
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure of the amorphous titania precursor phase of N-doped photocatalysts†
I. E. Grey *a,
P. Bordetb and
N. C. Wilsona
*a,
P. Bordetb and
N. C. Wilsona
aCSIRO Mineral Resources, Private Bag 10, Clayton South, Victoria 3169, Australia. E-mail: ian.grey@csiro.au
bUniversité Grenoble Alpes, CNRS, Institut Néel, Grenoble, 38000, France
First published on 24th February 2021
Abstract
Amorphous titania samples prepared by ammonia solution neutralization of titanyl sulphate have been characterized by chemical and thermal analyses, and with reciprocal-space and real-space fitting of wide-angle synchrotron X-ray scattering data. A model that fits both the chemical and structural data comprises small segments of lepidocrocite-type layer that are offset by corner-sharing as in the monoclinic titanic acids H2TinO2n+1·mH2O. The amorphous phase composition that best fits the combined chemical and scattering data is [(NH4)3H21Ti20O52]·14H2O, where the formula within the brackets is the cluster composition and the H2O outside the brackets is physically adsorbed. The NH4+ cations are an integral part of the clusters and are bonded to layer anions at the corners of the offset layers, as occurs in the alkali metal stepped-layer titanates. The stepped-layer model is shown to give a consistent mechanism for the reaction of aqueous ammonia with solid hydrated titanyl sulphate, in which the amorphous product retains the exact size and shape of the reacting titanyl sulphate crystals.
Introduction
The demonstration by Fujishima and Honda1 of photo-assisted water-splitting using titania as a photoanode, heralded a period of intensive research on the photocatalytic properties and applications of TiO2. Technological applications in areas ranging from solar cells to self-cleaning and anti-fogging surfaces to sunscreens and photonic devices require the titania to be in a nanocrystalline form. An immense volume of research has been focused on various synthesis procedures for controlling the size, morphology and structural form of nanocrystalline titania and on extending the light-harvesting ability of TiO2 from the ultraviolet into the visible region using dopants. This burgeoning field of research has been described in detail in numerous recent reviews.2–6 The most effective synthesis procedures for controlling the size, shape and phase-type are solution-based methods, including sol gel and hydrothermal/solvothermal techniques.7,8 These synthesis routes usually proceed through the formation of an amorphous phase, which then requires higher-temperature treatment and the use of additives to form nanocrystalline phases of titania.9The important role of amorphous titania in photocatalysis applications has been highlighted in a recent review,10 which also noted the comparative lack of published information on the structure and properties of amorphous titania relative to the common nanocrystalline forms, anatase, brookite and rutile. Published structure analyses using the Fourier transform of the total powder diffraction pattern (pair distribution function, PDF) coupled with reverse Monte Carlo (RMC) simulations have variously described the amorphous phase structure as resembling that of brookite,11,12 lepidocrocite13 or anatase.14 Molecular dynamics (MD) and Monte Carlo (MC) simulations have also been used to generate amorphous titania particles with particle sizes from 1 to 10 nm and containing up to 3000 atoms.15–19 The MD and MC studies do not identify particular known structure types that occur locally within the particles, but instead report partial radial distribution functions (Ti–Ti, Ti–O and O–O) together with coordination number and bond angle distributions. These are broadly consistent with a network of short staggered chains of edge- and corner-shared polyhedra, of which TiO6 octahedra dominate (∼70%) together with minor TiO5 and TiO7 polyhedra.
The nature of the amorphous titania precursor phase is of importance in helping to understand how non-metallic dopants are incorporated into nanocrystalline titania. Nitrogen is the most effective non-metallic dopant for enhancing photocatalytic activity of TiO2, but there is still considerable uncertainty about the nature of the N complex species that contribute to visible-light absorption of the doped titania.3 In their review of N-doped TiO2 photocatalysts these authors3 have noted that the formation of the N complex species most likely depends on the synthesis procedure used. Understanding how the N species are incorporated into the amorphous precursor phase is an important step.
In published studies on syntheses of nanocrystalline TiO2 photocatalysts via an amorphous phase, the relationships between the amorphous phase precursor and the crystalline phase-type formed from it have often been hampered by a lack of chemical characterization of the precursor phase. Most studies on amorphous titania do not provide chemical or thermal analyses on the samples that are being characterized by diffraction studies. A notable exception is a recent study on the characterization of amorphous titania formed by reaction of ammonia solution with solid titanyl sulphates20 for which chemical analyses, thermogravimetric and differential thermal analysis (TGA/DTA), and infrared (IR) and Raman spectroscopy results were given. These authors20 reported the interesting result that reaction of aqueous ammonia solution with solid titanyl sulphate crystals at 0 °C results in complete extraction of sulphate ions and their replacement with hydroxyl groups, forming amorphous products that preserve the size and shape of the original crystals. The chemical analyses showed that a significant amount of nitrogen, as NH4+, is bound in the amorphous phase.
In this study we report the structural characterization of the amorphous N-doped precursor phase formed during neutralization of titanyl sulphate with ammonia solution. Synchrotron wide-angle X-ray scattering data was used, with checking of models using the Debye function to fit the powder X-ray diffraction (PXRD) data directly and using the PDF to screen different structural models. The diffraction data was supported by chemical and thermal analyses to help determine how the N was incorporated into the amorphous phase.
Experimental methods
Synthesis
A variation of the synthesis procedure described by Ihara21 was used. This synthesis method was chosen because its hydrolysis product gives a vivid yellow, N-doped photocatalyst with visible light activity when calcined at 400 °C.21 A mixture of 2.0 g of Ti metal, 30 g of (NH4)2SO4 and 45 ml of concentrated H2SO4 was refluxed until the metal had dissolved and the solution started to become cloudy. The liquor was diluted with 400 ml of water and filtered to remove traces of residual Ti and precipitated titanyl sulphate. Concentrated ammonia solution (28%) was added dropwise to the filtrate with vigorous stirring until the solution pH reached 7. The white gel-like precipitate was repeatedly (×4) centrifuged and stirred with 500 ml of water, then filtered and evaporated to dryness at 60 °C.Analyses
The dried hydrolysis product from the neutralization reaction was chemically characterized by carbon, hydrogen, nitrogen and sulphur (CHNS) elemental analyses and by TGA/DTA, coupled with mass spectroscopy (MS). The latter were performed on ∼50 mg samples using a Setaram simultaneous instrument model 92. The samples were contained in platinum crucibles, and evacuated overnight before heating from 40 to 1000 °C in ultra-high purity (UHP) He and in HP oxygen at a heating rate of 10 °C min−1. The evolved gases were analysed using a Balzers Thermostar QS422 mass spectrometer.Wide-angle X-ray scattering and data processing
Synchrotron total scattering data on the dried hydrolysis product were collected at the Swiss Light Source Powder Diffraction beamline at the Paul Scherrer Institute, Switzerland using the multicrystal analyser at an energy of 28.6 keV (λ = 0.433 Å). The signal from an empty quartz capillary container was measured in the same condition and subtracted from the data. The pair distribution function (PDF) was obtained using PDFGetX3 (ref. 22) with the maximum Q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ value, Qmax = 21 Å−1 and refinements of the PDFs against structural models were carried out with PDFgui.23 For all refinements, isotropic atomic displacement parameters (a.d.p.'s) were used, constrained to be equal for atoms of the same species. Also, the symmetry constraints from the structure of the original compound were applied. The scale factor, cell parameters particle diameter, a.d.p.'s, and linear atomic correlation factor were refined. The Debye function calculations were done with the debyer software (https://github.com/wojdyr/debyer). Clusters of different sizes and shapes were cut from the periodic crystal structures for the Debye function calculations.
sinθ/λ value, Qmax = 21 Å−1 and refinements of the PDFs against structural models were carried out with PDFgui.23 For all refinements, isotropic atomic displacement parameters (a.d.p.'s) were used, constrained to be equal for atoms of the same species. Also, the symmetry constraints from the structure of the original compound were applied. The scale factor, cell parameters particle diameter, a.d.p.'s, and linear atomic correlation factor were refined. The Debye function calculations were done with the debyer software (https://github.com/wojdyr/debyer). Clusters of different sizes and shapes were cut from the periodic crystal structures for the Debye function calculations.
Results and discussion
Sample characterization
The CHNS analysis results are given in Table 1 where they are compared with results reported by Klementová.20 In both studies, the analyses confirm that the ammonia treatment completely removes sulphate. Nitrogen is retained in the amorphous phases as NH4+, as confirmed by IR spectroscopy results reported by Klementová20 and by Ihara.21 The presence of the NH4+ cation was identified by these authors from bands at ∼3145 and 1400 cm−1. Our N content is within the range reported by Klementová20 while our H content is only 20% lower. It is clear from the analyses in Table 1 that the amorphous phase precipitated from a titanyl sulphate solution is chemically quite similar to the amorphous phase formed from reaction of ammonia solution with solid titanyl sulphates.The TGA/DTA/MS results are summarised in Table 2. When heated in helium the DTA shows an endotherm centred at 145 °C corresponding to water loss and an exothermic peak at 417 °C corresponding to crystallization of anatase. When the TGA experiment was conducted in a flow of oxygen, a second exothermic peak was observed in the DTA trace, at 288 °C. From MS on the evolved gases it was confirmed that this exotherm corresponded to the oxidation of the ammonium ion to nitrogen oxides. The TGA/DTA/MS plots are reported as ESI data, Fig. S1(a)–(d).†
| Helium run | Oxygen run | |
|---|---|---|
| DTA peaks (°C) | ||
| Endotherm | 145 | 170 |
| Exotherm | 417 | 288 and 400 |
|
||
| TGA mass losses (%) | ||
| On evacuation | 11.9 | 13.0 |
| 20 to 130 °C | 3.2 | 1.9 |
| 130 to 1000 °C | 11.2 | 12.3 |
|
||
| Evolved gases temp. (°) | ||
| Water | 40–600 | 40–600 |
| NH3 | 100–600 | 100–280 |
| CO2 | 40–200 | 40–200 |
| NOx | — | 200–450 |
Thermal analyses conducted by Palkovská et al.24 on the Klementova sample D at different heating rates showed that at the slowest heating rate, of 2 °C min−1, the water release could be resolved into two steps, with losses below 130 °C corresponding to physically adsorbed waster and losses above 130 °C corresponding to more strongly bonded water. Our results are consistent with this observation, with 80% or more of the mass loss of our sample at temperatures below 130 °C occurring during evacuation of the sample. We corrected the mass loss at greater than 130 °C for loss of CO2 and NH3 to obtain the structurally bound water/hydroxyl content and combined this with the chemical analyses in Table 1 to obtain a formula for the amorphous phase. The derived formula is [(NH4)0.16H1.06TiO2.61]·(H2O)0.70(CO2)0.025, where the components outside the square brackets are physically adsorbed species. It is worth noting that the precursor phase is not a crystalline phase with a well-established periodic lattice, and so variations in the formula are expected both within the hydrolysis product and between products prepared in separate syntheses.
The formula inside the square brackets indicates that the titanate structure has a small negative charge, balanced by NH4+ cations. The titanate formula is close to H2Ti2O5, a member of the series of layered polytitanic acids, H2TinO2n+1.25 The titanate formula can also be written as ∼ TiO2·0.5H2O. It is interesting to note that Barringer and Bowen,26 who prepared amorphous titania subjected to PDF analysis by Wang11 gave the same formula TiO2·0.5H2O to account for the structurally bound water content.
PDF and Debye function results
The synchrotron PXRD pattern for the amorphous titania phase and the corresponding PDF are shown in Fig. 1. The PXRD pattern shows just three broad humps to a d-spacing of 1.5 Å, together with several much weaker humps at higher scattering angles, as labelled in Fig. 1(a). Using the FWHM of the strongest hump in the Scherrer equation27 gives a size of the domains of structural order of only ∼8 Å. A similar result is obtained from the PDF in Fig. 1(b), which shows well defined peaks only for r < 7 Å.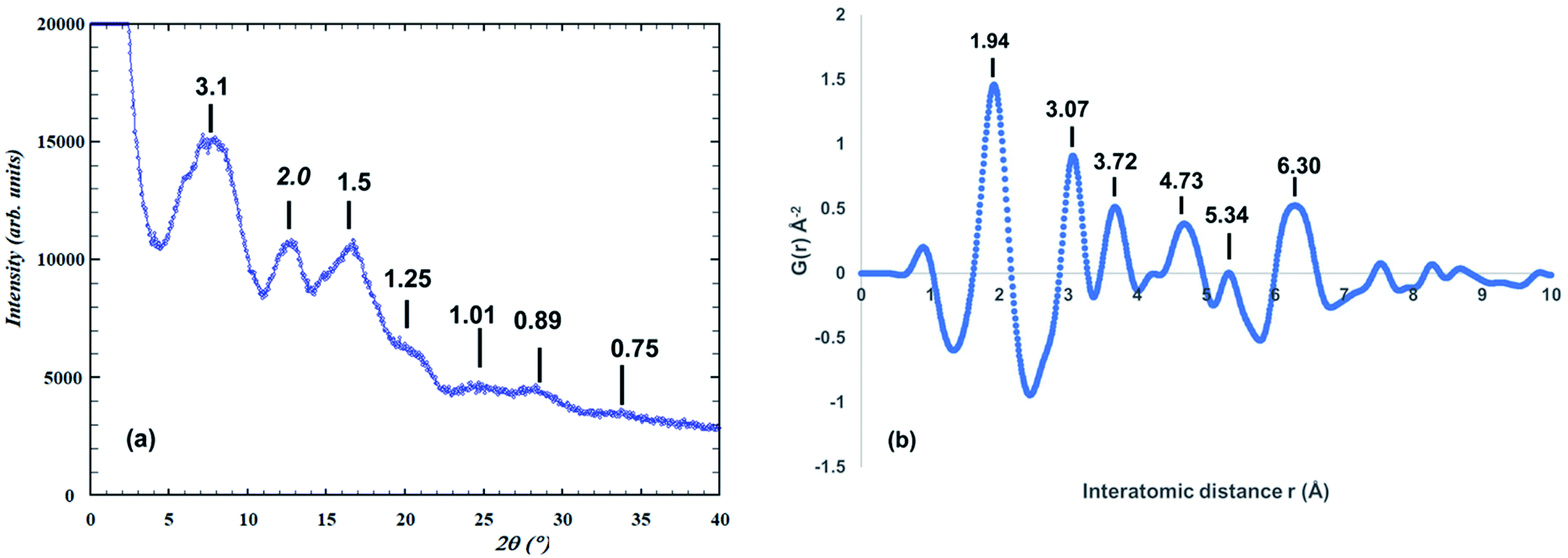 | ||
| Fig. 1 (a) Synchrotron PXRD pattern for amorphous precursor phase (λ = 0.4430 Å). d-Spacings shown. (b) Atomic PDF function for amorphous phase. d-Spacings are shown. | ||
The positions and relative intensities of the PDF peaks for our amorphous titania correspond closely to those reported by Gateshki,13 for amorphous titania prepared by reaction of hexamethylenetetramine with TiCl3 solution at 90 °C. These authors compared their experimental PDF with calculated PDFs for numerous phases; specifically rutile, brookite, anatase, two high-pressure forms of TiO2 (α-PbO2-type and baddeleyite-type structures), the reduced titanates Ti3O5 and Ti4O7, the titanic acid H2Ti3O7 and lepidocrocite (using the CsxTi2−x/4O4 structure28). They noted that the experimental PDF was sensitive enough to discriminate between the different models tested and they concluded that the one based on the lepidocrocite layer structure gave the best approximation to the precursor phase structure.
Protonated Ti-lepidocrocite, HxTi2−x/4O4, has a layer structure built from edge- and corner-shared octahedra as shown in Fig. 2(a), with a separation between the layers of ∼9 Å.29 Gateshki13 proposed that the precursor phase was composed of randomly orientated short segments of layers. They did not, however, check the validity of their model directly against the PXRD pattern.
 | ||
| Fig. 2 (a) 24-Ti atom segment of lepidocrocite structure used for Debye-function modelling of PXRD. (b) The segment rotated to show cubic-stacking of (111)rocksalt-like layers. | ||
Our fit to the experimental PDF using the lepidocrocite model is shown in Fig. 3(a).
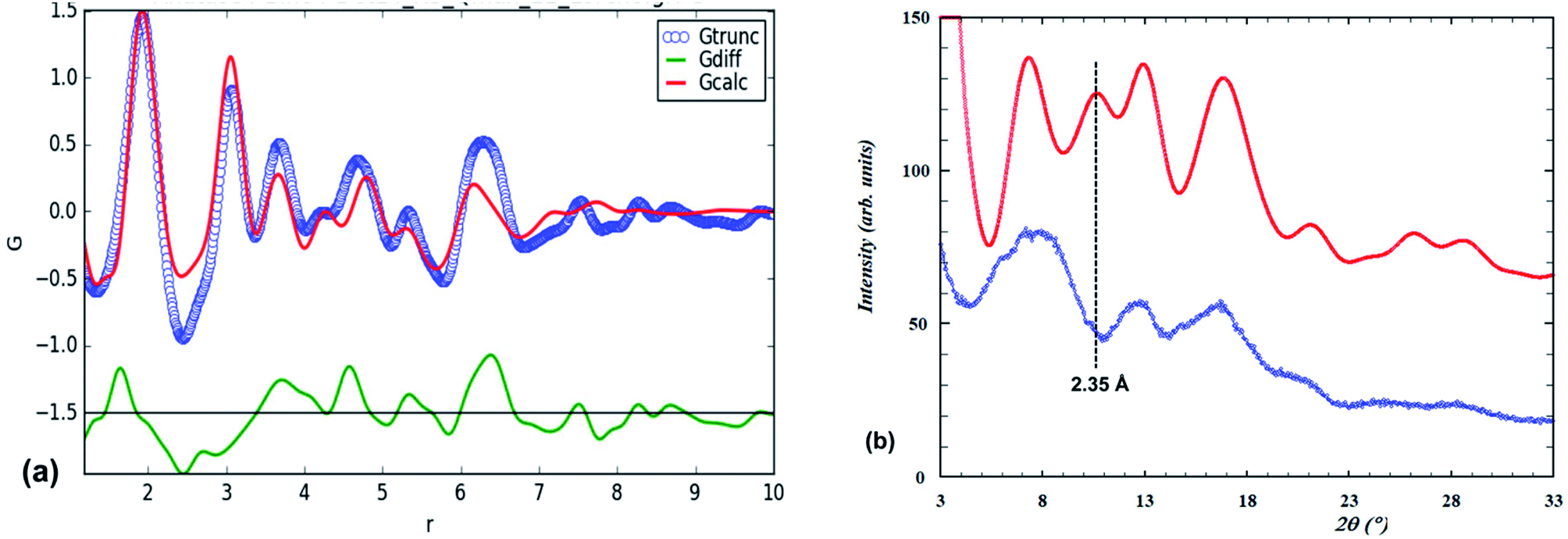 | ||
| Fig. 3 (a) Experimental PDF (blue points) and calculated PDF (red line) for the lepidocrocite structure. (b) Amorphous titania PXRD (blue points) and Debye-function calculated pattern (red line) based on the lepidocrocite-type cluster shown in Fig. 2(a). | ||
The fitting parameters that were refined were the scale, unit cell parameters and overall isotropic displacement parameters for Ti and O. Visually the match closely resembles that previously reported13 (their Fig. 5). In Fig. 3(b) the PXRD calculated using the Debye equation is compared with the experimental pattern. For the calculated pattern the segment of a lepidocrocite layer (24 Ti atoms) shown in Fig. 2(a) was used. The agreement is only fair; the position of the first calculated peak is displaced to a higher d-spacing of 3.35 Å compared to experiment (3.1 Å), and there is a relatively strong calculated peak at d = 2.35 Å that is not present in the experimental pattern. This peak can be explained if we consider that the lepidocrocite layer corresponds to a 2-octahedra-wide (110) slice of the face-centred cubic (fcc) rocksalt structure and the peak at d = 2.35 Å corresponds to diffraction from the cubic-stacked layers (=(111)rocksalt). In Fig. 2(b) the lepidocrocite layer shown in Fig. 2(a) has been rotated to show the cubic-stacked layers. We used the Debye equation to generate PXRD patterns for several lepidocrocite segments of varying size and shape but in all cases the 2.35 Å peak was a major feature and we conclude that lepidocrocite is not a good model for our amorphous titania phase. Debye function plots for different lepidocrocite segment sizes are given as ESI data, Fig. S2(a)–(d).†
In searching for alternative structural models to explain the diffraction data for amorphous titania we considered the possibility of titanium-oxo clusters. A variety of such clusters exist in solution depending on the pH and associated solution species and many clusters have been induced to crystallize and have had their structures determined.30,31 As described by Zhang,32 titanium-oxo clusters are “the trapped snapshots of intermediate structures in the sol–gel growth of TiO2 nanocrystals”. To date, however, there has been no direct confirmation that titanium-oxo clusters exist in amorphous precursor phases. For example Kozma33 conducted a laboratory study of the industrial sulphate process for titania pigment production and demonstrated that hydrated titanium oxysulphate clusters containing 18 Ti atoms per cluster exist in the strong sulphuric acid solution and can be crystallised, but when the solution was diluted and aged under ambient conditions, 2 nm particles of anatase precipitated without any indication of an amorphous phase precursor. On the other hand, Gautier-Luneau34 crystallized a hexanuclear titanium acetate complex by evaporation of an ethanolic solution of titanium tetraethoxide and acetic acid and noted that if the evaporation rate was increased an amorphous gel formed and a radial distribution function plot for the gel showed the same interatomic distances as for the crystalline cluster, leading them to postulate that the hexamer cluster exists in the gel.
We checked numerous published structures of titanium-oxo clusters for Ti–Ti distances consistent with the amorphous titania PDF and selected four clusters for PDF calculations, corresponding to widely different sizes and shapes of the clusters. The four clusters are shown in Fig. S3 of the ESI data.† Ti18O45 is a Keggin complex, Ti13O40, capped by five TiO5 square antiprisms;35 Ti12O32 has a hexameric belt of edge-shared square antiprisms, capped above and below by Ti3O13 edge-shared trimers;36 Ti6O28 is the Galy-cluster comprising two trimeric units of edge-and corner-connected octahedra34 and Ti15O30 is an optimised cluster obtained by simulated annealing and MC simulations.37 PDF calculations showed that none of the as-published cluster models fitted the experimental PDF as well as the lepidocrocite model. A cautionary lesson from the calculations was that unconstrained PDF refinements in which the Ti and O atom positions were allowed to move from the published values gave very good fits to the experimental PDF but resulted in unrealistically short Ti–O and O–O distances. Separate models as disparate as the planar Ti6O28 cluster and the spherical hedgehog-like Ti18O45 cluster gave equally good fits, emphasising the need to have careful control and appropriate constraints during PDF refinements. For the calculated PDFs reported in this study, the atoms were kept at the positions given for the published structures.
With the lepidocrocite model giving the closest match to the PDF of the models considered so far, we next considered derivates of this structure type. A family of related structures that has received considerable attention as precursors for controlled formation of different titania polymorphs and different nano-morphologies (nanotubes, nanobelts, nanowires) are the layered titanic acids of general formula H2TinO2n+1·mH2O, n = 2 to 5.4,25,38,39 The n = 2 member has an orthorhombic lepidocrocite-type structure whereas the other members have monoclinic symmetry and stepped-layer structures that comprise lepidocrocite strips that are n-octahedra wide and offset by corner-sharing. The series has been extended by monoclinic members with n = 6 (ref. 38) and n = 8 (ref. 40) in which 3- and 4-octahedra wide stepped layers are fused by corner-sharing. We tested the possibility of stepped layers being present in our amorphous titania by generating PDFs and Debye-function PXRDs.
The PDF for the member with n = 3 was calculated using the structural model reported for D2Ti3O7,41 shown in Fig. 4(a). The comparison with the experimental PDF after refining the same parameters used for the lepidocrocite-type PDF refinement is shown in Fig. 5(a).
 | ||
| Fig. 4 (a) [010] projection of the structure for H2Ti3O7. (b) Cluster of 20 Ti-centred octahedra from H2Ti3O7 stepped-layer used for Debye-function calculation of the PXRD pattern. | ||
 | ||
| Fig. 5 (a) Experimental PDF (blue points) and calculated PDF (red line) for the H2Ti3O7 structure. (b) Amorphous titania PXRD (blue points) and Debye-function calculated patterns for the H2Ti3O7 20 Ti atom cluster (red line) and with 3 NH4+ added (green line). | ||
It represents a significant improvement on the fit obtained with lepidocrocite-type structure (Fig. 3(a)), with the reduced chi-squared for the fit decreased from 0.031 to 0.019. A 20 Ti atom portion of the stepped layer was cut from the structure, Fig. 4(b), and used in a Debye-function calculation of the PXRD. The calculated pattern is compared to the experimental pattern in Fig. 5(b). There is a close match between the two, not only for the three strongest peaks to d = 1.5 Å, but also for the weaker peaks at higher scattering angle, suggesting that small segments of stepped-lepidocrocite layers occur in our amorphous titania sample.
We checked the effect of changing the size of the cluster on the calculated PXRD pattern, by increasing the cluster size from 20 to 30 and 40 Ti atoms per cluster. The clusters and the corresponding Debye-calculated PXRD patterns are given in the ESI data as Fig. S4.† The calculated PXRDs show increasing structure in the peaks relative to the experimental pattern and relative to the calculated pattern for the 20 Ti atom cluster, consistent with the interatomic Ti–Ti distances in these clusters extending beyond the ∼1 nm correlation length evident in the experimental PDF. The calculations suggest that the optimum size of aggregates in the amorphous phase corresponds to 20 Ti atoms clusters.
Incorporation of ammonium cations and structural formula of amorphous titania
The identification of stepped-lepidocrocite-layers as locally-ordered structure elements in our amorphous titania sample leads naturally to the location of likely sites for the incorporation of NH4+ cations. The published structures42,43 for monoclinic stepped-layer compounds A2TinO2n+1, where A are large monovalent cations like K+ and Tl+ that are comparable in size to NH4+, have the A cations occupying two independent sites, A1 and A2, shown in Fig. 6(a). The A1 cations occupy sites at the corners of the stepped layers whereas the A2 cations are located mid-way along the layer. In compounds like Na2Ti6O13, in which adjacent [Ti3O7]2− layers are interconnected by corner-sharing, only the A1 sites are occupied.44 Liu45 partially replaced protons by NH4+ in H2Ti3O7, giving (NH4)xH2−xTi3O7, and used Raman spectroscopy to determine the location of the NH4+ cations. Their study was consistent with NH4+ substituting at the A1 sites at the corners of the stepped layers.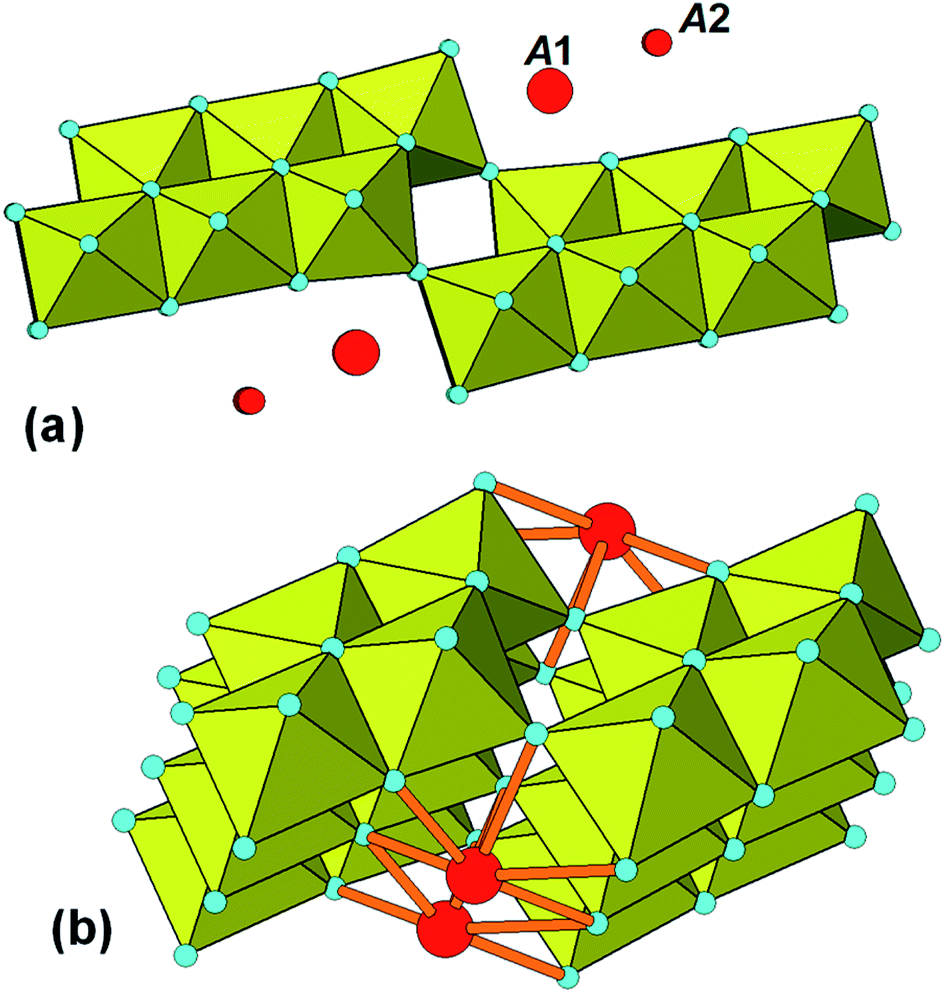 | ||
| Fig. 6 (a) Location of A cations in monoclinic A2TinO2n+1. (b) Bonding of A1 cations (NH4+) to 20-Ti atom stepped layer. | ||
Based on these studies the NH4+ cations in our amorphous titania sample are considered to be located at the A1 sites and coordinated to oxygen atoms of octahedra involved in corner-linking of the lepidocrocite segments. The bonding of the cations in the 20 Ti atom stepped layer cluster is shown in Fig. 6(b). The ratio of available A1 sites to Ti atoms is 3 to 20 = 0.15, which is close to the value of 0.16 obtained from the chemical analyses. The Debye-calculated PXRD pattern for the 20 Ti atom cluster with 3 NH4+ cations incorporated as in Fig. 6(b) is shown as the green line in Fig. 5(b). It gives a good match with the experimental PXRD and a PDF calculated with NH4+ added resulted in a small decrease in chi-squared (to 0.017) relative to the model without NH4+ (0.019).
The composition of the 20 Ti atom cluster, Fig. 4(b), with all Ti atoms octahedrally coordinated, is Ti20X63, X = anions O, OH, H2O. For comparison, chemical and thermal analyses give a formula [(NH4)0.16H1.06TiO2.61]·(H2O)0.70(CO2)0.025, where physically adsorbed species are shown outside the square brackets. Scaling the formula within the square brackets to 20 Ti and rounding to integers gives (NH4)3H21Ti20O52. To be compatible with this formula the 20 Ti atom cluster requires 11 fewer anions. This corresponds to 55% of the Ti atoms being 5-coordinated, giving a mean coordination number for Ti of 5.45.
Comparing a 30 Ti atom cluster, Ti30X87, to the scaled chemical formula of (NH4)5H31Ti30O78, indicates that this structural cluster requires 9 fewer anions, equivalent to 30% of the Ti atoms being 5-coordinated and giving a mean coordination number for Ti of 5.70. For comparison, Petkov12 reports a Ti–O first coordination number of 5.6 and Zhang14 reports a mean coordination number for Ti of 5.3 for their sol–gel derived amorphous titania samples. The 20-atom cluster, which gives the best fit to our scattering data, is in good agreement with both reported values. Accordingly, we can describe the amorphous titania prepared by ammonia neutralization of titanyl sulphate as being composed of small clusters of stepped-layer structure, with the average cluster size corresponding to a composition (NH4)3H21Ti20O52. The NH4+ cations are coordinated to the clusters at the corner-linkages between offset lepidocrocite-type segments. Aggregates of individual clusters are most likely held together through H-bonding and ∼14 molecules of H2O per cluster are physically adsorbed within the pore space of the nanoparticles.
Amorphous phase formation from reaction of solid titanyl sulphate with ammonia
As mentioned in the Introduction, Klementová20 reported that the reaction of solid titanyl sulphate hydrate with ammonia solution at 0 °C gave an amorphous hydrated ammonium titanate with identical morphology to the crystals of the starting compound. Their chemical, thermal and PXRD results for the amorphous phase correspond closely to our results so it is likely that the amorphous phase structures are similar. Their synthetic method, however, highly constrains the reaction pathway for the formation of the amorphous phase and makes it possible to develop a mechanism for the formation. With the reaction occurring at 0 °C, and with the size and shape of the original crystals preserved, there is unlikely to be significant diffusion of the Ti during the amorphization. If it occurs by a dissolution/reprecipitation mechanism, the structure of the amorphous product will be controlled by the topology of the undissolved substrate.In Fig. 7(a) is shown the structure of titanyl sulphate monohydrate. It has orthorhombic symmetry, with a = 9.818, b = 5.133, c = 8.614 Å.46 The structure comprises crankshaft-shaped ribbons of cis-corner-connected octahedra along [010] which are interlinked along [100] and [001] by corner-sharing with sulphate tetrahedra. The octahedral corner-linkages involve alternating very short (1.70 Å) and long (2.05 Å) Ti–O bonds and so the structure can be described in terms of chains of titanyl groups, [..O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ti–O..]n decorated with SO4 and H2O, running parallel to [010]. When TiOSO4·H2O is reacted with ammonia solution the sulphate groups are completely extracted giving TiO2·H2O with loss of SO3. Fig. 7(b) shows one of the resulting titanyl chains, in which each TiO5(H2O) octahedron has been altered to four-coordinated TiO3(H2O). Ti4+, a d0 transition element cation, is most stable when octahedrally coordinated. The octahedral coordination can be restored by folding of the polymeric chain so that the anions can be shared by two or three Ti atoms. In this folding the OTi–O backbone is retained, but the resulting shrinkage associated with the folding will result in the long chains being broken into shorter length chains. Fig. 7(c) shows an example of the folding of a 6-member chain to give a cluster of edge- and corner-shared octahedra. The location of the corner-sharing is expected to be influenced by the proximity of NH4+ cations. We have confirmed, using polyhedral models, that the titanyl backbone remains intact in the folding operation and that this is the structure-directing element.
Ti–O..]n decorated with SO4 and H2O, running parallel to [010]. When TiOSO4·H2O is reacted with ammonia solution the sulphate groups are completely extracted giving TiO2·H2O with loss of SO3. Fig. 7(b) shows one of the resulting titanyl chains, in which each TiO5(H2O) octahedron has been altered to four-coordinated TiO3(H2O). Ti4+, a d0 transition element cation, is most stable when octahedrally coordinated. The octahedral coordination can be restored by folding of the polymeric chain so that the anions can be shared by two or three Ti atoms. In this folding the OTi–O backbone is retained, but the resulting shrinkage associated with the folding will result in the long chains being broken into shorter length chains. Fig. 7(c) shows an example of the folding of a 6-member chain to give a cluster of edge- and corner-shared octahedra. The location of the corner-sharing is expected to be influenced by the proximity of NH4+ cations. We have confirmed, using polyhedral models, that the titanyl backbone remains intact in the folding operation and that this is the structure-directing element.
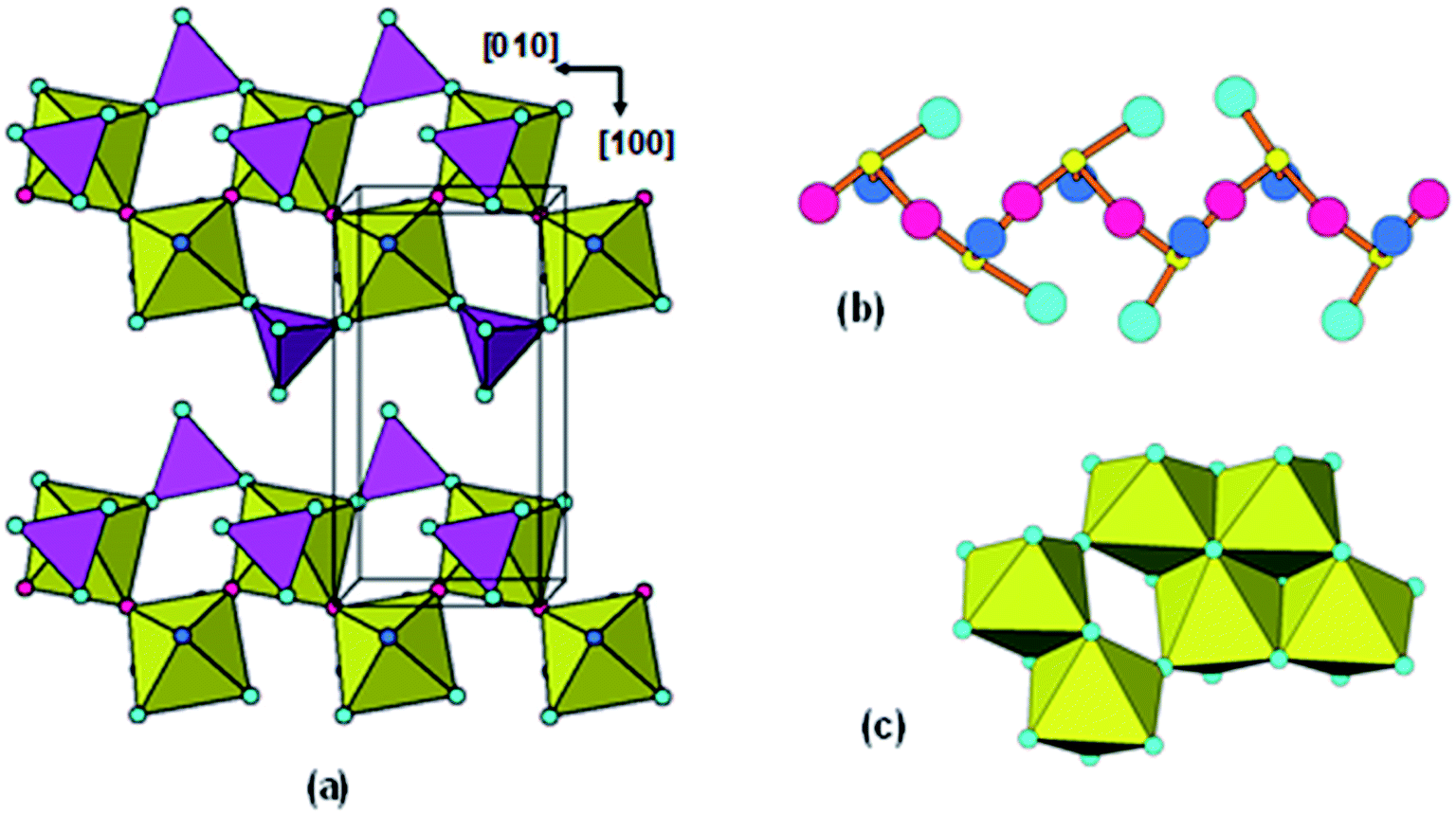 | ||
| Fig. 7 (a) TiOSO4·H2O structure projected along [001]. (b) Titanyl chain segment after extraction of sulphate. Red spheres are oxygen forming OTi–O backbone of chain. Light blue sphere = O, dark blue sphere = H2O, small yellow sphere = Ti. (c) Condensed cluster of edge- and corner-shared octahedra obtained by folding the 6-Ti chain segment in (b). | ||
In Fig. 8(a) the titanyl chains are viewed end-on along [010] after extraction of the sulphate groups. With the removal of interconnecting sulphate groups the chains are free to rotate and displace as shown by the arrow in Fig. 8(b). Condensation of adjacent chain segments, with loss of H2O gives the lepidocrocite-layer segment shown in Fig. 8(c). Thus the combination of sulphate extraction, folding and rupture of the titanyl chains and condensation of adjacent chains applied to the reaction of solid titanyl sulphate with aqueous ammonia, leads to a model for the amorphous phase reported by Klementová20 that matches the model we derived from X-ray scattering experiments on amorphous titania prepared by neutralization of titanyl sulphate solution with ammonia.
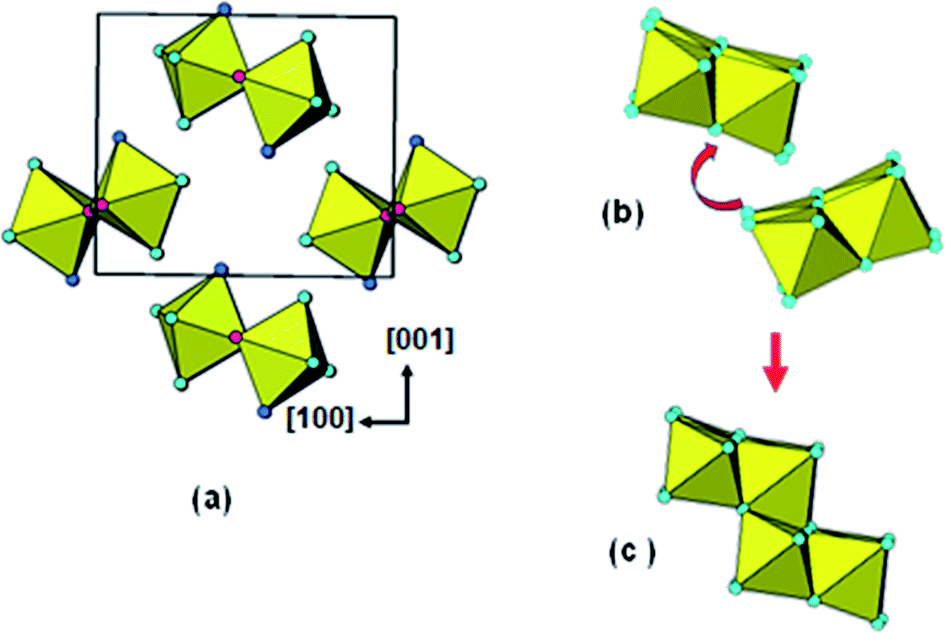 | ||
| Fig. 8 (a) TiOSO4·H2O structure projected along [010], after extraction of sulphate. (b) Movement of adjacent titanyl chains involved in condensation. (c) Segment of stepped-layer formed by condensation of two adjacent chains. | ||
Application to N-doped photocatalysts
The establishment of a structural model for the amorphous titania helps to give some insight into how nitrogen is incorporated into nanocrystalline titania. The stepped-lepidocrocite-layer model provides stable sites for NH4+ cations, at the corners of the stepped layers as shown in Fig. 6. Such sites are confirmed from published structures for monoclinic stepped-layer titanates.43,44 The NH4+ cations are strongly held by coordination to oxygen atoms of octahedra involved in the corner-linking of the lepidocrocite segments and are retained when the amorphous titania hydrolysis product is washed to remove sulphate ions. The TGA/MS experiment shows that oxidation of NH4+ to nitrogen oxides begins to occur when the hydrolysis product is heated above 200 °C in oxygen. Ihara21 has reported that the colour of the hydrolysis product starts changing from white to yellow when heated above 200 °C, suggesting that nitrogen is starting to coordinate to Ti at sites where H2O/OH− has been lost due to the heating. The yellow colour becomes intense at a temperature of 400 °C, where an exotherm due to anatase crystallisation occurs in the DTA (Table 2). Asahi47 attributes the bright yellow colour to direct bonding of N to Ti in TiO2−xNx. Ihara21 reports that 0.2 wt% N is retained in their sample heated at 400 °C. At this temperature, there remained about 0.5 wt% of H2O in our sample. We have previously shown, from density functional theory quantum mechanical (QM) modelling, that the N is preferentially incorporated into protonated titanium vacancy sites in the anatase,48 and we found49 that extra stability was obtained in models where the structure was relaxed to give bonding between N at the vacant Ti site and one of the surrounding Ti atoms, with a Ti–N distance of ∼2 Å.Conclusions
Amorphous titania prepared by neutralisation of titania solution with ammonia displays wide-angle scattering patterns that are consistent with a local structure based on short segments of monoclinic H2TinO2n+1 stepped-layer structure. Segments of pure lepidocrocite-type orthorhombic layer structures do not give a good fit to the PXRD pattern but the fit is improved if the layers are offset by corner-sharing. The correlation length for ordering is less than 1 nm, so the amorphous particles are essentially all surface-like with expected high concentrations of defects and lower coordination numbers than 6 for the Ti. The stepped nature of the layer segments provides sites at the corners for incorporation of NH4+. The bound ammonium cation is thus part of the structure and is held in place until heated to above 200 °C, when oxidation to nitrogen oxides is observed in TGA/MS experiments (Table 2).The stepped-layer model for the amorphous phase is consistent with a mechanism that we developed to explain the reaction of ammonia solution with solid titanyl sulphate hydrate as reported by Klementová.20 The ammonia solution completely extracts the sulphate groups, leaving titanyl chains. A model involving rupture, folding and condensation of the leached titanyl chains leads directly to the formation of small segments of stepped-layer structure. The operations involve only small local rotations and displacements of the chains and are thus consistent with the observation that the amorphous product retains the size and shape of the original titanyl sulphate crystals.
The stepped-layer model is different from a brookite-based model used by Petkov12 to describe the structure of amorphous titania prepared by controlled hydrolysis and condensation of tetra-iso-propoxytitanate in ethanol solution, and from a model based on a strained anatase core developed by Zhang14 to describe amorphous titania obtained by hydrolysis of titanium tetraethoxide in water at 0 °C with acetic acid added. It should be noted however that the PDFs presented by both groups differ significantly from our PDF in having lower ratios of the 3.0 Å peak (Ti–Ti edge-sharing) to the 3.6 Å peak (Ti–Ti corner-sharing). Evidently the local structure of amorphous titania precursor phase can differ depending on the synthesis conditions and this difference can result in different titania polymorphs being formed on crystallization of the precursor.
Author contributions
I. E. Grey. Planned and managed the study, interpreted the experimental results and wrote the manuscript. P. Bordet. Conducted the PDF and Debye function modelling, prepared the relevant diagrams and contributed to the writing. N. C. Wilson. Provided background checking of cluster models using QM calculations.Conflicts of interest
There are no conflicts of interest.Acknowledgements
We are indebted to Ian Madsen for the collection of the synchrotron X-ray data. Thanks to Christina Li for the syntheses and to Terry Hall for the thermal analyses.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- I. Ali, M. Suhail, Z. A. Alothman and A. Alwarthan, RSC Adv., 2018, 8, 30125–30147 RSC.
- R. Asahi, T. Morikawa and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS.
- S. Girish Kumar and K. S. R. Koteswara Rao, Nanoscale, 2014, 6, 11574–11632 RSC.
- Y. Nam, J. H. Lim, K. C. Ko and J. Y. Lee, J. Mater. Chem. A, 2019, 7, 13833–13859 RSC.
- M. T. Noman, M. A. Ashraf and A. Ali, Environ. Sci. Pollut. Res., 2019, 26, 3262–3291 CrossRef CAS.
- M. Cargnello, T. R. Gordon and C. B. Murray, Chem. Rev., 2014, 114, 9319–9345 CrossRef CAS.
- Y. Wang, Y. He, Q. Lai and M. Fan, J. Environ. Sci., 2014, 26, 2139–2177 CrossRef.
- (a) H. Yin, Y. Wada, T. Kitamura, S. Kambe, S. Murasawa, H. Mori, T. Sakata and S. Yanagida, J. Mater. Chem., 2001, 11, 1694–1703 RSC; (b) M. Strauss, C. M. Maroneze, J. M. Souza e Silva, F. A. Sigoli, Y. Gushikem and I. O. Mazali, Mater. Chem. Phys., 2011, 126, 188–194 CrossRef CAS.
- S. Sun, P. Song, J. Cui and S. Liang, Catal. Sci. Technol., 2019, 9, 4198–4215 RSC.
- Q. J. Wang, S. C. Moss, M. L. Shalz, A. M. Glaeser, H. W. Zandbergen and P. Zschack, Physics and Chemistry of Finite Systems: From Clusters to Crystals, ed. P. Jena et al., Kluwer Academic Publishers, Netherlands, 1992, vol. II, pp. 1287–1294 Search PubMed.
- V. Petkov, G. Holzhüter, U. Tröge, T. Gerber and B. Himmel, J. Non-Cryst. Solids, 1998, 231, 17–30 CrossRef CAS.
- M. Gateshki, S. Yin, Y. Ren and V. Petkov, Chem. Mater., 2007, 19, 2512–2518 CrossRef CAS.
- H. Zhang, B. Chen and J. F. Banfield, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 214106 CrossRef.
- Z. Guo, F. Ambrosio and A. Pasquarello, J. Mater. Chem. A, 2018, 6, 11804–11810 RSC.
- V. V. Hoang, Phys. Status Solidi B, 2007, 244, 1280–1287 CrossRef CAS.
- N. V. Kaliannan and K. Krishnamurthy, J. Nanopart. Res., 2017, 19, 346 CrossRef.
- H. H. Pham and L.-W. Wang, Phys. Chem. Chem. Phys., 2015, 17, 541–550 RSC.
- B. Prasai, B. Cai, M. K. Underwood, J. P. Lewis and D. A. Drabold, J. Mater. Sci., 2012, 47, 7515–7521 CrossRef CAS.
- M. Klementová, M. Motlochová, J. Bohácek, J. Kupcik, L. Palatinus, E. Plizingrová, L. Szatmáry and J. Subrt, Cryst. Growth Des., 2017, 17, 6762–6769 CrossRef.
- T. Ihara, M. Miyoshi, Y. Iriyama, O. Matsumoto and S. Sugihara, Appl. Catal., B, 2003, 42, 403–409 CrossRef CAS.
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef.
- C. L. Farrow, P. Juhás, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS.
- M. Palkovská, V. Slovák, J. Subrt, J. Bohácek, Z. Barbieriková, V. Brezová and R. Fajgar, J. Therm. Anal. Calorim., 2016, 125, 1071–1078 CrossRef.
- D. V. Bavykin, J. M. Friedrich and F. C. Walsh, Adv. Mater., 2006, 18, 2807–2824 CrossRef CAS.
- E. A. Barringer and H. K. Bowen, Langmuir, 1985, 1, 414–420 CrossRef CAS.
- B. D. Cullity, Elements of X-Ray Diffraction, Addison-Wesley, Reading, USA, 1959 Search PubMed.
- I. E. Grey, C. Li, I. C. Madsen and J. A. Watts, J. Solid State Chem., 1987, 77, 7–19 CrossRef.
- T. Sasaki, M. Watanabe, Y. Michiue, Y. Komatsu, F. Izumi and S. Takenouchi, Chem. Mater., 1995, 7, 1001–1007 CrossRef CAS.
- L. Rozes, N. Steunou, G. Fornasieri and C. Sanchez, Monatsh. Chem., 2006, 137, 501–528 CrossRef CAS.
- W.-H. Fang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2018, 47, 404–421 RSC.
- G. Zhang, J. Hou, C.-H. Tung and Y. Wang, Inorg. Chem., 2016, 55, 3212–3214 CrossRef CAS.
- K. Kozma, M. Wang, P. I. Molina, N. P. Martin, Z. Feng and M. Nyman, Dalton Trans., 2019, 48, 11086–11093 RSC.
- I. Gautier-Luneau, A. Mosset and J. Galy, Z. Kristallogr., 1987, 180, 83–95 CrossRef CAS.
- N. Steunou, F. Robert, K. Boubekeur, F. Ribot and C. Sanchez, Inorg. Chim. Acta, 1998, 279, 144–151 CrossRef CAS.
- V. W. Day, T. A. Eberspacher, W. G. Klemperer and C. W. Park, J. Am. Chem. Soc., 1993, 115, 8469–8470 CrossRef CAS.
- S. Hamad, C. R. A. Catlow, S. M. Woodley, S. Lago and J. A. Mejias, J. Phys. Chem. B, 2005, 109, 15741–15748 CrossRef CAS.
- J. C. Pérez-Flores, C. Baehtz, M. Hoelzel, A. Kuhn and F. Garcia-Alvarado, RSC Adv., 2012, 2, 3530–3540 RSC.
- H. G. Yang and H. C. Zeng, J. Am. Chem. Soc., 2005, 127, 270–278 CrossRef CAS.
- N. Bao, L. Shen and K. Yanagisawa, J. Phys. Chem. B, 2004, 108, 16739–16745 CrossRef CAS.
- T. P. Feist and P. K. Davies, J. Solid State Chem., 1992, 101, 275–295 CrossRef CAS.
- H. Izawa, S. Kikkawa and M. Koizumi, J. Phys. Chem., 1982, 86, 5023–5026 CrossRef CAS.
- A. Verbaere and M. Tournoux, Bull. Soc. Chim. Fr., 1973, 4, 1237–1241 Search PubMed.
- S. Andersson and A. D. Wadsley, Acta Crystallogr., 1962, 15, 194–201 CrossRef CAS.
- H. Liu, Z. Zheng, D. Yang, E. Waclawik, X. Ke, H. Zhu, S. Palmer and R. L. Frost, J. Raman Spectrosc., 2010, 41, 1601–1605 CrossRef.
- B. M. Gatehouse, S. N. Platts and T. B. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 39, 428–435 CrossRef.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS.
- I. E. Grey and N. C. Wilson, J. Solid State Chem., 2007, 180, 670–678 CrossRef CAS.
- S. P. Russo, I. E. Grey and N. C. Wilson, J. Phys. Chem. C, 2008, 112, 7653–7664 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08886b |
| This journal is © The Royal Society of Chemistry 2021 |