
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing the steric hindrance around the catalytic core of a self-assembled imine-based non-heme iron catalyst for C–H oxidation†
Federico Frateloreto a,
Giorgio Capocasaa,
Giorgio Olivoa,
Karim Abdel Hadya,
Carla Sappinoa,
Marika Di Berto Mancinia,
Stefano Levi Morterab,
Osvaldo Lanzalungaa and
Stefano Di Stefano*a
a,
Giorgio Capocasaa,
Giorgio Olivoa,
Karim Abdel Hadya,
Carla Sappinoa,
Marika Di Berto Mancinia,
Stefano Levi Morterab,
Osvaldo Lanzalungaa and
Stefano Di Stefano*a
aDipartimento di Chimica and Istituto CNR per i Sistemi Biologici (ISB-CNR), Sezione Meccanismi di Reazione, Università di Roma La Sapienza, P. le A. Moro 5, 00185 Rome, Italy. E-mail: stefano.distefano@uniroma1.it
bArea of Genetics and Rare Diseases, Unit of Human Microbiome, Bambino Gesù Children's, Italy
First published on 24th December 2020
Abstract
Sterically hindered imine-based non-heme complexes 4 and 5 rapidly self-assemble in acetonitrile at 25 °C, when the corresponding building blocks are added in solution in the proper ratios. Such complexes are investigated as catalysts for the H2O2 oxidation of a series of substrates in order to ascertain the role and the importance of the ligand steric hindrance on the action of the catalytic core 1, previously shown to be an efficient catalyst for aliphatic and aromatic C–H bond oxidation. The study reveals a modest dependence of the output of the oxidation reactions on the presence of bulky substituents in the backbone of the catalyst, both in terms of activity and selectivity. This result supports a previously hypothesized catalytic mechanism, which is based on the hemi-lability of the metal complex. In the active form of the catalyst, one of the pyridine arms temporarily leaves the iron centre, freeing up a lot of room for the access of the substrate.
Introduction
C–H functionalization is a prominent topic in contemporary research due to the opportunity of a late stage derivatization of organic compounds. The chance of a direct installation of a heteroatom on non-activated positions has opened new synthetic routes and methodologies.1 In particular, our attention was ultimately oriented to the sustainable oxygenation of aliphatic and aromatic C–H bonds carried out with green terminal oxidants such as hydrogen peroxide (H2O2) in the presence of non-heme iron complexes2 based on the imine function.3,4 The choice of this kind of catalyst was guided by a series of needs: (i) use of an abundant and environmentally friendly metal, (ii) easy and (iii) cheap preparation of the catalyst.Complex 1, which can be assembled in acetonitrile by simple addition of the cheap and commercially available Fe(CF3SO3)2, 2-picolylaldehyde and 2-picolylamine added in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2 ratio, respectively (see Scheme 1), was demonstrated to satisfy the above requirements. In a number of reports it was shown that complex 1 competes on equal terms with many of the most popular amine-based non-heme iron catalysts5 appearing in the literature so far, both in the oxidation of aliphatic and aromatic C–H bonds.
:2:2 ratio, respectively (see Scheme 1), was demonstrated to satisfy the above requirements. In a number of reports it was shown that complex 1 competes on equal terms with many of the most popular amine-based non-heme iron catalysts5 appearing in the literature so far, both in the oxidation of aliphatic and aromatic C–H bonds.
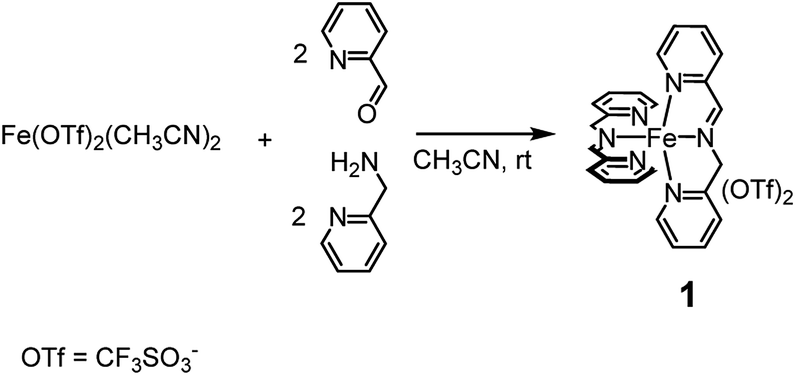 | ||
| Scheme 1 In situ preparation of complex 1 from cheap and commercially available precursors. Within 5 min from the addition of the precursors the complex is completely formed and the solution becomes deep violet. | ||
The reaction mechanism adopted by complex 1 in the H2O2 oxidation of both aliphatic and aromatic substrates was deeply studied2b,d but some features remain still poorly understood mostly due to the elusive character of the involved active species. The hemi-lability of at least one of the pyridine arms on the metal centre is believed to be essential for the iron activation of the H2O2, however, while the metal-based nature of the active species is given for certain, its precise structure is still unknown. An iron five oxo-complex N5FeV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O was hypothesized2b,d although an iron four (N5FeIVO) based pathway cannot be excluded.6
O was hypothesized2b,d although an iron four (N5FeIVO) based pathway cannot be excluded.6
In the recent literature supramolecular, non-covalent interactions have been employed in order to direct the selective oxidation of particular C–H bonds among many others present in the same substrate.7 In one case7a–c two crown-ether receptors were implanted in the ligand of a catalyst based on the Mn or Fe(pdp) core,1i,5b for the recognition of an ammonium anchoring group8 in the substrate (see Fig. 1, complex 2). Recognition of the ammonium head of the substrate by the crown-ether receptors allowed the selective oxidation of C–H bonds located at the right distance from the anchoring group (8–9 simple bonds). Application of the same strategy to the imine-based catalytic core 1 (see Fig. 1, complex 3), did not afford comparable results in terms of selectivity.9 In the latter case, the difference between the selectivity properties of complexes 1 and 3, devoid of and endowed with the crown-ether receptors, respectively, towards the oxidation of the C–H bonds present in the tested substrates was not evident enough, and when appreciable, it was ascribed to a mere steric hindrance rather than to recognition by the crown-ether moieties. In fact, steric hindrance around the catalyst is known to affect the reaction selectivity, favouring oxygenation of the most accessible sites (C–H or CC bonds) on the substrate.1k,10,11
 | ||
| Fig. 1 Catalysts endowed with crown-ether receptors for substrate decorated with ammonium head. Catalyst 2 is based on the M(pdp) catalytic core (amino-pyridine catalyst) while catalyst 3 on complex 1 (imino-pyridine catalyst). | ||
In order to shed light on the role and importance of steric hindrance on the action of catalyst 1 we investigated in detail the effect of the presence of bulky substituents in the catalyst structure. Here below we report the results of such investigation.
Results and discussion
Like complexes 1 and 3, new complexes 4 and 5, which are characterized by an increased steric hindrance around the catalytic core due to the presence of triisopropylsilyl (TIPS) groups, were prepared in situ by self-assembly of the parent compounds added in solution in the proper ratios (see Scheme 2), in acetonitrile at 25 °C.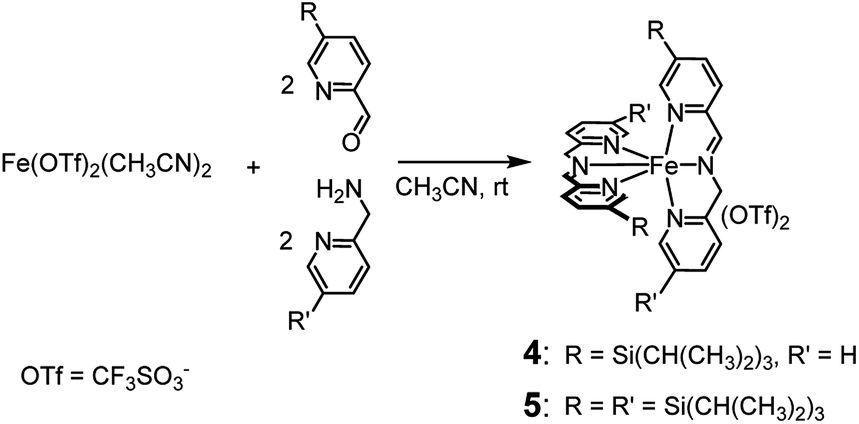 | ||
| Scheme 2 In situ preparation of complexes 4 and 5. Within 45 and 75 min, respectively, from the addition of the precursors, the complexes are completely formed and the solution becomes deep violet. | ||
1H NMR monitoring of the related solutions showed that complexes 4 (see ESI, Fig. S6†) and 5 (Fig. S18†) are completely formed within 45 min and 75 min, respectively, in contrast with complex 3, whose assembly requires 60 h at the same temperature. Formation of complex 1 under the same conditions is completed in 5 min.2a Characterization of the new complexes, based on 1H and 13C NMR monitoring, HSQC 2D NMR, HR-MS and UV-Vis spectroscopy, showed the high similarity of complexes 4 and 5 with the parent complex 1 (compare, for example, trace at t = 44 min in Fig. S6† with the 1H NMR spectrum of complex 1 reported in Fig. S1†). The UV-Vis spectra reported in Fig. S22† show the near resemblance among complexes 1, 4 and 5. Apart from a modest bathochromic effect due to the TIPS groups, the shape of the bands related to the iron core from 400 to 700 nm remains practically unchanged. Eventually, the 2:2:1 stoichiometry of the new complexes 4 and 5 is definitely demonstrated by the Job's plots reported in the ESI (Fig. S11 and S21,† respectively). Thus, the only difference among complexes 1, 4 and 5 should lie in the increasingly limited access to the metal centre.
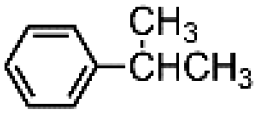
The results obtained in the H2O2 oxidation of a series of aromatic compounds carried out in the presence of complexes 4 and 5 are reported in Table 1 together with those obtained with catalyst 1 under the same conditions for the sake of comparison. In all cases hardly detectable trace amounts or no trace of products with oxidized lateral chain were found, in accordance with the well-known preference of this imine based catalytic core for aromatic C–H oxidation with respect to aliphatic C–H oxidation.2d
| Cat. | Substratec (recov.%) | orthoc (yield%) | meta + parac (yield%) | (m + p)/od |
|---|---|---|---|---|
| a Reaction conditions are: substrate 0.20 M, H2O2 2.5 mol eq. added in 30 min with a syringe pump, catalyst 2% mol, 25 °C, acetonitrile/H2O, 1 h 30 min total reaction time.b Yields from GC measurements calculated using nitrobenzene (0.5 mol eq.) as the internal standard.c Error is calculated from at least three independent runs.d Error from propagation applied to at least three independent runs. | ||||
 |
 |
 |
||
| 1 | 537 | 7.10.1 | 8.50.1 | 1.200.02 |
| 4 | 524 | 7.20.1 | 8.30.1 | 1.100.04 |
| 5 | 639 | 7.70.9 | 7.60.9 | 0.980.22 |
 |
 |
 |
||
| 1 | 699 | 8.10.9 | 162 | 20.5 |
| 4 | 649 | 7.40.3 | 131 | 1.80.3 |
| 5 | 743 | 7.40.1 | 111 | 1.50.1 |
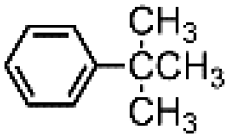 |
 |
 |
||
| 1 | 501 | 8.21 | 181 | 2.20.4 |
| 4 | 582 | 7.90.1 | 171 | 2.20.1 |
| 5 | 601 | 8.00.3 | 161 | 2.00.2 |
When catalyst 1 is taken into account, total yield of oxidation generally increases on increasing the size of the lateral alkyl chain of the substrate as previously observed.2d When complexes 4 and 5 are used as catalysts under the same conditions, quite astonishingly and in contrast with what was found with catalyst 3,9 the yields of phenol products remain more or less the same for each substrate in comparison with catalyst 1 in the limit of experimental errors. Even more surprising is the fact that the presence of two or four, very bulky TIPS groups in the catalyst backbone does not influence at any extent the (meta + para)/ortho ratio in the reaction products. Such ratio remains the same along each series 1, 4 and 5.
Although the exact nature of the active species involved in the reaction is still unknown, a series of clues collected in previous investigations2 prompted us to hypothesize a mechanism in which, after an initial outer-sphere oxidation of FeII to FeIII, the temporary detachment of one of the pyridine arms would unmask the iron centre, which could be available to take part to the oxidation process, generating an initial FeIII–OOH based species (Fig. S23†).
A possible evolution of the latter could be the heterolytic cleavage of the peroxidic bond leading to a FeVO species competent for the oxidative properties of the system.5a–d On the other hand, the homolytic cleavage of the same bond would originate an in-cage couple {FeIVO + HO˙}. The HO˙ radical would then attack the aromatic ring leading to a hydroxycyclohexadienyl radical, which in turn, would be oxidized by FeIVO to give the product upon deprotonation and to restore the FeIII species.6 However, whichever the active species is, many pieces of evidence have been collected pointing to a controlled oxidation mechanism where free radical oxidations do not play any role.2
The fact that in the series reported in Table 1 the (meta + para)/ortho selectivity remains practically untouched for each substrate no matter what the catalyst is, is a clear-cut proof of the absence of any steric interaction between the alkyl chain of the substrate and the TIPS groups mounted on the ligand. A little but appreciable substrate steric effect can be noted in the series ethylbenzene, cumene, tert-butylbenzene. Independently of the nature of the catalyst the (meta + para)/ortho ratio steadily increases along the series (1, 1.5, 2, respectively), however the access to the active core must be the same for all of three catalysts 1, 4 and 5.
A possible explanation for the above, quite unexpected results is that, although the catalytic core of 4 and particularly of 5 appears to be highly hindered in the close form (Fig. 2, left, CPK model of complex 5), when one of the pyridine arms is opened by H2O2 (see Fig. 2 right where a CPK model of the open FeVO putative active species is shown), the steric hindrance offered by the TIPS group decreases in such an extent as to become completely unimportant when the monosubstituted substrates reported in Table 1 are considered.
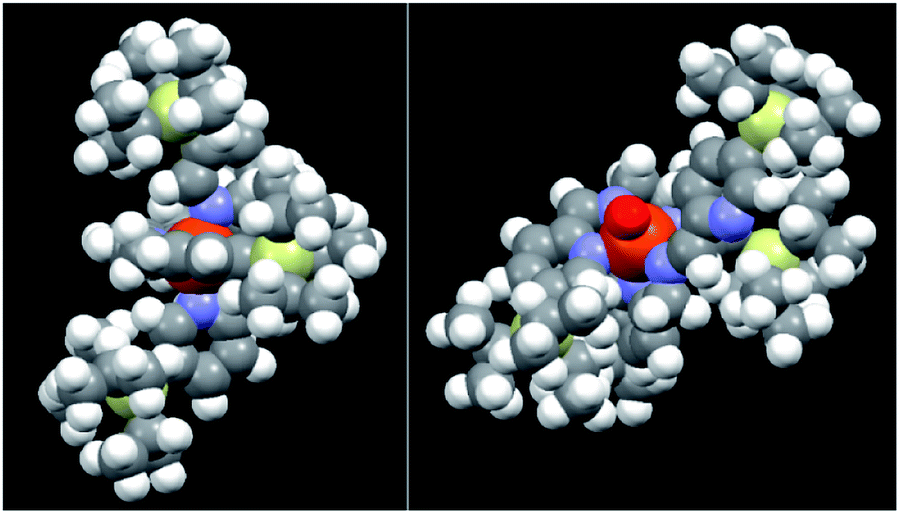 | ||
| Fig. 2 CPK models of complex 5 in its resting state (FeII, left) and in its putative active form (FeVO, right). | ||
In an attempt to exasperate the steric hindrance of the substrate, 1,3-di-tert-butylbenzene (Fig. 3) was employed in the study and oxidized in the presence of complex 1 and complex 5 which are the extreme limits of the series. In this case, with both catalysts phenol B was found to be the largely predominant product, with trace amounts of C and no trace of A. However, also in this case yield of phenol B, remains the same for catalysts 1 and 5, 7.0 ± 0.3% and 7.3 ± 0.1%, respectively. It has to be mentioned that under the same conditions but in the absence of any ligand (Fenton-like conditions), although the total yield was extremely low, also phenol A was found in the reaction crude.
 | ||
| Fig. 3 Oxidation of 1,3-di-tert-butylbenzene. The reported yields derive from GC analysis of the reaction crude after work-up and addition of nitrobenzene (0.5 mol eq.) as the internal standard. The oxidant was added over the course of 30 minutes, after which the mixture was allowed to react for 1 hour at 25 °C. | ||
Thus, to sum up, in the case of oxidation of simple aromatic substrates, we were not able to detect any effects due to the presence of the TIPS groups on the backbone of the catalyst.
Given that complex 1 is also known to be active in the catalysis of the oxidation of aliphatic C–H bonds,2a,b complexes 1 and 5 were also compared in the oxidation of menthyl acetate (see Fig. 4). Menthyl acetate is indeed often used as a benchmark substrate in order to evaluate steric effects on the regioselectivity of C–H oxidation. In both cases alcohols D and E were the major products. In the case of catalyst 1 total yield D + E was about 20% with a D/E ratio of 0.25. When catalyst 5 was used, total yield D + E was about 12% with a D/E ratio of 0.33. Thus, it is evident that for this substrate the steric effect of the TIPS groups is not unimportant and the D/E selectivity increases on increasing the steric hindrance on the catalyst, the peripheral isopropylic C–H bond being sterically more accessible than the tertiary endocyclic one.
 | ||
| Fig. 4 Oxidation of menthyl acetate. The reported yields are obtained by 1H NMR analysis of the reaction crude after work-up and addition of bibenzyl (0.5 mol eq.) as the internal standard. The oxidant was added over the course of 30 minutes, after which the mixture was allowed to react for 1 hour at 25 °C. | ||
Eventually, since complex 1 was proved to be an active catalyst also for alcohol oxidation2c (although this reaction cannot be properly considered a C–H oxidation but more properly an oxidation, from a steric standpoint it can serve to our purpose), we tested complexes 1 and 5 in the oxidation of cyclohexanol and its more sterically congested analogue 2,6-dimethylcyclohexanol (cis + trans mixture, see Fig. 5) to cyclohexanone and 2,6-dimethylcyclohexanone F, respectively.
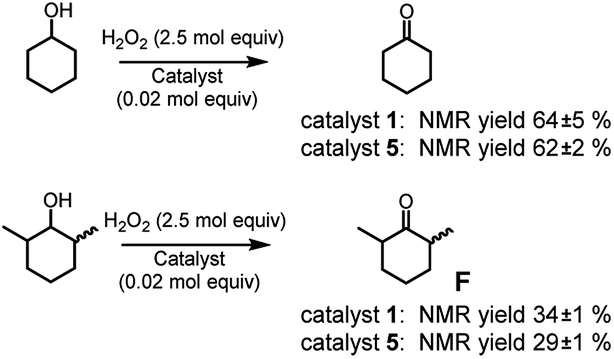 | ||
| Fig. 5 Oxidation of cyclohexanol and 2,6-dimethylcyclohexanol. The reported yields are obtained by 1H NMR analysis of the reaction crude after work-up and addition of bibenzyl (0.5 mol eq.) as the internal standard. The oxidant was added over the course of 30 minutes, after which the mixture was allowed to react for 1 hour at 25 °C. | ||
While no difference appears in the oxidation of cyclohexanol, complex 5 seems to be less active in the oxidation of 2,6-dimethylcyclohexanol with respect to complex 1. The effect is modest, but the origin of such difference has to be steric in nature. It should be noted that catalyst 1 was already found to be quite sensitive to steric effects in the oxidation of α-(poly)substituted cyclohexanols.2c Thus, the increased volume of complex 5, seems to moderately enhance the catalyst sensitivity to the steric bulk of the substrate.
Conclusions
In this report the synthesis of new complexes 4 and 5 is described. Self-assembly of both complexes is a relatively fast process taking place within 45 and 75 min, respectively, in CH3CN at 25 °C when the starting building blocks are added in solution in the proper ratios. Complexes 4 and 5 essentially differ from the parent complex for an increased steric hindrance around the metal ion.When complexes 4 and 5 were tested as catalysts for the H2O2 oxidation of a series of monoalkyl-substituted aromatic compounds, their catalytic activity was found to be nearly superimposable to that of the parent complex 1, within the experimental error. This behaviour was ascribed to a strong decrease of the steric hindrance when passing from the resting state to the active state of the catalyst, where one of the pyridine arms has temporarily left the iron centre, freeing up space for the access of the substrate. The absence of any steric effect was also found in the oxidation of a disubstituted aromatic substrate like 1,3-di-tert-butylbenzene.
Although moderate, appreciable differences between the efficiencies of catalysts 1 and 5 in term of yields and selectivity instead arise when substrates endowed with hindering groups around non-aromatic C–H oxidizable bonds are considered.
In conclusion it could be stressed that the insertion of large and hindering groups in the catalyst backbone does not necessary lead to the insurgence of steric effect on the catalysed reaction. The data collected in this report reinforce the mechanistic hypothesis depicted in Fig. S23,† remarking the importance of the hemi-lability of the complex in the explication of the catalytic action.
Experimental
Methods and material
GC analyses were carried out on a Varian CP-3800 gas chromatograph equipped with a capillary methylsilicone column (30 m × 0.25 mm × 25 μm) Chrompack CP-Sil 5 CB. NMR spectra were recorded on a Bruker AC300 (300 MHz) spectrometer and were internally referenced to the residual proton solvent signal. HR-MS were acquired with a Thermo Scientific Orbitrap Fusion Mass Spectrometer equipped with an ESI ion source. UV analyses were carried out with HP HEWLETT PACKARD 8453 spectrophotometer. All reagents and solvents were purchased at the highest commercial quality and were used without further purification unless otherwise stated. Fe(CH3CN)2(OTf)2 (−OTf = CF3SO3−) was prepared according to a literature procedure12 from Fe(II) chloride (Sigma Aldrich). Solvents were purchased from Sigma Aldrich and used as received.5-Triisopropylsilyl-pyridine-2-carboxaldehyde11,13 and 5-triisopropylsilyl-2-picolylamine11b were prepared according to literature procedure and spectral data were in accordance with those reported. Menthyl acetate14 and 2,6-dimethylcyclohexanol15 were prepared according to literature procedures and spectral data were in accordance with those reported.
Preparation (self-assembly) of complexes (1.0 × 10−2 M stock solutions in CH3CN)
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. D. Chiappini, J. B. C. Mack and J. Du Bois, Angew. Chem., Int. Ed., 2018, 57, 4956–4959 CrossRef CAS; (b) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. a. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS; (c) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS; (d) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230–234 CrossRef CAS; (e) M. M. Vita and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 5290–5292 CrossRef CAS; (f) H. Dai, A. F. Stepan, M. S. Plummer, Y. Zhang and J. Yu, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef CAS; (g) J. Borgel and T. Ritter, Chem, 2020, 6, 1–11 CrossRef; (h) W. K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef; (i) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS; (j) M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS; (k) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef CAS; (l) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS; (m) A. F. M. Noisier, M. J. Johansson, L. Knerr, M. A. Hayes, W. J. Drury, E. Valeur, L. R. Malins and R. Gopalakrishnan, Angew. Chem., Int. Ed., 2019, 52, 19096–19102 CrossRef; (n) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS; (o) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (p) L. J. Durak, J. T. Payne and J. C. Lewis, ACS Catal., 2016, 6, 1451–1454 CrossRef CAS; (q) R. Fasan, ACS Catal., 2012, 2, 647–666 CrossRef CAS.
- (a) G. Olivo, G. Arancio, L. Mandolini, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2014, 4, 2900–2904 RSC; (b) G. Olivo, M. Nardi, D. Vìdal, A. Barbieri, A. Lapi, L. Gómez, O. Lanzalunga, M. Costas and S. Di Stefano, Inorg. Chem., 2015, 54, 10141–10152 CrossRef CAS; (c) G. Olivo, S. Giosia, A. Barbieri, O. Lanzalunga and S. Di Stefano, Org. Biomol. Chem., 2016, 14, 10630–10635 RSC; (d) G. Capocasa, G. Olivo, A. Barbieri, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2017, 7, 5677–5686 RSC; (e) B. Ticconi, A. Colcerasa, S. Di Stefano, O. Lanzalunga, A. Lapi, M. Mazzonna and G. Olivo, RSC Adv., 2018, 8, 19144–19151 RSC; (f) B. Ticconi, G. Capocasa, A. Cerrato, S. Di Stefano, A. Lapi, B. Marincioni, G. Olivo and O. Lanzalunga, Catal. Sci. Technol., 10.1039/d0cy01868f.
- (a) M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. Di Stefano, Chem. Sci., 2013, 4, 2253–2261 RSC; (b) M. Ciaccia, S. Pilati, R. Cacciapaglia, L. Mandolini and S. Di Stefano, Org. Biomol. Chem., 2014, 12, 3282–3287 RSC; (c) M. Ciaccia and S. Di Stefano, Org. Biomol. Chem., 2015, 13, 646–654 RSC.
- G. Olivo, O. Lanzalunga and S. Di Stefano, Adv. Synth. Catal., 2016, 358, 843–863 CrossRef CAS.
- (a) L. Vicens, G. Olivo and M. Costas, ACS Catal., 2020, 10, 8611–8631 CrossRef CAS; (b) R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Molecules, 2016, 21, 1454 CrossRef; (c) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS; (d) G. Olivo, O. Lanzalunga, L. Mandolini and S. Di Stefano, J. Org. Chem., 2013, 78, 11508–11512 CrossRef CAS; (e) I. Prat, L. Gómez, M. Canta, X. Ribas and M. Costas, Chem.–Eur. J., 2012, 19, 1908–1913 CrossRef; (f) M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS; (g) P. Shejwalkar, N. P. Rath and E. B. Bauer, Dalton Trans., 2011, 40, 7617–7631 RSC; (h) M. Canta, D. Font, L. Gómez, X. Ribas and M. Costas, Adv. Synth. Catal., 2014, 365, 818–830 CrossRef; (i) L. Gómez, I. Garcia-Bosch, A. Company, J. Bernet-Buchholz, A. Polo, X. Sala, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2009, 48, 5720–5723 CrossRef; (j) S. Jana, M. Ghosh, M. Ambule and S. S. Gupta, Org. Lett., 2017, 19, 746–749 CrossRef CAS; (k) A. Raba, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Commun., 2014, 50, 11454–11457 RSC; (l) P. Liu, Y. Liu, E. L.-M. Wong, S. Xiang and C.-M. Che, Chem. Sci., 2011, 2, 2187–2195 RSC; (m) E. V. Kudrik and A. B. Sorokin, J. Mol. Catal. A: Chem., 2017, 426, 499–505 CrossRef CAS; (n) L. Carneiro and A. R. Silva, Catal. Sci. Technol., 2016, 6, 8166–8176 RSC; (o) O. Y. Lyakin, A. M. Zima, N. V. Tkachenko, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2018, 8, 5255–5260 CrossRef CAS.
- (a) A. Thibon, V. Jollet, C. Ribal, K. Sénéchal-David, L. Billon, A. B. Sorokin and F. Banse, Chem.–Eur. J., 2012, 18, 2715–2724 CrossRef CAS; (b) J.-N. Rebilly, W. Zhang, C. Herrero, H. Dridi, K. Sénéchal-David, R. Guillot and F. Banse, Chem.–Eur. J., 2020, 26, 659–668 CrossRef CAS.
- (a) G. Olivo, G. Farinelli, A. Barbieri, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2017, 56, 16347–16351 CrossRef CAS; (b) G. Olivo, G. Capocasa, O. Lanzalunga, B. Ticconi, S. Di Stefano and M. Costas, Chem. Commun., 2019, 55, 917–920 RSC; (c) G. Olivo, G. Capocasa, B. Ticconi, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2020, 59, 12703–12708 CrossRef CAS; (d) M. Knezevic, M. Heilmann, G. M. Piccini and K. Tiefenbacher, Angew. Chem., Int. Ed., 2020, 59, 12387–12391 CrossRef CAS; (e) D. Vidal, G. Olivo and M. Costas, Chem.–Eur. J., 2018, 24, 5042–5054 CrossRef CAS.
- S. Di Stefano, G. Capocasa and L. Mandolini, Eur. J. Org. Chem., 2020, 23, 3340–3350 CrossRef.
- G. Capocasa, M. Di Berto Mancini, F. Frateloreto, O. Lanzalunga, G. Olivo and S. Di Stefano, Eur. J. Org. Chem., 2020, 23, 3390–3397 CrossRef.
- (a) M. Milan, M. Costas, M. Otte and R. J. M. Klein Gebbink, Adv. Synth. Catal., 2017, 359, 2590–2595 CrossRef; (b) V. Dantignana, M. Milan, O. Cussó, A. Company, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 1350–1358 CrossRef CAS; (c) J. Zhao, T. Nanjo, E. C. de Lucca, Jr and M. C. White, Nat. Chem., 2019, 11, 213–221 CrossRef CAS.
- (a) D. Font, M. Canta, M. Milan, O. Cussò, X. Ribas, R. J. M. Klein Gebbink and M. Costas, Angew. Chem., Int. Ed., 2016, 55, 5776–5779 CrossRef CAS; (b) M. Borrell and M. Costas, J. Am. Chem. Soc., 2017, 139, 12821–12829 CrossRef CAS.
- A. Diebold, A. Elbouadili and K. S. Hagen, Inorg. Chem., 2000, 39, 3915–3923 CrossRef CAS.
- O. Cussó, M. Cianfanelli, X. Ribas, R. J. M. Klein Gebbink and M. Costas, J. Am. Chem. Soc., 2016, 138, 2732–2738 CrossRef.
- J. M. Álvarez-Calero, Z. D. Jorge and G. M. Massanet, Org. Lett., 2016, 18, 6344–6347 CrossRef.
- L. Chowdhury, C. J. Croft, S. Goel, N. Zaman, A. C. S. Tai, E. M. Walch, K. Smith, A. Page, K. M. Shea, C. D. Hall, D. Jishkariani, G. G. Pillai and A. C. Hall, J. Pharmacol. Exp. Ther., 2016, 357, 570–579 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full characterization of complexes 4 and 5. See DOI: 10.1039/d0ra09677f |
| ‡ Signals at 17.7, 17.4 and 17.3 ppm for 4 and at 17.6, 17.3 and 17.2 for 5, belong to the CH3 on the triisopropylsilyl groups as it can be inferred from the HSQC experiment (see ESI†). The existence of three distinct signals is very likely a consequence of the slowdown of the rotation around the Si–C bond due the increased steric hindrance upon complex formation. |
| This journal is © The Royal Society of Chemistry 2021 |