DOI:
10.1039/D0RA08902H
(Paper)
RSC Adv., 2021,
11, 1586-1594
Synthesis, characterisation and functionalisation of BAB-type dual-responsive nanocarriers for targeted drug delivery: evolution of nanoparticles based on 2-vinylpyridine and diethyl vinylphosphonate†
Received
19th October 2020
, Accepted 5th December 2020
First published on 5th January 2021
Abstract
The emerging field of nanomedicine gives new opportunities in the treatment of cancer. Aspects such as dosage, bioavailability or the application to the patient can be drastically improved. Previously our group reported an efficient route towards cross-linked nanospheres based on ABB′ block copolymers made from 2-vinylpyridine (2VP), diethyl vinylphosphonate (DEVP) and diallyl vinylphosphonate (DAlVP). Followed by thiol–ene click chemistry stable nanoparticles were formed. Herein, this promising concept was extended to copolymers with the analogous B′BABB′ architecture. In this context the new yttrium complex 5 was investigated in the rare-earth metal-mediated group transfer polymerisation (REM-GTP) and used for the generation of copolymers with different monomer feeds (2VP: 100–300 equiv.; DEVP: 200–300 equiv.; DAlVP: 6–20 equiv.) to explore the influence of the copolymer compositon on the nanoparticle properties. After successful cross-linking with various cross-linking agents, all nanoparticles were characterised via DLS and TEM. These size measurements revealed defined, almost spherical particles (dDLS = 17–52 nm; dTEM = 17–43 nm) and were mainly affected by the 2VP content and the cross-linking density. Zeta potential measurements resulted in values in the range from −6 mV to −22 mV and revealed an influence of the cross-linking agent on the surface charge. Studies on the release behaviour exhibited the fastest release at pH = 4.5. Temperature-wise best results were achieved at 42 °C. Furthermore, we aimed for the conjugation of folic acid as a model compound for a potential application in active drug targeting. The consecutive couplings of cysteamine and dithiol 6 enabled the formation of an amine-modified precursor which was reacted with a folic acid derivative. Zeta potential measurements and analysis by NMR spectroscopy corroborated a successful conjugation while DLS and TEM (dDLS = 44 nm; dTEM = 38 nm) indicated defined nanoparticles.
Introduction
In the present-day cancer can be considered one of the biggest challenges to our modern society. Around 18 million new cancer cases and 9.6 million cancer-related deaths have been estimated for 2018 alone.1 Common therapies can be divided into surgical removal, internal and external radiation and chemotherapy.2 However, radiation and chemotherapy can be accompanied by severe side effects damaging normal tissue and organs as well as causing systemic toxicity owing to their inability to distinguish between healthy and cancer cells. Furthermore, the efficacy of the therapy can be highly individual despite a strictly identical treatment.2–4
Consequently, the treatment remains a complex task and demands improved, patient-friendly approaches since, for example, a study of Schiller et al. in 2002 implied that a therapeutic plateau was reached regarding the use of chemotherapeutics.5,6 One promising development that aims to overcome this set of problems are nanosized drug-delivery vehicles. Currently, anticancer drugs are frequently limited owing to their high hydrophobicity and thus low solubility in aqueous media. Therefore, high tissue concentrations are required and can lead to systemic toxicity. Moreover, low molecular-weight agents are often eliminated by the liver or kidneys.2,7 Drug-delivery vehicles allow the encapsulation of such hydrophobic substances, increase their bioavailability substantially and benefit from the enhanced permeability and retention (EPR) effect which results in an improved accumulation at the tumour site. In addition, engineered nanoparticles are able to facilitate a controlled release of the drug.2,8–13 One widely investigated approach focuses on polymeric micelles. These are usually composed of a hydrophilic shell such as poly(ethylene glycol) (PEG) or poly(N-vinyl pyrrolidone) (PVP) and a hydrophobic core (e.g. poly(propylene oxide) (PPO), poly(D,L-lactic acid) (PDLLA), poly(ε-caprolactone) (PεCL), poly(L-aspartate)) (PASA), which enable a loading of the pharmacologically active agent.7,14 Exemplarily, the application of such a micellar carrier improved the concentration of paclitaxel significantly.15
From this broad basis, delivery vehicles made from smart materials emerge as next generation nanocarriers. Since cancerous tissue exhibits a slightly acidic environment (pH = 6.5–6.8) compared to healthy tissue (pH = 7.4) as well as elevated temperatures (hyperthermia), stimuli-responsive drug delivery vehicles can take advantage from the pathological conditions of the diseased tissue and use these triggers to enhance the drug release.2,7 In this context formulations with poly(2-vinylpyridine) (P2VP) demonstrated a pH-stimulated degradation of the micellar system while P2VP-comprising polymerosomes showed the release of the encapsulated cargo upon protonation of the pyridine motifs.16,17 Among the temperature-sensitive materials poly(N-isopropylacrylamide) (PNIPAAm) is commonly used and finds broad application in many biomedical fields. Consequently, PNIPAAm-based carriers were also investigated for their use as the hyperthermia-directed drug delivery vehicles.18,19 However, PNIPAAm features a lower critical solution temperature (LCST) of 32 °C demanding copolymerisation to reach physiological range and it can show functional loss in biological fluids.20,21 A rather new material class are polyvinylphosphonates, which are soluble in aqueous media, feature a tuneable LCST and exhibit high biocompatibility.22,23 Our group was able to establish an efficient and precise route towards well-defined, high molecular-weight polyvinylphosphonates via rare earth metal-mediated group transfer polymerisation.24–27 In 2016 this work concluded in the synthesis of BAB block copolymers consisting of P2VP and poly(diethyl vinylphosphonate) (PDEVP). These copolymers formed monodisperse and spherical micelles in aqueous solution and showed a pH as well as a temperature-triggered release of doxorubicin (DOX). Moreover in vitro studies revealed the internalisation of the DOX-loaded carriers into HeLa cells.28 However, the self-assembly of micelles remains a concentration-dependent process and is always affected by the equilibrium with free, non-associated polymer chains. As a result, micelles are subjected to high dilution in the human body upon injection which shifts the equilibrium-state towards the free polymer resulting in the dissociation of the micellar structures.29,30 This effect intensifies when the unimers bind to other constituents, e.g. proteins or membranes.30
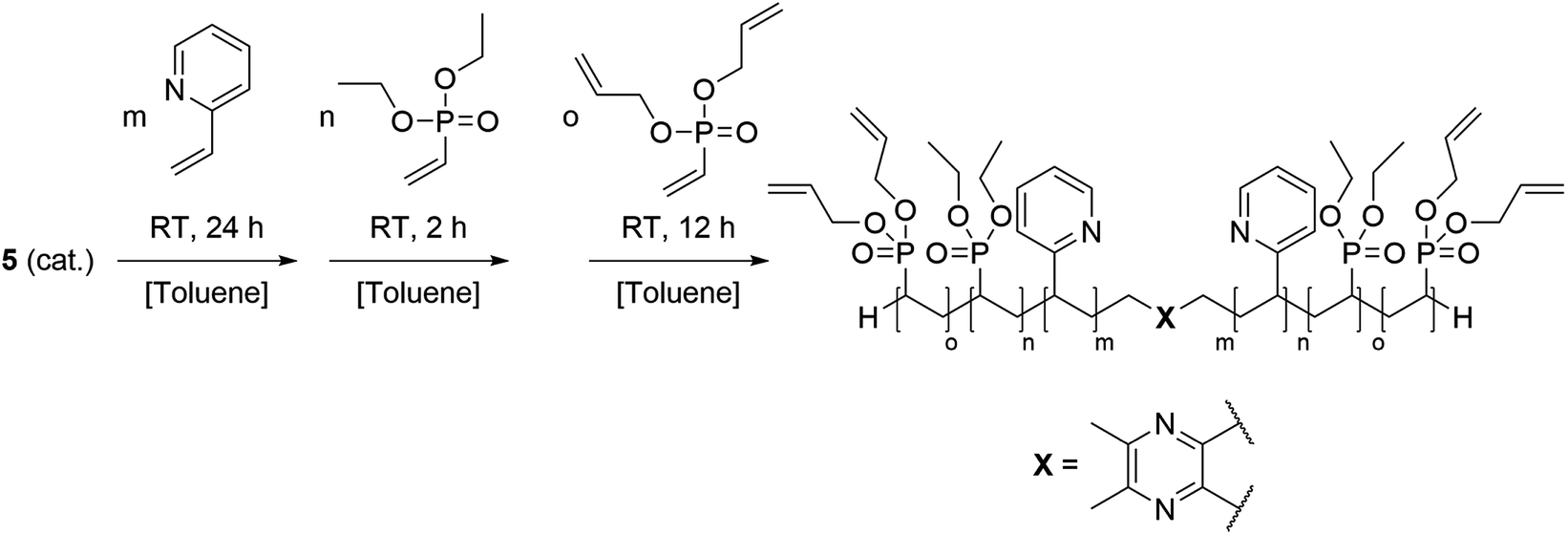
To avoid this issue, the basic concept of these nanocarriers was optimised by the introduction of diallyl vinylphosphonate (DAlVP) as a third block resulting in copolymers with an ABB′ block structure. The allyl groups facilitated the cross-linking of the copolymers via thiol–ene click chemistry and yielded stable, monodisperse and spherical particles. Most importantly the stimuli-triggered release was retained after cross-linking. In addition in vitro experiments gave evidence for the internalisation of the carriers into the cells and a statistic insight on the uptake level.31 In this context we adopted the cross-linking procedure for BAB-type block copolymers and investigated the effects of the changed polymer architecture on the particle formation and the release behaviour. Moreover, folic acid was conjugated to the particles as a targeting ligand.
Results and discussion
Complex synthesis
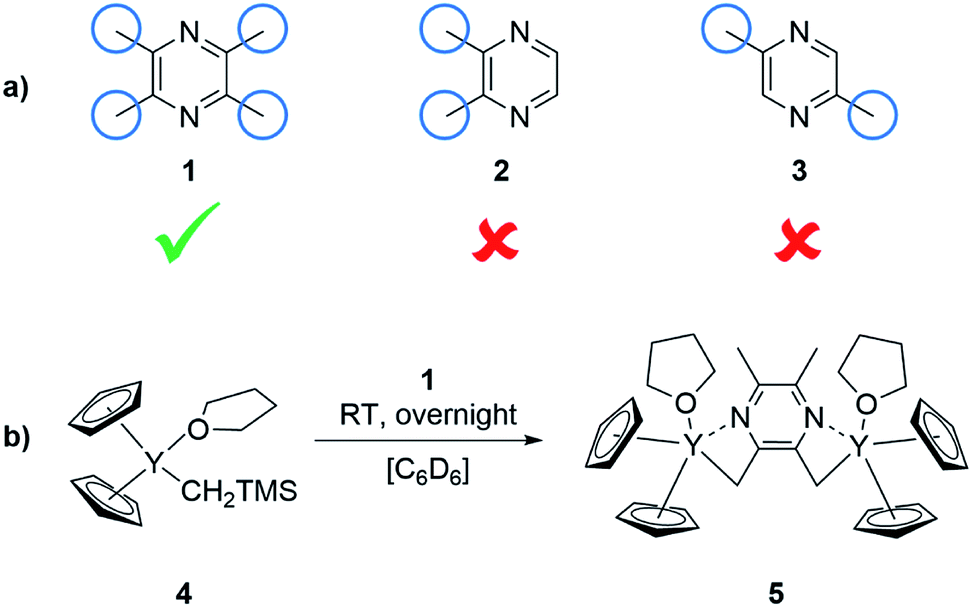
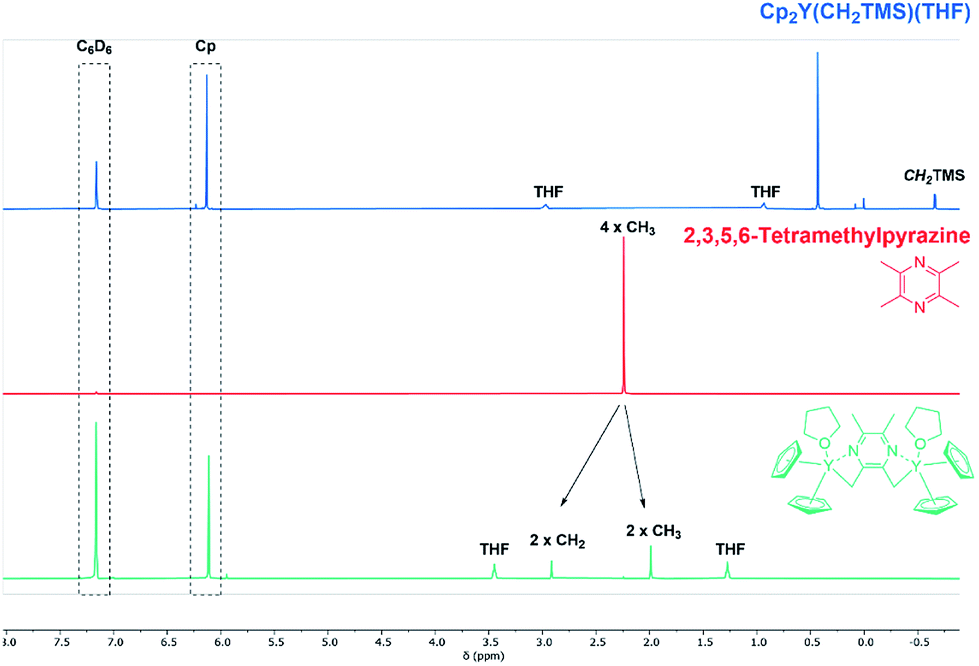
In order to change the copolymer architecture from the ABB′ structure (A: 2VP, B: DEVP, B′: DAlVP) to a B′BABB′ block structure, a binuclear catalyst is required. Such a structure can be achieved via performing C–H bond activations with bifunctional initiators such as 2,3,5,6-tetramethylpyrazine (TMPy) (1).32 Besides TMPy, 2,3-dimethylpyrazine (2) and 2,5-dimethylpyrazine (3) were also considered as possible initiators (Scheme 1a). The two opposing methyl groups of 1 located next to the heteroaromatic nitrogen presumably allow the C–H bond activation with Cp2Y(CH2TMS)(THF) (4) and resulted in a complex comprising two catalytically active sites. Surprisingly, the use of pyrazines 2 and 3 did not result in a successful activation and led only to the formation of degradation products. Fortunately, mixing of the Cp2Y complex 4 with TMPy immediately resulted in an intense orange colouration of the solution that indicated the σ-bond metathesis (Scheme 1b). The respective 1H-NMR spectrum of the isolated complex strongly suggests the presence of only one species (Fig. 1). Most importantly, the signals at 1.99 and 2.91 ppm can be attributed to the CH2Y and CH3 groups and exhibit a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio. Additionally, one THF molecule must be coordinated to each yttrium centre. These observations are supported by 13C-NMR spectroscopy as well as elemental analysis (EA). Moreover, NOESY NMR supports the formation of a single species with both methyl groups being spatially adjacent to each other (Fig. S3†). The kinetic analysis of the C–H bond activation via NMR revealed the full conversion after 18 hours (Fig. S4†).
:6 ratio. Additionally, one THF molecule must be coordinated to each yttrium centre. These observations are supported by 13C-NMR spectroscopy as well as elemental analysis (EA). Moreover, NOESY NMR supports the formation of a single species with both methyl groups being spatially adjacent to each other (Fig. S3†). The kinetic analysis of the C–H bond activation via NMR revealed the full conversion after 18 hours (Fig. S4†).
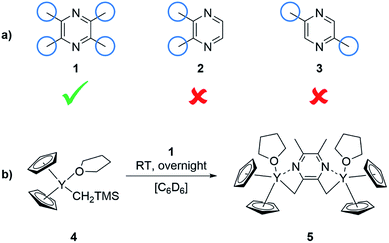 |
| | Scheme 1 (a) Screening of bifunctional pyrazines for the synthesis of a binuclear complex for REM-GTP. (b) Synthesis of the complex 5 with TMPy starting from Cp2Y(CH2TMS)(THF) (4). | |
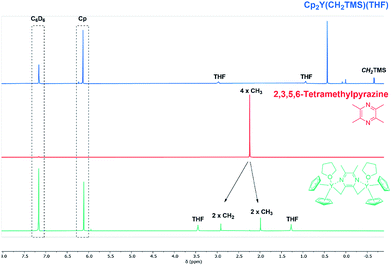 |
| | Fig. 1 Overview of C–H bond activation showing complex 4 (blue), TMPy (1) (red) and species 5 gained after completed C–H bond activation (green). 1H-NMR spectra were recorded in benzene-d6. | |
End-group analysis
Since the lanthanide-based complexes activated with sym-collidine showed the incorporation of the initiator in the polymer chain, evidence was needed to prove the covalent integration of TMPy into the polymer chain.25 By default, the characterisation of the end-group can be performed by electron spray ionisation mass spectrometry (ESI-MS) via analysis of the respective oligomer patterns. Three oligomer patterns were identified in the corresponding ESI-MS spectrum of the oligomers which were produced with complex 5 (Fig. S5†). The m/z values assigned to the first pattern show oligomer peaks ionised with H+ (452 and 557 u) with a difference equal to the molecular mass of 2VP (m2VP = 105 u). The second pattern with z = 2 was ionised by two protons and shows the respective oligomer signals (331, 384, 436, 489, and 541 u) with a difference of 52.5 u which is equivalent to the half of m2VP. In addition, a third series of oligomer signals (291, 326, 361, and 396 u) can be attributed to z = 3. Thus, the mass difference was determined to be 35 u. For completeness, the ESI-MS spectrum generated with Cp2Y(CH2(C5H2Me2N)) is illustrated in Fig. S6 in the ESI.†
Kinetic investigation of the TMPy-activated catalyst
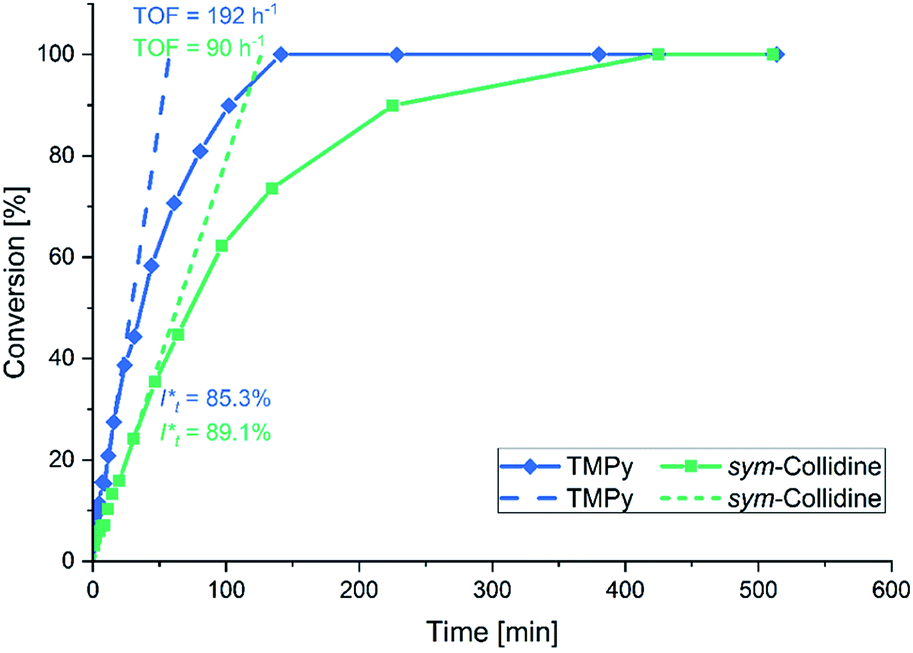
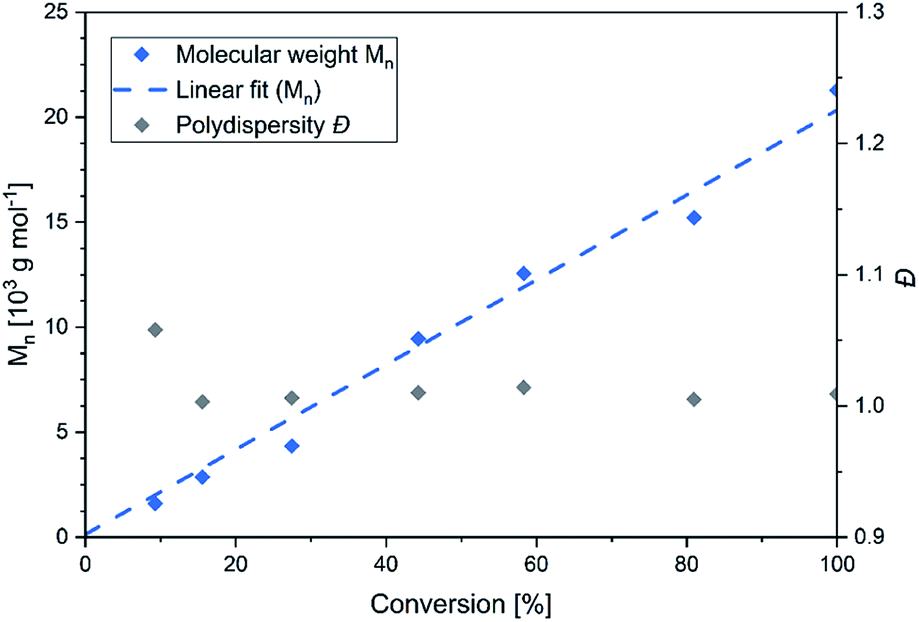
In this context sym-collidine or TMPy were already employed as initiators in the polymerisation of 2VP with bisphenolate-based yttrium complexes by our group and alkyl yttrium complexes from Mashima et al.33,34 As the C–H bond activated Cp2Y complexes have not been evaluated in kinetic experiments with this monomer, the polymerisation of 2VP was investigated with the new complex 5. Fig. 2 shows the conversion-time plot of complex 5 in blue. Hereby a turnover frequency (TOF) of 192 h−1 was determined while the corresponding initiator efficiencies (IE) were found to be 85% at the beginning and 88% at the end of the polymerisation. The living fashion of the polymerisation was able to be observed which is represented by the linear growth of the molecular weight. The corresponding polydispersites Đ remained low with values Đ < 1.10 (Fig. 3). For a better classification, the kinetic measurements were also performed with the sym-collidine activated yttrium complex. The respective conversion-time plot is illustrated by the green curve in Fig. 2. In this case a TOF of 90 h−1 was determined. Concerning the initiator efficiency, a value of 89% was calculated at the start of the polymerisation and 72% at the end. As expected, the growth was found to be linear and showed low polydispersities between Đ = 1.01 and 1.12 (Fig. S7†). The kinetic data on the turnover frequencies appear to be in good agreement with the molecular structures of 5 and the complex with sym-collidine. Since 5 has two active sites attached to one initiator group after C–H bond activation the polymer chain can grow in two directions while sym-collidine can only bear one yttrium centre. This results in a TOF twice as high for complex 5. Compared to the literature, the catalysts activated with TMPy or sym-collidine exhibit TOF values which are about three or 10 times lower than the bisphenolate system.28,35 This finding might be explained by an improvement of the electronic properties of the metal centre induced by the bisphenolate ligand.
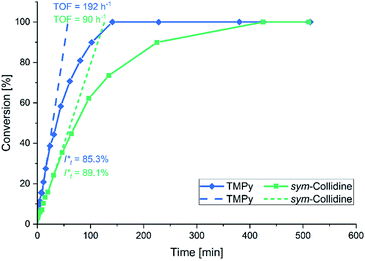 |
| | Fig. 2 Conversion-time plot for the polymerisation of 2VP with the TMPy activated complex 5 (blue) and with sym-collidine activated Cp2Y (green). | |
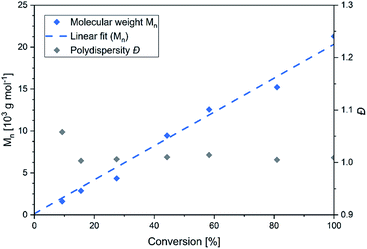 |
| | Fig. 3 Conversion-dependant plot of the molecular weights and corresponding polydispersities Đ of P2VP generated with complex 5. | |
Copolymerisation results
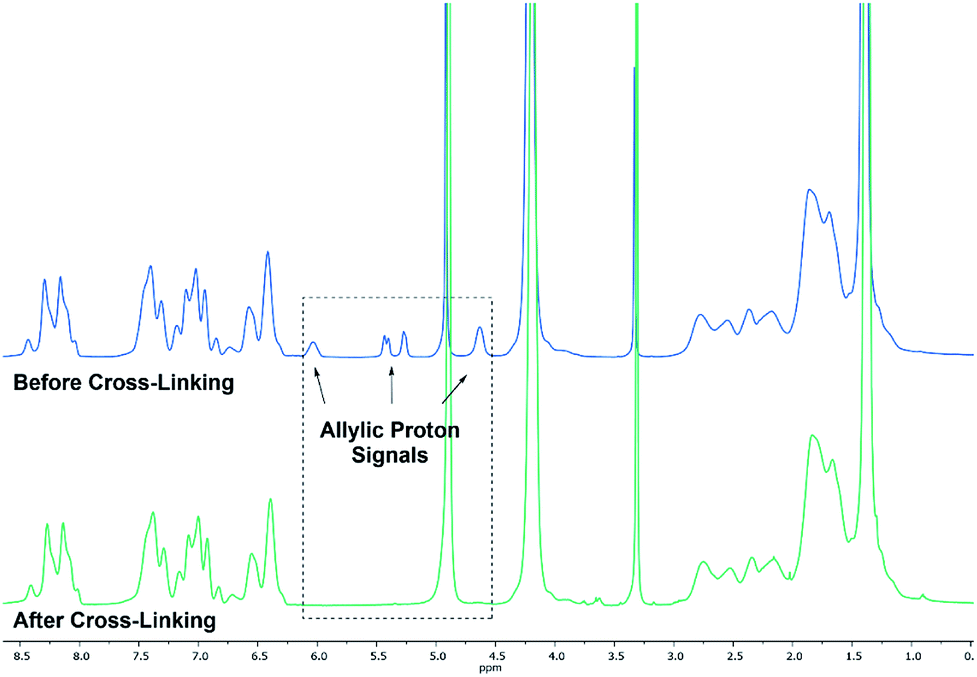
Based on the complex analysis and kinetic findings a variety of copolymers were synthesised that were later conducted to the cross-linking procedure (Scheme 2). In this work the monomer feed of 2VP and DEVP was varied between 100 equiv. and 300 equiv., while a feed ratio of 1:1 or 1:2 was adjusted. Furthermore, the DAlVP content was adjusted between six and 20 equiv. (Table 1). Each step of the copolymerisation was allowed to reach full conversion, which took approximately 24 hours in case of 2VP and two hours and 12 hours for DEVP and DAlVP, respectively (Scheme 2). Owing to the controlled nature of REM-GTP, all polymers showed narrow polydispersities ranging from Đ = 1.01 to 1.10 and had molecular weights between 48.0 and 81.2 kg mol−1, which exceeds the threshold for renal clearance of 40 kg mol−1. The initiator efficiencies were calculated after determination of the P2VP block length and ranged around 90%. In addition, 1H-NMR spectroscopy allowed the calculation of the experimental block ratios that match the theoretical block ratios very well. Here too, the signals of the allylic side groups were attributed via 1H-NMR spectra between 4.50 and 6.00 ppm and are in good agreement with the previously published ABB′ based copolymers (Fig. 4, blue spectrum).
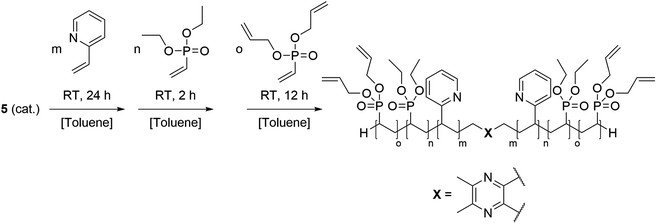 |
| | Scheme 2 Synthesis of the BAB block copolymers from 2VP, DEVP, and DAlVP via REM-GTP with catalyst 5. | |
Table 1 Feed composition, monomer conversions, molecular weights Mn of block (A) and polydispersities Đ for (A) and (BAB), initiator efficiencies IE of block (A) and composition of the copolymer
| |
Feed A/B/B′a/equiv. |
X (A)b/% |
Mn (A)c/104 g mol−1 |
Đ (A)c/- |
IE (A)d/% |
X (B)b/% |
Composition A/B [2VP/DEVP]b |
Đ (BAB)c/— |
Mn (B′BABB′)b/104 g mol−1 |
| By weighing the monomer, [M]/[cat.] = equiv. A: 2VP, B: DEVP, B′: DAlVP. Conversions X were calculated from the corresponding 1H-NMR or 31P-NMR spectra. Determined via GPC analysis in DMF. Initiator efficiency IE was determined for block (A), IE = Mn,theo/Mn,exp. |
| BAB1 |
100/200/10 |
>99 |
1.21 |
1.05 |
94 |
>99 |
1.0/1.9 |
1.08 |
4.80 |
| BAB2 |
200/200/10 |
>99 |
2.24 |
1.08 |
95 |
>99 |
1.0/1.1 |
1.10 |
5.90 |
| BAB3 |
300/300/10 |
>99 |
2.97 |
1.04 |
>99 |
>99 |
1.0/1.1 |
1.07 |
8.12 |
| BAB4 |
200/200/6 |
>99 |
2.76 |
1.06 |
80 |
>99 |
1.0/1.0 |
1.01 |
6.91 |
| BAB5 |
200/200/20 |
>99 |
2.10 |
1.03 |
>99 |
>99 |
1.0/1.0 |
1.01 |
5.65 |
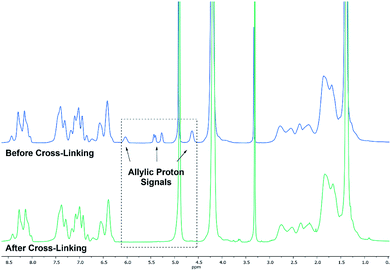 |
| | Fig. 4 Comparison of 1H-NMR spectra before (BAB2) (blue) and after cross-linking (NP2) with 3,6-dioxa-1,8-octanedithiol (6) (green). Spectra were recorded in MeOD-d4. | |
Nanoparticle formation via thiol–ene click chemistry
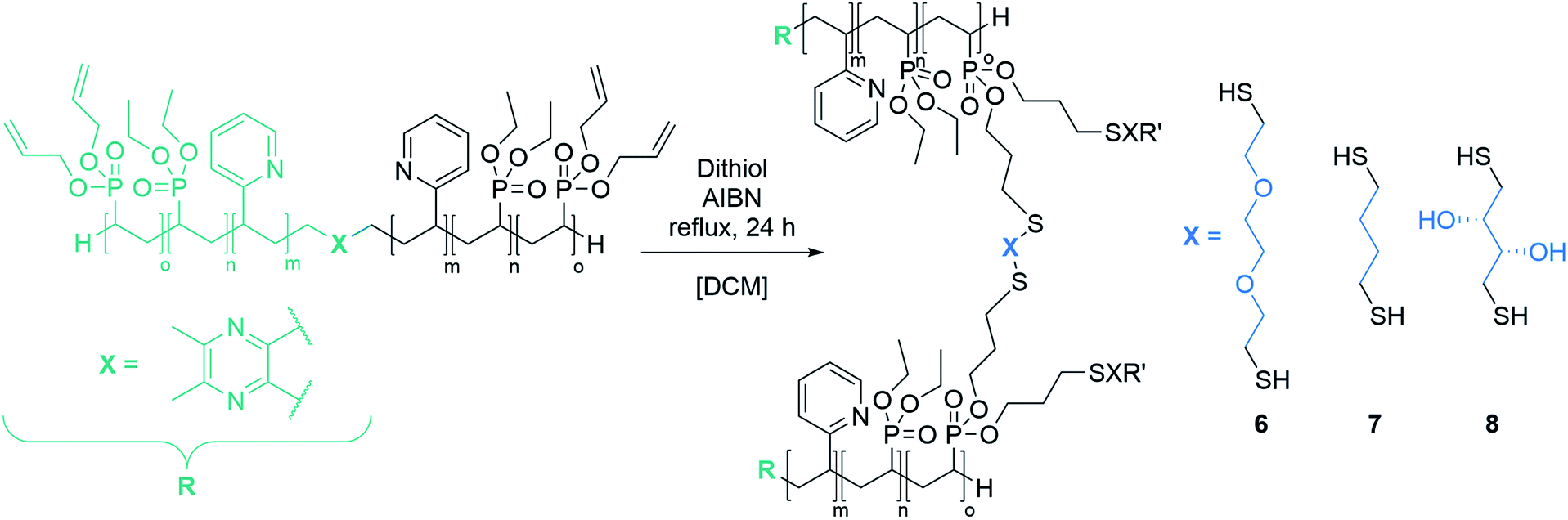
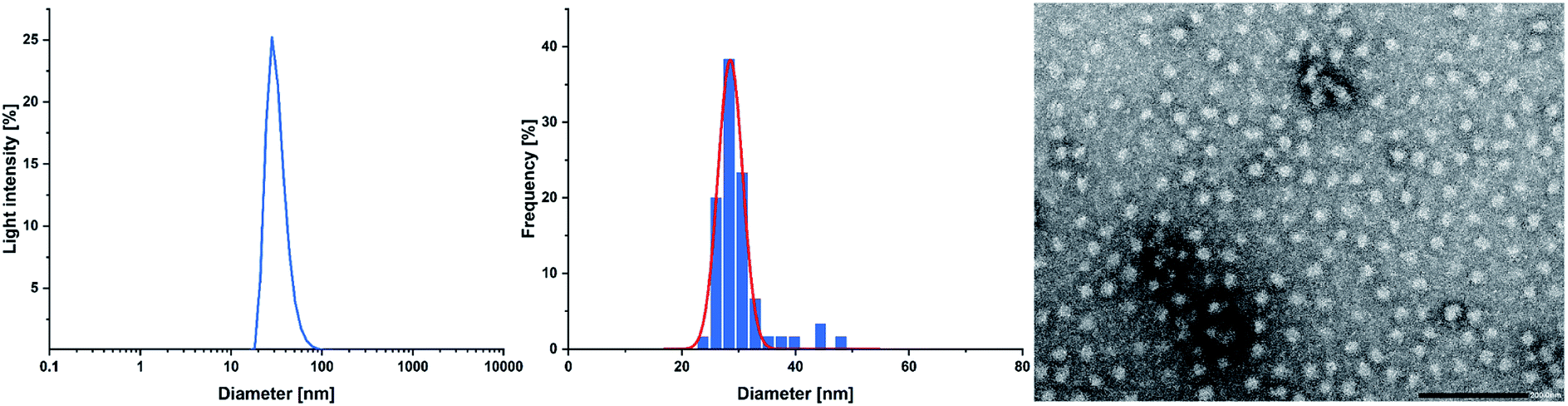
Cross-linking of the copolymers was performed via an established route using thiol–ene click chemistry with azobisisobutyronitrile (AIBN) as an initiator and 3,6-dioxa-1,8-octanedithiol (6),31 1,4-butanedithiol (7), or D,L-dithiothreitol (8) as a linking component (Scheme 3). As visualised in Fig. 4 the reaction was monitored via 1H-NMR and was considered to reach full conversion after complete disappearance of the allylic protons. Since the cross-linking of the ABB′ copolymers was performed with an excess of dithiol 6, we aimed for an optimisation by testing several dithiol amounts (3.0 equiv., 1.5 equiv., 1.0 equiv.) corresponding to the allyl group content of copolymer BAB2. As expected, an excess of three equivalents reached full conversion after 24 hours. But already a slight excess of the dithiol (1.5 equiv.) suffices to detect a complete reaction after the 24 hours. However, an amount of one equivalent appeared to be insufficient because traces of the allylic protons were still visible. After optimisation of the dithiol amount the successfully cross-linked samples were analysed via transmission electron microscopy (TEM) and dynamic light scattering (DLS) to verify the formation of the nanoparticles. Table 2 gives an overview of the experimental data of the generated nanoparticles. With the exception of NP1 and NP5, all cross-linked particles exhibit diameters larger than 30 nm with particle sizes between 32.6 and 51.9 nm. These values match with data of earlier reports that recommend a size range of 30–100 nm for an optimised targeting of the cancerous tissue because nanocarriers of this size benefit from the EPR effect and they are too large for renal clearance.36,37 Concerning the size distribution, PDIs ≤ 0.22 were found which slightly deviates from an optimal PDI below 0.10. Nevertheless, imaging via TEM indicates highly uniform and monodisperse, spherical particles. Furthermore, the size-related values gained with both methodologies are in good accordance with each other (Table 2). The DLS and TEM data of NP2 are illustrated in Fig. 5. The remaining data are summarized in the ESI (Fig. S26, S29, S31, S33, S35, S38).† As shown by NP1–3, the particle size is mainly influenced by the P2VP block while a change of the DEVP block only has minor influence on the diameter. Moreover, a decrease of the DAlVP amount resulted in a larger diameter for NP4 compared to NP2, while NP5 exhibited a much smaller diameter. Therefore, it can be concluded that the spatial extent of the particles can be tuned by the cross-linking density. The type of cross-linker has no significant influence on the particle size. Additional zeta potential measurements attribute a negative surface charge for the particles between −22 and −6.3 mV. A negative charge was also observed for the ABB′-type nanoparticles.31 However, NP6 and NP7 tend to have potentials closer to zero than the ones with 6 as cross-linker. Generally speaking, a positive surface charge is preferable because of the anionic character of the cellular membrane. However, this is contradicted by an increasing cytotoxicity and a higher affinity towards vascular endothelial cells and other anionic species which leads to aggregation and a corruption of the potential therapeutic effects.38–40 However, in 2008 Huang et al. demonstrated a slower clearance of neutral carriers compared to their anionic pendants, which indicated a surface charge near zero would be beneficial for drug delivery.31,41 Additionally, measurements in CHCl3 were performed to test the stability of the particles because both blocks are soluble in CHCl3 (Fig. S41†). In case of NP1–NP6 the particle diameters exceeded those values that were determined in water. Therefore, stable particles can be assumed since the free polymer chains should form random coils in solution which must be much smaller than the respective nanoparticles. However, the cross-linking of BAB3 to NP7 appeared not to be successful because a diameter of only 12.6 nm was determined compared to 46.9 nm in water.
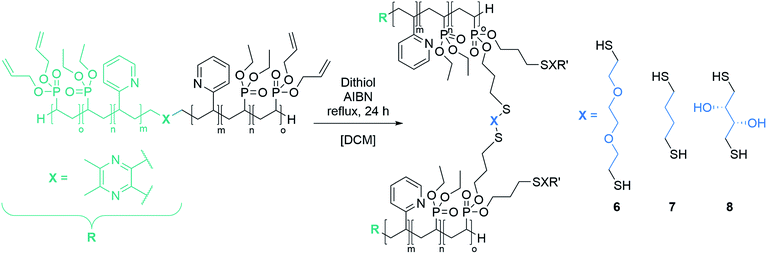 |
| | Scheme 3 Strategy for the cross-linking of the copolymers via thiol–ene click chemistry with dithiol 6, 7 and 8. | |
Table 2 Used copolymers and dithiols, diameters and polydispersities determined via DLS and TEM and zeta potential ζ of the nanoparticles
| |
Polymer |
Dithiol |
d (DLS)/nm |
PDI (DLS) |
ζ/mV |
d (TEM)/nm |
PDI (TEM) |
| NP1 |
BAB1 |
6 |
21.1 ± 0.9 |
0.17 |
−11 ± 0.3 |
17.1 ± 1.6 |
0.09 |
| NP2 |
BAB2 |
6 |
32.6 ± 0.4 |
0.19 |
−13 ± 0.2 |
31.0 ± 4.7 |
0.15 |
| NP3 |
BAB3 |
6 |
51.9 ± 3.9 |
0.16 |
−19 ± 1.0 |
42.7 ± 4.9 |
0.11 |
| NP4 |
BAB4 |
6 |
41.7 ± 5.6 |
0.17 |
−16 ± 0.6 |
34.2 ± 2.3 |
0.08 |
| NP5 |
BAB5 |
6 |
16.8 ± 0.6 |
0.22 |
−22 ± 0.5 |
26.1 ± 2.4 |
0.09 |
| NP6 |
BAB3 |
7 |
47.4 ± 3.8 |
0.16 |
−9.8 ± 0.4 |
41.1 ± 5.6 |
0.14 |
| NP7 |
BAB3 |
8 |
46.9 ± 1.8 |
0.13 |
−6.3 ± 0.2 |
41.0 ± 3.9 |
0.09 |
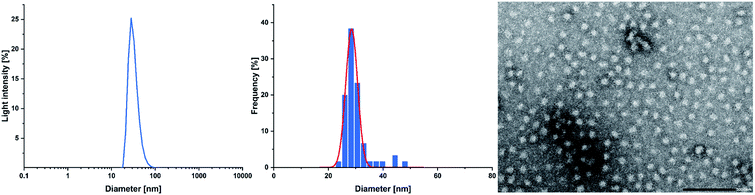 |
| | Fig. 5 Size distribution of NP2 determined via DLS measurements at a concentration of 2.5 mg mL−1 in water (left); histogram plot with a Gaussian regression fit (middle); and a TEM image of NP2 (right) with a scale bar of 200 nm. | |
Release behaviour of the nanoparticles
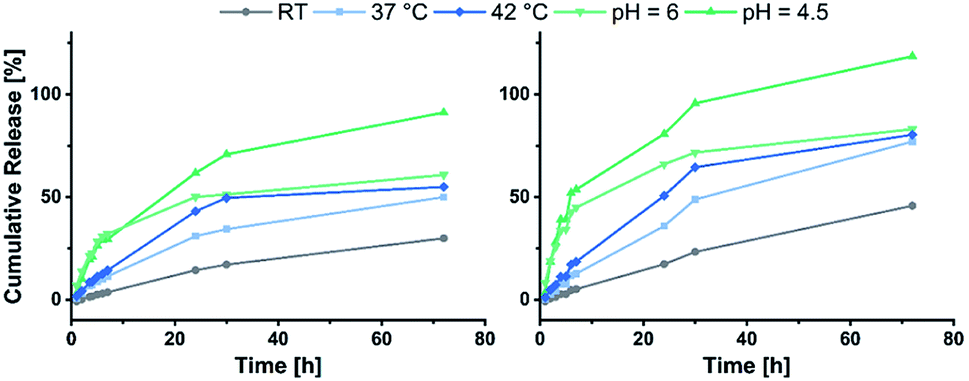
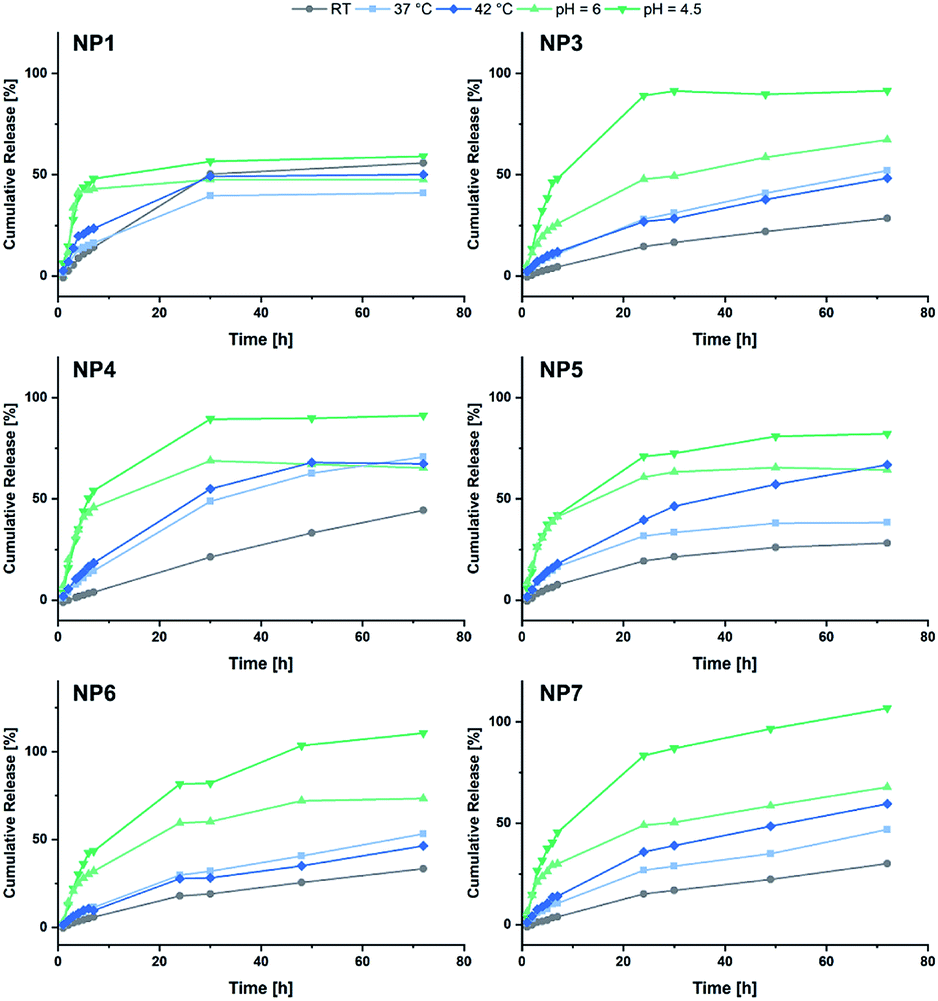
Eventually the loading and release behaviour of the nanoparticles was studied. After loading with fluorescein, the samples were analysed in respect of the cumulative release by dialysis. For this purpose, varying temperatures and pH-values were adjusted. For that purpose, room temperature and a neutral pH were adjusted as reference. A temperature of 37 °C was set to simulate the body temperature, while 42 °C was used to mimic hyperthermic conditions caused by cancer cells. With regard to the pH, acidic environments were adjusted to simulate the conditions in the endosomal environment (pH = 6.0) or the interior of the lysosome (pH = 4.5).31,42 In Fig. 6 the cumulative fluorescein release of BAB2 and NP2 is visualised. Compared to the micelles formed from BAB2 the release from the cross-linked nanoparticle NP2 was improved by approximately 20% after 30 hours. In both cases elevated temperatures as well as acidic conditions stimulated the release compared to the standard scenario at room temperature and a neutral environment. Regarding the temperature, a coil-to-globule-transition triggers the collapse of the DEVP-based shell. Unfortunately, there is no significant differentiation between 37 °C and 42 °C. Overall best results were obtained at pH = 4.5. But whereas BAB2 revealed a quantitative release after 70 hours, this was already achieved after 30 hours with NP2. The release data of nanoparticles NP1 and NP3–NP7 are illustrated in Fig. 7. Interestingly, NP1 shows inferior characteristics with a cumulative release limited to approximately 50% in all scenarios after 30 hours. This might be attributed to a relatively rigid and inflexible structure of BAB1 with its short P2VP core, which prevents the release of the cargo. NP3 exhibited best results at acidic conditions and showed a quantitative release of fluorescein after 24 hours at pH = 4.5 and 60% after 70 hours at pH = 6.0. Hence, NP3 must be most sensitive to conformational changes upon protonation of the pyridine units which promotes the release of the cargo as it has the largest P2VP block. NP4 and NP5 also showed best results at a pH of 4.5 with a cumulative release between 80 and 90%, but both nanoparticles also seem to be quite responsive to changes in temperature and outperform NP3 in this category. NP4 reaches a release of approximately 70% but shows no differentiation between 37 and 42 °C on the one hand. On the other hand, NP5 features a distinctive gap of the releases at 37 °C (30%) and 42 °C (70%). Therefore, a tightly cross-linked particle shell might be beneficial in terms of a temperature-triggered release. The release behaviour of NP6 and NP7 corresponds with the results of NP3 regarding the rate and extent of the release. However, the non-polar cross-linker 1,4-butanedithiol (7) seems to decrease the temperature-induced release of fluorescein.
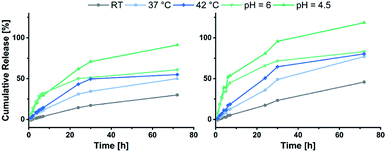 |
| | Fig. 6 Cumulative release of fluorescein from BAB2 (left) in comparison to NP2 (right). | |
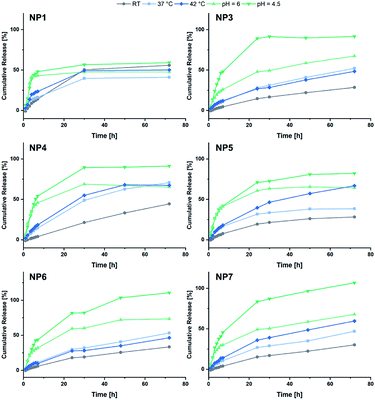 |
| | Fig. 7 Summary of the cumulative fluorescein release from NP1 and NP3–NP7. | |
Shell-functionalisation with folic acid
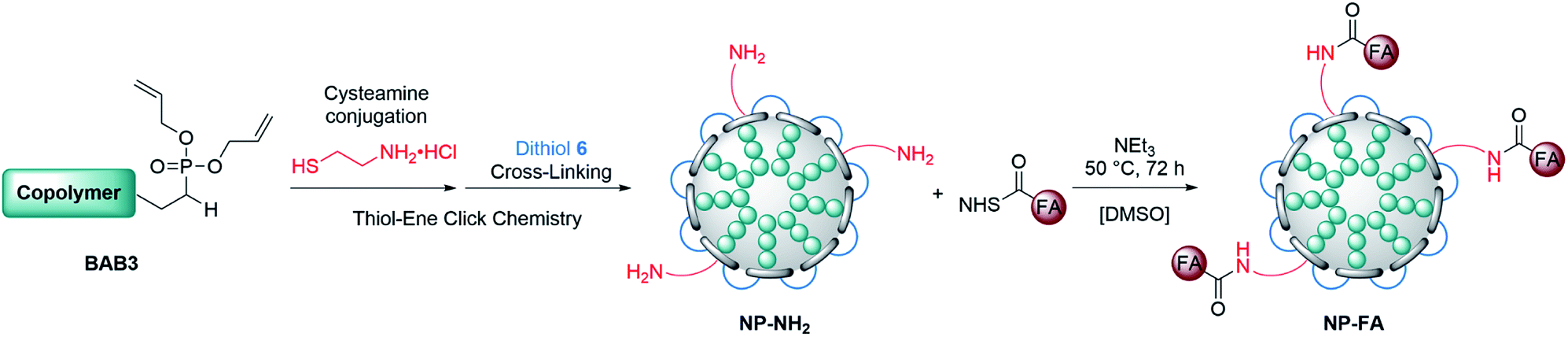
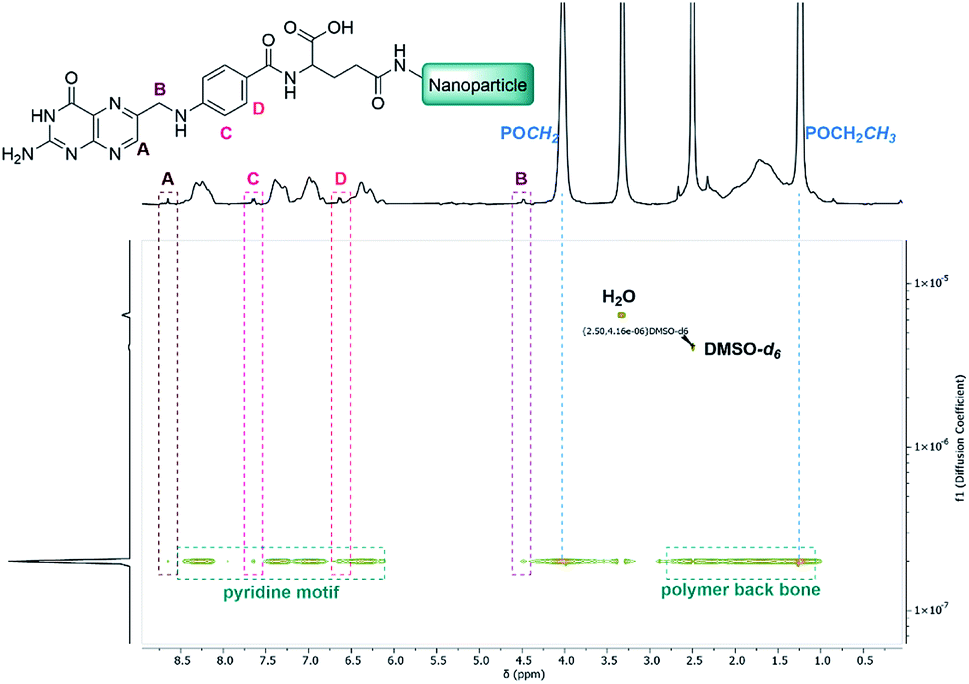
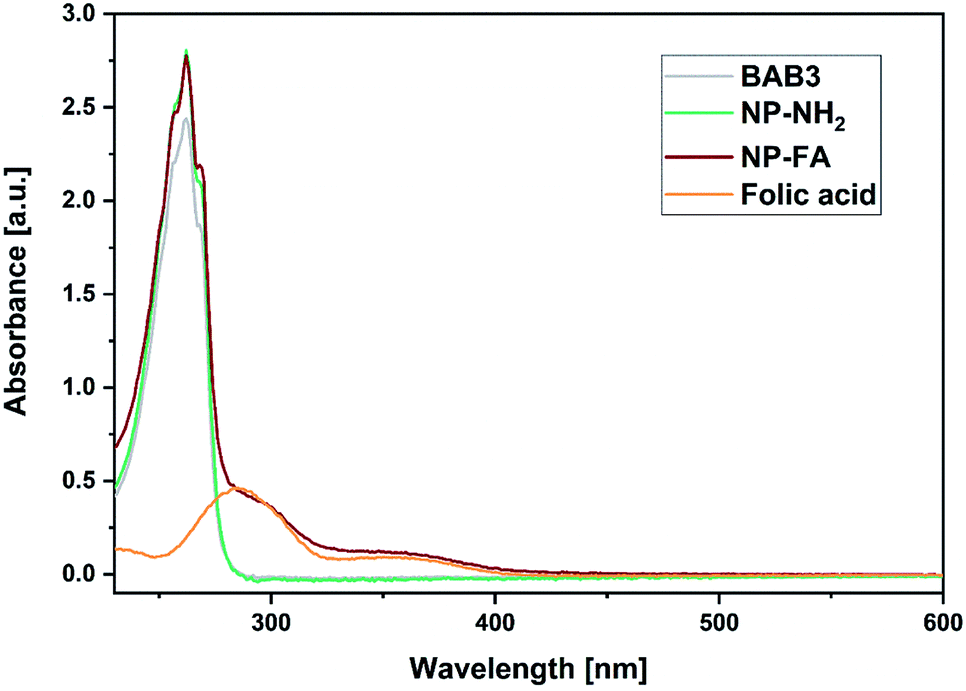
To broaden the scope of this concept the nanocarriers should be developed towards an application in active targeting. Such a type of nanoparticle requires the presence of superficial targeting ligands which can be recognised and bind to disease-specific cell features.8 Folic acid is a prominent example for such a targeting ligand and shows high affinity to its folate-receptor (Kd < 10−9) which is overexpressed on many cancer types,43 and is therefore a beneficial targeting motif for the development of novel therapeutic concepts.44–47 The conjugation of folic acid was achieved by the formation of a NH2-bearing nanoparticle NP-NH2. As depicted in Scheme 4 the allyl functions of BAB3 were functionalised with cysteamine-hydrochloride first and cross-linked with dithiol 6 via thiol–ene chemistry to yield an amino-group containing surface. This reaction sequence was monitored via 1H-NMR (Fig. S43†) and revealed a partial consumption of the allyl groups in the first step. Treatment of this precursor with the dithiol resulted in the quantitative conversion of the remaining allyl signals. With a zeta potential of −18 mV, NP-NH2 stayed in the same region as NP3 (−19 mV). Subsequently, the particles were converted with the N-hydroxysuccinimide (NHS) ester of folic acid to form NP-FA. The NHS ester was employed in large excess to ensure a reaction with the surface-bound amino groups. After purification by dialysis, the 1H-NMR exhibited characteristic signals of folic acid at 4.49 (CH2NH), 6.64, 7.65 (both p-aminobenzoic acid) and 8.65 ppm (pteridine) (Fig. 8 and S45†). The zeta potential increased to −11 mV, which indicates a modification of the surface features of the particle shell. Diffusion-ordered spectroscopy (DOSY) was also performed to verify further the conjugation of folic acid. In the corresponding spectrum the folic acid related signals share the same diffusion coefficient (D = 2.00 × 10−7 m2 s−1) with the polymer-related signals from the polymer blocks. Additional characterisation by DLS and TEM confirmed the formation of spherical, almost uniform particles (Fig. S44†). The diameters were found to be 44.4 ± 0.9 (PDI = 0.11) for DLS and 38.2 ± 3.1 (PDI = 0.08) for TEM, respectively. In comparison to the non-modified analogues NP3, NP6 or NP7, the diameters of NP-FA are slightly lower. Lastly, analysis by UV/Vis revealed the absorption band of folic acid at 360 nm, which was not visible for the copolymer BAB3 and the precursor particle NP-NH2 (Fig. 9).
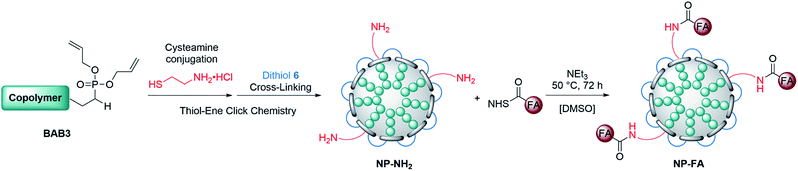 |
| | Scheme 4 Strategy for the introduction of shell-bound folic acid via partial conversion of the allyl groups with cysteamine hydrochloride. Folic acid was conjugated using an NHS-ester of folic acid after the cross-linking with dithiol 6. | |
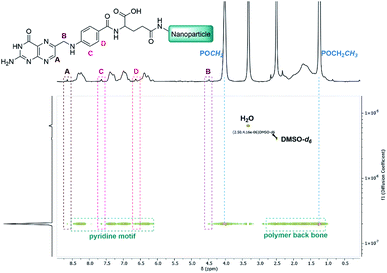 |
| | Fig. 8 DOSY-NMR spectrum of the folate-modified nanoparticle NP-FA in DMSO-d6. | |
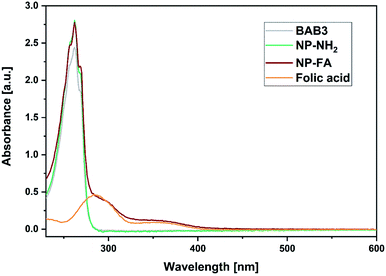 |
| | Fig. 9 UV/Vis spectra of BAB3 (1.25 mg mL−1), NP-NH2 (1.25 mg mL−1), NP-FA (1.25 mg mL−1), and free folic acid (100 μM) in methanol. | |
Conclusions
In summary, we expanded the scope of the ABB′-type (A: 2VP; B: DEVP, B′: DAlVP) nanoparticles towards a B′BABB′ architecture by using TMPy (1) as an initiator. The resulting complex 5 exhibited a TOF of 192 h−1 with 2VP and generated copolymers BAB1–BAB5 with high precision by a GTP-type propagation mechanism. The copolymers were then cross-linked to NP1–NP7 with three dithiols via thiol–ene click chemistry and formed defined particles in aqueous solution (d = 16.8–51.9 nm, PDI = 0.13–0.22), which was confirmed by transmission electron microscopy. The particle diameters were mostly determined by the copolymer composition, while the choice of the cross-linker influenced the zeta potential of the shell. Elevated temperatures and acidic conditions improved the release of fluorescein as the corresponding release studies have revealed. Throughout all experiments, the strongest response was observed at a pH = 4.5. Temperature-wise NP2, NP4 and NP5 were most promising with respect to hyperthermia treatments. Moreover, we presented a facile synthesis approach towards nanoparticles with surface-bound folic acid, which was accomplished by the incorporation of cysteamine on the particle surface. As a result, the shell-located amino groups enabled the coupling of the activated folic acid derivative. The conjugation of such a ligand is a prerequisite for the application as targeted drug delivery vehicles. In addition, this synthetic strategy can be used to form tailor-made nanoparticles with selected targeting ligands that consider the molecular characteristics of the respective disease.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Laura Fuchs and Simone Poprawa for their support with the experiments and kinetic measurements, Dr Carsten Peters for his efforts to get the best TEM images, Dr Friederike Adams for the productive discussions and her helpful input and Alina Denk and Christopher Thomas for revising the manuscript.
Notes and references
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- S. M. Bentzen, Nat. Rev. Cancer, 2006, 6, 702 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751 CrossRef CAS.
- J. H. Schiller, D. Harrington, C. P. Belani, C. Langer, A. Sandler, J. Krook, J. Zhu and D. H. Johnson, N. Engl. J. Med., 2002, 346, 92–98 CrossRef CAS.
- J. Zugazagoitia, C. Guedes, S. Ponce, I. Ferrer, S. Molina-Pinelo and L. Paz-Ares, Clin. Ther., 2016, 38, 1551–1566 CrossRef.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS.
- F. X. Gu, R. Karnik, A. Z. Wang, F. Alexis, E. Levy-Nissenbaum, S. Hong, R. S. Langer and O. C. Farokhzad, Nano Today, 2007, 2, 14–21 CrossRef.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS.
- S. Kalepu and V. Nekkanti, Acta Pharm. Sin. B, 2015, 5, 442–453 CrossRef.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- Y. Matsumura, Adv. Drug Delivery Rev., 2008, 60, 899–914 CrossRef CAS.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS.
- D. Sutton, N. Nasongkla, E. Blanco and J. Gao, Pharm. Res., 2007, 24, 1029–1046 CrossRef CAS.
- K. M. Huh, S. C. Lee, Y. W. Cho, J. Lee, J. H. Jeong and K. Park, J. Controlled Release, 2005, 101, 59–68 CrossRef CAS.
- A. Karanikolas, P. Tsolakis, G. Bokias and C. Tsitsilianis, Eur. Phys. J. E, 2008, 27, 335–343 CrossRef CAS.
- U. Borchert, U. Lipprandt, M. Bilang, A. Kimpfler, A. Rank, R. Peschka-Süss, R. Schubert, P. Lindner and S. Förster, Langmuir, 2006, 22, 5843–5847 CrossRef CAS.
- Y. Qu, J. Li, J. Ren, J. Leng, C. Lin and D. Shi, Nanoscale, 2014, 6, 12408–12413 RSC.
- H. Wei, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- L. Zha, B. Banik and F. Alexis, Soft Matter, 2011, 7, 5908–5916 RSC.
- E. Roux, M. Francis, F. M. Winnik and J.-C. Leroux, Int. J. Pharm., 2002, 242, 25–36 CrossRef CAS.
- C. Schwarzenböck, A. Schaffer, P. Pahl, P. J. Nelson, R. Huss and B. Rieger, Polym. Chem., 2018, 9, 284–290 RSC.
- N. Zhang, S. Salzinger and B. Rieger, Macromolecules, 2012, 45, 9751–9758 CrossRef CAS.
- U. B. Seemann, J. E. Dengler and B. Rieger, Angew. Chem., 2010, 122, 3567–3569 CrossRef.
- B. S. Soller, S. Salzinger, C. Jandl, A. Pöthig and B. Rieger, Organometallics, 2015, 34, 2703–2706 CrossRef CAS.
- S. Salzinger, U. B. Seemann, A. Plikhta and B. Rieger, Macromolecules, 2011, 44, 5920–5927 CrossRef CAS.
- S. Salzinger, B. S. Soller, A. Plikhta, U. B. Seemann, E. Herdtweck and B. Rieger, J. Am. Chem. Soc., 2013, 135, 13030–13040 CrossRef CAS.
- P. T. Altenbuchner, P. D. L. Werz, P. Schöppner, F. Adams, A. Kronast, C. Schwarzenböck, A. Pöthig, C. Jandl, M. Haslbeck and B. Rieger, Chem.–Eur. J., 2016, 22, 14576–14584 CrossRef CAS.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS.
- C. F. van Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- C. Schwarzenböck, P. J. Nelson, R. Huss and B. Rieger, Nanoscale, 2018, 10, 16062–16068 RSC.
- F. Adams, P. T. Altenbuchner, P. D. L. Werz and B. Rieger, RSC Adv., 2016, 6, 78750–78754 RSC.
- F. Adams, P. Pahl and B. Rieger, Chem.–Eur. J., 2018, 24, 509–518 CrossRef CAS.
- H. Kaneko, H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2011, 133, 19626–19629 CrossRef CAS.
- F. Adams, M. R. Machat, P. T. Altenbuchner, J. Ehrmaier, A. Pöthig, T. N. V. Karsili and B. Rieger, Inorg. Chem., 2017, 56, 9754–9764 CrossRef CAS.
- Y. Matsumura and K. Kataoka, Cancer Sci., 2009, 100, 572–579 CrossRef CAS.
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS.
- R. Elul, J. Physiol., 1967, 189, 351–365 CrossRef CAS.
- A. Musyanovych, J. Dausend, M. Dass, P. Walther, V. Mailänder and K. Landfester, Acta Biomater., 2011, 7, 4160–4168 CrossRef CAS.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef.
- S.-D. Li and L. Huang, Mol. Pharm., 2008, 5, 496–504 CrossRef CAS.
- Y.-B. Hu, E. B. Dammer, R.-J. Ren and G. Wang, Transl. Neurodegener., 2015, 4, 18 CrossRef.
- C. Chen, J. Ke, X. E. Zhou, W. Yi, J. S. Brunzelle, J. Li, E.-L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486–489 CrossRef CAS.
- C. M. Alexander, K. L. Hamner, M. M. Maye and J. C. Dabrowiak, Bioconjugate Chem., 2014, 25, 1261–1271 CrossRef CAS.
- B. Stella, S. Arpicco, M. T. Peracchia, D. Desmaële, J. Hoebeke, M. Renoir, J. D'Angelo, L. Cattel and P. Couvreur, J. Pharm. Sci., 2000, 89, 1452–1464 CrossRef CAS.
- X. Qiang, T. Wu, J. Fan, J. Wang, F. Song, S. Sun, J. Jiang and X. Peng, J. Mater. Chem., 2012, 22, 16078–16083 RSC.
- S. Dong, H. J. Cho, Y. W. Lee and M. Roman, Biomacromolecules, 2014, 15, 1560–1567 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, ESI analysis of the oligomers, GPC traces, release data for copolymers BAB1–3, DLS and TEM analysis of NP1, NP3–7, and NP-FA. See DOI: 10.1039/d0ra08902h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*