
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1D and 2D hybrid polymers based on zinc phenylphosphates: synthesis, characterization and applications in electroactive materials†
Maciej Dębowski *,
Zbigniew Florjańczyk,
Andrzej Ostrowski,
Piotr A. Guńka,
Janusz Zachara,
Anna Krztoń-Maziopa,
Jakub Chazarkiewicz,
Anna Iuliano and
Andrzej Plichta
*,
Zbigniew Florjańczyk,
Andrzej Ostrowski,
Piotr A. Guńka,
Janusz Zachara,
Anna Krztoń-Maziopa,
Jakub Chazarkiewicz,
Anna Iuliano and
Andrzej Plichta
Faculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: maciej.debowski@pw.edu.pl
First published on 18th February 2021
Abstract
The synthesis, structure and properties of three hybrid polymers based on zinc arylphosphates are described in this study. Zinc bis(diphenylphosphate) (ZnDPhP) was obtained as needle-like crystals containing hexagonally packed, homochiral 1∞[Zn(DPhP)2/2] helical chains. The XRD and DSC studies revealed that upon heating, ZnDPhP undergoes a reversible thermal transition at ca. 160 °C with expansion mainly perpendicular to its c-axis. Zinc phenylphosphate hydrate (ZnMPhP-H) formed plate-like particles with an average thickness of less than 1 μm and much thinner nanolayers with a basal spacing of 15.5 Å. ZnMPhP-H was easily and reversibly dehydrated to its anhydrous form, ZnMPhP-A, which exhibited a somewhat larger basal spacing of 16.5 Å and the capacity for amine intercalation. The thermal decomposition of ZnDPhP or ZnMPhP-A began around 250 °C, resulting in the formation of solid mixtures of zinc phosphates and electron-conducting carbonaceous phases. The bulk electrical conductivities of the poly(vinylidene fluoride)-based composites containing the ZnDPhP pyrolyzates reached 0.1–0.2 S cm−1. Upon mixing with silicone oil, all the synthesized hybrid polymers formed fluids that exhibit significant negative electrorheological effects and have potential for application in electroresponsive smart materials. The application of an electric field during the crosslinking of such systems affected the viscoelastic properties of the resultant solid composites, while the cured systems showed rather small electrorheological effects.
Introduction
Hybrid inorganic–organic polymers based on metal phosphates and phosphonates have been known for more than four decades.1,2 Most research in this area has focused on structural aspects of these compounds, including the mechanism of self-organization between metal cations and phosphoorganic anions that leads to the formation of crystalline domains with one-dimensional (1D), two-dimensional (2D), or three-dimensional (3D) architectures from inorganic linear chains or layers separated by organic groups.3–12In recent years, numerous studies have shown that polymers containing metal organophosphate, phosphonate, or phosphinate monomeric units can potentially be applied as effective flame retardants,13–17 reinforcing fillers14–19 and nucleating agents in polymer composites,19–25 single-source precursors for phosphate ceramics,6,26,27 useful substrates for the synthesis of mesoporous and zeolitic materials,28,29 and catalysts.30,31 In particular, organophosphate compounds containing rigid aromatic substituents show great promise due to their excellent stability and large variety of potential applications. These compounds are regarded as a useful platform for the synthesis of new metal–organic frameworks with improved air stability and high-temperature resistance.10,32,33 Moreover, due to the presence of aromatic groups in the phosphate ligands, these materials exhibit some interesting magnetic34 and optical35–37 properties that can be utilized in future applications.
Recently, we reported that the pyrolysis of aluminum tris(diphenylphosphate) (AlDPhP) leads to the formation of aluminum phosphate fibers containing electrically conductive deposits made of reduced graphene oxide.38 Moreover, the application of magnetic field to epoxy composites incorporating AlDPhP nanoparticles resulted in a reorientation of the nanoparticles,34 indicating that they can be utilized as components of so-called smart materials.
Today, smart materials play an important role in materials chemistry and engineering since their properties can be easily altered by the application of various external stimuli (e.g., temperature, pressure, and electric or magnetic field) as well as by the incorporation of specific molecules.39 The fast and reversible responses of smart materials can be useful in a wide range of practical applications. For example, smart fluids responding to electric fields (EFs), are formed by electrically-polarizable particles dispersed in a non-conducting liquid medium,40–43 and may be utilized in dampers, valves, engine mounts, hydraulic actuators, squeeze mounts, medicine (e.g., in drug delivery or haptic medical devices), and food processing.44–47 The industrial applications of currently known electrorheological (ER) fluids are based on a positive ER effect,44,47,48 resulting from the EF-controlled orientation of dispersed particles (e.g., metal oxides,41,42 metal salts,43 and modified polymeric particles40) into chain-like structures accompanied by a transition of fluid from liquid to viscoelastic system. However, there are many applications in which a controllable decrease in viscosity would be desirable (e.g., hydrostatic journal bearing for controlling rotor vibrations or squeeze film damper for actively controlling rotor dynamics).45 Therefore, in recent years, many efforts have been made to develop and characterize new liquid systems with negative ER effects.45,48 Various types of particles derived from either metal salts (e.g., calcium alginate) and (hydr)oxides (e.g., ZnO nanowires, WO3, and goethite particles) or organic polymers [e.g., polytetrafluoroethylene, PTFE; poly(methyl methacrylate), PMMA; amine-modified polyacrylonitrile, and urethane-based polyether systems] have been tested.45,46,48 The growing demand to find new systems exhibiting such properties prompted us to investigate the possibility of using organically modified metal phosphates to produce electro-stimulated smart materials.
In this paper, we describe two types of zinc derivatives obtained via the reaction of phenyl esters of phosphoric acid with water-soluble zinc acetate. We then discuss the structures, morphologies, and thermal transitions of those inorganic–organic hybrid polymers as well as the structures and electron-conducting properties of the products of their pyrolysis. Finally, we present the results of our studies aimed at synthesizing electroresponsive smart materials based on dispersions of zinc mono- and diphenylphosphates in silicone oil or crosslinked silicone rubber.
Experimental
Chemicals and materials
The following chemicals were purchased from commercial sources and, unless stated otherwise, were used without further purification: zinc acetate dihydrate (ZnOAc·2H2O; 99%, Merck KGaA), zinc sulfate heptahydrate (≥99.5%, Merck KGaA), triphenyl phosphate (TPhP; ≥99%, Merck Schuchardt OHG), diphenyl phosphate (HDPhP; 99%, Sigma-Aldrich), disodium phenylphosphate dihydrate (Na2MPhP·2H2O; ≥95%, Sigma-Aldrich), sodium bicarbonate (pure, POCH Gliwice), n-butyl amine (BuNH2; 99.5%, Sigma-Aldrich), benzyl amine (BnNH2; ≥99%, Fluka Analytical), cyclohexane (pure p.a., POCH), poly(vinylidene fluoride) (PVDF; powder, melt viscosity = 2.350–2.950 kPa s, Alfa Aesar), two-component, high-strength moldmaking silicone rubber (XIAMETER® RTV-4234-T4, shore hardness = 40 ShA, Dow Corning), and methyl-terminated polydimethylsiloxane (PDMS100; POLSIL®OM 100, viscosity = 1 × 10−4 m2 s−1 at 25 °C, Chemical Plant “Silikony Polskie” Ltd.). Before use, PDMS100 was vacuum-dried (1.33 × 10−6 mbar) at 120 °C for 10 h.Preparative procedures
TPhP method. The reaction of ZnOAc·2H2O with TPhP required a temperature above 100 °C and the use of a pressure vessel. A typical synthesis was performed as follows: ZnOAc·2H2O (6.00 g, 27.1 mmol) was dissolved in 30 mL of redistilled water followed by the addition of TPhP (18.92 g, 57.4 mmol). The mixture was placed in a 100 mL stainless-steel pressure reactor equipped with a PTFE liner (Parr Instrument Co.) and heated for 24 h in an oil bath (T = 130 °C). After cooling to room temperature, the crude solid product was isolated via decantation and purified by washing with distilled water and acetone. The final product (ZnDPhP) was obtained after drying at 60 °C in a vacuum oven (6.68 g, reaction yield = 71%).
HDPhP method. The reaction was carried out at room temperature according to the following procedure. In a beaker equipped with a magnetic stirrer, HDPhP (1.21 g, 4.8 mmol) was dissolved in 6 mL of methanol. A solution of ZnOAc·2H2O (0.50 g, 2.3 mmol) in 5 mL of methanol was added dropwise under vigorous stirring to the solution of HDPhP, resulting in the precipitation of a white solid. After 20 h of mixing at room temperature, the mixture was filtered off and washed with methanol; the solid component readily dissolved during this process. The clear and colorless filtrate was concentrated on a rotary evaporator at 60 °C, resulting in the formation of a large amount of needle-like crystals. The crystals were filtered off and dried at 60 °C in a vacuum oven to give ZnDPhP (0.53 g, reaction yield = 41%).
Unless stated otherwise, the analytical, spectroscopic, and X-ray diffraction (XRD) data presented in this paper for ZnDPhP refer to the sample prepared by the TPhP method.
Method M1. A sample of ZnMPhP-H (4.42 g, 17.3 mmol) was heated at 120 °C under vacuum (1 × 10−2 mbar) for 6 h, resulting in a loss of 0.29 g (6.56%) of its initial weight.
Method M2. A sample of ZnMPhP-H (4.12 g, 16.1 mmol) was placed for 6 h in an air-dryer heated to 120 °C, resulting in a loss of 0.28 g (6.80%) of its initial weight.
The spectral data and results of the elemental analysis (Table S1†) of the compounds investigated in the present study are presented in ESI.†
Characterization methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 666 reflections measured including 6906 independent (Rint = 0.0515), which were used in all calculations. The final R1/wR2 values were 0.0244/0.0553 for observed data [I > 2σ(I)] and 0.0253/0.0559 for all data; the Flack parameter was −0.011(4), and the goodness of fit on F2 was 1.023.
666 reflections measured including 6906 independent (Rint = 0.0515), which were used in all calculations. The final R1/wR2 values were 0.0244/0.0553 for observed data [I > 2σ(I)] and 0.0253/0.0559 for all data; the Flack parameter was −0.011(4), and the goodness of fit on F2 was 1.023.The crystallographic data of ZnDPhP have been deposited in the Cambridge Crystallographic Data Center with CCDC number 1472713.
Thermal degradation under an inert atmosphere and volatile thermal decomposition products were studied by thermogravimetry coupled with quadrupole mass spectrometry (TG-QMS) using a Netzsch STA 449 C Jupiter apparatus coupled with a Netzsch QMS 403C Aeolos quadrupole mass spectrometer. The samples were heated in a stream of argon (flow rate = 60 mL min−1) from 35 °C to 800 °C at a heating rate of 5 °C min−1.
The ER properties of the cured silicone rubber composites filled with 1 wt% of the prepared compounds were conducted at 20 °C on the same apparatus using the parallel plate measuring system equipped with a P-PTD/200E ER cell and PP25/E/Tl plate (diameter = 25 mm, measuring gap = 0.9–1.2 mm). Measurements with and without an applied EF were carried out in oscillatory mode with a constant angular frequency (10 rad s−1) and strain amplitude of 1%. The circular samples (diameter = 25 mm) were cut off with a special stainless-steel bore and placed in the measurement system, as depicted in Fig. S2 (ESI).†
Results and discussion
Synthesis, composition, and morphology of ZnDPhP, ZnMPhP-H, ZnMPhP-A, and ZnMPhP-A/amine intercalates
Scheme 1 summarizes all synthetic routes successfully leading to the formation of ZnMPhP-H, ZnMPhP-A, or ZnDPhP in the present study. ZnMPhP-H was easily obtained during the ion-exchange reaction between two commercially available water-soluble reagents: ZnSO4 and Na2MPhP·2H2O. Since this is a simple ion-exchange reaction between well-dissociated substrates, and ZnMPhP-H is the sole insoluble product that precipitates from the mixture, the synthetic process proceeds very quickly and almost quantitatively at 25 °C. | ||
| Scheme 1 Reaction pathways to ZnMPhP-H, ZnMPhP-A, and ZnDPhP applied in the present study. Abbreviations: Ac, acetyl group; Ph, phenyl group; RT, room temperature. | ||
In the case of ZnDPhP, there are two synthetic pathways based on the large difference in the acid strengths of acetic acid (AcOH) and diesters of H3PO4. Diesters of H3PO4 are known to have pKa values lower than that of phosphoric acid itself,49 making them very strong acids that can easily substitute for the much weaker acid AcOH in its zinc salt. The reaction utilizing HDPhP requires a proper organic solvent as the reaction medium and gives ZnDPhP in a yield of 41%. The main drawbacks of the HDPhP method are the high cost of the commercial HDPhP substrate and challenges isolating the product. One can avoid both of these issues by choosing the TPhP method, a one-pot, two-stage hydrolytic route in which HDPhP is generated in situ via the hydrolysis of one phosphoester bond of TPhP, which is relatively inexpensive. Since the efficient cleavage of the P–OPh bond requires prolonged heating above 100 °C with water as the reaction medium, the entire process must be carried out in a pressure vessel made of a corrosion-resistant material. However, this method gives ZnDPhP in a yield of 71%, which is ca. 1.6 times higher than the typical yield of the HDPhP route. It is worth noting that the Zn2+ cations produced by the dissociation of ZnOAc·2H2O play a dual role in this synthetic process: they catalyze the hydrolysis of TPhP in the same way as other divalent metal cations (e.g., Ca2+ and Mg2+);49 in the next stage of the reaction they act as complexing agents for the newly-formed [(PhO)2PO2]− anions, effectively removing these anions from the reaction mixture and preventing further hydrolysis. Accordingly, no traces of monoester species were detected, even after the longest reaction times. The results of elemental analysis (Table S1 in ESI†), FTIR spectroscopy (Fig. S3 in ESI†), and PXRD measurements (Fig. S4 in ESI†) indicate that regardless of the chosen synthetic path, only ZnDPhP was obtained. The SEM images presented in Fig. 1A show that ZnDPhP formed particles with needle- or rod-like shapes and hexagonal cross-sections (Fig. S5 in ESI†).
 | ||
| Fig. 1 SEM images of ZnDPhP (A), ZnMPhP-H (B), and ZnMPhP-A (C). ZnDPhP and ZnMPhP-A were prepared according to the TPhP and M2 methods, respectively. | ||
An insoluble product with a plate-like morphology and an average particle thickness below 1 μm precipitated during the synthesis of ZnMPhP-H (Fig. 1B and S6 in ESI†). The FTIR spectrum (Fig. S7a in ESI†) indicates that in addition to phenylphosphate groups, this compound also contained water molecules as auxiliary ligands. The FTIR spectrum includes a wide, bimodal absorption band between 3100 and 3300 cm−1 corresponding to stretching O–H vibrations and a signal located at 1654 cm−1 related to the H–O–H bending mode.50 This finding is supported by the results of TG-QMS results (Fig. S8 in ESI†), which indicate the evolution of water from ZnMPhP-H based on two characteristic signals with the m/z values of 17 and 18 at temperatures between 100 °C and 140 °C.51 Based on the elemental analysis results (Table S1 in ESI†) and the weight of water lost during heating (6.70–7.31% of the initial sample weight depending on the measurement conditions, see Table S2 in ESI†), it can be concluded that ZnMPhP-H is phenylphosphate monohydrate with the general formula Zn(O3POPh)·H2O and a theoretical water content of 7.05 wt%. It should be noted that the dehydration of ZnMPhP-H occurs easily without the need for sophisticated apparatus or conditions (e.g., reduced pressure). The complete disappearance of water absorption bands in the FTIR spectrum (Fig. S7b in ESI†) as well as the good agreement between the experimental and theoretical carbon and hydrogen contents (Table S1 in ESI†) suggest that even simple drying in hot air resulted in the conversion of ZnMPhP-H into its anhydrous form, ZnO3POPh. This new compound, ZnMPhP-A, partially retained the morphological characteristics of its substrate (Fig. 1C), although the high-magnification SEM images reveal that its plate-like particles were composed of much thinner nanolayers (Fig. S9 in ESI†). On the other hand, the loss of hydration water was accompanied by some rearrangement in the structure of zinc phenylphosphate, resulting in signals corresponding to two populations of phosphorus nuclei in the 31P CP/MAS NMR spectrum of ZnMPhP-A: one is shifted upfield by ca. 1.4 ppm, while the other is shifted downfield by ca. 0.5 ppm compared to the signal located at −2.66 ppm in the corresponding spectrum of ZnMPhP-H (Fig. S10 in ESI†).
It is worth noting that during prolonged exposure to air ZnMPhP-A reabsorbed a small amount of moisture, causing it to partly return to its hydrated form. However, this process was very slow (e.g., hydration was incomplete after 11 d). Hydration could be significantly accelerated by using liquid water, as indicated by both the FTIR spectra (see Fig. S7c and S7d in ESI†) and PXRD patterns (Fig. S11 in ESI†). The presence of well-defined reflections in the PXRD patterns indicates that ZnMPhP-H and ZnMPhP-A formed highly ordered crystalline structures. The striking similarities observed between the PXRD patterns of ZnMPhP-H and ZnMPhP-A, particularly the very small difference in the 2θ angle of the main diffraction signal (the difference in the basal spacings calculated for these signals was ca. 1.0 Å), suggesting that only a small degree of structural change occurred upon dehydration. Moreover, the similar shapes of the PXRD patterns of ZnMPhP-H, zinc ethylphosphate monohydrate,7 and zinc phenylphosphonate monohydrate,3 particularly the presence of a single, high-intensity diffraction peak at low 2θ value, might indicate that ZnMPhP-H possesses an analogous layered structure with the phenyl rings protruding from the surface of a compact inorganic layer of zinc atoms coordinated by oxygen ligands from water molecules and phosphate groups. However, the verification of this hypothesis was prevented by the insufficient number of diffraction peaks required for the reliable indexing of the PXRD patterns of ZnMPhP-H and ZnMPhP-A. Note that in the case of ZnMPhP-H, hydrothermal crystallization resulted in the complete hydrolysis of the phosphoester bond and the formation of zinc orthophosphate, as evidenced by a sharp peak at 3.79 ppm in the 31P CP/MAS NMR spectrum (Fig. S10 in ESI†)52,53 and the PXRD pattern characteristic of hopeite [Zn3(PO4)2·4H2O]53,54 (Fig. S12 in ESI†). Even synthesizing ZnMPhP-H at room temperature did not completely prevent this process, and traces of the inorganic phosphate were found in the product, as evidenced by the presence of a small peak at 3.94 ppm in the 31P CP/MAS NMR spectrum (Fig. S10 in ESI†).
Like its analogue containing phenylphosphonate ligand,55–58 ZnMPhP-A is able to absorb amines containing primary NH2 groups, namely BuNH2 and BnNH2. Upon the intercalation of amine into the ZnMPhP-A structure, a small shift in its main diffraction peak toward higher 2θ values was observed in the PXRD pattern (Fig. S13 in ESI†). This shift indicates that the incorporation of amine shortened the distance between Zn–O–P monolayers. Moreover, the basal spacing in these intercalates (ca. 15.1 Å) was even smaller than that estimated for ZnMPhP-H (15.5 Å). One can assume that the arrangements of the phenyl rings in the interlayer spaces of the zinc phenylphosphate/amine intercalates and ZnMPhP-H are different due to their interactions with amine alkyl groups. The formation of the zinc phenylphosphate/amine intercalates was also confirmed by their FTIR spectra (Fig. S14 in ESI†), which show absorption bands related to both the amine group (e.g., three N–H stretching modes between 3100 and 3300 cm−1 characteristic of metal-coordinated amine and NH2 bending modes that widen the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching band located around 1600 cm−1) and alkyl substituents (e.g., C–H stretching modes between 2850 and 3000 cm−1).55 It is worth noting that the results of elemental analysis (Table S1 in ESI†) suggest that the amount of amine intercalated into zinc phenylphosphate (x in the formula ZnO3POPh·xRNH2) depends strongly on the type of organic substituent in the amine; for example, x = 1.07 and 1.75 for ZnMPhP/BnNH2 and ZnMPhP/BuNH2, respectively. In this regard, our intercalates differ from their analogues containing the phenylphosphonate moiety.55
C stretching band located around 1600 cm−1) and alkyl substituents (e.g., C–H stretching modes between 2850 and 3000 cm−1).55 It is worth noting that the results of elemental analysis (Table S1 in ESI†) suggest that the amount of amine intercalated into zinc phenylphosphate (x in the formula ZnO3POPh·xRNH2) depends strongly on the type of organic substituent in the amine; for example, x = 1.07 and 1.75 for ZnMPhP/BnNH2 and ZnMPhP/BuNH2, respectively. In this regard, our intercalates differ from their analogues containing the phenylphosphonate moiety.55
ZnDPhP crystal structure and thermal transitions
The needle-like crystals of ZnDPhP obtained using the HDPhP method (see the Experimental section) were subjected to single-crystal X-ray analysis at 100 K. The results indicate that ZnDPhP crystallizes in a trigonal crystal system in chiral space group P32 with one zinc cation and two diphenylphosphate anions in the asymmetric unit (Fig. S15a in ESI†). In the crystal structure of ZnDPhP, all zinc(II) cations are crystallographically equivalent, whereas two types of phosphorus atoms (P1 and P2) can be distinguished. Zinc is coordinated in a tetrahedral arrangement by four oxygen atoms from four different phosphate anions, and the Zn–O bond lengths range from 1.910(3) Å to 1.941(2) Å. In every PO4 tetrahedron, the oxygen atoms of P–O are coordinated to Zn2+ with bond lengths in the range of 1.485(3) Å to 1.499(3) Å. These bonds are significantly shorter than the P–O bonds linking the phenyl rings, for which the bond lengths range from 1.578(3) Å to 1.588(2) Å.In the ZnDPhP structure, adjacent ZnO4 tetrahedra are connected by double bridges consisting of O2P(OPh)2 groups, forming infinite 1∞[Zn(DPhP)2/2] helical chains with p32 rod group symmetry (Fig. 2 and S15b in ESI†). The left-handed helices are packed closely in a hexagonal lattice with the organic groups directed outwards (Fig. S15b and S15c in ESI†). Since all the necessary conditions are fulfilled (e.g., the value of the Flack parameter xFlack is close to zero, its standard deviation u is less than 0.04, and the ratio |xFlack|/u is less than 3.0),59 one can conclude that the ZnDPhP coordination polymer is homochiral.
 | ||
| Fig. 2 Structure of a single ZnDPhP chain viewed along the a-axis. Zinc and phosphorus coordination tetrahedra are presented in blue and violet, respectively. For clarity, all hydrogen atoms are omitted. | ||
A detailed analysis based on the bond-valence vector (BVV) model60,61 indicated the presence of strain within the ZnDPhP structure. Considering both the estimated error of the BVV model and the data on the length of the resultant BVVs (|vP| and |vZn|) calculated based on the BVV parameters collected by Brown62 (see eqn (S1)–(S5) and Table S3 in ESI†), the strain can be ascribed solely to the slight deviation in the geometry of the PO4 tetrahedra from that of a regular tetrahedron. As shown in Fig. S16 (ESI),† the resultant BVVs for the phosphorus centers protrude outward from the chain. It should be noted that the small value of |vZn| (less than 0.04 valence units) excludes any distortion of the ZnO4 tetrahedra. Furthermore, it is reasonable to assume that such distortion of the coordination sphere may affect the local magnetic field experienced by phosphorus nuclei, resulting in different signals in the NMR spectrum. Indeed, two overlapping resonance peaks can be observed between −17 and −19 ppm (their maxima are separated by ca. 1 ppm) in the 31P CP/MAS NMR spectrum of ZnDPhP (Fig. S17 in ESI†). However, this is only a hypothesis that will be explored in detail in a separate study dealing with zinc dialkylphosphates.
Our previous studies on temperature-dependent structural changes in aluminum tris(diorganophosphates)38 prompted us to investigate whether similar changes occur in ZnDPhP. As shown in Fig. S18 (ESI),† the DSC profile of ZnDPhP contains an endothermic peak at 162.6 °C in the first heating curve, and the cooling curve shows the corresponding exothermic signal. The practical overlap of the endotherms of both heating curves implies the reversibility of the processes occurring within the ZnDPhP sample. Considering that all these effects were observed well below the starting point of ZnDPhP thermolysis (see Table S2 in ESI†) and that no change in the physical state of the sample was detected, one can assume that these effects arise from some type of structural rearrangement occurring in the solid state. To verify this hypothesis, we carried out VT-PXRD measurements of ZnDPhP at temperatures up to 235 °C. The collected diffraction data were used to estimate the ZnDPhP unit cell parameters. The results are shown in Fig. 3 as well as in Tables S4–S6 and eqn (S6)–(S13) (ESI).†
 | ||
| Fig. 3 Effects of temperature on the ZnDPhP unit cell parameters a and c and volume V based on VT-PXRD measurements carried out in heating (27 °C → 235 °C) or cooling (235 °C → 27 °C) mode. The temperature regions in which the low-temperature (A, A′) and high-temperature (C, C′) phases occur are indicated; B and B′ indicate the transition regions. Different phases of ZnDPhP are marked with colors: blue for LT-ZnDPhP and red for HT-ZnDPhP. | ||
Two different crystalline phases of ZnDPhP could be distinguished within the investigated temperature range. One ZnDPhP phase exists from room temperature to ca. 160 °C; within this temperature region (denoted as A in Fig. 3), ZnDPhP undergoes a thermal expansion characterized by a gradual increase in its unit cell volume (V). The corresponding volumetric thermal expansion coefficient βV (defined by eqn (14) in ESI†)63 was 17.5(4) × 10−5 K−1. This expansion was not isotropic and proceeded mainly along the a- and b-axes, whereas the lattice parameter c was less affected by temperature. Considering that βV is a sum of the linear thermal expansion coefficients along all crystallographic axes (αa, αb, and αc, which are defined by eqn (15) in ESI†)64 and the calculated αa and αc values of 8.05(21) × 10−5 K−1 and 13.3(8) × 10−6 K−1, respectively, for the low-temperature ZnDPhP phase (LT-ZnDPhP), one can conclude that thermal expansion coefficients along the a and b directions are the same (αb = αa).
A transition region (denoted as B in Fig. 3) in which another crystalline phase of ZnDPhP began to form was observed above 145 °C. Reflections related to both phases are clearly visible in the PXRD pattern of ZnDPhP in the middle of this region (ca. 160 °C). In contrast, starting at 175 °C (region C in Fig. 3), only the high-temperature phase of ZnDPhP (HT-ZnDPhP) can be detected (see Fig. S19 in ESI†). This phase is characterized by a lower degree of structural order, as evidenced by the widening of the highest-intensity diffraction peak, the disappearance of a large number of reflections at 2θ angles greater than 10°, and the presence of a broad halo with a maximum around 20°, which is typically observed in amorphous samples. In fact, for HT-ZnDPhP, a reliable estimate is only possible for the parameter a, which is ca. 6.5% higher than that of LT-ZnDPhP at the same temperature (see Table S4 in ESI†).
During cooling from 235 °C to room temperature virtually the same structural changes were observed within the ZnDPhP sample, although they occurred in the reverse order (i.e., a transition from HT-ZnDPhP to LT-ZnDPhP). Interestingly, HT-ZnDPhP was stable in a wider temperature range since the temperature window in which both crystal phases coexist (denoted as B′ in Fig. 3) was lowered by approximately 60 °C when the VT-PXRD measurements were carried out in cooling mode compared to in heating mode. In addition, a close correlation was observed between the location of both transition regions and the position of the previously mentioned peaks in the respective DSC profiles; the correlation was especially evident during the heating of ZnDPhP.
It should be noted that the values of both lattice parameters of the LT-ZnDPhP crystal phase (a = 12.70–12.72 Å and c = 12.53–12.61 Å) as well as its unit cell volume (1749–1766 Å3) calculated at 100 K using eqn (S6)–(S8) and (S10)–(S12) (ESI)† were close to those determined experimentally for ZnDPhP crystal (see the Experimental section); the differences between the calculated and experimental values did not exceed 2.5% of the experimental values. Moreover, the 31P CP/MAS NMR spectra recorded at temperatures between −50 °C and 75 °C (Fig. S17 in ESI†) are identical in terms of both the shapes and locations of the resonance peaks. Both these findings indicate that the LT-ZnDPhP phase had the same crystal structure as single-crystal ZnDPhP.
Considering all the presented XRD and DSC data, we conclude that upon heating from −173 °C to 160 °C, the ZnDPhP structure underwent a simple thermal expansion caused mainly by the spatial distancing of its helical chains in the a/b-direction parallel to the (001) lattice plane. The structure of the individual ZnDPhP chains changed only slightly with increasing temperature, resulting in a much slower expansion of the ZnDPhP unit cell in the c-axis direction. One can expect this phenomenon to continuously weaken the interchain interactions (e.g., the van der Waals interactions between phenyl groups in adjacent ZnDPhP chains). Around 145 °C, those interactions become sufficiently weak to allow the ZnDPhP chains to move in the direction parallel to the crystallographic c direction, and a discontinuous change in a was observed. These new degrees of freedom led to a partial loss of long-range order along the crystallographic Z axis, but they did not affect the hexagonal packing of the ZnDPhP chains, and no change in the physical state of the sample (e.g., melting) was observed. However, the reflections located at 2θ angles higher than 10° disappeared. A continuation of heating resulted in the further separation of ZnDPhP chains along the a-axis. Based on our calculations, this process was almost three times more pronounced in HT-ZnDPhP compared to LT-ZnDPhP; the αa value calculated for HT-ZnDPhP, 21.47(35) × 10−5 K−1, was ca. 2.7 times higher than that of LT-ZnDPhP. The above-described process can be reversed by cooling the sample. Since the LT-ZnDPhP phase has a lower energy than the HT-ZnDPhP phase, this leads to the occurrence of an exothermic peak in the DSC cooling curve. However, such a transformation is hindered and occurs much more slowly than the opposite process, likely due to the limitation of ZnDPhP chain movement, especially in the c-axis direction. Since the rate of temperature decrease during our VT-PXRD and DSC measurements exceeded that of the LT-ZnDPhP reconstruction, this phase was observed in a region of much lower temperatures (Fig. S20 in ESI†).
TGA and pyrolysis of ZnMPhP and ZnDPhP
According to the TGA data collected in air, ZnMPhP-A and ZnDPhP are thermally stable up to 250–260 °C, with the former being more resistant to heat than the latter (see Table S2 and Fig. S21–S22 in ESI†). Moreover, the relatively large temperature range (66–111 °C depending on the compound) between the beginning of weight loss (the temperature of the bend in the TGA curve, Tb) and the extrapolated onset temperature of decomposition (Tonset) observed for both of those compounds suggests that the initial step in their thermolysis is relatively slow. On the other hand, once initiated, the thermo-oxidative degradation of ZnMPhP-A proceeded very quickly and in a single step; only one peak is observed in the derivative thermogravimetry curve (Fig. S21 in ESI†) above 200 °C, and the Tonset of this process differs from the extrapolated endset temperature (Tendset) by less than 40 °C (see Table S2 in ESI†). In contrast, ZnDPhP lost its organic content in multiple steps, and its weight stabilized after thermolysis at approximately 712 °C, ca. 300 °C higher than the Tendset of ZnMPhP-A.Based on further TGA studies carried out in argon (Fig. S8A and S23A and Table S2 in ESI†) the elimination of oxygen from the atmosphere significantly increased the thermal stability of ZnDPhP and ZnMPhP-A; their Tb and Tonset values shifted toward higher temperature by a few tens of degrees Celsius compared to the measurements conducted in air. Another interesting phenomenon is the noticeable increase (ca. 5% of the initial mass of the sample taken for TGA) in residual mass estimated at 800 °C, which indicates the production of fewer volatile compounds during pyrolysis. The simultaneous analysis of the evolved gases by QMS revealed that common features in the thermal decomposition of ZnDPhP and ZnMPhP-A are the appearance of signals characteristic of phenol (m/z = 65, 66, and 94),51 carbon monoxide (m/z = 28),51 and carbon dioxide (m/z = 44)51 as well as the lack of any QMS peaks above the m/z value of 100 (Fig. S8C, S23B and S23C in ESI†). It should be noted that phenol was detected throughout the entire process, corresponding to the loss of the organic component in both hybrid polymers (Fig. S8E and S23D in ESI†). These findings indicate that the P–O bonds in the investigated zinc salts broke down during heating (hence the presence of phenol and carbon oxides),65 while the splitting of the C–O bonds did not occur. According to the literature data on TPhP pyrolysis,65 C–O bond splitting should produce phenyl radicals and the products of their transformations (recombination and condensation reactions), such as biphenyl, naphthalene, and biphenyl ether; no QMS signals with the m/z values above 100 (ref. 51) were detected in the evolved gases in this study. However, one cannot completely exclude the possibility of C–O bond cleavage followed by the formation of some polyaromatic moieties because a significant fraction of carbon appeared in the solid residues from ZnMPhP-H and ZnDPhP pyrolysis at 600 °C for 6 h (4.79 and 14.97 wt%, respectively).
The PXRD patterns of the pyrolysis residues agreed well with those of standards from the Powder Diffraction Database,54 confirming that the residues contained either zinc pyrophosphate and zinc phosphate (pyrolyzate from ZnMPhP-H, Fig. S24 in ESI†) or zinc pyrophosphate and zinc metaphosphate (pyrolyzate from ZnDPhP, Fig. S25 in ESI†) as an inorganic component. The Raman spectra (Fig. S26 and S27 in ESI†) suggest that the carbonaceous phase in the investigated pyrolyzates is composed of graphene layers, as evidenced by the appearance of the two distinctive graphene bands at ca. 1590 cm−1 (the primary in-plane Csp2–Csp2 vibrational mode, the G mode) and ca. 2700 cm−1 (the 2D mode).66,67 However, the simultaneous occurrence of two additional signals at approximately 2900 cm−1 (a combination D + G band) and 1350 cm−1 (the D vibrational mode) implies a highly distorted structure of this graphene deposit. In fact, the Raman spectra of the ZnDPhP and ZnMPhP pyrolyzates closely resemble those of nanographene66,68 and reduced graphene oxide (rGO),69 suggesting that one or both may be present in the investigated samples.
Electrical properties of the ZnDPhP pyrolyzate
It is well known that nanographene68 and rGO70 exhibit electron-conducting properties. Moreover, our study on aluminum tris(diorganophosphates) showed that even a small fraction of rGO may be sufficient to form pathways for electrical conduction in the metal diorganophosphate pyrolyzate.38 This prompted us to determine if the same phenomenon occurs in the ZnDPhP pyrolyzate since it contained a larger carbonaceous fraction than the ZnMPhP pyrolyzate. We estimated both the sheet electrical resistance (ρsh) and bulk electrical conductivity (σb) of composites of ZnDPhP pyrolyzate with PVDF using the 4PP method; the results are summarized in Table 1. The values of ρsh and σb were calculated according to eqn (1) and (2), respectively:|
ρsh = Rslope × π × (ln2)−1 × Fd × Ft
| (1) |
| σb = ρsh−1 × t−1 | (2) |
Based on the data presented in Table 1, the investigated ZnDPhP pyrolyzate indeed exhibited electrical conductivity, and conductive paths were formed after its incorporation into the PVDF matrix. The appearance of a somewhat unexpected maximum in the σb value at the filler concentration of 75 wt% can be plausibly explained by an increase in the brittleness of the composite at larger loadings of the ZnDPhP pyrolyzate. In fact, when the concentration of the filler exceeded 95 wt%, the measurements could not be carried out due to the destruction of the sample during probe insertion. One can expect that the formation of microcracks, while not destroying the sample, would effectively worsen the contact between the sample and the probe tips, thus artificially increasing the measured value of ρsh. Nevertheless, based on the above results, the bulk conductivity of pristine ZnDPhP pyrolyzate should not be less than 0.15 S cm−1, which is in between the values reported in the literature for rGO films (ca. 20–100 S cm−1)73 and partially reduced graphene oxide 3D assemblies (ca. 2.5 × 10−3 S cm−1).74
ZnDPhP and ZnMPhP as components of electro-stimulated smart materials
Considering the abovementioned structural features of ZnDPhP and zinc phenylphosphate, particularly the presence of electron-rich phenyl groups within their structures, we expected them to be susceptible to spatial reorientation and ordering under the influence of an EF. Since such a process should be reversible, it could provide the opportunity to apply these structures in so-called “smart materials”, in which a change in properties is triggered by some external factor. To verify this assumption we carried out some preliminary studies on the ER parameters of silicone-based systems containing our hybrid polymers.All the investigated compounds formed electrically active particles that, upon suspension in silicone oil, showed a negative ER effect. That is, the resistance to flow of the resultant fluids decreased under the applied EF, as indicated by smaller values of both shear stress (Fig. 4) and viscosity (η; Fig. S28–S30 in ESI†) at any given shear rate.45,46,75,76 By taking the results obtained for a sample not exposed to EF as reference data, one should be able to evaluate the degree of electro-stimulation for each of the studied dispersions. However, even for measurements carried out at a constant EF strength (E), the calculated change in the viscosity of the sample relative to the reference data (Δη) varied significantly with the shear rate, effectively hindering any quantitative analysis. In our opinion, a more reliable parameter is the average relative change in viscosity at a given E , the values of which are presented in Table 2.
, the values of which are presented in Table 2.
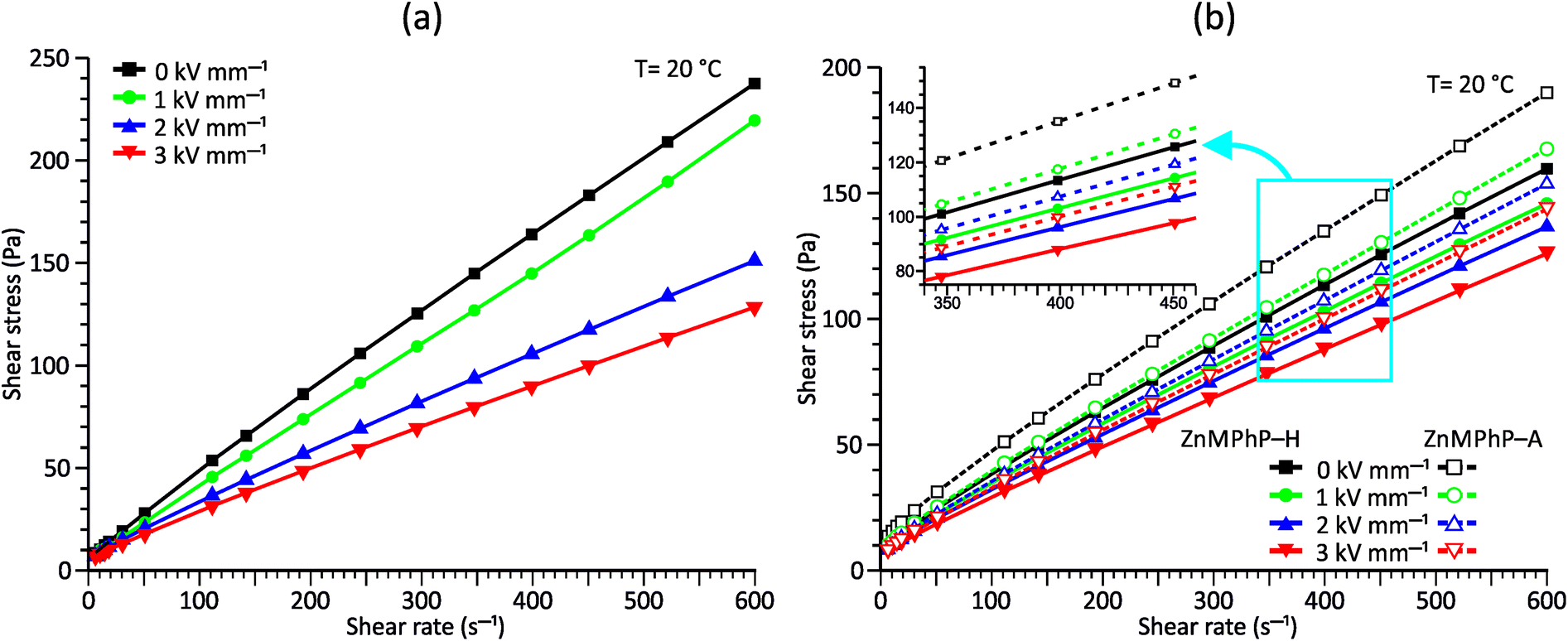 | ||
| Fig. 4 Flow curves measured under different E values for PDMS100 dispersions containing 20 wt% (a) ZnDPhP and (b) ZnMPhP-H or ZnMPhP-A. | ||
 in PDMS100-based dispersions containing 20 wt% ZnDPhP, ZnMPhP-H, or ZnMPhP-A
in PDMS100-based dispersions containing 20 wt% ZnDPhP, ZnMPhP-H, or ZnMPhP-A
|
|||
|---|---|---|---|
| E = 1 kV mm−1 | E = 2 kV mm−1 | E = 3 kV mm−1 | |
a  , where ηi,E is the EF-induced viscosity measured at a given shear rate i in a dispersion subjected to a specified E; ηi,ref is the reference viscosity determined at the same shear rate i but without any EF (E = 0 kV mm−1); and n is the total number of the shear rate values examined for a given E. , where ηi,E is the EF-induced viscosity measured at a given shear rate i in a dispersion subjected to a specified E; ηi,ref is the reference viscosity determined at the same shear rate i but without any EF (E = 0 kV mm−1); and n is the total number of the shear rate values examined for a given E. |
|||
| ZnDPhP | −12(3) | −29(7) | −38(9) |
| ZnMPhP-H | −7(4) | −14(4) | −22(4) |
| ZnMPhP-A | −17(5) | −26(6) | −31(6) |
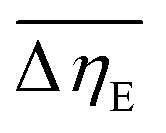
Based on the data shown in Table 2, the ZnDPhP- and ZnMPhP-A-containing dispersions were the most susceptible to EF, and their viscosities decreased by as much as 30–40% at the highest value of E. The presence of hydration water in ZnMPhP-H had an adverse effect on the magnitude of its response to EF, although the change in viscosity still reached ca. 20%. Generally, in all systems, the change in viscosity increased as the EF became stronger. However, for ZnDPhP and ZnMPhP-A, this relationship was not linear, and the response of viscosity to the unit change in electric voltage varied with E. The origin of the negative ER effect in the studied fluids remains unclear. Usually, two EF-related phenomena are proposed as the main driving forces behind the viscosity decrease: the electromigration and electrorotation (Quincke rotation) of particles suspended in a highly insulating liquid.45,46 Here, we cannot determine exactly which of these two options applies to our systems. However, the former should result in the electrophoretic accumulation and precipitation of solid particles on the surface of one or both electrodes, and we did not detect any sign of such a process; even after the measurements at the highest E values, our dispersions retained their homogeneity, and the electrodes were free of any solid depositions.
To gain further knowledge of the reversibility of the observed ER behavior, we recorded the curves of viscosity vs. time for our systems while periodically changing E from 0 to 2 kV mm−1 in constant time intervals. As shown in Fig. 5, all samples exhibited good stability and repeatability in their response to the EF, although some differences were observed in the rates of processes occurring in the systems when turning the electric voltage on or off. Generally, the decrease in viscosity caused by the activation of the EF occurs much faster than the opposite process occurring when the electric voltage is turned off. This was especially evident for the ZnMPhP-A-based dispersion, for which the return to the initial viscosity required nearly the whole E = 0 kV mm−1 interval and effectively slowed with time. In the other two systems, the rheological properties stabilized more quickly, with stabilization occurring the fastest in the dispersion containing ZnMPhP-A (ca. 42 s). The ZnDPhP-based dispersion required more than twice as much time (ca. 90 s) for rheological stabilization. As mentioned earlier, when activated by EF, the internal structures of all three fluids were rapidly reorganized, and a large drop in their viscosities occurred almost completely within 20 s (for ZnMPhP-A) to 40 s (for ZnDPhP) after the activation of the EF. Considering both the magnitude and rate of the observed transformations, one can conclude that the most promising “smart” system is the one based on ZnMPhP-H; its viscosity changed by about 20% upon switching the electric voltage on or off, and both operations proceeded at almost the same rate.
 | ||
Fig. 5 Impact of cyclic changes in the EF strength on the viscosity of PDMS100 (■) and its dispersions containing 20 wt% ZnDPhP ( ), ZnMPhP-H ( ), ZnMPhP-H ( ), or ZnMPhP-A ( ), or ZnMPhP-A ( ). The EF profile is depicted as a solid red line. For clarity of presentation, the viscosity axis is broken in the 0.5–1.0 Pa s region and the scale factor for the depicted axis regions (upper to lower) is ca. 0.4. ). The EF profile is depicted as a solid red line. For clarity of presentation, the viscosity axis is broken in the 0.5–1.0 Pa s region and the scale factor for the depicted axis regions (upper to lower) is ca. 0.4. | ||
The viscosity–temperature plots presented in Fig. 6 reveal an interesting feature that distinguishes the ZnDPhP-based dispersion from the dispersion containing ZnMPhP-A. When no EF was applied, the viscosity of the ZnDPhP-based dispersion decreased continuously during heating to approximately 140 °C. As heating continued, η began to vary, eventually leading to a drastic drop in η (ca. 66%) between 155 °C and 162 °C. No such behavior was observed in the other system indicating that this phenomenon is unique to ZnDPhP. Since the temperature window in which the drop in η occurs practically coincides with the phase transition from the LT- to HT-ZnDPhP crystal form, one can expect that HT-ZnDPhP contributes significantly to the reduction in viscosity. It is possible that the weak interchain interactions in this temperature region facilitate the destruction of large ZnDPhP particles, thereby decreasing the resistance of the system to flow. However, this is a mere hypothesis that requires further experimental verification. Interestingly, in the case of a dispersion subjected to an EF, no sudden drop in the viscosity was detected; in contrast, above ca. 140 °C, a slight increase in the measured η value was even observed.
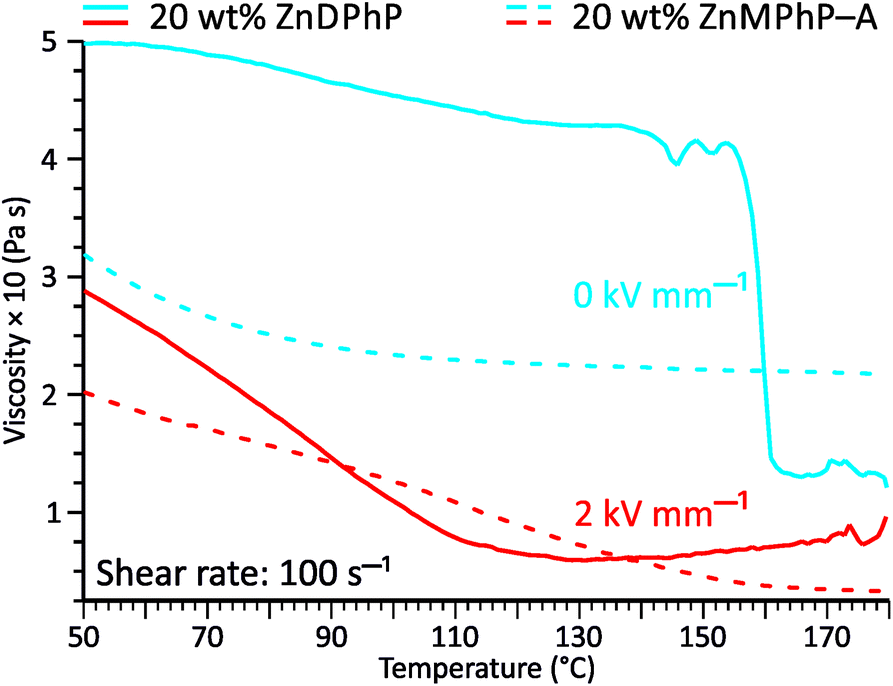 | ||
| Fig. 6 Effect of temperature on the viscosity curves of PDMS100 dispersions containing 20 wt% ZnDPhP (solid lines) and ZnMPhP-A (dashed lines). The measurements were carried out in a plate–plate geometry at E = 0 kV mm−1 (blue lines) and E = 2 kV mm−1 (red lines). | ||
The results of our further studies indicate that the investigated PDMS-based systems maintained their susceptibility to the EF stimulus to some extent, even in a solid state. Fig. S31 and S32 (ESI)† and Table 3 present selected viscoelastic properties of the cured PDMS composites containing 1 wt% of our hybrid polymers. The properties were measured in oscillatory mode with a periodic change in E. The moduli describing the elastic (storage modulus, G′) and viscous (loss modulus, G′′) components of the sample's behavior were greatly affected by EF, mainly during crosslinking. Even when a low-strength (0.5 kV mm−1) EF was applied, the G′ and G′′ values of the composite were easily changed by about 25–80% (Table 3). Interestingly, the effect on viscoelasticity depends on the type of filler present in the sample; when activated by EF during curing, the 1D ZnDPhP particles caused decreases in both moduli (with the elastic modulus being more strongly affected), whereas the 2D ZnMPhP-A particles gave rise to the opposite effect. Nevertheless, in both cases, the viscous properties of the samples increased since under the influence of EF, the values of the damping factor tan(δ), which is defined as the ratio of G′′ to G′ and describes the material's ability to viscous energy dissipation, were increased by ca. 15–30%.
| Filler typea |
|
ΔG′c (%) |
|
ΔG′′c (%) | tan(δ)0b × 102 | Δtan(δ)c (%) |
|---|---|---|---|---|---|---|
| a Abbreviations: 1D, ZnDPhP; 2D, ZnMPhP-A.b Value of a given parameter measured for the sample cured without any EF.c Changes in the value of a given parameter a caused by EF operating during the crosslinking of the sample and calculated from the equation Δa = 100 × (a0.5 − a0) × a0−1, in which the number in the index denotes the strength of the applied EF (in kV mm−1). | ||||||
| 1D | 50.228 | −35 | 9.352 | −26 | 18.6 | 15 |
| 2D | 29.923 | 41 | 4.712 | 82 | 15.7 | 30 |


Based on an analysis of the viscoelastic behavior of the cured PDMS-based composites subjected to periodic changes in EF (Fig. S31 and S32 in ESI†), one can conclude that constraining ZnDPhP or ZnMPhP-A particles within a covalently bound silicone 3D polymer network strongly diminished the susceptibility of the system to future activation by an EF. Surprisingly, even the reference sample (an unfilled PDMS matrix) showed some response to the applied EF (E = 3 kV mm−1), likely due to the presence of some polarizable functional groups incorporated into the 3D polymer network with the curing agent. Changes of similar type and magnitude were observed for the 1 wt% composites cured under EF but not in their analogues crosslinked without EF. This suggests that the ER effect of the silicone matrix was compensated by the opposite effect arising from the orientation of hybrid particles in the EF applied during the test. The preorientation of the particles at the curing stage could prevent such a phenomenon by making further movements in the solid matrix (activated by EF) less likely. This is especially true given that the lines of the EF were oriented in the same direction (perpendicular to the sample surface) during both the crosslinking of the silicone matrix and the subsequent ER test. It is interesting to note that the volumes of all tested samples changed reversibly in a direction perpendicular to the sample surface, as evidenced by the decrease in the normal force (FN) after EF activation (see Fig. S31 in ESI†). Moreover, this effect seems to accumulate in the ZnDPhP-containing composites, causing FN to get smaller with each subsequent test cycle. However, the source of this phenomenon remains unclear and requires further study.
Conclusions
We have reported simple synthetic procedures for the preparation of crystalline polymeric zinc phenylphosphates with 1D or 2D architectures. The 1D particles of ZnDPhP are constructed from homochiral helices of ZnO4 tetrahedra connected by double bridges composed of diphenylphosphate units. The linear chains are packed closely in a hexagonal lattice with the organic groups directed outwards. When heated, this structure undergoes a thermal transition at ca. 160 °C, resulting in its expansion, which mainly occurs perpendicular to the c-axis (the axis of symmetry of the ZnDPhP chain). A reverse process occurs during cooling, although it appears in a lower-temperature region (around 100–110 °C). ZnDPhP is thermally stable up to about 250 °C. The pyrolytic decomposition of this polymer results in the formation of an electrically conducting carbon deposit on the zinc phosphate core. The conductivity of this material is higher than 0.15 S cm−1, which is sufficient for its practical application as a conducting filler in polymer composites.The interaction of zinc cations with [PhOPO3]2− anions results in the precipitation of zinc phenylphosphate monohydrate with the general formula Zn(O3POPh)·H2O. The PXRD and SEM analyses of the precipitate suggest that this compound has a layered structure in which the phenyl groups separate adjacent sheets comprising ZnO4 tetrahedra and water molecules. We found that the water molecules can be easily removed from the zinc coordination sphere and replaced by primary amines. Although the exact binding mode of these auxiliary ligands is not known, we think that this type of ligand-exchange reaction may offer a simple synthetic path for the further functionalization of layered zinc phenylphosphate.
The particles of both linear and layered zinc phenylphosphates are reversibly polarized when subjected to an external EF. Their suspensions in silicone oil show negative ER effects, meaning that the rheological parameters of the fluid (e.g., viscosity and shear stress) decrease significantly under an applied EF. Preliminary observations indicate that the reorientation of the zinc phenylphosphate particles caused by the EF may deteriorate some of the mechanical properties and decrease the density of the 3D polymeric network. We think that these effects could be especially prominent in solid composite films with micrometer-scale thickness prepared by spin-coating or tape-casting methods. However, their successful preparation will require the careful optimization of the filler particle size, as well as some changes in how the particles are dispersed in silicone oil (e.g., preparation of the composite using mechanical or ultrasonic homogenizers).
Current work in our research group is aimed at understanding the effect of the types of metal centers and organic ligands on the structures formed by metal organophosphate hybrid polymers as well as the ER properties of polymeric matrices doped with their particles. We are also focused on finding new electrically conducting fillers that can be obtained via the controlled pyrolysis of metal organophosphates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Centre, Poland within the OPUS-11 research scheme, project number 2016/21/B/ST5/00126. This research was also financially supported by the Warsaw University of Technology (Faculty of Chemistry). The authors would like to thank Dr Eng. Paweł Falkowski and Dr Eng. Magdalena Zybert from the Faculty of Chemistry, Warsaw University of Technology, for carrying out the pyrolysis experiments and TG-QMS measurements, respectively.References
- G. Alberti, U. Costantino, S. Allulli and N. Tomassini, J. Inorg. Nucl. Chem., 1978, 40, 1113 CrossRef CAS.
- D. Cunningham, P. J. D. Hennelly and T. Deeney, Inorg. Chim. Acta, 1979, 37, 95 CrossRef CAS.
- K. J. Martin, P. J. Squattrito and A. Clearfield, Inorg. Chim. Acta, 1989, 155, 7 CrossRef CAS.
- W. T. A. Harrison, T. M. Nenoff, T. E. Gier and G. D. Stucky, J. Mater. Chem., 1994, 4, 1111 RSC.
- W. T. A. Harrison, T. M. Nenoff, T. E. Gier and G. D. Stucky, Inorg. Chem., 1992, 31, 5395 CrossRef CAS.
- C. G. Lugmair, T. D. Tilley and A. L. Rheingold, Chem. Mater., 1997, 9, 339 CrossRef CAS.
- Y. Ortiz-Avila, P. R. Rudolf and A. Clearfield, Inorg. Chem., 1989, 28, 2137 CrossRef CAS.
- K. Maeda, J. Akimoto, Y. Kiyozumi and F. Mizukami, Angew. Chem., Int. Ed. Engl., 1995, 34, 1199 CrossRef CAS.
- Z. Florjańczyk, A. Lasota, A. Wolak and J. Zachara, Chem. Mater., 2006, 18, 1995 CrossRef.
- R. Murugavel, A. Choudhury, M. G. Walawalkar, R. Pothiraja and C. N. R. Rao, Chem. Rev., 2008, 108, 3549 CrossRef CAS.
- Metal Phosphonate Chemistry: From Synthesis to Applications, ed. A. Clearfield and K. Demadis, RSC Publishing, Cambridge, England, 2011 Search PubMed.
- G. Yücesan, Y. Zorlu, M. Stricker and J. Beckmann, Coord. Chem. Rev., 2018, 369, 105 CrossRef.
- X.-B. Lin, S.-L. Du, J.-W. Long, L. Chen and Y.-Z. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 881 CrossRef CAS.
- J. J. Ma, J. X. Yang, Y. W. Huang and K. Cao, J. Mater. Chem., 2012, 22, 2007 RSC.
- Y. W. Huang, S. Q. Song, Y. Yang, K. Cao, J. X. Yang and G. J. Chang, J. Mater. Chem. A, 2015, 3, 15935 RSC.
- Y. Cao, Y. Ju, F. Liao, X. Jin, X. Dai, J. Li and X. Wang, RSC Adv., 2016, 6, 14852 RSC.
- Y.-X. Wei, C. Deng, H. Chen, L. Wan, W.-C. Wei and Y.-Z. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 28036 CrossRef CAS.
- L. S. Brandão, L. C. Mendes, M. E. Medeiros, L. Sirelli and M. L. Dias, J. Appl. Polym. Sci., 2006, 102, 3868 CrossRef.
- P. Pan, Z. Liang, A. Cao and Y. Inoue, ACS Appl. Mater. Interfaces, 2009, 1, 402 CrossRef CAS.
- Y.-H. Cai and L.-S. Zhao, Open Chem., 2017, 15, 248 CAS.
- S. Wang, C. Han, J. Bian, L. Han, X. Wang and L. Dong, Polym. Int., 2011, 60, 284 CrossRef CAS.
- Y. Chen, S. Wang, O. Chen, Z. Xi, C. Wang, X. Chen, X. Feng, R. Liang and J. Yang, Eur. Polym. J., 2015, 72, 222 CrossRef CAS.
- T. Xu, Y. Wang, D. He, Y. Xu, Q. Li and C. Shen, Polym. Test., 2014, 34, 131 CrossRef CAS.
- Y.-H. Zhang and Y.-H. Cai, Int. J. Polym. Sci., 2016, 2016, 3926876 Search PubMed.
- S.-H. Lin, H.-T. Wang, J.-E. Wang and T.-M. Wu, Materials, 2020, 13(2), 376 CrossRef CAS.
- G. A. Bhat, A. C. Kalita and R. Murugavel, CrystEngComm, 2017, 19, 5390 RSC.
- R. Murugavel, Emergent Mater., 2019, 2, 273 CrossRef CAS.
- P. Bhanja, J. Na, T. Jing, J. Lin, T. Wakihara, A. Bhaumik and Y. Yamauchi, Chem. Mater., 2019, 31, 5343 CrossRef CAS.
- S. K. Gupta, A. C. Kalita, A. A. Dar, S. Sen, G. Naresh Patwari and R. Murugavel, J. Am. Chem. Soc., 2017, 139(1), 59 CrossRef CAS.
- K. D. Demadis, Z. Anagnostou, A. Panera, G. Mezei, M. V. Kirillovac and A. M. Kirillov, RSC Adv., 2017, 7, 17788 RSC.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, RSC Adv., 2015, 5, 7628 RSC.
- M. Taddei, F. Costantino and R. Vivani, Eur. J. Inorg. Chem., 2016, 4300 CrossRef CAS.
- M. Taddei, S. J. I. Shearan, A. Donnadio, M. Casciola, R. Vivani and F. Costantino, Dalton Trans., 2020, 49, 3662 RSC.
- B. Mossety-Leszczak, B. Strachota, A. Strachota, M. Steinhart and M. Šlouf, Eur. Polym. J., 2015, 72, 238 CrossRef CAS.
- J.-G. Mao, Coord. Chem. Rev., 2007, 251, 1493 CrossRef CAS.
- R. M. P. Colodrero, A. Cabeza, P. Olivera-Pastor, D. Choquesillo-Lazarte, J. M. Garcia-Ruiz, A. Turner, G. Ilia, B. Maranescu, K. E. Papathanasiou, G. B. Hix, K. D. Demadis and M. A. G. Aranda, Inorg. Chem., 2011, 50, 11202 CrossRef CAS.
- K. Krekić, D. Klintuch, C. Lescop, G. Calvez and R. Pietschnig, Inorg. Chem., 2019, 58, 382 CrossRef.
- M. Dębowski, K. Łokaj, A. Ostrowski, J. Zachara, P. Wiecińska, P. Falkowski, A. Krztoń-Maziopa and Z. Florjańczyk, Dalton Trans., 2018, 47, 16480 RSC.
- F.-X. Coudert, Chem. Mater., 2015, 27, 1905 CrossRef CAS.
- W. L. Zhang, Y. D. Liu, H. J. Choi and Y. Seo, RSC Adv., 2013, 3, 11723 RSC.
- X. Guo, X. Yu, Y. Chen, Z. Feng, Q. Li, Z. Sun, G. Li, C. Hao and Q. Lei, RSC Adv., 2016, 6, 99072 RSC.
- Z. Wang, S. Xuan, W. Jiang and X. Gong, RSC Adv., 2017, 7, 25855 RSC.
- E. Kutalkova, T. Plachy, J. Osicka, M. Cvek, M. Mrlik and M. Sedlacik, RSC Adv., 2018, 8, 24773 RSC.
- Y. Z. Dong, Y. Seo and H. J. Choi, Soft Mater., 2019, 15, 3473 RSC.
- Y. G. Ko and U. S. Choi, J. Rheol., 2013, 57, 1655 CrossRef.
- M. Cabuk, in Smart and Functional Soft Materials, ed. X. Dong, IntechOpen, 2019, ch. 5, DOI:10.5772/intechopen.86455.
- S. Datta, R. Barua and J. Das, A Review on Electro-Rheological Fluid (ER) and Its Various Technological Applications, Extremophilic Microbes and Metabolites - Diversity, Bioprospecting and Biotechnological Applications, IntechOpen, 2020, DOI:10.5772/intechopen.90706, Available from: https://www.intechopen.com/online-first/a-review-on-electro-rheological-fluid-er-and-its-various-technological-applications.
- T. Do, Y. G. Ko, Y. Jung, Y. Chun and U. S. Choi, J. Mater. Chem. C, 2017, 5, 11683 RSC.
- D. E. C. Corbridge, Phosphorus: Chemistry, Biochemistry and Technology, CRC Press, Taylor & Francis Group, Boca Raton, USA, 6th edn, 2013 Search PubMed.
- L. Chuntonov, R. Kumar and D. G. Kuroda, Phys. Chem. Chem. Phys., 2014, 16, 13172 RSC.
- NIST Mass Spectrometry Data Center, W. E. Wallace, director, Mass Spectra, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69 [Online], ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, USA, can be found under, https://webbook.nist.gov/chemistry/, accessed, April 2020, NIST MS number: 7 (water), 133909 (phenol), 19 (carbon monoxide), 69 (carbon dioxide), 114218 (biphenyl), 6610 (diphenyl ether).
- M. Roming, C. Feldmann, Y. S. Avadhut and J. Schmedt auf der Günne, Chem. Mater., 2008, 20, 5787 CrossRef CAS.
- T. M. Nenoff, S. G. Thoma, P. Provencio and R. S. Maxwell, Chem. Mater., 1998, 10, 3077 CrossRef CAS.
- Cards: 33–1474 [hopeite, zinc phosphate tetrahydrate, Zn3(PO4)2·4H2O], 29–1390 [zinc phosphate, Zn3(PO4)2], 39–0711 (zinc pyrophosphate, Zn2P2O7), 30–1488 [zinc metaphosphate, Zn(PO3)3], Powder Diffraction File Database PDF-2, International Centre for Diffraction Data, Newtown Square, Pennsylvania, USA, 1998 Search PubMed.
- Y. Zhang, K. J. Scott and A. Clearfield, J. Mater. Chem., 1995, 5, 315 RSC.
- D. M. Poojary and A. Clearfield, J. Am. Chem. Soc., 1995, 117, 11278 CrossRef CAS.
- S.-H. Lin, J.-M. Wang, H.-T. Wang and T.-M. Wu, Eur. Polym. J., 2019, 116, 1 CrossRef CAS.
- Y.-A. Chen, E.-C. Chen and T.-M. Wu, J. Mater. Sci., 2015, 50, 7770 CrossRef CAS.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681 CrossRef CAS.
- J. Zachara, Inorg. Chem., 2007, 46, 9760 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- I. D. Brown, Accumulated Table of Bond Valence Parameters, https://www.iucr.org/__data/assets/file/0007/126574/bvparm2016.cif, accessed, July, 2020 Search PubMed.
- S. I. Sadovnikov, A. I. Gusev, A. V. Chukin and A. A. Rempel, Phys. Chem. Chem. Phys., 2016, 18, 4617 RSC.
- R. Hammond, K. Pencheva, K. J. Roberts, P. Mougin and D. Wilkinson, J. Appl. Crystallogr., 2005, 38, 1038 CrossRef CAS.
- V. Lhomme, C. Bruneau, N. Soyer and A. Brault, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 98 CrossRef CAS.
- J.-B. Wu, M.-L. Lin, X. Cong, H.-N. Liu and P.-H. Tan, Chem. Soc. Rev., 2018, 47, 1822 RSC.
- I. Childres, L. A. Jauregui, W. Park, H. Cao and Y. P. Chen, in New Developments in Photon and Materials Research, ed. J. I. Jang, Nova Science Publishers, Inc., New York, USA, 2013, ch. 19, p. 403 Search PubMed.
- H. Medina, Y.-C. Lin, C. Jin, C.-C. Lu, C.-H. Yeh, K.-P. Huang, K. Suenaga, J. Robertson and P.-W. Chiu, Adv. Funct. Mater., 2012, 22, 2123 CrossRef CAS.
- Y. Tanizawa, Y. Okamoto, K. Tsuzuki, Y. Nagao, N. Yoshida, R. Tero, S. Iwasa, A. Hiraishi, Y. Suda, H. Takikawa, R. Numano, H. Okada, R. Ishikawa and A. Sandhu, in Journal of Physics: Conference Series 352, IOP Publishing, Bristol, 2012, p. 012011, DOI:10.1088/1742-6596/352/1/012011.
- R. Tarcan, O. Todor-Boer, I. Petrovai, C. Leordean, S. Astilean and I. Botiz, J. Mater. Chem. C, 2020, 8, 1198 RSC.
- I. Miccoli, F. Edler, H. Pfnür and C. Tegenkamp, J. Phys.: Condens. Matter, 2015, 27, 223201 CrossRef CAS.
- F. M. Smits, Bell Syst. Tech. J., 1958, 37, 711 CrossRef.
- V. B. Mohan, R. Brown, K. Jayaraman and D. Bhattacharyya, Mater. Sci. Eng., B, 2015, 193, 49 CrossRef CAS.
- Z. Tang, S. Shen, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 4603 CrossRef CAS.
- M. M. Ramos-Tejada, F. J. Arroyo and A. V. Delgado, Langmuir, 2010, 26, 16833 CrossRef CAS.
- A. V. Agafonov, A. S. Kraev, T. V. Kusova, O. L. Evdokimova, O. S. Ivanova, A. E. Baranchikov, T. O. Shekunova and S. A. Kozyukhin, Molecules, 2019, 24, 3348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the ZnDPhP crystal structure analysis, results of elemental analysis; unit cell parameters estimated for ZnDPhP between room temperature and 235 °C; FTIR spectra and PXRD patterns of all investigated compounds and their amine intercalates; tabulated and graphical TGA and TG-QMS data for ZnDPhP and ZnMPhP-H; DSC profile of ZnDPhP; additional SEM images of ZnDPhP, ZnMPhP-H and ZnMPhP-A; 31P CP/MAS NMR spectra of the synthesized hybrid polymers; Raman spectra and PXRD patterns of ZnDPhP and ZnMPhP-H pyrolyzates; data and corrections factors concerning measurements of electrical conductivity of the PVDF-based composites carried out by means of the four-point probe (4PP) method; electrorheological data for the silicone-based liquid dispersions and cured composites. CCDC 1472713. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09493e |
| This journal is © The Royal Society of Chemistry 2021 |