
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-resolved detection of SDS-induced conformational changes in α-synuclein by a micro-stopped-flow system†
Shunki Takaramoto a,
Yusuke Nakasonea,
Kei Sadakaneb,
Shinsaku Marutab and
Masahide Terazima*a
a,
Yusuke Nakasonea,
Kei Sadakaneb,
Shinsaku Marutab and
Masahide Terazima*a
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Kyoto 606-8502, Japan. E-mail: mterazima@kuchem.kyoto-u.ac.jp
bDepartment of Bioinformatics, Graduate School of Engineering, Soka University, Hachioji, Tokyo 192-8577, Japan
First published on 4th January 2021
Abstract
An intrinsically disordered protein, α-synuclein (αSyn), binds to negatively charged phospholipid membranes and adopts an α-helical structure. This conformational change is also induced by interaction with sodium dodecyl sulfate (SDS), which is an anionic surfactant used in previous studies to mimic membrane binding. However, while the structure of the αSyn and SDS complex has been studied widely by various static measurements, the process of structural change from the denatured state to the folded state remains unclear. In this study, the interaction dynamics between αSyn and SDS micelles was investigated using time-resolved measurements with a micro-stopped-flow system, which has been recently developed. In particular, the time-resolved diffusion based on the transient grating technique in combination with a micro-stopped-flow system revealed the gradual change in diffusion triggered by the presence of SDS micelles. This change is induced not only by binding to SDS micelles, but also by an intramolecular conformational change. It was interesting to find that the diffusion coefficient decreased in an intermediate state and then increased to the final state in the binding reaction. We also carried out stopped-flow-kinetic measurements of circular dichroism and intramolecular fluorescence resonance energy transfer, and the D change was assigned to the formation of a compact structure derived from the helix bending on the micelle.
Introduction
α-Synuclein (αSyn) is a protein of 140 residues localized at the presynaptic terminals of neurons.1–3 The recombinant protein exists as a disordered monomer, and it is classified as an intrinsically disordered protein.4 αSyn is a principal component of Lewy bodies, which are accumulated in the brains of patients with Parkinson's disease.5,6 Recombinant human αSyn has been shown to form toxic oligomers and amyloid fibrils,7,8 and binds to lipid bilayers or vesicles.9,10 The conformation of αSyn depends on the environment; that is, it forms a random coil in aqueous solution, and mostly helical structures, particularly in the N-terminal region, upon association with negatively charged vesicles.11–13 It has been reported that αSyn also interacts with micelles of anionic surfactants, such as sodium dodecyl sulfate (SDS), and forms an α-helical structure.14 SDS has been widely used in biophysical studies to mimic the membrane interaction of αSyn.Previous studies have shown that the natively unfolded structure of αSyn exhibits multistate folding from a random coil structure to an α-helical folded state with increasing concentration of SDS ([SDS]).15,16 At a lower SDS/αSyn ratio, αSyn forms an incomplete helical form in the N-terminal region, and SDS molecules aggregate on αSyn. At this low concentration, αSyn tends to aggregate to ultimately form fibrils.16–18 With increasing [SDS], αSyn forms an extended helix in the N-terminal region by forming a complex with SDS molecules.19,20 At higher [SDS], αSyn interacts with micelles to form a bent (horseshoe) helix, which consists of two α-helices (Val3–Val37, Lys45–Thr92).21 Moreover, not only the secondary structure, but also the overall structure of the protein–SDS complex, including the size and shape of the micelle around the protein, changes depending on [SDS].16,18
Although a number of previous studies elucidated the complex structure of αSyn and SDS, most of them have focused on the static structures under equilibrium conditions. The dynamics of the binding process from the monomeric disordered state to the micelle-bound state has not been elucidated. To understand the interaction between αSyn and anionic surfactants and associated conformational change at the molecular level, the dynamical behavior of the binding process needs to be clarified. In this study, the dynamics of the conformational change from the unstructured state of αSyn to the α-helix formation with the SDS micelle was studied using time-resolved measurements.
For the time-resolved detection of the reaction, a solution mixing method using a stopped-flow (SF) system combined with some spectroscopic techniques should be appropriate. To detect the association states and intermolecular interaction between αSyn and SDS, we probe the diffusion coefficient (D) in particular because this property is sensitive to these changes.22,23 Therefore, the time-resolved diffusion (TRD) detection technique should be suitable for this work. However, with the traditional methods of D measurement, a longer time is required, and it has been impossible to trace the time course. The transient grating (TG) technique has changed the situation. Upon creating spatial modulations of concentrations of chemical species by pulsed light, D can be measured in a time-resolved manner. Utilizing high time resolution, this technique has been applied to studies on photochemical reaction mechanisms, including conformational changes during protein reactions.24–27 Although this technique is powerful, the target reaction must be photo-initiated in principle. This restriction has limited the targets to photochemical reactions by the TG method. Here, we applied a recently developed technique, stopped-flow TG (SF-TG)28 to detect the dynamics of αSyn-binding to the SDS micelle from the view point of diffusion. In the SF-TG technique, a reaction is triggered by mixing two solutions, and pulsed grating light is irradiated at various delay times to monitor D during reactions. There are two serious problems for applying this SF-TG to protein reactions by commercial SF systems; i.e., sample volume and time resolution. Usually, it is difficult to prepare a large volume of protein samples, so that the solution volume for the SF system has to be very small. Furthermore, the time resolution of the TRD measurement is determined by a speed of the turbulent flow ending in the mixing chamber. To solve these difficulties, we have developed a micro-stopped-flow (μ-SF) system.28 In this apparatus, the sample solution is introduced into the observation cell with high-pressure compressed air and mixed quickly in a micro-chamber. The total volume of the mixer and the observation window is about 3 μL, by which we can significantly reduce sample consumption. Furthermore, the small volume helps to stop the solution flow (turbulence) quickly.
Although we can start the reaction by solution mixing, photolabeling of the target protein is still necessary to detect diffusion of a protein by the TG method.29 For this purpose, we labeled αSyn with a photoresponsive molecule. It is desirable to use a small molecule to avoid disturbing the target reaction and protein diffusion. Furthermore, the probe molecule should give rise to a strong TG signal. In our previous work, we have reported a candidate for such a purpose, that is, one of the spiropyran30 derivatives, N-succinimidyl-3-[3,3-dimethyl-6′-nitrospiro[2′H-chromene-2,2'-(2,3,-dihydro-1H-indole)]-1 yl]-propionate (NSP) (Fig. SI-1A†).31 In this work, for site-specific labeling of αSyn, another type of spiropyran derivative, 3-{3′,3′-dimethyl-6-nitrospiro[chromene-2,2′-indole]-1′-yl}-N-[2-(2,5-dioxopyrrol-1yl)ethyl]propanamide (DSP) was used. Upon photoexcitation of these dyes at 308 nm, the ring-closed spiropyran (SP) form is converted to the ring-opened merocyanine (MC) form, which gives rise to a strong TG signal. This DSP dye can label Cys residues. Since there is no Cys residue in αSyn, a Cys residue is added at the end of the C-terminal to label αSyn only at a specific point. Hereafter, we refer to this Cys-added αSyn as αSynC and that with DSP labeling as αSynCD.
In this study, the SF system was used not only for TRD detection, but also for stopped-flow kinetic measurement of circular dichroism (SF-CD) and Förster resonance energy transfer (SF-FRET). Combining these data, we elucidated the dynamical process of the αSyn binding reaction on the SDS micelle. This work clarified the micelle-binding dynamics involved in the secondary structure and intramolecular tertiary structure change of αSyn that occurs upon SDS binding.
Materials
Protein preparation
A Cys residue was added as a 141 residue at the end of the wild type human αSyn sequence for the TG measurement (αSynC), or a G7C/G84C double mutation was introduced for the FRET measurement. These mutants were generated by site-directed mutagenesis using the pET-28a plasmid. Recombinant αSynC and G7C/G84C mutants were expressed in Escherichia coli BL21 (DE3) strain transformed with the pET-28a vector. These proteins were produced by a purification protocol described previously32 with some additional steps. Briefly, 1 L cultures (20 g tryptone, 10 g dried extract yeast, 5 g D-(+)-glucose, 5 g NaCl, 8.7 g K2HPO4, pH adjusted to 7.2) of E. coli were grown at 37 °C with shaking at 120 rpm and induced for expression of αSyn mutants when the OD600 reached 0.6–0.8 by 0.1 mM isopropyl-β-D-thiogalactopyranosid (IPTG). After 4–5 h of expression, the cells were pelleted by centrifugation. The mutants were released from the periplasm by the osmotic shock protocol. The supernatant was dialyzed overnight with 25 mM Tris–HCl, 5 mM dithiothreitol (DTT), pH 7.5, at 4 °C. The dialyzed solution was loaded on a Q-Sepharose column (Hitrap Q-HP, GE) equilibrated with 25 mM Tris, pH 7.5 running buffer, and eluted using a NaCl gradient. The purified fraction was boiled at 98 °C for 5 min. After the insoluble material was removed by centrifugation, the supernatant of αSynC was further purified by gel filtration with PBS buffer (pH 7.2). In the case of G7C/G84C, the supernatant was injected into a desalting column (Hitrap Desalting, GE) equilibrated with 25 mM Tris, pH 7.2. Before and after the ion exchange chromatography step, αSynC and G7C/G84C solutions were added to fresh DTT solution to prevent oxidization of the cysteine residues.SDS was purchased from Nacalai Tesque, Japan.
Labeling by NSP and DSP
For diffusion measurements in SDS solution without the proteins, NSP was dissolved in N,N-dimethylformamide (DMF) at an initial concentration of 20 mM and it was mixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 with 1 mM Tris in PBS buffer at pH 8.0. The mixed solution was incubated for 1 h, and the solution was filtered through a Cosmospin Filter G (0.2 μm, Nacalai Tesque, Japan) to remove precipitates. For DSP labeling of αSyn, DSP was dissolved in DMF, and 200 μM αSynC solution was mixed with an equimolar ratio of DSP. The mixed solutions were incubated for 3 h at 23 °C. After purification by size exclusion chromatography, the protein concentration was determined by DcAssay (Bio-Rad). The fraction of the labeling was determined by absorption measurement using a U-1900 Spectrophotometer (Hitachi) (ε350 = 11000 M−1 cm−1 for DSP and NSP31). The molar ratio of labeled DSP to αSyn was determined to be ∼0.8. NSP was synthesized as reported previously.31 DSP was synthesized by the coupling reaction of SP-COOH and N-(2-aminoethyl)maleimide with condensation reagent 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC). SP-COOH, 1-(β-carboxyethyl)-3,3-dimethyl-6-nitrospiro(indoline-2,2(2H-1)benzopyran) (57 μmol) was activated with EDC (68 μmol) in the presence of 3H-1,2,3-triazolo[4,5-b]pyridin-3-ol (68 μmol) in 0.8 mL DMF for 10 min at 25 °C. To this solution, N-(2-aminoethyl)maleimide (57 μmol) in 0.2 mL DMF was added and reacted for 10 min at 25 °C. The reactant was evaporated, and DSP was purified by flash column chromatography packed using a silica gel in a solvent of 20% methanol and 80% chloroform.
:40 with 1 mM Tris in PBS buffer at pH 8.0. The mixed solution was incubated for 1 h, and the solution was filtered through a Cosmospin Filter G (0.2 μm, Nacalai Tesque, Japan) to remove precipitates. For DSP labeling of αSyn, DSP was dissolved in DMF, and 200 μM αSynC solution was mixed with an equimolar ratio of DSP. The mixed solutions were incubated for 3 h at 23 °C. After purification by size exclusion chromatography, the protein concentration was determined by DcAssay (Bio-Rad). The fraction of the labeling was determined by absorption measurement using a U-1900 Spectrophotometer (Hitachi) (ε350 = 11000 M−1 cm−1 for DSP and NSP31). The molar ratio of labeled DSP to αSyn was determined to be ∼0.8. NSP was synthesized as reported previously.31 DSP was synthesized by the coupling reaction of SP-COOH and N-(2-aminoethyl)maleimide with condensation reagent 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC). SP-COOH, 1-(β-carboxyethyl)-3,3-dimethyl-6-nitrospiro(indoline-2,2(2H-1)benzopyran) (57 μmol) was activated with EDC (68 μmol) in the presence of 3H-1,2,3-triazolo[4,5-b]pyridin-3-ol (68 μmol) in 0.8 mL DMF for 10 min at 25 °C. To this solution, N-(2-aminoethyl)maleimide (57 μmol) in 0.2 mL DMF was added and reacted for 10 min at 25 °C. The reactant was evaporated, and DSP was purified by flash column chromatography packed using a silica gel in a solvent of 20% methanol and 80% chloroform.
Labeling for FRET experiments
For FRET experiments, G7C/G84C mutant was labeled with Alexa Fluor 488 C5 maleimide (F488, donor) and Alexa Fluor 594 maleimide (F594, acceptor) dyes (Fluoroprobes, Scottsdale, USA). The G7C/G84C mutant at 200 μM was first mixed with a 1.5-fold excess of F594 in a solution of 4 M guanidine hydrochloride, 500 μM tris(2-carboxyethyl)phosphine, and 25 mM Tris at pH 7.2, and the mixture was incubated at 4 °C overnight in the dark. The product was purified using a desalting column (HiTrap Desalting). The molar ratio of F594 to the protein was estimated to be approximately 1.4, based on the absorption (ε590 = 92000 M−1 cm−1). This ratio indicated that there were unlabeled sites in the protein. Then, this protein solution was mixed with a 5-fold excess concentration of F488 to produce a double-labeled protein. After unlabeled dye molecules were removed by desalting column chromatography (HiTrap Desalting), further purification was conducted using size exclusion chromatography (ENrich SEC 650 10 × 300 Column). The labeled protein concentration was determined by DcAssay. The molar ratio of labeled F594 to F488 was determined to be 1.5–2 using ε494 = 73000 M−1 cm−1 of F488. The sample should be a mixture of 7C-F488/84C-F594, 7C-F594/84C-F488, and 7C-F594/84C-F594 proteins. A double-F488-labeled protein may also exist in the sample. However, its fraction was expected to be smaller than those of others, because most of the protein was labeled by F594 in the first labeling step.
Measurements
In all the spectroscopic measurements, PBS buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl, pH 7.5) at 295 K was used. The concentration of αSynC, αSynCD, or G7C/G8 labeled with the fluorescent dyes was determined by DC assay.:1.In the SF-TG experiment, the signals were measured 20 times at each delay time, and the signals were averaged later.
In the SF-FRET experiment, laser light at 449 nm was focused on the mixing chamber of the μ-SF system. The fluorescence of F594 was detected by a photomultiplier tube with a 600 nm long-pass filter. The time-dependent fluorescence intensity after mixing solutions was detected 40 times for each [SDS], and the signals were averaged later. Background scattering light was detected for the PBS buffer without the labeled protein, and the signal was subtracted from the data. The dead time for the fluorescence measurement was ∼400 μs.28
Kinetic far-UV CD measurements (SF-CD) were carried out using an RX-2000 stopped-flow apparatus (Applied Photophysics, optical path length, 2 mm) and a CD spectrometer. In the SF-CD measurement, the reaction was initiated by mixing 40 μM protein solution and 6 mM SDS solution with a mixing ratio of 1:1. In the SF-CD measurement, the CD intensity at 222 nm (bandwidth, 5 nm) was monitored with a response time of 32 ms. The protein concentration was 20 μM. For each [SDS], the SF-CD signal was recorded 10 times, and the signals were averaged later.
Principle of TG method
The principle of the TG method has been reported before.34 Briefly, upon photoexcitation of the solution by the grating light, the sinusoidal modulations of the concentrations of the reactant and the product are created to lead the sinusoidal modulation in the refractive index (δn). Under the present experimental conditions, the signal solely consists of the thermal grating (δnth(t)) and created (or depleted) chemical species by the photoreaction (species grating). The species grating signal intensity is given by the difference of the refractive index changes from the reactant (δnR > 0) and the product (δnP > 0). If the photoreaction is fast enough and D is time-independent in the observation time window, the TG signal (ITG(t)) is expressed as|
ITG = α{δnthexp(−Dthq2t) + δnPexp(−DPq2t) − δnRexp(−DRq2t)}2
| (1) |
Micelle formation in SDS solution
The critical micelle concentration (cmc) of SDS under the present experimental conditions was measured using the diffusion detection of NSP. Since NSP is a hydrophobic probe, it partly exists in the SDS micelle. D was measured using the TG method. A typical TG signal of NSP at 20 μM in the PBS buffer upon photoexcitation of the SP form probed at 633 nm and q2 = 3.6 × 1012 m−2 is depicted in Fig. SI-1A.† The signal rose with a response time of our system, decayed to the baseline in the microsecond time region, and showed a slower decay profile. The faster decay component is the thermal grating signal, which decays with the rate constant of Dthq2. The slower one corresponds to the NSP diffusion signal, and it was well reproduced by a single exponential function, indicating DP = DR in eqn (1). From the decay rate constant, D of NSP (Dnsp) was determined to be (3.8 ± 0.1) × 10−10 m2 s−1.When SDS was added to the NSP solution, the decay rate of the diffusion signal did not change much until 1.0 mM SDS. With increasing [SDS] more than 1.3 mM, the decay became slower (Fig. SI-1B and C†). This slower decay indicates that the diffusion of NSP becomes slower due to micelle formation. The TG signals at higher [SDS] were fitted by eqn (2),
|
ITG = α{δnnspexp(−Dnspq2t) + δnmicexp(−Dmicq2t)}2
| (2) |
Result
SDS-induced helix formation of αSyn
First, the influence of the labeling on the secondary structure of αSyn was examined by the CD spectra under static conditions. In the absence and presence of SDS, the CD spectra of αSynC and αSynCD at 20 μM are shown in Fig. 1A. Both spectra are almost identical and very similar to those reported previously for αSyn,15 indicating that Cys addition and DSP-labeling do not introduce perturbations in structural properties of the protein. This observation is reasonable, because αSyn binds to SDS in the N-terminal region.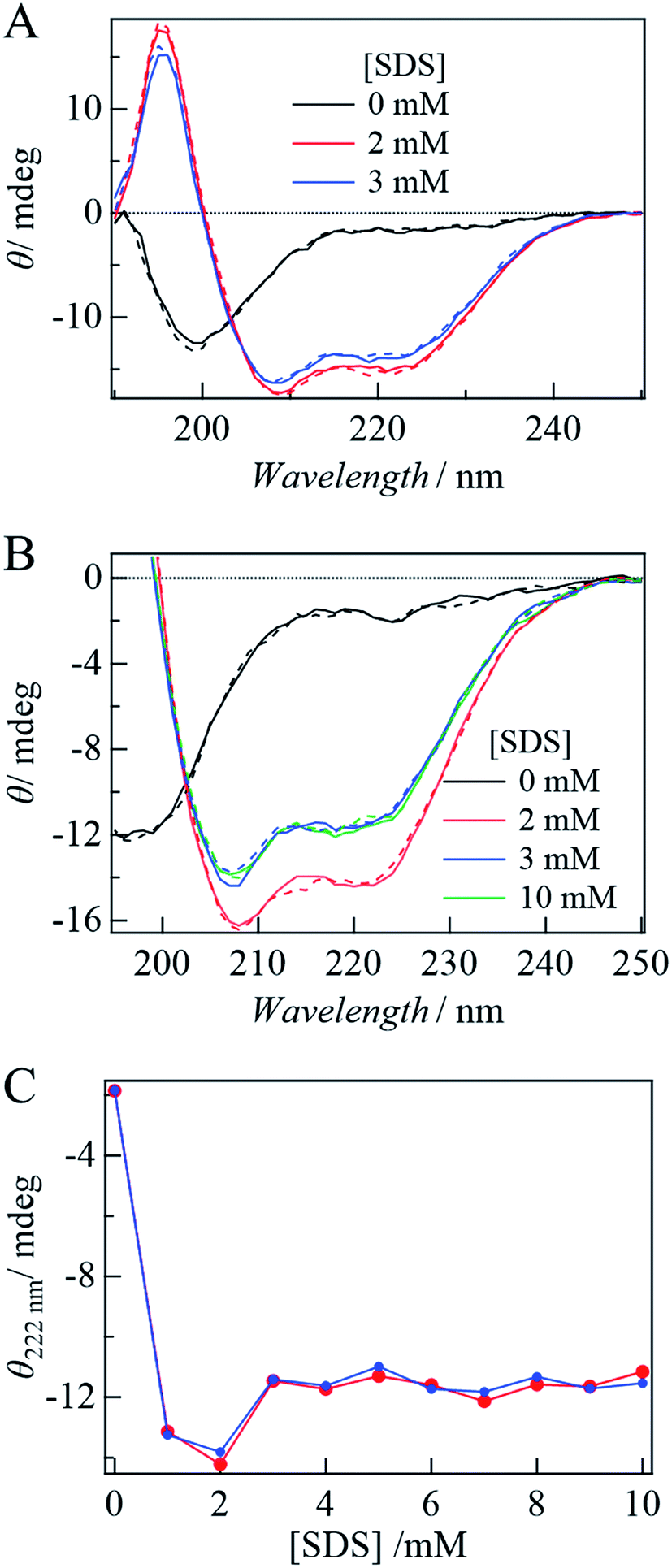 | ||
| Fig. 1 (A) CD spectra of αSynC (broken lines) and αSynCD (solid lines). (B) [SDS] dependence of CD spectra of αSynCD in the SP form (solid lines) and the MC form (broken lines). (C) Plot of ellipticities of αSynCD at 222 nm against [SDS] in the SP (blue) and MC forms (red). | ||
With the addition of SDS, the signal intensity of CD increases, indicating an increase in the α-helix content (Fig. 1A and B). This increase is independent of the state of DSP (MC or SP) (Fig. 1B), indicating that labeling by DSP and the photoconversion (SP to MC state) of DSP do not prevent αSyn adopting N-terminal helical conformation. Fig. 1C shows the [SDS] dependence of the CD intensity at 222 nm. The CD intensity is strong at 1–2 mM SDS, slightly decreases at 3 mM, and remains almost constant in the higher concentration region (Fig. 1C). This behavior agrees well with previous reports.15,16 The increase in the CD intensity at 1 mM can be explained by α-helix formation in the N-terminal region of αSyn. The subsequent decrease from 2 to 3 mM is assigned to the change of the α-helix from an elongated state to a bent horseshoe helix form.
Complex formation of αSyn with SDS by static diffusion measurement
We probed the effect of SDS on D of αSynCD (Fig. 2A) using the static TG technique. In the absence of SDS, the signal showed two decay components consisting of the thermal and species gratings (Fig. 2B). The species grating signal is fitted by a sum of two exponential functions;|
ITG = α{δnsexp(−Dsq2t) + δnfexp(−Dfq2t)}2
| (3) |
 | ||
| Fig. 2 (A) The domain structure of αSynCD. Added cysteine residue (141C) is shown by the yellow part. (B) A typical TG signals of αSynCD without SDS at q2 = 3.5 × 1012 m−2. An illustration of the photoconversion of the labeled DSP is inserted. (C) SDS dependence of the diffusion signals of αSynCD at q2 = 3.5 × 1012 m−2. The fitted curves by eqn (3) for [SDS] = 0 mM and eqn (4) for the others are shown by the broken lines. | ||
Addition of SDS to the solution changes the diffusion signal. A characteristic rise-decay profile appears and gradually increases with increasing [SDS] (Fig. 2C). The rise-decay curve implies that the DP and DR of αSyn in eqn (1) are different. Based on the fact that δnth is negative, we determine the signs of the decay and rise components to be positive and negative, respectively. Hence, the decay rate represents DPq2 (MC state), and the rise corresponds to DRq2 (SP state) in eqn (1).
To analyze the signal, the diffusion signal is measured at various q2 values at 3 mM SDS and shown in Fig. 3A. If the light-induced D change occurs on the observation time scale, this process should be considered for the analysis. However, the q2t plot of the diffusion signal is independent of q2 (Fig. 3B and SI-2†), indicating that the structural change associated with the photoreaction is completed before the observation time; that is, D is time-independent.
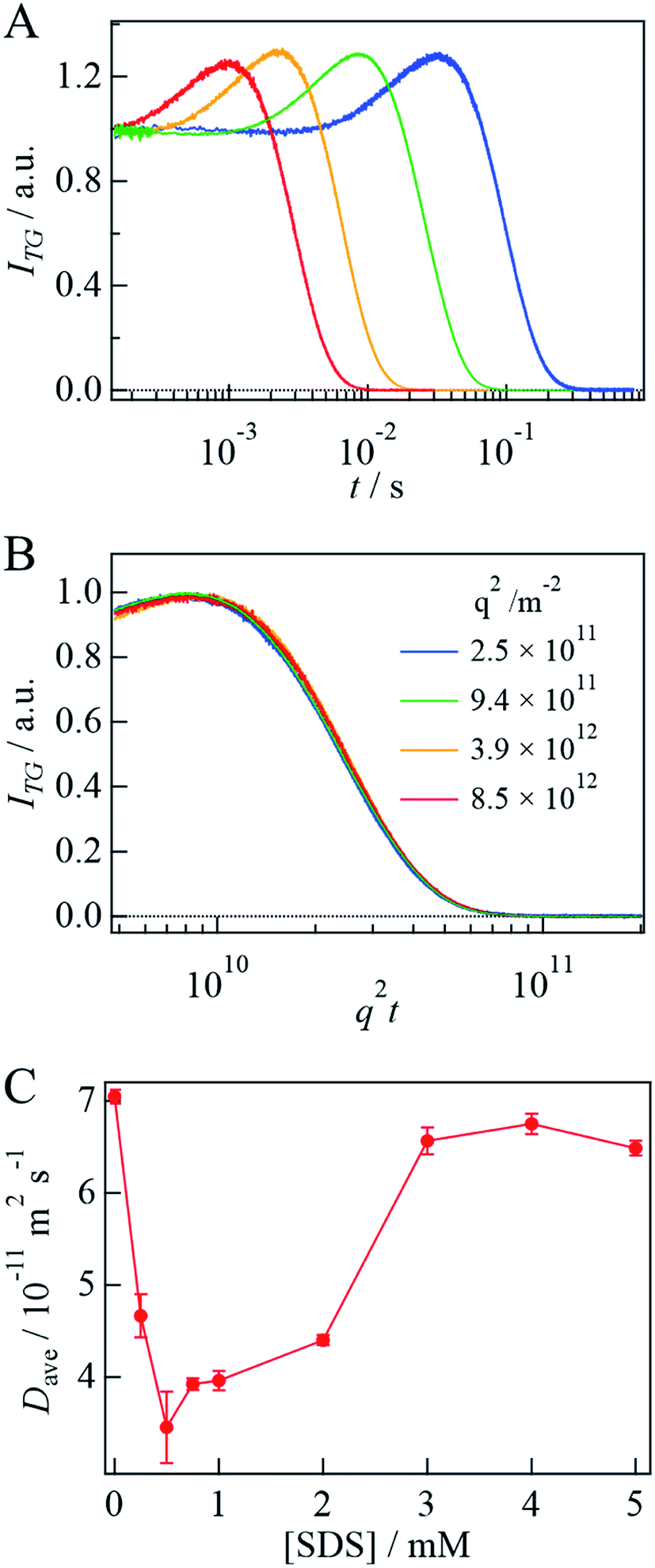 | ||
| Fig. 3 (A) The q2 dependence of the diffusion signal of αSynCD at [SDS] = 3 mM and at q2 = 8.5 × 1012 m−2, 3.9 × 1012 m−2, 9.4 × 1011 m−2, and 2.5 × 1011 m−2, from left to right. (B) q2t plot of the diffusion profiles of (A). (C) Plot of Dave of αSynCD against [SDS]. | ||
Based on these results, the rise-decay profiles should be fitted by eqn (4), which takes into account the diffusion of the reactant and the product;
|
ITG = α{δnPexp(−DPq2t) − δnRexp(−DRq2t) + δnfexp(−Dfq2t)}2
| (4) |
Dynamics of the complex formation by SF-TG
Before the evaluation of kinetic parameters by SF-TG, we determined the dead time of SF-TG under the present experimental conditions. When the delay time after solution mixing is shorter than 100 ms, the decay of the diffusion signal is faster than that in the static condition (Fig. SI-3†). This acceleration is due to the solution turbulence after mixing. At delay times longer than 100 ms, the diffusion signal is almost identical each other. Hence, the dead time is estimated to be 100 ms.Upon mixing equal volume of the two solutions of 40 μM αSynCD and 6 mM SDS, the final concentration of SDS is rapidly changed to 3 mM, which is sufficiently above the cmc (1.3 mM). Compared to the decay signal of the unfolded state (black line in Fig. 4A), a profile significantly changed at a delay time of 150 ms, at which a rise-decay profile was observed, and then the rise-decay curve gradually changes around 150–5000 ms (Fig. 4A). Since the diffusion time (∼10 ms) is sufficiently short compared with the delay time, the signals can be analyzed only by the diffusion process without considering the reaction. The diffusion signals are fitted by eqn (4), and the Dave are plotted against the delay time (Fig. 4B). The observed time dependence of Dave is fitted by a single exponential function. Referring to the intermediate state that appears just after the solution mixing within the dead time of the SF system as an I-state, the D change from the I-state to the final state at the longest delay time is determined to be (5.5 ± 0.1) × 10−11 m2 s−1 to (6.9 ± 0.1) × 10−11 m2 s−1 with a rate constant of 0.8(±0.3) s−1. The D of the I-state was smaller than that of the unfolded state ((7.0 ± 0.1) × 10−11 m2 s−1), and it increased to the final state of the complex. It should be noted that the initial process completes within the dead time (100 ms).
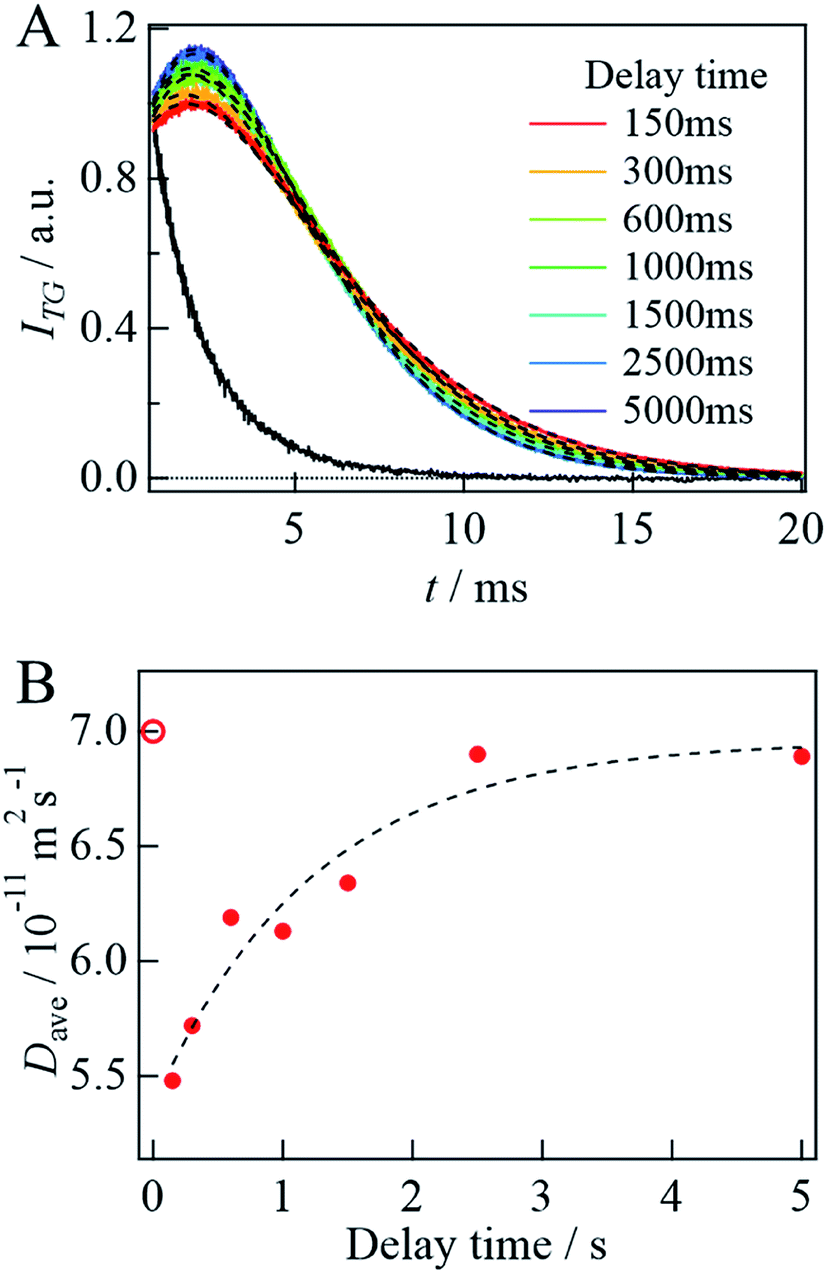 | ||
| Fig. 4 (A) The delay time dependence of the diffusion signal after mixing αSynCD with SDS solutions at q2 = 3.8 × 1012 m−2. A black solid line shows the signal obtained by mixing αSynCD with PBS buffer in the absence of SDS at delay time of 5000 ms. The best fitted curves by eqn (4) are shown by the broken lines. (B) Plot of Dave against the delay time (filled circles). The broken line shows a fitted curve by a single exponential function. The calculated D of the unfolded state is shown by an open circle. | ||
Detection of secondary structural changes by SF-CD
The secondary structural change dynamics is detected by the SF-CD measurement. When 40 μM αSynC and 6 mM SDS solutions were mixed at a 1:1 volume ratio, αSynC exhibited a significant increase in CD signal at 222 nm within a response time of approximately 50 ms (a red line in Fig. 5) compared to the unfolded state (a black line in Fig. 5). Hence, helix formation was completed within the dead time of the measurement. After this initial state, the CD intensity decreased, which suggested that the α-helix content decreased in this time domain. Since CD signal is not observed in the absence of αSynC, the change in CD intensity after mixing of αSynC and SDS is due to the structural change of αSynC. The time profile is fitted by a single exponential function with a rate constant of 0.55(±0.07) s−1. It is interesting to note that this rate is similar to that of the change in Dave obtained by the SF-TG measurement. This result suggests that the helical structure changes at the same time that the complex conformation changes in the transition from the I-state to the final state. The decrease in the helical content is about 20%, which is similar to the CD change from 2 mM to 3 mM SDS (Fig. 1C). Therefore, we consider that the observed change in the CD intensity corresponds to the transition of the N-terminal helical structure from the extended to the horseshoe helix.
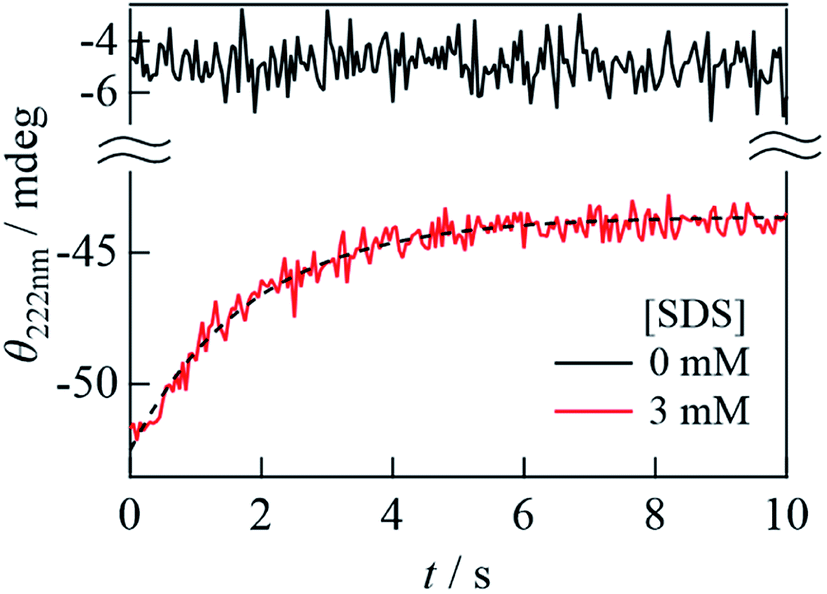 | ||
| Fig. 5 The time dependence of the CD intensity of αSynC at 222 nm after solution mixing by the SF system (red line). The broken line shows a fitted curve by a single exponential function. The solid black line shows the trace obtained by mixing equal volume of αSynC and PBS buffer in the absence of SDS under the same condition as the SDS mixing. | ||
Detection of intramolecular structural changes by SF-FRET
Previously, the decrease in the CD intensity at high [SDS] was explained in terms of the bent horseshoe helix formation in the N-terminal region.19 This suggestion was confirmed by a FRET measurement between two dye molecules located at both ends of the N-terminal helical region (3–92 residues).19,39 Here, we measured FRET of double-labeled protein at positions 7 and 84 with F488 (donor, λem = 517 nm) and F594 (acceptor, λem = 617 nm) (Fig. 6A) to examine the time-dependent conformational change detected by TRD and CD methods. If helix bending occurs in the observation time range, the distance between both ends of the helical region becomes short, so that the FRET efficiency increases.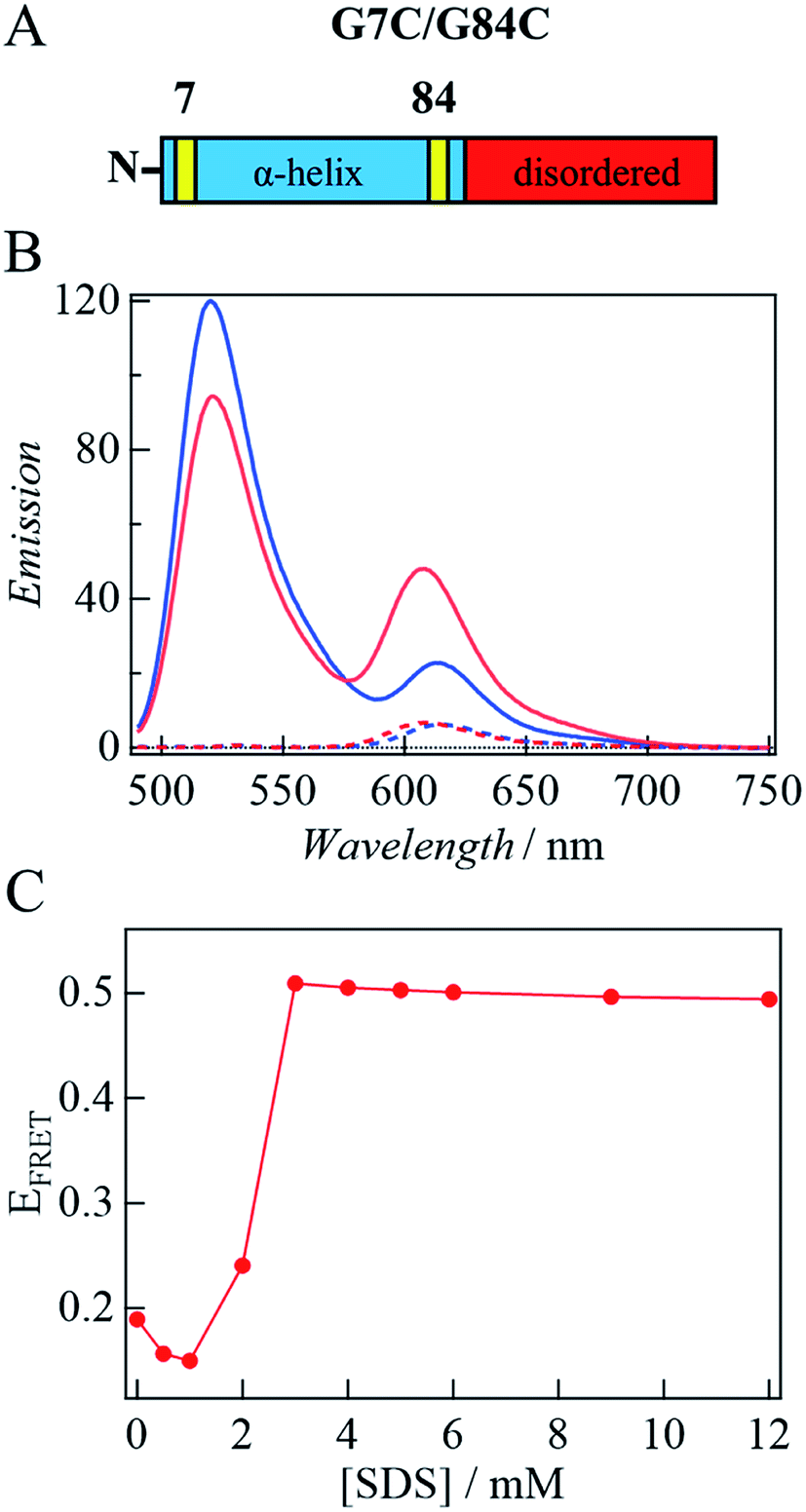 | ||
| Fig. 6 (A) The domain structure of G7C/G84C. Replaced cysteine residues are shown by the yellow parts. (B) Fluorescence spectrum of αSynAD without SDS (blue) and that at 3 mM SDS upon photoexcitation at 449 nm (red). Broken lines show the spectrum of αSynA upon photoexcitation at 449 nm. (C) Plot of EFRET against [SDS] estimated from the peak intensity. | ||
First, the fluorescence spectrum of αSyn labeled only by the acceptor (αSyn-A) is compared with αSyn labeled by the acceptor and donor (αSyn-AD) upon photoexcitation at 449 nm under the same conditions (Fig. 6B). The fluorescence from αSyn-A is very weak, because the absorbance of the acceptor at 449 nm is small. On the contrary, the fluorescence of the acceptor from αSyn-AD is strong. This observation indicates that the excited state energy of the donor is transferred to the acceptor by the FRET process in the buffer without SDS. This result is consistent with those of the previous studies, since the distance between residues of 7 and 84 in the unfolded state is calculated to be ∼5 nm and this distance is close enough for FRET (<10 nm).19,39,40 A similar behavior was also reported in other disordered proteins by single molecule FRET experiments.41,42
Next, SDS is added to the solution. For αSyn-A sample, the fluorescence intensity of the acceptor does not change by adding SDS. Contrastingly, for the αSyn-AD sample, the donor fluorescence intensity decreases and acceptor fluorescence increases with SDS addition (e.g., 3 mM SDS in Fig. 6B). This suggests that the distance between both ends of the helix decreases. The ratio of the fluorescence peak intensity of the acceptor to that of the donor (EFRET) is plotted against [SDS] (Fig. 6C). It shows that EFRET once decreases at 1 mM SDS and then increases around 2–3 mM, and remains almost constant in the higher concentration region. This trend agrees with previous reports showing the formation of a bent horseshoe helix at high [SDS].19,39
Fig. 7A depicts the time dependence of the acceptor fluorescence intensity after solution mixing. Within the dead time of this system (∼1 ms), the acceptor fluorescence intensity increases rapidly, and then gradually increase over a time range of several seconds. The gradual change is well reproduced by a single exponential function and the rate constant is determined to be 0.45(±0.03) s−1. With increasing [SDS], the rate of change is found to be faster (Fig. 7B). The fluorescence time trace is reproduced by a single exponential function, and the determined rate constant (kFRET) is plotted against [SDS], as shown in Fig. 7C. We determine the second-order rate constant from the slope of the plot to be 130 M−1 s−1. In the SDS solution at a concentration higher than the cmc, the SDS monomer concentration is considered to be constant and the micelle concentration increases.43 Since the cmc is 1.3 mM, the SDS dependence of the kinetic trace suggests that helix bending occurs with micelle binding. Assuming that the aggregation number of the micelle is 70,16 the second-order rate constant on the micelle concentration is 9 × 103 M−1 s−1.
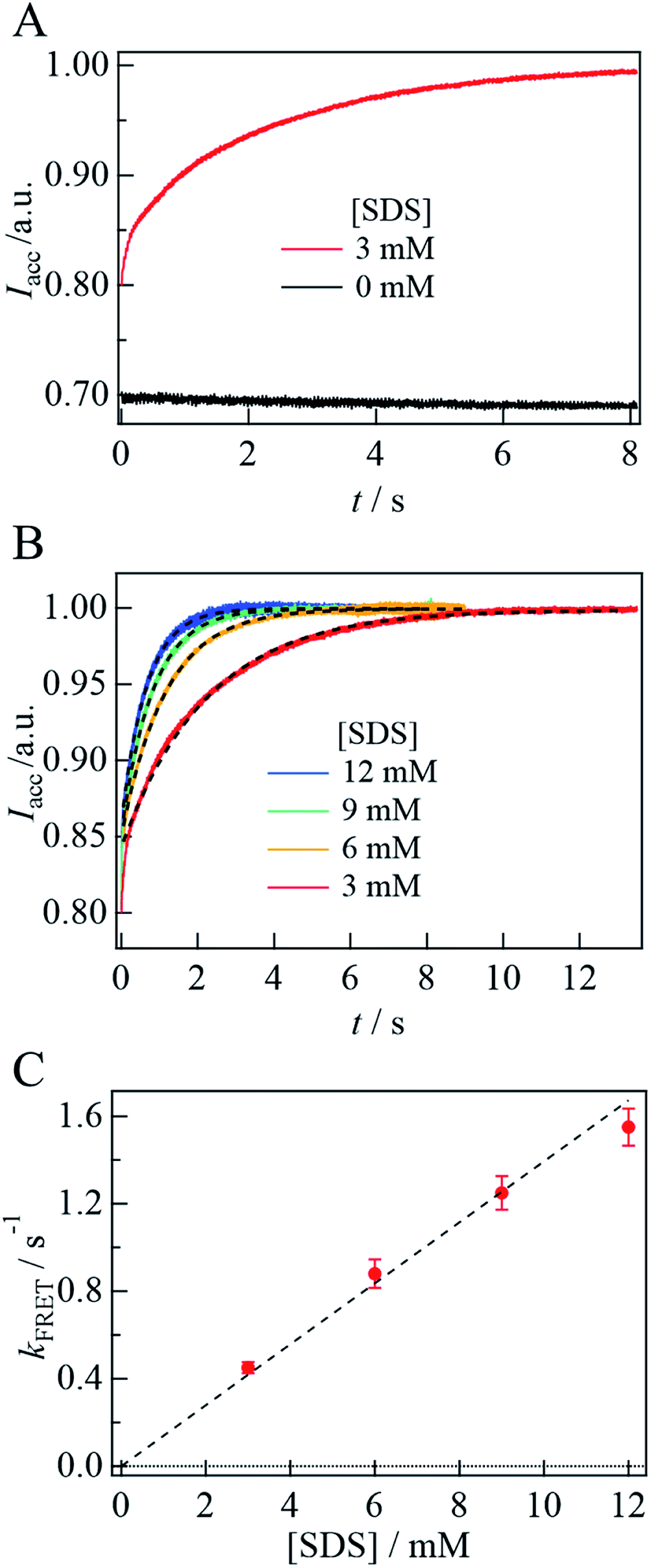 | ||
| Fig. 7 (A) The kinetic trace of the acceptor fluorescence (Iacc) of αSynAD upon photoexcitation at 449 nm after the SDS solution mixing (red). The acceptor fluorescence intensity after the mixing with the PBS buffer without SDS measured under the same condition is shown by the black line. (B) The time dependence of the acceptor fluorescence intensity after the SDS mixing at various [SDS] are fitted by a single exponential function (broken lines). The time traces were normalized by the intensity at the plateau regions. (C) The rate constant of the emission change of the acceptor (kFRET) is plotted against [SDS]. The plot was fitted by a linear function (broken line). | ||
Discussion
Before discussing the time-resolved measurements, let us summarize the conformational changes of αSyn interacting with SDS, investigated by TG, CD, and FRET measurements under static conditions. In the transition from the absence of SDS to the [SDS] range of 1–2 mM, the CD intensity increases significantly (Fig. 1C), D decreases by approximately half (Fig. 3C), and EFRET slightly decreases at 1 mM, followed by a slight increase at 2 mM (Fig. 6C). According to previous studies, these changes can be explained as follows. While the αSyn structure is unfolded in the buffer without SDS, the increase in the CD intensity indicates α-helix formation upon interaction with SDS. The decrease in D represents the increase in the size of αSyn due to the formation of the αSyn–SDS complex. Previously, it has been reported that D of an unfolded protein is about 1.5–2 times smaller compared with that of the folded protein44,45 because of the intermolecular hydrogen bonding between the protein and water.44 Hence, the decrease in D accompanying the formation of α-helix should be explained in terms of the increase in the size by the SDS complex formation to compensate for the decrease in the diffusion friction due to α-helix formation. An NMR structural analysis of the complex showed that while the N-terminal region is bound to the SDS micelle and forms a helical structure, the region after Gly93 is unbound to the micelle and unstructured, and the region of Asp98–Ala140 has highly mobile fluctuations.21 Therefore, the effect of the formation of the helical structure might be relatively smaller than in the case of complete folding of a protein. The slight decrease in EFRET at 1 mM indicates that the distance between residues 7 and 84 is longer than that in the unfolded state. We consider that this distance in the unfolded state is rather short due to a compact structure for FRET, whereas the distance is expected to be long in an extended α-helix form. Therefore, the origin of the slight decrease in EFRET is attributed to the extended α-helix formation. Even in this state, EFRET is not zero. This non-zero EFRET in the extended form has been observed previously19 and this observation implies that the distance between 7 and 84 is not long enough for negligible FRET. However, we cannot exclude another possibility, i.e., a conformational heterogeneity at 1 mM SDS. It may be reasonable to consider that αSyn is in equilibrium between the extended and the micelle-bound-horseshoe helix near the cmc. In the horseshoe helix, the distance should be shorter than that in the extended helix, and a small amount of the horseshoe component contributes to the non-zero FRET at 1 mM SDS. With increasing [SDS], the EFRET increases, suggesting that a bending of the α-helix is dominant.When [SDS] is increased to more than 3 mM, the CD intensity slightly decreases, D increases to almost that in the buffer solution, and EFRET also increases. The slight decrease in the CD intensity can be explained by the bent horseshoe α-helix structure. This interpretation is consistent with the increase in EFRET. The increase in D could be explained by two effects: D decreases due to complex formation with SDS micelles, and increases when the protein conformation on the SDS surface becomes compact. A previous small-angle X-ray scattering (SAXS) study has reported that the maximum diameter of αSyn having an elongated helix is 1.5–2 times larger than that of the complex with the bent helix on the micelle.16 In this study, Dave obtained from TG at 3 mM or higher [SDS] is 1.5 times larger than that in the 1–2 mM concentration range. Although there is no direct relationship between the maximum diameter estimated from SAXS measurements and the hydrodynamic radius from the diffusion measurement, it is interesting to find that these factors agree well. Furthermore, a previous study reported deformation of the globular micelle with protein binding.21 This deformation of the micelle shape may also contribute to the increase in D.
The time-resolved measurements, in combination with the SF system, revealed the kinetics of the conformational change and binding of αSyn on the SDS micelles. According to the SF-CD measurement, the CD intensity dramatically increased within the response time of 50 ms, followed by a slight decrease toward the final state. These changes seem to be similar to the changes observed in the [SDS] dependence experiment from the diluted to concentrated SDS conditions (Fig. 1C). Although this coincidence could be accidental, it may be reasonable to explain the observed time dependence by a process from the I-state possessing an extended α-helix in the N-terminal region to a final bent α-helix form (product).
The SF-FRET experiments showed that the FRET efficiency of the fluorescent molecules labeled on both ends of the N-terminal helix (position 7 and 84) increases within 1 ms of the addition of the SDS solution and further increased gradually. This result indicates that a rather compact α-helix formation is completed within 1 ms upon interaction with the SDS molecules and that the average distance between both ends of the N-terminal helix becomes shorter. Since EFRET at 1 ms is larger than that at 0 mM (Fig. 7A), the I-state could be slightly bent from the extended α-helix. Another possible origin is the heterogeneous contributions of the extended α-helix and the compact helix form, which is described above for the 1 mM SDS concentration. If both conformations are formed in the I-state, it may be reasonable to observe the slight increase in the FRET efficiency. After this process, the helix bent further by forming a complex with the SDS micelle on a time scale of several seconds, which is confirmed by the SF-CD signals. Interestingly, the rate of change is proportional to the micelle concentration, i.e., this step is the second-order reaction, suggesting that αSyn is transferred to the micelle to form the final product. Hence, αSyn does not interact with the micelle before this final state, but we speculate that αSyn interacts with the SDS monomers to form the extended α-helix conformation in the I-state.
The SF-TG experiments provide information on the change in the hydrodynamic radius. The observed time dependence indicates that D of the intermediate decreased from the unfolded state significantly, followed by an increase toward the micelle-bound state. As described above, D of the folded protein is larger than that of the unfolded protein in many cases. The decrease in D in the I-state may be an indication of the intermolecular interaction with many SDS monomers to form large aggregates. The increase in D after the formation of the final product, the αSyn–micelle complex, could be due to a compact (bend α-helix) form on the micelle after the transport from the SDS aggregates to the micelle.
There may be several explanations for the slow αSyn–micelle complex formation. It has been shown that at high [SDS], the N-terminal helix of αSyn is slightly embedded inside the micelle.21 The positively charged residues of the amphipathic N-terminal helix can interact with the negatively charged headgroups of SDS. Contrastingly, the inner helix, which is largely hydrophobic, interacts with the hydrophobic moiety of the SDS tails. If αSyn is surrounded by many SDS monomers in the I-state, as we suggested, these monomers must be released from αSyn before forming the complex with the micelle. This process could be slow. It has also been reported that the interaction of αSyn with the micelle leads to shape deformation of the micelle to a prolate ellipsoidal structure.21 Hence, the complex formation between αSyn and micelles results in not only structural changes in the conformation of αSyn, but also deformation of the micelle. These changes may also cause a slow transition from the intermediate to the final state in the binding reaction. The proposed reaction scheme based on the above considerations is shown in Fig. 8.
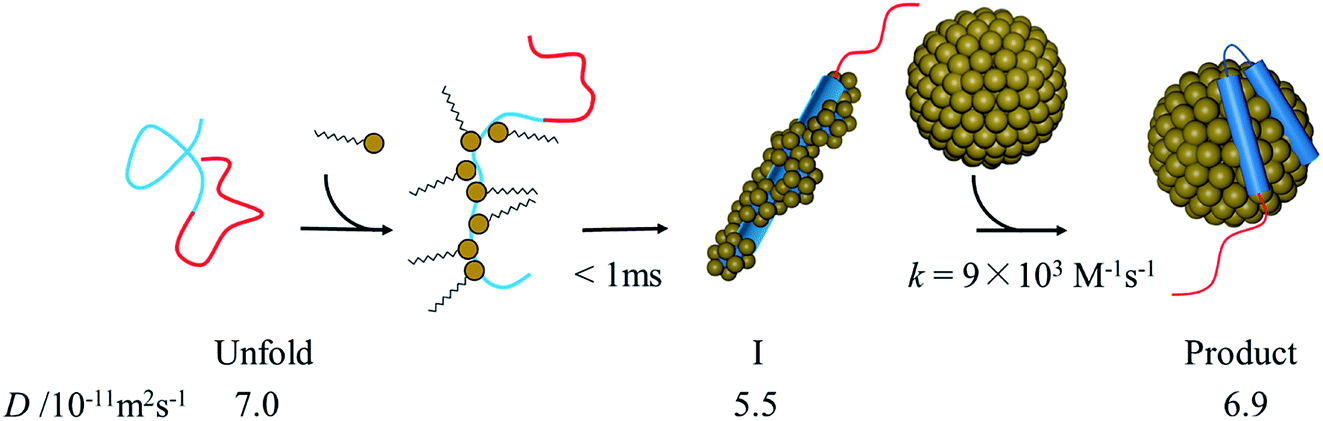 | ||
| Fig. 8 Illustration of proposed reaction scheme of αSyn–SDS interaction. | ||
Previous studies using molecular dynamics (MD) simulations suggested an assembly of SDS monomers in the presence of αSyn occurs within 100 ns.46 However, a stable helix formation was not observed within hundreds of nanoseconds simulation. Our results indicate that the formation of the extended helix is completed in the microsecond time scale. In the binding reaction, this intermediate interacts with the micelle, and the helix bending induced by binding to the micelle slowly proceeds in a time range of several seconds. In the MD simulation study, the αSyn–micelle complex formation was simulated, and the binding movement was observed within approximately 100 ns.46 However, in the present experimental study, the association of αSyn with the micelle occurred within a few seconds. This difference might be due to a simplified MD simulation, which was carried out by assuming no SDS monomers present in the initial state.
So far, SDS has been used to mimic interaction between αSyn and membrane in many studies. However, using lipid vesicles is also appropriate to understand the biological role of αSyn in cells. In fact, it has been reported that αSyn with lipid vesicles exhibits both the extended and the horseshoe helices and the extended form may be a major component.11 Our techniques presented here, in particular SF-TG method, can be used under such conditions.
Summary
The conformational change dynamics of αSyn induced by interaction with SDS micelles are studied in the time domain. The TRD measurement based on the TG detection as well as SF-CD, and SF-FRET experiments reveal the time course of the conformational change of αSyn upon interaction with SDS. Initially, the elongated helix was formed within 1 ms (the I-state) in the process of the interaction with many SDS monomers. The intermediate bound to the micelle with the second-order rate constant for [SDS]. We propose that the α-helix formation is completed by binding to the SDS monomers, followed by association with the micelle to form the bend structure.In this study, we also demonstrate that the μ-SF system we used for the detection by TG and FRET is very useful for the time-resolved studies of a sample that can be prepared only in minimal amounts, such as proteins. In particular, the time-resolved diffusion technique is a promising method for studies on protein reactions because D is sensitive to conformation changes, intermolecular interactions, and changes in size by forming protein complexes. It has been very difficult to detect the dynamics of these properties using other time-resolved measurements, such as CD and fluorescence measurements. For example, although the CD intensity reflects the secondary structure, the association/dissociation processes or intermolecular interaction cannot be detected by this method. The FRET experiment is powerful for detecting the distance change between the donor and acceptor, but it cannot detect again the other processes. In our previous paper, we demonstrated that the SF-TG technique is applicable to a pH jump experiment for a photoreactive protein. The present study is the first application of a reaction to a non-photoreactive protein. This novel method is a promising tool for studying protein interactions such as enzymatic reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-aid for Scientific Research on Innovative Areas (research in a proposed research area) (No. JP20107003, and JP25102004 to M. T.), a Grant-in-aid for Scientific Research from MEXT/JSPS (25288005, 17H03008 to M. T., 17H05001 and 20H04708 to Y. N.), and Research Foundation for Opto-Science and Technology (to M. T.)References
- P. H. Weinreb, W. Zhen, A. W. Poon, K. A. Conway and P. T. Lansbury Jr, NACP, A Protein Implicated in Alzheimer's Disease and Learning, Is Natively Unfolded, Biochemistry, 1996, 35, 13709–13715 CrossRef CAS.
- L. Maroteaux, J. T. Campanelli and R. H. Scheller, Synuclein: A Neuron-Specific Protein Localized to the Nucleus and Presynaptic Nerve Terminal, J. Neurosci., 1988, 8, 2804–2815 CrossRef CAS.
- L. Breydo, J. W. Wu and V. N. Uversky, α-Synuclein misfolding and Parkinson's disease, Biochim. Biophys. Acta, Mol. Basis Dis., 2012, 1822, 261–285 CrossRef CAS.
- B. Fauvet, M. K. Mbefo, M.-B. Fares, C. Desobry, S. Michael, M. T. Ardah, E. Tsika, P. Coune, M. Prudent, N. Lion, D. Eliezer, D. J. Moore, B. Schneider, P. Aebischer, O. M. El-Agnaf, E. Masliah and H. A. Lashuel, α-Synuclein in Central Nervous System and from Erythrocytes, Mammalian Cells, and Escherichia coli Exists Predominantly as Disordered Monomer, J. Biol. Chem., 2012, 287, 15345–15364 CrossRef CAS.
- M. H. Polymeropoulos, C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, E. S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T. Papapetropoulos, W. G. Johnson, A. M. Lazzarini, R. C. Duvoisin, G. Di Iorio, L. I. Golbe and R. L. Nussbaum, Mutation in the α-synuclein gene identified in families with Parkinson's disease, Science, 1997, 276, 2045–2047 CrossRef CAS.
- R. Kruger, W. Kuhn, T. Muller, D. Woitalla, M. Graeber, S. Kosel, H. Przuntek, J. T. Epplen, L. Schols and O. Riess, Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease, Nat. Genet., 1998, 18, 106–108 CrossRef CAS.
- H. A. Lashuel, C. R. Overk, A. Oueslati and E. Masliah, The many faces of α-synuclein: from structure and toxicity to therapeutic target, Nat. Rev. Neurosci., 2013, 14, 38–48 CrossRef CAS.
- N. Bengoa-Vergniory, R. F. Roberts, R. Wade-Martins and J. Alegre-Abarrategui, Alpha-synuclein oligomers: a new hope, Acta Neuropathol., 2017, 134, 819–838 CrossRef CAS.
- C. M. Pfefferkorn, Z. Jiang and J. C. Lee, Biophysics of α-synuclein membrane interactions, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 162–171 CrossRef CAS.
- A. Iyer and M. M. A. E. Claessens, Disruptive membrane interactions of alpha-synuclein aggregates, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 468–482 CrossRef CAS.
- S. Bekshe Lokappa and T. S. Ulmer, α-Synuclein populates both elongated and broken helix states on small unilamellar vesicles, J. Biol. Chem., 2011, 286, 21450–21457 CrossRef.
- C. C. Jao, A. Der-Sarkissian, J. Chen and R. Langen, Structure of membrane-bound-synuclein studied by site-directed spin labeling, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8331–8336 CrossRef CAS.
- M. Rovere, A. E. Powers, H. Jiang, J. C. Pitino, L. Fonseca-Ornelas, D. S. Patel, A. Achille, R. Langen, J. Varkey and T. Bartels, E46K-like α-synuclein mutants increase lipid interactions and disrupt membrane selectivity, J. Biol. Chem., 2019, 294, 9799–9812 CrossRef CAS.
- S. Chandra, X. Chen, J. Rizo, R. Jahn and T. C. Südhof, A broken α-helix in folded α-synuclein, J. Biol. Chem., 2003, 278, 15313–15318 CrossRef CAS.
- A. C. M. Ferreon and A. A. Deniz, α-Synuclein multistate folding thermodynamics: implications for protein misfolding and aggregation, Biochemistry, 2007, 46, 4499–4509 CrossRef CAS.
- L. Giehm, C. L. P. Oliveira, G. Christiansen, J. S. Pedersen and D. E. Otzen, SDS-Induced Fibrillation of α-Synuclein: An Alternative Fibrillation Pathway, J. Mol. Biol., 2010, 401, 115–133 CrossRef CAS.
- M. F. Ahmad, T. Ramakrishna, B. Raman and C. M. Rao, Fibrillogenic and Non-Fibrillogenic Ensembles of SDS-Bound Human α-Synuclein, J. Mol. Biol., 2006, 364, 1061–1072 CrossRef CAS.
- D. Ruzafa, Y. S. Hernandez-Gomez, G. Bisello, K. Broersen, B. Morel and F. Conejero-Lara, The influence of N-terminal acetylation on micelle-induced conformational changes and aggregation of α-Synuclein, PLoS One, 2017, 12, e0178576 CrossRef.
- A. C. M. Ferreon, Y. Gambin, E. A. Lemke and A. A. Deniz, Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5645–5650 CrossRef CAS.
- E. R. Georgieva, T. F. Ramlall, P. P. Borbat, J. H. Freed and D. Eliezer, The lipid-binding domain of wild type and mutant α-synuclein: Compactness and interconversion between the broken and extended helix forms, J. Biol. Chem., 2010, 285, 28261–28274 CrossRef CAS.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, Structure and dynamics of micelle-bound human α-synuclein, J. Biol. Chem., 2005, 280, 9595–9603 CrossRef CAS.
- M. Terazima, Time-dependent intermolecular interaction during protein reactions, Phys. Chem. Chem. Phys., 2011, 13, 16928–16940 RSC.
- T. Brand, E. J. Cabrita and S. Berger, Intermolecular interaction as investigated by NOE and diffusion studies, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 159–196 CrossRef CAS.
- M. Terazima, Studies of photo-induced protein reactions by spectrally silent reaction dynamics detection methods: applications to the photoreaction of the LOV2 domain of phototropin from Arabidopsis thaliana, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1093–1105 CrossRef CAS.
- K. Tanaka, Y. Nakasone, K. Okajima, M. Ikeuchi, S. Tokutomi and M. Terazima, Light-induced conformational change and transient dissociation reaction of the BLUF photoreceptor Synechocystis PixD (Slr1694), J. Mol. Biol., 2011, 409, 773–785 CrossRef CAS.
- M. Kondoh and M. Terazima, Conformational and Intermolecular Interaction Dynamics of Photolyase/Cryptochrome Proteins Monitored by the Time-Resolved Diffusion Technique, Photochem. Photobiol., 2017, 93, 15–25 CrossRef CAS.
- A. Takakado, Y. Nakasone and M. Terazima, Sequential DNA Binding and Dimerization Processes of the Photosensory Protein EL222, Biochemistry, 2018, 57, 1603–1610 CrossRef CAS.
- Y. Nakasone, S. Takaramoto and M. Terazima, Time-Resolved Diffusion Detection with Microstopped Flow System, Anal. Chem., 2019, 91, 11987–11993 CrossRef CAS.
- N. Baden and M. Terazima, A novel method for measurement of diffusion coefficients of proteins and DNA in solution, Chem. Phys. Lett., 2004, 393, 539–545 CrossRef CAS.
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- S. Takaramoto, Y. Nakasone, K. Sadakane, S. Maruta and M. Terazima, Spiropyran labeling for sensitive probing of protein diffusion by the transient grating method, Chem. Phys. Lett., 2020, 739, 136919 CrossRef CAS.
- C. Huang, G. Ren, H. Zhou and C. C. Wang, A new method for purification of recombinant human α-synuclein in Escherichia coli, Protein Expression Purif., 2005, 42, 173–177 CrossRef CAS.
- M. Terazima and N. Hirota, Measurement of the quantum yield of triplet formation and short triplet lifetimes by the transient grating technique, J. Chem. Phys., 1991, 95, 6490–6495 CrossRef CAS.
- M. Terazima, Diffusion coefficients as a monitor of reaction kinetics of biological molecules, Phys. Chem. Chem. Phys., 2006, 8, 545–557 RSC.
- R. M. Weinheimer, D. Fennell Evans and E. L. Cussler, Diffusion in Surfactant Solutions, J. Colloid Interface Sci., 1981, 80, 357–368 CrossRef CAS.
- G. Manzo, M. Carboni, A. C. Rinaldi, M. Casu and M. A. Scorciapino, Characterization of sodium dodecylsulphate and dodecylphosphocholine mixed micelles through NMR and dynamic light scattering, Magn. Reson. Chem., 2013, 51, 176–183 CrossRef CAS.
- Y. Wang, L. A. Benton, V. Singh and G. J. Pielak, Disordered protein diffusion under crowded conditions, J. Phys. Chem. Lett., 2012, 3, 2703–2706 CrossRef CAS.
- J. Bai, Z. Zhang, M. Liu and C. Li, α-Synuclein–lanthanide metal ions interaction: binding sites, conformation and fibrillation, BMC Biophys., 2016, 9, 1 Search PubMed.
- G. Veldhuis, I. Segers-Nolten, E. Ferlemann and V. Subramaniam, Single-molecule FRET reveals structural heterogeneity of SDS-bound α-synuclein, ChemBioChem, 2009, 10, 436–439 CrossRef CAS.
- S. S. Vogel, B. W. Van der Meer and P. S. Blank, Estimating the distance separating fluorescent protein FRET pairs, Methods, 2014, 66, 131–138 CrossRef CAS.
- S. Mukhopadhyay, R. Krishnan, E. A. Lemke, S. Lindquist and A. A. Deniz, A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2649–2654 CrossRef CAS.
- G. N. W. Gomes, M. Krzeminski, A. Namini, E. W. Martin, T. Mittag, T. Head-Gordon, J. D. Forman-Kay and C. C. Gradinaru, Conformational Ensembles of an Intrinsically Disordered Protein Consistent with NMR, SAXS, and Single-Molecule FRET, J. Am. Chem. Soc., 2020, 142, 15697–15710 CrossRef CAS.
- E. Kudryashov, T. Kapustina, S. Morrissey, V. Buckin and K. Dawson, The Compressibility of Alkyltrimethylammonium Bromide Micelles, J. Colloid Interface Sci., 1998, 203, 59–68 CrossRef CAS.
- S. Nishida, T. Nada and M. Terazima, Kinetics of intermolecular interaction during protein folding of reduced cytochrome c, Biophys. J., 2004, 87, 2663–2675 CrossRef CAS.
- K. Chattopadhyay, S. Saffarian, E. L. Elson and C. Frieden, Measuring unfolding of proteins in the presence of denaturant using fluorescence correlation spectroscopy, Biophys. J., 2005, 88, 1413–1422 CrossRef CAS.
- J. Tian, A. Sethi, D. Anunciado, D. M. Vu and S. Gnanakaran, Characterization of a disordered protein during micellation: Interactions of α-synuclein with sodium dodecyl sulfate, J. Phys. Chem. B, 2012, 116, 4417–4424 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Diffusion signal of NSP (Fig. SI-1), q2 dependence of the TG signal of αSyn (Fig. SI-2), diffusion signals of bovine serum albumin labeled with NSP (Fig. SI-3). See DOI: 10.1039/d0ra09614h |
| This journal is © The Royal Society of Chemistry 2021 |