
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the activity of transition metal M and heteroatom N4 co-doped in vacancy fullerene (M–N4–C64, M = Fe, Co, and Ni) towards the oxygen reduction reaction by density functional theory
Siwei Yang,
Chaoyu Zhao ,
Ruxin Qu,
Yaxuan Cheng,
Huiling Liu* and
Xuri Huang*
,
Ruxin Qu,
Yaxuan Cheng,
Huiling Liu* and
Xuri Huang*
Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Liutiao Road 2, Changchun 130023, China. E-mail: huiling@jlu.edu.cn; huangxr@jlu.edu.cn
First published on 22nd January 2021
Abstract
In this study, a novel type oxygen reduction reaction (ORR) electrocatalyst is explored using density functional theory (DFT); the catalyst consists of transition metal M and heteroatom N4 co-doped in vacancy fullerene (M–N4–C64, M = Fe, Co, and Ni). Mulliken charge analysis shows that the metal center is the reaction site of ORR. PDOS analysis indicates that in M–N4–C64, the interaction between Fe–N4–C64 and the adsorbate is the strongest, followed by Co–N4–C64 and Ni–N4–C64. This is consistent with the calculated adsorption energies. By analyzing and comparing the adsorption energies of ORR intermediates and activation energies and reaction energies of all elemental reactions in M–N4–C64 (M = Fe, Co, and Ni), two favorable ORR electrocatalysts, Fe–N4–C64 and Co–N4–C64, are selected. Both exhibited conduction through the more efficient 4e− reduction pathway. Moreover, PES diagrams indicate that the whole reaction energy variation in the favorable ORR pathways of Fe–N4–C64 and Co–N4–C64 is degressive, which is conducive to positive-going reactions. This study offers worthwhile information for the improvement of cathode materials for fuel cells.
1. Introduction
With the expansion of industrialization and urbanization, environmental pollution and energy consumption are being increasingly aggravated. Fuel cells (FCs) have become the most promising power generation technology, with the characteristics of sustainable, pollution-free and wide-ranging fuel sources.1–4 The oxygen reduction reaction (ORR) is carried out in the cathode through 2e− and 4e− pathways.5,6 The 4e− pathway directly reduces O2 to H2O and has high energy conversion efficiency; hence, it is the preferred pathway for FCs.7,8 However, the 2e− pathway (the less efficient pathway involving the formation of H2O2 as an intermediate) and slow kinetics on the cathode hinder the power generation efficiency of FCs.9–11 In addition, Pt-based catalysts, which are considered to be highly efficient,12,13 have limited commercial applications in FCs due to their high cost.14,15 Therefore, it is urgent to exploit high-performance and inexpensive ORR electrocatalysts to substitute the expensive Pt-based catalysts on the market.Because single-atom catalysts (SACs) have different activity, selectivity and stability from traditional nanocatalysts,16,17 they are proposed to exhibit remarkable behavior in various energy transformation reactions, such as HER,18,19 CO2RR20,21 and ORR.22,23 Moreover, the realization of single-atom dispersion of SACs usually requires a suitable substrate and dopant.24 Fullerenes are cage structures in which carbon atoms form a conjugated system through sp2 hybridization.25 As non-metallic materials with a wide range of sources, fullerenes have demonstrated excellent performance including biological applications,26 energy storage,27 thermoelectric materials,28 and optoelectronic devices.29 In addition, as a result of the curvature and pentagon defects of fullerenes,30,31 they can be used as promising candidates for electrocatalytic reactions. Vinu et al. investigated a polymeric mesoporous fullerene C60 material prepared at 130 °C (MFC60-130), which showed effective ORR behavior benefiting from its specific surface area of 680 m2 g−1 and steady and conductive C60 reticulation.32 Subsequently, Vinu et al. continued to report mesoporous fullerene C70 materials, which were endowed with good mesoporous framework, excellent conductivity and connectivity; further, materials exhibited good ORR performance.33 However, it is necessary to improve their electrocatalytic properties for practical applications.
Heteroatom doping is an effectual way to regulate the electronic and conductive properties of fullerenes and enhance their electrocatalytic behavior.34,35 Chen et al. discovered that nitrogen-doped fullerenes showed higher ORR properties than pure fullerenes.36 Wang et al. also reported that nitrogen-doped C60 cages (C59N) exhibited superior ORR electrocatalytic activity to other heteroatom-doped C60 cages,37 which was further experimentally proved by Gao et al.35
Furthermore, to facilitate the embedding of metal atoms, introducing vacancies in the substrate can change the coordination environment and electronic structure around the vacancies; thus, under-coordinated sites can act as bait and vacancies can act as traps to trap metal atoms.38,39 The co-doping of transition metal atoms and heteroatoms on vacant carbon materials has been widely studied both experimentally and theoretically. Fe-porphyrin-like carbon nanotubes synthesized by Lee et al. showed excellent oxygen reduction activity due to the characteristics of the Fe–N4 structure.40 Fei et al. found that the composite material obtained by embedding individual Ni atoms into the double vacancies of an N-doped graphene lattice displayed higher oxygen evolution reaction (OER) activity and stability.41 Chen et al. embedded individual iron atoms into N-pore carbon through the thermolysis approach of cage-encapsulated precursors, and the composite exhibited remarkable ORR properties and satisfactory methanol tolerance.42 Recently, Wang et al. reported synthesized Fe-anchored graphite carbonitride, which was used in CO2RR to improve the selectivity of CO and realize the high-efficiency transformation of CO2 due to the Fe–N4 sites in the composite.43 Theoretically, many studies have illuminated that metal–N4-vacancy carbon materials have attractive performance as cathode materials for FCs.44–47 Significantly, Brindaban Modak et al. found that transition metal-modified porous fullerene C24N24 can serve as an oxygen reduction electrocatalyst and carry out a more effectual 4e− reduction mechanism.48 These reports led us to wonder whether transition metal and heteroatom N could be co-doped in vacancy fullerenes as effective electrocatalysts for ORR. However, related reports are rare. Therefore, we systematically studied all possible intermediate steps involved in the ORR process of co-doping transition metal M and heteroatom N4 in vacancy fullerene (M–N4–C64, M = Fe, Co, and Ni). The promising ORR electrocatalysts were selected and the corresponding favorable ORR pathways were determined. This work offers worthwhile information for the improvement of cathode materials for FCs.
2. Computational methods
All calculations were carried out using the spin-polarized DFT implemented in Materials Studio DMol3 code.36,49 The generalized gradient approximation of the Perdew–Burke–Ernzerhof functional (GGA + PBE),48,50 together with all electrons and the double numerical basis set (DNP), was adopted throughout all calculations. To improve computational performance, the self-consistent field (SCF) was converged to 10−6 Ha and the smearing was set to 0.005 Ha. The convergence tolerance was 2 × 10−5 Ha for the energy change, 0.004 Ha Å−1 for the max force and 0.005 Å for the max displacement. The Grimme method for DFT-D correction was used to describe the van der Waals interactions. The energy difference between the transition state and the reactant was the activation barrier (Eact).The formation energy (Ef) of M–N4 co-doped in vacancy fullerene was calculated as follows:
| Ef = EM–N4–C64 + 6μC − (Efullerene + 4μN + EM) |
The adsorption energy (Eads) was calculated as follows:
| Eads = Eadsorbate@M–N4–C64 − EM–N4–C64 − Eadsorbate |
3. Results and discussion
3.1 Configuration and properties of M–N4–C64
To embed M-N4 in fullerene, as Fig. 1a shows, the no. 1 and 2 carbon atoms in the optimized fullerene C70 were removed. Then, four nitrogen atoms were utilized to substitute the no. 3, 4, 5 and 6 carbon atoms, which had insufficient coordination. Finally, the metal atom was incorporated in the center of the vacancy. We calculated the different multiplicities of M–N4–C64 (M = Fe, Co, and Ni) to obtain the most stable energies. The results show that the most stable states for Fe–N4–C64, Co–N4–C64, and Ni–N4–C64 are triplet, doublet, and singlet, correspondingly. The oxygen molecule is a triplet in our research system. The stable configurations of Fe–N4–C64, Co–N4–C64 and Ni–N4–C64 were optimized (see Fig. 1b–d), and the formation energies (Ef) of the three configurations were calculated as −3.82, −4.11, and −3.43 eV, correspondingly. This finding demonstrates that Fe–N4–C64, Co–N4–C64 and Ni–N4–C64 are thermodynamically stable. In addition, as can be seen from the Mulliken charges in Fig. 1e–g, nitrogen atoms in M–N4–C64 (M = Fe, Co, and Ni) show negative charge density, while the adjacent carbon atoms show positive charge density. This is caused by the fact that the electronegativity of N atoms is greater than that of C atoms. This porphyrin-like structure can stably fix M–N4 embedded on C64. The Mulliken charges of the Fe, Co and Ni atoms were calculated as 0.664, 0.483 and 0.540|e|, correspondingly. Because a positively charged site is more advantageous for O2 adsorption, the metal center was chosen as the reaction site for ORR. On the other hand, it was found that in the Fe–N4–C64 system, the electron transfer from the metal center to N4–C64 is the largest.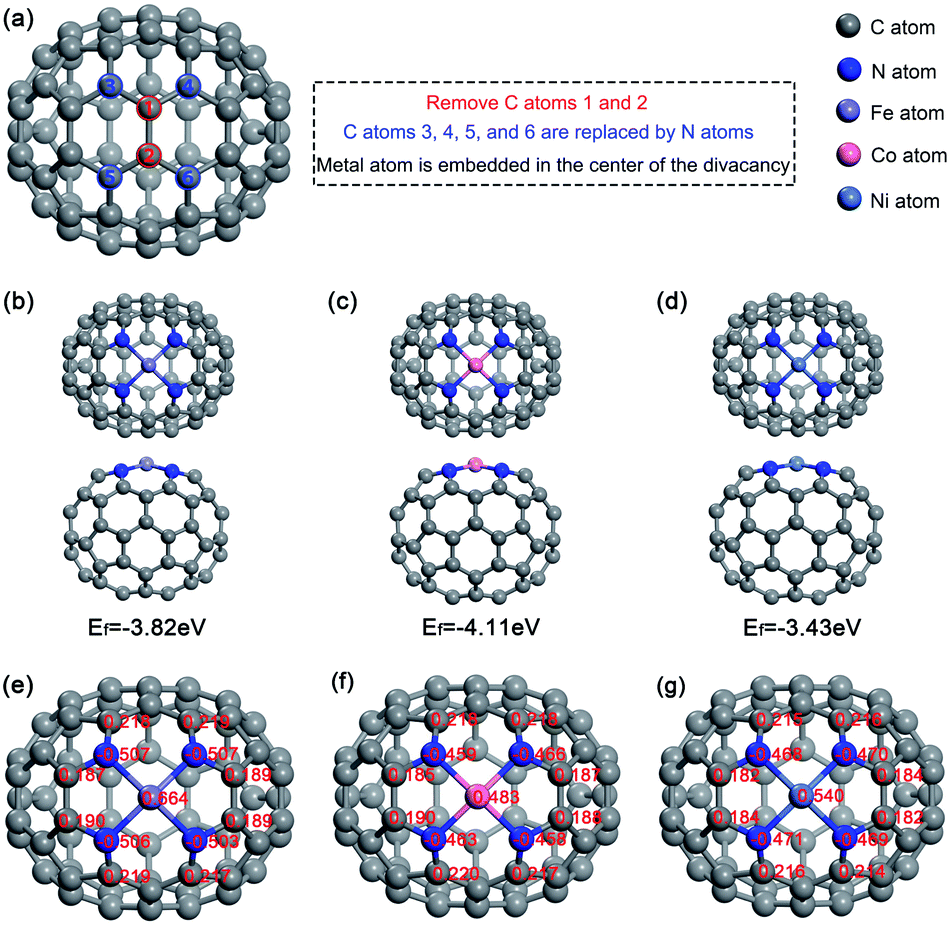 | ||
| Fig. 1 The optimized configurations of (a) C70, (b) Fe–N4–C64, (c) Co–N4–C64 and (d) Ni–N4–C64. Charge distributions on (e) Fe–N4–C64, (f) Co–N4–C64 and (g) Ni–N4–C64. | ||
3.2 Electronic structure of M–N4–C64
The calculated DOS (see Fig. 2a) display that the band gaps of Fe–N4–C64, Co–N4–C64 and Ni–N4–C64 are all zero, which reveals that they have the potential to be ORR electrocatalysts due to their excellent electrical conductivity.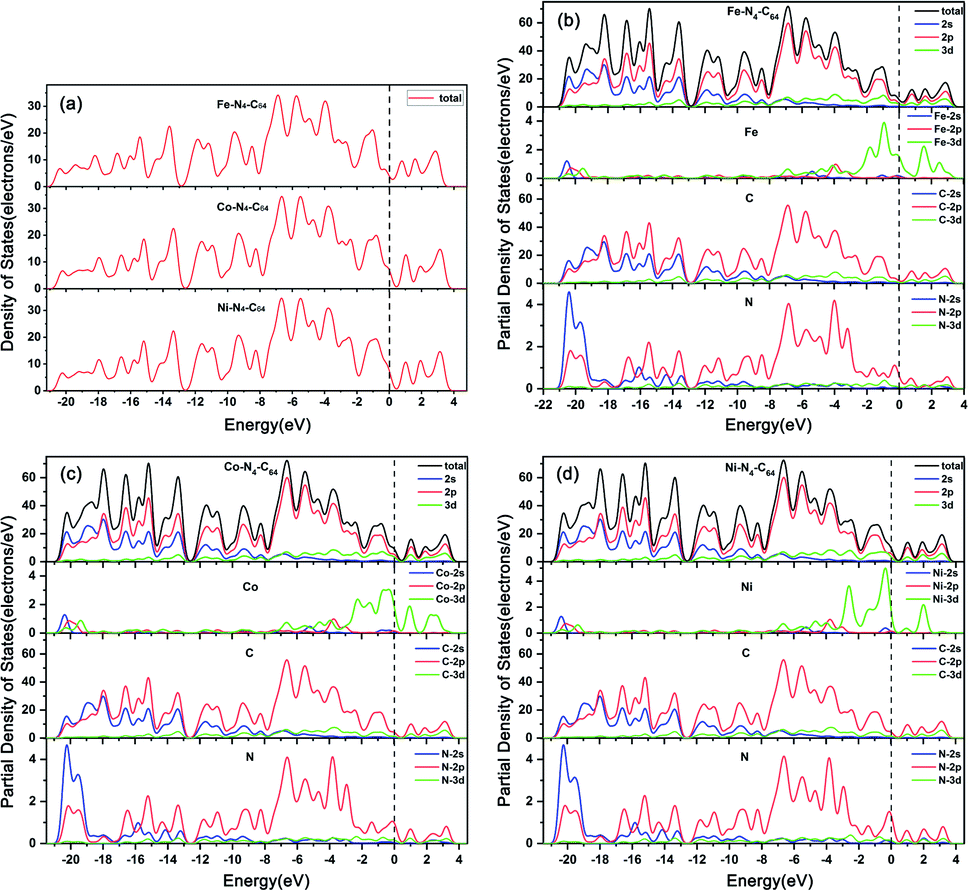 | ||
| Fig. 2 (a) The densities of states for Fe–N4–C64, Co–N4–C64 and Ni–N4–C64. The partial densities of states for (b) Fe–N4–C64, (c) Co–N4–C64 and (d) Ni–N4–C64. | ||
The calculated PDOS (see Fig. 2b–d) display that the C-2p and N-2p orbitals in M–N4–C64 (M = Fe, Co, and Ni) exhibit strong resonance regions below the Fermi level, which indicates that the interaction between C and N is very strong. Meanwhile, the PDOS display that in M–N4–C64, the Co-3d orbital contribution is the most average below the Fermi level, followed by Fe-3d and finally Ni-3d, which is consistent with stability of the calculated Ef for M–N4–C64. In addition, the calculated d-band centers in the range of −8.0 eV–4 eV are −1.24 eV for Fe–N4–C64, −1.33 eV for Co–N4–C64 and −1.61 eV for Ni–N4–C64, which demonstrates that the interaction between Fe–N4–C64 and the adsorbate is the strongest among the three, followed by Co–N4–C64 and finally Ni–N4–C64. This is attributable to the fact that the upward shift of the d-band center to the Fermi level will arouse the upward shift of the antibonding orbitals; thus, they are harder to fill, and in turn, the interaction between the catalyst and adsorbates is stronger.
3.3 The adsorption of ORR species on M–N4–C64
The adsorption of ORR species plays a formative role in the study of the ORR mechanism and electrocatalytic activity. For comparison, Fig. 3 exhibits the optimized adsorption configurations of the ORR species (O2, OH, OOH, O and H2O) on M–N4–C64 (M = Fe, Co, and Ni), and the corresponding adsorption energies (Eads) were calculated. The results manifest that Eads weakens in this sequence: Fe–N4–C64 > Co–N4–C64 > Ni–N4–C64, which is in line with the results of the PDOS analysis. The electrocatalytic capability of the catalyst can be exhibited visually by the chemisorption of O2 molecule on the substrate as the preliminary stage of ORR. The O2 molecule is stably adsorbed on the Fe center of Fe–N4–C64 by side-on configuration, and the Eads was calculated as −0.73 eV. After adsorption, dO–O is 1.345 Å (dA–B represents the distance between A and B), which is increased compared with that of the individual O2 molecule (1.225 Å). The O2 is stably adsorbed on the Co center of Co–N4–C64 by end-on configuration, and the Eads was calculated as −0.57 eV. After adsorption, dO–O is 1.270 Å, which is slightly larger than that of the individual O2 molecule (1.225 Å). However, in the Ni–N4–C64 system, the calculated Eads of the O2 is 0.33 eV, and the positive value of Eads indicates that it is unbound relative to the separated O2. This indicates that the ORR behavior of Ni–N4–C64 is disadvantageous. Atomic O is adsorbed on Fe–N4–C64 and Co–N4–C64 with the strongest Eads values of −3.86 and −2.62 eV, correspondingly. The Eads(OH) for M–N4–C64 (M = Fe, Co, and Ni) is larger than that of Eads(OOH). Furthermore, for the ORR product H2O, the Eads is −0.75 eV for Fe–N4–C64, −0.60 eV for Co–N4–C64 and −0.23 eV for Ni–N4–C64. These weak Eads values are close to the solvation energy (∼−0.40 eV) of massive H2O.45,51 Therefore, once H2O is formed, it can be effortlessly separated from M–N4–C64.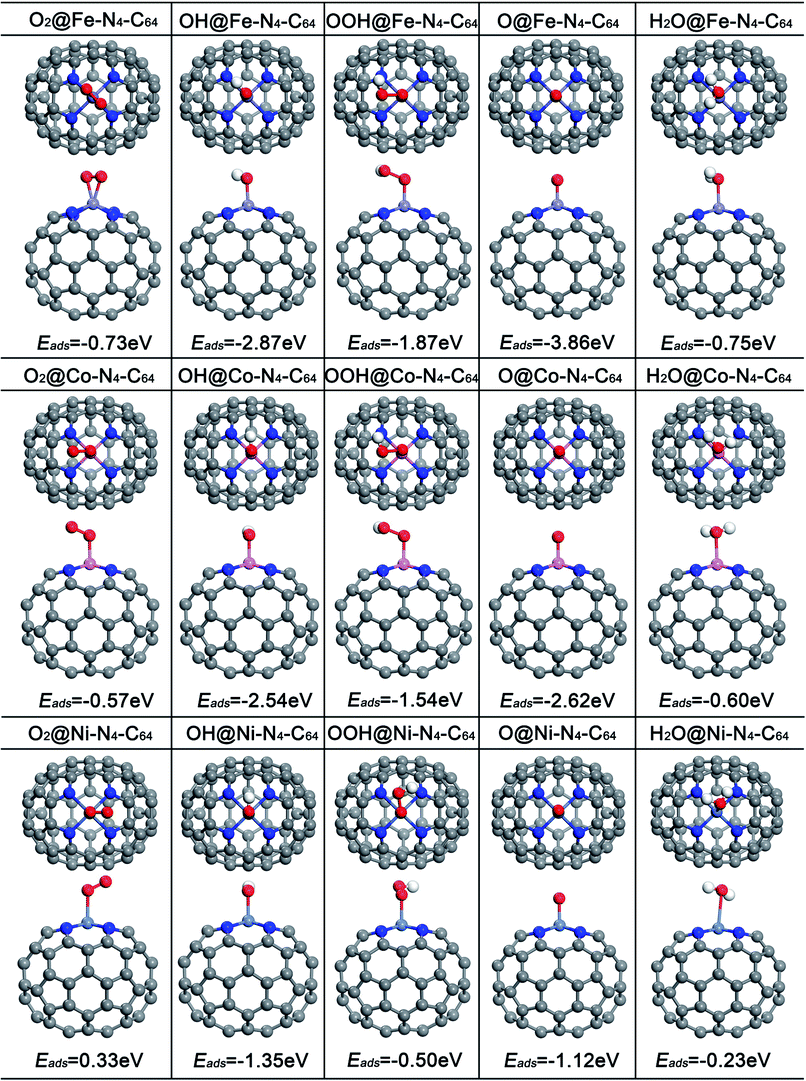 | ||
| Fig. 3 The optimized configurations of the adsorption of O2, OH, OOH, O and H2O on Fe–N4–C64, Co–N4–C64 and Ni–N4–C64. | ||
3.4 ORR mechanisms on M–N4–C64
The ORR mechanisms on M–N4–C64 (M = Fe, Co, and Ni) are complicated. To prove that the three catalysts can serve as effective ORR electrocatalysts, we will systematically study the ORR mechanisms of the three systems in the following sections. The activation barriers (Eact), reaction energies (ΔE) and virtual frequencies (Vfreq) of all the ORR basic reaction steps involved in M–N4–C64 (M = Fe, Co, and Ni) are listed in Table 1.| Catalysts | Reaction steps | Eact (eV) | ΔE (eV) | Vfreq (cm−1) |
|---|---|---|---|---|
| Fe–N4–C64 | *O2 → *O + *O | 2.25 | 0.40 | i439 |
| *O2 + *H → *OOH | 0.06 | −0.97 | i1514 | |
| *OOH + *H → *H2O2 | 2.77 | −0.31 | i1026 | |
| *OOH + *H → *O + *H2O | 0.32 | −1.21 | i1355 | |
| *O + *H → *OH | 0.34 | −1.35 | i1298 | |
| *OH + *H → *H2O | 0.47 | −0.60 | i1426 | |
| Co–N4–C64 | *O2 → *O + *O | 2.93 | 1.46 | i561 |
| *O2 + *H → *OOH | 0.13 | −0.69 | i981 | |
| *OOH + *H → *H2O2 | 0.80 | −0.46 | i1469 | |
| *H2O2 → *O + *H2O | 0.33 | −0.23 | i627 | |
| *OOH + *H → *O + *H2O | 0.78 | −0.41 | i768 | |
| *O + *H → *OH | 0.04 | −2.10 | i918 | |
| *OH + *H → *H2O | 0.22 | −0.86 | i1467 | |
| Ni–N4–C64 | *O2 + *H → *OOH | 0.08 | −0.36 | i1102 |
| *OOH + *H → *O + *H2O | 0.68 | 0.21 | i454 | |
| *O + *H → *OH | 3.25 | −2.43 | i468 | |
| *OH + *H → *H2O | 2.97 | −1.93 | i767 |
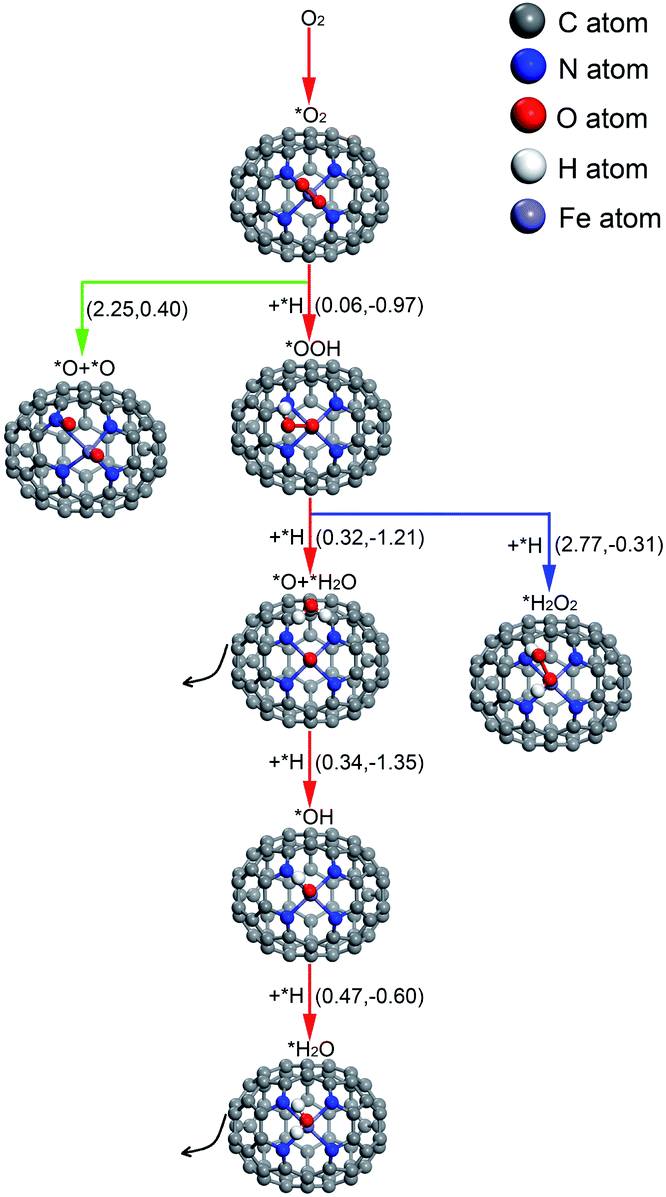 | ||
| Fig. 4 The conceivable ORR pathways on Fe–N4–C64. The value on the left in parentheses is the activation energy and the value on the right is the reaction energy (in eV). * indicates adsorbed species. | ||
For O2 chemisorbed on Fe–N4–C64, there are two conceivable pathways: dissociation (*O2 → *O + *O) and hydrogenation (*O2 + *H → *OOH) (* indicates adsorbed species). For *O2 → *O + *O, this reaction requires an endothermic reaction energy of 0.40 eV and is accompanied by a high activation barrier (Eact) of 2.25 eV. This indicates that the stage of *O2 → *O + *O on Fe–N4–C64 is disadvantageous in thermodynamics and kinetics. The dissociation details are presented in Fig. 5a. Conversely, the hydrogenation of *O2 to *OOH is more feasible because for *O2 + *H → *OOH, this reaction has an exothermic reaction energy of 0.97 eV. Moreover, this process is accompanied by an extremely low Eact of 0.06 eV, which is obviously lower than the Eads of *O2 (−0.73 eV) on Fe–N4–C64. The virtual frequency (Vfreq) of i1514 cm−1 in the transition state TS2 exactly shows the formation of the intermediate *OOH. The hydrogenation details (see Fig. 5b) indicate that dO–H decreases from 2.233 Å in IS2 to 1.401 Å in TS2 and to 0.981 Å in FS2. Meanwhile, the distance between the atomic O which is attacked by the atomic H and the Fe atom extends from 2.066 Å in IS2 to 2.455 Å in TS2 and to 2.671 Å in FS2, and the *OOH intermediate in FS2 is adsorbed on the Fe site by end-on configuration. Furthermore, dO–O extends from 1.335 Å in IS2 to 1.448 Å in FS2, which indicates that the interaction between the two O atoms is weakened and subsequent reactions will occur.
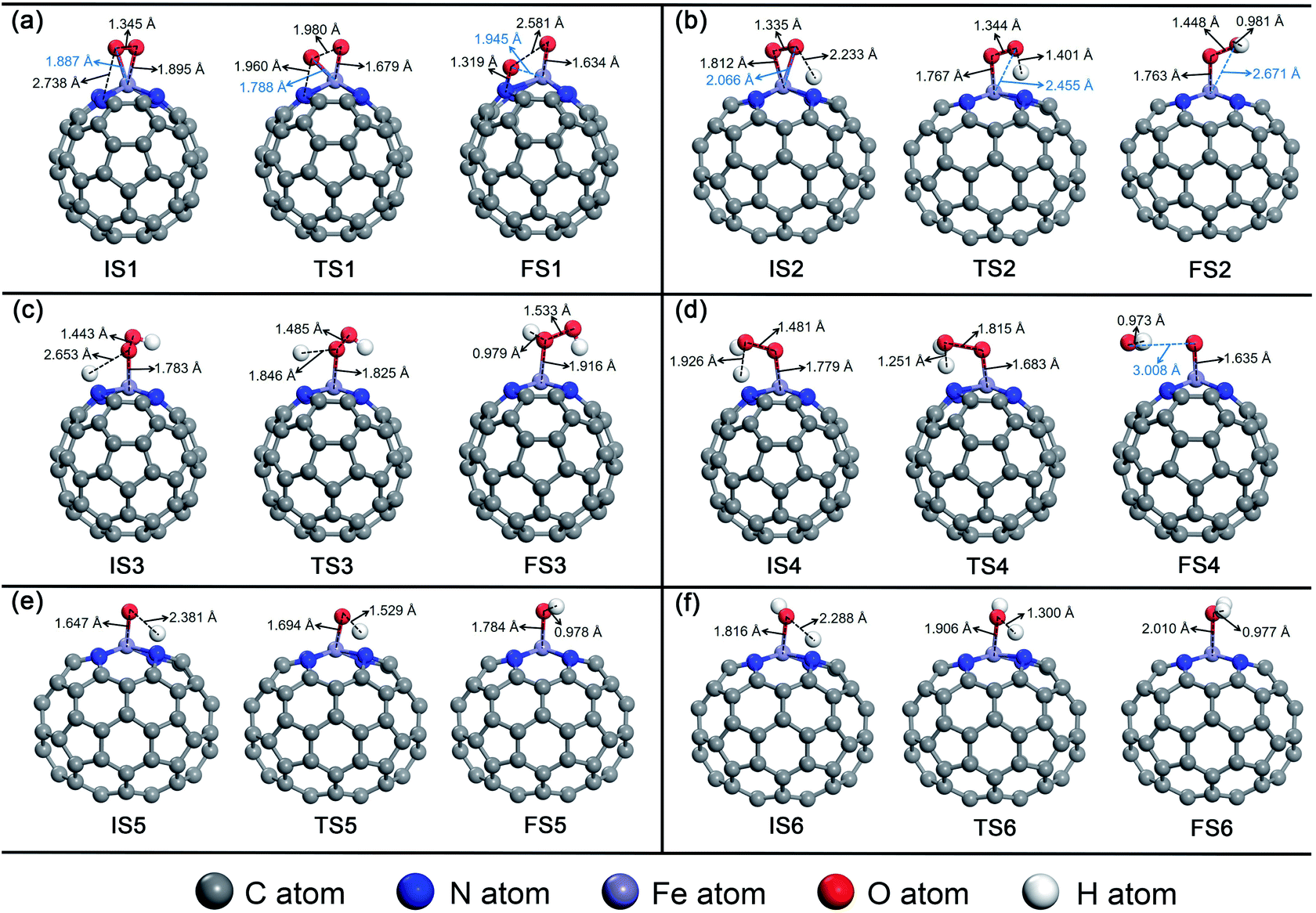 | ||
| Fig. 5 The detailed process of the ORR on Fe–N4–C64. (a) *O2 → *O + *O, (b) *O2 + *H → *OOH, (c) *OOH + *H → *H2O2, (d) *OOH + *H → *O + *H2O, (e) *O + *H → *OH, and (f) *OH + *H → *H2O. | ||
For the formed *OOH, there are two conceivable hydrogenation pathways: the *H attacks the O atom that bonds to Fe to afford *H2O2 (*OOH + *H → *H2O2), or *H attacks the hydroxyl oxygen in *OOH to afford *O and *H2O (*OOH + *H → *O + *H2O). For *OOH + *H → *H2O2, this reaction is accompanied by an insurmountable Eact of 2.77 eV, which manifests that the 2e− pathway to generate *H2O2 on Fe–N4–C64 is kinetically infeasible. This hydrogenation details are described in Fig. 5c. Conversely, the reaction *OOH + *H → *O + *H2O releases 1.21 eV of energy with a low Eact of 0.32 eV, which is more feasible under the support of thermodynamics and kinetics. Moreover, the energy required for this stage is notably lower than the Eads of *OOH (−1.87 eV) on Fe–N4–C64. The Vfreq of i1355 cm−1 in TS4 exactly exhibits the formation of *H2O, and the formed *H2O can be effortlessly detached from the Fe–N4–C64 surface, which only requires a very low energy of 0.27 eV. The hydrogenation details (see Fig. 5d) indicate that dFe-O shortens from 1.779 Å in IS4 to 1.635 Å in FS4 and dO–O increases from 1.481 Å in IS4 to 3.008 Å in FS4. This indicates that the interaction between Fe–O is strengthened and the O–O bond is broken.
Finally, the atomic O remaining on Fe experiences continuous hydrogenation to afford the second *H2O: *O + *H → *OH and *OH + *H → *H2O. The former reaction has a large exothermic reaction energy of 1.35 eV. Moreover, this process is accompanied by a low Eact of 0.34 eV, which is significantly lower than the Eads of *O (−3.86 eV) on Fe–N4–C64. The hydrogenation details (see Fig. 5e) indicate that dO–H diminishes from 2.381 Å in IS5 to 0.978 Å in FS5. Moreover, in TS5, the Fe–O bond is tilted to promote the formation of *OH. The dFe–O length increases from 1.647 Å in IS5 to 1.784 Å in FS5, which manifests that the interaction between Fe–O is weakened with the formation of *OH. Then, the *OH remaining on Fe experiences the succeeding hydrogenation: *OH + *H → *H2O. This stage has an exothermic reaction energy of 0.60 eV. Moreover, this process is accompanied by a low Eact of 0.47 eV, which is significantly lower than the Eads of *OH (−2.87 eV) on Fe–N4–C64. The hydrogenation details (see Fig. 5f) indicate that the dFe–O lengthens from 1.816 Å in IS6 to 2.010 Å in FS6, which indicates that the interaction between Fe–O is weakened, and the formed *H2O can be easily detached from the Fe–N4–C64 surface due to its low Eads of −0.75 eV. Vfreq is i1298 cm−1 for TS5 and i1426 cm−1 for TS6, respectively. This describes the formation of *OH and the second *H2O.
 | ||
| Fig. 6 The conceivable ORR pathways on Co–N4–C64. The value on the left in parentheses is the activation energy and the value on the right is the reaction energy (in eV). * means adsorbed species. | ||
For O2 chemisorbed on Co–N4–C64, there are two conceivable pathways: dissociation (*O2 → *O + *O) and hydrogenation (*O2 + *H → *OOH). For *O2 → *O + *O, this reaction requires a large endothermic reaction energy of 1.46 eV and is accompanied by an insurmountable Eact of 2.93 eV, which means that the stage of *O2 → *O + *O on Co–N4–C64 is extremely thermodynamically and kinetically disadvantageous. The dissociation details are illustrated in Fig. 7a. Conversely, the hydrogenation of *O2 to *OOH is more feasible because for *O2 + *H → *OOH, this reaction has an exothermic reaction energy of 0.69 eV. Moreover, this process is accompanied by a very low Eact of 0.13 eV, which is evidently lower than the Eads of *O2 (−0.57 eV) on Co–N4–C64. The Vfreq of TS8 is i981 cm−1. The hydrogenation details (see Fig. 7b) indicate that dO–H decreases from 1.993 Å in IS8 to 1.356 Å in TS8 and to 0.982 Å in FS8. Meanwhile, dCo–O shortens from 1.859 Å in IS8 to 1.838 Å in FS8, and dO–O increases from 1.295 Å in IS8 to 1.416 Å in FS8. This implies that the interaction between Co–O is strengthened and that between O–O is weakened.
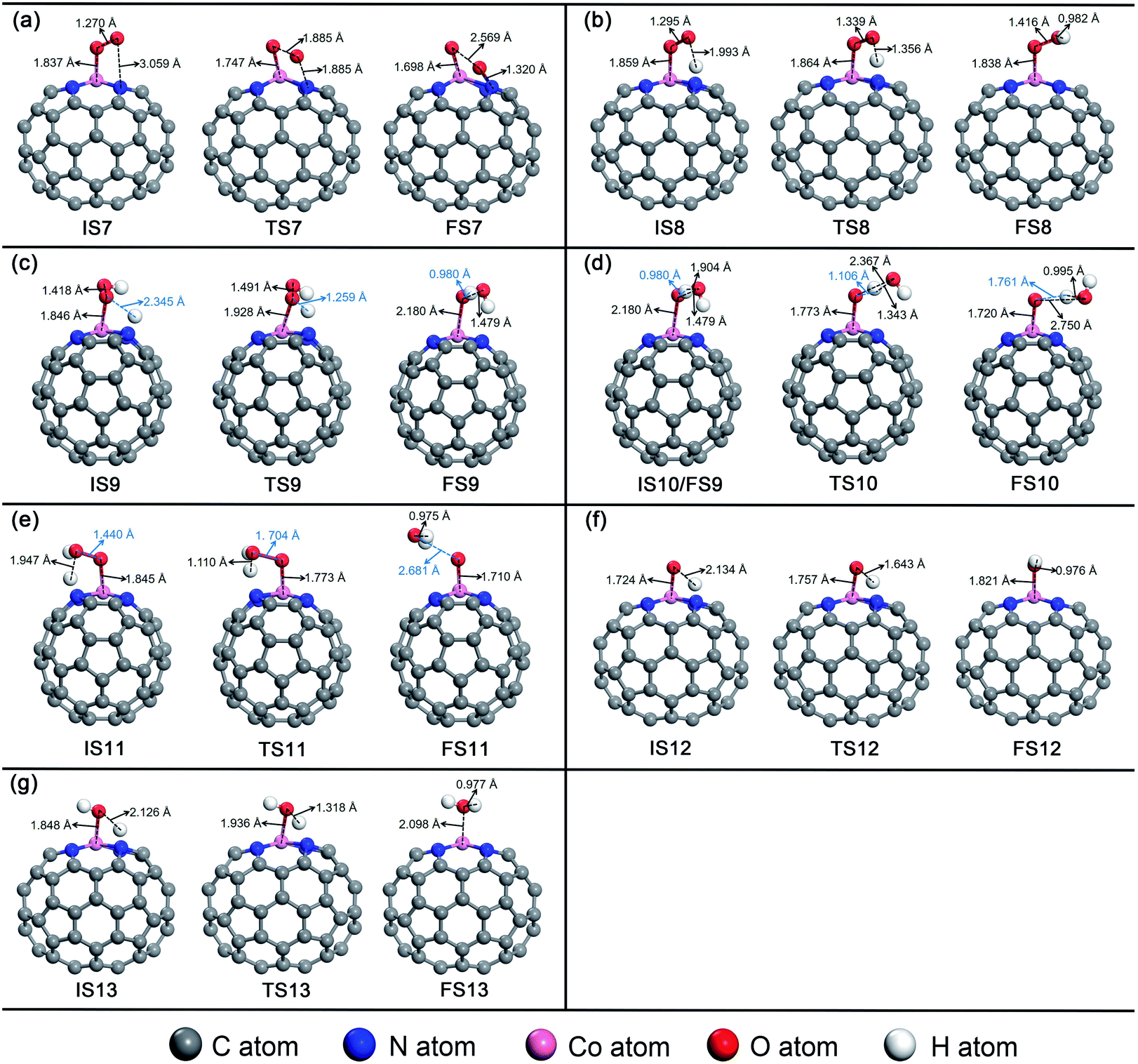 | ||
| Fig. 7 The detailed process of the ORR on Co–N4–C64. (a) *O2 → *O + *O, (b) *O2 + *H → *OOH, (c) *OOH + *H → *H2O2, (d) *H2O2 → *O + *H2O, (e) *OOH + *H → *O + *H2O, (f) *O + *H → *OH, and (g) *OH + *H → *H2O. | ||
For the formed *OOH, there are two conceivable hydrogenation pathways: the *H attacks the O atom that bonds to Co to afford *H2O2 (*OOH + *H → *H2O2), or *H attacks the hydroxyl oxygen in *OOH to afford *O and *H2O (*OOH + *H → *O + *H2O).
For *OOH + *H → H2O2, this reaction has an exothermic reaction energy of 0.46 eV. Moreover, this process is accompanied by an Eact of 0.80 eV, which is obviously lower than the Eads of *OOH (−1.54 eV) on Co–N4–C64. The hydrogenation details (see Fig. 7c) indicate that *H attacks the O atom that bonds to Co, and dO–H decreases from 2.345 Å in IS9 to 0.980 Å in FS9. Moreover, dCo–O increases from 1.846 Å in IS9 to 2.180 Å in FS9. The generated H2O2 is adsorbed on Co–N4–C64 with an Eads of −0.70 eV. Then, *H2O2 is instantly decomposed to *O + *H2O because the reaction of *H2O2 → *O + *H2O has an exothermic energy of 0.23 eV with a very low Eact of 0.33 eV, which means that the stage of *H2O2 → *O + *H2O on Co–N4–C64 is advantageous both thermodynamically and kinetically. Moreover, the energy required for this stage is lower than the Eads of *H2O2 (−0.70 eV) on Co–N4–C64. It can be seen that although the *H2O2 on Co–N4–C64 is formed by the 2e− path, both thermodynamics and kinetics character enable its decomposition reaction to pass through the favorable 4e− path again. The decomposition details (see Fig. 7d) indicate that the *H on the O atom that bonds to Co is transferred to the adjacent hydroxyl oxygen to generate the first *H2O, and the generated *H2O can be effortlessly separated from the Co–N4–C64 surface, which only requires 0.43 eV. Meanwhile, dCo–O shortens from 2.180 Å in IS10/FS9 to 1.720 Å in FS10 and dO–O increases from 1.479 Å in IS10 to 2.750 Å in FS10, which implies that the interaction between Co–O is strengthened and the O–O bond is broken. The Vfreq of i1469 cm−1 for TS9 and i627 cm−1 for TS10 respectively describe the formation and the decomposition of *H2O2.
For *OOH + *H → *O + *H2O, this reaction has an exothermic reaction energy of 0.41 eV. Moreover, this process is accompanied by an Eact of 0.78 eV, which is obviously lower than the Eads of *OOH (−1.54 eV) on Co–N4–C64. The Vfreq of i768 cm−1 in TS11 exactly exhibits the formation of *H2O, and the formed *H2O can easily detach from the Co–N4–C64 surface, which only requires 0.26 eV. The hydrogenation details (see Fig. 7e) indicate that dCo–O shortens from 1.845 Å in IS11 to 1.710 Å in FS11 and that dO–O increases from 1.440 Å in IS11 to 2.681 Å in FS11; these findings respectively indicate that the interaction between Co–O is strengthened and the O–O bond is broken.
The above results manifest that for *OOH, regardless of whether it is first hydrogenated to form *H2O2 and then decomposed to *O + *H2O or whether *OOH is directly hydrogenated to *O + *H2O, it is conducive to ORR behavior on Co–N4–C64.
After the *H2O is released, the atomic O remaining on Co experiences continuous hydrogenation to afford the second *H2O: *O + *H → *OH and *OH + *H → *H2O. The former reaction has a whopping exothermic reaction energy of 2.10 eV. Moreover, this process is accompanied by a very low Eact of 0.04 eV, which is significantly lower than the Eads of *O (−2.62 eV) on Co–N4–C64. The hydrogenation details (see Fig. 7f) indicate that dO–H diminishes from 2.134 Å in IS12 to 0.976 Å in FS12. dCo–O lengthens from 1.724 Å in IS12 to 1.821 Å in FS12, which manifests that the interaction between Co–O is weakened with the formation of *OH. Then, the *OH remaining on Co experiences the succeeding hydrogenation: *OH + *H → *H2O. This stage has an exothermic reaction energy of 0.86 eV. Moreover, this process is accompanied by a very low Eact of 0.22 eV, which is evidently lower than the Eads of *OH (−2.54 eV) on Co–N4–C64. The hydrogenation details (see Fig. 7g) indicate that dCo-O lengthens from 1.848 Å in IS13 to 2.098 Å in FS13; this indicates that the interaction between Co–O is weakened and the formed *H2O can be effortlessly detached from the Co–N4–C64 surface due to the low Eads of −0.60 eV. The Vfreq values of i918 cm−1 for TS12 and i1467 cm−1 for TS13 respectively describe the formation of *OH and the second *H2O.
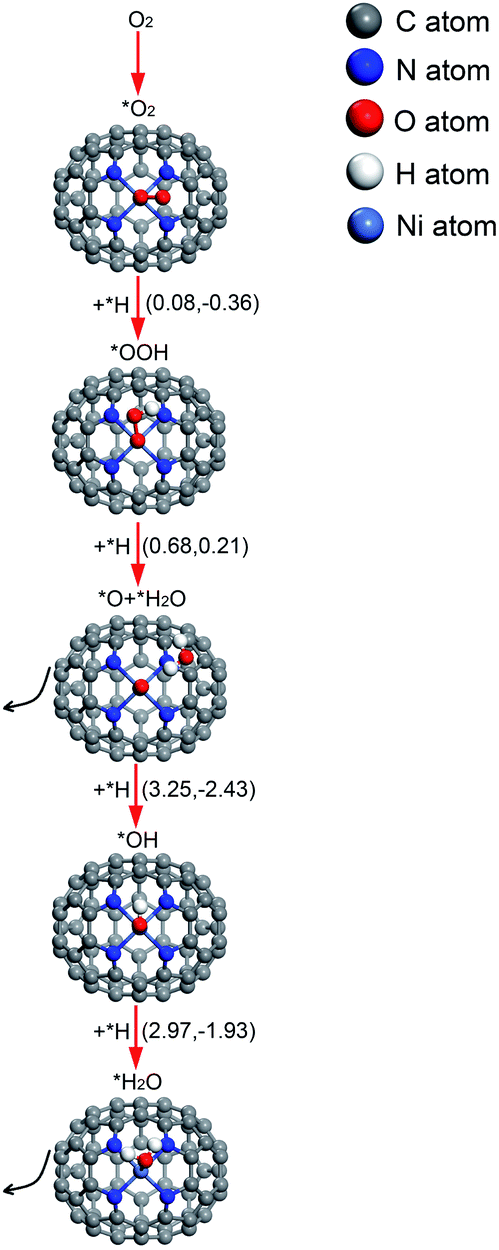 | ||
| Fig. 8 The conceivable ORR pathway on Ni–N4–C64. The value on the left in parentheses is the activation energy and the value on the right is the reaction energy (in eV). * indicates adsorbed species. | ||
3.5 PES and relative energy
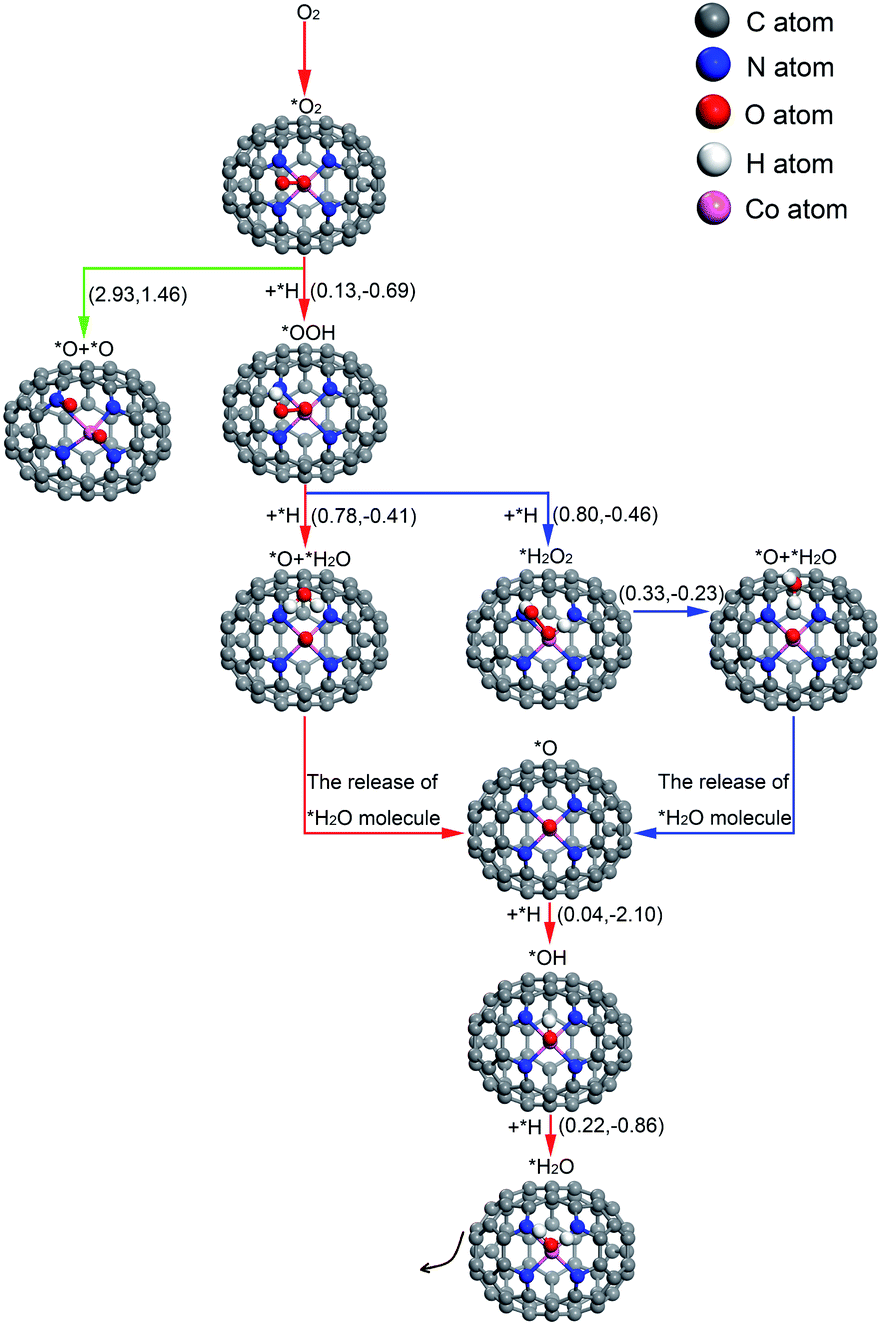
The relative energy curve can be utilised as an effective method to assess the ORR properties of M–N4–C64 (M = Fe, Co, and Ni). Fig. 9a–c exhibit the potential energy surface (PES) diagrams of Fe–N4–C64, Co–N4–C64, and Ni–N4–C64, correspondingly. In Fig. 9a and b, the green line denotes the stage of *O2 → *O + *O. The blue line in Fig. 9a denotes the stage of *OOH + *H → *H2O2 on Fe–N4–C64, and that in Fig. 9b denotes the two stages of *OOH + *H → *H2O2 and *H2O2 → *O + *H2O on Co–N4–C64. For the red line path that can promote ORR on Fe–N4–C64 (see Fig. 9a), the first stage is to hydrogenate *O2 to *OOH; *OOH is then hydrogenated to *O + *H2O, and finally the atomic O remaining on Fe is continuously hydrogenated. The rate-limiting step (RLS) for the favorable red line path on Fe–N4–C64 is *OH + *H → *H2O with an Eact of 0.47 eV. Moreover, the entire reaction energy variation of the red line path on Fe–N4–C64 is degressive, which is conducive to positive-going reactions. Co–N4–C64 can facilitate the ORR via two different paths (see Fig. 9b). One of the two paths is consistent with the aforementioned favorable ORR red line path on Fe–N4–C64. The RLS for this advantageous red line path on Co–N4–C64 is *OOH + *H → *O + *H2O with an Eact of 0.78 eV. For the other path, the difference from the red line path is that the intermediate *OOH in this path is hydrogenated to *H2O2. The generated *H2O2 is immediately decomposed into *O + *H2O to continue the 4e− path because this stage has an exothermic reaction energy of 0.23 eV with a much lower Eact (0.33 eV) than the Eads of *H2O2 (−0.70 eV); finally, the atomic O remaining on Co is continuously hydrogenated. Moreover, the RLS for this feasible path on Co–N4–C64 is *OOH + *H → *H2O2 with an Eact of 0.80 eV. The entire reaction energy variation of the two favorable ORR paths on Co–N4–C64 is degressive, which is conducive to positive-going reactions. Meanwhile, Fig. 9c intuitively shows that Ni–N4–C64 as an ORR electrocatalyst is an unfavorable choice because the two stages for *O + *H → *OH and *OH + *H → *H2O possess high energy barriers.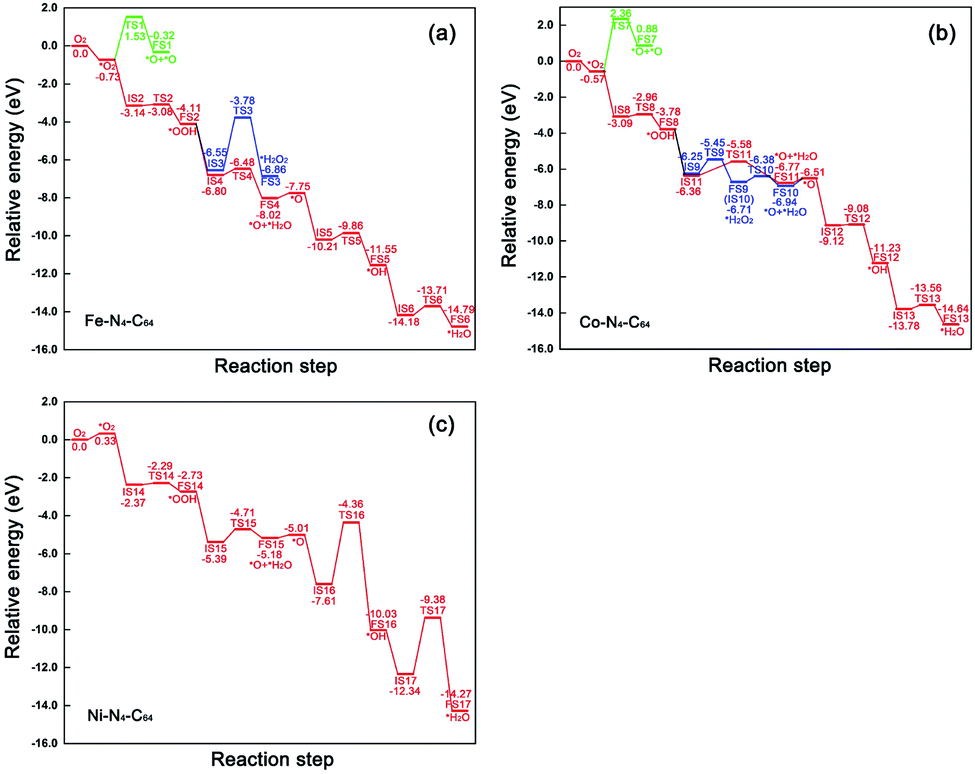 | ||
| Fig. 9 Relative energy profiles of the conceivable ORR pathways on (a) Fe–N4–C64, (b) Co–N4–C64 and (c) Ni–N4–C64. | ||
4. Conclusions
In this work, the density functional method was utilized to systematically study the co-doping of transition metal M and heteroatom N4 in vacancy fullerene (M–N4–C64, M = Fe, Co, and Ni) as a novel type of non-precious ORR electrocatalyst. The calculated formation energies indicated that Fe–N4–C64, Co–N4–C64 and Ni–N4–C64 are thermodynamically stable. Mulliken charge studies manifested that the metal center is the reaction site of the ORR and that the electron transfer from the metal center to N4–C64 is the largest in the Fe–N4–C64 system. PDOS revealed that the interaction between Fe–N4–C64 and the adsorbate is the strongest among the three, followed by Co–N4–C64 and finally Ni–N4–C64. The results of the calculated adsorption energies are consistent with the PDOS analysis. By analyzing and comparing the adsorption energies of the ORR species and the activation energies and reaction energies of all the basic ORR reaction steps involved in M–N4–C64, two favorable ORR electrocatalysts, Fe–N4–C64 and Co–N4–C64, were selected. Meanwhile, favorable ORR paths on Fe–N4–C64 and Co–N4–C64 were determined, and both catalysts used 4e− mechanisms to facilitate ORR. For Fe–N4–C64, the intermediate *OOH formed by hydrogenation of *O2 on Fe–N4–C64 continues to be hydrogenated to *O + *H2O, and finally, the *O remaining on Fe is continuously hydrogenated. The rate-limiting step (RLS) for the favorable path on Fe–N4–C64 is *OH + *H → *H2O, with an Eact of 0.47 eV. For Co–N4–C64, the intermediate *OOH formed by hydrogenation of *O2 on Co–N4–C64 can be directly hydrogenated to *O + *H2O or firstly hydrogenated to *H2O2 and then promptly decomposes to *O + *H2O; finally, the *O remaining on Co is continuously hydrogenated. The RLS of the two advantageous paths on Co–N4–C64 is the hydrogenation stage of the intermediate *OOH, and the corresponding activation energies are 0.78 eV for *OOH + *H → *O + *H2O and 0.80 eV for *OOH + *H → *H2O2, correspondingly. PES diagrams intuitively reflect that the whole reaction energy variation in the favorable ORR paths of Fe–N4–C64 and Co–N4–C64 is degressive, which is conducive to positive-going reactions. This study demonstrates that Fe–N4–C64 and Co–N4–C64 are high-efficiency ORR electrocatalysts. We expect that the proposed transition metal and heteroatom N4 co-doped in vacancy fullerene can contribute to the development of cathode materials for FCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21573090), and Scientific Research Fund of Jilin Provincial Education Department (2015437) for financial support of this research.References
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. B. Riffat and C. Liu, J. Mater. Chem., 2017, 5, 1808–1825 RSC.
- S. Wang and S. P. Jiang, Natl. Sci. Rev., 2017, 4, 163–166 CrossRef CAS.
- E. H. Majlan, D. Rohendi, W. R. W. Daud, T. Husaini and M. A. Haque, Renew. Sust. Energ. Rev., 2018, 89, 117–134 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. F. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal. B-Environ., 2005, 56, 9–35 CrossRef CAS.
- Y. Zheng, D. Yang, J. M. Kweun, C. Li, K. Tan, F. Kong, C. Liang, Y. J. Chabal, Y. Y. Kim and M. Cho, Nano Energy, 2016, 30, 443–449 CrossRef CAS.
- Z. Liang, H. Zheng and R. Cao, Sustain. Energy Fuels, 2020, 4, 3848–3870 RSC.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS.
- J. Liu, M. Jiao, B. Mei, Y. Tong, Y. Li, M. Ruan, P. Song, G. Sun, L. Jiang and Y. Wang, Angew. Chem., Int. Ed., 2019, 58, 1163–1167 CrossRef CAS.
- L. Liu, G. Zeng, J. Chen, L. Bi, L. Dai and Z. Wen, Nano Energy, 2018, 49, 393–402 CrossRef CAS.
- X. Ren, Q. Lv, L. Liu, B. Liu, Y. Wang, A. Liu and G. Wu, Sustain. Energy Fuels, 2020, 4, 15–30 RSC.
- J. Kim, H. Kim and H. Lee, Chemsuschem, 2018, 11, 104–113 CrossRef CAS.
- B. Lu, Q. Liu and S. Chen, ACS Catal., 2020, 10, 7584–7618 CrossRef CAS.
- H. Fei, J. Dong, M. J. Arellanojimenez, G. Ye, N. D. Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin and J. Bao, Nat. Commun., 2015, 6, 8668 CrossRef CAS.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu and C. Wu, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS.
- M. Xiao, J. Zhu, L. Ma, Z. Jin, J. Ge, X. Deng, Y. Hou, Q. He, J. Li, Q. Jia, S. Mukerjee, R. Yang, Z. Jiang, D. Su, C. Liu and W. Xing, ACS Catal., 2018, 8, 2824–2832 CrossRef CAS.
- X. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- T. Xu, W. Shen, W. Huang and X. Lu, Mater. Today Nano, 2020, 11, 100081 CrossRef.
- K. Minami, Y. Kasuya, T. Yamazaki, Q. Ji, W. Nakanishi, J. P. Hill, H. Sakai and K. Ariga, Adv. Mater., 2015, 27, 4020–4026 CrossRef CAS.
- Z. Tan, K. Ni, G. Chen, W. Zeng, Z. Tao, M. Ikram, Q. Zhang, H. Wang, L. Sun and X. Zhu, Adv. Mater., 2017, 29, 1603414 CrossRef.
- L. Rincongarcia, A. K. Ismael, C. Evangeli, I. Grace, G. Rubiobollinger, K. Porfyrakis, N. Agrait and C. J. Lambert, Nat. Mater., 2016, 15, 289–293 CrossRef CAS.
- K. Liu, S. Gao, Z. Zheng, X. Deng, S. Mukherjee, S. Wang, H. Xu, J. Wang, J. Liu, T. Zhai and Y. Fang, Adv. Mater., 2019, 31, e1808254 CrossRef.
- J. M. Hawkins, A. Meyer and M. A. Solow, J. Am. Chem. Soc., 1993, 115, 7499–7500 CrossRef CAS.
- K. Choho, W. Langenaeker, G. V. De Woude and P. Geerlings, J. Mol. Struct., 1995, 338, 293–301 CrossRef CAS.
- M. R. Benzigar, S. Joseph, H. Ilbeygi, D. Park, S. Sarkar, G. K. Chandra, S. Umapathy, S. Srinivasan, S. N. Talapaneni and A. Vinu, Angew. Chem., Int. Ed., 2018, 57, 569–573 CrossRef CAS.
- M. R. Benzigar, S. Joseph, A. V. Baskar, D. Park, G. K. Chandra, S. Umapathy, S. N. Talapaneni and A. Vinu, Adv. Funct. Mater., 2018, 28, 1803701 CrossRef.
- Y. Lin and D. S. Su, ACS Nano, 2014, 8, 7823–7833 CrossRef CAS.
- S. Gao, X. Wei, H. Fan, L. Li, K. Geng and J. Wang, Nano Energy, 2015, 13, 518–526 CrossRef CAS.
- X. Chen, J. Chang and Q. Ke, Carbon, 2018, 126, 53–57 CrossRef CAS.
- Y. Wang, M. Jiao, W. Song and Z. Wu, Carbon, 2017, 114, 393–401 CrossRef CAS.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- D. H. Lee, W. J. Lee, W. J. Lee, S. O. Kim and Y.-H. Kim, Phys. Rev. Lett., 2011, 106, 175502 CrossRef.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang and D. Wang, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS.
- X. Li, S. Xi, L. Sun, S. Dou, Z. Huang, T. Su and X. Wang, Adv. Sci., 2020, 7, 2001545 CrossRef.
- F. Callevallejo, J. I. Martinez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- S. Kattel and G. Wang, J. Mater. Chem., 2013, 1, 10790–10797 RSC.
- S. Kattel and G. Wang, J. Phys. Chem. Lett., 2014, 5, 452–456 CrossRef CAS.
- X. Chen, R. Hu and F. Bai, Materials, 2017, 10, 549 CrossRef.
- B. Modak, K. Srinivasu and S. K. Ghosh, Int. J. Hydrogen Energy, 2017, 42, 2278–2287 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- K. Liu, S. Kattel, V. Mao and G. Wang, J. Phys. Chem. C, 2016, 120, 1586–1596 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |