
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microfluidics for core–shell drug carrier particles – a review
Sepideh Yazdian Kashani ,
Amir Afzalian,
Farbod Shirinichi and
Mostafa Keshavarz Moraveji*
,
Amir Afzalian,
Farbod Shirinichi and
Mostafa Keshavarz Moraveji*
Department of Chemical Engineering, Amirkabir University of Technology (Tehran Polytechnic), 1591634311, Tehran, Iran. E-mail: moraveji@aut.ac.ir; Tel: +98 21 64543182
First published on 23rd December 2020
Abstract
Core–shell drug-carrier particles are known for their unique features. Due to the combination of superior properties not exhibited by the individual components, core–shell particles have gained a lot of interest. The structures could integrate core and shell characteristics and properties. These particles were designed for controlled drug release in the desired location. Therefore, the side effects would be minimized. So, these particles' advantages have led to the introduction of new methods and ideas for their fabrication. In the past few years, the generation of drug carrier core–shell particles in microfluidic chips has attracted much attention. This method makes it possible to produce particles at nanometer and micrometer levels of the same shape and size; it usually costs less than other methods. The other advantages of using microfluidic techniques compared to conventional bulk methods are integration capability, reproducibility, and higher efficiency. These advantages have created a positive outlook on this approach. This review gives an overview of the various fluidic concepts that are used to generate microparticles or nanoparticles. Also, an overview of traditional and more recent microfluidic devices and their design and structure for the generation of core–shell particles is given. The unique benefits of the microfluidic technique for core–shell drug carrier particle generation are demonstrated.
1. Introduction
Core–shell particles are a category of particles consisting of two or more distinct layers of material, usually a core and a shell by its name. Different or the same materials with various structures may be used for the core and the shell. This structure offers the features and properties that are not achievable by the core and shell's individual materials, providing a synergistic effect, stabilizing the active particles, and biocompatible properties.1The core could be solid, liquid, or gas. The shell is typically solid, which, depending on the design requirements and the intended application, may be produced using either organic or inorganic materials.2 The core–shell particles can have different core shapes, shell thickness, and surface morphology.3
Over the past few decades, the core–shell particles have been used more frequently in drug delivery, biomedical science, tumor therapy, food and cosmetic industry, medicine, material science, and so forth according to their different properties compared to the bulk materials.2,4,5
Nanoparticles (NPs) synthesis is a challenging process, and due to their usage in the advanced materials, new techniques have been made for these nanoparticles' synthesis.6 The development of characterization techniques has led to the production of structures for these different core–shell nanocomposites.7
Because of these nanoparticles' different chemical and physical properties, classical physics laws would fail to explain these properties. So, some new theoretical models in nanoscales were introduced to better understand these nanomaterials. Also, the use of these nanoscales' materials leads to new advanced technologies and strategies in different fields.8,9
A wide variety of methods, including polymerization, spray drying, solvent evaporation, and self-assembly, have been used to prepare core–shell structures. Among these physical and chemical methods, the controllable generation of monodisperse core–shell microparticles with a narrow size distribution is of great demand. Core–shell microparticle properties such as size, morphology, and structure significantly influence their applications. It has long been a significant challenge to produce core–shell microparticles with the desired size distribution using traditional methods. These methods typically lead to high polydispersity core–shell particles. Also, they have limited control over morphology and poor reproducibility.2
Recently, there are many improvements in making core–shell drug carrier particles due to their specific characteristics. Microfluidics has been developed and is a promising solution for the above problems.2,10
2. Microfluidics
Microfluidics is a science of studying the fluid manipulation on micro/nanoscale, increasing the surface area-to-volume ratio.11 In most microfluidic devices, nanoscale fluids pass through micro-dimensional channels.12 In these microfluidic systems, the pressure and flow must be adjustable.13,14Several kinds of materials, such as inorganic materials, metals, polymers, and plastics, are applicable for microfabrication. Microstructures and semiconductor devices have been fabricated using silicon and glass.15–17 Microfluidic devices are usually fabricated with polydimethylsiloxane (PDMS) polymer, glass, silicone, and polytetrafluoroethylene in the desired shape and dimensions.18–20
PDMS is the most common material used to fabricate microfluidic devices because of the ease of fabrication, optical transparency, gas permeability, low chemical reactivity, and inexpensiveness.17,21,22 It is also used in bioassays and targeted drug delivery systems (DDS) using implantable microfluidic devices.23 The microstructure is generally fabricated by soft lithography.24
Glass capillary microfluidic devices are typically made by pulling capillaries through a fine tip with an accurate size orifice, co-axially assembling them inside a wider square or circular capillary, and eventually gluing such capillaries onto a glass slide.20 There are a few drawbacks, considering the simplicity of glass-based microfluidics. First, the device geometry is limited, and device fabrication's technicality can be complicated, making reproducibility of devices a problem.25
Microfluidics is an interdisciplinary science that can be used in many scientific fields, including the pharmaceutical industry, biomedical and chemical sensors, drug delivery, cell growth, and the food industry.26
The main advantages of using microfluidic systems are high efficiency, integration capability, mass production, reduced response time, and parallel operations. These mentioned properties show the importance and breadth of research in the field of microfluidic.27–29 Fig. 1 shows the motivations for using microfluidic devices.
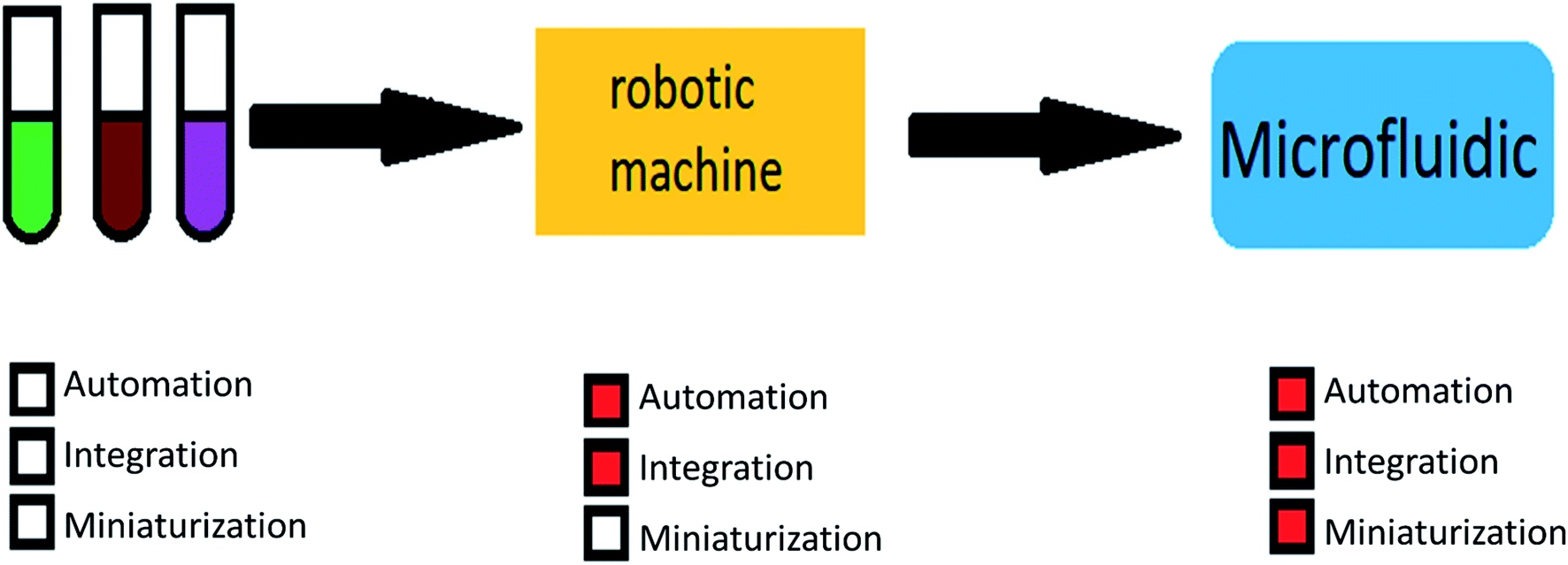 | ||
| Fig. 1 The motivation for using microfluidic devices. | ||
The presence of individual structures inside the microfluidic device, such as ducts, valves, mixers, and pumps, enables the device to allow one or more types of fluid to enter; move along the channels; if necessary, store them in a part of the chip for a while; mix them to create a specific reaction. Eventually, the original products and wastes are transferred to the outside of the device by the outputs. Also, it is worth mentioning that microfluidic systems made up of these components usually are not more than a few centimeters in size.13,23,30,31 All these processes can be monitored by various monitoring methods, such as optical and ultraviolet microscopes.32,33 In addition, the physical and chemical properties of fluids in small volumes and within capillary tubes differ from their macro-scale properties.34,35 In many cases, this makes it easier to work with fluids in this volume. These properties are also widely used to design chips and perform specific functions such as moving fluid inside a channel or mixing fluids.9,36
There is a lot of research and diagnostic tests in biology, chemistry, and medicine in which samples and soluble substances can be evaluated, so microfluidic devices have a wide range of applications in these areas. In microfluidic devices, each part of the chip can act as a part of the laboratory, and so these lab chips are called lab-on-chip.37,38
A lab-on-chip is a device used instead of a laboratory process, usually on a scale of square centimeters or even millimeters in size (Fig. 2). It will do everything have to be done in a laboratory on a small device called a chip, which has become more useful in recent years.39,40
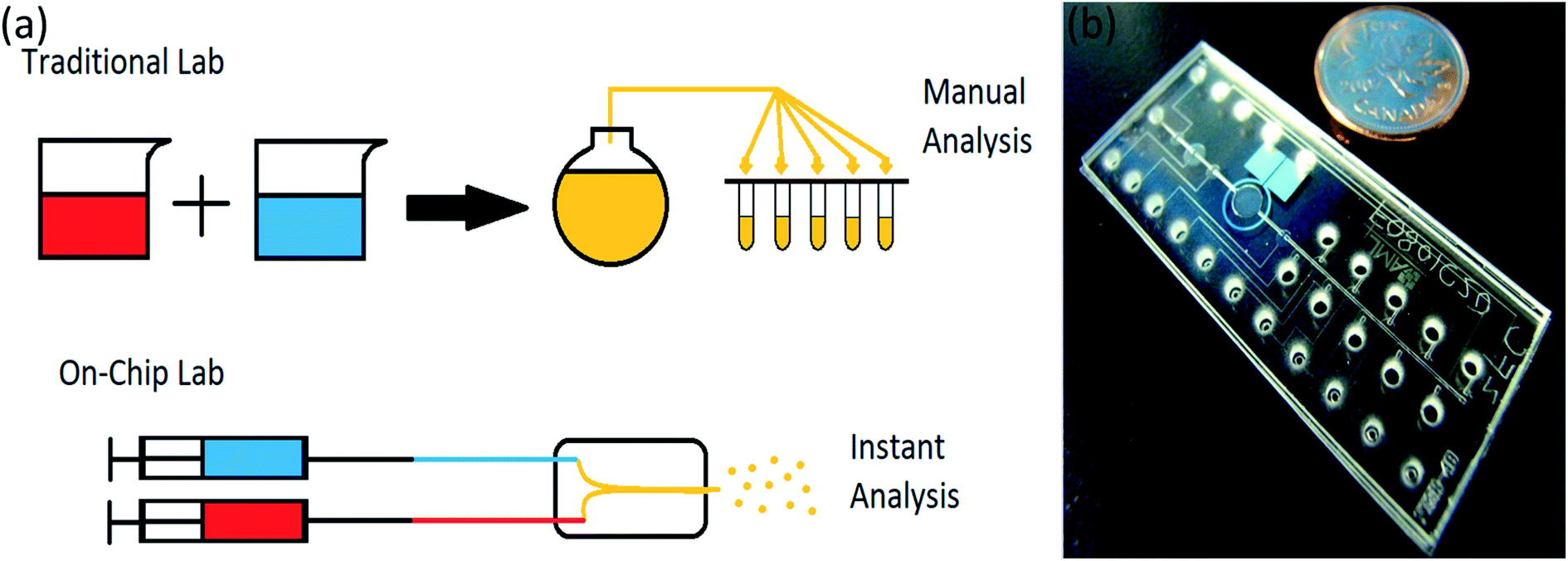 | ||
| Fig. 2 (a) Lab-on-chip in comparison with traditional lab, (b) a microfluidic chip compare to a coin, adapted from ref. 41. | ||
Another factor that has led to the use of lab-on-chips devices in recent years is the use of small amounts of fluid in these chips, which reduces costs and time of process.42,43 Also, the accuracy of these chips is very high. Because it is a continuous process and the operator is not involved in them, it causes less pollution to enter them and drastically reduces human errors due to high accuracy. Also, short execution time has gained a lot of attention.10,44,45
3. Drug delivery
Pharmaceutical systems use targeted transfer technology or control the release of therapeutic agents.46 The development of appropriate drug carriers in biomedical applications to reduce the side effects is desirable and has beneficial therapeutic effects.47–49 Nanoparticles are essential as drug carriers because of their ability to transport various types of drugs to different parts of the body and release them at the right time.50,51Drug delivery is the mechanism or process of delivering a pharmaceutical substance that involves releasing a bioactive agent at the optimal and required rate.51,52
Cancer is a leading cause of death, and despite tremendous efforts to fight cancer and the existence of multiple therapy modalities, cancer treatment still is a significant problem.53–55 Chemotherapy is a widely used treatment for cancers, and this has led to trials for a considerable number of chemotherapeutic anticancer drugs. The biggest drawback to the clinical use of these drugs is their broad bio-distribution and short half-life. The major disadvantage of traditional drug delivery methods is their weak selectivity. Also, healthy cells are exposed to drugs' cytotoxic effects, and an inadequate portion of the applied drug arrives at the tumor position in most cases.5,54–56 In addition, there is a need for high drug dosage, and it is another unwanted side effect. So, new drug delivery technologies need to be promoted to solve these limitations and improve cancer therapies' potency.5,54,57
Recent drug delivery approaches deliver the drug to the tumor position and reduce side effects. Different types of drug delivery methods are developed for multiple healing applications. Nanoparticles are one of the most widely used carriers that, due to their ability to achieve therapeutic targets at acceptable times and doses, have a great interest in their potential in drug delivery.5,56,58
4. Core–shell particles
Core–shell particles were initially introduced to make production methods in the field of biotechnology more effective. Nowadays, with the advent of encapsulation technologies using core–shell particles, medicines can stay active for longer periods of time.59,60 They protect the drug against harsh conditions and provide the possibility of release under specific temperature or pH conditions; that's why these micro and nano-capsulation methods have gained more attention.60–62Core-shelling can be defined as a process for trapping one substance, usually the drug, in a shell as a physical barrier to protect the core from adverse factors and conditions. The application of this method in the pharmaceutical industry has been increased recently. Also, microcapsule methods are widely used today. The application of microcapsules and nano-capsules has become very important in the pharmaceutical industry in the past few years. Therefore, a lot of research has been done in this specific field.63,64 The core-shelling method's properties include protection against moisture, heat, ultraviolet radiation, oxygen, and adverse conditions. Using these methods usually causes the drug to act under certain physical or chemical conditions, reducing drug usage. Also, taking these kinds of medication may reduce the drug's side effects and increase its effectiveness.65,66
Some types of core–shells particles are shown in Fig. 3. Different colors are used for the core and the shell. The core may be a single sphere (Fig. 3a) or have a hollow shell with a small sphere inside, a rattle-like or yolk–shell structure (Fig. 3b). It is also possible to have an aggregation of several small spheres (Fig. 3c). The shell structure can be a continuous layer (Fig. 3a–c) or attachment of smaller spheres onto a big core sphere (Fig. 3d and e) or aggregated core spheres (Fig. 3f).1
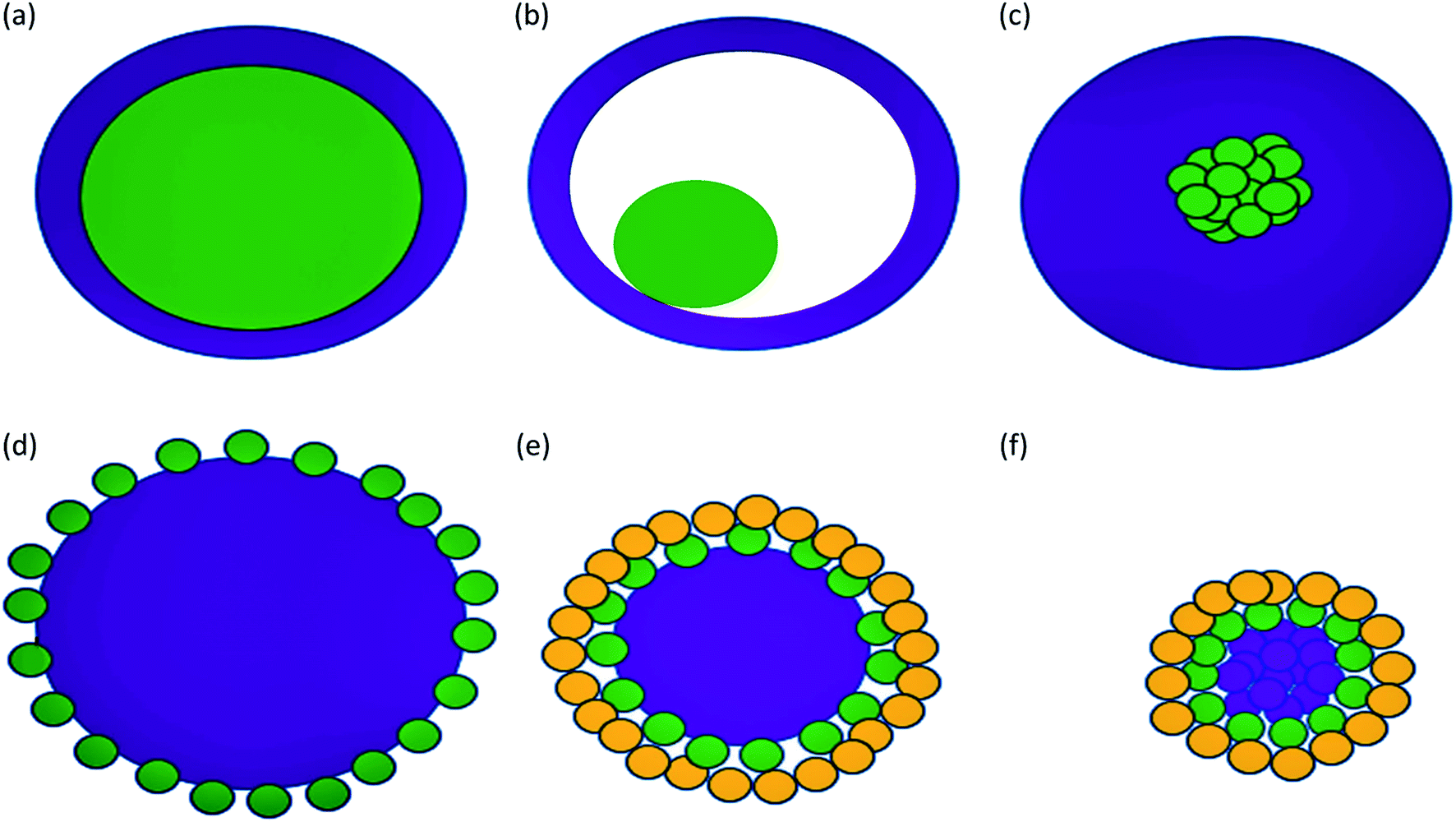 | ||
| Fig. 3 Some types of core–shell particles. The continuous shell structure (a–c): (a) single sphere core, (b) hollow shell with a small sphere inside, (c) core with the aggregation of several small spheres. (d–e) Shell with the attachment of smaller spheres onto a big core sphere. (f) Shell with the attachment of smaller spheres onto aggregated core spheres. | ||
4.1 Goals of using core–shell drug carrier particles
The first type referred to the release of active compounds with the delay in the desired location. There are many examples of delayed-release core–shell particles like encapsulated probiotics and tablets protected in the stomach against gastric acid by encapsulating them and releasing them in the small or large intestine.72,73 The other example of the delayed-release is using core–shell nanoparticles prepared using microfluidic chips for oral delivery of chemotherapeutics for colon cancer. They would only be released in the colon.54
The second type is a stable release, a mechanism designed to keep the release rate of compounds constant at the desired location. Gradually and evenly, the drug exits the shell and enters the body. The sweeteners can be encapsulated in chewing gum, which keeps the taste steady for a while.74
The appropriate selection of shell material for therapeutic delivery can improve controlled release, preservation, and responsiveness to stimuli.2
4.2 Core–shell drug carriers release methods
As mentioned in Section 4.1.2, core–shell particles could provide a controlled release of the drug. The shell may allow the core of which drug is in it, sustained-release at a steady rate, and avoid sudden release of the drug at specific parts in the human body.2,81
One of the most frequently employed methods to encapsulate, deliver, and release active ingredients in a sustained manner, is to trap active ingredients in polymer matrices in the shape of microspheres with a tunable degradation rate. Polylactic-co-glycolic acid (PLGA) is a biocompatible polymer with a degradation rate that can be tuned and is commonly used for the controlled release of drugs. However, according to the hydrophobic nature of PLGA, single PLGA microspheres are usually constrained by a low loading efficiency for hydrophilic active agents, and it remains difficult to customize their release kinetics and prevent undesired release patterns. These problems can be solved using composite microspheres with complex structures, such as core–shell composite microspheres.82
Deshpande et al.83 made core–shell nanogels with PNIPMAM shell and gold nanoparticles as the core for sustained and triggered release of doxorubicin (DOX).
Microparticles with a polymeric matrix with uniform size can release the encapsulated drugs more predictably. In addition, the formation of the microparticles with the porous matrix can provide more permeable sustained-release structures. In contrast, microparticle engineering with core–shell structures facilitated improved preservation. It decreased burst release, which is useful for the long-term treatment of several diseases such as asthma, angina, and psychiatric disorders.65
An encapsulated atorvastatin loaded porous silicon (PSi) NPs with a reactive oxygen species (ROS) was applied for diabetic wound healing.84 As it was mentioned, the polymeric shell formation can solve burst payload release, which is the main barrier for porous materials. The release kinetics can be easily adjusted by the shell material's choice, as the release of atorvastatin can be stimulated only by the coexistence of overproduced ROS. The release rate can be sustained for more than 24 h, making the core materials more appropriate for predicted biomedical applications.11
Core–shell microparticles can be obtained through evaporation of the oil from the middle layer and consolidation of the shell materials using water-in-oil-in-water (W/O/W) double emulsion with the middle oil phase containing biodegradable shell materials like PLA and PLGA as templates. For example, the PLA shell of the microparticles gradually degrades due to the hydrolysis of ester groups in the PLA chain, allowing the sustained-release of contents in the inner aqueous core.65
A DLPC shell and PLGA core were developed by Liu et al.85 for the sustained and controlled release of anticancer drugs with paclitaxel as a model drug. Paclitaxel's typically controlled release profile enables the use of NPs for the delivery of anticancer drugs. Sustained-release includes the ability to consistently fight cancer cells, resulting in a decline in cancer cells' viability.
Thi et al.81 showed that the sustained-release of DOX-loaded SPION@HP continuously lasted at a steady rate without an initial burst release up to 120 h, thereby retaining the therapeutic level of therapeutics for treatment over a long time.
4.3 Core–shell materials
The core part can be gas, liquid, or solid, and the shell is usually solid, but its nature depends on the targeted application.86 Table 1 shows some examples of core–shell drug carrier materials, their base, the loaded drug, and their drug delivery application.| Core | Shell | Base | Loaded drug | Application | Ref. |
|---|---|---|---|---|---|
| a MSN = mesoporous silica-based nanoparticle. | |||||
| Aqueous solution | Lipid | Polymer | Doxorubicin hydrochloride and paclitaxel, anticancer drugs | Simultaneous encapsulation of synergistic actives | 89 |
| Cholesterol | Chitosan | Polymer | Paclitaxel, an anticancer drug | Encapsulation of anticancer drug for drug delivery system | 112 |
| Ferrite impregnated acrylonitrile | Acrylamide | Polymer | Naproxen, a non-steroidal anti-inflammatory drug, and trimethoprim, a bacteriostatic antibiotic | Targeted drug delivery | 113 |
| PLLA | PLGA | Polymer | Paclitaxel and suramin, anticancer drugs | Sequential and parallel drug release | 114 |
| PLGA | DLPC | Polymer | Paclitaxel, an anticancer drug | Controlled drug release | 85 |
| Oxidized sodium alginate | Chitosan | Polymer | 5-Aminosalicylic acid (5-ASA), a model drug which is rapidly absorbed before entering the colon | Development of a colon-specific delivery | 115 |
| Pectin | Alginate | Polymer | Piroxicam (PRX) as a model non-steroidal anti-inflammatory drug (NSAID) | Delayed drug delivery | 116 |
| PMMA | PEI | Polymer | Ibuprofen (IB) | Intracellular drug delivery | 117 |
| PLGA | Casein | Polymer | Paclitaxel (Ptx) and epigallocatechin gallate (EGCG), anticancer drugs | Dual-drug-loaded nanomedicine | 118 |
| PLGA | Alginate | Polymer | Rifampicin | Controlled drug release | 119 |
| Fe2O3 | MSN | Silica | Fluorescein sodium, an anticancer drug | Magnetically triggered multidrug release | 120 |
| AuNRs | MSN | Silica | Doxorubicin (DOX), an anticancer drug | Targeted drug delivery to cancer cells | 121 |
| PEG | MSN | Silica | GSI (γ-secretase inhibitor) | Targeted inhibition of notch signaling in breast cancer | 122 |
| Pd–Ag | MSN | Silica | Doxorubicin (DOX), an anticancer drug | Photo- and pH-triggered release of anticancer drugs | 68 |
| Au | MSN | Silica | Rhodamine B, an anticancer cargo | Controlled cargo release activated by plasmonic heating | 123 |
| UCNPs–SiO2 | MSN | Silica | Doxorubicin (DOX), an anticancer drug | NIR-triggered anticancer drug delivery | 124 |
| Ag | Poly(N-isopropylacrylamide-co-acrylic acid) | Metal and metal oxide | Dipyridamole (DIP), an anticancer drug | pH-regulated drug delivery | 125 |
| Au | PEG | Metal and metal oxide | Temozolomide (TMZ), an anticancer drug | Optical temperature-sensing, targeted tumor cell imaging, and combined chemo-photothermal treatment | 126 |
| Ag | TiO2 | Metal and metal oxide | Doxorubicin (DOX) and LET, anticancer drugs | Biological applications such as drug delivery | 127 |
| Fe3O4 | Chitosan | Metal and metal oxide | Curcumin (Cur), an autofluorescent dye as well as an anti-tumor drug | Multimodal monitoring and drug targeting | 128 |
| Ag@SiO2 | mTiO2 | Metal and metal oxide | Doxorubicin (DOX), an anticancer drug | Simultaneous fluorescence-SERS bimodal imaging and drug delivery | 129 |
| Fe3O4 | PMAA | Metal and metal oxide | Ceftriaxone sodium (CTX), an anti-inflammatory drug | pH-controlled drug delivery | 130 |
Hollow microparticles have attracted increasing attention because of their specific properties such as higher surface area, lower density, and certain superior optical properties than bulk materials.87 Hollow internal microspheres are possibly used as an encapsulation vehicle to secure biologically active compounds such as proteins, enzymes, and DNA in controlled drug release.2
Core–shell microcapsules with an aqueous core could be applied to encapsulate and protect incompatible substances or active ingredients and drug delivery.88 Microparticles with an aqueous core and an oily core have appropriate space for hydrophilic and hydrophobic materials to be encapsulated in them, respectively.65 As can be seen in Table 1, the aqueous core can encapsulate a hydrophilic anticancer drug (doxorubicin hydrochloride). Simultaneously, the solid shell of it can encapsulate a hydrophobic anticancer drug (paclitaxel).89
Different materials, such as metal, metal oxides, silica, polystyrene, and polymers, could form the solid core based on their applications and production processes. One approach for producing a solid core core–shell particle is to the application of a hardcore template. Another approach is to generate core–shell particles directly by converting the emulsion droplets into solid core–shell particles, with a solid core and a solid shell. Solidification techniques include solvent evaporation, polymerization, and ionic crosslinking. A solid core structure coated with a shell layer has a great advantage for synergistic and regulated drug delivery. Although a burst release of drug is possible in a core–shell particle with a liquid core and a solid shell after the shell's breakage or degradation, the solid core structure solves this problem because it can release from the carriers only after the degradation of polymer layers.
Shell materials are commonly classified as organic and inorganic groups. There are many available materials for core and shell fabrication, and these materials specify the physical, chemical, and biological properties of them. This high versatility in the choice of core and shell materials enables core–shell microparticles with different functionalities and properties to be prepared. The shell materials can be chosen according to the application of core–shell particles. The shell also protects the core's chemically active components against corrosion, oxidative degradation, and erosion. Also, shell materials offer increased thermal stability and enhance the microparticles' electrical, optical, and magnetic properties.2
In the following, the most commonly used core and shell materials will be described.
There are various physically rigid polymeric materials such as polystyrene, PLGA, and isotactic polypropylene (iPP) used to produce solid core particles.2 As mentioned in the Section 4.2.3, PLGA has proved a useful polymer for drug delivery systems because of its high biocompatibility, biodegradability, wide range of erosion periods, and providing sustained-release of drug.82,93 Lukyanova et al.94 presented two microfluidic routes for producing solid-core/solid-shell particles. Poly(methyl methacrylate) (PMMA) was used as the solid core and encapsulated in the shell using a microfluidics device. Also, ethylene glycol dimethacrylate monomer was polymerized under UV light used to generate a rigid core.
In high-performance liquid chromatography, solid silica-core/porous-shell particles could be used for the separation with a high flow rate and relatively low backpressure. The small solid core protected by the porous shell results in a larger particle and a higher surface area, resulting in lower backpressure for the separation.1,97
Compared to polymer shells, a metal shell such as zeolite, titanium, and gold acts as a more powerful barrier and prevents small molecules' undesired release into the core. Furthermore, since inorganic materials' thermal conductivity is greater than that of polymers, microparticles' thermal conductivity can be greatly enhanced by inorganic additives such as metals in the shell. There may also be other special characteristics of these materials, such as magnetic properties.2
Metal and metal oxides are mostly used as cores and for drug delivery. Because with the help of a magnetic field, the drug can be easily transported to the desired location. Various metals and their oxides nanomaterials are unsuitable and toxic to the human body. However, MnO, TiO2, and ZnO nanoparticles have been considered in drug delivery.5,98–100
4.4 Materials for improving core–shell drug carriers
The core–shell drug carrier particles produced using magnetic nanoparticles could be led to the specific neighborhoods for releasing the drug in the body using an external magnetic field. For example, after nanocarriers' injection in magnetically induced systems, an extracorporeal magnetic field is used to concentrate drug-loaded nanocarriers at tumor sites. Some suitable magnetic stimulation candidates are structured core–shell nanoparticles coated with silica, polymer, or magnetoliposome (maghemite nanocrystals encapsulated in liposomes).103
Due to magnetic nanoparticles' instability in aqueous solutions, they cannot be used alone as drug carriers. A practical method to eliminate or minimize this problem is to use some coatings. Pharmaceutical magnetic nanoparticles with a magnetic core should be stabilized because these magnetic materials should be kept stable to use for drug delivery purposes.104 The magnetic core is used to release the drug to a specific location, and the polymer is used to load, transfer, and dispense the drug. In general, magnetic nanoparticles have high chemical activity and are easily oxidized in the presence of air.104–106 As a result, their magnetic properties will be lost. When these magnetic particles are coated with a suitable shell, they are protected from oxidation, which leads to a reduction in toxicity, and aggregation of these materials would be minimized. Also, the coating can enhance the stability of these magnetic drug carrier particles.107 In addition to protecting and stabilizing magnetic particles, suitable coatings can be used as a surface agent to make them more functional due to the presence of amino, hydroxyl, and carboxylate groups. One of the best coatings for this method is chitosan. Chitosan nanoparticles are one of the most promising protective coatings for magnetic nanoparticles due to their unique properties. As shown in Fig. 4, a multi-core or single-core magnetic structure can be used for chitosan nanoparticles.108
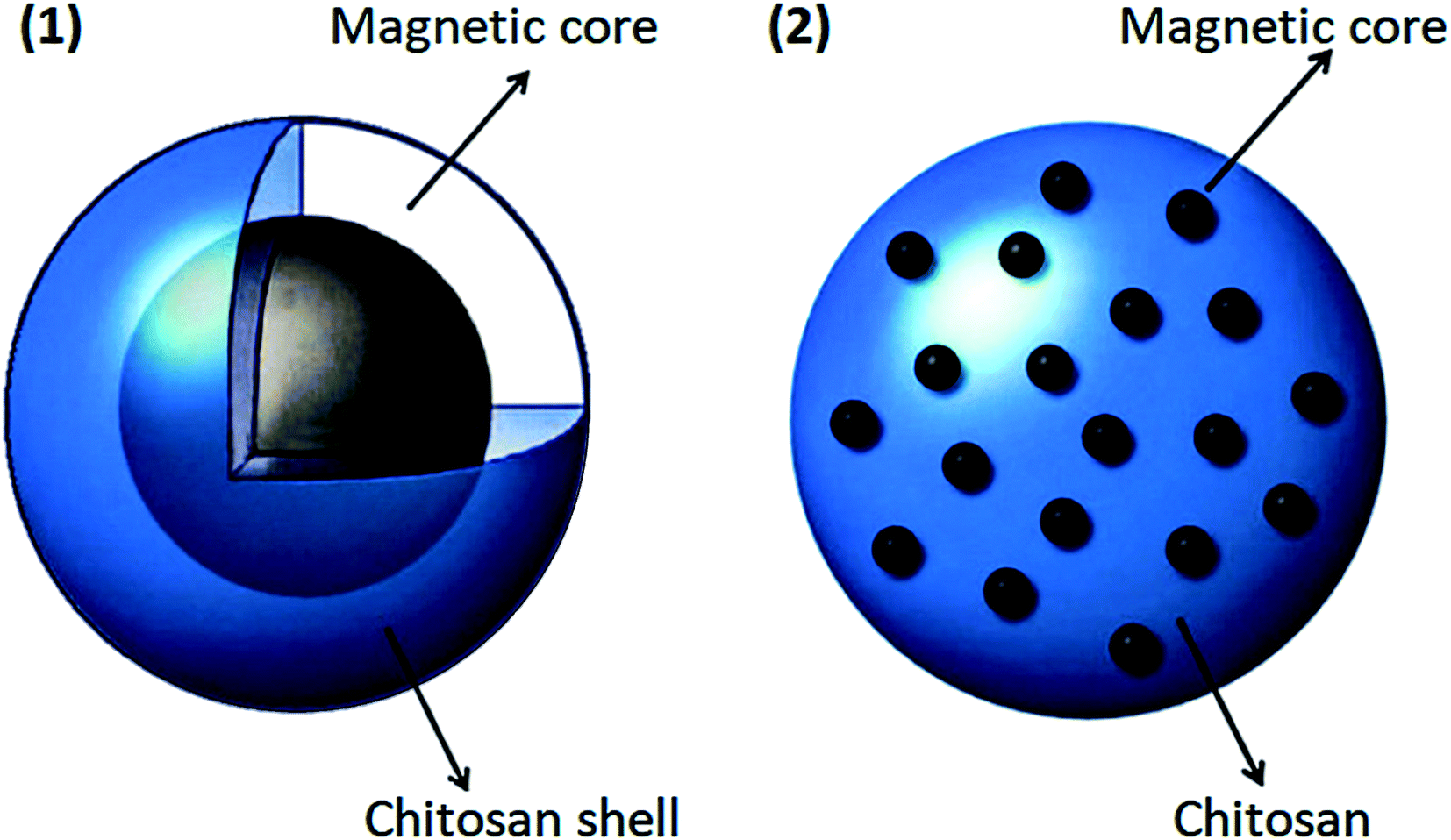 | ||
| Fig. 4 Two types of coating magnetic core in chitosan, this figure has been adapted from ref. 108 with permission from Elsevier B.V., copyright 2013. | ||
Silver nanoparticles have antibacterial properties, and this feature is the basis of their application.110 Mixing silver nanoparticles with polymer and making nanocomposites is one of their most-used application method.5,111
Silver nanoparticles with anti-fungal and anti-inflammatory effects are environmentally friendly, non-thermal, heat-resistant, and highly corrosion-resistant. As an example, in 2015, hydrogel granules of chitosan/silver nanocomposite were produced in the presence of NaBH4 as a reducing agent.5,111
Gold nanomaterials have attracted a great deal of attention in biomedical applications due to their high biocompatibility, low toxicity, and relatively low reactivity. They tend to accumulate in various forms of rods and prisms during synthesis. They have significant applications as sensors, solar cells, and in the field of pharmaceutical and tissue engineering. Gold nanoparticles and chitosan bound to glycolic acid were reported to be synthesized for pharmaceutical and engineering applications.131,132
While copper and its compounds have antibacterial properties already known in ancient times, they currently receive renewed attention due to copper's possible use in healthcare situations as an antibacterial material.133
Compared to other metals, copper is less toxic to bio cells and has a higher potency than micronutrients. Although copper nanoparticles are known for their unique characteristics, there is not much research on these nanoparticles. Anticancer activity of various metals such as Cu, Si, Se, Zn, Ag has been reported. In the meantime, copper has become more widespread due to its unique and excellent electronic configuration. Participation as a significant factor in the oxidation–reduction cycle of enzymes is one of copper's interesting properties. The anticancer role of copper compounds such as CuO, CuS has been reported in some research.5,131,132
The antibacterial effect of novel core–shell nanostructures based on copper and silver metals against Escherichia coli (E. coli) has been investigated. These nanostructures were prepared separately using the non-toxic, biodegradable, and biocompatible biopolymers chitosan and guar gum–polyvinyl alcohol (GG–PVA). A well-diffusion approach against E. coli analyzed the antibacterial property of the core–shell nanostructures. Due to the high ratio of NZVCu in the nanostructure, Cu/CuO@SiO2 nanostructures are very effective against E. coli.134
Also, recently, Fe3O4@copper(II) metal–organic framework Cu3(BTC)2 (Cu-BTC) as core–shell structured magnetic microspheres were investigated. The slowly released copper ions and improved production of reactive oxygen species (ROS) played a role in Fe3O4@Cu-BTC antibacterial activity by promoting the successful isolation and transfer of photoexcited electron–hole pairs.135
Meanwhile, chitosan polymer is a naturally occurring cationic polycarbonate with better biocompatibility and degradability properties than other cationic polymers. Compared to graphene oxide (GO), graphene oxide–chitosan (GO–CS) nanocomposites are smaller in size, positively charged, and less toxic. As an example, it was reported that the CpG oligodeoxynucleotides (ODNs) delivery system based on GO–CS nanocomposites significantly improved the loading capacity and cellular uptake. Therefore to increase the delivery efficiency of CpG ODNs, GO–CS nanocomposites will serve as efficient nanocarriers.140,141
In another research, the use of amine-containing core–shell nanoparticles has been studied as possible drug carriers for intracellular delivery. Thick poly(ethyleneimine) PEI shells (approximately 30 nm) greatly improved the drug loading potential of the complexed nanoparticle up to 23% (w/w).117
Also, silanes are available by different amine groups and can improve the functionality of the magnetite nanoparticle surface for protein conjugation. Therefore, silane-coated MNPs result in high-quality materials for magnetic drug delivery systems.5
5. Microfluidics devices in drug delivery systems
Microparticles with controllable structures are needed to be capable of quantitative encapsulation of drugs and regulated release of them to the desired location to ensure reduced side effects and optimized therapeutic efficacy. The traditional delivery methods, including oral, intravenous, sublingual, and intramuscular drug deliveries, have drawbacks, such as interactions with foods, low solubility and permeability, and irregular absorption, making steady-state dosing difficult to achieve in patients. Many drugs are also toxic to normal tissues or toxic when overdosed, but they are useless when underdosed. Consequently, control of the amount of encapsulation and drug release rate is needed. The fabrication of uniform microparticles with controllable and flexible sizes, compositions, and internal structures is needed to fulfill these demands.65,144 Most traditional bulk drug synthesis methods suffer from many drawbacks, such as the need to use a high quantity of valuable drugs or chemicals, the generation of polydisperse particles that influence the release profile, the limitation of the generation of multiple therapeutic agent-loaded carriers, and the difficulty associated with locating the delivery of drugs and in vivo investigation of the therapeutic or toxic effects that needs many animals.50New technologies, like microfluidics, can solve these problems. For the production of effective drug carrier particles, microfluidic devices offer specific advantages. Compared to bulk methods, microfluidic technology allows the production of highly stable, uniform, monodispersed particles with higher encapsulation efficiency by effectively regulating the fabricated chip's geometries and the flow rates of multiphase fluids.50,144
In traditional bulk synthesis methods, both the inertial and viscous effects govern mass transport in fluids, associated with nonlinearities that give rise to numerous instabilities, such as turbulence. In contrast, the inertial effect becomes negligible in microfluidics. This feature allows microfluidics to synthesize nanoparticles in a highly regulated and reproducible manner that has been difficult to achieve in traditional macroscale methods.68,123,145 Microfluidics-templated emulsions facilitate the controlled drug release by producing highly uniform microparticles with well-controlled sizes, shapes, and compositions.65
Microfluidics can be used for polymer synthesis with precise forms or chemicals for drug delivery application. For example, a technique developed by Nie et al.146 to use the capillary instability-driven break-up of a liquid jet made up of two immiscible fluids for producing polymer particles with various shapes and morphologies. The stated strategy allows the emulsification process to be precisely controlled, leading to monodispersed droplets with controlled morphologies ranging from 20 to 200 μm in size.144
In addition, some conventional delivery methods, including painful and harmful injections, can benefit from microscale technologies by fabrication of microneedles or needle-free injection devices. In order to improve the comfort and quality of life of patients, microfluidic systems have recently been developed for transdermal administration of drugs.51
Also, microfluidics' drug delivery systems have improved drug encapsulation efficiency, allowing the use of two or more drug molecules in the same carrier for combination therapy or dual function.12
Microfluidic systems can be used for the direct delivery of active molecules, in addition to the potential of producing complex drug carriers.147 In order to maximize the local availability of the drug and reduce the side effects induced by the drug's interaction with other organs and tissues, such systems are capable of efficiently transporting drugs to a targeted location. Furthermore, for so-called transdermal delivery, which is direct drug delivery through the skin, microfluidic systems have been successfully used. These systems, which utilize a needle or an array of microneedles, transfer the drug across the skin (epidermis) barrier.51
Recent advances in developing and utilizing such platforms for drug delivery systems have been discussed elsewhere12,20,49–51,65,144,145 are not reviewed here. In the following section, we will focus on core–shell drug carrier particles production in microfluidic devices.
In addition to basic solid microparticles, microfluidic devices offer a flexible approach to producing more complex functional microparticles with core–shell particles with multicompartmental structures for co-encapsulation and synergistic release of specific drugs. Also, they provide highly efficient encapsulation and regulated release of hydrophilic or hydrophobic drugs and. For biomedical and pharmaceutical applications, diverse controlled release types, such as sustained-release, triggered release, and combined release of both release styles can be accomplished based on their advanced shell functions and internal structures.65
By their name, core–shell particles are a category of particles that contain a core and a shell. Different materials or the same materials with different structures can be used as the core and the shell. Due to the combination of superior properties not exhibited by the individual components, core–shell particles have gained a lot of interest. The structures could integrate core and shell characteristics and properties, where the surface properties of the shell are passed to the core, bringing new features to the core–shell particles.86
Core–shell particles are typically synthesized by a two-step or multi-step process. First, the core particles are synthesized, and, based on the type of core and shell materials and their morphologies, the shell is then formed on the core particle through various methods.1 In the following section, we will describe microfluidic devices' application for core–shell drug carrier particle production and discuss various microfluidic systems that have been utilized for core–shell drug carrier particle preparation, characterization, and application.
6. Microfluidic devices for core–shell drug carrier particles preparation, characterization, and application
Microfluidics' adaptation to the development of micro-sized structures has become more important since the first theoretical work was carried out more than 10 years ago. The primary reasons for this technology's adoption originate from the emulsion homogeneity and the high degree of control over the operation. Because of the fluids' properties in the microfluidic channels, control over the entire production chain is possible. Therefore, these systems might be used for the production of single or double emulsions.11,12,20,148In recent years, a lot of research has been done about the application of microfluidic devices for making core–shell particles used in drug delivery. It is possible to use only a small proportion of samples in these microstructures, and controlled composition is one of the other significant advantages of this method. In microfluidic devices, drug-loaded particles' volume and permeability can be controlled more straightforwardly than other methods, making microfluidics more suitable for the fabrication of pharmaceutical drug carrier particles. So far, many microfluidic chips have been designed for the formation of core–shell nanoparticles drug carriers.24,32,63,142
The microfluidic method significantly reduces material costs and improves drug encapsulation efficiency. The most important advantage of using microfluidic devices is uniform core–shell droplets size. The polydispersity index (PDI) of these particles is usually less than 1%, which can be adjusted by the fluids' physical properties.20 Core–shell particles for drug delivery application are mainly fabricated by droplet microfluidics.12,20,49–51,65,144,145
6.1 Droplet microfluidics for core–shell drug carrier particles preparation
Droplet microfluidics has been increasingly developed in recent years. It allows precise control of droplet creation, encapsulations, and release kinetics using nonlinear channel geometries, such as T-junctions, co-flowing, and flow-focusing constrictions.149 A single emulsion is a droplet of one liquid dispersed in an immiscible fluid that forms the continuous phase.25 The common single emulsion includes oil-in-water (O/W) and water-in-oil (W/O) emulsions.20It is complicated to make core–shell droplets made of more than one immiscible liquid by other possible methods. However, it is possible to make these multi emulsion core–shell droplets more easily in the droplet-based microfluidic devices.5,150,151 The droplets can be generated from a single emulsion of two partially miscible or immiscible fluids or multiple emulsions (mostly the double emulsion) of three or more immiscible fluids.11,12,149 The single emulsion does not guarantee that multiple therapeutics are loaded simultaneously, and it is more complicated where the payloads have different solubility. Thus in drug delivery systems, double emulsions are also commonly used.20
Usually, if multi emulsion core–shell droplets are needed, a sequential process of producing a single emulsion should be continued to place the primary emulsified droplet in the secondary droplet.49,152 Double and multiple emulsions are frequently produced by a combination of two or more flow geometries.5 The most common geometries in 2D are T- and Y-junctions, while for the 3D design, flow-focusing, co-flow, and various combinations of the previous designs used for the production of double emulsions.11
The vital point in droplet-based microfluidics is that the two liquids must either be partially miscible or immiscible. If they are miscible in each other, the core–shell droplets that will carry the drug cannot be produced.
Fig. 5 shows different common geometries of droplet-based microfluidic devices for producing single and double emulsion.
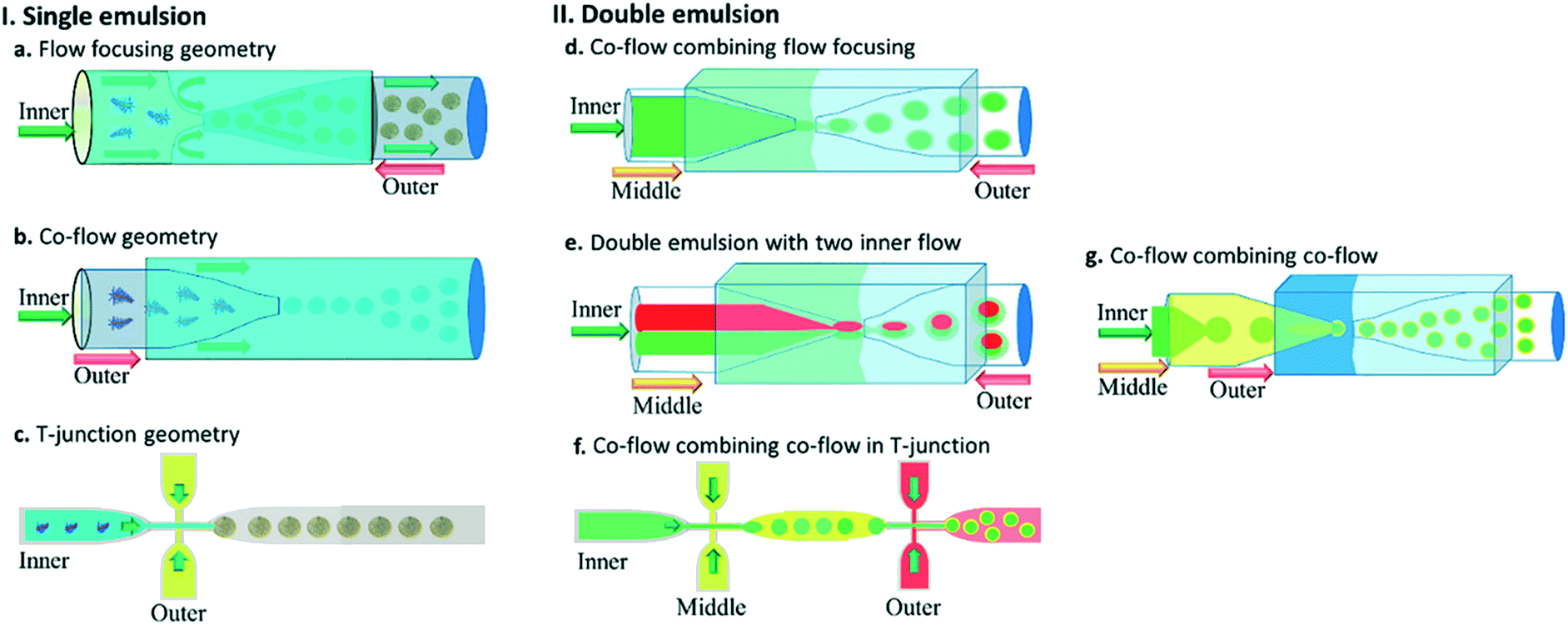 | ||
| Fig. 5 Different geometries of droplet-based microfluidic devices, this figure has been reproduced from ref. 20 with permission from Royal Society of Chemistry, copyright 2017. (a–c) The geometries for the preparation of single emulsions, (a) flow-focusing, (b) co-flow, and (c) T-junction (d–g) complex geometries to fabricate double emulsions, (d) co-flow combined with flow-focusing with one inner fluid, (e) co-flow combined with flow-focusing with two inner fluid (f) sequential T-junction, and (g) sequential co-flows for thin shell capsule production. | ||
We can classify microfluidic methods into single-step and sequential methods for double or multiple emulsion particle fabrications despite the different geometries of droplet microfluidic devices. First, we will introduce important dimensionless numbers associated with droplet-microfluidics. Then we will describe different geometries of droplet-based microfluidics, and then we will discuss single-step and sequential methods for core–shell particle preparation.
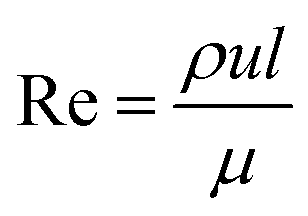 | (1) |
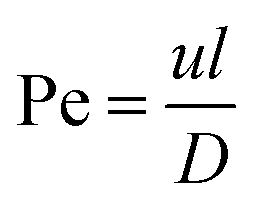 | (2) |
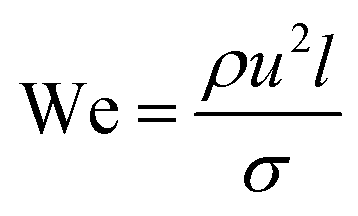 | (3) |
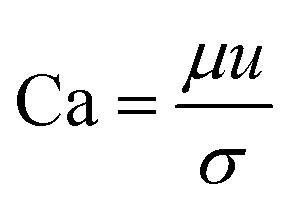 | (4) |
Reynolds number and Pe reflect the fluid flow characteristics and molecules' action within the fluid.20 The Reynolds number describes the relationship between inertial forces and viscous forces. A flow pattern of different streamlines that are parallel to the fluid direction (Fig. 6a) is reflected in the laminar flow (usually Re number < 1800). On the other hand, the fluid flow with a high Reynolds number (usually >2300) is categorized as turbulent flow, providing a chaotic pattern without distinct streamlines (Fig. 6b). Steady streamlines characterize the laminar flow, so the fluids and molecules within the fluids can be accurately manipulated to produce controllable and monodisperse droplets, reflecting the desirable characteristics of drug encapsulation in droplet microfluidics.11,20
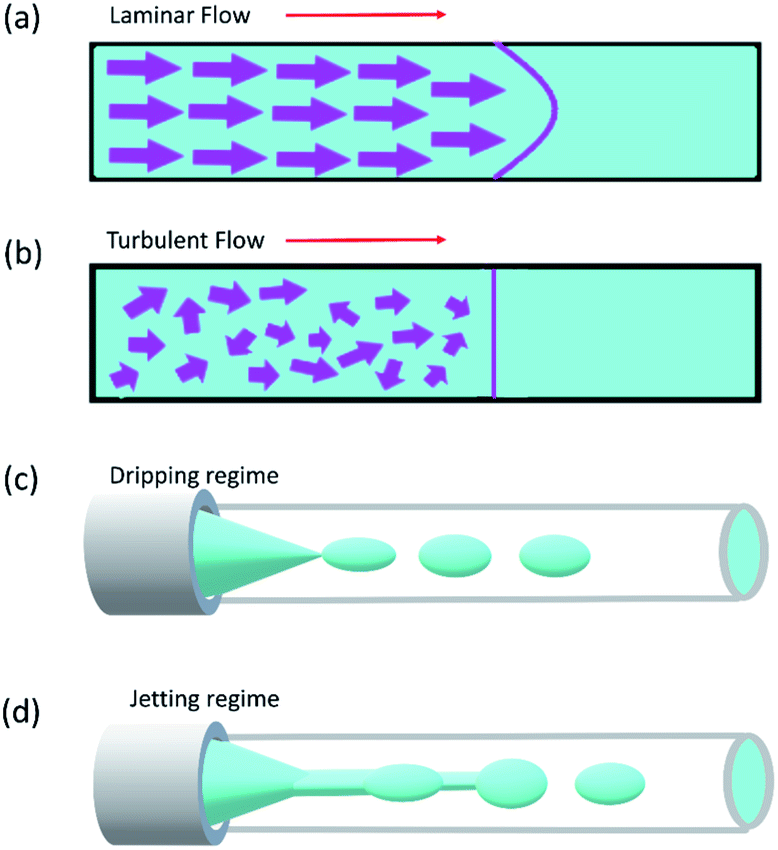 | ||
| Fig. 6 (a) Laminar flow, (b) turbulent flow, (c) dripping regime, and (d) Jetting regime. | ||
The Péclet number is a commonly applied dimensionless number for mass transfer processes, reflects the diffusion or convection of molecules in the fluids.11,20 Because of the small volumes and the laminar flow pattern, the transfer of molecules is slow in droplet microfluidics, primarily by diffusion instead of convection. Thus droplet microfluidics minimizes the transition of molecules from the dispersed phase to the continuous phase, allowing the high efficiency of drug encapsulation into the droplets.20
Two forms of instability in the fluid's behavior in a droplet-based microfluidic device are described: dripping and jetting regimen (Fig. 6c and d). The transition from one regimen to the other happens by increasing the flow rate and dimensionless parameters (e.g., capillary and Weber numbers).12 The Weber number, defined by eqn (3), is an essential descriptor of deformation in the droplets. The Weber number determines the relationship between surface tension and inertial forces. With the increase in deformation, this number increases; higher energy is therefore meant to generate smaller emulsions.11,12
The capillary number described by eqn (4) expresses the ratio between the viscous forces over the fluid's surface tension. In droplet-based microfluidics, the capillary number is of particular importance since it enables the investigation of various break-up patterns identified by different capillary number ranges. In the case of low values of the capillary number (range <10−2), the droplet formation is not influenced by the shear stress. In order to form a thread and finally squeeze out the droplet, it is only dependent on the accumulated pressure in the inner channel. According to Rayleigh–Plateau instability, droplet formation will occur when the droplet's maximum extension is greater than 1.11
Dripping and jetting regimen happens when the inner fluid is pumped into the secondary immiscible fluid, like in the formation of single emulsion droplets. Parameters related to fluids such as their viscosities, surface tensions, densities, flow rate, and the ratio between flow rates and the device characteristics such as geometry and surface chemistry control the formation of the droplet.153 The two-fluid system's behavior can be expressed according to the outer fluid's capillary number and the inner one's Weber number, i.e., the outer fluid's viscous forces and the inner one's inertial forces. Monodispersed drops result from dripping instability since the device disruptions that lead to drop formation are insensitive to any external intervention, whereas the jetting regimen creates polydispersed drops. The processes leading to the formation of the drops are identical in the case of double emulsions. The fluid stream breaks simultaneously when the inner and middle fluids' rates are equal, leading to the creation of a double emulsion presenting one internal drop. The outer fluid flow rate governs the transitions in the double or multiple emulsion devices between the two regimes; dripping happens while the outer fluid is slower, and vice versa for jetting.12
As mentioned, co-flow, flow-focusing, and T-junction, under a dripping or jetting regimen, are the most common geometries in droplet microfluidics. The junction shape defines the interface between the two immiscible flows. The droplet will be generated when the drag force is higher than the viscose force.20 We can control the droplet formation by understanding these theories, and droplet sizes can be perfectly controlled with passive methods by tuning the microfluidic device geometry, interfacial tension and viscosity of liquid phases, flow rates, and pressure, or with active methods by electrical forces, magnetic force, temperature, and acoustic force.5,12,143,154,155
A Y-junction has the same process, but only the branches' angle makes it different from a T-junction. Also, by adding more branches to these specific designs, they can produce multi emulsion core–shell particles.161–163
By its name single-step droplet formation method has only one step.164 A conventional double emulsion microfluidic device in a single-step method is made of two round capillaries with the orifices facing each other where the round capillaries are inserted into a square capillary (Fig. 5d). The inner phase goes through the inner capillary. In contrast, the middle and outer phases in the same and opposite direction of the inner flow go through the outer square capillary. This can be regarded as a co-flow that incorporates flow-focusing geometry. Once the three fluids enter the inner capillary facing the other inner inlet capillary, which operates as a collection tube, the double emulsion is formed. Water-in-oil-in-water (W/O/W) and oil-in-water-in-oil (O/W/O) are included in the double emulsions. In the production of drug delivery microcapsules with a hollow core and a polymer shell, the W/O/W emulsion has been commonly used. The inner and outer droplets' size can be controlled by adjusting the flow rates and ratios of the inner, middle, and continuous phases, thus controlling the size and shell thickness of the generated microcapsules. The double emulsion drops' size is mainly controlled by the continuous phase flow rate in the dripping mode, while the middle layer determines the shell thickness. Thus the greater the flow ratio between the inner and middle phases, the thicker the capsule layer, for a set flow rate of the continuous phase.165 The multiple-component emulsions can be produced by incorporating two inner fluids simultaneously (Fig. 5e).166
Kim et al.165 introduced a single-step emulsification method that creates monodisperse double-emulsion drops in a core–shell geometry with an ultra-thin wall as a middle layer using a co-axial capillary microfluidic device (Fig. 7a). A circular capillary with the tapered tip was inserted co-axially into a larger capillary and fixed into another square tube. A rounded capillary was located on the other side of the square capillary to limit the injection tip's exit flow. The aqueous fluid and oil, respectively, flow along the capillary wall and from the central capillary. The core–shell structured emulsion with a very thin shell with a thickness of 100 nanometers or even less was generated at the outlet of the injection tube after solidification. Despite the small shell thickness, these particles are very stable and have great encapsulation ability. It was demonstrated by developing biodegradable poly(lactic acid) microcapsules with a shell thickness of about 10 nanometers, potentially useful for drug delivery.
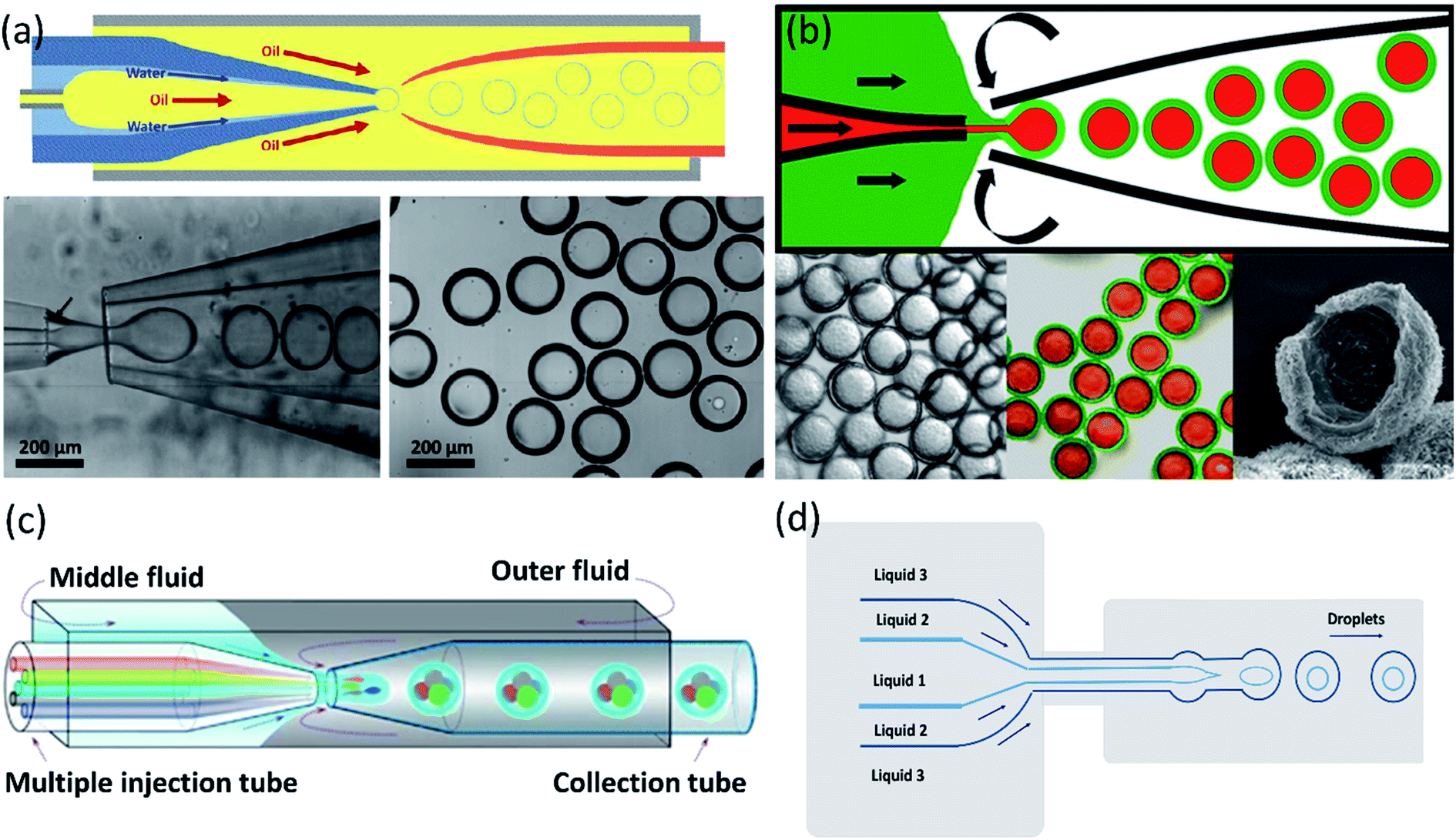 | ||
| Fig. 7 Single-step methods for a double-emulsion generation. (a) Schematic of the microfluidic device for generation of O/W/O double-emulsion droplet with an ultra-thin shell. The bottom images are optical microscope images showing the droplets in the dripping regime and the generated monodisperse double-emulsion droplets, this figure has been adapted from ref. 165 with permission from Royal Society of Chemistry, copyright 2011. (b) A microcapillary device used for the generation of double emulsion droplets for making core–shell particles. The bottom images are microscopy images of core–shell particles, fluorescence detection of the encapsulated drugs: paclitaxel (green) encapsulated within the lipid shell, and doxorubicin (red) in the liquid core, and electron microscopy image of a cracked lipid shell, respectively from left to right, this figure has been adapted from ref. 89 with permission from American Chemical Society, copyright 2013. (c) A schematic of the capillary microfluidic device used for the generation of the multiple core double emulsions, this figure has been adapted from ref. 167 with permission from Springer Nature Limited, copyright 2012, licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. (d) Schematic of 2D flow-focusing microfluidic device used to generate double emulsion droplets in a single-step method, this figure has been reproduced from ref. 146 with permission from American Chemical Society, copyright 2005. | ||
For synergistic combinations of drugs in therapeutic applications, simultaneous encapsulation of multiple active substances in a single carrier is important. However, conventional carrier systems frequently lack efficient encapsulation and release of integrated substances, mainly when drug combinations must be released at concentrations of a specified ratio. Windbergs et al.89 introduced a novel biodegradable core–shell carrier system produced in a solvent-free single-step microfluidic device. The aqueous core is encapsulated with a hydrophilic drug (doxorubicin hydrochloride), while the solid shell is encapsulated with a hydrophobic drug (paclitaxel). In this process, it is possible to control the particle size and composition precisely. Also, core–shell drug-carrier particles can be dried and stored as a powder. Two tapered cylindrical capillary tubes nested inside a square capillary whose inner dimensions correspond to the cylindrical capillaries' outer diameters were used in the microfluidic device (Fig. 7b).
A multiple-core double emulsion composed of multiple oil cores was developed by Zhao et al.167 using a glass capillary microfluidic system with multiple injection tubes (Fig. 7c). There are five separate internal channels in the injection tube, allowing four different oil phase fluids (indicated in red, green, blue, and gray colors) and one aqueous phase fluid (indicated in the center of the four oil fluids as cyan color) to enter the devices separately. The multiple core double emulsion was used as a template to create the photonic crystal barcodes. Barcodes are made of polyethylene glycol (PEG) hydrogel shells and multiple photonic crystals or magnetic-tagged ethoxylated trimethylolpropane triacrylate (ETPTA) cores. Under magnetic fields, the presence of magnetism in the barcodes provides their controllable motion. So they have a unique characteristic that makes them a perfect choice as encoded microcarriers for biomedical applications.
In addition to 3D and capillary microfluidic devices used in the single-step method for producing core–shell particles, 2D devices can also be used. Nie et al.146 presented a 2D flow-focusing microfluidic device to produce core–shell droplets in a single-step method. A double flow-focusing unit in this system forces three immiscible fluids into an orifice and then forms droplets in the downstream chamber (Fig. 7d).
Through a sequential process in two steps (Fig. 5f), a double emulsion may be formed by T-junction geometry. The inner fluid is encapsulated in the first step by the middle fluid, creating a single emulsion. The droplets then flow into the second drop maker and are encapsulated, and the double emulsions are produced by the outer fluid.20
The basis for the generation of core–shell droplets is the same in both 2D and 3D devices,171 except that 3D devices remove the wettability constraints imposed by 2D devices. The droplets in 3D microfluidic systems have minimum interaction with the channel wall in comparison to 2D devices. This ability will prevent the fragile shell from rupturing and the channels from wetting during early interfacial polymerization.2,164
A microfluidic double emulsion geometry (Fig. 5g) has been developed to obtain very thin shells: the inner capillary is placed into a middle capillary. The middle capillary is then placed into the square outer capillary facing the collection capillary orifice within the outer capillary. In this case, the inner, middle, and outer phases flow through multiple capillaries but in the same direction, combining co-flow and co-flow geometry.165
Wang et al.174 developed a hierarchical and flexible microfluidic device fabricated from a combination of three building blocks, including a drop maker, a connector, and a liquid extractor that allows multiple emulsions to be strongly controlled by multicomponent generation (Fig. 8a). Droplets are made in the drop maker and then merged using the connectors. The liquid extractor removes excess continuous phase. The size, number, and ratio of the co-encapsulated droplets could be precisely tuned. This combination also enables the scale-up of the device to produce higher-order multicomponent multiple emulsions with extremely different structures. These multicomponent multiple emulsions offer a flexible and promising system for synergistic delivery of precisely encapsulated incompatible actives or chemicals.
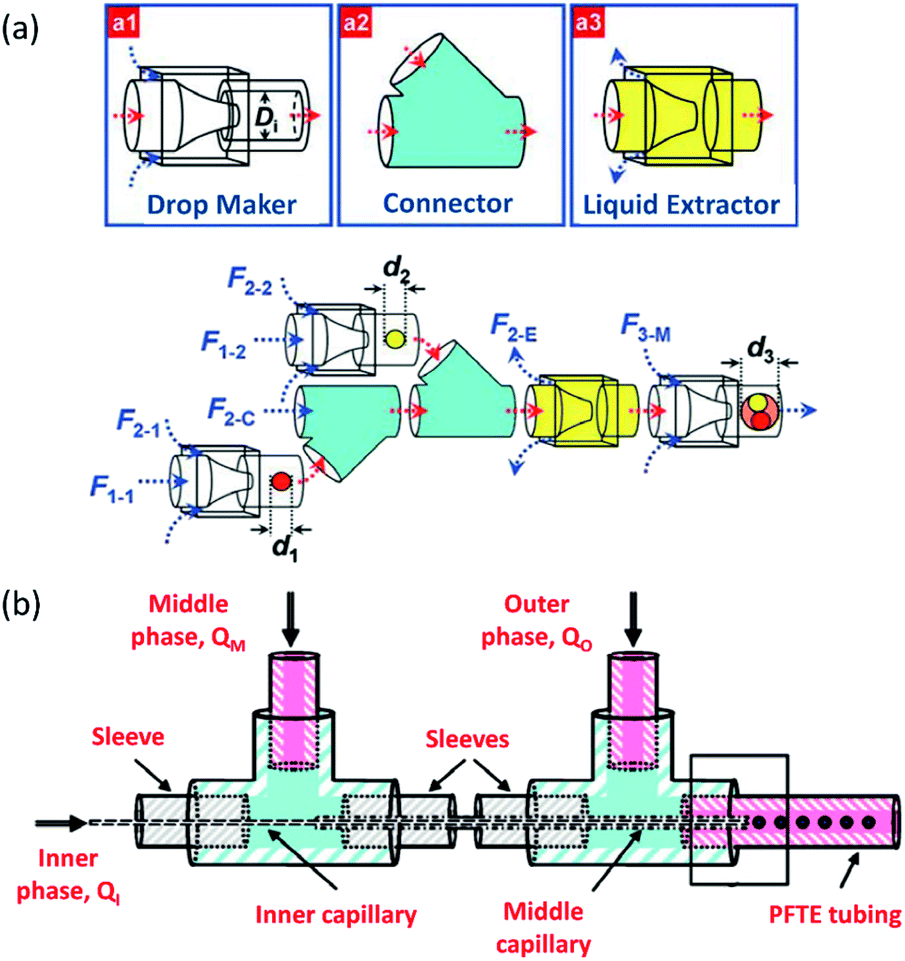 | ||
| Fig. 8 Sequential methods for a double-emulsion generation (a) microfluidic device for the controlled generation of quadruple-component double emulsions and its functional building blocks, this figure has been adapted from ref. 174 with permission from Royal Society of Chemistry, copyright 2011. (b) Schematic of the various co-axial capillaries microfluidic devices for the polymer core–polymer shell particles production, this figure has been adapted from ref. 175 with permission from Royal Society of Chemistry, copyright 2009. | ||
Chang et al.175 presented a 3D microfluidic device with two co-axial capillaries for the polymer core–polymer shell particles' tuneable generation in two sequential steps (Fig. 8b). As can be seen, the device consists of two capillaries with hydrophilic or hydrophobic inner walls co-axially placed inside a T-junction along its main axis. The core and shell droplets were generated in the inner and middle capillaries, respectively, and core–shell droplets then were dispersed in an outer continuous aqueous phase.
Development in the microfluidic field is not limited to the creation of liquid-in-liquid-in-liquid (L/L/L) microemulsion; researchers have demonstrated the generation of emulsions of gas-in-water-in-oil (G/W/O) and gas-in-oil-in-water (G/O/W). In preparing G/W/O and G/O/W emulsions that could be used as a template for producing hollow microparticles, this technology has shown great advantages. A few academic researchers have attempted to use various devices such as flow-focusing, double flow-focusing, co-flowing, T-junction, and dual-coaxial geometry to prepare G/L/L emulsion.2
6.2 Recent advances of microfluidic devices for core–shell particles preparation
In addition to common microfluidic devices for the fabrication of core–shell particles, some complicated microstructures have been introduced recently. In order to produce microcapsules with a core–shell structure, Jin et al.176 showed the application of focused surface acoustic wave (FSAW) microfluidics with one or two focused interdigital transducers (FIDTs) and a bonded polydimethylsiloxane (PDMS) microfluidic channel on a lithium niobate (LiNbO3) substrate (Fig. 9a). The FIDTs are placed on both sides of the flow channel to generate opposing FSAWs. It drives the particles back and forth across the oil/water interface, ideal for generating solid core–shell microcapsules and coating an aqueous microdroplet core with the oil shells. In comparison with previous methods, including T-junction, flow-focusing, co-flowing microfluidic devices, and pillar-based microfluidic devices, solid particles or liquid microdroplets in multiphase laminar flow are generated by the acoustic radiation force resulting from the FSAW without any special modification using one or two FIDTs on a microfluidic device with a simple configuration. This work provides a new active technique for producing a structure of the core–shell on the solid particles. More FIDTs can be added to the device to create more microcapsule layers if necessary. Thus it is possible to synthesize single-layer, two-layer, or even multi-layer microcapsules as desired.176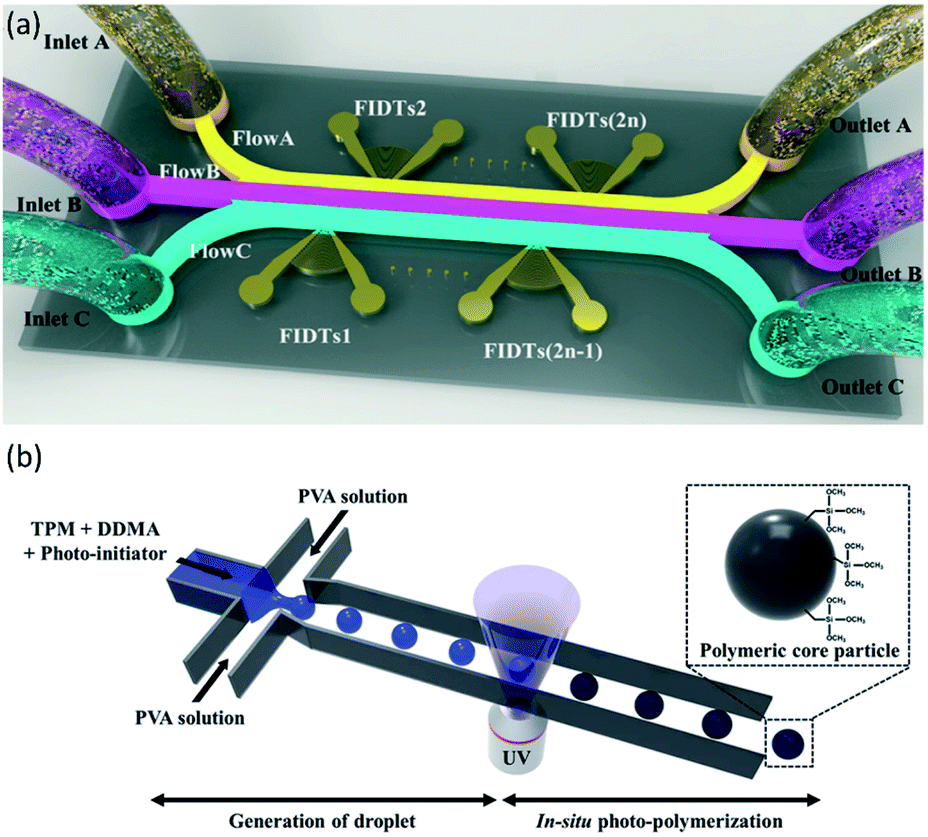 | ||
| Fig. 9 (a) Schematic of FSAW-based microfluidic device to generate core–shell microcapsules, this figure has been adapted from ref. 176 with permission from Royal Society of Chemistry, copyright 2020. (b) Schematic of monodisperse hybrid particle production, a shear force guided pinch-off mechanism shapes droplets containing the monomers (TPM and DDMA) and the photoinitiator in the continuous process (PVA solution). Photopolymerization is initiated by UV-irradiation, this figure has been reproduced from ref. 177 with permission from Springer Nature Limited, copyright 2018, licensed under a Creative Commons Attribution 4.0 International License. | ||
Kim et al.177 proposed a microfluidic method for the fabrication of organic–inorganic hybrid core–shell microparticles in which the core is from poly(1,10-decanediol dimethacrylate-co-trimethoxysilyl propyl methacrylate) (P(DDMA-co-TPM)) shell is from silica nanoparticles. In this method, in combination with in situ photopolymerization, the droplet-based microfluidic method generates highly monodisperse organic microparticles from P(DDMA-co-TPM) in a simple way (Fig. 9b). The silica nanoparticles gradually develop on the surface of the microparticles prepared by hydrolysis and tetraethoxysilane (TEOS) condensation in a simple ammonium hydroxide medium without excessive surface treatment. This approach leads to a decrease in the number of processes and, compared to traditional approaches, facilitates significantly improved size uniformity.177
Ahrberg et al.178 demonstrated an automated microfluidic capillary droplet reactor for the multi-step iron oxide/gold core–shell nanoparticles synthesis. Synthesis outcomes can be monitored in real-time by incorporating a transmission measurement at the outlet of the reactor.
Recently a novel fabrication technique for core–shell structure nanoparticles was created by combining microfluidic chip and electrohydrodynamic atomization to resolve the drawbacks of drug-loaded nanoparticles, such as high initial burst release and wide size distribution. The mixture solution of the surfactant (1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol) and the polymeric coating material (polylactic-glycolic-acid) was injected into the microfluidic chip's outer microchannel as the shell of the particle in this experiment. The encapsulated drug (paclitaxel) was injected into the inner microchannel as the core. Then by applying an electric field on the laminar flow that was developed in the microfluidic chip, the particles with a nanoscale-sized core–shell structure were created. The drug release of these nanoparticles may be extended for more than ten days over a considerable period of time. It can be anticipated that this innovative technology will provide a useful platform for the production of drug-loaded core–shell nanoparticles.179
6.3 Microfluidic devices for core–shell drug carrier particles application in drug delivery
Core–shell drug-carrier particles are known for their unique features. Due to the combination of superior properties not exhibited by the individual components, core–shell particles have gained a lot of interest. As drug delivery carriers, core–shell particles have many benefits, including the reduction in initial burst release, sustained and controlled drug release rate, and the ability to carry a wide variety of biomolecules.2 Core–shell particles could be used in cancer treatment because they could encapsulate multiple ingredients and release them during a multistage process. In contrast, core–shell particles with a broad size distribution produced by conventional techniques are not appropriate for drug delivery.180 The core–shell droplets generated using the microfluidics technique allow for high encapsulation and loading efficiency. The microparticles have a narrow size distribution profile, uniform morphology, and composition resulting in a steady and controlled drug release. The microparticle size is an essential factor in selecting a suitable drug delivery method.80 For example, microparticles with a size distribution from a few to hundreds of microns are more appropriate for oral drug delivery.181Li et al.10 produced a new form of core–shell particles for synergistic and sustained drug delivery. They were made from gelatin methacrylate (GelMa) aqueous solution as core and PLGA oil solution as shell in which different hydrophilic and hydrophobic drugs, such as doxorubicin hydrochloride (DOX) and camptothecin (CPT) could be loaded, respectively. Since the inner cores were polymerized in the microfluidics when the double emulsions were generated, the hydrophilic actives could be trapped with high efficiency in the cores. During the solidification of the microparticle shells with other actives, the cores' rupture or fusion could be avoided. During microfluidic emulsification, the microparticles' size and components can be easily and precisely controlled by adjusting the flow solutions. The encapsulated actives were only released from the delivery systems with the degradation of the biopolymer layers due to the solid nature of the resulting microparticles. Thus the burst release of the actives was prevented. These characteristics of the microparticles make them suitable for drug delivery applications.2
Xu et al.182 developed a doxorubicin-loaded core–shell structured microsphere through a coaxial electro-hydrodynamic atomization process. A PLGA core and a poly(D,L-lactic acid) (PDLLA) shell were included in the microspheres. As a model drug, doxorubicin, a hydrophilic chemotherapy drug, was used and encapsulated within the core. Doxorubicin was effectively encapsulated and lead to an approximately drug-free shell. Doxorubicin release was a two-stage operation, with a steady rate of release.
Nie et al.180 also used the same approach for the development of a distinct core–shell structure of microparticles. In a single step, two different hydrophilic drugs were encapsulated in microparticles with enhanced encapsulation efficiency. They showed that different drug release profiles were affected by varying the outer and inner flow ratio. In addition, the performance of different microspheres in cytotoxicity, cellular apoptosis were analyzed in vitro. Also, tumor inhibition against subcutaneous U87 glioma xenograft was performed in vivo. The benefits and potential applications of this kind of multi-drug release system in the treatment of brain tumors were demonstrated.
Ho et al.183 defined a flexible technique for developing monodisperse polymersomes with biocompatible and biodegradable diblock copolymers for the efficient encapsulation of active substances, which is a good demonstration of core–shell droplets where a dewetting transition occurred. Due to osmotic shock, the release was triggered. The double emulsion droplets were used as a template to generate PEG–PLA polymersomes by using amphiphilic diblock copolymers. The polymersomes were used for the encapsulation of a hydrophilic fluorescent solution. An osmotic pressure difference caused the polymersomes' breakage by adding salt, and the solutes were released. This easy and efficient release mechanism could be used to design encapsulation and controlled release in various biomedical applications.
6.4 Microfluidic devices for core–shell drug carrier particles characterization
Size, shape, drug loading, and stability are the most important properties of nanoparticles to be characterized before probing their interaction with biological systems. Also, the development of novel particle characterization tools for drug delivery greatly affects the probability of an effective therapeutic translation. An inability to verify the drug delivery system's safety in vivo is one real obstacle to the clinical-scale application of nanoparticles.145 This section presents microfluidic techniques capable of characterizing drug delivery nanoparticles.Microfluidic-based liquid chromatography (LC) has attracted a lot of attention because of its improved sensitivity, reduced sample utilization, and ability to multiplex measurements. Gao et al.186 developed an integrated microfluidic device with mass spectrometry (MS) detection for high-throughput drug screening. A concentration gradient generator, cell culture chamber, and solid-phase extraction columns were incorporated into this microfluidic device into a PDMS chip. The method of drug absorption and cytotoxicity assessment may be achieved simultaneously with the use of combination systems.
With a low amount of reagent usage, integrated devices may provide a means of high-throughput drug analysis. In addition, many microfluidic-based nanospray emitters187,188 and microfluidic-based LC-MS analysis189,190 have been introduced.
7. Conclusions
In this review, various kinds of core–shell microparticles are first discussed based on the core and shell structures' materials. The motivation of using core–shell particles is to combine the desired properties of different materials and structures to offer a synergistic effect, stabilize the active particles, or provide biocompatible properties. In addition, using core–shell drug carrier particles aims to reduce the drug's side effects with protection against environmental conditions, deliver it to the desired location, reduce its production cost, and increase its efficacy and controlled release. The choice of shell material and sometimes core for pharmaceutical systems is very complicated and should be chosen depending on the conditions. For example, suppose the desired area for releasing the drug is close to the skin's surface. In that case, it is possible to use a shell destroyed by the temperature. It is possible to direct it to the target position using a magnetic field if metal or metal oxide is used.This review also provides an overview of microfluidic techniques for the generation of core–shell drug carrier particles. Conventional methods have some drawbacks which have considerably limited their applications. The main disadvantages of these methods are low monodispersity and high material usage. Microfluidic devices have been developed to generate core–shell particles with controlled features and providing many advantages such as cheapness, uniformity of particle size and shape, and simplicity of application compared to the other alternatives. Different microfluidic chip designs can be used based on the desired type of core–shell drug carrier particle, so there are more alternatives than the other available methods. Microfluidic devices are classified based on their geometry, and they could be designed to generate core–shell particles using single-step or sequential emulsification methods.
On the other hand, nanoparticles can be easily made in nanoscale by applying microfluidic devices, which is very difficult or impossible to achieve in the other methods. In some cases, particles made with microfluidic devices reach a few hundred nanometers in diameter, making this method popular. The microfluidic method has some more advantages, such as automation, integration, miniaturization, and the possibility of reducing human error. Their coherence makes these chips the best choice for the pharmaceutical industry. All of the above is evidence that microfluidic chips are the perfect choice for making core–shell drug carriers particle.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Iran National Science Foundation (INSF) [grant no. 96016453].Notes and references
- R. Hayes, A. Ahmed, T. Edge and H. Zhang, J. Chromatogr. A, 2014, 1357, 36–52 CrossRef CAS.
- F. M. Galogahi, Y. Zhu, H. An and N. T. Nguyen, J. Sci. Adv. Mater. Devices, 2020, 5, 417–435 CrossRef.
- K. S. Kumar, V. B. Kumar and P. Paik, J. Nanopart., 2013, 2013, 1–24 CrossRef.
- D. He, S. Wang, L. Lei, Z. Hou, P. Shang, X. He and H. Nie, Chem. Eng. Sci., 2015, 125, 108–120 CrossRef CAS.
- Z. Mahdavi, H. Rezvani and M. Keshavarz Moraveji, RSC Adv., 2020, 10, 18280–18295 RSC.
- S. Xu and T. Nisisako, Sci. Rep., 2020, 10, 1–10 CrossRef.
- T. Ogi, A. B. D. Nandiyanto and K. Okuyama, Adv. Powder Technol., 2014, 25, 3–17 CrossRef CAS.
- W. M. Tolles and B. B. Rath, Curr. Sci., 2003, 85, 1746–1759 CAS.
- T. Chavan, P. Muttil and N. K. Kunda, in AAPS Advances in the Pharmaceutical Sciences Series, Springer, 2020, vol. 41, pp. 3–26 Search PubMed.
- Y. Li, D. Yan, F. Fu, Y. Liu, B. Zhang, J. Wang, L. Shang, Z. Gu and Y. Zhao, Sci. China Mater., 2017, 60, 543–553 CrossRef CAS.
- Z. Liu, F. Fontana, A. Python, J. T. Hirvonen and H. A. Santos, Small, 2020, 16, 2070048 CrossRef.
- F. Fontana, M. P. A. Ferreira, A. Correia, J. Hirvonen and H. A. Santos, J. Drug Delivery Sci. Technol., 2016, 34, 76–87 CrossRef CAS.
- A. Al-Halhouli, S. Demming, A. Waldschik and S. Büttgenbach, Micromachines, 2014, 5, 442–456 CrossRef.
- K. Smistrup, O. Hansen, H. Bruus and M. F. Hansen, J. Magn. Magn. Mater., 2005, 293, 597–604 CrossRef CAS.
- K. Ren, J. Zhou and H. Wu, Acc. Chem. Res., 2013, 46, 2396–2406 CrossRef CAS.
- Q. Zhao, H. Cui, Y. Wang and X. Du, Small, 2020, 16, 1903798 CrossRef CAS.
- S. Sugiura, K. Nakazawa, T. Kanamori and K. Ohnuma, in Advances in Microfluidics - New Applications in Biology, Energy, and Materials Sciences, InTech, 2016 Search PubMed.
- J. Garra, T. Long, J. Currie, T. Schneider, R. White and M. Paranjape, J. Vac. Sci. Technol., A, 2002, 20, 975–982 CrossRef CAS.
- A. R. Abate, D. Lee, T. Do, C. Holtze and D. A. Weitz, Lab Chip, 2008, 8, 516–518 RSC.
- D. Liu, H. Zhang, F. Fontana, J. T. Hirvonen and H. A. Santos, Lab Chip, 2017, 17, 1856–1883 RSC.
- D. Lin, B. Li, J. Qi, X. Ji, S. Yang, W. Wang and L. Chen, Sens. Actuators, B, 2020, 303, 127213 CrossRef CAS.
- Z. Isiksacan, M. T. Guler, B. Aydogdu, I. Bilican and C. Elbuken, J. Micromech. Microeng., 2016, 26, 035008 CrossRef.
- M. Kiran Raj and S. Chakraborty, J. Appl. Polym. Sci., 2020, 137, 48958 CrossRef.
- Y. Zhou and F. Amirouche, Micromachines, 2011, 2, 345–355 CrossRef.
- J. Guerrero, Y. Chang, A. A. Fragkopoulos and A. Fernandez-Nieves, Small, 2020, 16, 1904344 CrossRef CAS.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS.
- L. Martín-Banderas, M. Flores-Masquera, P. Riesco-Chueca, A. Rodríguez-Gil, Á. Cebolla, S. Chávez and A. M. Gañán-Calvo, Small, 2005, 1, 688–692 CrossRef.
- X. Lü, J. Jiang, H. Wang, Q. Gao, S. Zhao, N. Li, J. Yang, S. Wang, W. Bao and R. Chen, Sensors, 2018, 19, 72 CrossRef.
- A. J. DeMello, Nature, 2006, 442, 394–402 CrossRef CAS.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS.
- L. Mahler, K. Wink, R. J. Beulig, K. Scherlach, M. Tovar, E. Zang, K. Martin, C. Hertweck, D. Belder and M. Roth, Sci. Rep., 2018, 8, 13087 CrossRef.
- J. Evans, D. Liepmann and A. P. Pisano, Proc. - IEEE Micro Electro Mech. Syst., 1997, 96–101 Search PubMed.
- V. E. Kalyonov, Y. D. Orekhov, A. N. Shahabdin, A. P. Broyko and D. O. Testov, in Proceedings of the 2020 IEEE Conference of Russian Young Researchers in Electrical and Electronic Engineering, EIConRus 2020, Institute of Electrical and Electronics Engineers Inc., 2020, pp. 1531–1534 Search PubMed.
- M. Jender, P. Novo, D. Maehler, U. Münchberg, D. Janasek and E. Freier, Anal. Chem., 2020, 92, 6764–6769 CrossRef CAS.
- A. L. Forget, C. C. Dombrowski, I. Amitani and S. C. Kowalczykowski, Nat. Protoc., 2013, 8, 525–538 CrossRef CAS.
- C. J. Pipe and G. H. McKinley, Mech. Res. Commun., 2009, 36, 110–120 CrossRef.
- M. J. Jebrail, M. S. Bartsch and K. D. Patel, Lab Chip, 2012, 12, 2452–2463 RSC.
- R. Amin, A. Joshi and S. Tasoglu, J. 3D Print. Med., 2017, 1, 85–89 CrossRef CAS.
- D. Erickson and D. Li, Anal. Chim. Acta, 2004, 507, 11–26 CrossRef CAS.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS.
- Microfluidics: a general overview of microfluidics definition of microfluidics, https://www.elveflow.com/microfluid%0Aic-reviews/general-microfluidics/ageneral-%0Aoverview-of-microfluidics Search PubMed.
- V. Narayanamurthy, Z. E. Jeroish, K. S. Bhuvaneshwari, P. Bayat, R. Premkumar, F. Samsuri and M. M. Yusoff, RSC Adv., 2020, 10, 11652–11680 RSC.
- A. Podwin, D. Lizanets, D. Przystupski, W. Kubicki, P. Śniadek, J. Kulbacka, A. Wymysłowski, R. Walczak and J. A. Dziuban, Micromachines, 2020, 11, 1–11 CrossRef.
- R. R. Niedl and C. Beta, Lab Chip, 2015, 15, 2452–2459 RSC.
- S. W. Dutse and N. A. Yusof, Sensors, 2011, 11, 5754–5768 CrossRef CAS.
- R. Ran, Q. Sun, T. Baby, D. Wibowo, A. P. J. Middelberg and C.-X. Zhao, Chem. Eng. Sci., 2017, 169, 78–96 CrossRef CAS.
- M. Piergiovanni, G. Casagrande, F. Taverna, I. Corridori, M. Frigerio, E. Bianchi, F. Arienti, A. Mazzocchi, G. Dubini and M. L. Costantino, Ann. Biomed. Eng., 2020, 48, 236–246 CrossRef.
- G. Bruno, N. Colistra, G. Melle, A. Cerea, A. Hubarevich, L. Deleye, F. De Angelis and M. Dipalo, Front. Bioeng. Biotechnol., 2020, 8, 626 CrossRef.
- A. I. Shallan and C. Priest, Chem. Eng. Process., 2019, 142, 107559 CrossRef CAS.
- S. Damiati, U. B. Kompella, S. A. Damiati and R. Kodzius, Genes, 2018, 9, 103 CrossRef.
- R. Riahi, A. Tamayol, S. A. M. Shaegh, A. M. Ghaemmaghami, M. R. Dokmeci and A. Khademhosseini, Curr. Opin. Chem. Eng., 2015, 7, 101–112 CrossRef.
- G. Tiwari, R. Tiwari, S. Bannerjee, L. Bhati, S. Pandey, P. Pandey and B. Sriwastawa, Int. J. Pharm. Invest., 2012, 2, 2 CrossRef.
- J. M. Chan, L. Zhang, K. P. Yuet, G. Liao, J.-W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS.
- M. M. Hasani-Sadrabadi, S. Taranejoo, E. Dashtimoghadam, G. Bahlakeh, F. S. Majedi, J. J. VanDersarl, M. Janmaleki, F. Sharifi, A. Bertsch, K. Hourigan, L. Tayebi, P. Renaud and K. I. Jacob, Adv. Mater., 2016, 28, 4134–4141 CrossRef CAS.
- A. Zargar, S. Chang, A. Kothari, A. M. Snijders, J.-H. Mao, J. Wang, A. C. Hernández, J. D. Keasling and T. G. Bivona, Chronic Dis. Transl. Med., 2019, 5, 258–266 Search PubMed.
- E. Davaa, J. Lee, R. Jenjob and S. G. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 71–79 CrossRef CAS.
- I. U. Khan, C. A. Serra, N. Anton and T. Vandamme, J. Controlled Release, 2013, 172, 1065–1074 CrossRef CAS.
- A. R. Fernandes, J. Dias-Ferreira, M. C. Teixeira, A. A. M. Shimojo, P. Severino, A. M. Silva, R. Shegokar and E. B. Souto, Drug Delivery Trends, Elsevier, 2020, pp. 1–13 Search PubMed.
- C. Han, S. Zhang, H. Huang, Y. Dong, X. Sui, B. Jian and W. Zhu, J. Pharm. Sci., 2019, 108, 3225–3232 CrossRef CAS.
- V. Cauda, C. Argyo and T. Bein, J. Mater. Chem., 2010, 20, 8693–8699 RSC.
- K. S. Soppimath, D. C. W. Tan and Y. Y. Yang, Adv. Mater., 2005, 17, 318–323 CrossRef CAS.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS.
- A. Geraili, M. Janmaleki, A. Sanati-Nezhad and K. Mequanint, Biofabrication, 2020, 12, 045007 CrossRef CAS.
- A. Z. M. Badruddoza, P. D. Godfrin, A. S. Myerson, B. L. Trout and P. S. Doyle, Adv. Healthcare Mater., 2016, 5, 1960–1968 CrossRef CAS.
- F. He, M. Zhang, W. Wang, Q. Cai, Y. Su, Z. Liu, Y. Faraj, X. Ju, R. Xie and L. Chu, Adv. Mater. Technol., 2019, 4, 1800687 CrossRef.
- X.-T. Sun, R. Guo, D.-N. Wang, Y.-Y. Wei, C.-G. Yang and Z.-R. Xu, J. Colloid Interface Sci., 2019, 553, 631–638 CrossRef CAS.
- H. Wang, J. Yi, S. Mukherjee, P. Banerjee and S. Zhou, Nanoscale, 2014, 6, 13001–13011 RSC.
- W. Fang, J. Yang, J. Gong and N. Zheng, Adv. Funct. Mater., 2012, 22, 842–848 CrossRef CAS.
- S. K. Tiwari, R. Tzezana, E. Zussman and S. S. Venkatraman, Int. J. Pharm., 2010, 392, 209–217 CrossRef CAS.
- S. Seiffert, ChemPhysChem, 2013, 14, 295–304 CrossRef CAS.
- Y. Du, L. Mo, X. Wang, H. Wang, X. hui Ge and T. Qiu, Microfluid. Nanofluid., 2020, 24, 1–11 CrossRef.
- S. Gai, P. Yang, C. Li, W. Wang, Y. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166–1172 CrossRef CAS.
- L. M. Medina and R. Jordano, J. Food Prot., 1994, 57, 731–733 CrossRef CAS.
- B. F. Gibbs, S. Kermasha, I. Alli and C. N. Mulligan, Int. J. Food Sci. Nutr., 1999, 50, 213–224 CrossRef CAS.
- B. Mandal, H. Bhattacharjee, N. Mittal, H. Sah, P. Balabathula, L. A. Thoma and G. C. Wood, Nanomedicine, 2013, 9, 474–491 CrossRef CAS.
- O. V. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef.
- L. Wang, W. Liu, Y. Wang, J. C. Wang, Q. Tu, R. Liu and J. Wang, Lab Chip, 2013, 13, 695–705 RSC.
- W. F. Sewell, J. T. Borenstein, Z. Chen, J. Fiering, O. Handzel, M. Holmboe, E. S. Kim, S. G. Kujawa, M. J. McKenna, M. M. Mescher, B. Murphy, E. E. Eary Swan, M. Peppi and S. Tao, Audiol. Neurotol., 2009, 14, 411–422 CrossRef CAS.
- M. L. Focarete and A. Tampieri, Core-Shell Nanostructures for Drug Delivery and Theranostics, Elsevier, 2018, pp. 3–5 Search PubMed.
- W. Li, L. Zhang, X. Ge, B. Xu, W. Zhang, L. Qu, C. H. Choi, J. Xu, A. Zhang, H. Lee and D. A. Weitz, Chem. Soc. Rev., 2018, 47, 5646–5683 RSC.
- T. T. Hoang Thi, D. H. Nguyen Tran, L. G. Bach, H. Vu-Quang, D. C. Nguyen, K. D. Park and D. H. Nguyen, Pharmaceutics, 2019, 11, 120 CrossRef.
- T. Kong, J. Wu, K. W. Yeung, M. K. To, H. C. Shum and L. Wang, Biomicrofluidics, 2013, 7, 044128 CrossRef.
- S. Deshpande, S. Sharma, V. Koul and N. Singh, ACS Omega, 2017, 2, 6455–6463 CrossRef CAS.
- Z. Liu, Y. Li, W. Li, W. Lian, M. Kemell, S. Hietala, P. Figueiredo, L. Li, E. Mäkilä, M. Ma, J. Salonen, J. T. Hirvonen, D. Liu, H. Zhang, X. Deng and H. A. Santos, Mater. Horiz., 2019, 6, 385–393 RSC.
- Y. Liu, J. Pan and S.-S. Feng, Int. J. Pharm., 2010, 395, 243–250 CrossRef CAS.
- R. A. Ramli, W. A. Laftah and S. Hashim, RSC Adv., 2013, 3, 15543–15565 RSC.
- M. C. Neves, T. Trindade, A. M. B. Timmons and J. D. Pedrosa de Jesus, Mater. Res. Bull., 2001, 36, 1099–1108 CrossRef CAS.
- S. Mytnyk, I. Ziemecka, A. G. L. Olive, J. W. M. Van der Meer, K. A. Totlani, S. Oldenhof, M. T. Kreutzer, V. Van Steijn and J. H. Van Esch, RSC Adv., 2017, 7, 11331–11337 RSC.
- M. Windbergs, Y. Zhao, J. Heyman and D. A. Weitz, J. Am. Chem. Soc., 2013, 135, 7933–7937 CrossRef CAS.
- S. Cammas, K. Suzuki, C. Sone, Y. Sakurai, K. Kataoka and T. Okano, J. Controlled Release, 1997, 48, 157–164 CrossRef CAS.
- L. Ribeiro de Souza, Design and synthesis of microcapsules using microfluidics for autonomic self-healing in cementitious materials, Doctoral thesis, University of Cambridge, 2017 DOI:10.17863/CAM.16673.
- S. Chakraborty, I. C. Liao, A. Adler and K. W. Leong, Adv. Drug Delivery Rev., 2009, 61, 1043–1054 CrossRef CAS.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS.
- L. Lukyanova, L. Séon, A. Aradian, O. Mondain-Monval, J. Leng and R. Wunenburger, J. Appl. Polym. Sci., 2013, 128, 3512–3521 CrossRef CAS.
- Y. Liang, J. Ouyang, H. Wang, W. Wang, P. Chui and K. Sun, Appl. Surf. Sci., 2012, 258, 3689–3694 CrossRef CAS.
- Y. Long, B. Vincent, D. York, Z. Zhang and J. A. Preece, Chem. Commun., 2010, 46, 1718–1720 RSC.
- A. Ahmed, K. Skinley, S. Herodotou and H. Zhang, J. Sep. Sci., 2018, 41, 99–124 CrossRef CAS.
- E. J. Guidelli, O. Baffa and D. R. Clarke, Sci. Rep., 2015, 5, 14004 CrossRef CAS.
- M. I. Shukoor, F. Natalio, M. N. Tahir, M. Wiens, M. Tarantola, H. A. Therese, M. Barz, S. Weber, M. Terekhov, H. C. Schroder, W. E. G. Muller, A. Janshoff, P. Theato, R. Zentel, L. M. Schreiber and W. Tremel, Adv. Funct. Mater., 2009, 19, 3717–3725 CrossRef CAS.
- M. Rahimi, A. Wadajkar, K. Subramanian, M. Yousef, W. Cui, J.-T. Hsieh and K. T. Nguyen, Nanomedicine, 2010, 6, 672–680 CrossRef CAS.
- K. J. Widder, A. E. Senyei and D. G. Scarpelli, Exp. Biol. Med., 1978, 158, 141–146 CrossRef CAS.
- K. E. Albinali, M. M. Zagho, Y. Deng and A. A. Elzatahry, Int. J. Nanomed., 2019, 14, 1707–1723 CrossRef CAS.
- S. Hossen, M. K. Hossain, M. K. Basher, M. N. H. Mia, M. T. Rahman and M. J. Uddin, J. Adv. Res., 2019, 15, 1–18 CrossRef CAS.
- Y. Kim, F. Fay, D. P. Cormode, B. L. Sanchez-Gaytan, J. Tang, E. J. Hennessy, M. Ma, K. Moore, O. C. Farokhzad, E. A. Fisher, W. J. M. Mulder, R. Langer and Z. A. Fayad, ACS Nano, 2013, 7, 9975–9983 CrossRef CAS.
- L.-L. Li, X. Li and H. Wang, Small Methods, 2017, 1, 1700140 CrossRef.
- A. Abou-Hassan, in Microfluidics: Fundamental, Devices and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018, pp. 405–418 Search PubMed.
- H. Elmizadeh, M. Khanmohammadi, K. Ghasemi, G. Hassanzadeh, M. Nassiri-Asl and A. B. Garmarudi, J. Pharm. Biomed. Anal., 2013, 80, 141–146 CrossRef CAS.
- D. H. K. Reddy and S. M. Lee, Adv. Colloid Interface Sci., 2013, 201–202, 68–93 CrossRef CAS.
- L. Wang, J. Hu, H. Zhang and T. Zhang, Chem. Commun., 2011, 47, 6837–6839 RSC.
- M. Yadollahi, S. Farhoudian and H. Namazi, Int. J. Biol. Macromol., 2015, 79, 37–43 CrossRef CAS.
- S. Kumari and R. P. Singh, Int. J. Biol. Macromol., 2012, 50, 878–883 CrossRef CAS.
- M.-K. Jang, Y.-I. Jeong and J.-W. Nah, Colloids Surf., B, 2010, 81, 530–536 CrossRef CAS.
- N. Sahiner and P. Ilgin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5239–5246 CrossRef CAS.
- H. Nie, Z. Dong, D. Y. Arifin, Y. Hu and C. Wang, J. Biomed. Mater. Res., Part A, 2010, 95, 709–716 CrossRef.
- C. Chen, C. Gao, M. Liu, L. Shaoyu, C. Yu, S. Ma, J. Wang and G. Cui, J. Biomater. Sci., Polym. Ed., 2013, 24, 1127–1139 CrossRef CAS.
- P. Del Gaudio, G. Auriemma, P. Russo, T. Mencherini, P. Campiglia, M. Stigliani and R. P. Aquino, Eur. J. Pharm. Biopharm., 2014, 87, 541–547 CrossRef CAS.
- M. Feng and P. Li, J. Biomed. Mater. Res., Part A, 2007, 80, 184–193 CrossRef.
- S. Narayanan, M. Pavithran, A. Viswanath, D. Narayanan, C. C. Mohan, K. Manzoor and D. Menon, Acta Biomater., 2014, 10, 2112–2124 CrossRef CAS.
- J. Wu, T. Kong, K. W. K. Yeung, H. C. Shum, K. M. C. Cheung, L. Wang and M. K. T. To, Acta Biomater., 2013, 9, 7410–7419 CrossRef CAS.
- A. Baeza, E. Guisasola, E. Ruiz-Hernández and M. Vallet-Regí, Chem. Mater., 2012, 24, 517–524 CrossRef CAS.
- X. Yang, X. Liu, Z. Liu, F. Pu, J. Ren and X. Qu, Adv. Mater., 2012, 24, 2890–2895 CrossRef CAS.
- V. Mamaeva, J. M. Rosenholm, L. T. Bate-Eya, L. Bergman, E. Peuhu, A. Duchanoy, L. E. Fortelius, S. Landor, D. M. Toivola, M. Lindén and C. Sahlgren, Mol. Ther., 2011, 19, 1538–1546 CrossRef CAS.
- J. Croissant and J. I. Zink, J. Am. Chem. Soc., 2012, 134, 7628–7631 CrossRef CAS.
- J. Liu, W. Bu, L. Pan and J. Shi, Angew. Chem., Int. Ed., 2013, 52, 4375–4379 CrossRef CAS.
- W. Wu, T. Zhou, A. Berliner, P. Banerjee and S. Zhou, Chem. Mater., 2010, 22, 1966–1976 CrossRef CAS.
- W. Wu, J. Shen, P. Banerjee and S. Zhou, Biomaterials, 2010, 31, 7555–7566 CrossRef CAS.
- D. Mangalaraj and D. Nithya Devi, in Springer Proceedings in Physics, Springer Science and Business Media, LLC, 2017, vol. 189, pp. 9–17 Search PubMed.
- L. D. Tran, N. M. T. Hoang, T. T. Mai, H. V. Tran, N. T. Nguyen, T. D. Tran, M. H. Do, Q. T. Nguyen, D. G. Pham, T. P. Ha, H. Van Le and P. X. Nguyen, Colloids Surf., A, 2010, 371, 104–112 CrossRef CAS.
- Y. Wang, L. Chen and P. Liu, Chem.–Eur. J., 2012, 18, 5935–5943 CrossRef CAS.
- L. Zhao, H. Liu, F. Wang and L. Zeng, J. Mater. Chem. A, 2014, 2, 7065–7074 RSC.
- A. Pramanik, D. Laha, S. K. Dash, S. Chattopadhyay, S. Roy, D. K. Das, P. Pramanik and P. Karmakar, Mater. Sci. Eng., C, 2016, 68, 327–337 CrossRef CAS.
- Q. Wei, J. Ji and J. Shen, Macromol. Rapid Commun., 2008, 29, 645–650 CrossRef CAS.
- G. Grass, C. Rensing and M. Solioz, Appl. Environ. Microbiol., 2011, 77, 1541–1547 CrossRef CAS.
- S. Karakus, E. Tan, M. Ilgar, I. S. Basdemir and A. Kilislioglu, in New Trends in Ion Exchange Studies, InTech, 2018 Search PubMed.
- S. Zhang, J. Ye, Z. Liu, H. Lu, S. Shi, Y. Qi and G. Ning, Dalton Trans., 2020, 49, 13044–13051 RSC.
- H. P. Feng, L. Tang, G. M. Zeng, J. Tang, Y. C. Deng, M. Yan, Y. N. Liu, Y. Y. Zhou, X. Y. Ren and S. Chen, J. Mater. Chem. A, 2018, 6, 7310–7337 RSC.
- Z. Assadi, G. Emtiazi and A. Zarrabi, J. Mol. Liq., 2018, 250, 375–380 CrossRef CAS.
- Y. Sameenoi, M. M. Mensack, K. Boonsong, R. Ewing, W. Dungchai, O. Chailapakul, D. M. Cropek and C. S. Henry, Analyst, 2011, 136, 3177–3184 RSC.
- D. Yang, M. Tayebi, Y. Huang, H. Y. Yang and Y. Ai, Sensors, 2016, 16, 1911 CrossRef.
- H. Zhang, T. Yan, S. Xu, S. Feng, D. Huang, M. Fujita and X. D. Gao, Mater. Sci. Eng., C, 2017, 73, 144–151 CrossRef.
- R. P. Singh, G. Sharma, Sonali, S. Singh, S. Bharti, B. L. Pandey, B. Koch and M. S. Muthu, Mater. Sci. Eng., C, 2017, 77, 446–458 CrossRef CAS.
- V. P. Padmanabhan, R. Kulandaivelu and S. N. T. S. Nellaiappan, Mater. Sci. Eng., C, 2018, 92, 685–693 CrossRef CAS.
- W. J. Pietro, J. Nanosci, Adv. Technol., 2016, 1, 25–31 CrossRef.
- E. I. Mancera-Andrade, A. Parsaeimehr, A. Arevalo-Gallegos, G. Ascencio-Favela and R. Parra-Saldivar, Front. Biosci., Elite Ed., 2018, 10, 74–91 CrossRef.
- J. Ahn, J. Ko, S. Lee, J. Yu, Y. Kim and N. L. Jeon, Adv. Drug Delivery Rev., 2018, 128, 29–53 CrossRef CAS.
- Z. Nie, S. Xu, M. Seo, P. C. Lewis and E. Kumacheva, J. Am. Chem. Soc., 2005, 127, 8058–8063 CrossRef CAS.
- D. A. LaVan, T. McGuire and R. Langer, Nat. Biotechnol., 2003, 21, 1184–1191 CrossRef CAS.
- W. J. Duncanson, T. Lin, A. R. Abate, S. Seiffert, R. K. Shah and D. A. Weitz, Lab Chip, 2012, 12, 2135–2145 RSC.
- Z. Lian, Y. Chan, Y. Luo, X. Yang, K. S. Koh, J. Wang, G. Z. Chen, Y. Ren and J. He, Electrophoresis, 2020, 41, 891–901 CrossRef CAS.
- X. Zhao, F. Bian, L. Sun, L. Cai, L. Li and Y. Zhao, Small, 2020, 16, 1901943 CrossRef CAS.
- I. U. Khan, L. Stolch, C. A. Serra, N. Anton, R. Akasov and T. F. Vandamme, Int. J. Pharm., 2015, 478, 78–87 CrossRef CAS.
- N. Teo, C. Jin, A. Kulkarni and S. C. Jana, J. Colloid Interface Sci., 2020, 561, 772–781 CrossRef CAS.
- A. S. Utada, A. Fernandez-Nieves, H. A. Stone and D. A. Weitz, Phys. Rev. Lett., 2007, 99, 094502 CrossRef.
- J. Li, D. K. Baxani, W. D. Jamieson, W. Xu, V. G. Rocha, D. A. Barrow and O. K. Castell, Adv. Sci., 2020, 7, 1901719 CrossRef CAS.
- T. Watanabe, I. Motohiro and T. Ono, Langmuir, 2019, 35, 2358–2367 CrossRef CAS.
- P. Zhu and L. Wang, Lab Chip, 2017, 17, 34–75 RSC.
- E. Chiarello, L. Derzsi, M. Pierno, G. Mistura and E. Piccin, Micromachines, 2015, 6, 1825–1835 CrossRef.
- S. C. Kim, D. J. Sukovich and A. R. Abate, Lab Chip, 2015, 15, 3163–3169 RSC.
- J. H. Xu, S. W. Li, J. Tan, Y. J. Wang and G. S. Luo, Langmuir, 2006, 22, 7943–7946 CrossRef CAS.
- S.-Y. Teh, R. Lin, L.-H. Hung and A. P. Lee, Lab Chip, 2008, 8, 198–220 RSC.
- K. Muijlwijk, W. Huang, J. E. Vuist, C. Berton-Carabin and K. Schroën, Soft Matter, 2016, 12, 9025–9029 RSC.
- T. Nisisako and T. Hatsuzawa, Sens. Actuators, B, 2016, 223, 209–216 CrossRef CAS.
- E. E. Ekanem, Z. Zhang and G. T. Vladisavljević, J. Colloid Interface Sci., 2017, 498, 387–394 CrossRef CAS.
- S. A. Nabavi, G. T. Vladisavljević and V. Manović, Chem. Eng. J., 2017, 322, 140–148 CrossRef CAS.
- S. H. Kim, J. W. Kim, J. C. Cho and D. A. Weitz, Lab Chip, 2011, 11, 3162–3166 RSC.
- L. L. A. Adams, T. E. Kodger, S. H. Kim, H. C. Shum, T. Franke and D. A. Weitz, Soft Matter, 2012, 8, 10719–10724 RSC.
- Y. Zhao, Z. Xie, H. Gu, L. Jin, X. Zhao, B. Wang and Z. Gu, NPG Asia Mater., 2012, 4, 25 CrossRef.
- D. Yoon, K. Hasegawa, Y. Kaneko, T. Arakawa, J. Go, T. Sekiguchi and S. Shoji, Micromachines, 2015, 6, 622–633 CrossRef.
- Y. Hennequin, N. Pannacci, C. P. De Torres, G. Tetradis-Meris, S. Chapuliot, E. Bouchaud and P. Tabeling, Langmuir, 2009, 25, 7857–7861 CrossRef CAS.
- F. Cellesi, W. Weber, M. Fussenegger, J. A. Hubbell and N. Tirelli, Biotechnol. Bioeng., 2004, 88, 740–749 CrossRef CAS.
- C. X. Zhao, Adv. Drug Delivery Rev., 2013, 65, 1420–1446 CrossRef CAS.
- T. Nisisako, S. Okushima and T. Torii, Soft Matter, 2005, 1, 23–27 RSC.
- D. T. Chong, X. S. Liu, H. J. Ma, G. Y. Huang, Y. L. Han, X. Y. Cui, J. J. Yan and F. Xu, Microfluid. Nanofluid., 2015, 19, 1071–1090 CrossRef CAS.
- W. Wang, R. Xie, X. J. Ju, T. Luo, L. Liu, D. A. Weitz and L. Y. Chu, Lab Chip, 2011, 11, 1587–1592 RSC.
- Z. Chang, C. A. Serra, M. Bouquey, L. Prat and G. Hadziioannou, Lab Chip, 2009, 9, 3007–3011 RSC.
- S. Jin, X. Wei, J. Ren, Z. Jiang, C. Abell and Z. Yu, Lab Chip, 2020, 20, 3104–3108 RSC.
- D.-Y. Kim, S. H. Jin, S.-G. Jeong, B. Lee, K.-K. Kang and C.-S. Lee, Sci. Rep., 2018, 8, 8525 CrossRef.
- C. D. Ahrberg, J. Wook Choi and B. Geun Chung, Sci. Rep., 2020, 10, 1737 CrossRef CAS.
- W. Zeng, P. Guo, P. Jiang, W. Liu, T. Hong and C. Chen, Nanotechnology, 2020, 31, 145301 CrossRef CAS.
- H. Nie, Y. Fu and C. H. Wang, Biomaterials, 2010, 31, 8732–8740 CrossRef CAS.
- A. Choi, K. D. Seo, D. W. Kim, B. C. Kim and D. S. Kim, Lab Chip, 2017, 17, 591–613 RSC.
- Q. Xu, H. Qin, Z. Yin, J. Hua, D. W. Pack and C. H. Wang, Chem. Eng. Sci., 2013, 104, 330–346 CrossRef CAS.
- C. S. Ho, J. W. Kim and D. A. Weitz, J. Am. Chem. Soc., 2008, 130, 9543–9549 CrossRef.
- E. J. Cho, H. Holback, K. C. Liu, S. A. Abouelmagd, J. Park and Y. Yeo, Mol. Pharm., 2013, 10, 2093–2110 CrossRef CAS.
- T. L. Sounart, P. A. Safier, J. A. Voigt, J. Hoyt, D. R. Tallant, C. M. Matzke and T. A. Michalske, Lab Chip, 2007, 7, 908–915 RSC.
- D. Gao, H. Li, N. Wang and J.-M. Lin, Anal. Chem., 2012, 84, 9230–9237 CrossRef CAS.
- L. Sainiemi, T. Sikanen and R. Kostiainen, Anal. Chem., 2012, 84, 8973–8979 CrossRef CAS.
- P. Mao, R. Gomez-Sjoberg and D. Wang, Anal. Chem., 2013, 85, 816–819 CrossRef CAS.
- J. Op De Beeck, M. Callewaert, H. Ottevaere, H. Gardeniers, G. Desmet and W. De Malsche, Anal. Chem., 2013, 85, 5207–5212 CrossRef CAS.
- S.-L. Lin, H.-Y. Bai, T.-Y. Lin and M.-R. Fuh, Electrophoresis, 2012, 33, 635–643 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |