
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and stability of single-phase chalcopyrite – a potential reference material for key investigations in chemistry and metallurgical engineering†
Ninett Frenzel a,
Marcel Mehneb,
Sebastian Bettec,
Sven Kuretib and
Gero Frisch*a
a,
Marcel Mehneb,
Sebastian Bettec,
Sven Kuretib and
Gero Frisch*a
aTU Bergakademie Freiberg, Institute of Inorganic Chemistry, Leipziger Str. 29, 09599 Freiberg, Germany. E-mail: frisch@tu-freiberg.de
bTU Bergakademie Freiberg, Institute of Energy Process Engineering and Chemical Engineering, Chair of Reaction Engineering, Fuchsmühlenweg 9, 09599 Freiberg, Germany
cMax-Planck-Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany
First published on 15th January 2021
Abstract
Single-phase chalcopyrite (CuFeS2) is a key reference material in the development of new metallurgical processes to ensure a reliable copper supply. Here, we report on the successful synthesis of single-phase chalcopyrite and its phase behaviour. We further rationalise different opinions previously expressed in the literature. Chalcopyrite synthesis has been studied at 450 °C with varying sulfur contents and analysed using X-ray powder diffraction (XRPD) and 57Fe-Mössbauer spectroscopy. With stoichiometric amounts (Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:S = 25:25:50) the main chalcopyrite phase is contaminated with pyrite (FeS2) and bornite (Cu5FeS4). Single-phase chalcopyrite was only found in samples containing around 49.7 at% sulfur in the reactant mixture. Mößbauer spectroscopy confirmed that chalcopyrite contains trivalent iron. Temperature dependent XRPD measurements detected an order–disorder phase transition starting at 485 °C. At temperatures above 535 °C, samples only contained intermediate solid solutions. These adopt the sphalerite structure with the lattice constant slightly varying with Cu:Fe ratio.
:Fe:S = 25:25:50) the main chalcopyrite phase is contaminated with pyrite (FeS2) and bornite (Cu5FeS4). Single-phase chalcopyrite was only found in samples containing around 49.7 at% sulfur in the reactant mixture. Mößbauer spectroscopy confirmed that chalcopyrite contains trivalent iron. Temperature dependent XRPD measurements detected an order–disorder phase transition starting at 485 °C. At temperatures above 535 °C, samples only contained intermediate solid solutions. These adopt the sphalerite structure with the lattice constant slightly varying with Cu:Fe ratio.
Introduction
Chalcopyrite is one of the most abundant copper minerals and the most significant for copper production worldwide. Depletion of high-grade chalcopyrite ores has made pyrometallurgical processing less efficient. Hence, copper producers are seeking hydrometallurgical routes for copper extraction which may be more suitable for currently exploited deposits.1 Hydrometallurgical leaching processes are well established for copper oxides, however, these cannot be applied to chalcopyrite due to its poor solubility in acid, where it requires an oxidation step.2 Numerous approaches, from the use of chemical oxidising agents to bioleaching techniques, are the subject of current research.1,3,4 To optimize leaching processes, both reaction mechanisms and kinetics must be understood.5–11 It is also known that trace elements12,13 and by-phases such as pyrite (FeS2)14,15 or bornite (Cu5FeS4)16,17 can have a significant influence on the leaching mechanism. The first step towards elucidating the chemistry behind chalcopyrite leaching must hence be to study the oxidation of pure chalcopyrite, for which synthetic methods described in the literature are often ambiguous or inaccurate. Subsequently, this should be compared to chalcopyrite which has been specifically doped with relevant trace elements such as silver, lead, indium or zinc.18In this article we report the successful synthesis of single-phase chalcopyrite. We further try to rationalise contradicting statements found in the literature regarding the phase behaviour and composition of this mineral and closely related phases.
The following summary highlights key issues which may have led to diverse and sometimes contradicting information regarding chalcopyrite's leaching behaviour, composition, metal ion oxidation states, phase stability and transition temperature:
– The most significant point is likely the use of natural minerals for the investigation of chalcopyrite.19–21 It is well known that impurities and by-phases have a significant effect, not only on the leaching mechanisms, but also on properties such as phase transition temperature,21 which are key to synthetic pathways. Few investigations have been conducted with synthetic chalcopyrite, however, there are good reasons to doubt that all these studies were performed using the single-phase compound. This may be in part due to analytical techniques not being available to the investigators, or not having the necessary accuracy at the time. For example in 1980, Conard et al.22 found pyrite and bornite contamination in what, in 1973, was believed to be single-phase synthetic chalcopyrite.23
– In some investigations, samples were not sufficiently analysed and the phase composition was assumed to be the same as in other sources24 with some citations referring to private communications.25 At the same time, important experimental parameters, such as heating rates or exact sulfur contents, are sometimes not provided.22,24,26 We will highlight below that the phase composition of the sample is very sensitive to minor changes in sulfur contents. Furthermore, several sources report on chalcopyrite synthesis at temperatures which are unreasonably high according to the phase behaviour discussed below.22,27–29 Therefore, the actual phase composition may deviate significantly from the expected composition.
– The name “chalcopyrite” is not used consistently. In particular, there is confusion between tetragonal chalcopyrite and the structurally related cubic high-temperature phase, often termed “intermediate solid solution” (iss) crystallising in the sphalerite structure.30 A large range of compositions have been reported for this phase,26,30 including the stoichiometries of mooihoekite (Cu9Fe9S16), haycockite (Cu4Fe5S8), talnakhite (Cu18Fe16S32) and even of cubanite (CuFe2S3). These minerals are structurally related, exhibit similar X-ray powder diffraction (XRPD) patterns and have sometimes been summarized as chalcopyrite. This adds to the variety of contradicting information about chalcopyrite's composition, structure, and properties.
– Attempts to explore the Cu–Fe–S phase diagram have to take into account that some high-temperature phases cannot be isolated via quenching.30 It is hence essential to analyse the phase composition in situ at the temperatures suggested by the phase diagram.
– At elevated temperature, a significant amount of sulfur can evaporate and is found as elemental sulfur after quenching the sample, even if this is not suggested by the phase diagram.‡ The composition of the reactants must hence not be confused with the composition of the target phase.
Many of these issues have been highlighted by other authors27,31,32 but persist in the literature regardless.
Structures and phase diagram
Chalcopyrite crystallises in the tetragonal space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d (122). This was first published by Pauling and Brockway in 193233 and has been confirmed by multiple authors. A detailed description of the structure determined on a natural chalcopyrite single crystal was published by Hall and Stewart in 197334 with lattice parameters a = 5.289(1) Å and c = 10.423(1) Å.
2d (122). This was first published by Pauling and Brockway in 193233 and has been confirmed by multiple authors. A detailed description of the structure determined on a natural chalcopyrite single crystal was published by Hall and Stewart in 197334 with lattice parameters a = 5.289(1) Å and c = 10.423(1) Å.
In 1973, Barton35 published the pseudo-binary phase diagram (Cu,Fe)–S (Fig. 1) which is arguably the most commonly accepted literature reference today. It shows that chalcopyrite is stable at the stoichiometric composition of 50 at% sulfur up to 532 °C. With increasing temperature, the chalcopyrite crystallisation field extends to slightly sulfur deficient compositions. Recently, a sulfur deficient mixture was used for chalcopyrite synthesis.36 It is further proposed that chalcopyrite decomposes at 557 °C into the iss and pyrite. This is based on the rationale that MacLean et al.19 observed no bornite in the breakdown of chalcopyrite, and Pankratz et al.24 found an upper stability limit of 557 °C using thermal analysis. In other literature sources, the upper stability limit of chalcopyrite varies between 500 and 560 °C.21,37 It is further debated whether the observed transformation should be classified as a phase transition27 or a decomposition.19,21,22,24
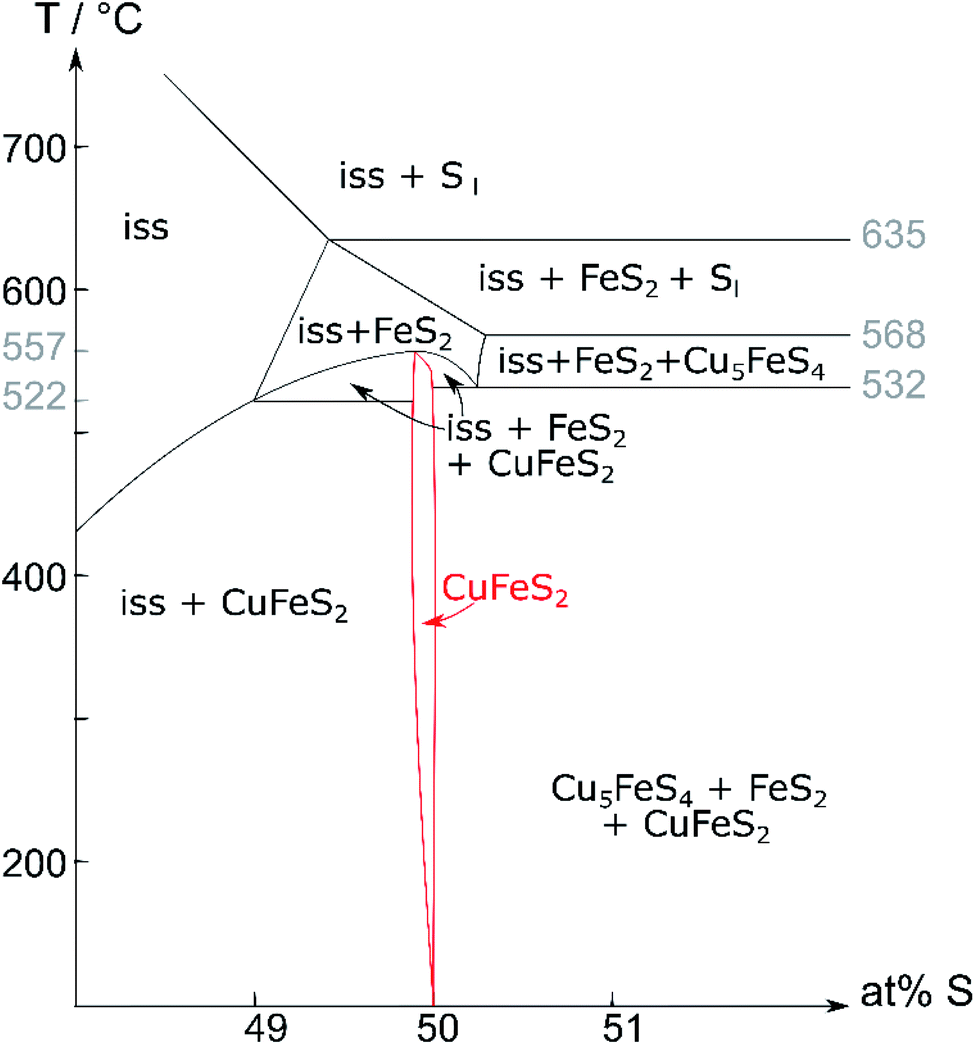 | ||
| Fig. 1 Pseudo-binary (Cu,Fe)–S phase diagram (recreated from Barton35); Cu:Fe ratio 1:1; iss = intermediate solid solution. | ||
Contrary to Barton's proposed decomposition reaction, recent literature27 shows a phase transition from a tetragonal to a cubic phase. The high-temperature phase was identified as the cubic iss, which crystallises in the sphalerite structure (F3m (216)) with a statistical disorder of Cu and Fe atoms.27 The same structure was found by Burdick and Ellis in 191738 but was mistaken for chalcopyrite at the time.
Talnakhite (Cu18Fe16S32) is a metal-rich phase,32 which is commonly confused with chalcopyrite and the iss.20 It crystallises in the cubic space group I3m (217) with a = 10.593 Å. The talnakhite structure is closely related to that of chalcopyrite and exhibits a very similar XRPD pattern.
Cubanite (CuFe2S3) also adopts a structure related to that of chalcopyrite but differs in the occupation of the metal ion positions. It crystallises in the orthorhombic space group Pcmn (62) with a = 6.4679(5) Å, b = 11.1201(8) Å and c = 6.2336(4) Å.39,40 Orthorhombic cubanite performs an irreversible phase transition to metastable isocubanite41 when heated above 210 °C.30 Like the iss, isocubanite crystallises in the sphalerite structure (a = 5.2949(3) Å) with a statistical disorder on the metal ion positions, but a different Fe:Cu ratio.39,42 Accordingly, the diffraction pattern of isocubanite structure is closely related to the iss. Further, the reflections of this structure appear at the same angles as those of talnakhite.
Hence, a mixture of chalcopyrite, talnakhite and isocubanite is difficult to analyse qualitatively and quantitatively with XRPD. Due to distinct Mößbauer spectra of these minerals, we chose Mößbauer spectroscopy as an additional method to analyse our samples.
Experimental
Synthetic methods
Copper, iron and sulfur (99.999%, <149 μm copper powder from AlfaAesar; 99.99+%, 0.7–1.2 μm iron powder from chemPUR; 99.998% sulfur flakes from SigmaAldrich) were used as received after confirming their purity using XRPD and Mößbauer spectroscopy.The required masses of Cu, Fe and S (element ratios see Table 1) were ground together in an agate mortar until a homogeneous mixture was produced and filled into a quartz-glass ampule. The ampoules were approximately 8–10 cm long with an inner diameter of 1.8 cm and an outer diameter of 2.0 cm. The ampules were evacuated, flushed with argon several times, and finally evacuated to a pressure of ca. 10−3 mbar, sealed and placed vertically in a muffle furnace. The samples were heated to 450 °C at 0.3 K min−1. Samples were left at this temperature for the annealing times given in Table 1 and subsequently quenched by removing the sample from the oven. Selected samples were also cooled with a rate of 10 K h−1 and compared to the quenched products. No difference was found between slow cooling and quenching.
| Cu/at% | Fe/at% | S/at% | Annealing time/d |
|---|---|---|---|
| 26.00 | 26.00 | 48.00 | 35 |
| 25.75 | 25.75 | 48.50 | 64 |
| 25.50 | 25.50 | 49.00 | 36 |
| 25.375 | 25.375 | 49.25 | 48 |
| 25.24 | 25.27 | 49.49 | 40 |
| 25.190 | 25.186 | 49.624 | 25 |
| 25.125 | 25.127 | 49.758 | 42 |
| 25.06 | 25.06 | 49.88 | 39 |
| 25.05 | 25.04 | 49.91 | 43 |
| 24.98 | 25.01 | 50.01 | 43 |
| 24.749 | 24.746 | 50.505 | 28 |
Additionally, a bornite sample was prepared in the same way as the chalcopyrite samples by mixing the elements in an atomic ratio of Cu:Fe:S of 50:10:40. XRPD data confirmed bornite as the single-phase product in this sample.
The synthesis of pyrite for reference purposes was not successful. Therefore, a natural sample of pyrite from Navajún (Spain) was used as reference material for Mößbauer spectroscopy. ICP analysis of the pyrite sample detected contaminations of: Ca (5319 mg kg−1), Al (546 mg kg−1), Ti (518 mg kg−1), Ni (433 mg kg−1), K (314 mg kg−1), Co (286 mg kg−1), Si (280 mg kg−1), Na (177 mg kg−1), Cu (74 mg kg−1), Mg (45 mg kg−1), Mn (38 mg kg−1), Ba (mg kg−1), Zn (11 mg kg−1), Sr (8 mg kg−1) and Cr (3 mg kg−1). XRPD data showed that pyrite is the main phase in this sample.
Analytical methods and results
XRPD patterns were measured at room temperature on a diffractometer D5000 (Siemens) in Bragg–Brentano geometry with Cu-Kα1/2 radiation (λK-α1 = 1.5406 Å, λK-α2 = 1.54439 Å). The generator was set to 40 kV and 40 mA. After the X-ray tube and a 1.0 mm entrance slit, a primary Göbel mirror is placed in front of an 0.6 mm exit slit and a 2.5° primary axial soller slit. After the rotating flat-plate sample (rotation rate of 15 rpm with a diameter measuring plane of ≈2.5 cm) an 0.6 mm antiscatter slit is followed by a secondary monochromator (Göbel mirror) in front of a 0.2 mm detector slit and a scintillation counter. The XRPD data were recorded within a range from 15 to 65 or 91° 2θ, using a step size of 0.02° 2θ and a measurement time of 20 or 25 seconds per step.Temperature dependent XRPD for in situ phase analysis and quantification were measured on a diffractometer D8 Advance (Bruker) with Mo-Kα radiation (λ = 0.7093 Å), equipped with a Ge 222 Johan type monochromator and a Lynx Eye point sensitive detector. A water-cooled furnace (mri Ofenbau) was used. Samples were sealed in quartz glass capillaries under argon atmosphere and heated with a heating rate of 2 K min−1. XRPD data were recorded every 5 K with a delay time of ten minutes in order to ensure thermal equilibration. The patterns were recorded with a scan range from 10 to 42° 2θ, using a step size of 0.003° 2θ and a total scan time of 4 h. During the measurements, the capillaries were spun around the horizontal axis in order to avoid preferred orientation effects. For a crystal structure refinement of chalcopyrite, the sample was heated up to 600 °C and measured with a scan range from 10 to 90° 2θ, using a step size of 0.003° 2θ and a total scan time of 12 h. The program TOPAS 6.0 was used to refine the XRPD pattern of chalcopyrite at 545 °C.43 Chebychev polynomial of 6th order were used to model the background and the peak profile was described by the fundamental parameter approach implemented into TOPAS.44 The crystal structure of the cubic intermediate solid solution39 was used as a starting model for a fully weighted Rietveld45 refinement by adjusting the site-occupancy factor (Cu and Fe) to the prepared element mixture. The lattice parameters, isothermal displacement parameters and crystallite size broadening parameters were released iteratively. The refinement converged quickly. A graphical result of the final Rietveld refinement is presented in Fig. 2, in Table S2 (ESI†) and were submitted to the CCDC database under the deposition number 2033752. For quantitative XRPD analysis of crystalline components, the powders were filled into 0.5 mm borosilicate glass capillaries (Hilgenberg Glass 0140). Diffraction patterns were recorded on a Stoe Stadi P powder diffractometer in Debye–Scherrer geometry with Mo-Kα radiation (λ = 0.7093 Å), equipped with a Ge 111 Johan type monochromator and a triple array of Mythen 1K detectors (Dectris). The patterns were collected in a 2θ range from 0.0° to 110° applying a total scan time of 3 hours. The crystal structures of chalcopyrite,34 bornite46 and pyrite47 were used for the quantitative phase analyses (Table 3).
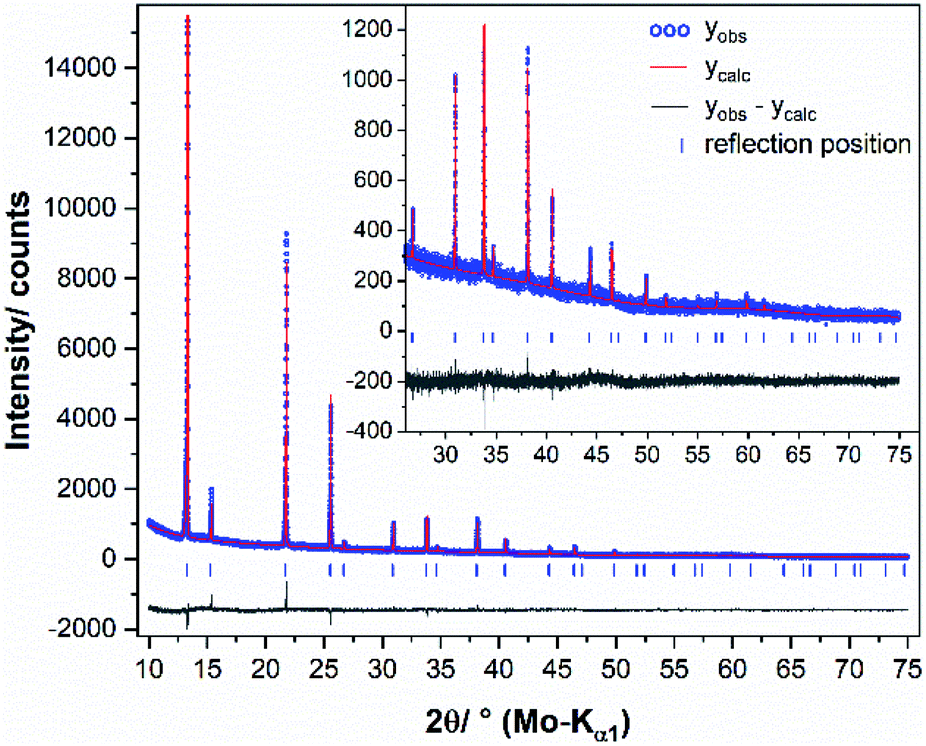 | ||
| Fig. 2 Graphical result of the final Rietveld refinement of chalcopyrite (synthesised with 49.50 at% S) at 545 °C. The high angle part starting at 25° 2θ is enlarged for clarity. | ||
57Fe Mößbauer spectra were taken on a spectrometer (WissEl GmbH) with movable 100 mCi 57Co source implemented in a Rh matrix and fixed sample holder. The analyses were carried out in constant acceleration mode, at ambient conditions and without an external magnetic field. Transmitted radiation was measured by a proportional counter. The collected spectra were analysed by WinNormos software using least-square fitting procedure assuming Lorentzian peak shapes. The obtained correlation coefficients were always above 0.95 indicating appropriate accuracy of the deconvolution. Isomer shift (ISO), quadrupole splitting (QUA) and hyperfine field (BHF) are reported relative to an α-Fe reference. Special fitting procedure for selected samples: due to very low amounts of side products next to chalcopyrite in some samples the Mößbauer spectra were fitted by fixing selected parameters (ESI, Table S1†). For the samples containing bornite as side product the WID, ISO and QUA parameters were fixed for this mineral. For orientation, the parameters from the results with synthetic bornite were used. This procedure was based on the phase composition obtained from XRPD. The 49.88 at% sample only shows minor contents of bornite and pyrite. Therefore, Mößbauer parameters of pyrite were also kept fixed. With samples containing talnakhite in addition to chalcopyrite, the talnakhite's WID was fixed. Mößbauer spectra of isocubanite containing samples were fitted by fixing the WID, ISO and QUA parameters for both doublets.
Results and discussion
Using Barton's35 phase diagram (Fig. 1), our attempts to synthesise single-phase chalcopyrite were based on the following assumptions:○ Chalcopyrite forms a stable phase below a certain temperature, which still needs to be determined, but is probably below 550 °C. Hence, we choose 450 °C for solid-state synthesis.
○ Chalcopyrite's crystallisation field extends to areas with a slight sulfur deficit. To determine the stability range of chalcopyrite with respect to sulfur content, all samples were prepared with an equimolar Cu:Fe ratio and varied metal:sulfur ratio. Sample names are referring to the sulfur content in at% used in the reactant mixture.
In the following, we discuss the results of XRPD and 57Fe-Mössbauer spectroscopy experiments with samples synthesised at 450 °C. Sulfur content was varied between 48.0 and 50.5 at%. Annealing times of at least 25 days were necessary to achieve full conversion, i.e. constant sample composition. Whilst XRPD allowed us to analyse the phase composition and conduct experiments at high temperature, there was limited accuracy in quantification of microcrystalline phases, especially in case of bornite. Additionally, quantitative XRPD analysis of isocubanite next to chalcopyrite and talnakhite was not possible, because reflections of isocubanite overlap with those of talnakhite. Therefore, we choose a second analysis method: Mößbauer spectroscopy was very sensitive to small amounts of impurities and allowed the quantification of all phases, although with limited accuracy.
A comparison of XRPD patterns for products synthesised with equimolar Cu:Fe ratio and sulfur content between 48.0 and 50.5 at% is shown in Fig. 3. Detailed Mößbauer parameters for these samples are listed in Table S1 in the ESI.†
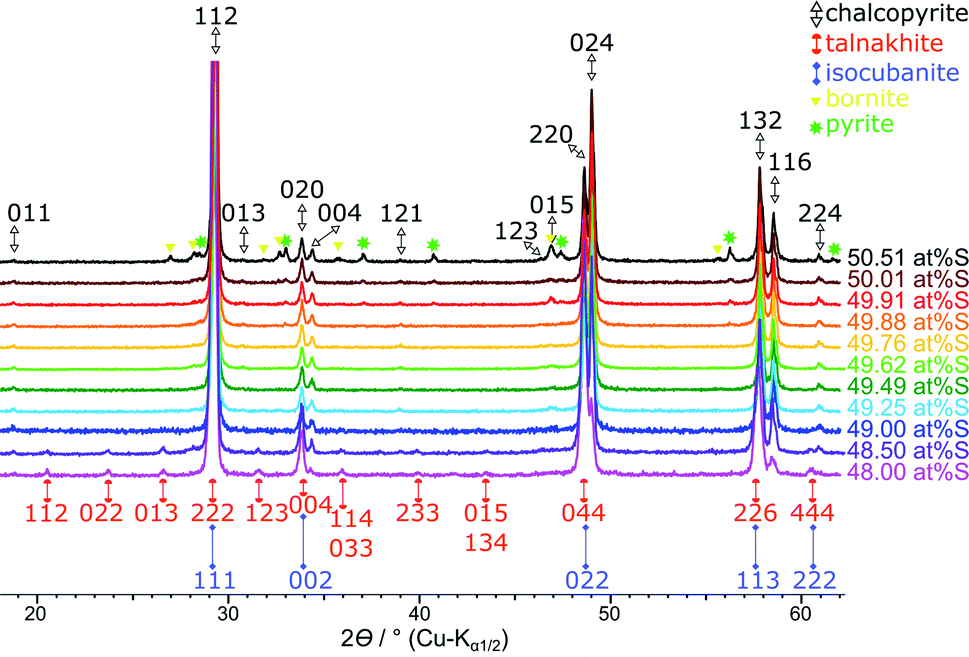 | ||
| Fig. 3 Stacked XRPD patterns of the products obtained when sulfur content was varied from 48.0 to 50.5 at% S. Reflections for the chalcopyrite cell are indexed in black from Hall and Stewart,34 the talnakhite cell indexed in orange from Hall and Gabe32 and the isocubanite cell indexed in violet from Szymanski.42 | ||
Synthesis of single-phase chalcopyrite
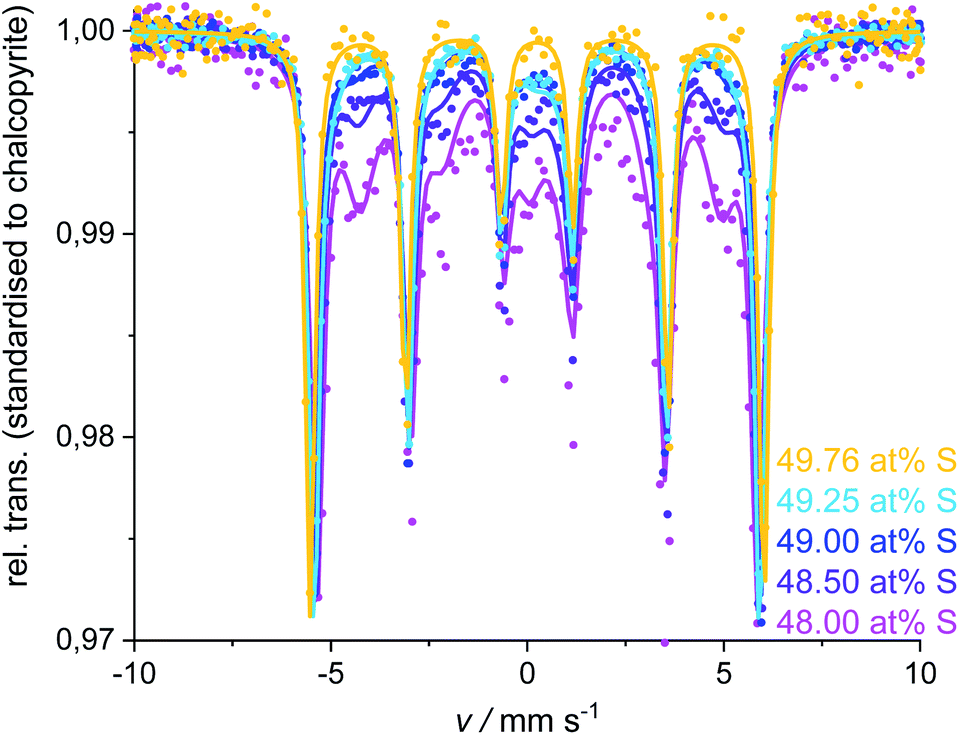
Single-phase chalcopyrite could only be obtained when a sub-stoichiometric amount around 49.7 at% S was used. This is evident from the XRPD data shown in Fig. 3 (49.62 and 49.76 at% S). Mößbauer spectroscopy was used to confirm the purity of these samples: data could be fitted with a single sextet at room temperature (Fig. 4, 49.76 at% S). Fitted parameters are consistent with literature data on chalcopyrite.48–51 Average parameters for chalcopyrite from fits of all samples synthesised with ≥49.49 at% S are shown in Table 2 and were typical for high-spin Fe3+ ions in an undistorted tetrahedral coordination.52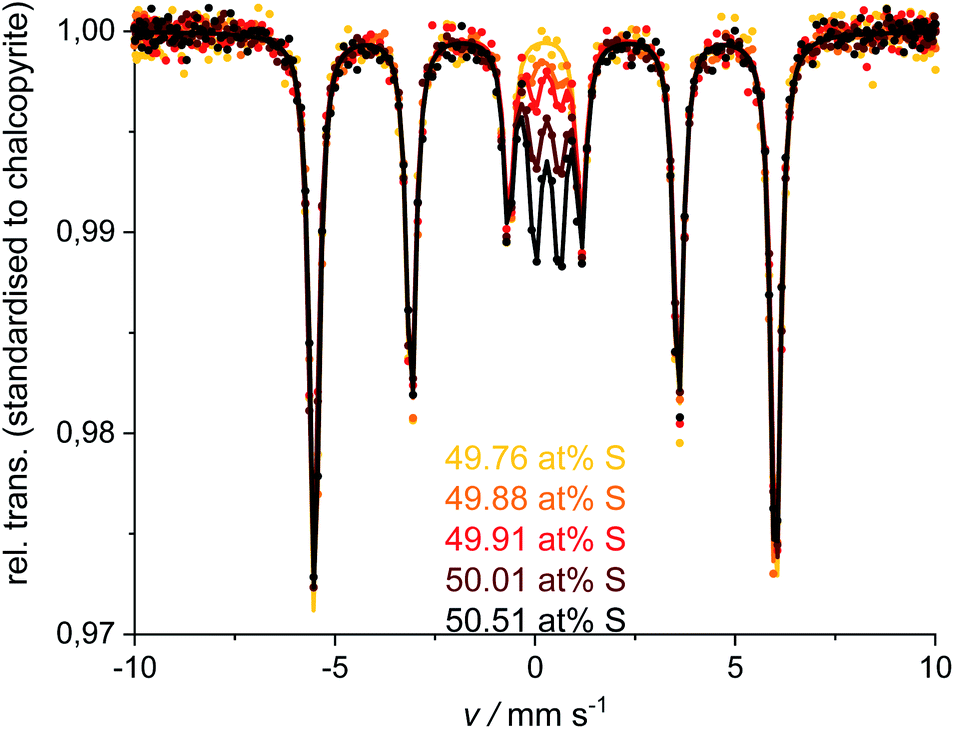 | ||
| Fig. 4 Mößbauer spectra and fits for the single-phase chalcopyrite (49.76 at% S) and measurements for the ≥49.88 at% S compositions. Spectra and fits were standardised to the chalcopyrite-sextet regarding the 49.76 at% S measurement. | ||
| WID/mm s−1 | ISO/mm s−1 | QUA/mm s−1 | BHF/T |
|---|---|---|---|
| 0.29(1) | 0.25(1) | 0.00(1) | 35.7(1) |
Products obtained with 49.5 at% S in the reactant mixture show no clear phase composition. Some products were single-phase material and some contained up to approx. 1.5 at% bornite. We suspect that at this composition the reaction is very sensitive to small amounts of oxide in the reactants but could not quantify this effect so far.
Phase composition with >49.76 at% S
XRPD and Mößbauer data show that a stoichiometric sulfur content (50 at%) does not lead to single-phase chalcopyrite. In samples with more than 49.76 at% S in the reactant mixture, XRPD patterns indicate small amounts of pyrite and bornite (Fig. 3). Mößbauer spectra of these samples exhibit an additional doublet (Fig. 4) which increases in intensity with sulfur content.Mößbauer spectroscopy has proven to be very sensitive to small amounts of impurities: for the 49.88 at% S composition, the amount of side products (especially bornite) is so small that its barely detectable with XRPD but is clearly observed in the Mößbauer spectrum. With this method, quantification of bornite and pyrite is possible in the presence of a dominant chalcopyrite phase, even though it results in a relatively high uncertainty due to the overlapping spectra of pyrite and bornite (Fig. 5, left). The Mößbauer spectrum for pyrite exhibits a doublet and the fitted parameters are in good agreement with literature data.53–56 The Mößbauer effect of bornite has been the subject of extensive discussions in the literature. While older sources have fitted spectra with a singlet, recent sources confirm that this signal is actually a doublet with very small quadrupole splitting.57–63 Our Mößbauer data are in good agreement with this literature (Fig. 5, right).
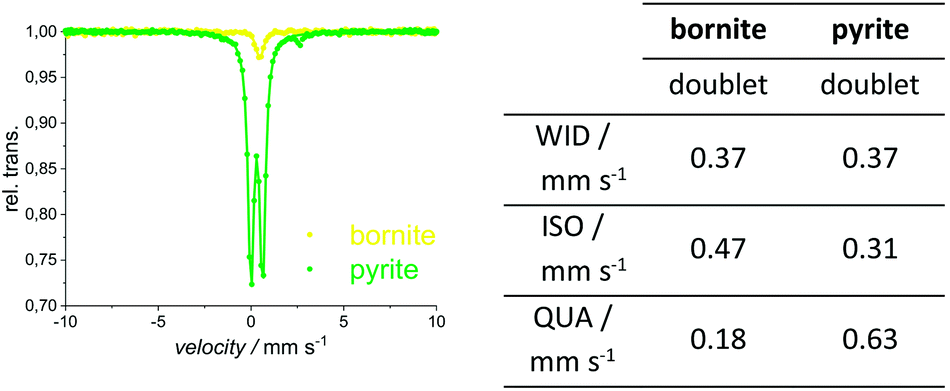 | ||
| Fig. 5 Mößbauer spectra (left) and fitted parameters (right) for the natural pyrite and synthetic bornite. More detailed values in ESI.† | ||
During reference measurements to calibrate the quantitative analysis of Mößbauer spectra with mixtures of natural pyrite, synthetic bornite and single-phase chalcopyrite, we observed a difference between the measured and the actual chalcopyrite content of less than 3 at%. Pyrite and bornite contents differed by approximately 9 and 6 at% respectively, where pyrite tends to be over-quantified, and bornite is rather under-quantified.
Table 3 shows the quantification of chalcopyrite, bornite and pyrite in samples synthesised with >49.76 at% S in the reactant mixture. In these samples, Mößbauer quantification results for the bornite content appears nearly constant within the experimental uncertainty (ESI, Table S1†). However, XRPD indicates clearly that both pyrite and bornite contents are increasing with sulfur content (Fig. 6). Results for the phase composition of Mößbauer and XRPD are in good agreement except, for those samples where pyrite and bornite reach the Mößbauer quantification limits. Samples synthesised from a stoichiometric reactant composition with respect to the chalcopyrite formula (i.e., 50 at% S) already contain less than 90 at% of the target mineral. Despite the high amount of side products, their reflections only became clearly visible in long-term XRPD measurements (Fig. 6). Considering the very intense and sharp reflections of chalcopyrite in XRPD, it is understandable that impurities may have been overlooked in previous research.
| at% S | Phase | Mößbauer | XRPD | |
|---|---|---|---|---|
| Sub-band | Portion/at% | Portion/at% | ||
| 49.88 | Chalcopyrite | Sextet | 96.2 | 98.2 |
| Pyrite | Doublet1 | 3.4 | 1.8 | |
| Bornite | Doublet2 | 0.4 | — | |
| 49.91 | Chalcopyrite | Sextet | 93.6 | 95.6 |
| Pyrite | Doublet1 | 6.2 | 3.5 | |
| Bornite | Doublet2 | 0.4 | 0.9 | |
| 50.01 | Chalcopyrite | Sextet | 86.5 | 89.9 |
| Pyrite | Doublet1 | 13.2 | 8.4 | |
| Bornite | Doublet2 | 0.3 | 1.7 | |
| 50.51 | Chalcopyrite | Sextet | 80.8 | 87.0 |
| Pyrite | Doublet1 | 17.8 | 11.1 | |
| Bornite | Doublet2 | 1.4 | 1.9 | |
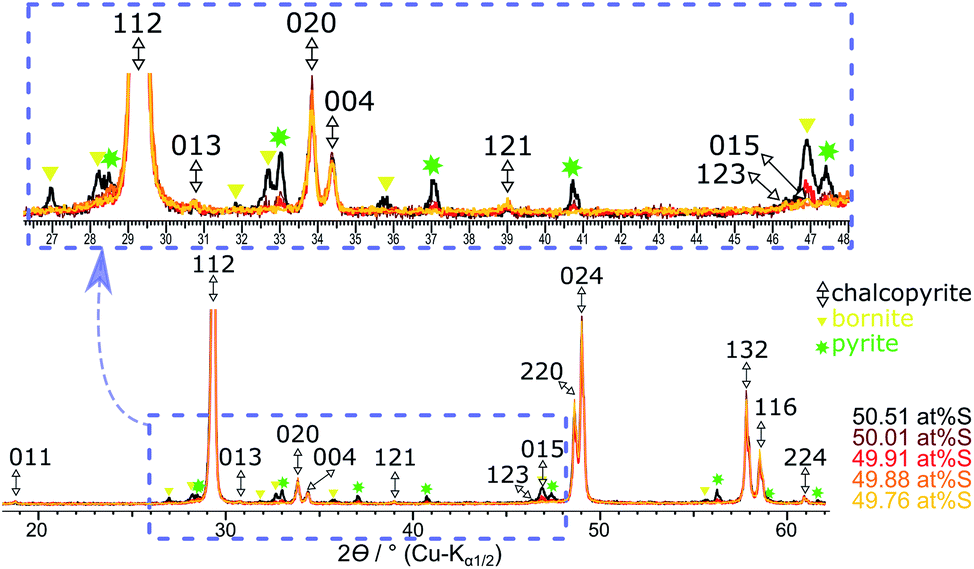 | ||
| Fig. 6 Overlaid XRPD patterns of the products obtained with a sulfur content from ≥49.76 at% S. Reflection patterns are standardised to the 112 reflection of chalcopyrite. | ||
Phase composition with <49.62 at% S
As discussed above, the phase purity for the 49.5 at% S samples is still questionable and may depend on trace amounts of oxide in the reactants. But, for all samples prepared with less than 49.5 at% sulfur in the reactant mixture, XRPD and Mößbauer spectroscopy clearly detected a small but constant amount of bornite (ca. 0.5 at%). The poor crystallinity or small crystal size of bornite in these samples results in a high uncertainty for XRPD quantification whilst Mößbauer spectroscopy also detects microcrystalline or amorphous phases. The intensity of bornite's doublet next to chalcopyrite's sextet is very small, due to the low iron content in bornite resulting in a relatively high uncertainty. Therefore, the quantification results for these samples should be used with care (ESI, Table S1†).The Mößbauer spectra for these samples could be fitted with several sub-bands (Fig. 7). Next to the chalcopyrite sextet, a second sextet can be identified as talnakhite. Mößbauer parameters (ESI, Table S1†) are in the range of literature data for talnakhite (ISO = 0.425 or 0.50 mm s−1, QUA = 0.0 mm s−1 and BHF = 35.5 or 35.3 T).51,64 Deviations from literature values can be explained by the broad splitting of talnakhite's sextet. Fits with only chalcopyrite, bornite and talnakhite for the spectra of these samples created high χ2 values (>8). Hence, there must be another iron-containing phase next to chalcopyrite and talnakhite. As we will discuss below, the residual can be explained with the presence of isocubanite in these samples. Isocubanite consists of tetrahedrally coordinated Fe2+ and Fe3+-ions. Hence, two doublets will be obtained in the Mößbauer spectra. Literature data for isocubanite Mößbauer parameters vary. Lyubutin et al.65 showed different models for possible fitting procedures. Due to the small amount of isocubanite in the synthesised samples, parameters for this phase could not be refined freely. Parameters were taken from Wintenberger et al.66 and kept fixed during the refinement. Including isocubanite into the Mößbauer fitting procedure resulted in a significant drop of the χ2 values (ESI, Table S1†).
 | ||
| Fig. 7 Mößbauer spectra and fits of the products obtained with ≤49.25 at% S in the reactant mixture. The 49.76 at% S sample is insert for comparison purposes. Spectra and fits were standardised to the chalcopyrite-sextet regarding the 49.76 at% S measurement. | ||
In agreement with Mößbauer results, XRPD clearly detects other phases next to chalcopyrite if less than 49.5 at% S is used in the reactant mixture (Fig. 8). Besides the reflections attributed to the target mineral remaining reflections can be assigned to talnakhite (Fig. 8, orange indices32). Mößbauer spectra showed, the products must contain more phases than only chalcopyrite and talnakhite. As XRPD does not show additional reflections, we believe that the remaining phase in these products must be isocubanite. The reflections of isocubanite coincide with the main reflections of talnakhite. Hence, the existence of isocubanite next to talnakhite could not be proven with XRPD but only with Mößbauer spectroscopy. Nevertheless, the appearance of the cubic phases can be clearly seen with XRPD, as evidence by a shift from chalcopyrite's 112 to talnakhite's 222 and isocubanite's 111 reflections around 29° 2θ (Cu-Kα1/2) with decreasing S contents (inset Fig. 8). The crystal structures of these compounds are closely related, which is also reflected in their lattice parameters and reflection positions. Pairs of reflections of chalcopyrite's tetragonal cell merge into a single reflection of the cubic cells of talnakhite and isocubanite: chalcopyrite's 020 and 004 to talnakhite's 004 and isocubanite's 002; 220 and 024 to 044 and 022; and 132 and 116 to 226 and 113. A reasonable XRPD quantification of a mixture of talnakhite, isocubanite and chalcopyrite is not possible, due to the direct overlap of the reflections of talnakhite and isocubanite. Therefore, the obtained quantities of the cubic phase are highly correlated and the result is in consequence meaningless. So, we do not show the XRPD quantification results for these sample compositions.
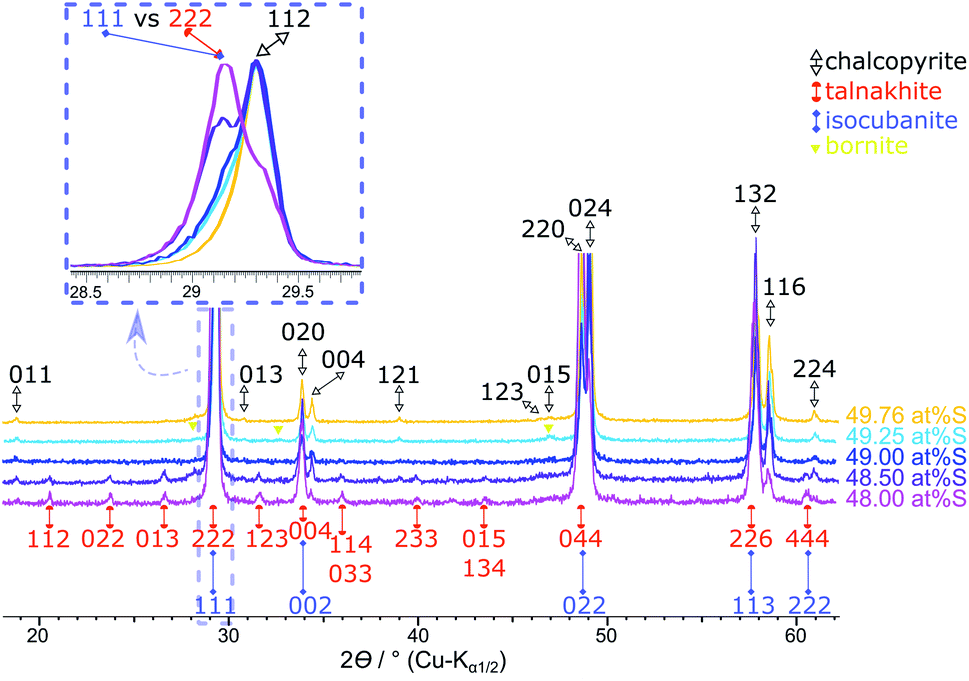 | ||
| Fig. 8 Results of the products obtained with <49.62 at% S. The 49.76 at% S sample is insert for comparison purposes. Stacked XRPD with zoomed inset (overlaid and standardised pattern) shows the shift of the main reflections of chalcopyrite 112 and talnakhite 222 with sulfur content. Reflections for the chalcopyrite cell are indexed in black from Hall and Stewart,34 for the talnakhite cell are indexed in orange from Hall and Gabe32 and the isocubanite cell are indexed in violet from Szymanski.42 | ||
Even though Mößbauer and XRPD quantifications have their specific drawbacks, a clear trend is evident from both methods: with decreasing sulfur content, the amount of cubic by-phases increases.
As the results of XRPD and Mößbauer analysis are consistent, it can be concluded that single-phase chalcopyrite can be synthesised at 450 °C using a slightly sulfur deficient reactant mixture around 49.7 at% and an annealing time of at least 25 days.
Phase transition temperature of chalcopyrite
We highlighted in the introduction that the temperature of the upper stability limit of chalcopyrite is still subject to discussion. It is further debated whether it undergoes a phase transition into the cubic iss or decomposition into iss and pyrite.We investigated the thermal stability of chalcopyrite by temperature dependent in situ XRPD measurements in sealed capillaries under an inert atmosphere and low heating rates (2 K min−1). Around the expected transition temperature, XRPD patterns were collected every 5 K.
Fig. 9 shows XRPD patterns recorded for single-phase chalcopyrite (49.76 at% S) between 450 °C and 540 °C. Due to the thermal expansion of the unit cell, a shift to lower diffraction angles is observed with increasing temperature. At 480 °C the intensity of the 112 chalcopyrite reflection decreases slightly. At 485 °C a phase change is indicated by the formation of a shoulder which could be identified as the 111 reflection of the cubic iss. The intensity of this shoulder increases with temperature. The transition from chalcopyrite to the iss can be described as an order–disorder phase transition with tetragonal chalcopyrite representing an ordered superstructure of cubic iss (sphalerite structure). Accordingly, pairs of reflections merge, e.g. 220 and 024 to 022, whilst small superstructure reflections disappear in the phase transition. The diffraction patterns of the cubic iss cells are identical with those of the cubic structure found by Engin et al.27 At 520 °C a second cubic phase can be detected by the formation of a shoulder next to the iss reflection pattern. This phase displays an identical diffraction pattern with slightly different cell parameter in comparison to the cubic iss. With ongoing phase transition, it seems that there is a phase separation in two variants of the iss, which differ in lattice constant and hence probably Fe:Cu ratio. As discussed above, the stability range of the iss extends over a large Cu:Fe:S ratio30 and, depending on the definition of iss, includes even the cubanite composition. Hence, even isocubanite could be regarded as a variant of the iss.30 Under the experimental conditions applied here, the transition was complete at 530 °C and chalcopyrite reflections were no longer visible. At 535 °C the distinct iss phases can be observed best in XRPD. At this temperature, lattice parameters of the iss phases were found to be a = 5.3281(13) and 5.3447(16) Å. The iss phases are stable up to at least 700 °C, which was the highest temperature we investigated in these experiments.
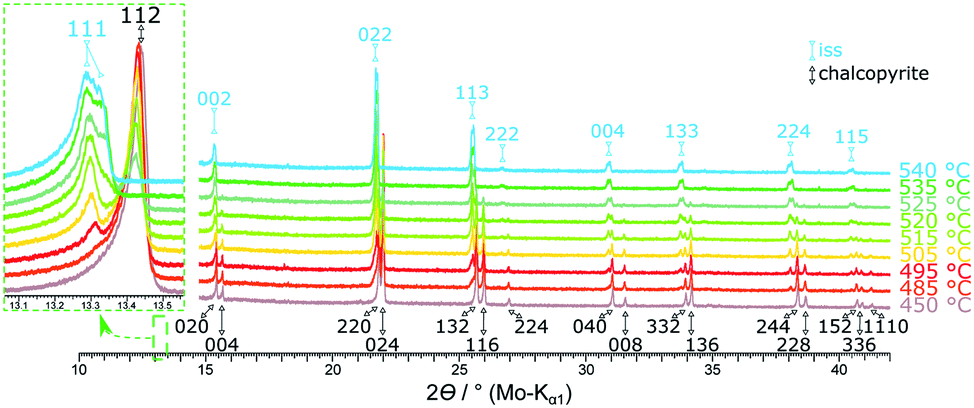 | ||
| Fig. 9 Stacked XRPD patterns of the 49.76 at% S composition at different temperatures. Reflections for the tetragonal chalcopyrite cell are indexed black,34 for the cubic high-temperature phase indexed in light blue.27 A more detailed version of this figure can be found in the ESI.† | ||
In contrast to the phase diagram (Fig. 1), we could not detect any pyrite in these measurements. This result agrees with the observation of Engin et al.27 and confirms the hypothesis that chalcopyrite undergoes an order–disorder phase transition and not a decomposition.
To assess the influence of minor amounts of impurities on the thermal stability of chalcopyrite, we repeated the above experiment with a chalcopyrite sample containing 1.2 at% bornite (49.50 at% S used in the reactant mixture). Here, the chalcopyrite phase transition started already at 475 °C and was completed at 540 °C. Fig. 10 shows a comparison of the XRPD patterns of the single-phase chalcopyrite and the impure chalcopyrite at 535 °C. At this temperature, the phase transition is complete in the single-phase sample but not in the sample containing bornite impurities. In this impure sample the phase transition resulted in only one cubic iss phase with a lattice parameter of a = 5.3269(9) Å. In contrast to the two co-existing iss phases described above, the composition, and in particular the Cu:Fe ratio, for this single iss phase is known. We hence refined the iss structure using data recorded with this sample at 545 °C. Crystallographic data are shown in Table S2 (ESI†) and have been submitted to the CCDC database (deposition number 2033752).
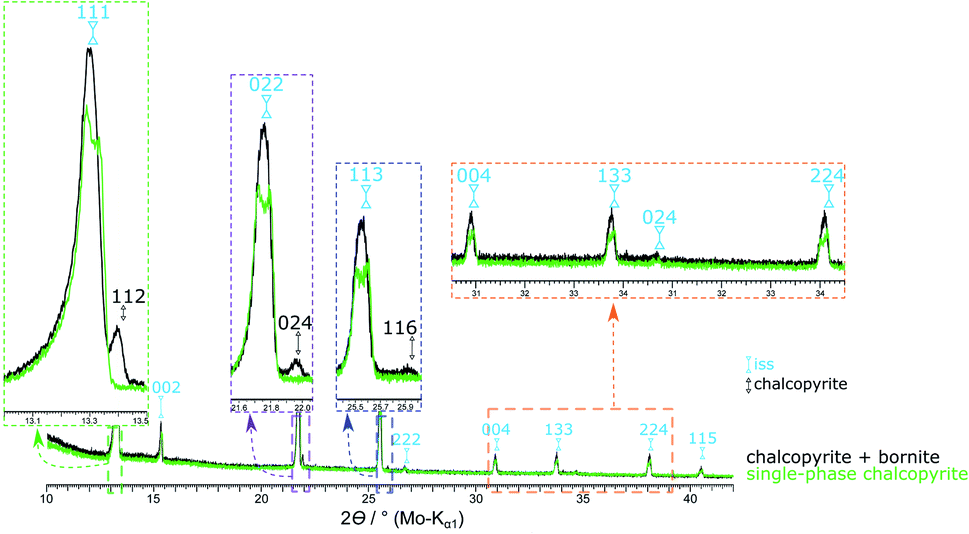 | ||
| Fig. 10 XRPD patterns of the single-phase chalcopyrite (49.76 at% S) and a chalcopyrite sample contaminated with 1.2 at% bornite (49.50 at% S) at 535 °C. At this temperature, the phase transition is not completed for the impure sample. By heating to 540 °C the chalcopyrite related reflections disappear (Fig. 2). | ||
Choice of temperature for chalcopyrite synthesis
To underline the importance of synthesising chalcopyrite at temperatures below the transition temperature, selected experiments were carried out at higher temperatures. Reactant mixtures annealed at 550 °C resulted in a significant increase in side products, e.g. in a sample synthesised with 49.5 at% S the amount of bornite was 2.3%. This could not be reversed by subsequent annealing at lower temperature: if the same mixture was heated to 700 °C followed by annealing at 450 °C, long-term XRPD and Mößbauer spectroscopy still indicated approximately 1.8 at% bornite next to chalcopyrite as the main phase.We hence conclude that during the synthesis of chalcopyrite from the elements, the phase transition temperature must not be exceeded at any time.
Conclusions
In our investigations, we could show that single-phase chalcopyrite can be synthesised from Cu, Fe and S by annealing at 450 °C in quartz glass ampules, using a slightly sub-stoichiometric amount of sulfur.X-ray powder diffraction and 57Fe-Mössbauer spectroscopy showed that small amounts of pyrite and bornite form next to chalcopyrite when >49.76 at% S is used in the reactant mixture. For samples with <49.62 at% S, small amounts of bornite and talnakhite are detected next to the target mineral. We could further show that, contrary to several literature sources, chalcopyrite does not decompose at its upper stability temperature. It rather undergoes an order–disorder phase transition starting at 485 °C and forming the intermediate solid solutions (iss). These phases crystallise in the sphalerite structure (space group F3m (216)) with a statistical disorder of Fe and Cu on the cation positions. Lattice constants observed here varied between a = 5.3269(9) and 5.3447(16) Å. Mößbauer spectroscopy confirmed the iron in chalcopyrite to be trivalent.
These results may help to disperse contradictions in the literature about the synthesis, phase behaviour and stability of chalcopyrite. By providing a facile and reliable synthetic route to an appropriate reference material, these results may further contribute to the elucidation of reaction mechanisms in hydrometallurgical chalcopyrite processing – a key step in making low-grade chalcopyrite deposits an economically viable source of copper.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Sächsische Aufbaubank (SAB) for financial support in the framework of the BioTechMetal project (Project-ID: 100316012). Funding for the Mößbauer spectrometer by the European Regional Development Fund, European Social Fund and Federal State of Saxony is gratefully acknowledged (Project-ID: 100323456). C. Mühle and C. Stefanie from the Max-Planck-Institute for Solid State Research are gratefully acknowledged for the assistance in XRPD sample preparation and measurement. Open Access Funding was provided by the Publication Fund of TU Bergakademie Freiberg.Notes and references
- H. R. Watling, Hydrometallurgy, 2014, 146, 96 CrossRef CAS.
- M. J. King, M. E. Schlesinger, W. G. I. Davenport and A. K. Biswas, Extractive Metallurgy of Copper, Elsevier Science Ltd, Oxford, 4th edn., 2002, pp. 289–302 Search PubMed.
- H. R. Watling, Hydrometallurgy, 2006, 84, 81 CrossRef CAS.
- Y. Li, N. Kawashima, J. Li, A. P. Chandra and A. R. Gerson, Adv. Colloid Interface Sci., 2013, 197–198, 1 CAS.
- D. Lu, W. Wang, Y. Chang, F. Xie and K. Jiang, Metals, 2016, 6, 303 CrossRef.
- M. Nicol, H. Miki and L. Velásquez-Yévenes, Hydrometallurgy, 2010, 103, 86 CrossRef CAS.
- N. Hiroyoshi, H. Miki, T. Hirajima and M. Tsunekawa, Hydrometallurgy, 2000, 57, 31 CrossRef CAS.
- N. Hiroyoshi, H. Miki, T. Hirajima and M. Tsunekawa, Hydrometallurgy, 2001, 60, 185 CrossRef CAS.
- J. E. Dutrizac, Metall. Trans. B, 1981, 12B, 371 CrossRef CAS.
- 5th Copper-Cobre International Conference, The Role of Non-oxidative Processes in the Leaching of Chalcopyrite,ed. M. J. Nicol and I. Lazaro, 2003 Search PubMed.
- E. L. Recalde Chiluiza and P. Navarro Donoso, J. Mex. Chem. Soc., 2016, 60, 238 Search PubMed.
- H. R. Watling, Hydrometallurgy, 2013, 140, 163 CrossRef CAS.
- A. Parker, C. Klauber, A. Kougianos, H. R. Watling and W. van Bronswijk, Hydrometallurgy, 2003, 71, 265 CrossRef CAS.
- O. G. Olvera, L. Quiroz, D. G. Dixon and E. Asselin, Electrochim. Acta, 2014, 127, 7 CrossRef CAS.
- H. Zhao, J. Wang, X. Gan, M. Hu, L. Tao, W. Qin and G. Qiu, Hydrometallurgy, 2016, 164, 159 CrossRef CAS.
- R. G. Acres, S. L. Harmer and D. A. Beattie, Int. J. Miner. Process., 2010, 94, 43 CrossRef CAS.
- T. Hidalgo, L. Kuhar, A. Beinlich and A. Putnis, Hydrometallurgy, 2019, 188, 140 CrossRef CAS.
- C. Yang, W. Qin, H. Zhao, J. Wang and X. Wang, Ind. Eng. Chem. Res., 2018, 57, 1733 CrossRef CAS.
- W. H. MacLean, L. J. Cabri and J. E. Gill, Can. J. Earth Sci., 1972, 9, 1305 CrossRef CAS.
- J. E. Hiller and K. Probsthain, Z. Kristallogr., 1956, 108, 108 CrossRef CAS.
- J. E. Dutrizac, Can. Mineral., 1976, 14, 172 Search PubMed.
- B. R. Conard, R. Sridhar and J. S. Warner, J. Chem. Thermodyn., 1980, 12, 817 CrossRef CAS.
- J. E. Dutrizac and R. J. C. MacDonald, Mater. Res. Bull., 1973, 8, 961 CrossRef CAS.
- L. B. Pankratz and E. G. King, United States Department of the Interior, Bureau of Mines; report of investigation, 1970, 7435, 1 Search PubMed.
- C. L. Herzenberg, Nuovo Cimento B, 1968, 53, 516 CrossRef.
- R. A. Yund and G. Kullerud, J. Petrol., 1966, 7, 454 CrossRef CAS.
- T. E. Engin, A. V. Powell and S. Hull, J. Solid State Chem., 2011, 184, 2272 CrossRef CAS.
- H. N. Ok and C. S. Kim, Nuovo Cimento B, 1975, 28, 138 CrossRef.
- H. Xie, X. Su, Y. Yan, W. Liu, L. Chen, J. Fu, J. Yang, C. Uher and X. Tang, NPG Asia Mater., 2017, 9, e390 CrossRef CAS.
- L. J. Cabri, Econ. Geol., 1973, 68, 443 CrossRef CAS.
- L. J. Cabri and S. R. Hall, Am. Mineral., 1972, 57, 689 CAS.
- S. R. Hall and E. J. Gabe, Am. Mineral., 1972, 57, 368 CAS.
- L. Pauling and L. O. Brockway, Z. Kristallogr. - Cryst. Mater., 1932, 82, 188 CAS.
- S. R. Hall and J. M. Stewart, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 579 CrossRef CAS.
- P. B. Barton, Econ. Geol., 1973, 68, 455 CrossRef CAS.
- B. V. Korzun, A. V. Pushkarev, A. L. Zhaludkevich, N. M. Olekhnovich, M. Rusu and M. C. Lux-Steiner, Phys. Status Solidi B, 2014, 251, 224 CrossRef CAS.
- H. E. Merwin and R. H. Lombard, Econ. Geol., 1937, 32, 203 CrossRef CAS.
- C. L. Burdick and J. H. Ellis, Proc. Natl. Acad. Sci. U. S. A., 1917, 3, 644 CrossRef CAS.
- M. E. Fleet, Z. Kristallogr. - Cryst. Mater., 1970, 132, 276 CrossRef CAS.
- M. J. Buerger, Am. Mineral., 1947, 32, 415 CAS.
- A. Putnis, Phys. Chem. Miner., 1977, 1, 335 CrossRef CAS.
- T. Szymański, Z. Kristallogr. - Cryst. Mater., 1974, 140, 240 Search PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210 CrossRef CAS.
- R. W. Cheary and A. Coelho, J. Appl. Crystallogr., 1992, 25, 109 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- K. Koto and N. Morimoto, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2268 CrossRef.
- S. L. Finklea, L. Cathey and E. L. Amma, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 529 CrossRef.
- E. Frank, Nuovo Cimento B, 1968, 58, 407 CrossRef CAS.
- C. I. Pearce, R. A. D. Pattrick, D. J. Vaughan, C. M. B. Henderson and G. van der Laan, Geochim. Cosmochim. Acta, 2006, 70, 4635 CrossRef CAS.
- N. N. Greenwood and H. J. Whitfield, J. Chem. Soc. A, 1968, 1697 RSC.
- L. J. Cabri and R. H. Goodman, Geochem. Int., 1970, 7, 453 Search PubMed.
- E. Kuzmann, S. Nagy and A. Vértes, Pure Appl. Chem., 2003, 75, 801 CAS.
- B. J. Evans, R. G. Johnson, F. E. Senftle, C. B. Cecil and F. Dulong, Geochim. Cosmochim. Acta, 1982, 46, 761 CrossRef CAS.
- P. A. Montano and M. S. Seehra, Solid State Commun., 1976, 20, 897 CrossRef CAS.
- A. A. Temperley and H. W. Lefevre, J. Phys. Chem. Solids, 1966, 27, 85 CrossRef CAS.
- J. A. Morice, L. V. C. Rees and D. T. Rickard, J. Inorg. Nucl. Chem., 1969, 31, 3797 CrossRef CAS.
- M. Borgheresi, F. Di Benedetto, M. Romanelli, M. Reissner, W. Lottermoser, R. R. Gainov, R. R. Khassanov, G. Tippelt, A. Giaccherini, L. Sorace, G. Montegrossi, R. Wagner and G. Amthauer, Phys. Chem. Miner., 2018, 45, 227 CrossRef CAS.
- M. G. Townsend, J. R. Gosselin, R. J. Tremblay, L. G. Ripley, D. W. Carson and W. B. Muir, J. Phys. Chem. Solids, 1977, 38, 1153 CrossRef CAS.
- D. J. Vaughan and J. R. Craig, Mineral chemistry of metal sulfides, Cambridge Univ. Press, Cambridge, 1978, p. 143 Search PubMed.
- M. S. Jagadeesh, H. M. Nagarathna, P. A. Montano and M. S. Seehra, Phys. Rev. B, 1981, 23, 2350 CrossRef CAS.
- M. F. Collins, G. Longworth and M. G. Townsend, Can. J. Phys., 1981, 59, 535 CrossRef CAS.
- H. N. Oak, K. S. Baek and Y. Jo, Solid State Commun., 1996, 100, 467 CrossRef CAS.
- R. R. Gainov, F. G. Vagizov, V. A. Golovanevskiy, V. A. Ksenofontov, G. Klingelhöfer, V. V. Klekovkina, T. G. Shumilova and I. N. Pen’kov, Hyperfine Interact., 2014, 226, 51 CrossRef CAS.
- M. G. Townsend, J. L. Horwood, S. R. Hall and L. J. Cabri, AIP Conf. Proc., 1972, 5, 887 Search PubMed.
- I. S. Lyubutin, C.-R. Lin, S. S. Starchikov, Y.-J. Siao and Y.-T. Tseng, J. Solid State Chem., 2015, 221, 184 CrossRef CAS.
- M. Wintenberger, G. André, C. Garcin, P. Imbert, G. Jéhanno, Y. Fouquet and A. Wafik, J. Magn. Magn. Mater., 1994, 132, 31 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2033752. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09700d |
| ‡ For example, a sample consisting of 24.5 at% Cu + 24.5 at% Fe + 51.0 at% S and annealed at 550 °C for 50 days with subsequent quenching in ice water, exhibited a thin sulfur layer on the entire inner surface of the ampoule. However, the phase diagram does not show elemental sulfur existing at this element ratio and temperature. |
| This journal is © The Royal Society of Chemistry 2021 |