
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly economical and direct amination of sp3 carbon using low-cost nickel pincer catalyst†
Andrew Brandt‡
a,
Ambar B. RanguMagar‡a,
Peter Szwedoa,
Hunter A. Waylanda,
Charlette M. Parnella,
Pradip Munshi *b and
Anindya Ghosh*a
*b and
Anindya Ghosh*a
aDepartment of Chemistry, University of Arkansas at Little Rock, 2801 South University Avenue, Little Rock, AR 72204, USA. E-mail: axghosh@ualr.edu; Fax: +1 501 569 8838; Tel: +1 501 569 8827
bResearch Center, Reliance Industries Limited, Vadodara, Gujarat 391346, India. E-mail: pradip.munshi@ril.com; Tel: +91 265 261 6066
First published on 6th January 2021
Abstract
Developing more efficient routes to achieve C–N bond coupling is of great importance to industries ranging from products in pharmaceuticals and fertilizers to biomedical technologies and next-generation electroactive materials. Over the past decade, improvements in catalyst design have moved synthesis away from expensive metals to newer inexpensive C–N cross-coupling approaches via direct amine alkylation. For the first time, we report the use of an amide-based nickel pincer catalyst (1) for direct alkylation of amines via activation of sp3 C–H bonds. The reaction was accomplished using a 0.2 mol% catalyst and no additional activating agents other than the base. Upon optimization, it was determined that the ideal reaction conditions involved solvent dimethyl sulfoxide at 110 °C for 3 h. The catalyst demonstrated excellent reactivity in the formation of various imines, intramolecularly cyclized amines, and substituted amines with a turnover number (TON) as high as 183. Depending on the base used for the reaction and the starting amines, the catalyst demonstrated high selectivity towards the product formation. The exploration into the mechanism and kinetics of the reaction pathway suggested the C–H activation as the rate-limiting step, with the reaction second-order overall, holding first-order behavior towards the catalyst and toluene substrate.
Introduction
The great prevalence of carbon–nitrogen (C–N) frameworks in biologically active natural products, pharmaceuticals, and electroactive materials1–4 places a high value on streamlined synthetic processes that can produce these compounds reliably on a commercial scale with minimal wastage. The desire for amine compounds as drug molecules and the versatility of functionalization make it more attractive in synthetic chemistry.5–7 Amine compounds can also be made very flexible to adopt in polar, non-polar, or in aqueous media through quaternization or by introducing structural features, which has taken a particular interest to prodrug synthesis8–10 in order to overcome blood–brain barriers or other transport challenges. Strong nucleophilic capabilities and efficient binding strengths of heterogenic amines drive major biological processes and are a part of synthetic organic, inorganic, and medicinal chemistry.11,12 Thus a high demand exists for amine derivatives, and to uncover the hidden mysteries of the synthetic fallacies involving trivalent nitrogen have recently directed remarkable developments in the catalytic routes of preparation of amine compounds.13,14 Still, the synthesis of amines leaves tremendous opportunities to rejuvenate synthetic methodologies for a more straightforward, environmentally, and economically admirable approach for large-scale industrial preparation.Commonly available methods are multistep and involve ammoxidation, reduction, or the use of highly reactive and costly reagents and large amounts of catalyst, making these processes highly uneconomic. Ullmann reported a C–N coupling reaction by carbon–hydrogen (C–H) activation for the first time in the early 1900s using stoichiometric amounts of copper salt for coupling aryl halides with aryl amines in refluxing aniline to give diarylamines.15 Later, Buchwald and Hartwig led explorations for the full scope of C–N couplings utilizing palladium, nickel, and copper catalysts.16–19 Over the decade to follow, the teams succeeded in synthesizing even previously difficult to produce arylamine using palladium-based phosphine catalysts20,21 leading to research targeting novel amination approaches ranging from microwave-assisted methods to synergistic multi-ligand catalyst systems and also the use of ammonia as a nitrogen substrate.22–24 N-Alkylation of alcohol in supercritical carbon dioxide (CO2) using Pd nanoparticles was also reported via cascade oxidation, condensation, and reduction steps.25
We report here (Scheme 1), for the first time, direct alkylation of aliphatic amines via activation of sp3 C–H bond of pure hydrocarbons, e.g., toluene, producing secondary, tertiary, cyclic and unsaturated amines using only 0.2 mol% of an amide-based nickel pincer type catalyst (1) in the presence of base with quantitative yields reported. With 100% atom economy and an 0.62 E factor,26,27 the present discovery brings a paradigm change in amine synthesis, inviting huge promises for large scale industrial preparation, the dream of industrial chemists.
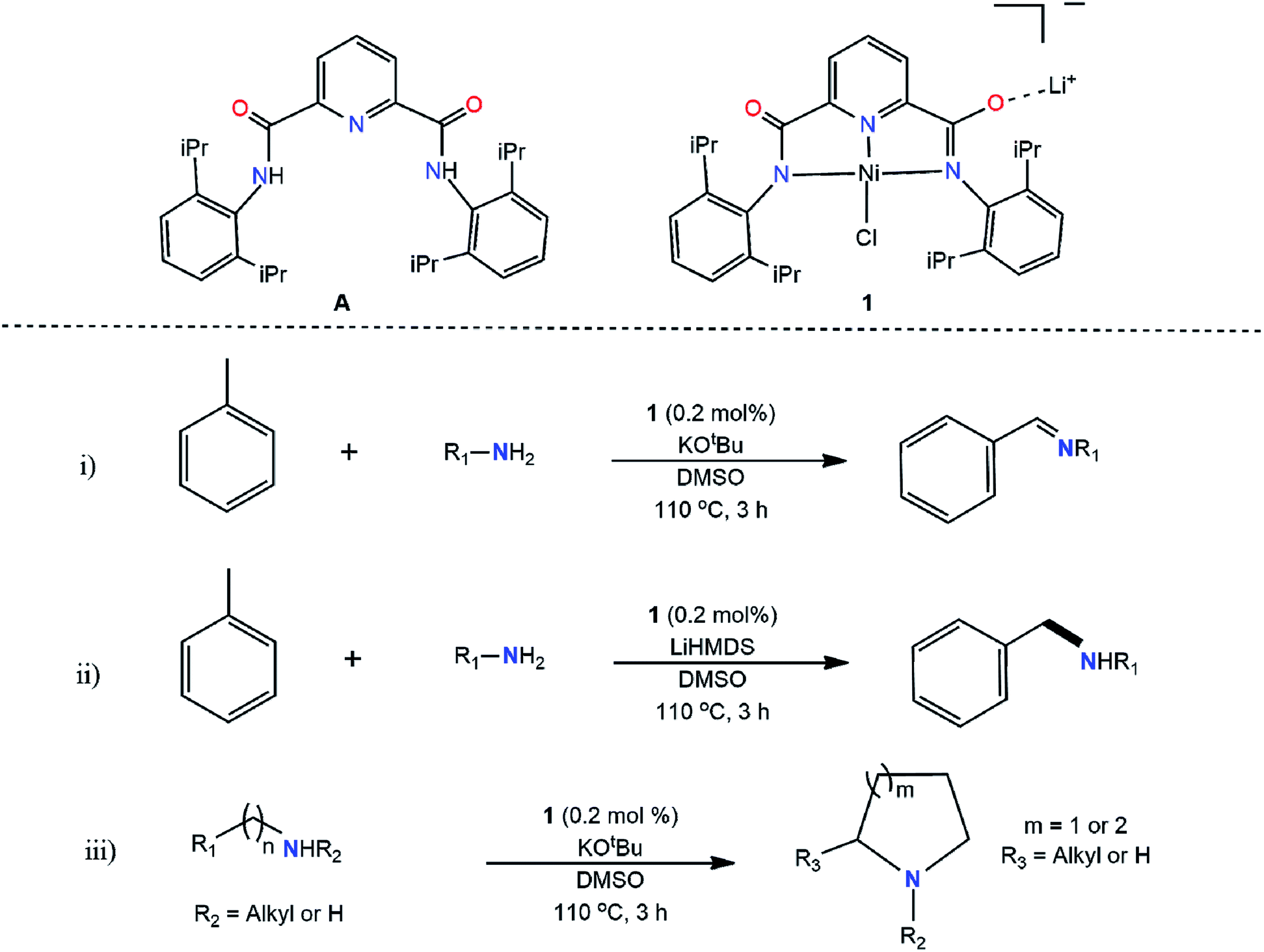 | ||
| Scheme 1 Structures of ligand (A) and catalyst (1) and C–N functionalization reactions: (i) toluene with amines to form imine products. (ii) toluene with amines to form amine products. (iii) intramolecular cyclization of amines to develop the cyclic products. | ||
First-row transition metals such as manganese, cobalt, iron, and nickel have been investigated for catalyzing various C–N bond-forming reactions, and in many instances, have been found suitable;28–32 however, pincer catalysts have drawn special interest due to their superiority of binding by satisfying the metal electronic environment.33–38 Pincer-type catalysis progressed from phosphine-based to nitrogen-incorporated structures with less expensive and commercially viable transitional metals instead of expensive transition metals.39–43 Closely resembling nickel and iron coordinated pincer catalysts have been previously investigated for C–C coupling of Grignard reagents,44–46 but direct C–N coupling arising from activation of sp3 C–H hydrocarbon and primary amine has never been investigated. Thus, the potential of the pincer catalyst is furthered in the present work opening a new area of application in C–N bond formation.
The C–H bonds are very stable by nature. While biological life has evolved processes for selective activation using moieties such as in cytochrome p450, it can be challenging to mimic this activity synthetically without using high catalyst loads and accessory oxidants.47,48 In comparison to traditional approaches, alkylation of nitrobenzene with toluene moieties has been reported using a heterogeneous cobalt catalyst in the presence of peroxide and hydrogen.49 Aziridine based amination of toluene via activation of toluene sp3 carbon through H-atom transfer process using an iron(II) complex has been reported.50 Furthermore, investigations for a novel metal-free oxidative amination of benzylic C–H bonds using benzotriazole and nBu4NI have been demonstrated.51 However, these processes are mostly reagent assisted and thus differ from this present work of direct amination. The present finding of alkylation of amines via C–H activation has been developed as a direct coupling method that overcomes challenges such as poor atom economy, the necessity of pre-functionalization, and requisite of harsh conditions associated with the traditional alkyl halide method.52–54
Results and discussions
Optimization of reaction conditions: solvent and base
To begin exploring C–N or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N cross-coupling using 1, various solvents and bases were tested to determine the optimal conditions of which the results are displayed in Table S1.† As demonstrated, several solvents were incompatible for the reaction due to the insolubility of 1, including methyl tert-butyl ether (MTBE), iso-butanol, trifluorotoluene, cyclohexane and acetonitrile (entries 3, 4, 6, 9, and 10, Table S1†). Toluene, which is the substrate used for all subsequent reactions, showed limited solubility for 1, and, therefore, was infeasible to use as a solvent (entry 2, Table S1†). Propylene carbonate, dimethylacetamide (DMA), and dimethylformamide (DMF) performed similarly to toluene and were thus also deemed as unsuitable candidates (entries 5, 7, and 8, Table S1†). Dimethyl sulfoxide (DMSO) produced the highest turnover number (TON) of 46 among the solvents tested (entry 1, Table S1†). Next, C–N coupling reactions were performed in the presence of DMSO using various bases. In this study, all bases showed capable of facilitating C–N cross-coupling to some extent, although the range varied greatly. More alkaline bases generated low yields, most likely due to the base strength of the reagents. Stronger bases were able to deprotonate the amine hydrogen readily to facilitate C–N cross-coupling more readily. However, some bases considered useful in cross-coupling (e.g. 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and 1,4-diazabicyclo(2.2.2)octane (DABCO)) were still inefficient in proton removal. Thus, a relatively bulky but strong base, potassium tertiary butoxide (KOtBu), was used and revealed the highest TON of 46 recorded among the bases tested (entry 16, Table S1†).
N cross-coupling using 1, various solvents and bases were tested to determine the optimal conditions of which the results are displayed in Table S1.† As demonstrated, several solvents were incompatible for the reaction due to the insolubility of 1, including methyl tert-butyl ether (MTBE), iso-butanol, trifluorotoluene, cyclohexane and acetonitrile (entries 3, 4, 6, 9, and 10, Table S1†). Toluene, which is the substrate used for all subsequent reactions, showed limited solubility for 1, and, therefore, was infeasible to use as a solvent (entry 2, Table S1†). Propylene carbonate, dimethylacetamide (DMA), and dimethylformamide (DMF) performed similarly to toluene and were thus also deemed as unsuitable candidates (entries 5, 7, and 8, Table S1†). Dimethyl sulfoxide (DMSO) produced the highest turnover number (TON) of 46 among the solvents tested (entry 1, Table S1†). Next, C–N coupling reactions were performed in the presence of DMSO using various bases. In this study, all bases showed capable of facilitating C–N cross-coupling to some extent, although the range varied greatly. More alkaline bases generated low yields, most likely due to the base strength of the reagents. Stronger bases were able to deprotonate the amine hydrogen readily to facilitate C–N cross-coupling more readily. However, some bases considered useful in cross-coupling (e.g. 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and 1,4-diazabicyclo(2.2.2)octane (DABCO)) were still inefficient in proton removal. Thus, a relatively bulky but strong base, potassium tertiary butoxide (KOtBu), was used and revealed the highest TON of 46 recorded among the bases tested (entry 16, Table S1†).
It is worthwhile to mention the low yields of products in some instances. The low yield is attributed to low concentrations (0.2 mol%) of catalyst used in the reactions. Though it is expected that yield will increase with higher catalyst loading, the reaction was carried out in a vial where higher catalyst loading was avoided due to safety concerns and the formation of a viscous solution that leads to catalyst and product deposition on the sides of the vial wall. Therefore, in this case, more emphasis on being placed on TON to validate catalyst efficiency.
Optimization of reaction conditions: temperature and time
After determining the optimal solvent and base for the reaction, optimizations for time and temperature were performed. In both reactions, DMSO and KOtBu were respectively used as solvent and base for the reaction of benzylamine and toluene at varying temperatures or times (Fig. S1†). As shown in Fig. S1a,† the temperature played a crucial role in yields. As the temperature increased, the TON values dramatically increased up to 110 °C. However, when the temperature exceeded 120 °C, TON values decreased. This is potentially related to the boiling point of toluene, which is around 111 °C. Thus, upon reaching 120 °C, the toluene is under reflux, and with no coolant in the system, a reduced amount of accessible toluene is available to perform C–N coupling. As a result, 110 °C was chosen as the ideal temperature to avoid any possible degradation of the substrate. C–N cross-coupling was conducted at a range of reaction times from 1 to 12 h (Fig. S1b†). Reaction yields at 1 h gave a TON of approximately 31, while 3 h reaction time dramatically increased over six times (TON = 207). Increased reaction times of up to 12 h exhibited a negligible effect on final TON values. It is apparent that after 3 h, the catalyst became saturated, and only a slight increase in TON was observed. Thus, 3 h was determined to be the optimal reaction time.Amination of toluene with different 1° amines
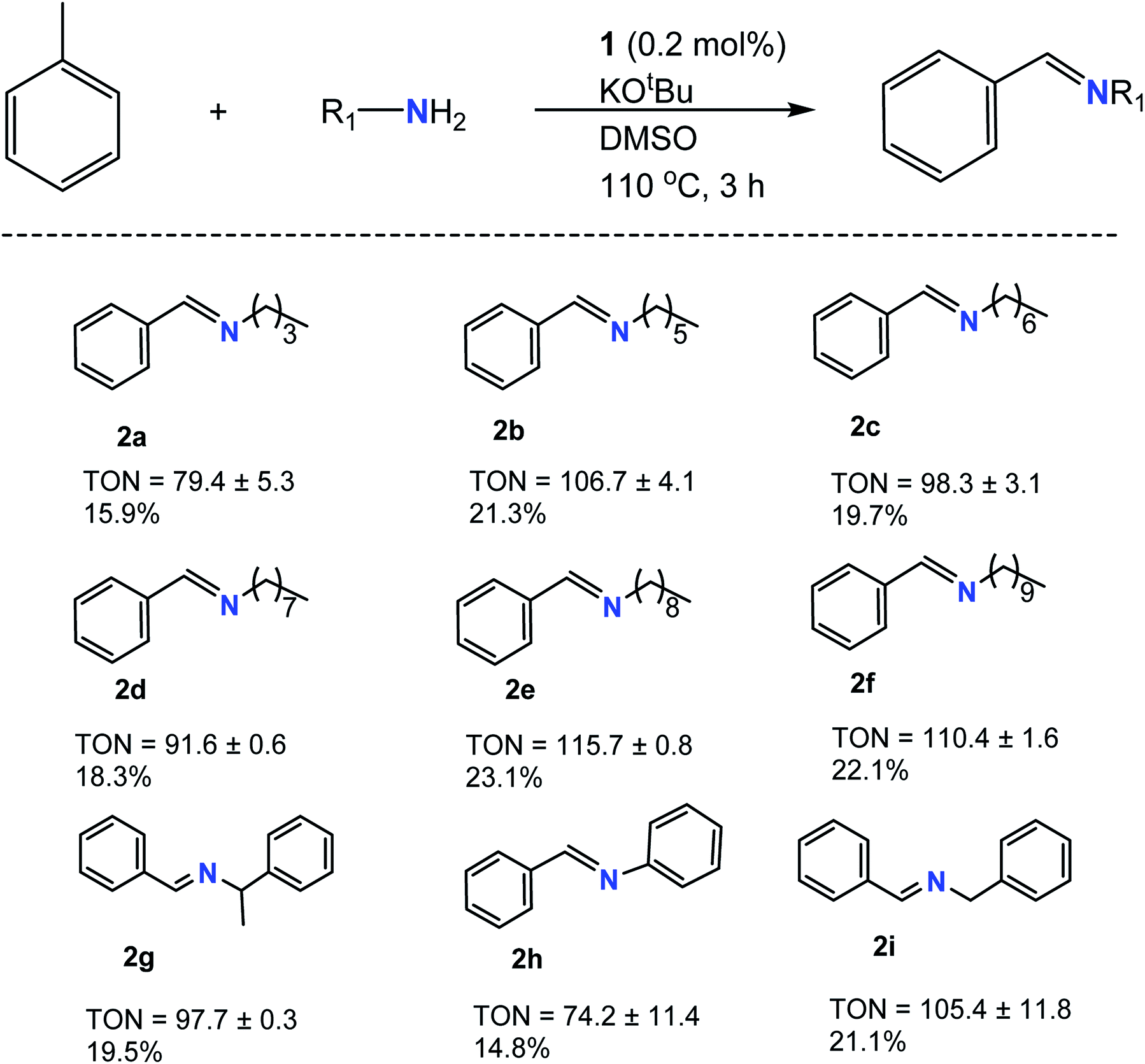
Following optimization studies, various 1° amine derivatives were analyzed for C–N cross-coupling, and the results are tabulated in Tables 1 and 2. Initially, CN formation was observed for a variety of 1° amines when using KOtBu as a base (Table 1). Yields ranged from TON of 74 (2h) to 116 (2e), showing relatively similar TON values, even with varying degrees of steric hindrance in the molecule. 2h had the lowest TON value, most likely due to delocalization of the nitrogen lone pair in the benzene ring via resonance, leading to less accessibility, preventing further reaction. The longer aliphatic amines showed better yields due to the increased electron releasing ability in line with the amine's overall nucleophilicity trend. Smaller aliphatic amines such as 2a showed relative difficulty reacting under such conditions due to lower boiling points below the optimized reaction conditions (Fig. S1†) as constant refluxing effects in the system attenuated final product formation. More so, shorter-chained aliphatic amines are less reactive due to lower electron releasing abilities.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| a TONs and GC yields represent an average of two runs. TONs and percent GC yields were calculated by gas chromatography/mass spectrometry (GC/MS) using decane as an internal standard. The GC/MS data of the products are given in the ESI. |
|---|
 |
a
| a TONs and GC yields represent an average of two runs. TONs and percent GC yields were calculated by gas chromatography/mass spectrometry (GC/MS) using decane as an internal standard. The GC/MS data of the products are given in the ESI. |
|---|
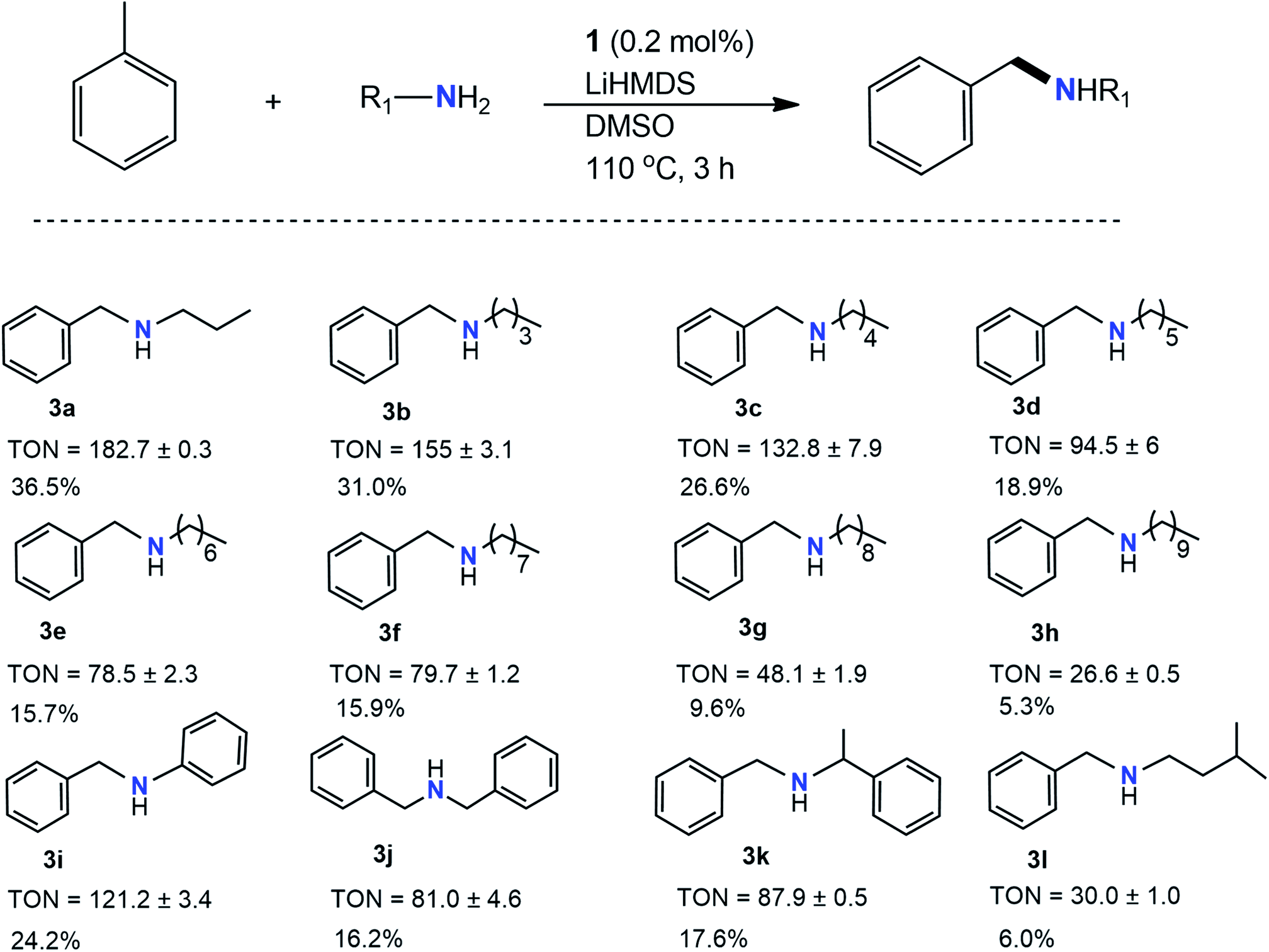 |
Once the initial imine formation studies were completed, investigations began into the pathways resulting in C–N products. Further optimization reactions were performed to study the effects of solvent and base on the formation of products. As shown in Table 2, lithium bis(trimethylsilyl)amide (LiHMDS) was quite efficient at not only forming the amine cross products but also to maintain similar yields to those produced in the presence of KOtBu. Most notably, various new products were formed (3a, 3b, 3c, 3k, and 3l), although they were unable to be successfully produced to their imine counterparts in the presence of KOtBu due to their chemical characteristics. Interestingly, several products were achieved in higher yields compared to those in Table 1 when using LiHMDS (3i vs. 2h) while other products (3g, 3h, 3l) exhibited attenuated yields. Products 3g and 3h exhibited lower yields (TON = 48 and 27, respectively), most likely due to the increasingly long carbon chains present. Longer chains spread out the charge of the entire molecule, possibly contributing to decreased reactivity. Notably, 3l (TON = 30) produced minimal amounts of product likely attributed to the lower boiling point, difficulty in the deprotonating, and possible steric hindrance of the amine. 3a and 3b showed the largest TONs of 183 and 155, respectively. The highest and lowest yields were demonstrated by aliphatic amines, mentioned later, whereas the bulky, aromatic amines resulted in more moderate yields. Quite remarkably, only LiHMDS produced secondary amines while the rest of the bases produced imines with a variation of yields. While the pKa of bases in DMSO plays an essential role in controlling the product yield, in the case of LiHMDS, the nature of the product reflects that nucleophilicity is likely more pronounced than basicity55 LiHMDS is primarily used as a strong nucleophile rather than a base as the pKa of the conjugate base is ∼26 less basic than lithium diisopropylamide (LDA, pKa 36).56,57 The pKa of LiHMDS in DMSO is 30,58 and that of KtOBu is 32.2.59 This clearly dictates that KOtBu acts as a stronger base, whereas LiHMDS may act as more of a nucleophile.
Cyclization of aliphatic amines
A variety of C–N and CN products obtained in this study indicated that aliphatic amines followed a different reaction pathway than the one initially recognized in optimization studies. Table 3 lists long-chain amines that are found to be cyclized during the reaction in the absence of toluene. Markedly, cyclic products formed best for reactions involving aliphatic amines four to seven carbons long. Generally, amines capable of forming either 5 or 6-membered rings demonstrated this effortlessly; however, several larger amines did not cooperate in this fashion. Piperidine (4e) was the product of the highest yield (TON = 131), and this can be attributed to the stability of 6-membered rings. Hexylamine produced the lowest yield (TON = 85) in the formation of 2-methylpiperdine (4f). Octylamine, nonylamine, and decylamine all preferred C–N cross-coupling over intramolecular cyclization and were unaccommodating for cyclic products likely due to steric hindrance and other difficulties associated with cyclization reactions. Several 2° amines were used to determine formations of target cyclized 3° amines. Several 3° amines (4b, 4c, 4d, 4g) were produced, and these results are shown in Table 3. Yields were largely moderate to good (TON = 85–131). The lowest TON of 85 (4f) is attributed to the varied products resulting from the use of hexylamine, which reacts at multiple sites along its carbon chain. The highest TON of 131 occurred for piperidine (4e), which enigmatically contains only one less carbon than 2-methylpiperdine (4f). Some 2° amines were unable to perform cyclization successfully (i.e., dihexylamine).
| a TONs and GC yields represent an average of two runs. TONs and percent GC yields were calculated by gas chromatography/mass spectrometry (GC/MS) using decane as an internal standard. The GC/MS data of the products are given in the ESI. |
|---|
 |
Amination of toluene derivatives and other hydrocarbons with different 1° amines
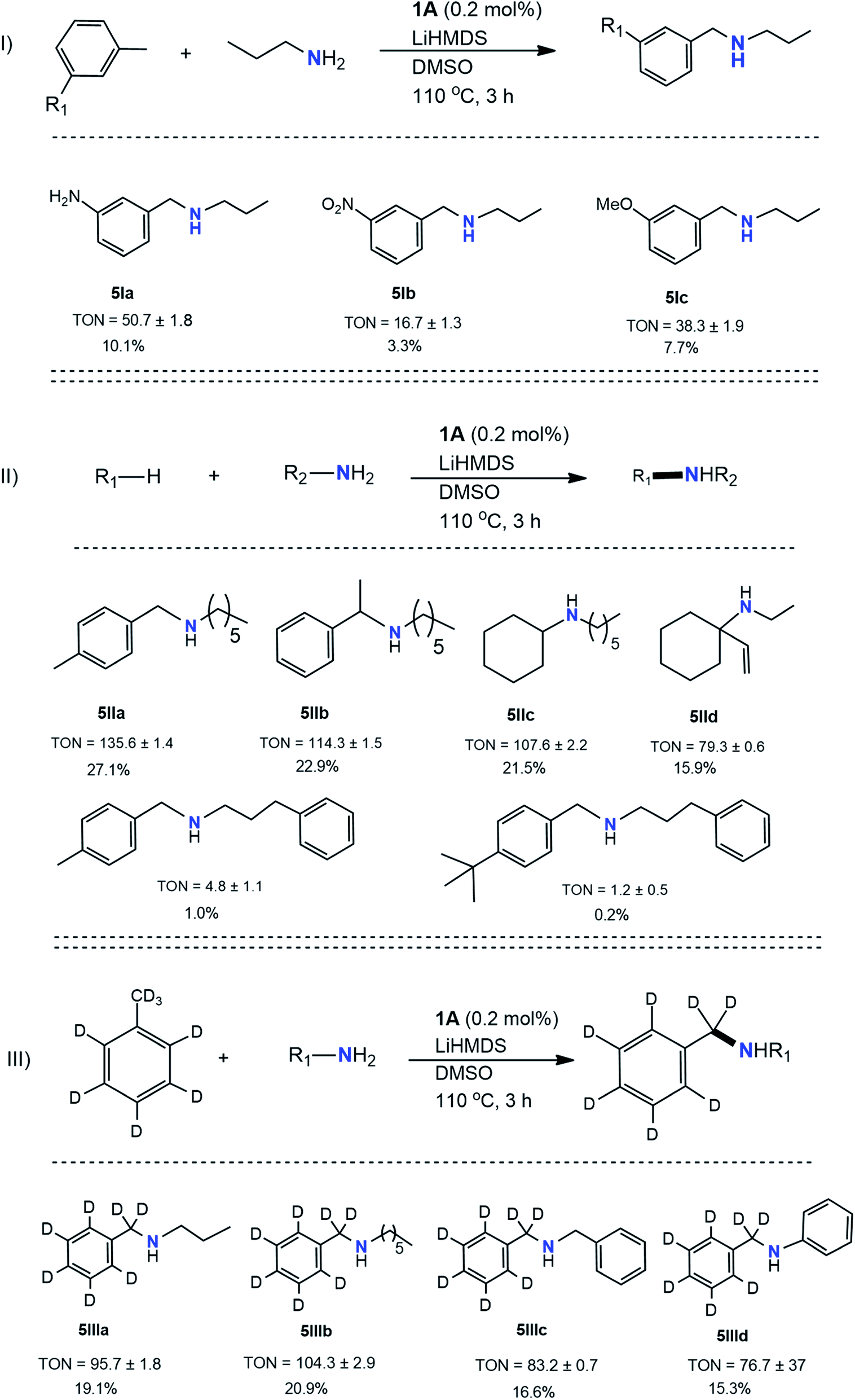
The results in Table 3 indicate the production of pyrrolidine, piperidine, and their derivatives instead. Further investigation revealed toluene was a nonfactor in formations of cyclized products. Incorporating LiHMDS in the presence of KOtBu for previously attempted reactions accentuated the results. Aside from toluene itself, substituted toluene had difficulty reacting in the presence of KOtBu, mostly when electron-donating groups were present in the para position. However, LiHMDS facilitated cross-couplings for these derivatives, particularly in the formation of single bonds for these derivatives. In general, such derivatives (5I) still exhibited low yields (TON = 17–51). The lowest TON is attributed to m-nitrotoluene (5Ib) directly resulting from the utility of 1. In several situations, reactants prone to reduction often underwent such a process in the presence of 1. Specifically, for reactions containing m-nitrotoluene and propylamine, reduction of m-nitrotoluene to m-toluidine was experienced before the C–N cross-coupling occurred with partner propylamine. While it is also possible that further reduction of the nitro groups occurred after the cross-coupling occurred, ultimately, this still diminished target product yields due to the propensity of reduced product formation. Reactions involving toluene derivatives containing para and ortho substitutions were also examined but were unsuccessful in forming any C–N cross-coupled products.Next, investigations were performed for C–N cross-couplings using hydrocarbon sources other than toluene (5II). The hydrocarbons were selected based on low C–H bond energies. The yields for these reactions ranged in TON values from 79 to 136, of which the lowest was attributed to vinyl cyclohexane (5IId) due to the many side reactions inhibiting target product formation. On the other hand, p-xylene (5IIa) showed a better TON than its toluene counterpart (3d, Table 2) under identical conditions. Due to the promise it showed in earlier studies, and because it avoided cyclization, hexylamine was chosen as the ideal amine coupling partner. In the reactions involving ethylbenzene and vinyl cyclohexane, the amine reacted at the 2° carbon of the ethyl group. Surprisingly, p-xylene (5IIe) and p-tert-butyltoluene (5IIf) perform poorly with long-chain benzylic amine (3-phenylpropylamine). Electronic or steric effects may play a role in lowering the trend of yield from p-methyl to p-tert butyl.
Next, several additional amines were reacted in the presence of toluene-d8. These reactions allowed us to establish toluene's role in the formation of imines and amines. Slightly decreased yields were observed during isotope labeling as exhibited (5III) for all participating amines. 5IIIa and 5IIId showed TON values almost half of the original non-deuterated toluene counterparts (3a and 3i, respectively). For product 5IIIb, however, which exhibited the highest TON of 104, the product yield was similar to the original reaction involving non-deuterated toluene (3d). Although 5IIIc showed the lowest TON of 77, this yield was also similar to what was previously observed (3j). Further, GC/MS analysis of deuterated versus non-deuterated dibenzylamine (Fig. S11 and S40†) was studied in order to determine whether the product was retaining the hydrogens or receiving them from another source (e.g., tetrahydrofuran (THF)). Both the chromatograms and mass spectrum of benzylamine (Fig. S11†) and labeled benzylamine (Fig. S40†) showed some variance. Most notably, the labeled dibenzylamine showed two distinct peaks that were attributed to a slight variation in molar mass (203 m/z, 204 m/z) of the compound due to the presence of both hydrogen and deuterium atoms. Additionally, using catalyst 1, pharmaceutically active molecules such as cyclizine – an anti-nausea drug that requires a multistep synthesis on the industrial scale60 – can be synthesized in one step by activating C–H bond and coupling to an amine. Cyclizine was synthesized using diphenylmethane and 1-methylpiperizine in the presence of catalyst 1, and this was confirmed using GC/MS analysis (Fig. S44†).
Kinetic studies
To determine whether the mechanism consisted of radical intermediates, (2,2,6,6,-tetramethylpiperidin-1-yl)oxyl (TEMPO) and galvionxyl, radical quenchers, were introduced into the reaction of propylamine with toluene in the presence of LiHMDS at 110 °C for 3 h in two separate reactions. It was noted, however, that the reactions proceeded with similar yields to those observed in 3a (Table 2). The rate of the reactions was determined under identical conditions to the previously conducted C–N cross-coupling reactions by using various concentrations of the base, toluene, 1, and amine during the amination of toluene with benzylamine to determine the order of each component with respect to the overall reaction (Fig. S45†). The variation on the concentration of a particular reagent simultaneously retaining other parameters unchanged was achieved by modifying the reagents stoichiometrically. The base was first investigated by varying concentrations from 0.16–0.47 M while maintaining all other reaction conditions identically. As shown in Fig. S45a,† the changes in base concentration had small overall effects on the final TON, as demonstrated by the horizontal slope of the plot. This is indicative the base played only a slight role in the actual formation of the C–N coupled product that the kinetic behavior of the base was likely zero order with respect to the overall reaction.Next, the reactions were repeated by changing toluene concentrations from 0.45–1.80 M. In Fig. S45b,† the pronounced increase in the rates shows a relatively linear relationship with respect to the toluene concentration, indicating that toluene was first order with respect to the overall reaction. Next, in Fig. S45c,† changing the concentration of the catalyst was examined for its effect on the reaction. The pronounced increased rates ranging from concentrations of 1.2–4.0 mol% also showed a linear relationship with respect to product yield leading to the conclusion that 1 was first order with respect to the overall reaction. Lastly, upon examination of amine concentration from 0.16–0.49 M, Fig. S45d† exhibits a similar effect to that of the base indicative of a likely zero-order process with respect to the overall reaction. Further, the effects of varying the concentrations of the base, benzylamine, toluene, and 1 on the formation of the dibenzylamine product can be seen in Fig. S46.† As the plots exhibit, the effects are almost the same validating the previous results. Therefore, changing the concentrations of toluene and 1 affected overall reaction rates, yet, doing so for the concentrations of base and amine did not affect the overall rate of product yield.
Next, the reactions were repeated in the presence of toluene-d8 as the evaluation of kH/kD can help to determine the rate-determining step (RDS) of the reaction. Intuitively, C–H activation was studied as the possible RDS in the reaction since toluene was being examined. The reaction was repeated and monitored at 20 min intervals to establish the rate of the reaction (Fig. S47†), which upon comparison revealed a kH/kD value of approximately 2.7 indicative that the C–H bond activation in toluene was indeed the RDS. Furthermore, the reduction reactions were also performed to trace the isotopic hydrogens in the reaction. As previously established, with the reduction of m-nitrotoluene to m-toluidine in the presence of 1 before C–N cross-coupling with the amine (5Ia, Table 4), we introduced toluene-d8 into the system to observe if the reduction was explicitly related to toluene or another source. Isotopic versions of m-toluidine were observed using GC/MS, further strengthening the idea that toluene was donating its hydrogens during the reduction process. The reduction of benzophenone to phenylbenzene methanol was also attempted; however, benzophenone reduced directly to diphenylmethane, completely removing the oxygen group present.
| a TONs and GC yields represent an average of two runs. TONs and percent GC yields were calculated by gas chromatography/mass spectrometry (GC/MS) using decane as an internal standard. The GC/MS data of the products are given in the ESI. |
|---|
 |
Proposed mechanism
A mechanism is proposed for respective amine and imine products. By changing the base affected product formation. KOtBu promoted iminations (CN) with no formation of amine (C–N) products. On the other hand, LiHMDS promoted aminations with no imine products. This occurs because the catalyst exhibits high specificity in the presence of different bases. Therefore, base selection plays a significant role in each case. Evaluations of several control reactions along with kinetic isotopic studies were conducted to elucidate mechanistic pathways. Based on controls, our findings indicate that when two different bases were used, the system responded along two distinct pathways. Both processes are summarized below in Fig. 1.
 | ||
| Fig. 1 Schematic representation of amine and imine formation by means of C–N cross of toluene and an amine catalyzed by 1. | ||
In one of the cycles, we believe that KOtBu triggers the action. The substrate activated catalyst (2ab) easily deprotonates in the presence of KOtBu (2ad) to release the imine product (2af). It has been reported that DMSO can hold roles in chemical reactions aside from solvent only.61,62 An aldehyde is formed and combines with the amine, ultimately producing the imine. The base will choose either the proton or carbon required for bonding. In the case of KOtBu, this is simply accomplished through proton abstraction, ultimately paving the way to produce the imine (2af). DMSO's high polarizability potential creates a negative charge around the oxygen, enabling it to coordinate to electron-deficient centers. The substrate activated carbon enters the reaction cycle upon oxidative addition, and DMSO's sacrificial oxygen facilitates reductive elimination. The reaction cycle is then recommenced as DMSO is reduced to dimethyl sulfide (DMS), replanting 2aa from 2ac.
As confirmed by electrospray ionization mass spectrometer (ESI-MS) analysis of 1 (Fig. S48†), a chloride ion attachment exists on the Ni(II) complex. In the presence of a base, the amine is likely to reduce 1 to 2aa, removing the labile Cl−.63 Upon oxidative addition, toluene is substituted, producing 2ab. We hypothesize that the toluene is then oxidized to the aldehyde in the presence of DMSO. Catalyst 1 was treated with DMSO & toluene to demonstrate the formation of benzaldehyde, and the product benzaldehyde was detected via GC/MS analysis (Fig. S49†). Here, we believe that DMSO is acting as the primary oxidant. Furthermore, GC/MS analysis also revealed the formation of DMS (Fig. S49†).
The second cycle, also demonstrated in Fig. 1 is primed by the base as in the first cycle, but this time LiHMDS initiates the action. Again, oxidative addition is facilitated by ligand substitution as toluene enters the reaction cycle (2ab). The cycle then propagates differently, as previously illustrated. LiHMDS deprotonates the amine directing the reaction cycle to 2ag. Before reductive elimination, another amine molecule further drives the reaction forward as the amine product is expelled in a concerted rearrangement leading to 2ac. Again, the catalyst returns from 2ac to 2aa as DMSO is reduced to DMS. Unlike the reactions involving KOtBu, no benzaldehyde formation was detected via GC/MS analysis.
While it is possible that a high-valent Ni species, as two deprotonated σ-donating amides, make up 1, in the presence of DMSO, which may be responsible for the oxidation of toluene, it is unlikely that such a high-valent Ni species64–68 is stabilized. Dibenzylamine was treated with KOtBu and 1 to prove imine formation occurred before the amine (Fig. 1). Yet, no change in amine formation was observed via GC/MS analysis. However, catalyst 1 facilitates the reaction in the presence of LiHMDS. The catalyst was synthesized using lithium salt, and additional Li+ was added in the form of LiHMDS. It is assumed that LiHMDS may play a dual role. Due to excess lithium, 1 is more likely to sustain the imine type bonding coordinated to Ni(II). Thus, it may be possible that both amide bonds in 1 are in the imine form, and such structures have been previously reported.69
Due to its small size, Li+ has a higher ionic potential (charge/radius) compared to other alkali metal ions and is known to coordinate with oxygen donor atoms. Therefore, it is likely that Li+ coordinated to the amide oxygen of 1, forcing it to conform to the imine-like functionality. NMR evidence showed asymmetry in the ligand structure, and, therefore, we believe such a structure manifested.44
Single-electron transfer (SET) has been proposed in nickel-catalyzed organic transformations, and we believe such reactions were also occurring in our case.70,71 So, a low-valent Ni species (2aa) is formed after the addition of amine or LiHMDS through SET. When amination reactions were conducted using toluene and benzylamine in the presence of radical quenchers such as TEMPO and galvinoxyl, the isolated product indicated this possibility. The imine bond in the structure of the catalyst (1), along with the pyridinic ligand, may be suitable for stabilizing Ni in a low-valent state such as Ni(I).72 Once reduced through SET, toluene was oxidatively added to the Ni catalyst leading to 2ab. As indicated by kH/kD studies, this may be the rate-limiting step. Some control reactions involving toluene and amines were attempted and characterized via ESI-MS. Upon the introduction of the amine, a new species resembling 2ag was observed. Upon reductive elimination, the final product was furnished. Furthermore, it is believed that 2ac was oxidized by DMSO to its original structure, which further reacted with toluene completing the catalytic cycle. When the reaction was performed in the presence of either air or oxygen, no product was observed. It can also be possible that an imine product underwent further reduction to the amine product.
Multiple control reactions were performed to elucidate the mechanism involving LiHMDS. Since LiHMDS is stabilized in THF, it is hypothesized that perhaps the reduction of imines to amines occurred in the presence of THF. Initially, an imine was synthesized by reacting benzaldehyde and benzylamine in equivalent amounts in methanol. Then, the imine was used to perform a few control reactions in the presence of 1. Two reactions were carried out to test the potential roles of THF and Li+. Firstly, the imine was reacted in a reaction vessel in the presence of lithium chloride (LiCl) without THF, and secondly, in the presence of both LiCl and THF. Both reactions were run for 3 h at 110 °C in the presence of KOtBu. GC/MS analysis confirmed that in both cases, no reduction of the imine to amine occurred. Furthermore, toluene's role was scrutinized as it can act as the hydrogen source necessary for the reduction of imine to the amine. In one set of reactions, toluene was present, and in the other, no toluene was used. GC/MS analysis confirmed the system lacking toluene did not reduce the imine to the amine. Whereas, the reaction in the presence of toluene reduced the imine to the amine. This suggests that toluene likely donated hydrogens in the reduction of the imine intermediate rather than THF within LiHMDS. When the reaction was done without LiHMDS while keeping all other reagents intact, no product was obtained. Therefore, LiHMDS is essential for our reactions and is presumably the source of SET.
Lastly, for coupling reactions involving m-nitrotoluene, reduction of the nitro group was observed; however, it is unclear what the hydrogen source may be. Therefore, the reaction was repeated in the presence of equivalent amounts of toluene-d8 for 3 h at 110 °C with LiHMDS. After purification, GC/MS analysis revealed a m-toluidine product with a heavy mass spectrum (Fig. S41†). This indicates that the hydrogen is responsible for the nitro group reduction originated from toluene or some sort of hydride species consequential of the toluene substrate rather than from any other hydrogen source. Thus, it is surmised that the imine is formed before the amine, and further reduction occurs due to possible hydride intermediate production within the reaction.
Repeated experiments and careful GC/MS analyses confirm the absence of any byproducts and impurities. The reaction seems to stop after the first substitution of N–H functionality for amine production. Calculations of thermodynamic parameters indicate that the reaction is thermodynamically controlled with Gibbs free energy change (ΔG) of the stoichiometric reaction under experimental conditions is nearly zero (Table S2†). Therefore, such an inherent limitation of yield can only be overcome by higher catalyst loading.
Conclusion
In summary, 1 was used successfully to activate C–H bonds and directly form C–N and CN bonds, as well as cyclic amines, under mild reaction conditions. Depending on the base used for the reaction and the starting amines, the catalyst demonstrated high selectivity towards the product formation. The catalyst also showed great potential for product formation using aromatic and non-benzylic substrates. 1 was noted to have several competing functionalities ranging from C–H activation to direct C–N cross-coupling as well as intramolecular C–N coupling. Various bases produced vastly different results in the reactions studied, and notable product formations were observed. Kinetics insight demonstrated that C–H activation in toluene was the RDS in the overall reaction; however, the hydrogens removed from toluene were possibly not used for reduction as indicated in the several examined cases. Furthermore, both oxidation and reduction reactions meeting specific criteria were performed. This included base and oxygen-based substituent requirements. The nickel(II) pincer complex (1) exhibited tremendous versatility, and many other facets were discovered in the course of this study. As a result, other interesting pathways have been uncovered and are currently being further investigated. We believe the methodology opens up an avenue for direct synthesis of amine-based drugs eliminating multistep organic synthesis that is environmentally responsible.
Materials and methods
General
All chemicals were purchased from Aldrich Chemical Company, Fischer Scientific Company, and VWR International and were used without further purification unless stated otherwise. Solvents were purified according to literature procedures73 and purged under argon gas (Ar) prior to use. Amines were passed through neutral alumina and purged with Ar prior to use. Reaction products were analyzed by GC/MS using the Shimadzu model QP2010, equipped with a DB-5 column. Samples were run at an initial ramp temperature of 70 °C with a ramp climb of 15 °C min−1 until 200 °C was reached and then a ramp increase of 5 °C min−1 until 300 °C was reached where the temperatures were held for 5 minutes. The injection temperature was 250 °C, with an ionization temperature of 300 °C, and samples were injected 3 times. Catalyst characterization was performed using a JEOL 400 NMR and an Agilent 100 series MSD VL ESI-MS.Synthesis of A and 1
Both A and 1 were synthesized according to the literature procedure with slight modifications.44–46 A was synthesized in the following manner: acid chloride of 2,6-dicarboxylic acid pyridine was reacted with 2,6-diisopropylaniline in the presence of triethylamine and purified via crystallization. To produce 1, A (0.297 g, 0.612 mmol) was dissolved in THF (15 mL) under nitrogen atmosphere in a Schlenk flask, and the temperature was adjusted to 0 °C in an ice bath. N-butyllithium (0.80 mL, 1.26 mmol, 1.6 M in hexanes) was added to the solution and stirred for 30 min. Nickel(II) chloride glyme (0.137 g, 0.612 mmol) was added to the reaction. The reaction was slowly brought back to room temperature and allowed to stir overnight. THF was removed by a liquid nitrogen trap to yield 1 (345 mg). It is believed that chlorometallated form (1) may get converted to hydroxymetallated catalyst in the presence of moisture, which may also show catalytic activity.74,75Synthesis of amine and imine coupled products
All reactions were performed under similar conditions unless stated otherwise. 0.2 mol% of 1 (2.0 mg, 3.46 μmol) and KOtBu (55.0 mg, 0.49 mmol) were added to a 10 mL screw-top vial (previously purged with Ar for 10 min) and purged under Ar atmosphere for an additional 30 min. DMSO (1.0 mL) was added to the system via a gas-tight syringe to prevent any additional exposure to the external environment. After 10 min of purging, amine (1.71 mmol) and toluene (1.71 mmol) were added using gas-tight syringes. The system was then sealed and vigorously mixed to dissolve any catalyst and base. The temperature was brought to 110 °C in an oil bath, and the reaction was allowed to react for 3 h. After the allotted time, the system was removed from the heat source and allowed to cool to room temperature for 1 h. The reaction mixture was passed through a small filtration column to remove any precipitate followed by dilution with ethanol in preparation for GC-MS analysis. Various amines and imines were analyzed in the presence of a base (KOtBu or LiHMDS) depending on desired products. Yields ranged from 74–184 TON, where TON = (concentration of standard/AUC of standard) × (AUC of product) × (dilution factors) × (1/moles of catalyst) and AUC is the area under the curve. Dilution factors included GC/MS vial dilution plus any additional dilution done during purification before GC/MS preparation. An internal standard of 3.00 mL decane in 10.00 mL of ethanol was used to quantify the product formation.Synthesis of cyclic products
All reactions were performed under similar conditions unless stated otherwise. 0.2 mol% of 1 (2.0 mg, 3.46 μmol) and KOtBu (55.0 mg, 0.49 mmol) were added to a 10 mL screw-top vial (previously purged with Ar for 10 min) and purged under Ar atmosphere for an additional 30 min. DMSO (1.0 mL) was added to the system via a gas-tight syringe to prevent any additional exposure to the external environment. After 10 min of purging, amine (1.71 mmol) was added using gas-tight syringes. The system was then sealed and vigorously mixed to dissolve any catalyst and base. The temperature was brought to 110 °C in an oil bath, and the reaction was allowed to react for 3 h. After the allotted time, the system was removed from the heat source and allowed to cool to room temperature for 1 h. The reaction mixture was passed through a small filtration column to remove any precipitate followed by dilution with ethanol in preparation for GC-MS analysis. TON ranged from 85–131.Synthesis of amine coupled products with toluene derivatives and other hydrocarbons
0.2 mol% of 1 (2.0 mg, 3.46 μmol) and LiHMDS (83.0 mg, 0.49 mmol) were added to a 10 mL screw-top vial (previously purged with Ar for 10 min) and purged under Ar atmosphere for an additional 30 min. DMSO (1.0 mL) was added to the system via a gas-tight syringe to prevent any further exposure to the external environment. After 10 min of purging, amine (1.71 mmol) and toluene derivatives or other hydrocarbons (1.71 mmol) were added using gas-tight syringes. The system was then sealed and vigorously mixed to dissolve any catalyst and base. The temperature was brought to 110 °C in an oil bath, and the reaction was allowed to react for 3 h. After the allotted time, the system was removed from the heat source and allowed to cool to room temperature for 1 h. The reaction mixture was passed through a small filtration column to remove any precipitate followed by dilution with ethanol in preparation for GC-MS analysis. The TON ranged from 16–137.Kinetic studies
In order to determine the roles of 1, base, and each substrate in the reaction, kinetic studies were conducted in duplicates. The effect of each reaction parameter was checked by varying it within a specific range keeping other conditions remains constant. Firstly, the base was investigated by varying its concentration (0.16–0.47 M). Next, 1 was varied from 1.2–4.0 mol%. Then, the effect of each substrate was explored. Toluene concentration was varied from 0.45–1.80 M. Lastly, the concentration of amine was varied from 0.16–0.49 M.Author contributions
A. B., A. B. R., P. S., H. A. W., and C. M. P. contributed to writing the manuscript, performing the experiments and interpreting the data. A. B. and A. B. R. contributed to catalyst synthesis. A. G. conceived the research idea, designed the experiments, interpreted the data, and wrote the manuscript. P. M. contributed to writing the manuscript, designing the experiments, interpreting the data, and establishing the reaction mechanism. All authors reviewed and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Foundation (grant no. IIA-1457888) and the Arkansas EPSCoR Program, ASSET III (CASE).References
- R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS.
- A. O. King and N. Yasuda, in Topics in Organometallic Chemistry, Springer, Berlin, Heidelberg, 2004, pp. 205–245 Search PubMed.
- T.-H. Le, Y. Kim, H. Yoon, T.-H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef.
- J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS.
- A. Seayad, M. Ahmed, H. Klein, R. Jackstell, T. Gross and M. Beller, Science, 2002, 297, 1676–1678 CrossRef CAS.
- S. A. Lawrence, Amines: synthesis, properties, and applications, Cambridge University Press, Cambridge, UK, 2004 Search PubMed.
- H. Wittcoff, B. G. Reuben and J. S. Plotkin, Industrial organic chemicals, John Wiley & Sons, Ltd, New Jersey, USA, 3rd edn, 2013 Search PubMed.
- I. H. Pitman, Med. Res. Rev., 1981, 1, 189–214 CrossRef CAS.
- A. Simplício, J. Clancy, J. Gilmer, A. L. Simplício, J. M. Clancy and J. F. Gilmer, Molecules, 2008, 13, 519–547 CrossRef.
- A. R. Brown, D. C. Rees, Z. Rankovic and J. R. Morphy, J. Am. Chem, Soc., 1997, 119, 3288–3295 CrossRef CAS.
- L. Emmanuvel, R. K. Shukla, A. Sudalai, S. Gurunath and S. Sivaram, Tetrahedron Lett., 2006, 47, 4781–4791 CrossRef.
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7811 CrossRef CAS.
- S. A. I. Quadri, T. C. Das, S. Jadhav and M. Farooqui, Synth. Commun., 2018, 48, 267–277 CrossRef CAS.
- C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 12494 CrossRef.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382–2384 CrossRef.
- F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS.
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed., 1995, 34, 1348–1350 CrossRef CAS.
- A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS.
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049–11061 CrossRef CAS.
- B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS.
- R. E. Tundel, A. K. W. Anderson and S. L. Buchwald, J. Org. Chem., 2005, 71, 430–433 CrossRef.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 15914–15917 CrossRef CAS.
- R. A. Green and J. F. Hartwig, Org. Lett., 2014, 16, 4388–4391 CrossRef CAS.
- B. Cacciuttolo, O. Pascu, C. Aymonier and M. Pucheault, Molecules, 2016, 21, 1042 CrossRef.
- R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS.
- R. A. Sheldon, Chem. Ind., 1997, 1, 12–15 Search PubMed.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
- J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283 RSC.
- K. D. Jones, D. J. Power, D. Bierer, K. M. Gericke and S. G. Stewart, Org. Lett., 2018, 20, 208–211 CrossRef CAS.
- X. Huang, Y. Chen, S. Zhen, L. Song, M. Gao, P. Zhang, H. Li, B. Yuan and G. Yang, J. Org. Chem., 2018, 83, 7331–7340 CrossRef CAS.
- W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153–12156 CrossRef CAS.
- P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 14373–14377 CrossRef CAS.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, J. Catal., 2017, 347, 57–62 CrossRef CAS.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef.
- P. J. Donoghue, J. Tehranchi, C. J. Cramer, R. Sarangi, E. I. Solomon and W. B. Tolman, J. Am. Chem. Soc., 2011, 133, 17602–17605 CrossRef CAS.
- M. R. Halvagar and W. B. Tolman, Inorg. Chem., 2013, 52, 8306–8308 CrossRef CAS.
- D. Dhar and W. B. Tolman, J. Am. Chem. Soc., 2015, 137, 1322–1329 CrossRef CAS.
- L. B. Junquera, F. E. Fernández, M. C. Puerta and P. Valerga, Eur. J. Inorg. Chem., 2017, 2017, 2547–2556 CrossRef CAS.
- J. Tehranchi, P. J. Donoghue, C. J. Cramer and W. B. Tolman, Eur. J. Inorg. Chem., 2013, 2013, 4077–4084 CrossRef CAS.
- E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. C. MacMillan, Science, 2016, 353, 279–283 CrossRef CAS.
- D. Huang, O. V Makhlynets, L. L. Tan, S. C. Lee, E. V Rybak-Akimova and R. H. Holm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1222–1227 CrossRef CAS.
- K. J. Szabó, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Berlin, Germany, 2013, pp. 203–241 Search PubMed.
- Y. Gartia, P. Ramidi, S. Cheerla, C. M. Felton, D. E. Jones, B. C. Das and A. Ghosh, J. Mol. Catal. A: Chem., 2014, 392, 253–259 CrossRef CAS.
- Y. Gartia, S. Pulla, P. Ramidi, C. C. Farris, Z. Nima, D. E. Jones, A. S. Biris and A. Ghosh, Catal. Lett., 2012, 142, 1397–1404 CrossRef CAS.
- Y. Gartia, P. Ramidi, D. E. Jones, S. Pulla and A. Ghosh, Catal. Lett., 2014, 144, 507–515 CrossRef CAS.
- N. M. Camasso and M. S. Sanford, Science, 2015, 347, 1218–1220 CrossRef CAS.
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS.
- S.-F. Pang, Y. Hang-Kong, Y.-J. Wu and S. Feng, J. Mol. Catal., 2017, 31(2), 105–120 CAS.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS.
- Q. Xue, J. Xie, H. Li, Y. Cheng and C. Zhu, Chem. Commun., 2013, 49, 3700–3702 RSC.
- D. Dhar, G. M. Yee, T. F. Markle, J. M. Mayer and W. B. Tolman, Chem. Sci., 2017, 8, 1075–1085 RSC.
- L. Ackermann, R. Sandmann and W. Song, Org. Lett., 2011, 13, 1784–1786 CrossRef CAS.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- R. R. Fraser and T. S. Mansour, J. Org. Chem., 1984, 49, 3442–3443 CrossRef CAS.
- R. R. Fraser, T. S. Mansour and S. Savard, J. Org. Chem., 1985, 50, 3232–3234 CrossRef CAS.
- F. G. Bordwell, D. Algrim and N. R. Vanier, J. Org. Chem., 1977, 42, 1817–1819 CrossRef CAS.
- P. Munshi, A. D. Main, J. C. Linehan, A. Chih-Cheng Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963–7971 CrossRef CAS.
- S. Hishinuma, K. Sugawara, Y. Uesawa, H. Fukui and M. Shoji, Biochem. Pharmacol., 2014, 91, 231–241 CrossRef CAS.
- T. T. Tidwell, Synthesis, 1990, 1990, 857–870 CrossRef.
- M. Calligaris, Coord. Chem. Rev., 2004, 248, 351–375 CrossRef CAS.
- Y. M. Albkuri, A. B. RanguMagar, A. Brandt, H. A. Wayland, B. P. Chhetri, C. M. Parnell, P. Szwedo, A. Parameswaran-Thankam and A. Ghosh, Catal. Lett., 2019, 1–10 Search PubMed.
- E. Chong, J. W. Kampf, A. Ariafard, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 6058–6061 CrossRef CAS.
- N. A. Weires, D. D. Caspi and N. K. Garg, ACS Catal., 2017, 7, 4381–4385 CrossRef CAS.
- J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS.
- M. Mohadjer Beromi, A. Nova, D. Balcells, A. M. Brasacchio, G. W. Brudvig, L. M. Guard, N. Hazari and D. J. Vinyard, J. Am. Chem. Soc., 2017, 139, 922–936 CrossRef CAS.
- S. Shi, G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 6959–6963 CrossRef CAS.
- J.-C. Wasilke, G. Wu, X. Bu, G. Kehr and G. Er, Organometallics, 2005, 24(17), 4289–4297 CrossRef CAS.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS.
- P. Zimmermann and C. Limberg, J. Am. Chem. Soc., 2017, 139, 4233–4242 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of laboratory chemicals, Butterworth Heinemann, Oxford, UK, 4th edn, 1997 Search PubMed.
- X. Lefèvre, D. M. Spasyuk and D. Zargarian, J. Organomet. Chem., 2011, 696, 864–870 CrossRef.
- D. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693–4701 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The study of the effect of base and solvent (Table S1) and temperature and time (Fig. S1) on C–N coupling reaction, GC/MS data of the products (Fig. S2–S44), effect of the base, toluene, 1, and benzylamine concentrations on the rate of dibenzylamine product yield (Fig. S45), the formation of product in increasing concentrations of the base, benzylamine, toluene substrate and catalyst loads (Fig. S46), the plot of product concentration versus time using toluene and toluene-d8 (Fig. S47), ESI-MS plot of catalyst (Fig. S48) and GC/MS analysis of benzaldehyde and DMSO formed during the reaction of toluene in the presence of 1 and base (Fig. S49). See DOI: 10.1039/d0ra09639c |
| ‡ Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2021 |