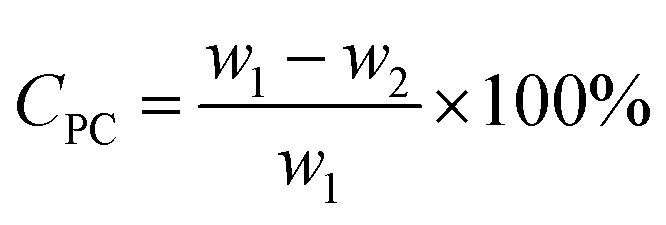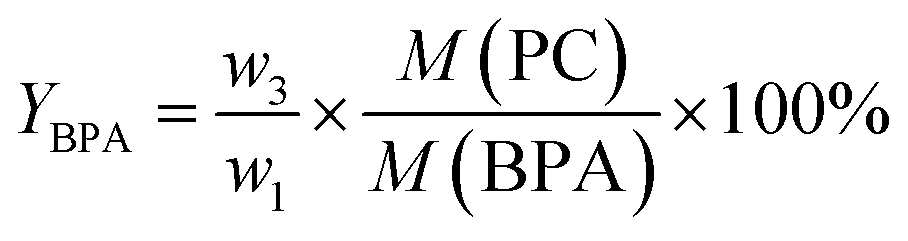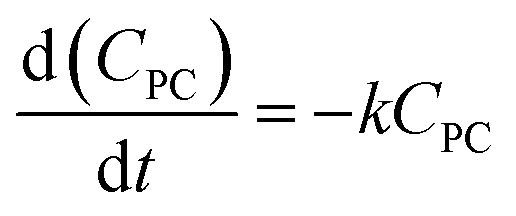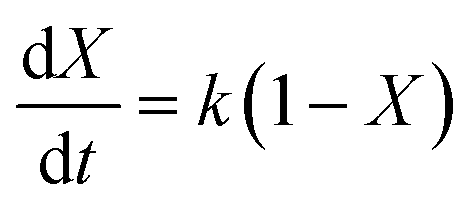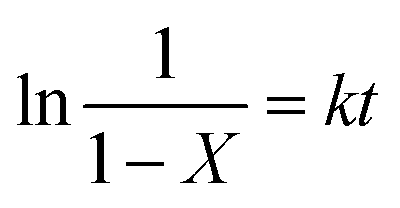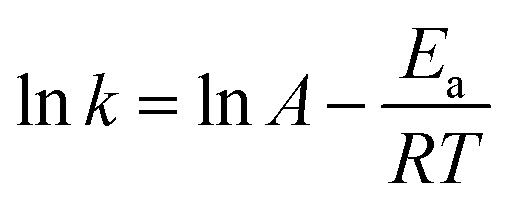DOI:
10.1039/D0RA09215K
(Paper)
RSC Adv., 2021,
11, 1595-1604
Degradation of polycarbonate to produce bisphenol A catalyzed by imidazolium-based DESs under metal-and solvent-free conditions†
Received
29th October 2020
, Accepted 7th December 2020
First published on 5th January 2021
Abstract
Bisphenol A (BPA) is an important chemical raw material, but the traditional preparation process of BPA is costly and complicated, so it is necessary to find an efficient and environmentally friendly method for the production of BPA. Deep eutectic solvents (DESs) have attracted widespread attention due to their low cost, low toxicity, low melting point, non-volatilization, easy preparation, recyclablility and biodegradability. In this work, a series of imidazolium-based DESs were synthesized and used for the degradation of polycarbonate (PC), and BPA was obtained from the methanolysis of PC catalyzed by DESs under metal- and solvent-free conditions. It was found that imidazolium-based DES [EmimOH]Cl-2Urea showed excellent catalytic activity and reusability. Under the optimized reaction conditions (the mass ratio of DES to PC is 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the molar ratio of CH3OH to PC is 5:1, 120 °C, reaction time 2 h), the PC conversion and BPA yield were almost 100% and 98%, respectively. Moreover, the kinetics of methanolysis catalyzed by [EmimOH]Cl-2Urea was investigated in the temperature range 100–120 °C, and the results indicated that it is a pseudo-first order reaction with an activation energy of 133.59 kJ mol−1. In addition, a possible catalytic mechanism of PC methanolysis is proposed.
:1, the molar ratio of CH3OH to PC is 5:1, 120 °C, reaction time 2 h), the PC conversion and BPA yield were almost 100% and 98%, respectively. Moreover, the kinetics of methanolysis catalyzed by [EmimOH]Cl-2Urea was investigated in the temperature range 100–120 °C, and the results indicated that it is a pseudo-first order reaction with an activation energy of 133.59 kJ mol−1. In addition, a possible catalytic mechanism of PC methanolysis is proposed.
1. Introduction
Polycarbonate (PC), as a high-performance engineering plastic, has been widely used in compact disk manufacturing, glass assembly industry, automotive industry, medical equipment, food packaging, etc.1 With the rapid growth of PC production and sales, more and more PC waste has been generated. Although this PC waste is non-toxic and harmless, it is bulky and difficult to degrade naturally, which may cause serious environmental pollution and waste resources. From the perspective of resource conservation and sustainable development, chemical recycling of this discarded PC is still an important issue and has attracted widespread attention.2,3
Several chemical methods, including thermal pyrolysis,4–6 glycolysis,7,8 hydrolysis9–11 and aminolysis12,13 have been studied by many researchers. Some good results with high conversion and high yield have been obtained, but there are still many disadvantages, such as low purity of BPA, slow reaction rate, unrecyclable catalyst and harsh reaction conditions, resulting in high cost and high energy consumption. Compared with these methods, alcoholysis, especially methanolysis, is a more important approach for PC recycling due to its mild reaction conditions and relatively easy recovery of raw material monomers. Various catalysts, including concentrated base or superbases (NaOH,14,15 DBU16 and TBD17), ZnO-NPs/NBu4Cl nanoparticles,18 mesoporous molecular sieves (CaO, SrO, BaO)/SBA-15 (ref. 19) and CaO–CeO2/SBA-15 (ref. 20) and some ionic liquids ([Bmim]Cl,21 [Bmim]OAc,22 [Bmim]Cl-2.0FeCl3,23 [HDBU][LAc]24 and [HDBU][Im]25) have been used to catalyze the PC methanolysis (Table S1†) and have several advantages, such as high PC conversion and yield of BPA,14,21–25 easy to separate solid catalyst,18,20 low temperature (60, 70 °C)14,22,25 and short reaction time.24 However, there are also some disadvantages, such as equipment corrosion,14,15 cumbersome operation, waste water generation,14,15 large amount of solvent (toluene,14 or THF18,20) or catalyst,21,22 and the synthesis process of ionic liquids are very complicated, costly, metal ions contaminate the product23 and cannot be biodegraded. As a result, finding a new simple, environmentally benign and low-cost synthetic process is still an unsolved issue in this field as the methods mentioned above are not green.
To develop a cost-effective and environmentally friendly method, we focus on exploring a new and affordable catalyst for the methanolysis of PC under “green” conditions. Deep eutectic solvents (DESs) were first described by Abbott26 et al. in 2003, and are usually synthesized by simply mixing hydrogen bond acceptors (HBAs) and hydrogen bond donors (HBDs). As an alternative, DESs not only have similar properties to ionic liquids, but also have many other advantages, including simple preparation, cheap raw materials, low toxicity, biodegradability and no further purification.27 Because of these properties, DESs have gradually become a new type of green solvents and catalysts, and have been widely used in biocatalysis,28,29 extraction,30,31 carbon dioxide capture,32–34 biomedical applications,35 material synthesis36–38 and other fields. In recent decades, DESs as a reaction medium have attracted widespread attention, making them have broad application prospects in industrial production.39
Although their application as catalysts in PC degradation has not been reported, DESs such as urea/metal salt, ChCl/Zn(Ac)2 and 1,3-dimethylurea/Zn(OAc)2 have been successfully used for the alcoholysis of polyethylene terephthalate (PET),40–42 and achieved some good results. At present, DESs are mainly made of choline chloride (ChCl) and urea or certain metal salts, but almost no other types of DESs have been reported. In our previous work, ChCl-2Urea43 was successfully used to catalyze the PC methanolysis, with 100% CPC and 99% YBPA at 130 °C for 2.5 h. Compared with ChCl-2Urea, imidazolium-based DES has lower viscosity, lower melting point, wider liquid phase range, higher electrical conductivity and better thermal stability. Therefore, this kind of DESs will have a broader application fields in the feature.
In order to obtain an efficient and green catalyst for PC methanolysis, a series of [EmimOH]Cl/urea DESs (molar ratio from 4:1 to 1:5) was synthesized by using [EmimOH]Cl and urea as HBA and HBD in this work. The effects of reaction conditions, such as reaction temperature, reaction time, PC/methanol molar ratio, and DESs/PC mass ratio were further examined in detail. In addition, the recycling of [EmimOH]Cl/urea, the mechanism of PC methanolysis and the kinetics of this reaction were also investigated.
2. Experimental
2.1 Material and instruments
Pure PC pellets (3 mm length and 2.5 mm diameter) were used as model plastics and purchased from Idemitsu Kosan Co., Ltd. of Japan. The weight average molecular weight (Mw) and number average molecular weight (Mn) were 23563 g mol−1 and 8993 g mol−1, respectively. N-Methylimidazole, 2-chloroethanol and urea were obtained from Shanghai McLean Biochemical Technology Co., Ltd., China. Methanol, oxalic acid (OA), succinic acid (SA), benzoic acid (BA), p-toluenesulfonic acid (PTSA), aniline (AN), acetamide (AA), glycerol (Gly), glucose (Glc), anhydrous manganese chloride (MnCl2) and anhydrous cupric chloride (CuCl2) were purchased from Sinopharm Chemical Reagent Beijing Co., Ltd., China. All chemicals (AR) were utilized directly without any further treatment.
Infrared (IR) spectra of the main degradation product and DES were recorded on a BRUKER FT-IR VERTEX 70 spectrometer in the range of 4000–400 cm−1 by liquid film or KBr discs. 1H and 13C Nuclear magnetic resonance (NMR) spectra were measured on a BRUKER AV 500 Digital NMR spectrometer in d6-DMSO solution. The TGA curves were recorded with a NETZSCH STA 409 PC/PG thermogravimetric analyzer in a nitrogen atmosphere at a flow rate of 100 mL min−1 over a temperature range from room temperature to 600 °C at a heating rate of 20 °C min−1. The Mw and Mn of fresh PC and residues were checked by Gel permeation chromatography (GPC) (TOSOH-8320) under the condition of room temperature, solvent tetrahydrofuran (THF) as the mobile phase at a flow rate of 1.0 mL min−1. The GPC is equipped with a differential refraction detector and TSK gel Super Mutipore HZ-M*2 columns. Calibration of GPC was performed with standard polystyrene samples.
2.2 Synthesis of imidazolium-based DESs
2.2.1 Preparation of precursor [EmimOH]Cl. In a 250 mL three-necked flask equipped with a reflux condenser, a thermometer and a magnetic stirrer, adding (32.843 g, 0.4 mol) N-methylimidazole, (32.330 g, 0.4 mol) 2-chloride ethanol and 150 mL ethyl acetate. Under nitrogen atmosphere, the mixture was heated to reflux and stirred for 48 h, then cooled to room temperature, and a large amount of white precipitate appeared at the bottom of the flask. The precipitate was collected by filtration, washed three times with ethyl acetate, and dried for 8 h under vacuum at 80 °C to obtain a white solid powder, 1-hydroxyethyl-3-methylimidazolium chloride [EmimOH]Cl. The product weighed 50.54 g with the yield of 77.5%. 13C NMR (126 MHz, d6-DMSO), δ = 136.8 (s), 123.2 (s), 122.6 (s), 59.2 (s), 51.5 (s), 35.6 (s) (see Fig. S1†).
2.2.2 Preparation of [EmimOH]Cl-2Urea. Take (8.187 g, 0.05 mol) precursor [EmimOH]Cl and (6.004 g, 0.1 mol) urea, and put into a 100 mL round bottom flask equipped with a reflux condenser and a thermometer. Under a nitrogen atmosphere, the reaction was heated and stirred at 80 °C for 2.0 h, until a colorless, transparent and homogeneous liquid was completed, the reaction was over and the product [EmimOH]Cl-2Urea (13.8 g) was collected with the yield of 97.5%. [EmimOH]Cl-2Urea 13C NMR (126 MHz, d6-DMSO), δ = 159.7 (s), 136.8 (s), 123.3 (s) 122.6 (s), 59.3 (s), 51.5 (s), 35.6 (s) (see Fig. S2†).According to the above method, DES of [EmimOH]Cl and urea with a molar ratio of 4:1 to 1:5 were synthesized, and other DESs were synthesized by mixing [EmimOH]Cl with different HBAs, such as [EmimOH]Cl-OA, [EmimOH]Cl-SA, [EmimOH]Cl-BA, [EmimOH]Cl-PTSA, [EmimOH]Cl-2CuCl2, [EmimOH]Cl-2MnCl2, [EmimOH]Cl-2AN, [EmimOH]Cl-2AA, [EimmOH]Cl-2Gly and [EMIMOH]Cl-2Glc.
2.3 General procedure for catalytic methanolysis of PC
In each experiment, (4.0 g, 15.7 mmol) PC (w1) was degraded, using methanol catalyzed by urea or one kind of synthesized DESs. The mass ratio of catalyst to PC ranges from 0.05:1 to 0.1:1, and the amount of methanol ranges from 0.047 mol to 0.11 mol. A 50 mL autoclave equipped with a thermometer and a stirrer was put into an oil bath for all the methanolysis experiments. The reaction was carried out under autogenous pressure at reaction temperatures ranging from 110–125 °C for reaction time of 1.0–2.5 h. After the methanolysis reaction was completed, the autoclave was cooled to room temperature. The unreacted PC residue (w2) was collected by filtration and dried at 100 °C for 4 h, weighed (w2) to calculate the conversion of PC (CPC), which is determined by eqn (1):| |
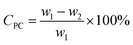 | (1) |
where w1 refers to the initial weight of PC, w2 refers to the weight of unreacted PC. Additionally, a light yellow viscous liquid was obtained by removing methanol and dimethyl carbonate (DMC) from the distilled filtrate under atmospheric pressure, which was then dissolved in an equal volumes of ethyl acetate and distilled water till the mixture separated into two phases. The upper liquid was removed ethyl acetate by rotary evaporation to obtain a white solid, which was dried for 8 h under vacuum to obtain the product BPA (w3). The lower liquid was mainly DES and water, which was treated by vacuum distillation to remove water. The residue was mainly DES, can be reused for the next experiment without any further treatment. Yield of BPA (YBPA) is defined by eqn (2):| |
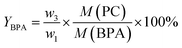 | (2) |
where w3 represents the weight of the obtained BPA, M(PC) and M(BPA) are the molar mass of the repeating unit of PC and molecular weight of BPA, which are 254 g mol−1 and 228 g mol−1, respectively.
The reaction formula is shown in Scheme 1.
 |
| | Scheme 1 Reaction formula of PC methanolysis catalyzed by [EmimOH]Cl-2Urea. | |
3. Results and discussion
3.1 Selection of catalyst for PC methanolysis
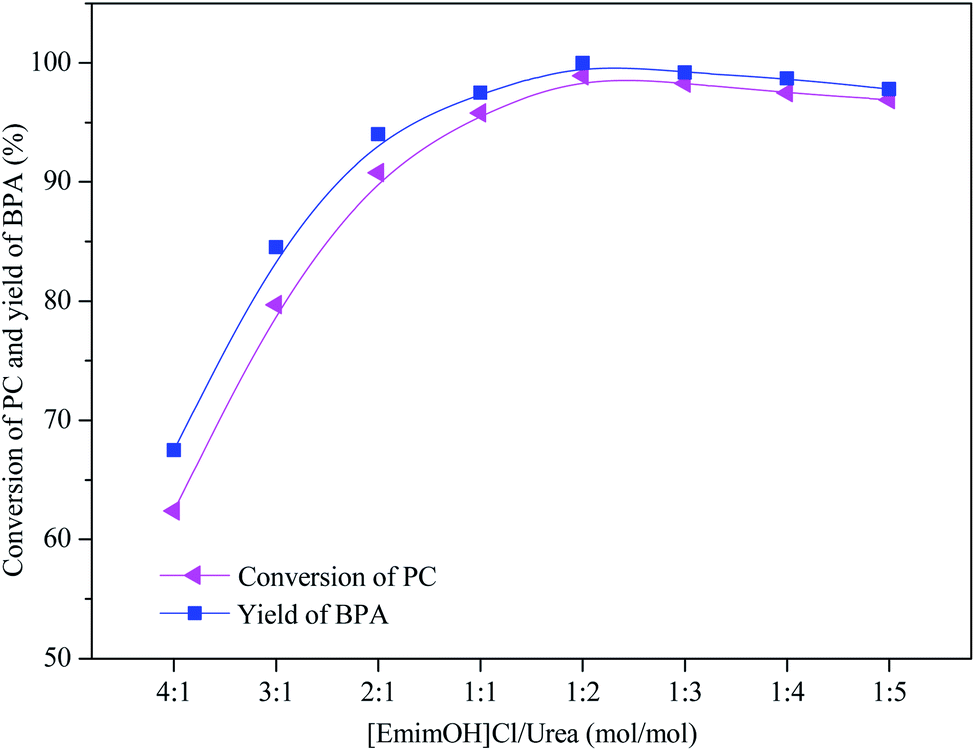
The effect of different molar ratio of DESs on the methanolysis of PC was investigated, and the results are shown in Fig. 1. When n([EmimOH]Cl):n(urea) was added from 4:1 to 1:2, the CPC increased from 62% to 100%, and the YBPA increased from 62% to 99%, respectively. In other words, when the molar ratio of [EmimOH]Cl/urea is 1:2, PC is completely degraded into products. The reason may be due to the formation of hydrogen bonds between HBD and HBA, when n([EmimOH]Cl):n(urea) = 1:2, the synergistic effect between them is better, improving the catalytic performance of DES. Continue to increase the molar ratio to 1:5, the CPC and YBPA decreased slightly, the reason may be caused by the operation process.
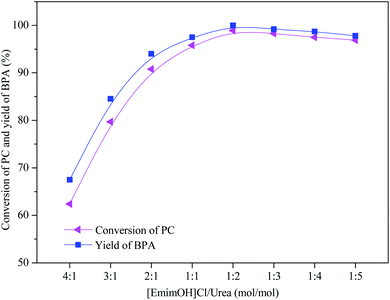 |
| | Fig. 1 Catalytic performance of DESs with different molar ratio of [EmimOH]Cl and urea (reaction conditions: PC (4.0 g), DES (0.4 g), n(PC):n(CH3OH) = 1:5, 120 °C and 2.0 h). | |
In addition, the catalytic performance of synthesized DESs in the PC methanolysis was evaluated and the results were presented in Table 1. It was indicated that this reaction cannot occur without any catalyst (entry 1). Some acidic DESs like [EmimOH]Cl-OA, [EmimOH]Cl-SA, [EmimOH]Cl-BA, [EmimOH]Cl-PTSA (entries 2–5), and some neutral DESs like [EmimOH]Cl-2Gly, [EmimOH]Cl-2Glc (entries 8 and 9) had no catalytic effect on the PC methanolysis. The Lewis acidic DESs like [EmimOH]Cl-2MnCl2 and [EmimOH]Cl-2CuCl2 (entries 6 and 7), although they can catalyze this reaction, the CPC is very low and no BPA is obtained. However, basic DESs like [EmimOH]Cl-2Urea, [EmimOH]Cl-2AN and [EmimOH]Cl-2AA (entries 10–12) can catalyze the methanolysis of PC. The reason may be that PC methanolysis is a transesterification reaction, basic catalysts are beneficial to this reaction. It was also found that when [EmimOH]Cl and urea were used as catalyst alone (entries 13 and 14), the methanolysis cannot take place. However, when adding the DES formed by the same quality of [EmimOH]Cl and urea (molar ratio is 1:2), [EmimOH]Cl-2Urea exhibited very good catalytic activity with 100% CPC and 99% YBPA (entry 12). This high catalytic performance may be caused by the synergistic effect between [EmimOH]Cl and urea. Compare with ChCl-2Urea,43 the catalyst not only decreased the reaction temperature but also shorted reaction time.
Table 1 Catalytic activities of different catalysts in the methanolysis of PCa
| Entry |
Catalyst |
YBPA/% |
CPC/% |
| Experimental conditions: PC (4.0 g), catalyst (0.4 g), n(PC):n(CH3OH) = 1:5, T = 120 °C, t = 2 h. m([EmimOH]Cl) = 0.234 g. m(Urea) = 0.166 g. |
| 1 |
Blank |
0 |
0 |
| 2 |
[EmimOH]Cl-OA |
0 |
0 |
| 3 |
[EmimOH]Cl-SA |
0 |
0 |
| 4 |
[EmimOH]Cl-BA |
0 |
0 |
| 5 |
[EmimOH]Cl-PTSA |
0 |
0 |
| 6 |
[EmimOH]Cl-2MnCl2 |
0 |
4 |
| 7 |
[EmimOH]Cl-2CuCl2 |
0 |
2 |
| 8 |
[EmimOH]Cl-2Gly |
0 |
0 |
| 9 |
[EmimOH]Cl-2Glc |
0 |
0 |
| 10 |
[EmimOH]Cl-2AN |
95 |
99 |
| 11 |
[EmimOH]Cl-2AA |
0 |
6 |
| 12 |
[EmimOH]Cl-2Urea |
99 |
100 |
| 13 |
[EmimOH]Clb |
0 |
0 |
| 14 |
Ureac |
0 |
0 |
3.2 Characterization of [EmimOH]Cl-2Urea
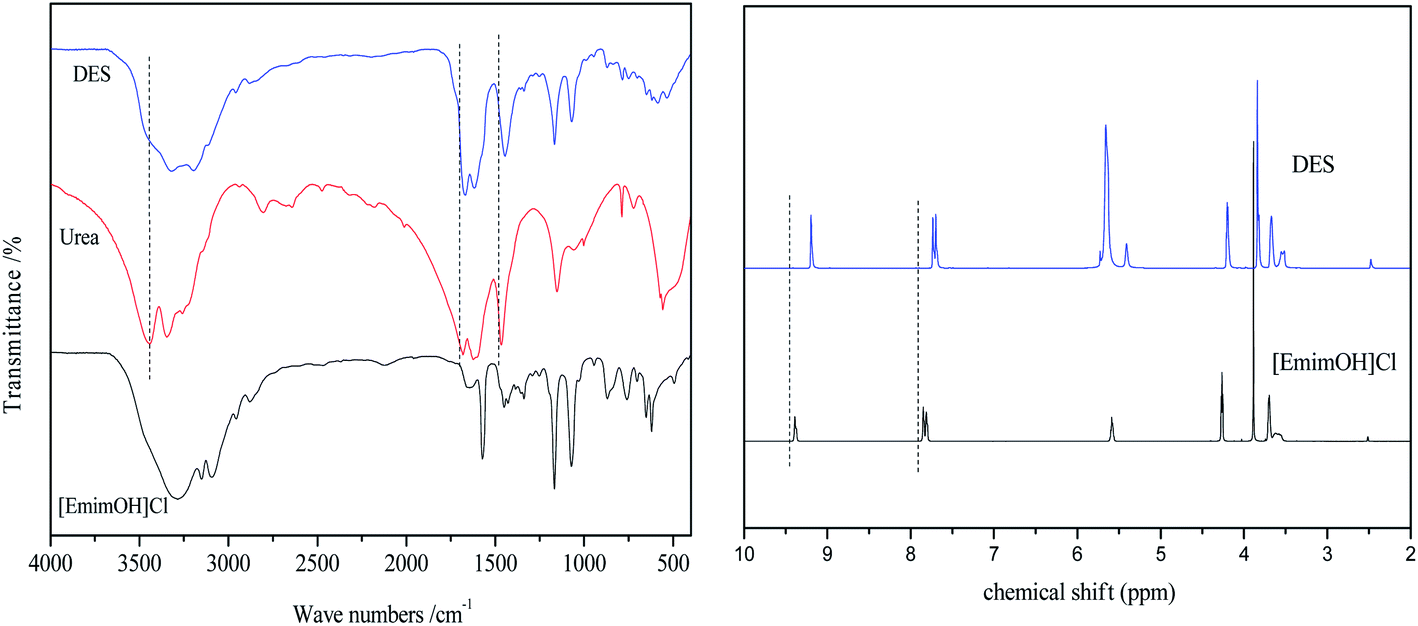
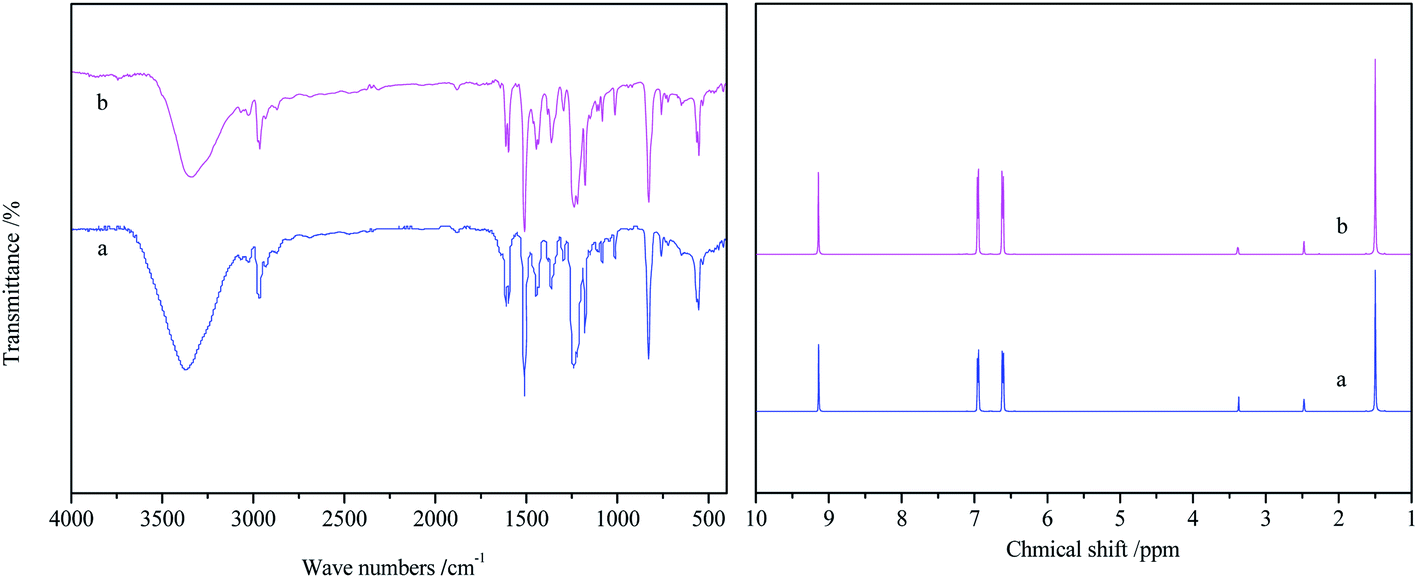
The structure of [EmimOH]Cl-2Urea was characterized by FT-IR and 1H NMR, and the results are shown in Fig. 2. The [EmimOH]Cl characteristic absorption peaks at 3285, 3093, 1642, 1572, 1167, 1072 cm−1, and peaks of urea at 3443, 3346, 1642, 1572, 1167 and 1072 cm−1. As can be seen that the IR spectrum of DES contains almost all the functional groups of [EmimOH]Cl and urea, such as the peaks of –NH2, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–Cl. But the peak at 3336 cm−1 belongs to the N–H stretching vibration, is wider than that of [EmimOH]Cl and urea, and moved to a low wave number, showing a significant red shift. In addition, the peaks of CO and N–H also have obvious red shifts.
O and C–Cl. But the peak at 3336 cm−1 belongs to the N–H stretching vibration, is wider than that of [EmimOH]Cl and urea, and moved to a low wave number, showing a significant red shift. In addition, the peaks of CO and N–H also have obvious red shifts.
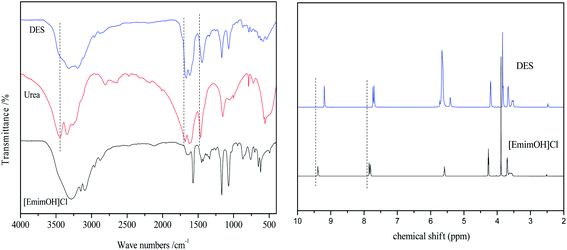 |
| | Fig. 2 FT-IR and 1H NMR spectra of DES, [EmimOH]Cl and urea. | |
In the 1H NMR of [EmimOH]Cl, the peaks at δ = 9.3–9.5, 7.84 and 7.81 ppm are assigned to the H of the imidazole ring. The multiple peaks at δ = 5.5–5.6 ppm attributed to the H of –OH. The triplet peak appeared at δ = 4.2–4.3 ppm belongs to 2H of –C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2CH2OH, another triplet peak at δ = 3.6–3.7 ppm is ascribed to 2H of –CH2C2OH. The singlet peak at δ = 3.8–3.9 ppm belongs to 3H of N–C3 in the imidazole ring. Compared with the 1H NMR peaks of [EmimOH]Cl, DES shows a single peak at δ = 5.6–5.7 ppm, which is attributed to –N2 in urea. From 1H NMR, DES basically contains all the peaks of the raw material.
2CH2OH, another triplet peak at δ = 3.6–3.7 ppm is ascribed to 2H of –CH2C2OH. The singlet peak at δ = 3.8–3.9 ppm belongs to 3H of N–C3 in the imidazole ring. Compared with the 1H NMR peaks of [EmimOH]Cl, DES shows a single peak at δ = 5.6–5.7 ppm, which is attributed to –N2 in urea. From 1H NMR, DES basically contains all the peaks of the raw material.
From the above results, it can be seen that due to the interaction between HBD and HBA, a hydrogen bond is formed between [EmimOH]Cl and urea, resulting in obvious red shifts shown at the peaks of N–H and CO in the IR spectrum. In the 1H NMR of DES, some characteristic peaks, C–H for instance move toward high field, and the chemical shift values decrease.
3.3 Qualitative analysis of methanolysis product
When the methanolysis was over, an equal volume of ethyl acetate and distilled water was put into the mixture. After removing the ethyl acetate in the upper phase by rotary evaporation, the product was mainly BPA. The FT-IR and 1H NMR of the obtained product and standard BPA were shown in Fig. 3. In the FT-IR of the obtained product, the peak at 3335 cm−1 corresponds to the O–H stretching vibration of phenolic hydroxy group, the peak at 2964 cm−1 corresponds to the C–H stretching vibration of methyl, the peaks at 1446–1612 cm−1 corresponds to the CC stretching vibration of the benzene ring, and the peaks at 1177–1237 cm−1 corresponds to the C–O stretching vibration. The peak at 827 cm−1 corresponds to C–H out-of-plane bending vibration of benzene ring, indicating it was para-substituted benzene.
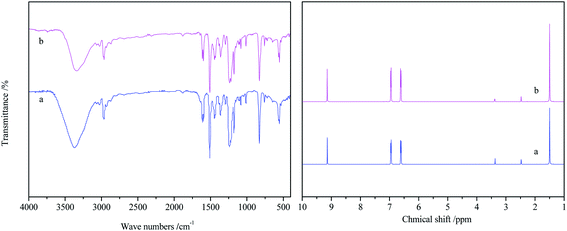 |
| | Fig. 3 FT-IR and 1H NMR spectra of obtained product and standard BPA ((a) standard BPA; (b) obtained product) (reaction conditions: 4.0 g PC, 0.4 g DES, n(PC):n(CH3OH) = 1:5, 120 °C and 2.0 h). | |
From the 1H NMR of the obtained product, it can be seen that the peak at δ = 1.50 ppm (s, 6H) belongs to the hydrogen of –C3, The symmetrical peaks appears at δ = 6.61 ppm (d, 4H), 6.95 ppm (d, 4H) attribute to the hydrogen of benzene ring, indicating a para-substitution. The peak at δ 9.14 ppm (s, 2H) represents the hydrogen of –O. Regardless of IR or 1H NMR, the spectrum of obtained product is almost consistent with that of standard BPA, which proves that the obtained product is BPA.
3.4 Effect of experimental conditions on PC methanolysis
The influences of reaction conditions on PC methanolysis were investigated and the results are shown in Table 2. Obviously, reaction temperature is the main influencing factor on the PC methanolysis. When the temperature increased from 110 to 120 °C, the CPC and YBPA increased from 14%, 8% to 100%, 99%, respectively (entries 1–3). However, when the reaction temperature was raised to 125 °C (entry 4), the CPC and YBPA did not increase significantly. The reason may be that the methanolysis of PC is a transesterification reaction, and it reached equilibrium at 125 °C. When the reaction time was extended from 1.25 h to 2.0 h (entries 3 and 5–6), the CPC increased from 24% to 100%, meanwhile, the YBPA reached the maximum 99% at 2.0 h. When m(DES):m(PC) was added from 0.05:1 to 0.1:1, both CPC and YBPA increased clearly, the CPC from 56% to 100% and the YBPA from 53% to 99%, respectively. Moreover, as the molar ratio of methanol to PC changed from 3:1 to 7:1 (entries 3 and 9–10), the CPC and YBPA increased slightly. When the molar ratio of methanol to PC was 5:1, the CPC and YBPA reached the maximum values, which were 100% and 99%, respectively. When n(CH3OH):n(PC) increased from 5:1 to 7:1 (entries 3 and 10), the CPC and YBPA had a slight decrease, which may be due to excessive methanol addition, resulting in a decrease in the reactant concentration.
Table 2 Effects of reaction conditions on methanolysis of PCa
| Entry |
Temperature/°C |
Time/h |
m(DES):m(PC) |
n(CH3OH):n(PC) |
CPC/% |
YBPA/% |
| Experimental conditions: PC (4.0 g). |
| 1 |
110 |
2.0 |
0.1:1 |
5:1 |
14 |
8 |
| 2 |
115 |
2.0 |
0.1:1 |
5:1 |
55 |
48 |
| 3 |
120 |
2.0 |
0.1:1 |
5:1 |
100 |
99 |
| 4 |
125 |
2.0 |
0.1:1 |
5:1 |
100 |
98 |
| 5 |
120 |
1.25 |
0.1:1 |
5:1 |
24 |
19 |
| 6 |
120 |
1.5 |
0.1:1 |
5:1 |
73 |
66 |
| 7 |
120 |
2.0 |
0.05:1 |
5:1 |
56 |
53 |
| 8 |
120 |
2.0 |
0.075:1 |
5:1 |
86 |
81 |
| 9 |
120 |
2.0 |
0.1:1 |
3:1 |
98 |
97 |
| 10 |
120 |
2.0 |
0.1:1 |
7:1 |
99 |
97 |
3.5 Reusability of [EmimOH]Cl-2Urea
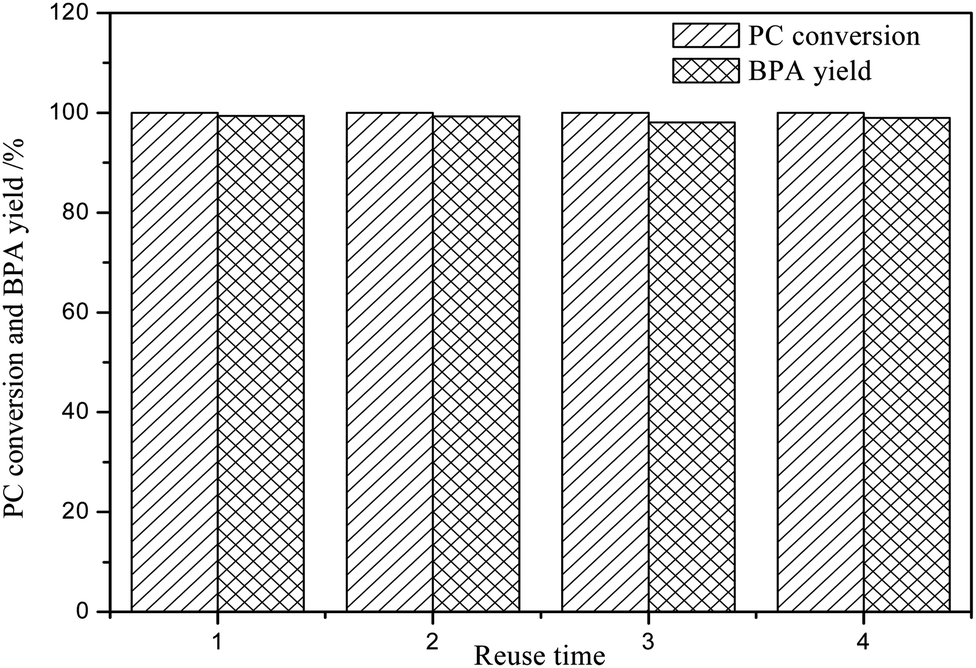
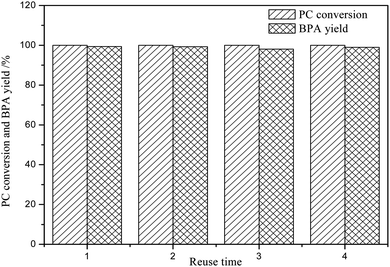
From the perspective of environmental protection and sustainable development, it is necessary to study the reusability of DES in PC methanolysis. Under the following conditions: PC (4.0 g), DES (0.4 g), n(PC):n(CH3OH) = 1:5, 120 °C and 2.0 h, the reusability of DES was investigated and the results were shown in Fig. 4. It can be clearly seen that even if the DES is reused for 4 times, the CPC and YBPA are not apparently reduced, indicating that [EmimOH]Cl-2Urea has good reusability performance. After reused for 4 times, the CPC and YBPA will decrease slightly due to the loss of catalyst during operation.
 |
| | Fig. 4 Reusability results of [EmimOH]Cl-2Urea (reaction conditions: PC (4.0 g), DES (0.4 g), n(PC):n(CH3OH) = 1:5, 120 °C and 2.0 h). | |
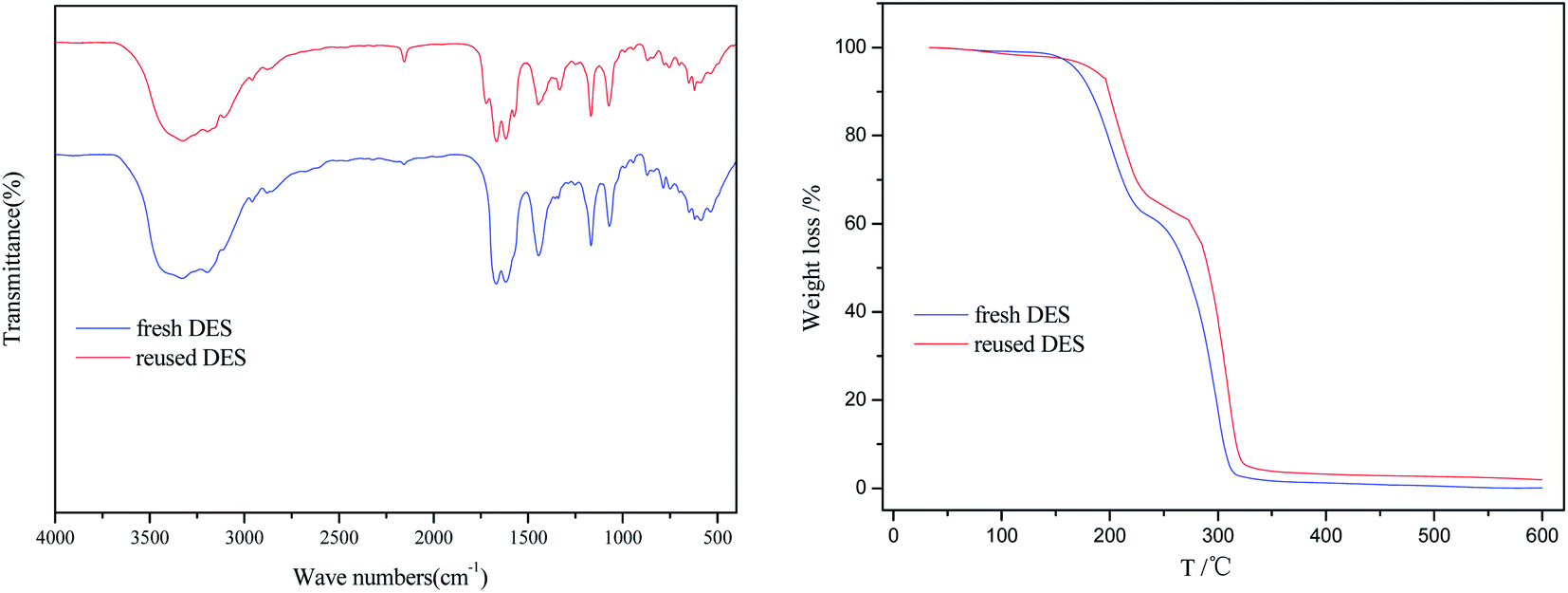
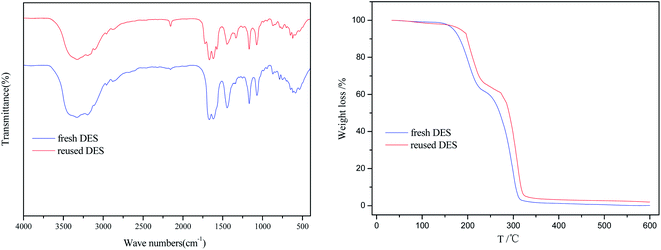
The fresh and reused [EmimOH]Cl-2Urea were characterized by FT-IR and TGA, and results were shown in Fig. 5. Comparing with the IR spectrum of fresh [EmimOH]Cl-2Urea, the reused one basically contained all the characteristic absorption peaks, indicating that there was no change in chemical structure of [EmimOH]Cl-2Urea after reused for 4 times. In addition, the TGA curve of fresh DES is almost the same as that of reused one, demonstrating that the [EmimOH]Cl-2Urea has an excellent thermal stability. Moreover, the TGA curves also shows that the decomposition process of DES is mainly divided into two stages. The weight loss was nearly 38% in the range of 175–252 °C, another weight loss is about 58% in the scope of 252–320 °C. When the temperature exceeds 320 °C, the mass of the sample gradually approaches zero, meaning the sample is completely decomposed. However, the methanolysis temperature is only 120 °C, lower than the decomposition temperature of [EmimOH]Cl-2Urea, which shows that DES can stably exist and hard to decompose under the above conditions.
 |
| | Fig. 5 FT-IR spectra and TGA curves of fresh and reused [EmimOH]Cl-2Urea. | |
3.6 Mechanism of PC methanolysis catalyzed by [EmimOH]Cl-2Urea
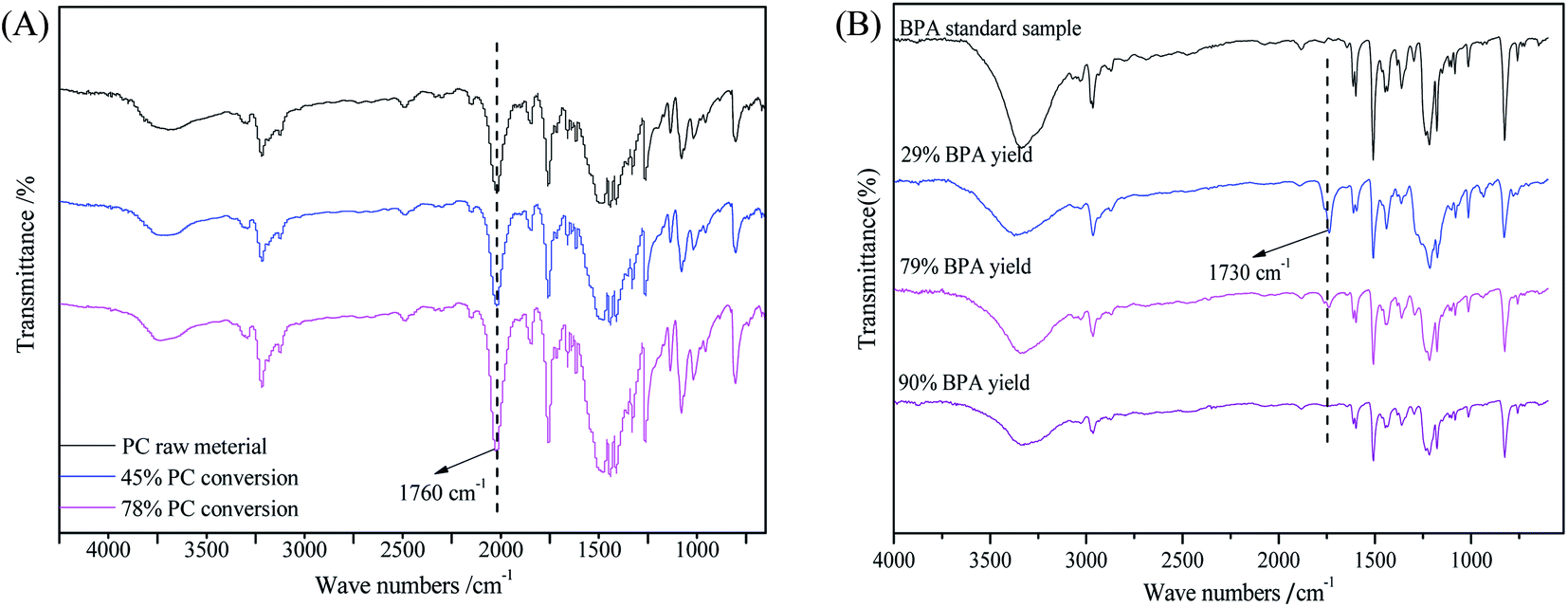
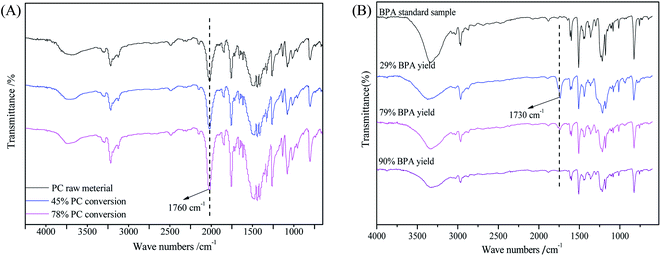
To study the mechanism of PC methanolysis, FT-IR was used to characterize the PC residues and BPA products at different conversions. As shown in Fig. 6(A), the PC residue at low conversion of 46% showed characteristic peaks similar to that of PC raw material. With increasing PC conversion, the –OH peak at about 3500 cm−1 increases slightly, and the –CO peak at about 1760 cm−1 becomes stronger. This might be due to the breakage of long chains into short chains during the reaction, resulting in more –OH and –CO being exposed. The BPA product obtained at low yield of 29% showed an obvious difference in the carbonyl group at about 1730 cm−1, indicating there are some oligomers in BPA product. As the YBPA increased to 78% and 90%, the carbonyl peak gradually weakened until completely disappeared. The structure of BPA obtained at a yield of 90% is almost identical with that of the BPA standard sample (Fig. 6(B)).
 |
| | Fig. 6 FT-IR spectra of PC residues (A) and BPA (B) obtained at different conversions. | |
The PC residues were also determined by GPC and the results are shown in Table 3. When the CPC increased from 0 to 46% and 78%, the Mn of PC residue decreased from 8993 to 5589, 5423 g mol−1, and the Mw of PC residue reduced from 23563 to 14130, 13933 g mol−1. The results indicate that with the increase of PC, the long-chain PC decomposed into some short-chain oligomers, which reduced the degree of polymerization and this was more favorable for further degradation.
Table 3 Molecular weights of PC residues obtained at different conversions (a 0%; b 46%; c 78%)
| Entry |
Mn |
Mw |
Mw/Mn |
| a |
8993 |
23563 |
2.620 |
| b |
5589 |
14130 |
2.528 |
| c |
5423 |
13933 |
2.569 |
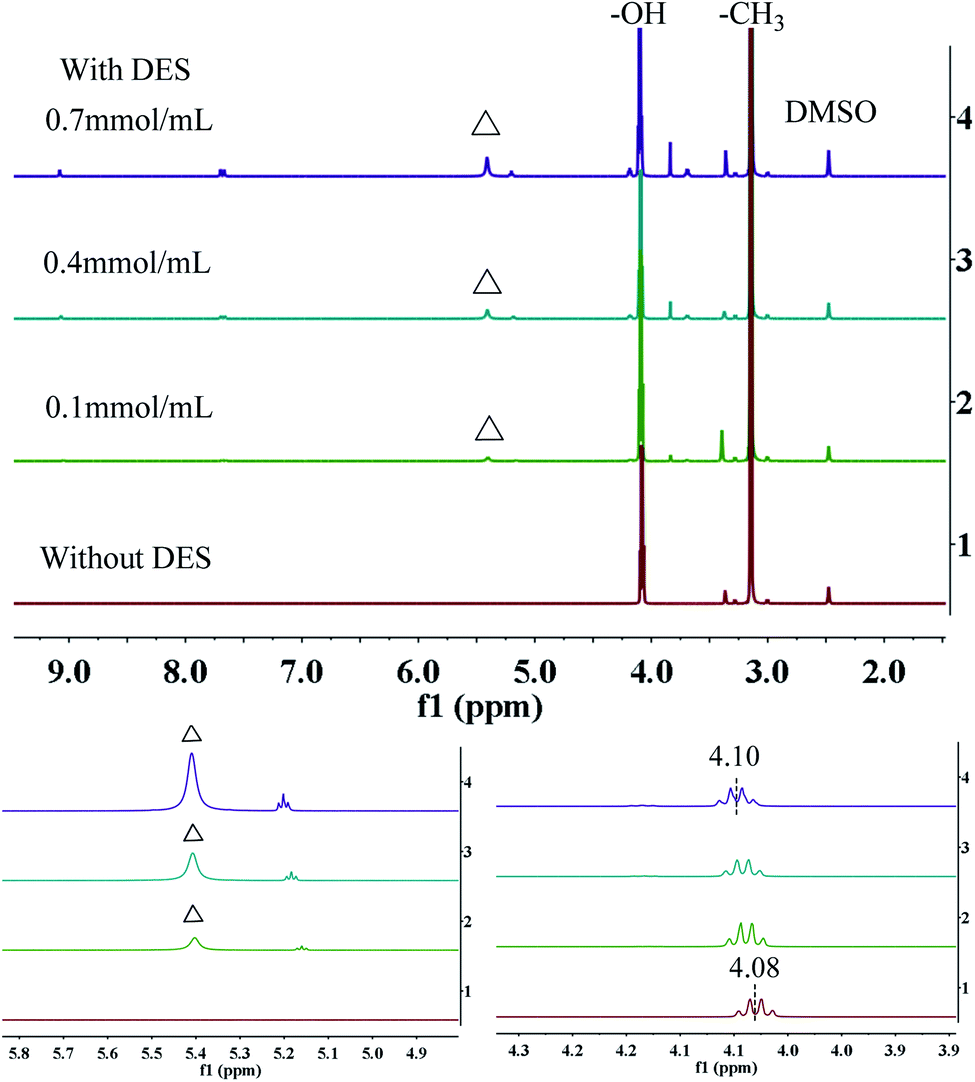
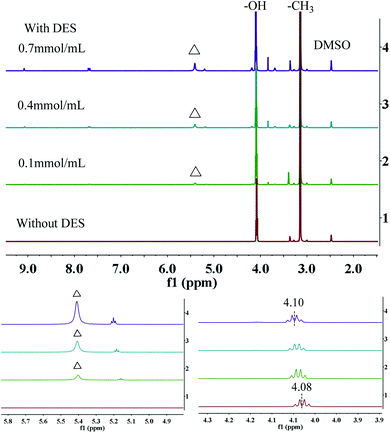
To further investigate this mechanism, 1H NMR spectroscopy analysis was performed in d6-DMSO. As shown in Fig. 7, in the absence of DES, the chemical shift of hydroxyl proton in methanol is 4.08 ppm. As the amount of DES increases (from curve 1 to 4), the signals of hydrogens in –OH move towards the lower field. When the amount of DES is 0.7 mmol mL−1, the chemical shift of hydrogen in –OH reached 4.10 ppm (Δδ = +0.02 ppm). In addition, as the concentration of methanol increased (from curve 4 to 1), the N– signal shifted from 5.43 to 5.42 ppm. However, as the concentration of DES increased from 0.1 to 0.4, 0.7 mmol mL−1, the signals of –C3 (δ = 3.14 ppm) do not change obviously. Therefore, it can be reasonably speculated that the change of chemical shift is due to the strong hydrogen bond interaction between the hydroxyl group in methanol and the amino group in DES, which is similar to what reported by Zhang's group.40–42
 |
| | Fig. 7 1H NMR spectra of [EmimOH]Cl-2Urea DES and CH3OH mixture with different molar ratio; the symbols in the 1H NMR spectra identify the N–H (triangle) of urea. | |
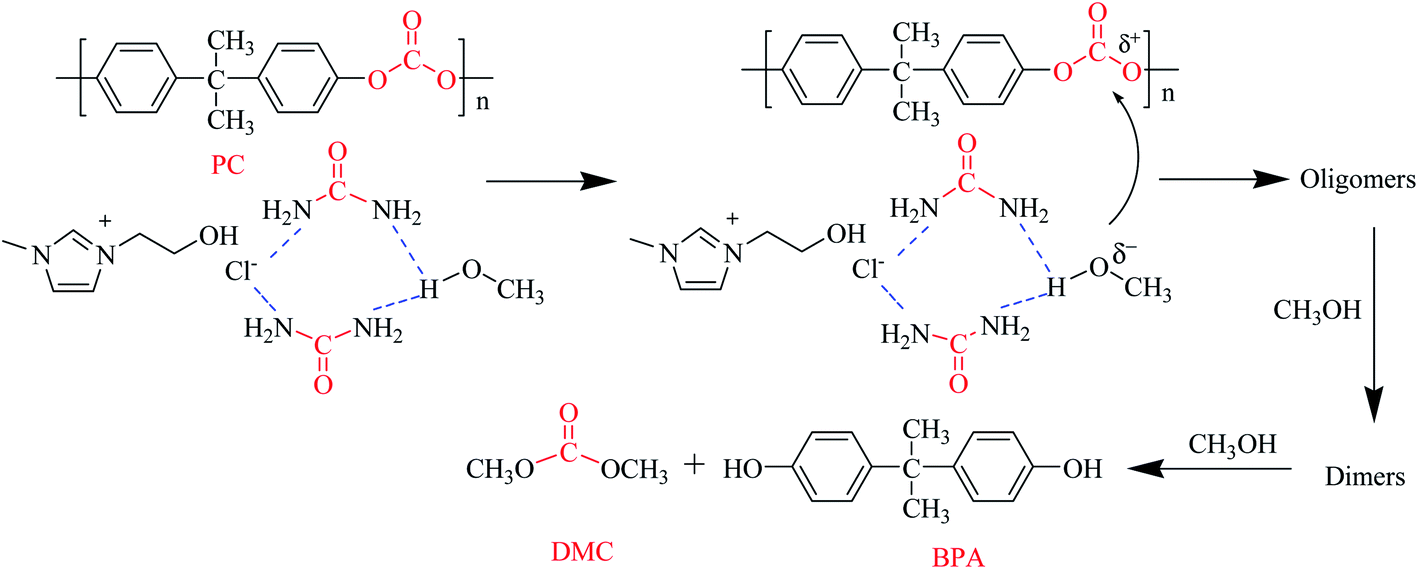
Based on the above experimental results, a probable mechanism of PC methanolysis catalyzed by [EmimOH]Cl-2Urea is proposed in Scheme 2. First, the H-bond is formed between the amino group of urea and the hydroxyl group of methanol, improving the electronegativity of the oxygen atom in CH3OH, which is conducive to –OCH3 attacking the carbon atom in the ester group of PC, resulting in the breaking of the ester bond. PC gradually decomposes into different molecular weight oligomers. These oligomers then reacted with methanol repeatedly to form smaller oligomers and dimers until BPA and dimethyl carbonate were produced.
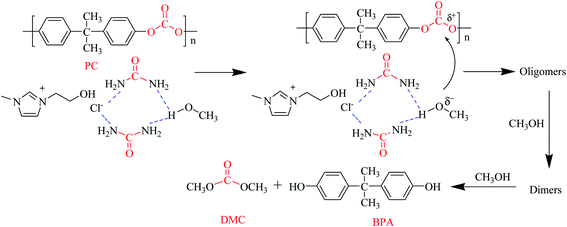 |
| | Scheme 2 Proposed mechanism of PC methanolysis catalyzed by [EmimOH]Cl-2Urea. | |
3.7 Kinetics of PC methanolysis
The methanolysis of PC previously reported in our group20–25,43 and the glycolysis of PET40–42 reported by Zhang groups. Kinetic studies of chemical degrading polymers were generally considered to be a pseudo-first order reaction. Therefore, the methanolysis of PC catalyzed by the DES [EmimOH]Cl-2Urea is also assumed to be a pseudo-first order, and the kinetic equation is as follows:| |
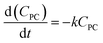 | (3) |
where k represents the rate constant of the reaction, and CPC is the concentration of PC at time t.where X represents the conversion of PC, so eqn (3) can be converted to eqn (5):| |
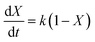 | (5) |
Eqn (5) is integrated against time to get eqn (6).
| |
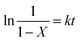 | (6) |
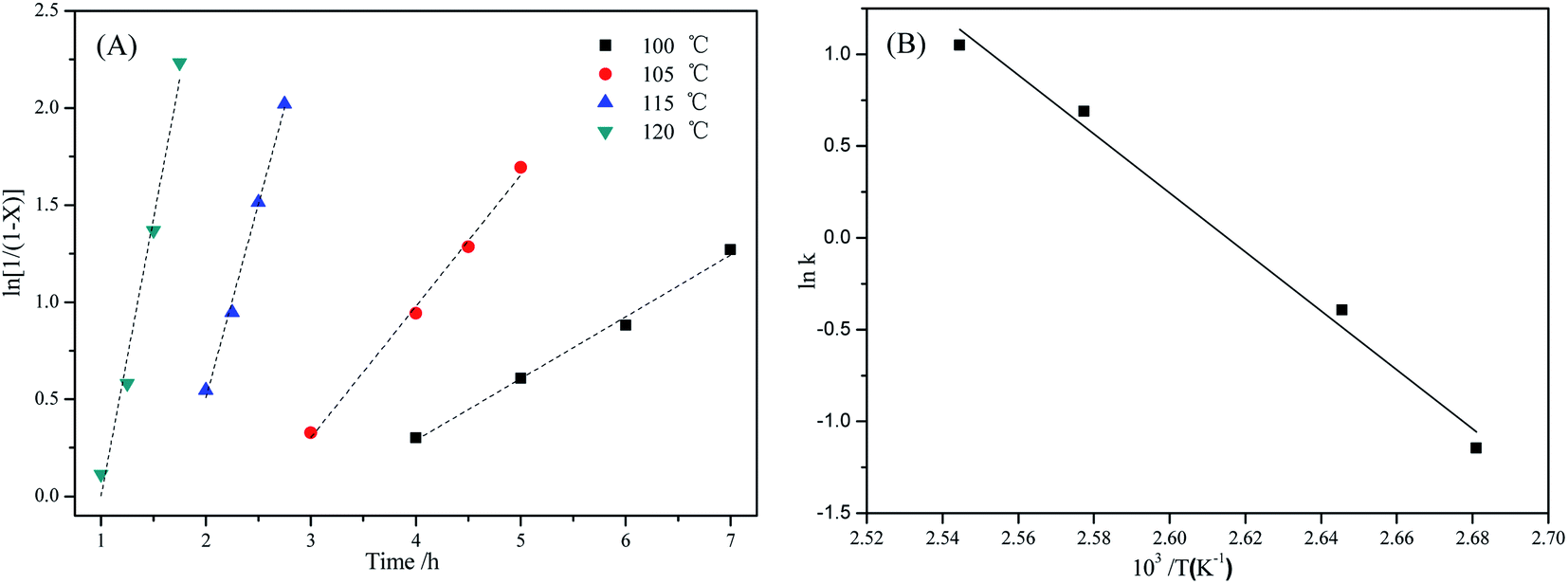
Under the conditions of PC (4.0 g), m(DES):m(PC) = 0.1:1, and n(CH3OH):n(PC) = 5:1, the effects of reaction temperature on the rate of PC methanolysis were shown in Fig. 8(A), and the corresponding linear regression results were shown in Table 4. It can be seen from Table 4 that all of the linear correlative coefficients were almost 0.98, indicating that ln1/(1 − X) was proportional to time (t) at different reaction temperatures. In other words, the rate of PC methanolysis was proportional to PC concentration. So it can prove that the methanolysis of PC is also a pseudo-first order reaction. This reaction rate constants can be calculated from the slope values of straight line in Fig. 8(A), and they were 2.859 h−1, 1.994 h−1, 0.676 h−1 and 0.318 h−1, respectively, which represented the rate constants of PC methanolysis reaction at 120 °C, 115 °C, 105 °C and 100 °C.
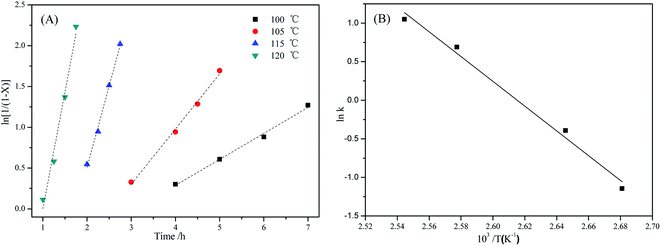 |
| | Fig. 8 (A) Effect of reaction temperature on methanolysis rate of PC catalyzed by [EmimOH]Cl-2Urea; (B) Arrhenius plot of rate constant of PC methanolysis (reaction conditions: PC (4.0 g), m(DES):m(PC) = 0.1:1, n(CH3OH):n(PC) = 5:1). | |
Table 4 Linear regression results of the data in Fig. 8A
| Reaction temperature/°C |
Regressive equation |
Rate constant |
Linear correlation coefficient |
| 120 |
y = −2.857 + 2.859x |
2.859 |
0.976 |
| 115 |
y = −3.478 + 1.994x |
1.994 |
0.993 |
| 105 |
y = −1.725 + 0.676x |
0.676 |
0.993 |
| 100 |
y = −0.984 + 0.318x |
0.318 |
0.992 |
Moreover, the rate constant increases rapidly with the reaction temperature, showing that the PC methanolysis is significantly influenced by reaction temperature, which is consistent with the previous experimental results (Table 2, entries 1–4).
Using the above rate constant, the reaction activation energy (Ea) can be calculated by the Arrhenius eqn (7).
| |
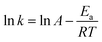 | (7) |
where
A is pre-exponential factor,
R is molar gas constant (8.314 J k
−1 mol
−1) and
T is the Kelvin temperature. The Arrhenius plot of the rate constant was shown in
Fig. 8(B), and
Ea of PC methanolysis calculated from the slope of the Arrhenius plot is 133.59 kJ mol
−1, which is lower than that of methanolysis of PC catalyzed by ChCl-2Urea
43 (141.47 kJ mol
−1).
4. Conclusions
Under metal- and solvent-free conditions, imidazolium-based DES [EmimOH]Cl-2Urea can effectively catalyze the PC methanolysis to obtain BPA, without using any traditional acid or base catalyst. Under the optimal reaction of [EmimOH]Cl-2Urea (0.4 g), PC (4.0 g) and n(CH3OH):n(PC) = 5:1 for 2.0 h at 120 °C, the CPC and YBPA reached 100% and 99%, respectively. Moreover, [EmimOH]Cl-2Urea had good thermal stability and can be reused for 5 times without any apparent decrease in the CPC and YBPA. The kinetics results suggest that the methanolysis of PC catalyzed by [EmimOH]Cl-2Urea is a pseudo-first order reaction with Ea of 133.59 kJ mol−1. This study can provide important guidance for the design of effective DESs used to promote the degradation reaction of polymer wastes and other transesterification.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported financially by the National Natural Science Foundation of China (No. 51673106), the Science and Technology Research Project of Shandong Province (No. 2019GSF109015), the Taishan Scholars Projects of Shandong (ts201511033) and Shandong Key Laboratory of Reactions and Separation of Muti-phase Liquid (No. 2019MFRSE-B03).
References
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, Recycling of polymers: a review, ChemSusChem, 2014, 7, 1579–1593, DOI:10.1002/cssc.201300898.
- K. Ragaert, L. Delva and K. Van Geem, Mechanical and chemical recycling of solid plastic waste, Waste Manage, 2017, 69, 24–58, DOI:10.1016/j.wasman.2017.07.044.
- H. Dahlbo, V. Poliakova, V. Mylläri, O. Sahimaa and R. Anderson, Recycling potential of post-consumer plastic packaging waste in Finland, Waste Manage, 2018, 71, 52–61, DOI:10.1016/j.wasman.2017.10.033.
- T. Yoshioka, K. Sugawara, T. Mizoguchi and A. Okuwaki, Chemical recycling of polycarbonate to raw materials by thermal decomposition with calcium hydroxide/steam, Chem. Lett., 2015, 34, 282–283, DOI:10.1246/cl.2005.282.
- G. Grause, K. Sugawara, T. Mizoguchi and T. Yoshioka, Pyrolytic hydrolysis of polycarbonate in the presence of earth-alkali oxides and hydroxides, Polym. Degrad. Stab., 2009, 94, 1119–1124, DOI:10.1016/j.polymdegradstab.2009.03.014.
- M. N. Siddiqui, H. H. Redhwi, E. V. Antonakou and D. S. Achilias, Pyrolysis mechanism and thermal degradation kinetics of poly (bisphenol A carbonate)-based polymers originating in waste electric and electronic equipment, J. Anal. Appl. Pyrolysis, 2018, 132, 123–133, DOI:10.1016/j.jaap.2018.03.008.
- M. M. A. Nikje, Glycolysis of polycarbonate wastes with microwave irradiation, Polimery, 2011, 56, 381–384, DOI:10.14314/polimery.2011.381.
- D. Kim, B. K. Kim, Y. Cho, M. Han and B. S. Kim, Kinetics of polycarbonate glycolysis in ethylene glycol, Ind. Eng. Chem. Res., 2009, 48, 685–691, DOI:10.1021/ie8010947.
- G. P. Tsintzou, E. V. Antonakou and D. S. Achilias, Environmentally friendly chemical recycling of poly(bisphenol-A carbonate) through phase transfer-catalysed alkaline hydrolysis under microwave irradiation, J. Hazard. Mater., 2012, 241, 137–145, DOI:10.1016/j.jhazmat.2012.09.027.
- G. P. Tsintzou and D. S. Achilias, Chemical recycling of polycarbonate based wastes using alkaline hydrolysis under microwave irradiation, Waste Biomass Valorization, 2013, 4, 3–7, DOI:10.1007/s12649-012-9125-7.
- M. Taguchi, Y. Ishikawa, S. Kataoka, T. Naka and T. Funazukuri, CeO2 nanocatalysts for the chemical recycling of polycarbonate, Catal. Commun., 2016, 84, 93–97, DOI:10.1016/j.catcom.2016.06.009.
- C. Zimmerer, J. Nagel, G. Steiner and G. Heinrich, Nondestructive molecular characterization of polycarbonate-polyvinylamine composites after thermally induced aminolysis, Macromol. Mater. Eng., 2016, 301, 648–652, DOI:10.1002/mame.201500419.
- C. H. Wu, L. Y. Chen, R. J. Jeng and S. A. Dai, 100% atom-economy efficiency of recycling polycarbonate into versatile intermediates, ACS Sustainable Chem. Eng., 2018, 6, 8964–8975, DOI:10.1021/acssuschemeng.8b01326.
- L. C. Hu, A. Oku and E. Yamada, Alkali-catalyzed methanolysis of polycarbonate. A study on recycling of bisphenol A and dimethyl carbonate, Polymer, 1998, 39, 3841–3845 CrossRef CAS.
- A. Oku, S. Tanaka and S. Hata, Chemical conversion of poly (carbonate) to bis (hydroxyethyl) ether of bisphenol A. An approach to the chemical recycling of plastic wastes as monomers, Polymer, 2000, 41, 6749–6753, DOI:10.1016/S0032-3861(00)00014-8.
- E. Quaranta, D. Sgherza and G. Tartaro, Depolymerization of poly (bisphenolA carbonate) under mild conditions by solvent-free alcoholysis catalyzed by 1,8-diazabicyclo[5.4.0]undec-7-ene as a recyclable organocatalyst: a route to chemical recycling of waste polycarbonate, Green Chem., 2017, 19, 5422–5434, 10.1039/C7GC02063E.
- T. Do, E. R. Baral and J. G. Kim, Chemical recycling of poly (bisphenol A carbonate): 1,5,7-triazabicyclo[4.4.0]-dec-5-ene catalyzed alcoholysis for highly efficient bisphenol A and organic carbonate recovery, Polymer, 2018, 143, 106–114, DOI:10.1016/j.polymer.2018.04.015.
- F. Iannone, M. Casiello, A. Monopoli, P. Cotugno, M. C. Sportelli, R. A. Picca and A. Nacci, Ionic liquids/ZnO nanoparticles as recyclable catalyst for polycarbonate depolymerization, J. Mol. Catal. A: Chem., 2017, 426, 107–116, DOI:10.1016/j.molcata.2016.11.006.
- Y. J. Zhao, X. Zhang, X. Y. Song and F. S. Liu, Highly active and recyclable mesoporous molecular sieves CaO(SrO,BaO)/SBA-15 with base sites as heterogeneous catalysts for methanolysis of polycarbonate, Catal. Lett., 2017, 147, 2940–2949, DOI:10.1007/s10562-017-2193-3.
- Y. C. Yang, C. Y. Wang, F. S. Liu, X. X. Sun, G. H. Qin, Y. T. Liu and J. Gao, Mesoporous electronegative nanocomposites of SBA-15 with CaO-CeO2 for polycarbonate depolymerization, J. Mater. Sci., 2019, 54, 9442–9455, DOI:10.1007/s10853-019-03560-2.
- F. S. Liu, Z. Li, S. T. Yu, X. Cui and X. P. Ge, Environmentally benign methanolysis of polycarbonate to recover bisphenol A and dimethyl carbonate in ionic liquids, J. Hazard. Mater., 2010, 174, 872–875, DOI:10.1016/j.jhazmat.2009.09.007.
- F. S. Liu, L. Li, S. T. Yu, Z. G. Lv and X. P. Ge, Methanolysis of polycarbonate catalysed by ionic liquid [Bmim][Ac], J. Hazard. Mater., 2011, 189, 249–254, DOI:10.1016/j.jhazmat.2011.02.032.
- J. Guo, M. S. Liu, Y. Q. Gu, Y. C. Wang, J. Gao and F. S. Liu, Efficient Alcoholysis of polycarbonate catalyzed by recyclable lewis acidic ionic liquids, Ind. Eng. Chem. Res., 2018, 57, 10915–10921, DOI:10.1021/acs.iecr.8b02201.
- M. S. Liu, J. Guo, Y. Q. Gu, J. Gao, F. S. Liu and S. T. Yu, Pushing the limits in alcoholysis of waste polycarbonate with DBU-based ionic liquids under metal-and solvent-free conditions, ACS Sustainable Chem. Eng., 2018, 6, 13114–13121, DOI:10.1021/acssuschemeng.8b02650.
- M. S. Liu, J. Guo, Y. Q. Gu, J. Gao and F. S. Liu, Versatile imidazole-anion-derived ionic liquids with unparalleled activity for alcoholysis of polyester wastes under mild and green conditions, ACS Sustainable Chem. Eng., 2018, 6, 15127–15134, DOI:10.1021/acssuschemeng.8b03591.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Novel solvent properties of choline chloride/urea mixtures, Chem. Commun., 2003, 1, 70–71, 10.1039/B210714G.
- T. N. Gu, M. L. Zhang, T. Tan, J. Chen, Z. Li, Q. H. Zhang and H. D. Qiu, Deep eutectic solvents as novel extraction media for phenolic compounds from model oil, Chem. Commun., 2014, 50, 11749–11752, 10.1039/c4cc04661g.
- P. Xu, G. W. Zheng, M. H. Zong and W. Y. Lou, Recent progress on deep eutectic solvents in biocatalysis, Bioresour. Bioprocess., 2017, 4, 34, DOI:10.1186/s40643-017-0165-5.
- T. X. Yang, L. Q. Zhao, J. Wang and G. L. Song, Improving whole-cell biocatalysis by addition of deep eutectic solvents and natural deep eutectic solvents, ACS Sustainable Chem. Eng., 2017, 5, 5713–5722, DOI:10.1021/acssuschemeng.7b00285.
- J. Cao, M. Yang, F. L. Cao and J. H. Wang, Well-designed hydrophobic deep eutectic solvents as green and efficient media for the extraction of artemisinin from Artemisia annua leaves, ACS Sustainable Chem. Eng., 2017, 5, 3270–3278, DOI:10.1021/acssuschemeng.6b03092.
- M. H. Shafie, R. Yusof and C. Y. Gan, Deep eutectic solvents (DES) mediated extraction of pectin from Averrhoa bilimbi: optimization and characterization studies, Carbohydr. Polym., 2019, 216, 303–311, DOI:10.1016/j.carbpol.2019.04.007.
- S. Sarmad, J. P. Mikkola and X. Ji, Carbon dioxide capture with ionic liquids and deep eutectic solvents: a new generation of sorbents, ChemSusChem, 2017, 10, 324–352, DOI:10.1002/cssc.201600987.
- K. Zhang, Y. C. Hou, Y. M. Wang, K. Wang, S. Ren and W. Wu, Efficient and reversible absorption of CO2 by functional deep eutectic solvents, Energy Fuels, 2018, 32, 7727–7733, DOI:10.1021/acs.energyfuels.8b01129.
- G. Cui, M. Lv and D. Z. Yang, Efficient CO2 absorption by azolide-based deep eutectic solvents, Chem. Commun., 2019, 55, 1426–1429, 10.1039/C8CC10085C.
- T. N. Gu, M. L. Zhang, J. Chen and H. D. Qiu, A novel green approach for the chemical modification of silica particles based on deep eutectic solvents, Chem. Commun., 2015, 51, 9825–9828, 10.1039/c5cc02553b.
- E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Z. Slawin and R. E. Morris, Ionothermal materials synthesis using unstable deep-eutectic solvents as template-delivery agents, Angew. Chem., Int. Ed., 2006, 45, 4962–4966, DOI:10.1002/anie.200600290.
- D. Carriazo, M. C. Serrano, M. C. Gutiérrez, M. L. Ferrer and F. Del Monte, Deep-eutectic solvents playing multiple roles in the synthesis of polymers and related materials, Chem. Soc. Rev., 2012, 41, 4996–5014, 10.1039/c2cs15353j.
- P. Zamani and A. R. Khosropour, A combination of natural deep eutectic solvents and microflow technology: a sustainable innovation for the tandem synthesis of 3-aminohexahydrocoumarins, Green Chem., 2016, 18, 6450–6455, 10.1039/C6GC02642G.
- Q. H. Zhang, K. D. O. Vigier, S. Royer and F. Jérôme, Deep eutectic solvents: syntheses, properties and applications, Chem. Soc. Rev., 2012, 41, 7108–7146, 10.1039/c2cs35178a.
- Q. Wang, X. Q. Yao, Y. R. Geng, Q. Zhou, X. M. Lu and S. J. Zhang, Deep eutectic solvents as highly active catalysts for the fast and mild glycolysis of poly(ethylene terephthalate) (PET), Green Chem., 2015, 17, 2473–2479, 10.1039/c4gc02401j.
- L. Zhou, X. M. Lu, Z. Y. Ju, B. Liu, H. Y. Yao, J. L. Xu, Q. Z. Zhou, Y. F. Hu and S. J. Zhang, Alcoholysis of polyethylene terephthalate to produce dioctyl terephthalate using choline chloride-based deep eutectic solvents as efficient catalysts, Green Chem., 2019, 21, 897–906, 10.1039/C8GC03791D.
- B. Liu, W. Z. Fu, X. M. Lu, Q. Zhou and S. J. Zhang, Lewis acid-base synergistic catalysis for polyethylene terephthalate degradation by 1,3-dimethylurea/Zn(OAc)2 deep eutectic solvent, ACS Sustainable Chem. Eng., 2019, 7, 3292–3300, DOI:10.1021/acssuschemeng.8b05324.
- X. Y. Song, W. Y. Hu, W. W. Huang, H. Wang, S. H. Yan, S. T. Yu and F. S. Liu, Methanolysis of polycarbonate into valuable product bisphenol A using choline chloride-based deep eutectic solvents as highly active catalysts, Chem. Eng. J., 2020, 388, DOI:10.1016/j.cej.2020.124324.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09215k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shitao Yuc,
Fusheng Liuc and
Xiuyan Song
a,
Shitao Yuc,
Fusheng Liuc and
Xiuyan Song