persulfate-mediated the convenient synthesis of sulfonyl indenones via a ring-closing radical sulfonylation of 2-alkynyl-benzonitriles with sodium arylsulfinates, as was demonstrated by Liang and co-workers (Scheme 221).330 Various 2-alkynylbenzonitriles were explored with sodium arylsulfinates trifluoromethanesulfinate for the construction of C–C and C–S bonds to form a wide range of sulfonated indenones in moderate to good yields. The trend of the reactivity of 2-alkynylbenzonitriles having the electron-donating group was superior to the electron-withdrawing group.
 | ||
| Scheme 221 Na2S2O8-mediated ring-closing radical sulfonylation of 2-alkynylbenzonitriles with sodium arylsulfinates. | ||
The electrochemical sulfonylation/semipinacol rearrangement of allylic alcohols with sodium sulfinates for the preparation of α,α-disubstituted cyclopentanones with moderate to excellent yields (Scheme 222).331 The transformations involved a 1,2-migration process under oxidant/metal-free mild reaction conditions. A variety of electronic substituents on the benzene ring of cycloalkanol-derived styrenes reacted with arylsulfinates to afford the corresponding products in good yields. Subsequently, spiro 1-indanone was also afforded in good yield with excellent diastereoselectivity and underwent a cation 1,2-migration process.
 | ||
| Scheme 222 Electrochemical sulfonylation/semipinacol rearrangement of allylic alcohols with sodium sulfinates. | ||
AgNO3 catalyzed the ring-opening/closing-sulfonylation of methylenecyclopropanes with sodium sulfinates for the synthesis of sulfone-derived 1,2-dihydronaphthalenes in the presence of K2S2O8 as the oxidant (Scheme 223).332 A wide range of mono-, di- and trisubstituted or unsubstituted methylenecyclopropanes reacted smoothly with sodium p-tolylsulfinate through difunctionalization to obtain the desired products in good to high yields. Moreover, a series of arylsubstituted sodium sulfinates was successfully converted into substituted 3-sulfonyl-1,2-dihydro-naphthalenes in moderate to good yields, whereas sodium methanesulfinate was afforded in only a trace amount.
 | ||
| Scheme 223 Sodium persulfate-mediated ring-closing radical sulfonylation of 2-alkynylbenzonitriles with sodium arylsulfinates. | ||
Mn(III) promoted the radical sulfonylation/dearomatization reaction between biaryl ynones and sodium sulfinates to construct spiro[5.5]trienones, which was explored by Liu and co-workers (Scheme 224).333 The oxidative ipso-annulation reaction was explored with a variety of biaryl ynones with different aromatic and aliphatic sulfinates to provide a range of sulfonyl-substituted spiro[5.5]trienones in moderate to high yields. The biaryl ynone substrate bearing two methoxy groups were placed in the ortho-positions and two meta-methoxy groups proceeded an alternative pathway under same conditions and delivered seven-membered ring products in variant yields.
 | ||
| Scheme 224 The Mn(III)-promoted radical sulfonylation/dearomatization reaction between biaryl ynones and sodium sulfinates. | ||
3.4.11.1. Three-component reactions. The Deng group developed the iron-catalyzed oxidative coupling of sodium sulfinates, methyl ketones and dimethylacetamide (DMA as a solvent) in a sequential one-pot manner through C–H functionalization for two C–C and C–S bonds (Scheme 225).334 A highly chemoselective, three-starting material four-component reaction (3SM-4CR) strategy was carried out for the synthesis of β-acyl allylic sulfones from readily available starting materials. Various aromatic/aliphatic sulfinates and the effect of the substituents on the ketone moiety in DMA gave the desired products in low to high yields. Similarly, the reaction of o-hydroxyacetophenone with sodium benzenesulfinate in DMA proceeded smoothly to produce the corresponding sulfonyl chroman-4-one derivatives in moderate to good yields.
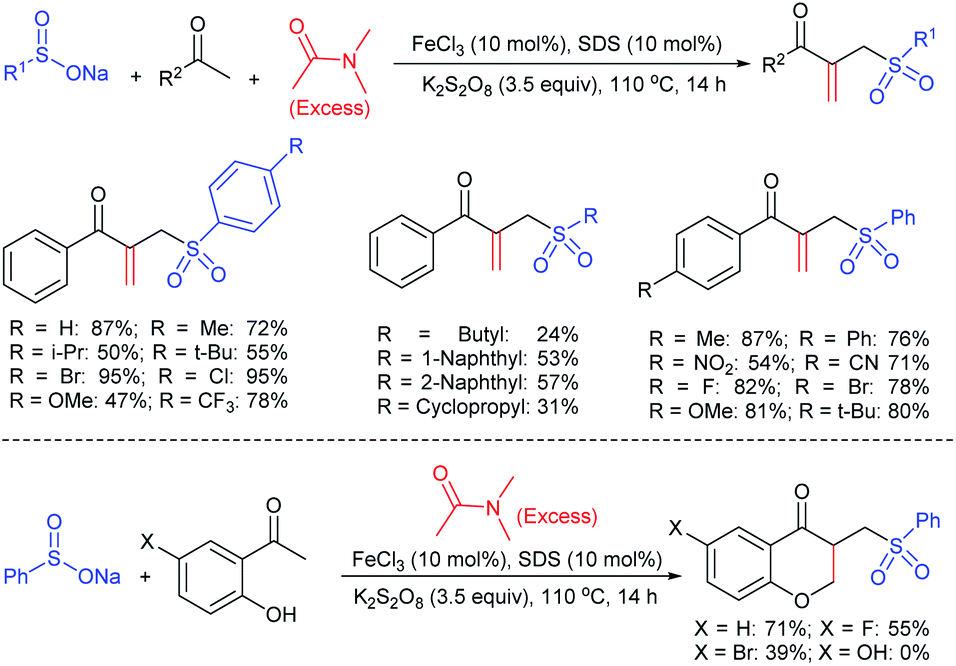 | ||
| Scheme 225 Fe-catalyzed oxidative coupling of sodium sulfinates, methyl ketones and dimethylacetamide. | ||
The same authors conducted the iron-catalyzed oxidative benzylic C–H functionalization of 2-methylazaarenes, sodium sulfinates and dimethylacetamide (DMA) under mild conditions (Scheme 226).335 The broad functional group tolerance makes this protocol attractive for the synthesis of highly functionalized quinoline and benzothiazole derivatives. The 2-methylquinolines, 1-methylisoquinoline, 4-methyl-quinoline, 2-methylquinoxaline and 2-methylbenzothiazoles were smoothly coupled with a range of sodium arylsulfinates and provided the corresponding products in moderate to good yields. However, the substrate was limited to the above methyl aza-heterocycles. Other methylheterocycles such as 8-methylquinoline, 2-methylbenzo[d]imidazole, 2-methyl-benzo[d]oxazole and 2-methylpyrazine failed to react with sodium benzenesulfinate.
 | ||
| Scheme 226 Fe-catalyzed oxidative coupling of sodium sulfinates, 2-methylazaarenes and dimethylacetamide. | ||
The Anderson group developed an unprecedented remote C to N arene migration induced by the in situ generation of iminyl radicals from vinyl azides. Ag catalyzed the multicomponent reaction of homopropargylic alcohols, trimethylsilyl azide and sodium sulfinates to access allyl sulfones in good to high yields (Scheme 227).336 The symmetrical homopropargylic alcohol migration products are consistent, whereas the non-symmetric homopro-pargylic alcohols showed interesting chemoselectivity of the migration of the arene groups. The electronic properties of the arylsulfinates revealed a relatively low influence, and alkyl sulfinates also showed comparable efficiency. A range of non-symmetric biaryl propargylic alcohols was subjected to TMS-azide and sodium p-toluenesulfinate under the same rearrangement conditions to afford a mixture of allyl sulfones in moderate to good yields. The competition between two different aryl groups revealed a clear trend for the preferential migration of the more electron-rich arene. The migratory aptitude was deliberated based on the linear free energy relationships that could provide valuable insight into nature's rearrangements. The reaction was conducted using TEMPO or BHT, and the product formation was completely inhibited, resulting in a plausible mechanism outlined as the radical process.
 | ||
| Scheme 227 Ag-catalyzed multicomponent reaction of homopropargylic alcohols, trimethylsilyl azide and sulfinate salts. | ||
Zhang and co-workers described an efficient method for the synthesis of β-azidosulfonates through the CuCl-catalyzed radical oxidative azido-sulfonylation of alkenes with TMSN3 and sodium sulfinate (Scheme 228).337 The combination of the CuCl/TBPB reagent system is widely applicable to a variety of sodium sulfinates and a broad range of alkenes in the vicinal difunctionalization to generate valuable divergent β-azidosulfonate products in moderate to high yields with high selectivity. More interestingly, 1,3-butadiene and cyclohexa-1,3-diene preferred to form thermodynamically more stable 1,4-addition regioisomers.
 | ||
| Scheme 228 CuCl-catalyzed radical oxidative azido-sulfonylation of alkenes with TMSN3and sodium sulfinates. | ||
The Han group developed an efficient electrochemical oxidative vicinal alkoxysulfonylation of aryl alkenes with alcohols and sodium sulfinates for the synthesis of β-alkoxy sulfones. The reaction was conducted in an undivided cell at room temperature using various electron-donating and electron-withdrawing arylsulfinates, a variety of styrenes bearing different substituents on the aromatic ring and MeOH, and proceeded smoothly to give the corresponding β-methoxy sulfones in moderate to good yields (Scheme 229).338 Additionally, different α-substituted styrene derivatives participated and no obvious effect was observed on the outcome (63–78%). Other aliphatic alcohols were also tolerated, however, owing to the low solubility, water was used as a co-solvent and afforded the corresponding alkoxy-sulfonylation products in 29–60% yields. The sodium methanesulfinate was a poor substrate for the transformation.
 | ||
| Scheme 229 Electrochemical oxidative vicinal alkoxysulfonylation of aryl alkenes with alcohols and sodium sulfinates. | ||
The photoredox-catalyzed net-redox neutral tandem arylation/sulfonylation of styrene derivatives with sodium sulfinates and cyanopyridines under visible light irradiation was developed by Opatz and co-workers (Scheme 230).339 The vicinal aryl-sulfonylation allowed significant variation within each of the three components and showed high diastereoselectivity. A broad variety of substituents were present at the styrene's aromatic core, including electron-donating and electron-withdrawing groups, and the products were obtained in good to high yields. Additionally, α-methylstyrene and β-substituted styrenes were well-tolerated to give desired products in good to excellent yields. The range of aromatic and aliphatic sulfinate salts give higher yields than their branched counterparts. Subsequently, the different substituents on cyanopyridines, 2-cyano-isoquinoline and 2-cyanoquinoline successfully furnished arylsulfonylated products; however, cyanopyridines bearing strong electron-donating substituents afforded only moderate yields. Noteworthily, the styrene-functionalized biomolecules, such as peptide, carbohydrate, cholic acid and estrone-derived alkenes led to the corresponding products without significant impact on the outcome. The visible light-mediated aryl-sulfonylation was also successfully proved at a large scale reaction without any great difference in the yield.
 | ||
| Scheme 230 Photoredox-catalyzed vicinal aryl-sulfonylation of styrene derivatives, cyanopyridines with sulfinic acid salts. | ||
Manolikakes and co-workers developed iron-catalyzed vicinal oxysulfonylation of enamides and enecarbamates with sulfinic acid salts and alcohols for the preparation of oxysulfonylated products. The three-component coupling of various (E/Z)-mixture enamides and enecarbamates proceeded smoothly with different aryl/alkyl sulfinates in MeOH at room temperature and furnished diverse β-amidosulfones in moderate to high yields with a satisfactory diastereomeric mixture (Scheme 231).340 The EtOH or i-PrOH was used as solvent to deliver the expected β-amidosulfones in low yields, however, other aliphatic alcohols cyclohexanol, phenols or water proved unsuitable. Reactions with cyclic enamides or encarbamates, as well as pyridine sulfinate or trifluoromethane sulfinate, did not afford the desired products. Moreover, the 1,2-oxysulfonylation process was amendable to the gram-scale synthesis of the amidosulfone products.
 | ||
| Scheme 231 The Fe-catalyzed vicinal oxysulfonylation of enamides and enecarbamates with sulfinic acid salts and alcohols. | ||
N-Heterocyclic carbene (NHC-37) catalyzed the three-component β-sulfonylation-esterification of β-bromoenals and sodium sulfinates with alcohols for the synthesis of functionalized sulfonyl esters (Scheme 232).341 The Du group used a variety of 3-(substituted phenyl)-2-bromoenals with electron-withdrawing or electron-donating groups at different positions to afford the corresponding products in good to high yields. Subsequently, aliphatic 2-bromoenals were also applicable, giving rise to sulfone esters in moderate yields. Several aryl, hetereoaryl and alkyl sulfinates were also compatible and the desired products were obtained in moderate to good yields. The alcohols, such as, MeOH, EtOH, BnOH, i-PrOH, PhOH and 6-bromonaphthol were also accommodated to give different esters in good to high yields. Unfortunately, sulfonylation of β,β-disubstituted β-bromoenal was unsuccessful. The reaction of different nucleophiles like cyclohexanethiol and aniline worked equally well to afford the anticipated products in high yields. Disappointingly, aliphatic amines like n-butylamine were not suitable reactants for this transformation.
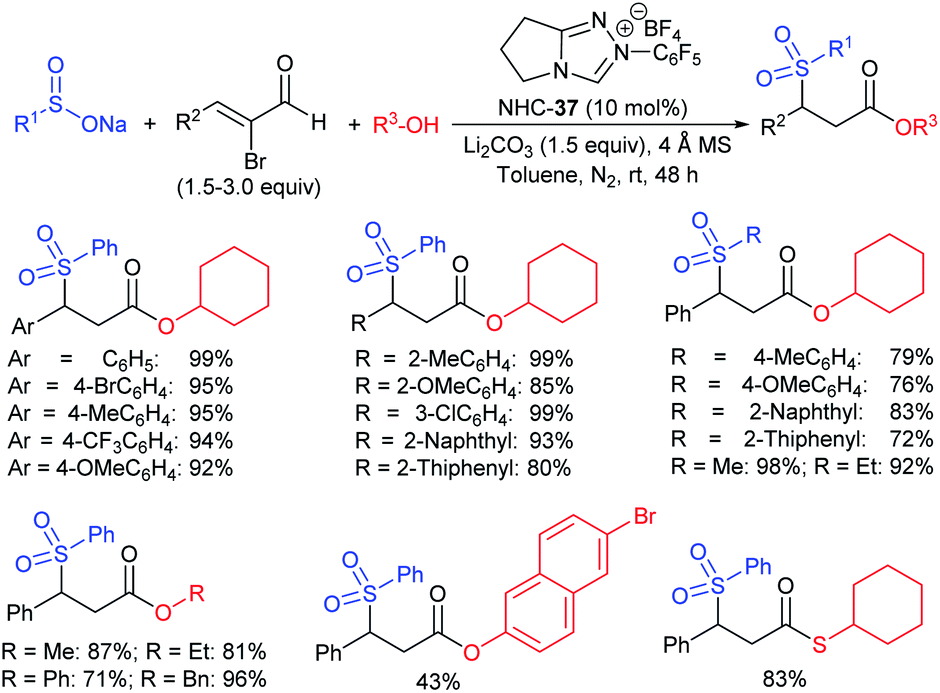 | ||
| Scheme 232 NHC (37)-catalyzed three-component coupling of β-bromoenals and sodium sulfinates with alcohols. | ||
A plausible mechanism for the generation of functionalized sulfonyl esters is rationalized in Scheme 233. The deprotonation of the precatalyst NHC salt with Li2CO3 generated the NHC catalyst, which reacted with (Z)-2-bromoenal to give the Breslow intermediate A. The umpolung of A affording intermediate B would be tautomerized to 2-bromoacylazolium C. Subsquently, the loss of the bromide of C prompted the formation of the more stable (E)-α,β-unsaturated product. The conjugate-addition of acyl azolium D at the more electrophilic β-position with sodium sulfinate led to the formation of E, which underwent protonation to form F. Then, the tautomerization of F to afford G was followed by nucleophilic substitution with alcohol, giving rise to the desired product.
 | ||
| Scheme 233 A plausible mechanism for the NHC-catalyzed three-component coupling of β-bromoenals, sodium sulfinates with alcohols. | ||
Kumar and co-workers described the vicinal phenoxy-sulfonylation of alkynes with sodium sulfinates and phenols under mild reaction conditions using I2/K2CO3 for the synthesis of (Z)-β-aryloxy vinylsulfones. The three-component regioselective difunctionalization of various terminal and internal alkynes, sulfinates and a range of phenols, thiophenols and naphthols provided a series of vinyl sulfone products in good to high yields (Scheme 234).342 Although several sulfinates were compatible, however, 2- or 4-nitrobenzenesulfinates and trifluoromethanesulfinate unsuccessfully participated due to the instability of the corresponding sulfone radicals. Moreover, the practical gram-scale (20 mmol) reaction was also demonstrated as a valuable potential for the process.
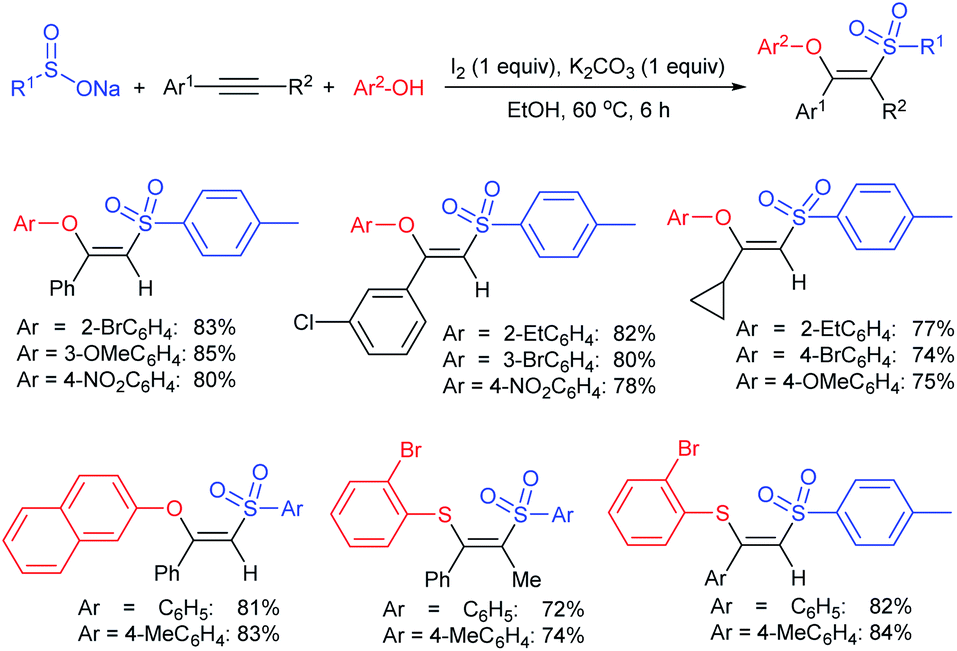 | ||
| Scheme 234 I2-catalyzed vicinal phenoxysulfonylation of alkynes with sodium sulfinates and phenols. | ||
A direct three-component vicinal difunctionalization of alkynes, sodium sulfinates and Togni reagent through trifluoro-methylation and sulfonylation under catalyst- and additive-free, extremely mild conditions was reported. A wide range of (E)-β-trifluoro-methyl vinyl sulfones were obtained in moderate to good yields with excellent stereoselectivity by using various terminal alkynes, different benzenesulfinates, heteroarenesulfinate or alkanesulfinates and Togni reagent (Scheme 235).343 The internal alkyne, phenylpropyne, was employed for the synthesis of tetrasubstituted vinylsulfones in low yield with excellent stereoselectivity.
 | ||
| Scheme 235 I2-catalyzed vicinal phenoxysulfonylation of alkynes with sodium sulfinates and phenols. | ||
A convenient three-component reaction of terminal alkynes, TMSN3 and sodium sulfinates was realized by Bi and co-workers.344 A wide range of aryl- and heteroaryl-functionalized terminal alkynes suitable for reacting with sodium p-toluenesulfinate and TMSN3 for the silver-catalyzed vicinal aminosulfonylation reaction afforded the corresponding β-sulfonyl enamines in good to excellent yields (Scheme 236). The robust nature of this method indicates that the estrone-derived terminal alkyne was successfully transformed into the corresponding β-sulfonyl enamine in 79% yield. The electron-rich or electron-deficient sulfinates as well as alkyl sulfinate salts afforded the corresponding enamines in high yields.
 | ||
| Scheme 236 Silver-catalyzed vicinal aminosulfonylation of terminal alkynes, TMSN3 and sodium sulfinates. | ||
Recently, an electrochemical three-component intermolecular vicinal aminosulfonylation of alkenes with sulfinates and amines was achieved by the Li group (Scheme 237).345 A wide range of terminal alkenes were all smoothly converted into 2-sulfonylethan-1-amines in moderate to good yields and the electronic nature of substituents had some impact on the reactivity. The aliphatic alkene had no reactivity, whereas 1,1-disubstituted alkene afforded the desired product. The internal alkene also succeeded in accessing the corresponding product with 61% yield in a 4:1 diastereomeric mixture. An array of sodium sulfinates, including aryl and alkylsulfinates, were able to deliver 2-sulfonylethan-1-amines in good to high yields. Moreover, a series of primary and secondary amines were compatible in 1,2-aminosulfonylation, thus leading to diverse 2-sulfonylethan-1-amines in satisfactory yields. Unfortunately, the use of NH3 or NH3·H2O had no target product.
 | ||
| Scheme 237 Electrochemical oxidative vicinal alkoxysulfonylation of aryl alkenes with alcohols and sodium sulfinates. | ||
Copper catalyzed the vicinal difunctionalization of styrenes with sodium arylsulfinates and t-BuONO for the selective synthesis of diverse β-oxime sulfones (Scheme 238).346 The intermolecular three-component method enabled the one-step formation of C–N and C–S bonds. A wide range of terminal styrenes and internal alkenes were reacted with sodium arylsulfinates, bearing either electron-donating or electron-withdrawing groups to produce the desired products in good to high yields.
 | ||
| Scheme 238 Cu-catalyzed vicinal difunctionalization of styrenes with sodium arylsulfinates and t-BuONO. | ||
The Hu and Li group reported two independent methods for the synthesis of functionalized 2H-azirines via the azirido-disulfonylation protocol. A tandem annulation of terminal alkynes with sodium sulfinates and tert-butyl nitrite as the nitrogen source has been described under catalyst-free conditions (Scheme 239A).347 A wide range of aryl and alkyl alkynes were efficiently converted to give the targeted 2H-azirine in moderate to good yields. In general, an array of substituents on the aryl rings of arylalkynes were tolerated well, and both the electronic nature and position affected the reactivity. The use of electron-deficient alkynes delivered in diminishing yields. Alkylalkynes bearing different functional groups were also accommodated perfectly, albeit with lower yields. Notably, menthol, glycine, D-proline, adamantine, 3-oxo-androstene and estrone-derived alkynes were also viable for the annulation protocol affording the valuable modified bioactive molecules in acceptable yields. Further, a broad spectrum of aryl and alkylsulfinates reacted smoothly to produce the 2,2-disulfonyl-2H-azirineproducts in good to high yields. The same group successfully extended the use of alkynyl carboxylic acids instead of terminal alkynes (Scheme 239B).348 TBAF catalyzed the decarboxylative heteroannulation of various arylpropiolic acids bearing electron-donating or -withdrawing substituents on the aryl ring in their reaction with different sodium sulfinates and tert-butyl nitrite in the presence of TBHP to produce a series of 2,2-disulfonylated-2H-azirines including bioactive analogs. Despite the vast substrate scope, the ethyl propiolate and propiolic acid showed no reactivity for these transformations.
 | ||
| Scheme 239 Tandem annulation of alkynes with sodium sulfinates and tert-butyl nitrite. | ||
A combination of NiCl2, Ru-photo (38) and ligand (39) catalyzed the vicinal difunctionalization of alkynes with sodium sulfinates and aryl halides under blue light-emitting diode (7.4 W LED) strips for 24 hours, thus enabling the generation of tri-substituted alkenes (Scheme 240).349 The Rueping group successfully demonstrated a broad variety of aryl bromides and aryl iodides bearing electron-withdrawing, neutral and donating substituents involving common sensitive functional groups and provided a series of highly functionalized vinyl sulfones in good to high yields. The protocol was readily extended to various nitrogen-containing heteroaromatic halides and thiophene-derived halides to give the corresponding products in good to high yields. Further, the scope of arylalkynes was well illustrated by the tolerance of a series of functional groups that had no obvious effects on the reaction efficiency. Notably, cyclohexenyl alkyne and internal alkynes were also suitable substrates that gave the desired product in moderate yields. Overall, the size of the alkyne substituent increased and the product yield significantly decreased. Structurally different arenesulfinates bearing electron-donating as well as electron-withdrawing functional groups gave the desired products in moderate to high yields. Methyl, ethyl, cyclohexyl and cyclopropyl-derived sulfinates were also suitable substrates that afforded the corresponding products in acceptable yields. In particular, a series of bioactive molecules having galactopyranose, probenecid, pregnenolone, cholestanol and adamantine carboxylic acid motifs bearing aryl halides as well as oestrone and δ-tocopherol-derived alkynes effectively underwent vicinal aryl-sulfonylation to give the anticipated products in moderate to high yields.
 | ||
| Scheme 240 Photoredox/nickel dual-catalyzed vicinal difunctionalization of alkynes with sodium sulfinates and aryl halides. | ||
A green and recyclable Amberlyst-15 ion-exchange resin (50 wt%)-catalyzed one-pot three-component reaction in water was developed by Zeng and co-workers (Scheme 241).350 The coupling of different aldehydes (aryl/heteroaryl/alkyl) with aromatic and aliphatic sulfinate salts and trimethoxy- and dimethoxy-benzenes produced diarylmethyl sulfones in good to high yields. The recyclability of the Amberlyst-15 ion-exchange resin was inspected for up to four cycles and there was a significant impact on the outcome.
 | ||
| Scheme 241 Amberlyst-15-catalyzed the three-component reaction of tri-/dimethoxy-benzenes and aldehydes with sulfinate salts. | ||
Cao and co-workers351 developed a reagent-controlled efficient transition-metal-free highly regioselective strategy for the synthesis of two different sulfonylated imidazoles via a three-component reaction of amidines, ynals, and sodium sulfinates as presented in Scheme 242. AcOH mediated various electron-rich and electron-poor N-substituted benzimidamides and formed the corresponding imidazoles in 56–83% yields. Similarly, electron-withdrawing and electron-donating groups on the other benzene ring of N-substituted amidines and multi-substituted amidines also reacted smoothly in this cascade annulation process to generate the target products in moderate to high yields. Unfortunately, the alkyl amidines were not suitable substrates and the desired products were not detected. A series of aromatic and aliphatic sulfinates smoothly underwent cascade annulations to give the desired products in good yields. On the other hand, the multi-component ring-closing sulfonylation of various N-substituted benzimidamides and amidines with different substituents reacted smoothly with different sulfinate salts in the presence of TBHP in MeCN at 70 °C. As a result, other kinds of sulfonylated imidazoles were successfully achieved in good to high yields.
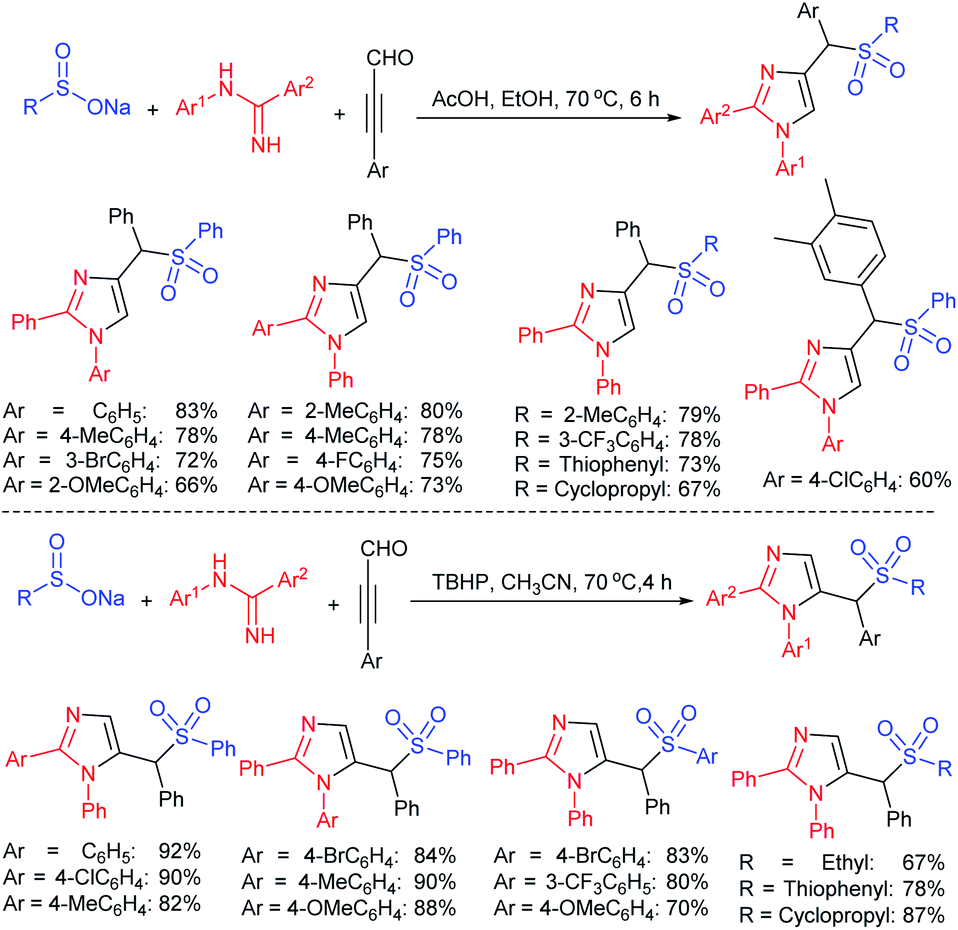 | ||
| Scheme 242 The three-component reaction of amidines, ynals, and sodium sulfinates. | ||
Yang and co-workers developed the Pd-catalyzed regio- and stereoselective three-component reaction via sequence decarboxylative allylic substitution/N–H carbene insertion to access allylic sulfone-containing amino acids (Scheme 243).352 The domino reaction was applicable to a broad range of coupling partners of N-unprotected vinyl benzoxazinones, aryl and alkyl sulfinates and aryl-substituted diazo esters to furnish a series of amino acid-derived allyl sulfones in synthetically acceptable yields with excellent regioselectivity as an E-isomer. Subsequently, DBU mediated the cycloannulation of allylic sulfones for the construction of quaternary carbon centers including privileged functionalities, sulfones and amino acid esters of 1-benzaazepines in moderate yields with excellent diastereoselectivity.
 | ||
| Scheme 243 The Pd-catalyzed three-component reaction of vinyl benzoxazinones, diazo esters and sodium sulfinates. | ||
Wang and co-workers353 have utilized DMSO as a one-carbon synthon for the radical coupling reaction of sodium arylsulfinates in the presence of 3,4-dibromo-5- hydroxy-2(5H)-furanone as a brominating agent at 100 °C. A series of arylsulfonyl dibromomethanes was obtained in satisfactory yields (69–90%) using different substituents on the benzene ring of arylsulfinates as well as 2-naphthyl sulfinate and 2-thiophenyl sulfinate sodium salts (Scheme 244). The impacts of both electronic effects and steric hindrance on the reaction outcome were observed.
 | ||
| Scheme 244 The three-component reaction of 3,4-dibromo-5-hydroxy-2(5H)-furanone, DMSO and sodium sulfinates. | ||
Nematpour et al.354 established the CuI-catalyzed three-component reaction involving sodium arylsulfinates, terminal alkynes and sulfonyl azides under microwave irradiation for the synthesis of N-sulfonylketenimine derivatives in good yields (75–89%) as shown in Scheme 245. It was observed that the reaction with phenylacetylene gave better yields when compared with aliphatic acetylenes.
 | ||
| Scheme 245 CuI-catalyzed three-component reaction of sodium sulfinates, terminal alkynes and sulfonyl azides under MW irradiation. | ||
3.4.11.2. Four-component reactions. Opatz and co-workers developed the highly efficient photoredox-catalyzed four-component sulfonylation/aminoalkylation of styrenes. There were a large variety of substituents on the styrene cores with a series of sodium sulfinates as a result of adding various arylaldehydes and anilines under visible light irradiation to give widespread γ-sulfonylamine derivatives in medium to good yields with an almost 1
:1 diastereomeric mixture (Scheme 246).355 The developed 4-CR protocol is widely applicable to the modification of complex biomolecules; for instance, styrene-functionalized cholic acid and biotin derivatives are tolerated. Additionally, the aldehyde-substituted dipeptide as well as a styrene-modified peptide resulted and the corresponding products were obtained in satisfactory yields. Even complex carbohydrates such as di- and trisaccharide derivatives reacted smoothly and provided synthetically useful yields of the desired coupling products.
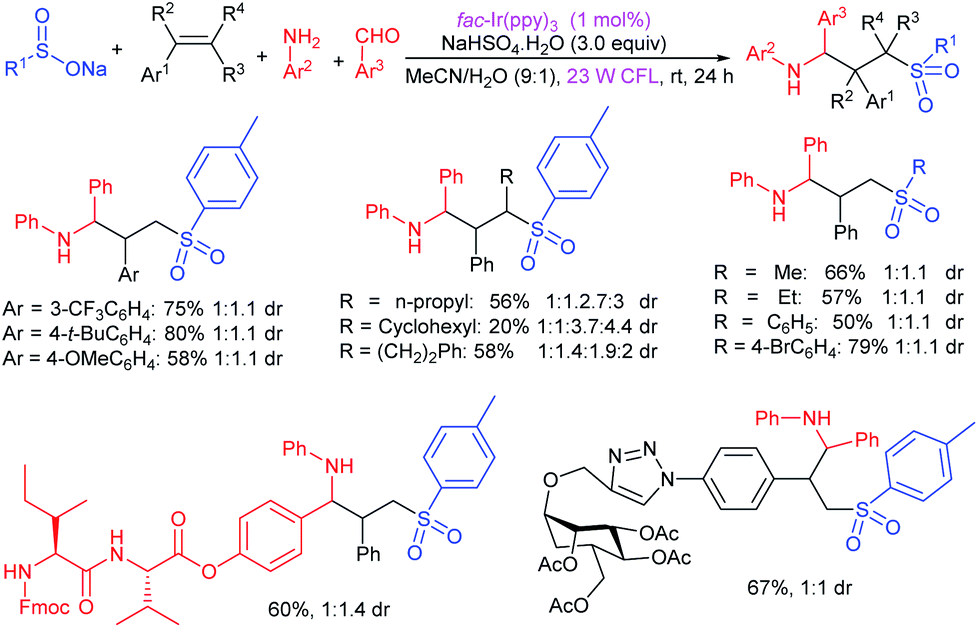 | ||
| Scheme 246 Photoredox-catalyzed sulfonylation/aminoalkylation of styrenes, sodium sulfinates, arylaldehydes and anilines. | ||
Copper catalyzed a multi-component reaction employing alkenes, sodium sulfinates and diazonium salt through tandem radical oxysulfonylation-diazenylation under aerobic conditions to access α-arylhydrazo-β-keto sulfones (Scheme 247).356 A broad range of functional groups allowed the formation of C–O, C–S, and C–N bonds in a one-pot operation. A series of styrene derivatives with different substituents were obtained, including two naturally derived alkenes from coumarin and L-tyrosine to give the corresponding products in moderate to high yields. Unfortunately, aliphatic alkenes or β-methylstyrene were not compatible substrates for yielding the corresponding products. The scope of sulfinates and diazonium salts was also carefully investigated. Several sodium sulfinates bearing electron-donating or -withdrawing groups on the phenyl ring as well as sodium methanesulfinate were smoothly transformed into the desired products in moderate to good yields. Furthermore, various aryl diazonium salts with different substituents on the phenyl ring also proceeded smoothly and provided the α-arylhydrazo-β-keto sulfones in 62–70% yields.
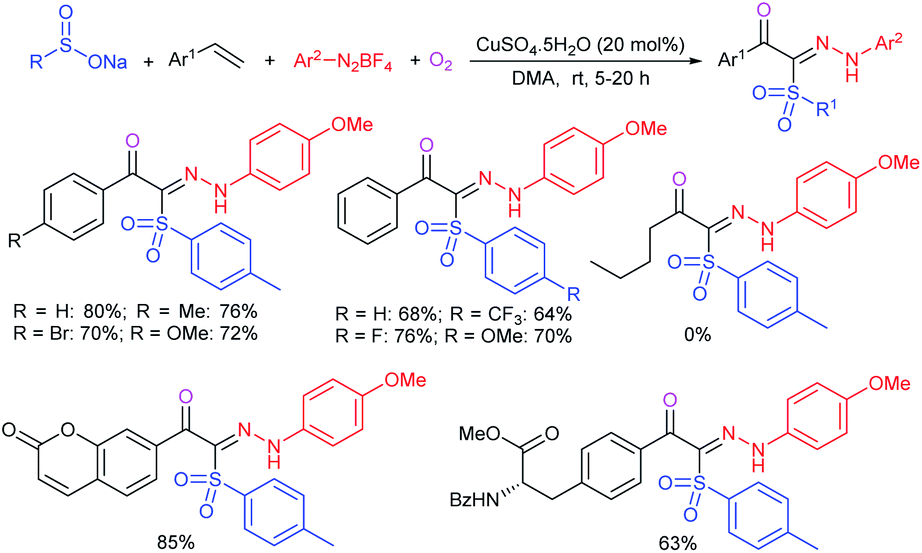 | ||
| Scheme 247 Copper-catalyzed multi-component reaction of alkenes, sodium sulfinates, diazonium salt and molecular oxygen. | ||
The preparation of sulfonyl-imidazolone derivatives in 73–90% yields occurred via a four-component reaction between sodium arylsulfinates, trichloroacetonitrile, benzylamines and acetylene dicarboxylates in H2O at room temperature (Scheme 248).357 Alternatively, alkyl propiolate was used instead of acetylenedicarboxylates to obtain sulfonyl-pyrimidinones in 72–82% yields. The scope and generality of four-component substrates are limited for the synthesis of sulfone-containing imidazolones and pyrimidinones.
 | ||
| Scheme 248 Four-component reaction of trichloroacetonitrile, benzylamines, activated alkynes and sodium arylsulfinates. | ||
Subsequently, the same group reported the tandem four-component reaction of sodium arylsulfinates, trichloro-acetonitrile, benzylamines and heterocumulenes (aryl isothiocyanate and aryl isocyanate) in DMF at room temperature (Scheme 249).358 The reaction procedure was described is mild and provides a useful path for the synthesis of functionalized sulfonyl-derived phenylcarbamimidic(thio) anhydrides in 75–88% yields.
 | ||
| Scheme 249 Four-component reaction of trichloroacetonitrile, benzylamines, activated alkynes and sodium arylsulfinates. | ||
Chang and co-workers359 developed the trifunctionalization of alkenes involving three-steps in one operation to generate oxygenated sulfonylcumenes. As shown in Scheme 250, the NBS mediated the allylic bromination of 2-arylpropenes to form styryl bromides, followed by nucleophilic substitution with sodium sulfinates to provide in situ allylic sulfones in the ROH/H2O solvent system. Further, V2O5/H2O2 mediated the alkoxyhydroxylation of allylic sulfones for the 1,2,3-tricarbo-functionalized products. Various α-methylstyrenes were smoothly reacted with aryl and alkylsulfinates in different alcohols and provided the corresponding products in a range of (5–86%) yields. The 4-methoxy, 4-methyl, 2-furyl-derived 2-arylpropenes, CF3SO2Na and t-BuOH led to a complex mixture.
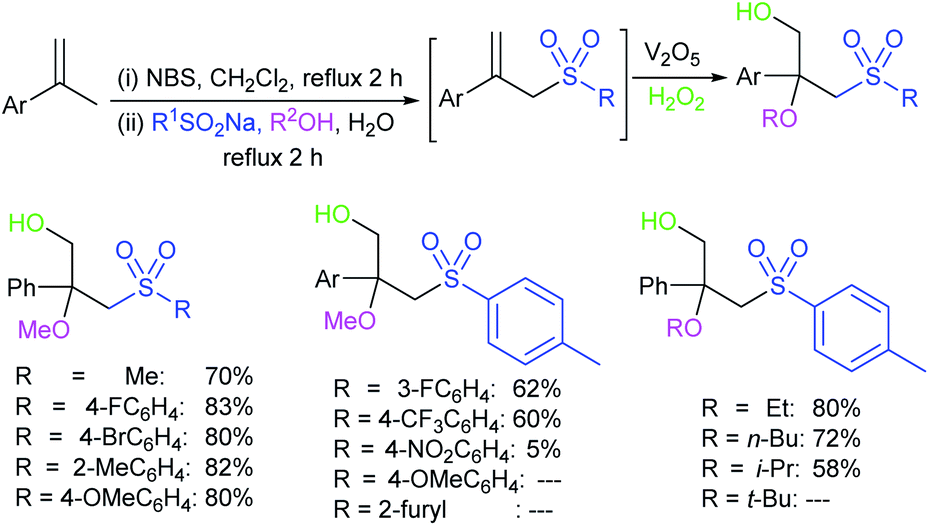 | ||
| Scheme 250 Trifunctionalization of 2-arylpropenes with sodium sulfinates and ROH. | ||
3.5. Miscellaneous
In 1971, Middelbos et al., described the reaction between sodium sulfinates and haloforms in the presence of aqueous base (KOH) to give the dihaloromethyl sulfones. A few aryl and alkyl sulfinates reacted with both chloroform or bromoform, affording dichloromethyl and dibromomethyl sulfones, respectively, in moderate to good yields (Scheme 251).360 Sodium benzylsulfinate in chloroform resulted in 5% yield only of the desired dichloromethyl sulfone. | ||
| Scheme 251 KOH-mediated coupling of sodium sulfinates with haloforms. | ||
LiBr catalyzed the regioselective sulfonylation of aziridines with sodium sulfinates in water at 80–90 °C for the synthesis of β-amino sulfones as described by the Yadav group.361 A range of aziridines were sulfonylated using two sulfinate salts for the synthesis of various β-amino sulfones in moderate to high yields (Scheme 252). The author stated that the p-toluenesulfinate gave better yields than benzenesulfinate and the regioselectivity ring-opening of aziridines depends on the substituents. The sulfonylation of aziridines bearing an aryl substituent preferably occurred at the benzylic carbon; in contrast, the nucleophilic attack took place exclusively at the terminal carbon atom of the aziridine ring. The cyclohexene-derived aziridine also participated in the ring-opening sulfonylation, resulting in the trans-cyclohexane-1,2-aminosulfone in good yield.
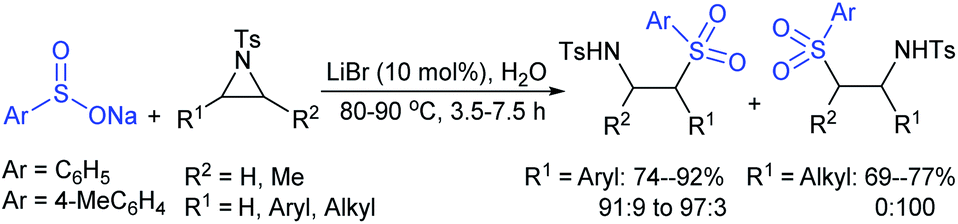 | ||
| Scheme 252 LiBr catalyzed the regioselective sulfonylation of aziridines with sodium sulfinates. | ||
Zhou et al., disclosed a simple and efficient sulfonyl-Michael addition of activated alkenes with sodium sulfinates in water at room temperature. Various α,β-unsaturated acyclic ketones, acrylamide and acrylates were successfully sulfonylated with sodium 4-toluenesulfinate to access a series of β-sulfonyl carbonyl compounds in good to high yields (Scheme 253).362 Sterically hindered crotonates/crotonamides or cinnamates/cinnamides failed to yield the desired products, albeit, β-nitrostyrene was well accommodated. Besides, different N-substituted maleimides and cyclic enones were also employed to afford the desired β-sulfonylated products in acceptable yields. Likewise, a range of aryl-substituted sulfinates were fairly reacted with chalcones, providing the desired products in varied yields. Unfortunately, sodium alkylsulfinates such as methylsulfinate and benzylsulfinate were unsuccessful in this transformation.
 | ||
| Scheme 253 Sulfonyl-Michael addition of activated alkenes with sodium sulfinates. | ||
In 2002, Murakami and Furusawa demonstrated a one-pot synthesis of aryl sulfones involving the bromination of alcohols, followed by the sulfonylation of primary alcohols with sodium arenesulfinates (Scheme 254).363 Allylic and benzylic alcohols were treated with N-bromosuccinimide and triphenylphosphine, followed by tetrabutylammonium iodide (TBAI)-catalyzed nucleophilic substitution with PhSO2Na and 4-MeC6H4SO2Na to afford the aryl sulfones in good to high yields. This was further extended to the less reactive aliphatic alcohols bearing a functional group such as ether, ester and nitrile, which were smoothly converted into alkyl aryl sulfones in good yields under NaI instead of TBAI in DMF at 70 °C.
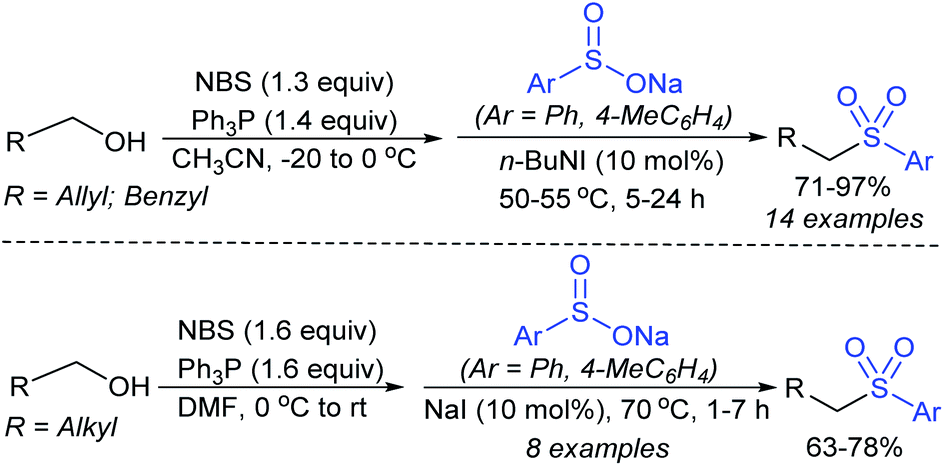 | ||
| Scheme 254 One-pot bromination-sulfonylation of primary alcohols with sodium arenesulfinates. | ||
In 2010, Sreedhar and co-workers described the FeCl3-catalyzed direct sulfonylation of benzylic, allylic and homoallylic alcohols with sodium arenesulfinates in the presence of TMSCl (Scheme 255).364 Diverse sulfones were obtained in moderate to good yields via the nucleophic sulfonylation of various secondary alcohols, benzyl alcohols and allyl alcohols with sodium p-toluenesulfinate. In contrast, the sulfonylation of homoallyl alcohols occurs at the terminal double bond instead of nucleophic substitution at the alcohol and the desired allyl sulfones were obtained in 52–60% yields.
 | ||
| Scheme 255 FeCl3 catalyzed the direct sulfonylation of benzylic, allylic and homoallylic alcohols with sodium p-toluenesulfinate. | ||
An efficient electrochemical alkynylation/1,2-sulfonylation of alkenes via the radical 1,4-alkynyl migration of alkynyl-substituted tertiary alcohols with sodium sulfinates was established by Han, Pan and co-workers (Scheme 256).365 Various aryl-substituted alkynyl-derived tertiary alcohols were reacted with para-methylbenzenesulfinate to provide the desired products in moderate to high yields. The cyclohexyl alkynyl-substituted tertiary alcohol did not produce the desired product because the radical adjacent to the cyclohexyl group was not stable. It is worth noting that variations at other positions of the substrate were also suitable and afforded the desired products. Several arylsulfinates worked well to afford the corresponding α-sulfonyl-β-alkynylation products in good yields. Unfortunately, sodium para-nitrophenylsulfinate was a challenging substrate; however, the alkyl-substituted sulfinates were also compatible and obtained high yields of desired products. The electrochemical 1,2-sulfonylation/alkynylation was scalable at 5 mmol, which was conducted using an inexpensive graphite plate and led to the corresponding product in moderate yield.
 | ||
| Scheme 256 Electrochemical alkynylation/1,2-sulfonylation of alkynyl-substituted tertiary alcohols with sodium sulfinates. | ||
A plausible mechanism involves sodium sulfinate being oxidized at the anode to give the oxygen-entered radical, which resonates to the stable sulfonyl radical as shown in Scheme 257. The sulfonyl radical attacks the terminal position of the alkene to give the radical A and subsequent 1,5-radical cyclization of A leads to the cyclopentane vinyl radical B. Further, the regioselective C–C bond cleavage of B to generate the radical C and dehydrogenation will produce the ketyl radical intermediate D, which is oxidized at the anode to form the anticipated sulfonated product.
 | ||
| Scheme 257 Mechanistic representation of the electrochemical alkynylation/1,2-sulfonylation. | ||
Palladium catalyzed the sulfonylation of benzylic carbonates with sodium sulfinates under the influence of DPEphos (40) ligand in DMSO at 80 °C to give benzylic sulfones. Kuwano and co-workers366 successfully used a variety of benzylic carbonates and 1-naphthylmethyl carbonate with arenesulfinates and methanesulfinate to afford a broad spectrum of sulfones in high yields (Scheme 258). Both electron-rich and electron-poor benzylic carbonates reacted rapidly, whereas two ortho-methyl groups sluggishly proceeded to the sulfonylation.
 | ||
| Scheme 258 Palladium-catalyzed sulfonylation of benzylic carbonates with sodium sulfinates. | ||
Very recently, Cs2CO3 promoted the direct sulfonylation of benzylic ammonium salts with sodium sulfinates in DMF at 130 °C as presented by Tu and co-workers (Scheme 259).367 A wide variety of benzylic ammonium iodides were smoothly coupled with various aryl and heteroaryl sulfinates, leading to structurally varied benzyl sulfones in moderate to good yields. Disappointingly, sodium methanesulfinate did not afford the targeted product under the same conditions.
 | ||
| Scheme 259 Cs2CO3 promoted the direct sulfonylation of benzylic ammonium salts with sodium sulfinates. | ||
Miura and co-workers described a Pd/(R)-BINAP (41)-catalyzed enantioselective sulfonylation of racemic secondary benzylic carbonates with sodium sulfinates. The reaction proceeded in a dynamic kinetic asymmetric transformation (DYKAT) manner to form the enantiomerically enriched benzylic sulfones (Scheme 260).368 As the authors observed, the addition of H2O was found to be critical for high enantioselectivity. Several secondary benzylic carbonates with sodium benzenesulfinate delivered the corresponding chiral diarylmethyl sulfones in good yields with high enantioselectivity. The highly fused phenanthrene derivative demonstrated a good substrate for producing a high enantiomeric ratio (98:2 er). A few arylsulfinates were coupled with 1-naphthylmethy carbonate to form the chiral benzylic sulfone with a good enantiomeric ratio, but aliphatic methanesulfinate gave somewhat lower enantioselectivity.
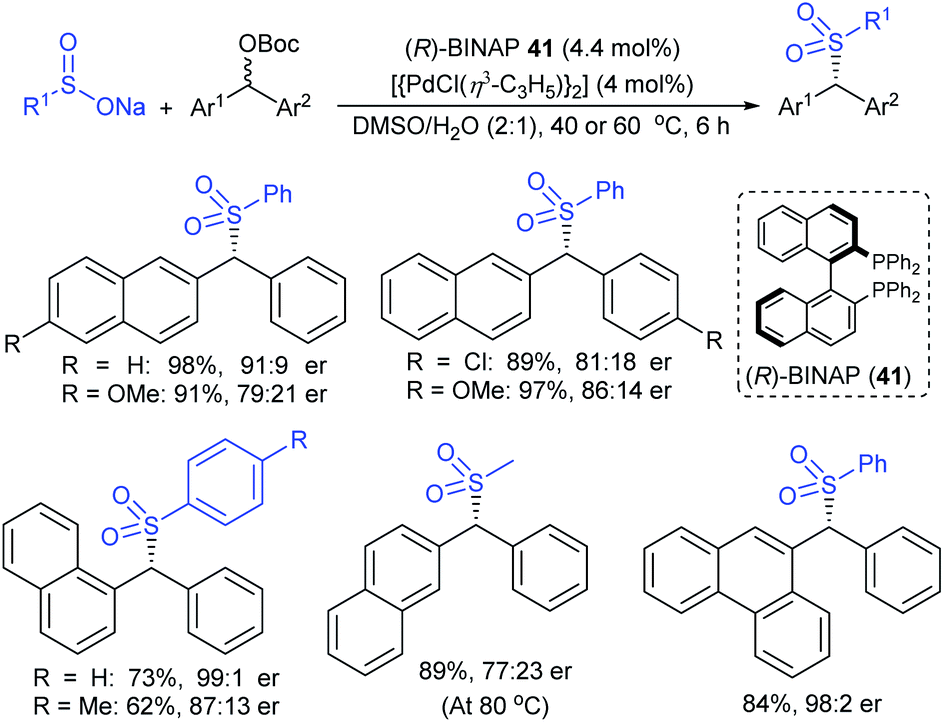 | ||
| Scheme 260 Pd/(R)-BINAP (41)-catalyzed enantioselective sulfonylation of racemic secondary benzylic carbonates with sodium sulfinates. | ||
Yamamoto and Nakata established the SnBr2-catalyzed sulfonylation of chiral auxiliary derived racemic diarylmethanols with sodium sulfinates in the presence of trimethylsilyl chloride (TMSCl) for the synthesis of diastereoselective diarylmethyl sulfones (Scheme 261).369 Various diarylmethanols with different substituents on the aromatic rings of the substrates were explored systematically with PhSO2Na and afforded diarylmethyl sulfones with high diastereoselectivity. The yields and diastereoselectivity were substantially improved by lowering the temperature (−40 °C) or decreasing the catalyst loading for diastereo-convergent sulfonylation. The authors also considered increasing the catalyst loading for poorly reactive substrates bearing electron-withdrawing groups. A variety of sodium arylsulfinates for the sulfonylation reacted smoothly and gave good results, regardless of steric effects. In contrast, the reaction using MeSO2Na showed a prolonged reaction time and the selectivity slightly decreased.
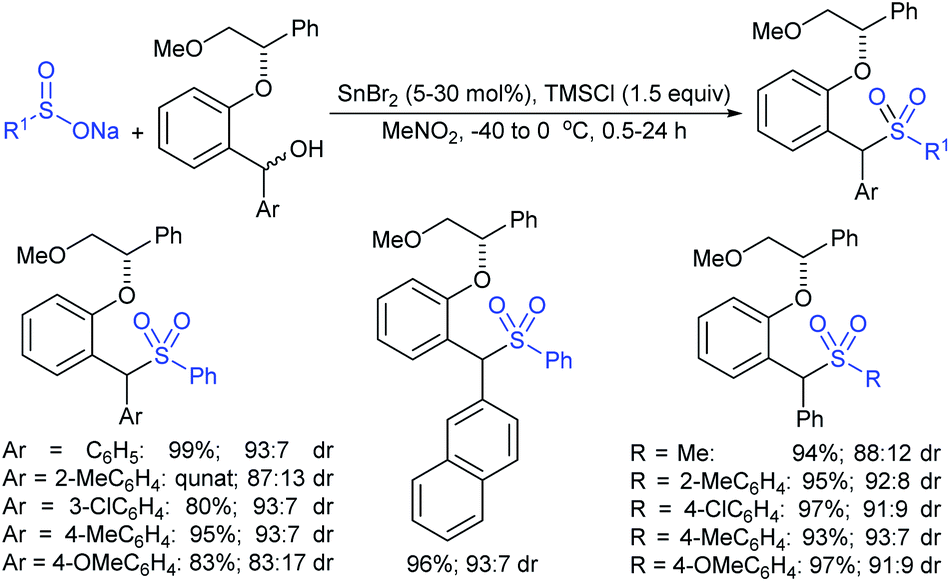 | ||
| Scheme 261 The SnBr2-catalyzed sulfonylation of the chiral auxiliary-derived racemic diarylmethanols with sodium sulfinates. | ||
Yuan and co-workers described the iodine-induced O-sulfonylation of phenols with sodium sulfinates for the synthesis of sulfonate esters in the presence of K2CO3 (Scheme 262).370 A series of ortho-, meta-, and para-substituted phenols reacted well with sodium p-toluenesulfinate to afford the corresponding sulfonylated products in good to high yields. Besides, naphthol and quinolin-8-ol as well as aliphatic alcohols (used NaOMe as a strong base) all proceeded smoothly in CH3CN gto give the target products with good yields. Different types of aryl and benzyl sulfinates are compatible to react with 4-chlorophenol and obtain the desired sulfonate esters in high yields.
 | ||
| Scheme 262 Iodine-induced O-sulfonylation of phenols with sodium sulfinates for the synthesis of sulfonate esters. | ||
Hypervalent iodine-promoted O-sulfonylation of alkyl alcohols with sodium sulfinates was developed by the Canesi group.371 A series of alkyl sulfonate esters were obtained in moderate to good yields using various functionalized primary and secondary alcohols with 2-methylbenzenesulfinate in the presence of (diacetoxyiodo)benzene (PIDA) as described in Scheme 263. Disappointingly, sterically hindered tertiary alcohols, such as tert-butanol, were unsuccessful.
 | ||
| Scheme 263 PIDA-promoted O-sulfonylation of alkyl alcohols with sodium 2-methylbenzenesulfinate. | ||
Xiong and co-workers372 investigated a convenient BF3·OEt2-mediated sulfination of alcohols via carbocation with sodium sulfinates for the synthesis of O-alkyl sulfinates. A series of primary and secondary benzylalcohols smoothly reacted with sodium p-toluenesulfinate to give the desired O-alkyl sulfinates in good to high yields (Scheme 264). Surprisingly, the sulfination with 4-methoxybenzyl alcohol was unsuccessful, however, in the case of allylic alcohols, low to moderate yields were obtained. Additionally, various linear and branched aliphatic alcohols proceeded cleanly with sodium p-toluenesulfinate to give the corresponding sulfinates in acceptable yields. Complex compounds, such as dihydroepiandrosterone and testosterone also afforded the sulfinates effectively. Furthermore, various aryl/alkylsulfinates reacted smoothly to furnish the desired steroidal sulfinates in moderate to good yields.
 | ||
| Scheme 264 The BF3·OEt2-mediated sulfination of alcohols with sodium sulfinates. | ||
The Wu group employed the sulfination of alcohols with sodium arenesulfinates in the presence of TMSCl for the synthesis of O-substituted sulfinates (Scheme 265).373 A wide range of primary and secondary alcohols including benzyl, allyl and alkyl alcohols as well as L-menthol reacted well with sodium p-toluenesulfinate and obtained the desired O-substituted sulfinates in good to high yields. A series of aryl and heteroarylsulfinates were readily coupled with benzyl alcohol, ethanol, phenethyl alcohol and 2,3-dihydro-1H-inden-2-ol and afforded the corresponding sulfinates in variable yields.
 | ||
| Scheme 265 TMSCl-mediated sulfination of alcohols with sodium sulfinates. | ||
Wei and co-workers performed the molecular iodine-catalyzed preparation of thiocarbamates from isocyanides, sodium sulfinates, and water under the influence of HP(O)(OMe)2 as the reducing agent. A variety of isocyanides, such as cyclohexyl isocyanide, tert-butyl isocyanide, isocyanoacetate and tosylmethyl isocyanide were thiolated with a series of aryl/heteroaryl sulfinates and a widespread thiocarbamates were obtained in moderate to high yields (Scheme 266).374 However, the use of methylsulfinate and trifluoromethane-sulfinate as well as 2,6-dimethylphenyl isocyanide could not detect the desired products.
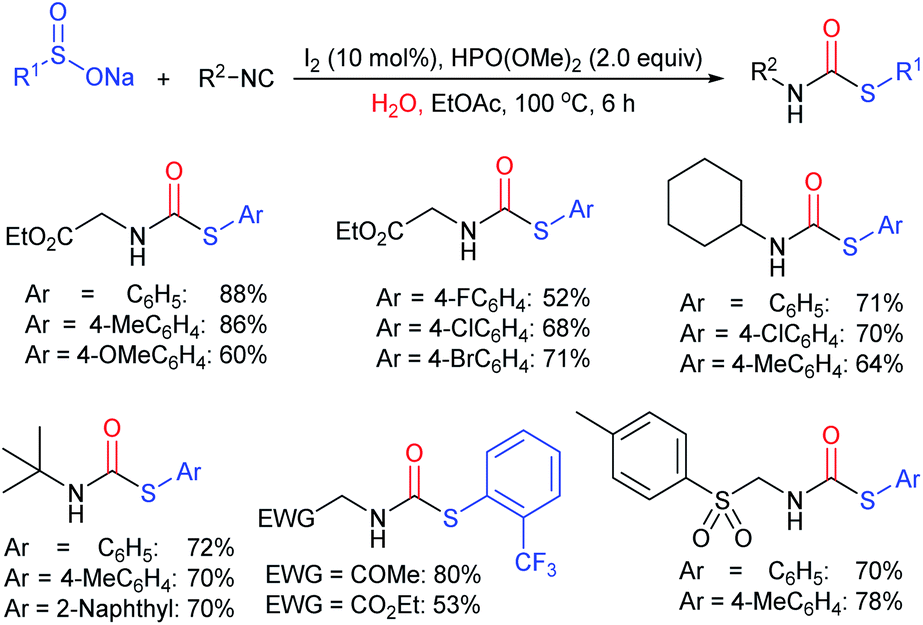 | ||
| Scheme 266 I2-catalyzed synthesis of thiocarbamates from sodium sulfinates, isocyanides and water. | ||
Visible-light photoredox-catalysis of a new radical–radical coupling of acid chlorides and sodium sulfinates for the synthesis of thioesters was successfully employed by the Kim and Oh group (Scheme 267).375 The sulfenylation of various benzoyl chlorides with sodium p-toluenesulfinate was conducted to afford the desired thioesters in good to high yields. The electron-donating groups provided rather low yields as compared to benzoyl chlorides bearing electron-withdrawing groups. Besides, naphthoyl, heteroaroyl and aliphatic acid chlorides obtained good yields of the corresponding products. Moreover, different types of aryl and heteroaryl sulfinates acted as sulfenyalting agents and provided the corresponding thioesters in 50–85% yields.
 | ||
| Scheme 267 Photoredox-catalyzed radical–radical coupling of acid chlorides and sodium sulfinates. | ||
TMSOTf-promoted the sulfinylation of electron-rich aromatics such as pyrroles, thiophenes, indoles, and electron-rich arenes, with sodium sulfinates to form the corresponding sulfoxides (Scheme 268).376 The C3-sulfinylation of N-alkyl pyrroles and thiophenes with a variety of aromatic and aliphatic sulfinates was successfully achieved and gave the corresponding 3-sulfinylpyrroles in acceptable yields. The C2-sulfinylation of pyrrole takes place when the C3-substituted pyrrole is used. Next, thiophene and 3-hexylthiophene reacted smoothly with sodium p-toluenesulfinate to afford sulfoxides in 23% and 63% yields, respectively. Moreover, the sulfinylation protocol was extended to a wide variety of indoles with arenesulfinates to afford 3-indolyl aryl sulfoxides in moderate to good yields. Additionally, diaryl sulfoxides were obtained in variable yields from various arenes bearing electron-donating groups with sodium arylsulfinates. As expected, the electron-withdrawing groups of arenes, nitrobenzene and cyanobenzene were unsuccessful.
 | ||
| Scheme 268 TMSOTf-promoted sulfinylation of electron-rich aromatics with sodium sulfinates. | ||
Chen and co-workers reported the copper-catalyzed radical cross-coupling of cycloketone oxime esters and sodium sulfinate salts (Scheme 269).377 Representative sodium (hetero)aryl sulfinates reacted well with the O-acyl oxime to furnish the corresponding sulfones in good to high yields. There was no obvious influence on the reaction efficiency on using different substitution patterns and steric hindrance on the phenyl ring. Although the reaction with sodium benzylsulfinate proceeded smoothly, methane and ethane sulfinates only participated under modified reaction conditions; however, sodium trifluoromethyl sulfinate is not suitable for the reaction. A wide range of cycloketone oxime esters reacted well with sodium p-toluenesulfinate under modified reaction conditions and typically formed the corresponding products in good yields. Additionally, in the case of the oxetan-3-one-derived oxime ester, it did not afford the expected product under the same conditions. A possible reaction mechanism could involve a SET reduction of oxime ester by the LCuI complex, followed by fragmentation to afford the cyclic iminyl radical (A) and oxidized [Cu(II)] complex. The regioselective ring-opening of the cyclic iminyl radical A occurred via the C–C bond cleavage to form the cyanoalkyl radical B. Nucleophilic sodium sulfinate was reacted with the LCu(I) complex to form LCu(II)SO2Ar species, which reacted with B to furnish the high-valent Cu(III) complex C. A reductive elimination of the C complex resulted in the desired product with the regeneration of the [Cu(I)] catalyst.
 | ||
| Scheme 269 Copper-catalyzed radical cross-coupling of redox-active cycloketone oxime esters and sodium sulfinates. | ||
K2S2O8 mediated the ring-opening sulfonylation of methylene-cyclopropanes with sodium sulfinates and H2O for constructing (E)-4-sulfonylbut-1-enes. The Tang group successfully established that the difunctionalization of C–C σ-bonds in a variety of aryl-substituted methylenecyclopropanes was carried out with sodium trifluoromethanesulfinate to generate a series of corresponding functionalized (E)-4-sulfonylbut-1-enes in moderate to good yields (Scheme 270).378 Based on the outcome, the electronic and steric effects had almost no influence on the transformation. Although wide ranges of arylsulfinates also readily participated, however, 2-thiophenyl sulfinate and aliphatic sulfinates did not afford the desired products. A possible mechanistic pathway involves the electrophilic addition of a proton to the C–C double bond in methylenecyclopropanes to form the onium ion intermediate (I). Subsequently, generated aryl group(s) stabilized carbocation II, which underwent a ring-opening to access the (E)-alkenyl carbocation III in the presence of K2S2O8. Sodium sulfinate formed the sulfonyl anion IV, which is prone to react with the alkyl carbocation intermediate III to give the corresponding sulfone products.
 | ||
| Scheme 270 K2S2O8-mediated ring-opening and sulfonylation of methylene-cyclopropanes with sodium sulfinates. | ||
4. Conclusion and outlook
We have presented a comprehensive overview of the preparation of sodium sulfinates. Their extensive applications have witnessed remarkable expansion over the past decade. Sodium sulfinates are recognized as potent sulfonylative, sulfenylative and sulfinylative agents, depending on the reaction conditions. Careful analysis of the published data indicates that the sodium sulfinates play multiple roles as nucleophiles, electrophiles, and radical reagents. Many reliable methods have increasingly attracted interest in the synthesis of various sulfur-containing valuable precursors, such as thiosulfonates, sulfonamides, thioethers (sulfides), sulfones, allyl & vinyl sulfones, β-iodovinyl sulfones, β-keto sulfones, etc.Although significant achievements have been made in ring-closing sulfonylation, multicomponent reactions, and ortho C–H sulfonylation using sodium sulfinate salts, many exciting methods need to be realized. Moreover, photoredox sulfonylation and electrochemical transformations also emerged in recent years and are less explored. Therefore, the exploitation of sulfonyl and sulfenyl radicals in photoredox and electrochemical methods will attract more and more research attention. Notwithstanding substantial advances, sodium sulfinates have been proven to be versatile coupling partners in organic synthesis. However, several challenges still need to be addressed for the further exploration of the rapidly developing fields.
We hope this review will be beneficial to the synthetic community at large scale and will provide a new perspective in medicinal chemistry. We strongly believe that this review will encourage synthetic organic chemists to carry out future research endeavours. We would like to express our sincere regrets in advance for any references that may have been overlooked during the search.
General procedure for the preparation of sodium arenesulfinates346,379
The aryl sulfonyl chloride (10.0 mmol, 1.0 equiv.) was dissolved in 30 mL water. Sodium sulfite (16.0 mmol, 1.6 equiv.) and sodiumbicarbonate (16.0 mmol, 1.6 equiv.) were added, and the reaction mixture was refluxed for 3 h. The water was evaporated, and ethanol was added to the residue. The suspension was heated for 10 min, cooled, and filtered through a 20 μm polyethylene frit. This was repeated twice with the residue from the filtration. The ethanol fractions were combined, the solvent was evaporated under vacuum, and the sodium arylsulfinates were isolated as a white powder.Conflicts of interest
The authors declare there is no conflict of interest.Acknowledgements
We thank CSIR-EMR-II [02(0340)/18/EMR-II] and SERB-ECR [ECR/2015/000053], New Delhi for financial assistance. RJR thanks to UGC for faculty position under Faculty Recharge Programme and Start-up grant (F.4-5/2006/BSR). ARK thanks DST-WOSA [SR/WOS-A/CS-14/2019] for financial assistance.Notes and references
- (a) S. Krishna and H. Singh, J. Am. Chem. Soc., 1928, 50, 792–798 CrossRef CAS; (b) C. E. Grothaus and F. B. Dains, J. Am. Chem. Soc., 1936, 58, 1334–1336 CrossRef CAS; (c) F. O'Hara, R. D. Baxter, A. G. O'Brien, M. R. Collins, J. A. Dixon, Y. Fujiwara, Y. Ishihara and P. S. Baran, Nat. Protoc., 2013, 8, 1042–1047 CrossRef.
- J. M. Smith, J. A. Dixon, J. N. DeGruyter and P. S. Baran, J. Med. Chem., 2019, 62, 2256–2264 CrossRef CAS.
- S. Liang, K. Hofman, M. Friedrich and G. Manolikakes, Eur. J. Org. Chem., 2020, 4664–4676 CrossRef CAS.
- (a) Organosulfur Chemistry I & II, ed. P. C. B. Page, Springer, Berlin, 1999 Search PubMed; (b) S. R. Dubbaka and P. Vogel, Angew. Chem., Int. Ed., 2005, 44, 7674–7684 CrossRef CAS; (c) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS.
- (a) C.-F. Lee, R. S. Basha and S. S. Badsara, Top. Curr. Chem., 2018, 376, 25 CrossRef; (b) C. F. Lee, Y. C. Liu and S. S. Badsara, Chem.–Asian J., 2014, 9, 706–722 CrossRef CAS.
- O. M. Mulina, A. I. Ilovaisky, V. D. Parshin and A. O. Terent'ev, Adv. Synth. Catal., 2020, 362, 4579–4654 CrossRef CAS.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS.
- J. Aziz, S. Messaoudi, M. Alami and A. Hamze, Org. Biomol. Chem., 2014, 12, 9743–9759 RSC.
- K. Yuan, J.-F. Soulé and H. Doucet, ACS Catal., 2015, 5, 978–991 CrossRef CAS.
- D. H. Ortgies, A. Hassanpour, F. Chen, S. Woo and P. Forgione, Eur. J. Org. Chem., 2016, 408–425 CrossRef CAS.
- J. Aziz and A. Hamze, Org. Biomol. Chem., 2020, 18, 9136–9159 RSC.
- P. Mampuys, C. R. McElroy, J. H. Clark, R. V. A. Orru and B. U. W. Maes, Adv. Synth. Catal., 2020, 362, 3–64 CrossRef CAS.
- O. M. Mulina, A. I. Ilovaisky and A. O. Terent'ev, Eur. J. Org. Chem., 2018, 4648–4672 CrossRef CAS.
- R. Wu, K. Huang, G. Qiu and J.-B. Liu, Synthesis, 2019, 51, 3567–3587 CrossRef CAS.
- D. Q. Dong, S. H. Hao, D. S. Yang, L. X. Li and Z. L. Wang, Eur. J. Org. Chem., 2017, 6576–6592 CrossRef CAS.
- N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939–1973 CrossRef CAS.
- D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793–814 CrossRef CAS.
- Y. Fang, Z. Luo and X. Xu, RSC Adv., 2016, 6, 59661–59676 RSC.
- Y. M. Markitanov, V. M. Timoshenko and Y. G. Shermolovich, J. Sulfur Chem., 2014, 35, 188–236 CrossRef CAS.
- S. Ghosh, S. Samanta, A. K. Ghosh, S. Neogi and A. Hajra, Adv. Synth. Catal., 2020, 362, 4552–4578 CrossRef CAS.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS.
- S. Shaaban, S. Liang, N. W. Liu and G. Manolikakes, Org. Biomol. Chem., 2017, 15, 1947–1955 RSC.
- J. Liu and L. Zheng, Adv. Synth. Catal., 2019, 361, 1710–1732 CrossRef CAS.
- A. Wimmer and B. König, Beilstein J. Org. Chem., 2017, 14, 54–83 CrossRef.
- H. Mei, R. Pajkert, L. Wang, Z. Li, G.-V. Röschenthaler and J. Han, Green Chem., 2020, 22, 3028–3059 RSC.
- W. E. Truce and F. E. Roberts, J. Org. Chem., 1963, 28, 593–594 CrossRef CAS.
- J. F. C. Whitmore, F. H. Hamilton, J. B. Conant and P. Allen, Org. Synth., 1922, 2, 89 CrossRef.
- L. K. Liu, Y. Chi and K. Y. Jen, J. Org. Chem., 1980, 45, 406–410 CrossRef CAS.
- L. Field and R. B. Barbee, J. Org. Chem., 1969, 34, 1792–1798 CrossRef CAS.
- P. K. Srivastava, L. Field and M. M. Grenan, J. Med. Chem., 1975, 18, 798–802 CrossRef CAS.
- Y. Ueno, A. Kojima and O. Makoto, Chem. Lett., 1984, 2125–2128 CrossRef CAS.
- Z. He, P. Tan, C. Ni and J. Hu, Org. Lett., 2015, 17, 1838–1841 CrossRef CAS.
- C. Tran, B. Flamme, A. Chagnes, M. Haddad, P. Phansavath and V. Ratovelomanana-Vidal, Synlett, 2018, 29, 1622–1626 CrossRef CAS.
- G. K. S. Prakash, C. Ni, F. Wang, J. Hu and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 2559–2563 CrossRef CAS.
- Q. Zhou, A. Ruffoni, R. Gianatassio, Y. Fujiwara, E. Sella, D. Shabat and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 3949–3952 CrossRef CAS.
- R. Gianatassio, S. Kawamura, C. L. Eprile, K. Foo, J. Ge, A. C. Burns, M. R. Collins and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 9851–9855 CrossRef CAS.
- A. C. M. A. Nassoy, P. Raubo and J. P. A. Harrity, Chem. Commun., 2015, 51, 5914–5916 RSC.
- M. G. Johnson, M. W. Gribble, J. B. Houze and N. A. Paras, Org. Lett., 2014, 16, 6248–6251 CrossRef CAS.
- B. Skillinghaug, J. Rydfjord and L. R. Odell, Tetrahedron Lett., 2016, 57, 533–536 CrossRef CAS.
- T. Wang, F. Wang, J. Shen, T. Pang, Y. Ren, B. Wu and X. Zhang, Tetrahedron Lett., 2018, 59, 1183–1187 CrossRef CAS.
- R. Sakamoto, T. Yoshii, H. Takada and K. Maruoka, Org. Lett., 2018, 20, 2080–2083 CrossRef CAS.
- Y. Zhao and F. Liu, Tetrahedron Lett., 2018, 59, 180–187 CrossRef CAS.
- M. Tordeux, B. Langlois and C. Wakselman, J. Org. Chem., 1989, 54, 2452–2453 CrossRef CAS.
- H. P. Cao and Q. Y. Chen, J. Fluorine Chem., 2007, 128, 1187–1190 CrossRef CAS.
- B. R. Langlois, T. Billard, J. C. Mulatier and C. Yezeguelian, J. Fluorine Chem., 2007, 128, 851–856 CrossRef CAS.
- M. D. Bentley, I. B. Douglass and J. A. Lacadie, J. Org. Chem., 1972, 37, 333–334 CrossRef CAS.
- Y. Abe and J. Tsurugi, Chem. Lett., 1972, 441–442 CrossRef CAS.
- G. Liang, J. Chen, J. Chen, W. Li, J. Chen and H. Wu, Tetrahedron Lett., 2012, 53, 6768–6770 CrossRef CAS.
- U. Claude, B. Lyon, T. Billard, B. R. Langlois, S. Large, D. Anker, N. Roidot and P. Roure, J. Org. Chem., 1996, 61, 7545–7550 CrossRef.
- K. Fujiki, N. Tanifuji, Y. Sasaki and T. Yokoyama, Synthesis, 2002, 343–348 CrossRef CAS.
- G. Liang, M. Liu, J. Chen, J. Ding, W. Gao and H. Wu, Chin. J. Chem., 2012, 30, 1611–1616 CrossRef CAS.
- N. Taniguchi, Eur. J. Org. Chem., 2014, 5691–5694 CrossRef CAS.
- T. Keshari, R. Kapoorr and L. D. S. Yadav, Synlett, 2016, 27, 1878–1882 CrossRef CAS.
- N. Taniguchi, J. Org. Chem., 2015, 80, 1764–1770 CrossRef CAS.
- X. Shao, C. Xu, L. Lu and Q. Shen, J. Org. Chem., 2015, 80, 3012–3021 CrossRef CAS.
- L. Cao, S. H. Luo, K. Jiang, Z. F. Hao, B. W. Wang, C. M. Pang and Z. Y. Wang, Org. Lett., 2018, 20, 4754–4758 CrossRef CAS.
- D. W. Chen and Z. C. Chen, Tetrahedron Lett., 1994, 35, 7637–7638 CrossRef CAS.
- X. Tang, L. Huang, C. Qi, X. Wu, W. Wu and H. Jiang, Chem. Commun., 2013, 49, 6102 RSC.
- W. Wei, C. Liu, D. Yang, J. Wen, J. You and H. Wang, Adv. Synth. Catal., 2015, 357, 987–992 CrossRef CAS.
- K. Yang, M. Ke, Y. Lin and Q. Song, Green Chem., 2015, 17, 1395–1399 RSC.
- X. Pan, J. Gao, J. Liu, J. Lai, H. Jiang and G. Yuan, Green Chem., 2015, 17, 1400–1403 RSC.
- C. Buathongjan, D. Beukeaw and S. Yotphan, Eur. J. Org. Chem., 2015, 1575–1582 CrossRef CAS.
- W. Liu, W. Deng, S. Sun, C. Yu, X. Su, A. Wu, Y. Yuan, Z. Ma, K. Li, H. Yang, X. Peng and J. Dietrich, Org. Lett., 2019, 21, 9909–9913 CrossRef CAS.
- J. Zhao, J. Xu, J. Chen, X. Wang and M. He, RSC Adv., 2014, 4, 64698–64701 RSC.
- Y. Fu, Q. Z. Li, Q. S. Xu, H. Hügel, M. P. Li and Z. Du, Eur. J. Org. Chem., 2018, 6966–6970 CrossRef CAS.
- S. X. Wu, Y. K. Zhang, H. W. Shi and J. Yan, Chin. Chem. Lett., 2016, 27, 1519–1522 CrossRef CAS.
- S. Wu, Y. Zhang and J. Yan, Synth. Commun., 2016, 46, 1432–1437 CrossRef CAS.
- L. Fu, X. Bao, S. Li, L. Wang, Z. Liu, W. Chen, Q. Xia and G. Liang, Tetrahedron, 2017, 73, 2504–2511 CrossRef CAS.
- Y. Y. Jiang, Q. Q. Wang, S. Liang, L. M. Hu, R. D. Little and C. C. Zeng, J. Org. Chem., 2016, 81, 4713–4719 CrossRef CAS.
- C. Zhang, Y. Chen and G. Yuan, Chin. J. Chem., 2016, 34, 1277–1282 CrossRef CAS.
- J. Lai, L. Chang and G. Yuan, Org. Lett., 2016, 18, 3194–3197 CrossRef CAS.
- S. Wu, Y. Zhang, M. Zhu and J. Yan, Synlett, 2016, 27, 2699–2704 CrossRef CAS.
- Z. Zhou and X. He, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 195, 280–284 CrossRef CAS.
- R. J. Reddy, A. Shankar, M. Waheed and J. B. Nanubolu, Tetrahedron Lett., 2018, 59, 2014–2017 CrossRef CAS.
- W. Y. Chan and C. Berthelette, Tetrahedron Lett., 2002, 43, 4537–4540 CrossRef CAS.
- H. Zhu, Y. Shen, Q. Deng and T. Tu, Chem. Commun., 2015, 51, 16573–16576 RSC.
- X. Bao, X. Rong, Z. Liu, Y. Gu, G. Liang and Q. Xia, Tetrahedron Lett., 2018, 59, 2853–2858 CrossRef CAS.
- A. Rahimi, D. Habibi, A. Rostami, M. A. Zolfigol and S. Mallakpour, Tetrahedron Lett., 2018, 59, 383–387 CrossRef CAS.
- W. Zhang, J. Xie, B. Rao and M. Luo, J. Org. Chem., 2015, 80, 3504–3511 CrossRef CAS.
- N. Eid, I. Karamé and B. Andrioletti, Eur. J. Org. Chem., 2018, 5016–5022 CrossRef CAS.
- S. Liu, R. Chen and J. Zhang, Molecules, 2019, 24, 1407–1418 CrossRef.
- F. Xiao, H. Xie, S. Liu and G. J. Deng, Adv. Synth. Catal., 2014, 356, 364–368 CrossRef CAS.
- P. Katrun, S. Hongthong, S. Hlekhlai, M. Pohmakotr, V. Reutrakul, D. Soorukram, T. Jaipetch and C. Kuhakarn, RSC Adv., 2014, 4, 18933 RSC.
- H. Rao, P. Wang, J. Wang, Z. Li, X. Sun and S. Cao, RSC Adv., 2014, 4, 49165–49169 RSC.
- L. Jiang, J. Qian, W. Yi, G. Lu, C. Cai and W. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14965–14969 CrossRef CAS.
- M.-J. Bu, G.-P. Lu and C. Cai, Org. Chem. Front., 2017, 4, 266–270 RSC.
- D. W. W. Sun, X. Jiang, M. Jiang, Y. Lin and J. T. T. Liu, Eur. J. Org. Chem., 2017, 3505–3511 CrossRef CAS.
- Q. Yan, L. Jiang, W. Yi, Q. Liu and W. Zhang, Adv. Synth. Catal., 2017, 359, 2471–2480 CrossRef CAS.
- X. Ge, F. Sun, X. Liu, X. Chen, C. Qian and S. Zhou, New J. Chem., 2017, 41, 13175–13180 RSC.
- X. Luo, Q. Liu, H. Zhu and H. Chen, R. Soc. Open Sci., 2018, 5, 180170 CrossRef.
- C. Liu, J. Fan, M. Wu, J. Chen, Y. Zhao and M. Xie, Chin. J. Chem., 2018, 36, 819–825 CrossRef CAS.
- Y.-Z. Ji, H.-J. Li, J.-Y. Zhang and Y.-C. Wu, Chem. Commun., 2019, 55, 11864–11867 RSC.
- D. W. Sun, X. Jiang, M. Jiang, Y. Lin and J. T. Liu, Eur. J. Org. Chem., 2018, 2078–2081 CrossRef CAS.
- X. L. He, S. Majumder, J. Wu, C. Di Jin, S. R. Guo, Z. P. Guo and M. Yang, Org. Chem. Front., 2019, 6, 2435–2440 RSC.
- X. Huang, S. Wang, B. Li, X. Wang, Z. Ge and R. Li, RSC Adv., 2015, 5, 22654–22657 RSC.
- Y. J. Guo, S. Lu, L. L. Tian, E. L. Huang, X. Q. Hao, X. Zhu, T. Shao and M. P. Song, J. Org. Chem., 2018, 83, 338–349 CrossRef CAS.
- Y. Ding, W. Wu, W. Zhao, Y. Li, P. Xie, Y. Huang, Y. Liu and A. Zhou, Org. Biomol. Chem., 2016, 14, 1428–1431 RSC.
- D. Wang, R. Zhang, S. Lin, Z. Yan and S. Guo, RSC Adv., 2015, 5, 108030–108033 RSC.
- F. Xiao, S. Chen, J. Tian, H. Huang, Y. Liu and G.-J. Deng, Green Chem., 2016, 18, 1538–1546 RSC.
- Z. Xu, G. Lu and C. Cai, Org. Biomol. Chem., 2017, 15, 2804–2808 RSC.
- Y. Lin, G. Lu, G. Wang and W. Yi, Adv. Synth. Catal., 2016, 358, 4100–4105 CrossRef CAS.
- S. Liu, L. Tang, H. Chen, F. Zhao and G. J. Deng, Org. Biomol. Chem., 2014, 12, 6076–6079 RSC.
- Y. Lin, G. Lu, C. Cai and W. Yi, Org. Lett., 2015, 17, 3310–3313 CrossRef CAS.
- Y. M. Lin, G. P. Lu, G. X. Wang and W. Bin Yi, J. Org. Chem., 2017, 82, 382–389 CrossRef CAS.
- Y. Gao, Y. Gao, X. Tang, J. Peng, M. Hu, W. Wu and H. Jiang, Org. Lett., 2016, 18, 1158–1161 CrossRef CAS.
- W. Wu, Y. An, J. Li, S. Yang, Z. Zhu and H. Jiang, Org. Chem. Front., 2017, 4, 1751–1756 RSC.
- C. Liu, B. Wang, Z. Guo, J. Zhang and M. Xie, Org. Chem. Front., 2019, 6, 2796–2800 RSC.
- S. Guo, W. He, J. Xiang and Y. Yuan, Tetrahedron Lett., 2014, 55, 6407–6410 CrossRef CAS.
- F. Zhao, Q. Tan, D. Wang, J. Chen and G. Deng, Adv. Synth. Catal., 2019, 361, 4075–4081 CrossRef CAS.
- Y. Chen, F. Xiao, H. Chen, S. Liu and G. J. Deng, RSC Adv., 2014, 4, 44621–44628 RSC.
- C. Hydroxy, S. Approach, G. L. Olson, H. Cheung, K. D. Morgan, C. Neukom and G. Saucy, J. Org. Chem., 1976, 41, 3287–3293 CrossRef.
- K. Hiroi and K. Makino, Chem. Lett., 1986, 617–620 CrossRef CAS.
- H. Eichelmann and H.-J. Gais, Tetrahedron: Asymmetry, 1995, 6, 643–646 CrossRef CAS.
- H. J. Gais, T. Jagusch, N. Spalthoff, F. Gerhards, M. Frank and G. Raabe, Chem.–Eur. J., 2003, 9, 4202–4221 CrossRef CAS.
- S. Chandrasekhar, V. Jagadeshwar, B. Saritha and C. Narsihmulu, J. Org. Chem., 2005, 70, 6506–6507 CrossRef CAS.
- F. X. Felpin and Y. Landais, J. Org. Chem., 2005, 70, 6441–6446 CrossRef CAS.
- Y. Uozumi and T. Suzuka, Synthesis, 2008, 1960–1964 CrossRef CAS.
- M.-Y. Chang, H.-Y. Chen and H.-S. Wang, J. Org. Chem., 2017, 82, 10601–10610 CrossRef CAS.
- M. Jegelka and B. Plietker, Org. Lett., 2009, 11, 3462–3465 CrossRef CAS.
- X. S. Wu, Y. Chen, M. B. Li, M. G. Zhou and S. K. Tian, J. Am. Chem. Soc., 2012, 134, 14694–14697 CrossRef CAS.
- X.-T. Ma, R.-H. Dai, J. Zhang, Y. Gu and S.-K. Tian, Adv. Synth. Catal., 2014, 356, 2984–2988 CrossRef CAS.
- M. Ueda and J. F. Hartwig, Org. Lett., 2010, 12, 92–94 CrossRef CAS.
- A. Cai and A. W. Kleij, Angew. Chem., Int. Ed., 2019, 58, 14944–14949 CrossRef CAS.
- J. E. Gómez, À. Cristòfol and A. W. Kleij, Angew. Chem., Int. Ed., 2019, 58, 3903–3907 CrossRef.
- A. Khan, H. Zhao, M. Zhang, S. Khan and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 1340–1345 CrossRef CAS.
- M. Ke, G. Huang, L. Ding, J. Fang and F. Chen, ChemCatChem, 2019, 11, 4720–4724 CrossRef CAS.
- L. Jiang, Y.-G. Li, J.-F. Zhou, Y.-M. Chuan, H.-L. Li and M.-L. Yuan, Molecules, 2015, 20, 8213–8222 CrossRef CAS.
- L. Jiang, Y.-G. Li, H. Li, M. Yuan, Y. Chuan, H. Li and M.-L. Yuan, J. Chem. Res., 2017, 41, 160–164 CrossRef CAS.
- R. J. Reddy, M. Waheed, T. Karthik and A. Shankar, New J. Chem., 2018, 42, 980–987 RSC.
- J. Liao, W. Guo, Z. Zhang, X. Tang, W. Wu and H. Jiang, J. Org. Chem., 2016, 81, 1304–1309 CrossRef CAS.
- J. Ni, B. Nishonov, A. Pardaev and A. Zhang, J. Org. Chem., 2019, 84, 13646–13654 CrossRef CAS.
- M. Ochiai, Y. Kitagawa, M. Toyonari, K. Uemura, K. Oshima and M. Shiro, J. Org. Chem., 1997, 62, 8001–8008 CrossRef CAS.
- R. Chawla, R. Kapoor, A. K. Singh and L. D. S. Yadav, Green Chem., 2012, 14, 1308–1313 RSC.
- M. Bian, F. Xu and C. Ma, Synthesis, 2007, 2951–2956 CAS.
- S. Liang, R. Y. Zhang, G. Wang, S. Y. Chen and X. Q. Yu, Eur. J. Org. Chem., 2013, 7050–7053 CrossRef CAS.
- D. Zvezdova, S. Stoeva and D. Aleksiev, J. Chin. Chem. Soc., 2016, 63, 247–253 CrossRef CAS.
- Z. H. Guan, W. Zuo, L. B. Zhao, Z. H. Ren and Y. M. Liang, Synthesis, 2007, 1465–1470 CAS.
- M. Y. Chang, Y. C. Cheng and P. P. Sun, Synthesis, 2017, 49, 2411–2422 CrossRef CAS.
- M.-Y. Chang, Y.-S. Wu and Y.-T. Hsiao, Synthesis, 2018, 50, 4651–4658 CrossRef CAS.
- D. C. Reeves, S. Rodriguez, H. Lee, N. Haddad, D. Krishnamurthy and C. H. Senanayake, Tetrahedron Lett., 2009, 50, 2870–2873 CrossRef CAS.
- M. Y. Chang and M. H. Wu, Synlett, 2014, 25, 411–416 CrossRef CAS.
- L. Cao, J.-X. Li, H.-Q. Wu, K. Jiang, Z.-F. Hao, S.-H. Luo and Z.-Y. Wang, ACS Sustainable Chem. Eng., 2018, 6, 4147–4153 CrossRef CAS.
- V. Nair, A. Augustine, T. G. George and L. G. Nair, Tetrahedron Lett., 2001, 42, 6763–6765 CrossRef CAS.
- V. Nair, A. Augustine and T. D. Suja, Synthesis, 2002, 2259–2265 CrossRef CAS.
- P. Katrun, S. Chiampanichayakul, K. Korworapan, M. Pohmakotr, V. Reutrakul, T. Jaipetch and C. Kuhakarn, Eur. J. Org. Chem., 2010, 5633–5641 CrossRef CAS.
- B. Das, M. Lingaiah, K. Damodar and N. Bhunia, Synthesis, 2011, 2941–2944 CrossRef CAS.
- T. Sawangphon, P. Katrun, K. Chaisiwamongkhol, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, Synth. Commun., 2013, 43, 1692–1707 CrossRef CAS.
- N. Taniguchi, Synlett, 2011, 1308–1312 CrossRef CAS.
- N. Taniguchi, Synlett, 2012, 23, 1245–1249 CrossRef CAS.
- N. Taniguchi, Tetrahedron, 2014, 70, 1984–1990 CrossRef CAS.
- G. M. Shelke, V. Kameswara Rao, K. Pericherla and A. Kumar, Synlett, 2014, 25, 2345–2349 CrossRef CAS.
- G. Fang, J. Liu, W. Shang, Q. Liu and X. Bi, Chem.–Asian J., 2016, 11, 3334–3338 CrossRef CAS.
- A. U. Meyer, K. Straková, T. Slanina and B. König, Chem.–Eur. J., 2016, 22, 8694–8699 CrossRef CAS.
- Q. Gui, K. Han, Z. Liu, Z. Su, X. He, H. Jiang, B. Tian and Y. Li, Org. Biomol. Chem., 2018, 16, 5748–5751 RSC.
- G. Khalili, J. Sulfur Chem., 2013, 34, 532–538 CrossRef CAS.
- Y. C. Luo, X. J. Pan and G. Q. Yuan, Tetrahedron, 2015, 71, 2119–2123 CrossRef CAS.
- Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657–4661 CrossRef CAS.
- L.-N. Hua, H. Li, F.-L. Qing, Y. Huang and X.-H. Xu, Org. Biomol. Chem., 2016, 14, 8443–8447 RSC.
- W. Li, G. Yin, L. Huang, Y. Xiao, Z. Fu, X. Xin, F. Liu, Z. Li and W. He, Green Chem., 2016, 18, 4879–4883 RSC.
- J. Jiang, H. Zou, N. Yi, R. Wang, H. Zhang, L. Lan and J. Xiang, Chin. J. Chem., 2016, 34, 1245–1250 CrossRef CAS.
- W. Zhang, G. M. Johnson, Z. Guan and Y.-H. He, Adv. Synth. Catal., 2018, 360, 4562–4570 CrossRef CAS.
- S. Jia, Z. Chen, N. Zhang, Y. Tan, Y. Liu, J. Deng and H. Yan, J. Am. Chem. Soc., 2018, 140, 7056–7060 CrossRef CAS.
- C. Dai, J. Wang, S. Deng, C. Zhou, W. Zhang, Q. Zhu and X. Tang, RSC Adv., 2017, 7, 36112–36116 RSC.
- Z. Liu, L. Yang, K. Zhang, W. Chen, T. Yu, L. Wang, W. Gao and B. Tang, Org. Lett., 2020, 22, 2081–2086 CrossRef CAS.
- M. Miao, Y. Luo, H. Xu, Z. Chen, J. Xu and H. Ren, Org. Lett., 2016, 18, 4292–4295 CrossRef CAS.
- K. Luo, L. Zhang, J. Ma, Q. Sha and L. Wu, J. Org. Chem., 2017, 82, 6978–6985 CrossRef CAS.
- O. A. Storozhenko, A. A. Festa, G. I. Detistova, V. B. Rybakov, A. V. Varlamov, E. V. Van Der Eycken and L. G. Voskressensky, J. Org. Chem., 2020, 85, 2250–2259 CrossRef CAS.
- P. Kramer, S. C. Krieg, H. Kelm and G. Manolikakes, Org. Biomol. Chem., 2019, 17, 5538–5544 RSC.
- D. Sun and R. Zhang, Org. Chem. Front., 2017, 5, 92–97 RSC.
- H. Jiang, X. Tang, Z. Xu, H. Wang, K. Han, X. Yang, Y. Zhou, Y.-L. Feng, X.-Y. Yu and Q. Gui, Org. Biomol. Chem., 2019, 17, 2715–2720 RSC.
- Q. Jiang, B. Xu, J. Jia, A. Zhao, Y.-R. Zhao, Y.-Y. Li, N.-N. He and C.-C. Guo, J. Org. Chem., 2014, 79, 7372–7379 CrossRef CAS.
- B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2014, 79, 8110–8117 CrossRef CAS.
- R. Guo, Q. Gui, D. Wang and Z. Tan, Catal. Lett., 2014, 144, 1377–1383 CrossRef CAS.
- Y. Xu, X. Tang, W. Hu, W. Wu and H. Jiang, Green Chem., 2014, 16, 3720–3723 RSC.
- P. Katrun, S. Hlekhlai, J. Meesin, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, Org. Biomol. Chem., 2015, 13, 4785–4794 RSC.
- J. Chen, J. Mao, Y. Zheng, D. Liu, G. Rong, H. Yan, C. Zhang and D. Shi, Tetrahedron, 2015, 71, 5059–5063 CrossRef CAS.
- J. Gao, J. Lai and G. Yuan, RSC Adv., 2015, 5, 66723–66726 RSC.
- P. Qian, M. Bi, J. Su, Z. Zha and Z. Wang, J. Org. Chem., 2016, 81, 4876–4882 CrossRef CAS.
- G. Rong, J. Mao, H. Yan, Y. Zheng and G. Zhang, J. Org. Chem., 2015, 80, 7652–7657 CrossRef CAS.
- N. Xue, R. Guo, X. Tu, W. Luo, W. Deng and J. Xiang, Synlett, 2016, 27, 2695–2698 CrossRef CAS.
- J. Meesin, P. Katrun, C. Pareseecharoen, M. Pohmakotr, V. Reutrakul, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2016, 81, 2744–2752 CrossRef CAS.
- P. Li and G.-W. Wang, Org. Biomol. Chem., 2019, 17, 5578–5585 RSC.
- T. Keshari, R. Kapoorr and L. D. S. Yadav, Eur. J. Org. Chem., 2016, 2695–2699 CrossRef CAS.
- G. Nie, X. Deng, X. Lei, Q. Hu and Y. Chen, RSC Adv., 2016, 6, 75277–75281 RSC.
- G.-F. Hong, J.-W. Yuan, Z.-H. Dong, Y.-M. Xiao, P. Mao and L.-B. Qu, Phosphorus, Sulfur Silicon Relat. Elem., 2018, 193, 771–779 CrossRef CAS.
- X. Lei, L. Zheng, C. Zhang, X. Shi and Y. Chen, J. Org. Chem., 2018, 83, 1772–1778 CrossRef CAS.
- Y. Gao, W. Wu, Y. Huang, K. Huang and H. Jiang, Org. Chem. Front., 2014, 1, 361–364 RSC.
- Y. Sun, A. Abdukader, D. Lu, H. Zhang and C. Liu, Green Chem., 2017, 19, 1255–1258 RSC.
- W. Bi, C. Ren, L. Jia, X. Xia, X. Chen, X. Chen and Y. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 391–396 CrossRef CAS.
- R. Kumar, V. Dwivedi and M. Sridhar Reddy, Adv. Synth. Catal., 2017, 359, 2847–2856 CrossRef CAS.
- J. Zhang, Z. Liang, J. Wang, Z. Guo, C. Liu and M. Xie, ACS Omega, 2018, 3, 18002–18015 CrossRef CAS.
- N. Taniguchi, Tetrahedron, 2018, 74, 1454–1460 CrossRef CAS.
- K. Zeng, L. Chen, Y. Chen, Y. Liu, Y. Zhou, C. T. Au and S. F. Yin, Adv. Synth. Catal., 2017, 359, 841–847 CrossRef CAS.
- S. Handa, J. C. Fennewald and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 3432–3435 CrossRef CAS.
- A. K. Singh, R. Chawla and L. D. S. Yadav, Tetrahedron Lett., 2014, 55, 2845–2848 CrossRef CAS.
- N. Kumar and A. Kumar, ACS Sustainable Chem. Eng., 2019, 7, 9182–9188 CrossRef CAS.
- R. Chawla, A. K. Singh and L. D. S. Yadav, Eur. J. Org. Chem., 2014, 2032–2036 CrossRef CAS.
- A. K. Singh, R. Chawla and L. D. S. Yadav, Tetrahedron Lett., 2014, 55, 4742–4746 CrossRef CAS.
- N. R. Yalavarthi, N. Gundoju, R. Bokam and M. G. Ponnapalli, J. Chem. Sci., 2019, 131, 1–8 CrossRef CAS.
- X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205–4208 CrossRef CAS.
- T. A. To, C. B. Tran, N. T. H. Nguyen, H. H. T. Nguyen, A. T. Nguyen, A. N. Q. Phan and N. T. S. Phan, RSC Adv., 2018, 8, 17477–17485 RSC.
- J. Xia, X. Huang, S. You and M. Cai, Appl. Organomet. Chem., 2019, 33, e5001 CrossRef.
- Y.-Y. Xie and Z.-C. Chen, Synth. Commun., 2001, 31, 3145–3149 CrossRef CAS.
- S. Deng, E. Liang, Y. Wu and X. Tang, Tetrahedron Lett., 2018, 59, 3955–3957 CrossRef CAS.
- M. Muneeswara, N. Sundaravelu and G. Sekar, Tetrahedron, 2019, 75, 3479–3484 CrossRef CAS.
- X.-W. Lan, N.-X. Wang, C.-B. Bai, W. Zhang, Y. Xing, J.-L. Wen, Y.-J. Wang and Y.-H. Li, Sci. Rep., 2015, 5, 18391–18399 CrossRef.
- B. Lin, J. Kuang, J. Chen, Z. Hua, V. Khakyzadeh and Y. Xia, Org. Chem. Front., 2019, 6, 2647–2653 RSC.
- V. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Tetrahedron Lett., 2016, 57, 2236–2238 CrossRef CAS.
- F. Han, B. Su, P. Song, Y. Wang, L. Jia, S. Xun, M. Hu and L. Zou, Tetrahedron, 2018, 74, 5908–5913 CrossRef CAS.
- R. Chawla, R. Kapoor and L. D. S. Yadav, Tetrahedron Lett., 2019, 60, 150964–150969 CrossRef CAS.
- Y. Li, D. Liang, Y. Chang, X. Li, S. Fu, Y. Yuan and B. Wang, Synth. Commun., 2017, 47, 2044–2052 CrossRef CAS.
- W.-C. Gao, J.-J. Zhao, H.-H. Chang, X. Li, Q. Liu and W.-L. Wei, RSC Adv., 2014, 4, 49329–49332 RSC.
- W. C. Gao, J. J. Zhao, F. Hu, H. H. Chang, X. Li and W. L. Wei, RSC Adv., 2015, 5, 25222–25228 RSC.
- P. Katrun, T. Songsichan, D. Soorukram, M. Pohmakotr, V. Reutrakul and C. Kuhakarn, Synthesis, 2017, 49, 1109–1121 CAS.
- O. M. Mulina, D. A. Pirgach, G. I. Nikishin and A. O. Terent'ev, Eur. J. Org. Chem., 2019, 4179–4188 CrossRef CAS.
- X.-J. Pan, J. Gao and G.-Q. Yuan, Tetrahedron, 2015, 71, 5525–5530 CrossRef CAS.
- I. Yavari and S. Shaabanzadeh, Org. Lett., 2020, 22, 464–467 CrossRef CAS.
- H. Wang, P. Lian, Y. Zheng, J. Li and X. Wan, Org. Biomol. Chem., 2020, 18, 2163–2169 RSC.
- N. Taniguchi, J. Org. Chem., 2015, 80, 7797–7802 CrossRef CAS.
- Q. Freitas, R. Lira, J. Freitas, G. Zeni and P. Menezes, J. Braz. Chem. Soc., 2018, 29, 1167–1174 CrossRef CAS.
- J. Lai and G. Yuan, Tetrahedron Lett., 2018, 59, 524–527 CrossRef CAS.
- N. Suryakiran, T. Srikanth Reddy and Y. Venkateswarlu, J. Sulfur Chem., 2007, 28, 513–518 CrossRef CAS.
- C.-K. K. Chan, N.-C. C. Lo, P.-Y. Y. Chen and M.-Y. Y. Chang, Synthesis, 2017, 49, 4469–4477 CrossRef CAS.
- D. Zhang, T. Cheng, Q. Zhao, J. Xu and G. Liu, Org. Lett., 2014, 16, 5764–5767 CrossRef CAS.
- P. Cui, Q. Liu, J. Wang, H. Liu and H. Zhou, Green Chem., 2019, 21, 634–639 RSC.
- A. Ulman and E. Urankar, J. Org. Chem., 1989, 54, 4691–4692 CrossRef CAS.
- H. Suzuki and H. Abe, Tetrahedron Lett., 1995, 36, 6239–6242 CrossRef CAS.
- J. M. Baskin and Z. Wang, Org. Lett., 2002, 4, 4423–4425 CrossRef CAS.
- S. Cacchi, G. Fabrizi, A. Goggiamani and L. M. Parisi, Org. Lett., 2002, 4, 4719–4721 CrossRef CAS.
- W. Zhu and D. Ma, J. Org. Chem., 2005, 70, 2696–2700 CrossRef CAS.
- K. M. Maloney, J. T. Kuethe and K. Linn, Org. Lett., 2011, 13, 102–105 CrossRef CAS.
- Y. Yuan and S. Guo, Synlett, 2011, 2750–2756 Search PubMed.
- S. Liang, R.-Y. Zhang, L.-Y. Xi, S.-Y. Chen and X.-Q. Yu, J. Org. Chem., 2013, 78, 11874–11880 CrossRef CAS.
- M. Yang, H. Shen, Y. Li, C. Shen and P. Zhang, RSC Adv., 2014, 4, 26295–26300 RSC.
- C. Shen, J. Xu, W. Yu and P. Zhang, Green Chem., 2014, 16, 3007–3012 RSC.
- J. D. Smith, T. N. Ansari, M. P. Andersson, D. Yadagiri, F. Ibrahim, S. Liang, G. B. Hammond, F. Gallou and S. Handa, Green Chem., 2018, 20, 1784–1790 RSC.
- J. Zhao, S. Niu, X. Jiang, Y. Jiang, X. Zhang, T. Sun and D. Ma, J. Org. Chem., 2018, 83, 6589–6598 CrossRef CAS.
- X. Ge, F. Sun, X. Liu, X. Chen, C. Qian and S. Zhou, Mol. Catal., 2018, 449, 72–78 CrossRef CAS.
- F. Chen, F. Chacón-Huete, H. El-Husseini and P. Forgione, Synthesis, 2018, 50, 1914–1920 CrossRef CAS.
- H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371–1375 CrossRef CAS.
- N. W. Liu, K. Hofman, A. Herbert and G. Manolikakes, Org. Lett., 2018, 20, 760–763 CrossRef CAS.
- M. J. Cabrera-Afonso, Z.-P. Lu, C. B. Kelly, S. B. Lang, R. Dykstra, O. Gutierrez and G. A. Molander, Chem. Sci., 2018, 3186–3191 RSC.
- N.-W. Liu, S. Liang, N. Margraf, S. Shaaban, V. Luciano, M. Drost and G. Manolikakes, Eur. J. Org. Chem., 2018, 1208–1210 CrossRef CAS.
- L. Chen, J. Liang, Z. yu Chen, J. Chen, M. Yan and X. jing Zhang, Adv. Synth. Catal., 2019, 361, 956–960 CAS.
- V. D. Nguyen, V. T. Nguyen, G. C. Haug, H. T. Dang, H. D. Arman, W. C. Ermler and O. V. Larionov, ACS Catal., 2019, 9, 4015–4024 CrossRef CAS.
- C. Beaulieu, D. Guay, Z. Wang and D. A. Evans, Tetrahedron Lett., 2004, 45, 3233–3236 CrossRef CAS.
- F. Huang and R. A. Batey, Tetrahedron, 2007, 63, 7667–7672 CrossRef CAS.
- A. Kar, L. A. Sayyed, W. F. Lo, H. M. Kaiser, M. Beller and M. K. Tse, Org. Lett., 2007, 9, 3405–3408 CrossRef CAS.
- M. L. Kantam, B. Neelima, B. Sreedhar and R. Chakravarti, Synlett, 2008, 1455–1458 CrossRef CAS.
- B. T. V. Srinivas, V. S. Rawat, K. Konda and B. Sreedhar, Adv. Synth. Catal., 2014, 356, 805–817 CrossRef CAS.
- G. C. Nandi, Synth. Commun., 2017, 47, 319–323 CrossRef CAS.
- X. Yang, Y. Cao, E. Li, P. Zhang and Z. Hou, Appl. Organomet. Chem., 2014, 28, 785–788 CrossRef CAS.
- L. A. Smyth, E. M. Phillips, V. S. Chan, J. G. Napolitano, R. Henry and S. Shekhar, J. Org. Chem., 2016, 81, 1285–1294 CrossRef CAS.
- R. Chawla and L. D. S. Yadav, Org. Biomol. Chem., 2019, 17, 4761–4766 RSC.
- Y. Yu, Q. Wu, D. Liu, L. Yu, Z. Tan and G. Zhu, J. Org. Chem., 2019, 84, 11195–11202 CrossRef CAS.
- N. Umierski and G. Manolikakes, Org. Lett., 2013, 15, 188–191 CrossRef CAS.
- S. C. Cullen, S. Shekhar and N. K. Nere, J. Org. Chem., 2013, 78, 12194–12201 CrossRef CAS.
- V. G. Pandya and S. B. Mhaske, Org. Lett., 2014, 16, 3836–3839 CrossRef CAS.
- J. Liu, L. Yu, S. Zhuang, Q. Gui, X. Chen, W. Wang and Z. Tan, Chem. Commun., 2015, 51, 6418–6421 RSC.
- W.-H. Rao and B.-F. Shi, Org. Lett., 2015, 17, 2784–2787 CrossRef CAS.
- S.-L. Liu, X.-H. Li, S.-S. Zhang, S.-K. Hou, G.-C. Yang, J.-F. Gong and M.-P. Song, Adv. Synth. Catal., 2017, 359, 2241–2246 CrossRef CAS.
- W. H. Rao, B. B. Zhan, K. Chen, P. X. Ling, Z. Z. Zhang and B. F. Shi, Org. Lett., 2015, 17, 3552–3555 CrossRef CAS.
- C. Xia, K. Wang, J. Xu, Z. Wei, C. Shen, G. Duan, Q. Zhu and P. Zhang, RSC Adv., 2016, 6, 37173–37179 RSC.
- P. Bai, S. Sun, Z. Li, H. Qiao, X. Su, F. Yang, Y. Wu and Y. Wu, J. Org. Chem., 2017, 82, 12119–12127 CrossRef CAS.
- Y. Du, L. Wei and Y. Liu, Heteroat. Chem., 2017, 28, 1–6 CrossRef.
- S. Liang, N. W. Liu and G. Manolikakes, Adv. Synth. Catal., 2016, 358, 159–163 CrossRef CAS.
- S. Liang, M. Bolte and G. Manolikakes, Chem.–Eur. J., 2017, 23, 96–100 CrossRef CAS.
- F. Xiao, S. Chen, Y. Chen, H. Huang and G.-J. Deng, Chem. Commun., 2015, 51, 652–654 RSC.
- U. Karmakar and R. Samanta, J. Org. Chem., 2019, 84, 2850–2861 CrossRef CAS.
- M. Sattar, N. Kumar, P. Yadav, Y. Mandhar and S. Kumar, Chem.–Asian J., 2019, 14, 4807–4813 CrossRef CAS.
- W. C. Yang, P. Dai, K. Luo and L. Wu, Adv. Synth. Catal., 2016, 358, 3184–3190 CrossRef CAS.
- M. Singh, L. D. S. Yadav and R. K. P. Singh, Tetrahedron Lett., 2019, 60, 810–813 CrossRef CAS.
- R. J. Griffiths, W. C. Kong, S. A. Richards, G. A. Burley, M. C. Willis and E. P. A. Talbot, Chem. Sci., 2018, 1, 2295–2300 RSC.
- J. I. Higham and J. A. Bull, Chem. Commun., 2020, 56, 4587–4590 RSC.
- B. Du, P. Qian, Y. Wang, H. Mei, J. Han and Y. Pan, Org. Lett., 2016, 18, 4144–4147 CrossRef CAS.
- L. Sumunnee, C. Buathongjan, C. Pimpasri and S. Yotphan, Eur. J. Org. Chem., 2017, 1025–1032 CrossRef CAS.
- W. K. Fu, K. Sun, C. Qu, X. L. Chen, L. B. Qu, W. Z. Bi and Y. F. Zhao, Asian J. Org. Chem., 2017, 6, 492–495 CrossRef CAS.
- L.-Y. Xie, S. Peng, F. Liu, G.-R. Chen, W. Xia, X. Yu, W.-F. Li, Z. Cao and W.-M. He, Org. Chem. Front., 2018, 5, 2604–2609 RSC.
- P. Li, Y. Jiang, H. Li, W. Dong, Z. Peng and D. An, Synth. Commun., 2018, 48, 1909–1918 CrossRef CAS.
- S. Peng, Y. X. Song, J. Y. He, S. S. Tang, J. X. Tan, Z. Cao, Y. W. Lin and W. M. He, Chin. Chem. Lett., 2019, 30, 2287–2290 CrossRef CAS.
- M. Jiang, Y. Yuan, T. Wang, Y. Xiong, J. Li, H. Guo and A. Lei, Chem. Commun., 2019, 55, 13852–13855 RSC.
- F. Xiao, H. Chen, H. Xie, S. Chen, L. Yang and G.-J. Deng, Org. Lett., 2014, 16, 50–53 CrossRef CAS.
- P. Katrun, C. Mueangkaew, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2014, 79, 1778–1785 CrossRef CAS.
- Y. Yang, W. Li, C. Xia, B. Ying, C. Shen and P. Zhang, ChemCatChem, 2016, 8, 304–307 CrossRef CAS.
- H. Li, X. Wang and J. Yan, New J. Chem., 2017, 41, 4277–4280 RSC.
- M.-L. Feng, L.-Y. Xi, S.-Y. Chen and X.-Q. Yu, Eur. J. Org. Chem., 2017, 2746–2750 CrossRef CAS.
- Y. Zhou, W. Bin Cao, L. L. Zhang, X. P. Xu and S. J. Ji, J. Org. Chem., 2018, 83, 6056–6065 CrossRef CAS.
- C. D. Prasad, M. Sattar and S. Kumar, Org. Lett., 2017, 19, 774–777 CrossRef CAS.
- W. Wei, H. Cui, D. Yang, X. Liu, C. He, S. Dai and H. Wang, Org. Chem. Front., 2017, 4, 26–30 RSC.
- L.-X. Li, D.-Q. Dong, S.-H. Hao and Z.-L. Wang, Tetrahedron Lett., 2018, 59, 1517–1520 CrossRef CAS.
- T. C. Johnson, B. L. Elbert, A. J. M. M. Farley, T. W. Gorman, C. Genicot, B. Lallemand, P. Pasau, J. Flasz, J. L. Castro, M. MacCoss, D. J. Dixon, R. S. Paton, C. J. Schofield, M. D. Smith and M. C. Willis, Chem. Sci., 2018, 9, 629–633 RSC.
- Y.-C. Wu, S.-S. Jiang, S.-Z. Luo, R.-J. Song and J.-H. Li, Chem. Commun., 2019, 55, 8995–8998 RSC.
- F. Lu, J. Li, T. Wang, Z. Li, M. Jiang, X. Hu, H. Pei, F. Yuan, L. Lu and A. Lei, Asian J. Org. Chem., 2019, 8, 1838–1841 CrossRef CAS.
- J. Nikl, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Chem.–Eur. J., 2019, 25, 6891–6895 CrossRef CAS.
- J. Nikl, D. Ravelli, D. Schollmeyer and S. R. Waldvogel, ChemElectroChem, 2019, 6, 4450–4455 CrossRef CAS.
- F. Xie, G.-P. Lu, R. Xie, Q.-H. Chen, H.-F. Jiang and M. Zhang, ACS Catal., 2019, 9, 2718–2724 CrossRef CAS.
- S. Liang, Y. Ren and G. Manolikakes, Eur. J. Org. Chem., 2017, 4117–4120 CrossRef CAS.
- D. Habibi, A. Rahimi, A. Rostami and S. Moradi, Tetrahedron Lett., 2017, 58, 289–293 CrossRef CAS.
- P. Halder, V. T. Humne and S. B. Mhaske, J. Org. Chem., 2019, 84, 1372–1378 CrossRef CAS.
- A. P. Avdeenko, S. A. Konovalova, O. N. Mikhailichenko, S. V. Shelyazhenko, V. V. Pirozhenko and L. M. Yagupol'skii, Russ. J. Org. Chem., 2012, 48, 221–233 CrossRef CAS.
- S. A. Konovalova, A. P. Avdeenko and I. L. Marchenko, Russ. J. Org. Chem., 2014, 50, 973–985 CrossRef CAS.
- S. A. Konovalova, A. P. Avdeenko, A. A. Santalova, V. V. D'yakonenko, G. V. Palamarchuk and O. V. Shishkin, Russ. J. Org. Chem., 2014, 50, 1757–1762 CrossRef CAS.
- W. Wu, S. Yi, Y. Yu, W. Huang and H. Jiang, J. Org. Chem., 2017, 82, 1224–1230 CrossRef CAS.
- W. Wu, S. Yi, W. Huang, D. Luo and H. Jiang, Org. Lett., 2017, 19, 2825–2828 CrossRef CAS.
- H. S. Li and G. Liu, J. Org. Chem., 2014, 79, 509–516 CrossRef CAS.
- M. Rajesh, M. K. R. Singam, R. K. Gadi and M. Sridhar Reddy, ACS Omega, 2018, 3, 17155–17163 CrossRef CAS.
- Y. Wan, J. Zhang, Y. Chen, L. Kong, F. Luo and G. Zhu, Org. Biomol. Chem., 2017, 15, 7204–7211 RSC.
- M. Miao, H. Xu, Y. Luo, M. Jin, Z. Chen, J. Xu and H. Ren, Org. Chem. Front., 2017, 4, 1824–1828 RSC.
- Y.-S. Xiong, B. Zhang, Y. Yu, J. Weng and G. Lu, J. Org. Chem., 2019, 84, 13465–13472 CrossRef CAS.
- T. Yang, P. Kou, F. Jin, X.-R. Song, J. Bai, H. Ding, Q. Xiao and Y.-M. Liang, Org. Chem. Front., 2019, 6, 3162–3166 RSC.
- H. Ren, Y. Mu, M. Zhang and A. Q. Zhang, J. Chem. Res., 2018, 42, 515–520 CrossRef CAS.
- Q. Q. Han, G. H. Li, Y. Y. Sun, D. M. Chen, Z. L. Wang, X. Y. Yu and X. M. Xu, Tetrahedron Lett., 2020, 61, 151704 CrossRef CAS.
- R. S. Rohokale, S. D. Tambe and U. A. Kshirsagar, Org. Biomol. Chem., 2018, 16, 536–540 RSC.
- Y.-Y. Jiang, S. Liang, C.-C. Zeng, L.-M. Hu and B.-G. Sun, Green Chem., 2016, 18, 6311–6319 RSC.
- L.-J. Wang, J.-M. Chen, W. Dong, C.-Y. Hou, M. Pang, W.-B. Jin, F.-G. Dong, Z.-D. Xu and W. Li, J. Org. Chem., 2019, 84, 2330–2338 CrossRef CAS.
- W.-H. Rao, L.-L. Jiang, X.-M. Liu, M.-J. Chen, F.-Y. Chen, X. Jiang, J.-X. Zhao, G.-D. Zou, Y.-Q. Zhou and L. Tang, Org. Lett., 2019, 21, 2890–2893 CrossRef CAS.
- Y. Wang, J. Ding, J. Zhao, W. Sun, C. Lian, C. Chen and B. Zhu, Org. Chem. Front., 2019, 6, 2240–2244 RSC.
- W. Dong, L. Qi, J. Y. Song, J. M. Chen, J. X. Guo, S. Shen, L. J. Li, W. Li and L. J. Wang, Org. Lett., 2020, 22, 1830–1835 CrossRef CAS.
- H. Fu, S.-S. S. Wang and Y.-M. M. Li, Adv. Synth. Catal., 2016, 358, 3616–3626 CrossRef CAS.
- Q. Tang, P. Xie, J. Wang, J. Lin, C. Feng, C. U. Pittman and A. Zhou, Tetrahedron, 2017, 73, 5436–5443 CrossRef CAS.
- K. L. Zuo, Y. H. He and Z. Guan, Eur. J. Org. Chem., 2019, 939–948 CrossRef CAS.
- T.-H. Chuang, Y.-Y. Chen, Y.-J. Huang and C.-P. Chuang, ChemistrySelect, 2016, 1, 6762–6767 CrossRef CAS.
- L. Li, X. Zhang, B. Hu and X. Zhang, Chem.–Asian J., 2019, 14, 4358–4364 CrossRef CAS.
- J.-M. Yuan, J. Li, H. Zhou, J. Xu, F. Zhu, Q. Liang, Z. Liu, G. Huang and J. Huang, New J. Chem., 2020, 44, 3189–3193 RSC.
- J. Tang, P. Sivaguru, Y. Ning, G. Zanoni and X. Bi, Org. Lett., 2017, 19, 4026–4029 CrossRef CAS.
- M. Singh, A. K. Yadav, L. D. S. Yadav and R. K. P. Singh, Tetrahedron Lett., 2018, 59, 3198–3201 CrossRef CAS.
- O. Khaikate, J. Meesin, M. Pohmakotr, V. Reutrakul, P. Leowanawat, D. Soorukram and C. Kuhakarn, Org. Biomol. Chem., 2018, 16, 8553–8558 RSC.
- L.-J. J. Wang, M. Chen, L. Qi, Z. Xu and W. Li, Chem. Commun., 2017, 53, 2056–2059 RSC.
- J. Wu, Y. Zong, C. Zhao, Q. Yan, L. Sun, Y. Li, J. Zhao, Y. Ge and Z. Li, Org. Biomol. Chem., 2019, 17, 794–797 RSC.
- B. Zhou, W. Chen, Y. Yang, Y. Yang, G. Deng and Y. Liang, Org. Biomol. Chem., 2018, 16, 7959–7963 RSC.
- J.-C. Kang, Y.-Q. Tu, J.-W. Dong, C. Chen, J. Zhou, T.-M. Ding, J.-T. Zai, Z.-M. Chen and S.-Y. Zhang, Org. Lett., 2019, 21, 2536–2540 CrossRef CAS.
- Y. Liu, Q. L. Wang, Z. Chen, Q. Zhou, C. S. Zhou, B. Q. Xiong, P. L. Zhang, C. A. Yang and K. W. Tang, Org. Biomol. Chem., 2019, 17, 1365–1369 RSC.
- L. Zhou, Y. Xia, Y.-Z. Wang, J.-D. Fang and X.-Y. Liu, Tetrahedron, 2019, 75, 1267–1274 CrossRef CAS.
- F. Xiao, C. Liu, D. Wang, H. Huang and G.-J. Deng, Green Chem., 2018, 20, 973–977 RSC.
- F. Xiao, C. Liu, S. Yuan, H. Huang and G.-J. Deng, J. Org. Chem., 2018, 83, 10420–10429 CrossRef CAS.
- Y. Ning, A. Mekareeya, K. R. Babu, E. A. Anderson and X. Bi, ACS Catal., 2019, 9, 4203–4210 CrossRef CAS.
- Y. Xiong, Y. Sun and G. Zhang, Org. Lett., 2018, 20, 6250–6254 CrossRef CAS.
- H. Mei, J. Liu, Y. Guo and J. Han, ACS Omega, 2019, 4, 14353–14359 CrossRef CAS.
- B. Lipp, L. M. Kammer, M. Kücükdisli, A. Luque, J. Kühlborn, S. Pusch, G. Matulevičiūtė, D. Schollmeyer, A. Šačkus and T. Opatz, Chem.–Eur J., 2019, 25, 8965–8969 CrossRef CAS.
- P. Kramer, M. Halaczkiewicz, Y. Sun, H. Kelm and G. Manolikakes, J. Org. Chem., 2020, 85, 3617–3637 CrossRef CAS.
- S. Jin, S. Fang, R. Ma, Z. Liang, Y. Xu, T. Lu and D. Du, Org. Chem. Front., 2019, 6, 3392–3396 RSC.
- M. Y. Ansari, N. Kumar and A. Kumar, Org. Lett., 2019, 21, 3931–3936 CrossRef CAS.
- Y. Xiang, Y. Li, Y. Kuang and J. Wu, Adv. Synth. Catal., 2017, 359, 2605–2609 CrossRef CAS.
- Y. Ning, Q. Ji, P. Liao, E. A. Anderson and X. Bi, Angew. Chem., Int. Ed., 2017, 56, 13805–13808 CrossRef CAS.
- M. Luo, B. Liu, Y. Li, M. Hu and J. Li, Adv. Synth. Catal., 2019, 361, 1538–1542 CrossRef CAS.
- J. Yang, Y. Y. Liu, R. J. Song, Z. H. Peng and J. H. Li, Adv. Synth. Catal., 2016, 358, 2286–2292 CrossRef CAS.
- X. He, X. Yue, L. Zhang, S. Wu, M. Hu and J.-H. Li, Chem. Commun., 2019, 55, 3517–3520 RSC.
- X. Yue, X. He, Y. Wu, M. Hu, S. Wu, Y.-X. Xie and J.-H. Li, ChemCatChem, 2020, 12, 2690–2694 CrossRef CAS.
- C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, Nat. Catal., 2019, 2, 678–687 CrossRef CAS.
- R. R. Kuchukulla, F. Li, Z. He, L. Zhou and Q. Zeng, Green Chem., 2019, 21, 5808–5812 RSC.
- W. Liu, Y. Zhang, J. He, Y. Yu, J. Yuan, X. Ye, Z. Zhang, L. Xue and H. Cao, J. Org. Chem., 2019, 84, 11348–11358 CrossRef CAS.
- J. Hao, Y. Xu, Z. Xu, Z. Zhang and W. Yang, Org. Lett., 2018, 20, 7888–7892 CrossRef CAS.
- J. Shi, X.-D. Tang, Y.-C. Wu, J.-F. Fang, L. Cao, X.-Y. Chen and Z.-Y. Wang, RSC Adv., 2016, 6, 25651–25655 RSC.
- M. Nematpour and E. Abedi, J. Sulfur Chem., 2017, 38, 76–82 CrossRef CAS.
- L. M. Kammer, M. Krumb, B. Spitzbarth, B. Lipp, J. Kühlborn, J. Busold, O. M. Mulina, A. O. Terentev and T. Opatz, Org. Lett., 2020, 22, 3318–3322 CrossRef CAS.
- W.-C. Gao, Y.-F. Cheng, Y.-Z. Shang, H.-H. Chang, X. Li, R. Zhou, Y. Qiao and W.-L. Wei, J. Org. Chem., 2018, 83, 11956–11962 CrossRef CAS.
- M. Nematpour, S. R. Koohi, E. Abedi and M. Lotfi, J. Chem. Res., 2016, 40, 652–654 CrossRef CAS.
- M. Nematpour, E. Abedi and M. Lotfi, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 427–430 CrossRef CAS.
- H. S. Wang, Y. S. Wu and M. Y. Chang, Tetrahedron, 2017, 73, 6465–6470 CrossRef CAS.
- W. Middelbos, J. Strating and B. Zwanenburg, Tetrahedron Lett., 1971, 351–352 CrossRef CAS.
- R. Chawla, A. K. Singh and L. D. S. Yadav, Tetrahedron, 2013, 69, 1720–1724 CrossRef CAS.
- G. Lu, C. Cai, F. Chen, R. Ye and B. Zhou, ACS Sustainable Chem. Eng., 2016, 4, 1804–1809 CrossRef CAS.
- T. Murakami and K. Furusawa, Synthesis, 2002, 479–482 CrossRef CAS.
- M. A. Reddy, P. S. Reddy and B. Sreedhar, Adv. Synth. Catal., 2010, 352, 1861–1869 CrossRef CAS.
- Y. Gao, H. Mei, J. Han and Y. Pan, Chem.–Eur. J., 2018, 24, 17205–17209 CrossRef CAS.
- R. Kuwano, Y. Kondo and T. Shirahama, Org. Lett., 2005, 7, 2973–2975 CrossRef CAS.
- Y. Liu, H. Zhu, L. Yang, Z. Xie, G. Jiang, Z. G. Le and T. Tu, Asian J. Org. Chem., 2020, 9, 247–250 CrossRef CAS.
- A. Najib, K. Hirano and M. Miura, Chem.–Eur. J., 2018, 24, 6525–6529 CrossRef CAS.
- H. Yamamoto and K. Nakata, Eur. J. Org. Chem., 2019, 4906–4910 CrossRef CAS.
- J. Gao, X. Pan, J. Liu, J. Lai, L. Chang and G. Yuan, RSC Adv., 2015, 5, 27439–27442 RSC.
- E. Deruer, V. Hamel, S. Blais and S. Canesi, Beilstein J. Org. Chem., 2018, 14, 1203–1207 CrossRef CAS.
- M. Huang, L. Hu, H. Shen, Q. Liu, M. I. Hussain, J. Pan and Y. Xiong, Green Chem., 2016, 18, 1874–1879 RSC.
- Y. Z. Ji, H. J. Li, J. Y. Zhang and Y. C. Wu, Eur. J. Org. Chem., 2019, 1846–1855 CrossRef CAS.
- P. Bao, L. Wang, H. Yue, Y. Shao, J. Wen, D. Yang, X. Zhao, H. Wang and W. Wei, J. Org. Chem., 2019, 84, 2976–2983 CrossRef CAS.
- G. Bogonda, Di. V. Patil, H. Y. Kim and K. Oh, Org. Lett., 2019, 21, 3774–3779 CrossRef CAS.
- Y. Z. Ji, H. J. Li, H. R. Yang, Z. Y. Zhang, L. J. Xie and Y. C. Wu, Synlett, 2020, 31, 349–354 CrossRef CAS.
- X. S. Zhou, Y. Cheng, J. Chen, X. Y. Yu, W. J. Xiao and J. R. Chen, ChemCatChem, 2019, 11, 5300–5305 CrossRef CAS.
- Q. Wang, Z. Chen, Q. Zhou, C. Zhou, B. Xiong, P. Zhang, C. Yang, Y. Liu and K. Tang, Adv. Synth. Catal., 2019, 361, 2315–2320 CrossRef CAS.
- B. Skillinghaug, C. Sköld, J. Rydfjord, F. Svensson, M. Behrends, J. Sävmarker, P. J. R. Sjöberg and M. Larhed, J. Org. Chem., 2014, 79, 12018–12032 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |