
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmunomodulatory and antimicrobial non-mulberry Antheraea mylitta silk fibroin accelerates in vitro fibroblast repair and regeneration by protecting oxidative stress†
Sohini Sena,
Shaunak Ghosh‡
 b,
Sayantan De‡b,
Piyali Basak*a,
Praveen Mauryec,
Nandan Kumar Janab and
Tapan Kumar Mandald
b,
Sayantan De‡b,
Piyali Basak*a,
Praveen Mauryec,
Nandan Kumar Janab and
Tapan Kumar Mandald
aSchool of Bioscience and Engineering, Jadavpur University, 188, Raja S.C. Mullick Road, Kolkata-700032, West Bengal, India. E-mail: piyalibasak@gmail.com; Tel: +91-33-2414-6666 ext. 2958
bDepartment of Biotechnology, Heritage Institute of Technology, Chowbagha Road, Anandapur, PO: East Kolkata Township, Kolkata 700107, West Bengal, India
cCentral Inland Fisheries Research Institute, Barrackpore, Monirampore, Kolkata 700120, India
dVeterinary Pharmacology & Toxicology, West Bengal University of Animal & Fishery Sciences, 37&68 Kshudiram Bose Sarani, Kolkata 700037, India
First published on 28th May 2021
Abstract
The antimicrobial nature of Antharaea mylitta silk-fibroin (SF) is reported but antioxidant potential and the immunomodulatory role towards the fibroblast cell repair process is not explored. Polyurethane is reported to have inflammatory potential by mononuclear cells directed cytokine release, which can guide fibroblast repair. Present study demonstrates the conjunctive effect of inflammatory PU/SF to regulate the favorable shift from pro-inflammatory to anti-inflammatory cytokine stimulation for accelerated fibroblast repair. Minimal inhibitory concentration of SF was determined against pathogenic strains and the effect of SF was investigated for fibroblast NIH3T3 cell adhesion. SF doses (8, 8.5, 9 mg mL−1) were found to be greater than both the IC50 of DPPH scavenging and the ED50 for NIH3T3 proliferation. Anti-lipid peroxidase (ALP) activity of SF doses and citric acid-treated NIH3T3 cells were compared under hydrogen peroxide (H2O2) induced oxidative stress. 9 mg mL−1 SF showed greater ALP activity than the citric acid standard. SF-driven protection to oxidative damage was measured by viable cell fraction in trypan blue dye exclusion assay where 9 mg mL−1 SF showed the highest viability (p ≤ 0.05). 9 mg mL−1 SF was blended with PU for scaffold (w/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 2:7, 2:9) fabrication. The protective effect of PU/SF (2:5, 2:7, 2:9) against oxidative stress was verified by damaged cell survival in MTT assay and DNA quantification. The highest number of cells survived on PU/SF (2:9) at all intervals (p ≤ 0.01) upon oxidative damage; PU/SF (2:9) was also fabricated by employing the immobilization technique. Immobilized PU/SF (2:9) exhibited a greater zone of microbial inhibition, a higher extent of inhibition to microbial adherence, and caused more LDH release from bacterial cell membrane due to membrane rupture, resulting in bacterial cell death (E. coli, K. pneumoniae, P. aeruginosa, S. aureus) compared to the experimental results shown by blended PU/SF (2:9). The protective nature of PU/SF (2:9) against oxidative stress was ensured through the LDH activity of damaged NIH3T3 cells. Initial raised IL-6, TNF-alpha (pro-inflammatory cytokines) and lowered IL-8, IL-10 (anti-inflammatory cytokine) profiles coupled with fallen IL-6, TNF-alpha, and elevated IL-8, IL-10 at later hours synergistically progress the inflammatory phase of in vitro scratch wound repair in mononuclear culture treated by PU/SF (2 : 9).
:5, 2:7, 2:9) fabrication. The protective effect of PU/SF (2:5, 2:7, 2:9) against oxidative stress was verified by damaged cell survival in MTT assay and DNA quantification. The highest number of cells survived on PU/SF (2:9) at all intervals (p ≤ 0.01) upon oxidative damage; PU/SF (2:9) was also fabricated by employing the immobilization technique. Immobilized PU/SF (2:9) exhibited a greater zone of microbial inhibition, a higher extent of inhibition to microbial adherence, and caused more LDH release from bacterial cell membrane due to membrane rupture, resulting in bacterial cell death (E. coli, K. pneumoniae, P. aeruginosa, S. aureus) compared to the experimental results shown by blended PU/SF (2:9). The protective nature of PU/SF (2:9) against oxidative stress was ensured through the LDH activity of damaged NIH3T3 cells. Initial raised IL-6, TNF-alpha (pro-inflammatory cytokines) and lowered IL-8, IL-10 (anti-inflammatory cytokine) profiles coupled with fallen IL-6, TNF-alpha, and elevated IL-8, IL-10 at later hours synergistically progress the inflammatory phase of in vitro scratch wound repair in mononuclear culture treated by PU/SF (2 : 9).
1. Introduction
Tissue engineering implies tissue regeneration within the biomimetic scaffold to implant functional tissue where tissue functionality depends on a suitable microenvironment for an optimum cellular response. Such an environment is provided by a 3-dimensional tissue graft, which behaves as an architectural template for tissue growth-functionality.1 The choice of suitable biomaterials is a major concern for tissue repair. Few biomaterials are naturally immunomodulatory and fewer are designed to attain it.2 It is reported that the tissue-engineered graft transforms into a mature tissue via an inflammation-mediated process of remodelling.3,4 In this context, silk fibroin (SF) has attracted attention in various tissue engineering fields5–9 considering the immunogenicity and bio-resorbability aspects. Non-mulberry SF from different varieties shows distinct species specificity.9 In an earlier study, the in vivo inflammatory effects of non-mulberry SF (Antheraea mylitta) were reported10 but no in vitro study has been reported to date on its prospective antioxidant role and immunomodulatory potential and precise balancing shift between pro-inflammatory and anti-inflammatory cytokines. Both mulberry and non-mulberry SF support the growth and proliferation of fibroblasts and keratinocytes.6 Non-mulberry SF lack light (L) chain and contains only heavy (H) chain molecular weight of ∼380 kDa (ref. 11) and richer RGD sequences compared to a mulberry variety.4,5 However, to our knowledge, no study has been carried out to evaluate the precise immunomodulatory role played by A. mylitta SF upon in vitro scratch wound (it is an in vitro model to demonstrate wound healing under in vitro conditions) assay. Since polymeric properties can be tailored by surface functionalization, formulating with bioactive molecules and developing newer strategies are a major focus for current tissue regenerative research. Polyurethane (PU) has already been used as a wound-healing agent in commercial dressings such as Tegaderm™ and Bioclusive™ (ref. 12–16) due to its high biocompatibility, cell adhesiveness, and non-immunogenicity. Commercial PU dressings have few limitations such as less antimicrobial nature, and lack of regulated inflammatory response to both in vitro hyperglycemic and normal wound microenvironment. This study focuses on SF incorporation to exploit its antioxidant, immunomodulatory, and antimicrobial efficiency into PU. The antioxidant activity of non-mulberry silk sericin is known but the antioxidant property of non-mulberry (A. mylitta) SF towards oxidative stress is not investigated. As A. mylitta silk sericin17 contains 40% polar hydroxy amino acids residue such as serine and threonine that are responsible for antioxidant activity and A. mylitta SF has a high content of hydroxy amino acids, peptide length, and molecular weight (10–400 kDa), it is anticipated to have antioxidant property. Increased reactive oxygen species (ROS) can damage various cellular processes. Hydrogen peroxide (H2O2) is an established oxidant model as its cellular actions and fate is well studied. Hydrogen peroxide generates hydroxyl groups, reacting with different transition metals that damage DNA, leading to mutagenesis and carcinogenesis.18 Dose-dependent antioxidant effect of A. mylitta SF against H2O2-induced oxidative damage has been investigated in this study. Non-mulberry SF has well established antimicrobial properties;19 the antimicrobial potential of varying concentration of SF in different matrices was investigated in terms of the microbial adherence inhibition and zone of inhibition study and further verified by the amount of LDH released in the corresponding microbial culture. PU/SF was fabricated in different weight ratios [(w/w) = 2:5, 2:7, 2:9] to identify the cell proliferation effect with varying SF concentration. The most effective SF dose was incorporated in PU/SF (w/w = 2:9) by the immobilization technique. In vitro cell proliferation, free radical scavenging activity, and inflammatory response were collectively investigated by both blended and immobilized PU/SF to implement it in wound healing or fibroblast repair application. A strategy for modulating fibroblast phenotype is choosing potential inflammatory biomaterials. The effect of Moxifloxacin on the production of proinflammatory cytokines from Human Peripheral Blood Mononuclear Cells (PBMC) has been reported.20 Potential inflammatory biomaterials can attenuate the pro-inflammatory response to promote in vitro cell growth, migration, as well as positive tissue remodeling outcomes in vivo.21,22 It is common practice to use cytokine-induced proliferation23,24 by modifying the inflammatory scaffolds. Monocytes are known to secrete cytokines and regulate the neo-vessel formation; thus, monocytes are often seeded for in vivo wound healing.25 The pro-and anti-inflammatory cytokine profile released from monocytes is biomaterial dependent,26,27 which regulates cellular response. Polyurethane alone supports interactions with immune cells;28,29 hence PU was strategically coupled with SF with the aim of enhancing the monocyte/macrophage cytokine activity and promoting further fibroblast proliferation and repair. This study highlights the importance of PU/SF directed IL-6 and IL-8 release by the monocytes, which are important contributors to cellular regeneration. PU/SF elicited IL-8 showed mitogenic activity on keratinocyte proliferation, which is supported by Rennekampff et al.30 When mononuclear cells are cultured on PU, the conditioned media secretes IL-6, IL-8, IL-10, and TNF-alpha, which would have a positive impact on fibroblast or keratinocytes infiltration and the inflammation phase of wound healing, as corroborated by the results of Battiston et al.31 No study is available on how SF contributes towards the immune response of IL-6, IL-8, IL-10, and TNF-alpha profile by mononuclear cells cultured on inflammatory PU/SF under in vitro wound stimulus. Pro-inflammatory cytokines such as IL-6 and TNF-alpha initiate the repair process by recruiting inflammatory cells to the scratched fibroblasts cell membrane acting as mitogens. The absence or low levels of these cytokines is associated with impaired healing, while excessive levels result in enhanced fibrotic scarring.32 Pro-inflammatory cytokines such as TNF-alpha, IL-6, and MCP-1 promote proliferative, migratory fibroblast, and keratinocyte phenotype,33–35 while the anti-inflammatory cytokine IL-10 inhibits proliferation.36 In our results, up to 48 h, TNF-alpha and IL-6 showed hiked release, which is supported by findings of Thomay et al.37 and Shinozaki et al.38 Afterwards, a desired balanced shift was required from pro-inflammatory towards anti-inflammatory cytokine production for successful cell repair. Monocyte adherence to PU/SF favors the initial hike in the inflammatory cytokines, which are beneficial as long as the biomaterial supports cytokine-mediated phenotypic transition towards repair and regeneration that enables neo-tissue formation and not chronic inflammation.39–42
2. Materials and methods
2.1 Extraction and identification of SF
Silk fibroin was extracted following the standard procedure described earlier from non-mulberry Antheraea mylitta.10 Reconstituted SF was stored at 4 °C and the total SF protein was identified by molecular weight confirmation using 12% resolving gel standard procedure in SDS–PAGE.432.2 Estimation of minimum inhibitory concentration (MIC) for dose selection
Though the antimicrobial property of non-mulberry SF is known against commonly found Gram-positive and Gram-negative pathogenic strains, its minimum inhibitory concentration MIC has been reported in our earlier for these bacterial strains such as Escherichia coli (ATCC 25922), Staphylococcus aureus (ATCC 6538), Pseudomonas aeruginosa (ATCC 15442), and Klebsiella pneumoniae (ATCC 6538). The detailed procedure of obtaining minimum inhibitory concentrations (MIC) for K. pneumoniae (ATCC 6538), E. coli (ATCC 25922), S. aureus (ATCC 6538), and P. aeruginosa (ATCC 15442) have been described in our earlier work.10 To prevent microbial invasion, the MIC of SF for these strains were determined and then SF dose fixation was performed, choosing above the highest MIC values among these four strains. This fixed-dose of SF (above highest MIC) was selected for scaffold fabrication.2.3 Preparation of polyurethane–SF composite scaffold
The dose selection for SF to incorporate into the scaffold by minimum inhibitory concentration (MIC) of SF against Escherichia coli (ATCC 25922), Staphylococcus aureus (ATCC 6538), Pseudomonas aeruginosa (ATCC 15442), Klebsiella pneumoniae (ATCC 6538), and isophorone diisocyanate (IPDI, Sigma Aldrich)-based PU scaffold fabrication procedure has already been described elaboratively in our earlier study.10 Briefly, the PU/SF porous matrices were prepared by lyophilization in three different weight ratio of PU:SF (2:5, 2:7, and 2:9) with 9 mg mL−1 SF concentration of the standard blending procedure. Detailed synthesis mechanism is shown in Fig. 1a. Another bare Isophorone diisocyanate (IPDI, Sigma Aldrich)-based PU was lyophilized, vacuum-dried under room temperature, and subjected to surface modification44 to obtain the immobilized PU/SF matrix (2:9). The lyophilized bare PU membranes were taken out for surface aminolysis. PU matrices (12.14 g, circular diameter 4.12 cm) were reacted with 7 wt% 1,6-hexane diamine solution (60 mg mL−1) in 2-propanol for 10–15 min at 37 °C to allow aminolysis, followed by extensive washing in distilled water for a day to remove the unbound free 1,6-hexane diamine. The washed matrices were dried and reacted for 3 h in glutaraldehyde [1.5% (w/v)] solution to turn the surface-exposed amino groups into aldehyde groups at room temperature. Again, extensive rinsing was done with a large amount of deionized water to remove free glutaraldehyde. The solution was discarded keeping the membrane intact. Then, it was rinsed with de-ionized water for 24 h at room temperature to remove the extra 1,6-hexane diamine. Then, the membranes were dried by a vacuum dryer. Further, the aminolyzed PU membranes were immersed in 1 wt% glutaraldehyde for 3 h at room temperature to transform the amino groups into aldehyde groups, followed by rinsing with a large amount of deionized water to remove free glutaraldehyde. Then, the aminolyzed PU was immersed overnight in 9 mg mL−1 SF at 4 °C by the immobilization mechanism (Fig. 1b).
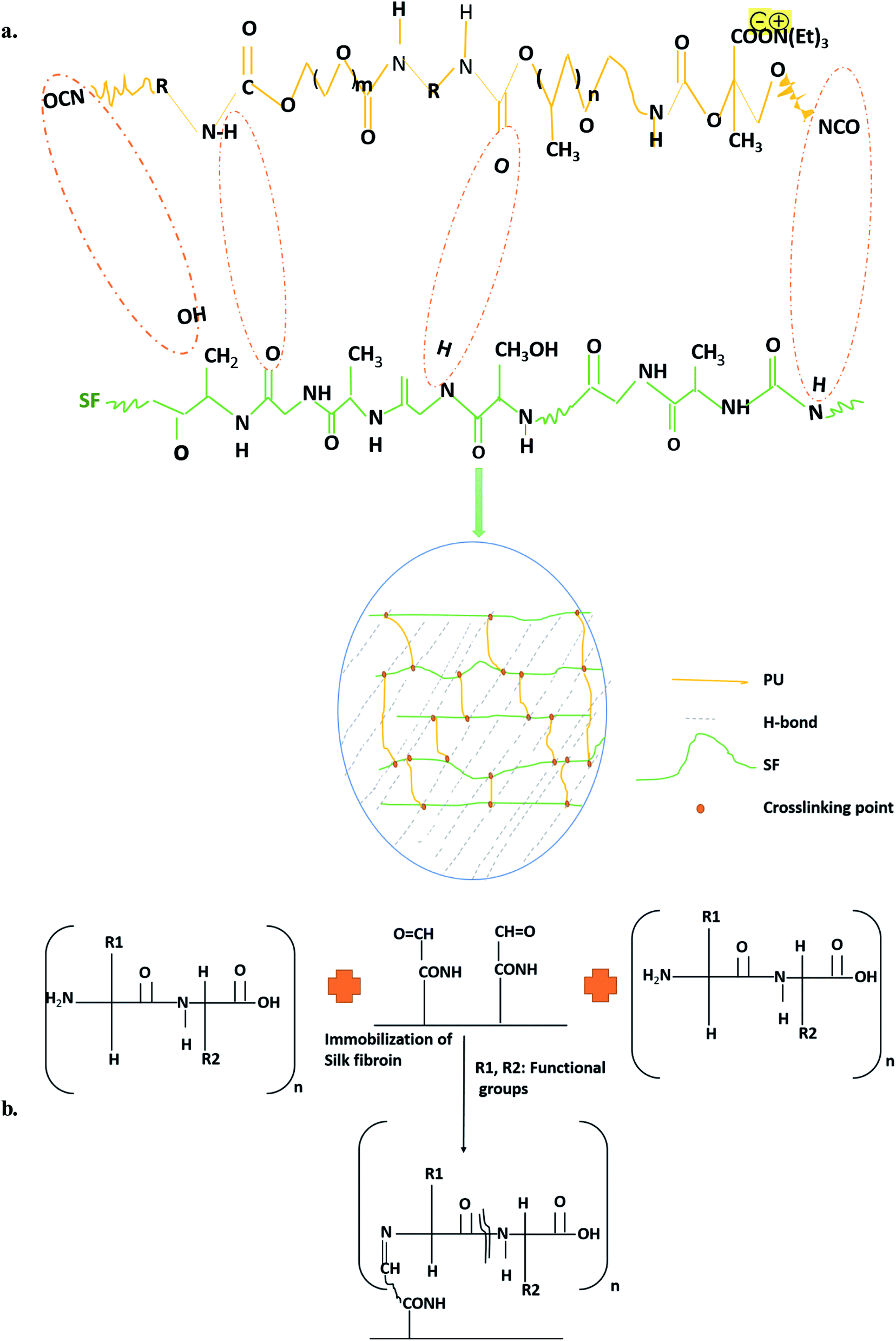 | ||
| Fig. 1 Schematics of synthesis of (a) blended and (b) immobilized PU/SF scaffold. | ||
2.4 Physico-chemical characterization
Fourier transform infrared spectroscopy of the extracted silk fibroin, bare PU scaffold (2 × 2 cm), and PU/SF scaffolds (2 × 2 cm) was carried out (FT-IR Prestige-21 Shimadzu make, Japan) in the 4000–400 cm−1 range in the attenuated total reflectance mode. The presence of urethane (PU) and amide linkages was investigated to confirm the presence of both SF and PU in the blended and immobilized PU/SF. The absorbance spectra were measured at room temperature and 50% relative humidity.45 The data were analyzed using Origin 9.0 software. The average pore size of the scaffolds was evaluated using Image analysis 2.1 software (NIH, USA). For each scaffold, 30 different measurements were considered to calculate the pore size through microcomputed tomography. Briefly, dried nanofibers were placed on a sample holder and gold-coated for 30 s with a plasma coater under high vacuum. The diameter of the scaffold pores was measured using the ImageJ software. The overall number of micrographs that were analyzed was 100 counts. Images were captured at three different magnifications: 1 mm, 100 μm, and 500 μm. Clear images of the pores were observed in 1 mm magnification. The pore size, diameter, and density were calculated from the microscopy images with the ImageJ software. Atomic force microscopic (AFM) studies evaluated the surface roughness and microstructure, including the three-dimensional structures of bare PU (2 × 2 cm), blended PU/SF (2 × 2 cm), and the immobilized PU/SF (2 × 2 cm) scaffold. Vacuum-dried scaffolds were placed on a holder with the aid of carbon-coated tape and scanned under AFM (Multiview 3000, Nanonix Imaging Limited, Israel) using the tapping mode.2.5 Microbial experiments
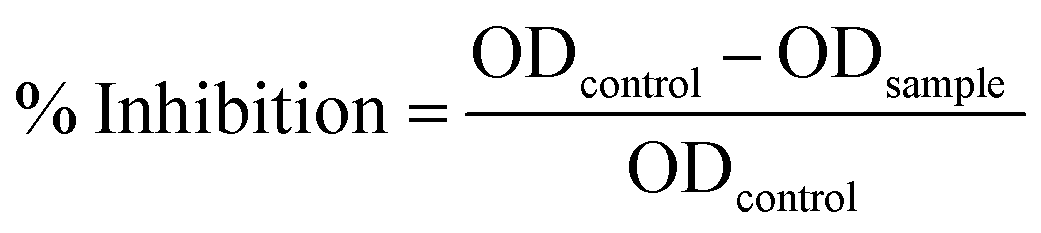 :7, 2:9) matrices was only performed for this study. Blended PU/SF matrices were sterilized and tailored into a circular sterile disk of 1 cm diameter. The antimicrobial sensitivity of these PU/SF matrices along with bare PU (as control) was verified using the standard disk diffusion method47–49 against E. coli, S. aureus, P. aeruginosa, and K. pneumoniae. Zones exhibited by the PU/SF matrices (circular disks of 1 cm diameter) were measured at 48 h of incubation at 37 °C.
:7, 2:9) matrices was only performed for this study. Blended PU/SF matrices were sterilized and tailored into a circular sterile disk of 1 cm diameter. The antimicrobial sensitivity of these PU/SF matrices along with bare PU (as control) was verified using the standard disk diffusion method47–49 against E. coli, S. aureus, P. aeruginosa, and K. pneumoniae. Zones exhibited by the PU/SF matrices (circular disks of 1 cm diameter) were measured at 48 h of incubation at 37 °C.2.6 Determination of effective dose (ED50) of SF for cell proliferation
The ED50 value of SF was calculated by NIH3T3 mouse fibroblast cell viability through the MTT assay to further assess the final concentration to be incorporated while fabricating the scaffolds. In this assay, 24-well plates were coated with varying concentrations ranging from 1 mg mL−1 to 9 mg mL−1 SF. SF-coated well plates were pre-incubated overnight and mouse foreskin fibroblast NIH3T3 cells were seeded at a seeding density of 1 × 105 cells per well on SF-coated 24-well plates and incubated overnight. Confluent cell growth was attained in a controlled atmosphere (Leishmen Shanghai CO2 Incubator 37 °C, 5% CO2) using Dulbecco's Modified Eagle's Medium (DMEM, Hi-Media, India), 10% Fetal Bovine Serum (FBS, Hi-Media, India), and penicillin–streptomycin antibiotic solution (Hi-Media, India). Confluent monolayers were harvested by trypsinization (0.25% Trypsin and 0.02% EDTA, HiMedia, India). MTT reagent was added to 10% of the total volume of cell suspension in each well and incubated in dark condition for 4 h. Then, 100 μL of the solubilizing agent was poured into each well and kept in a gyratory shaker for 30 min to enhance the dissolution of the crystals. The solution in each well was mixed well to ensure the proper mixing of the crystals. The percent cell viability of the cell was calculated using the following equation.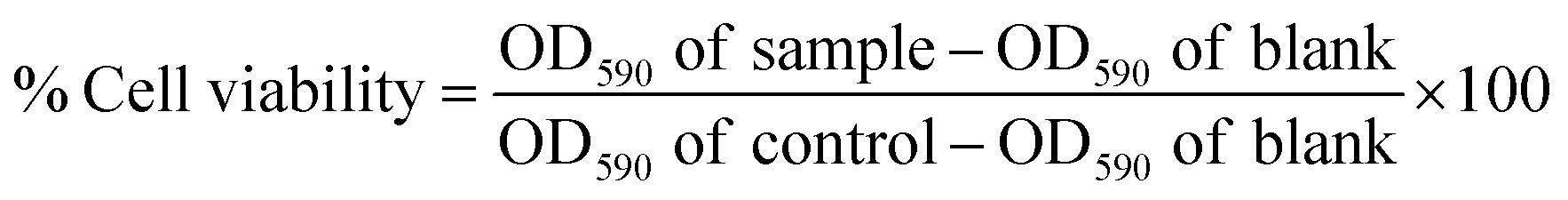
ED50 was calculated as the SF concentration corresponding to 50% viable cells.
Cell proliferation was determined through MTT and was performed on four types of scaffolds, namely, PU/SF blended (2:5), PU/SF blended (2:7), PU/SF blended (2:9), and PU/SF immobilized (2:9) in triplicate; 5 mm scaffolds discs were sterilized and pre-incubated in the media at the bottom of 96-well plates overnight. At the third passage of cell maintenance, the cells were seeded on the pre-incubated scaffolds at a seeding density of 2 × 105 cells per well, followed by incubation up to 72 h. In vitro, MTT assay is a metabolic indicator of the mitochondrial activity of live cells where MTT tetrazolium dye turns formazan into purple color, indicating the functioning of mitochondria in live cells. This phenomenon is an indirect notification of cell viability on PU/SF matrices.
To check the NIH3T3 cell adhesion in SF concentration-dependent manner, after seeding the cells at a density of 5 × 104 cells per well in serum-starved media, 8 mg mL−1, 8.5 mg mL−1, and 9 mg mL−1 SF was coated into well plates and incubated up to 24 h. Then, the wells were washed with PBS thrice, the adhered cells were fixed with 4% paraformaldehyde in PBS, washed with PBS, treated with 0.1% Triton X-100 to permeabilize the cell membranes for 10 min, and subsequently stained with DAPI for nuclear staining. The cells were observed under a fluorescence microscope and counted in three independent fields.
2.7 Immunostaining for fibroblast proliferation and DNA quantification assay
After 5 and 7 days of culture, cell suspension scaffolds were fixed using formaldehyde 3.7% (v/v) for 15 min, followed by 2–5 min 0.1% Triton X-100 in PBS treatment and washing twice with PBS. 1% BSA was added into each well to block them at 4 °C in a humid chamber for 3 h. Ki-67 primary antibody (Abcam, ab16667) (1:15000) was added to each well and incubated overnight. HRP conjugated secondary antibody (Elabscience, China) (1:200) in 1% goat serum was added and incubated for 2 h at room temperature and washed in PBS thrice. The cells were stained with DAB (brown) and the nuclei were counterstained with hematoxylin (blue). Ki-67 expression detected in NIH3T3 cells on two types of PU/SF was visualized using the following manufacturer's protocol of DAKO Envision Plus-HRP (DAB) kit.
Cell proliferation was further verified by DNA quantification following the earlier described protocol.50 The total DNA content was harvested from all the varieties of scaffolds. Scaffolds were frozen in ultrapure water (1.5 mL), thawed, and the sonicated cell pellet was washed with ice-cold PBS (pH 7.4) and centrifuged (1000 rpm, 10 min). Cell lysates (1 × 106 cells/0.4 mL PBS) were prepared by sonication over ice for 20 s–30 s to facilitate DNA extraction into the solution. The resultant solution was centrifuged at 500g for 5 min to eliminate the scaffold debris. From the supernatant, DNA was extracted (850 μL) and heated in 10× TE buffer (95 μL) with 1.5 M NaCl and 5% SDS was added to the DNA portion. To eliminate the residual RNA and protein part, the solution was treated with RNase (20 mg) and Proteinase K (800 mg). The solution was chloroform extracted and DNA was precipitated with isopropanol, followed by washing with 70% ethanol and finally dissolving in 10× TE buffer. The DNA content extracted from the scaffold was determined by absorbance readings at 260 and 280 nm.
2.8 Free radical scavenging activity of silk fibroin
where A0 is the absorbance of the control (water) and A1 is the absorbance of SF at various concentrations. Obtained % DPPH inhibition is indicative of whether these three concentrations are above or below IC50. If the obtained % DPPH radical scavenging activity is more than 50%, then it indicates the chosen concentration to be more than IC50 and vice versa. Then, the %inhibition was plotted against concentration, and from the graph, IC50 was calculated.
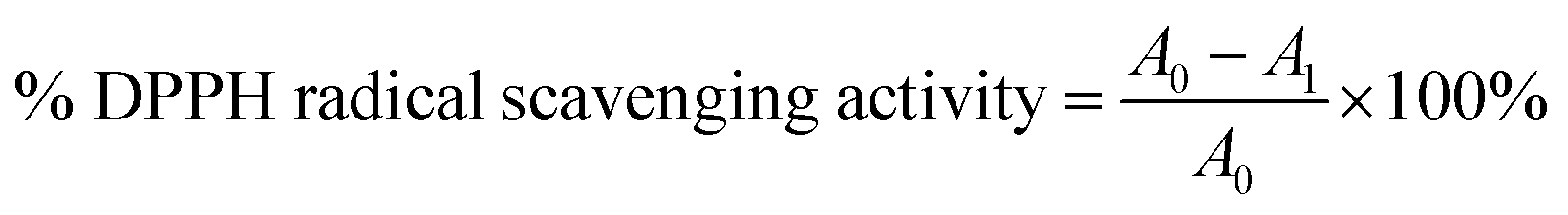 000 rpm, 10 min). The supernatant (200 μL) was taken in a 96-well plate to record the absorbance at 535 nm. The same experiment was performed to determine the absorbance in peroxidation (with H2O2) induced cells and the control. Citric acid was used as the positive control in this study. The percentage of ALP was estimated using the following formula.
000 rpm, 10 min). The supernatant (200 μL) was taken in a 96-well plate to record the absorbance at 535 nm. The same experiment was performed to determine the absorbance in peroxidation (with H2O2) induced cells and the control. Citric acid was used as the positive control in this study. The percentage of ALP was estimated using the following formula.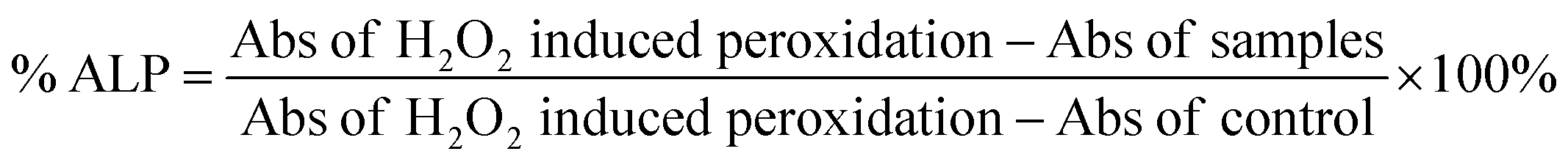
2.9 Protective activity assessment against hydrogen peroxide-induced oxidative stress
:5, 2:7, 2:9) and incubated for 24 h, which was used for carrying out the MTT assay as described in the previous section.:9) matrices (2 × 2 cm) were selected for hydrogen peroxide-induced oxidative stress protection experiment as only these two scaffolds offered the best proliferation (from cell viability assay) and the corresponding SF concentration (9 mg mL−1) showed the highest DPPH free radical scavenging potential. For this biochemical experiment, NIH3T3 cells were pre-incubated in blended and immobilized PU/SF (2:9) matrices for 24 h, followed by 24 h oxidative stress induction by 0.5 mM hydrogen peroxide treatment. The cells were washed with ice-cold PBS and harvested with 0.025% Trypsin–0.02% EDTA solution. The cells were re-suspended in ice-cold PBS and homogenized. The cell homogenates were centrifuged for 5 min at 5000g and the supernatant collected from each sample was taken for lactate dehydrogenase (LDH) tests.52 Fibroblast damage caused by hydrogen peroxide was quantitatively assessed from the ratio of the lactate dehydrogenase enzyme released from the damaged cells to the activity in undamaged/intact cells, monitored 24 h after the induction of stress. The medium collected after stress induction was centrifuged, and the supernatant was treated with LDH reaction solution (LDH kit, Biotechnology Co., Ltd., Wuhan, China) and incubated for 30 min. After incubation, the optical density of the samples was taken at 450 nm for the assay of the LDH enzyme released from the damaged cells.512.10 Isolation of PBMCs
Blood was collected by retro-orbital puncture from healthy rats, following the guidelines of Institutional Animal Ethical Committee (CPCSEA; IAEC vide letter no: AEC/PHARM/1702/22/2017) of Jadavpur University. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Jadavpur University. Animals were maintained with utmost care, strictly adhering to the Institutional Animal Ethical Committee's guidelines at the animal house of Jadavpur University and experiments were approved by the Animal Ethics Committee of Jadavpur University. Blood collection was performed in compliance with relevant Institutional guidelines. PBMCs were separated by density gradient separation method. Rat blood (3 mL) was mixed with anticoagulant agent EDTA and was diluted with normal saline solution (0.9%) to 1:3 (w/w) ratio. Diluted blood (7.5 mL) was added to Ficoll-Paque (2.5 mL) (Sigma Aldrich) to develop a density gradient in the solution. The resultant solution was centrifuged at 5000 rpm for 30 min to filter the blood mononuclear cells with a white ring-like appearance. The cells were washed twice with PBS and serum-starved RPMI-1640 medium. The cells were re-suspended in two different media, i.e., RPMI 1640 with high glucose (A1049101, Thermofisher scientific) and RPMI 1640 (Gibco) without the glucose media. For both the variants of the RPMI 1640 media, 10% Fetal Bovine Serum (FBS) and 1% penicillin–streptomycin were supplemented. PBMC were cultured in a humidified incubator with 5% CO2 at 37 °C at a density of 106 cells/mL and seeded onto bare PU, blended PU/SF (2:9), and immobilized PU/SF (2:9) scaffolds in triplicate (n = 3) at a seeding density of 106 cells per well after the 2nd passage.
2.11 In vitro cytokine release
In our earlier work,53,54 pro-inflammatory and anti-inflammatory cytokine response by two types of PU/SF treated diabetic and non-diabetic wound in vivo model was recorded compared to the control. In reference to that, the in vivo inflammatory response elicited by two varieties of PU/SF, namely, in vitro pro-inflammatory and anti-inflammatory cytokine response by scratch wound created on PBMC (individually cultured on blended and immobilized PU/SF) in both high and glucose deprived media, were investigated. In vitro scratch wound study in high and glucose deprived media was carried out to mimic the inflammatory status in a hyperglycemic and non-hyperglycemic wound in vivo. After 24 h of seeding on PU, blended PU/SF (2:9) matrices (2 × 2 cm), and immobilized PU/SF (2:9) matrices (2 × 2 cm), a scratch was made with a micropipette (10 μL) onto the PBMC grown on each variety of scaffolds in a culture dish, both maintained individually in RPMI 1640 medium supplemented with high glucose (A1049101, Thermofisher scientific) and RPMI medium 1640 (Gibco) without glucose media to investigate the elicited response of IL-6, IL-8, IL-10, and TNF-alpha. The study was performed following the earlier described method.59 After making the scratch, the cells were precisely collected from the edge of the scratches and harvested from the corresponding scaffolds with Trypsin–EDTA. Cells were washed with PBS twice, lysed by RIPA lysis buffer, and incubated for 1 h at 4 °C to obtain the cell lysates. IL-6, IL-8, and IL-10 levels were quantified by cell lysates using the manufacturer's instructions of rat specific IL-6, IL-10, TNF-alpha ELISA kit (Elabscience, China), and IL-8 (Raybiotek, China).
2.12 Statistical analysis
Statistical analysis for all the relevant experiments was performed using Origin (version 9) by one-way ANOVA followed by Tukey's post hoc test. All the in vitro experiments were evaluated in triplicates and expressed as mean values ± standard deviation. In one-way ANOVA, the significance level of 95%, 99% and 99.9% was determined at p ≤ 0.05, p ≤ 0.01 and p ≤ 0.001.3. Results
3.1 Antheraea mylitta SF identification
In SDS–PAGE, only 390 kDa SF protein bands were obtained, which indicates the presence of an SF heavy chain and the confirmation of proper extraction of non-mulberry (Antheraea mylitta) SF variety (ESI Fig. 1†).3.2 Minimal inhibitory concentration and dose selection, antimicrobial sensitivity, microbial adherence preventing potential, and lactate dehydrogenase activity
To prepare the antimicrobial PU/SF scaffold, the inherent antimicrobial potential of SF was utilized and not exploited in vitro drug loading. Thus, it has been strategized that the amount or SF concentration will be higher than their minimal inhibitory concentrations against individual strains. Microbial sensitivity of SF concentration was first verified at 4 mg mL−1 against all the four strains by measuring the OD600 values but no inhibition was obtained at this SF concentration (Table 2). Secondly, 4.8 mg mL−1 inhibited only E. coli growth completely but could not inhibit the growth of S. aureus, P. aeruginosa, and K. pneumoniae [Fig. 2(a1)]. In the third trial, 5.6 mg mL−1 SF completely inhibited S. aureus growth consistently up to 72 h [Fig. 2(a2)]. Since this dose was not sufficient to completely cease the other Gram-negative strains growth, increasing the SF concentration to 6.4 mg mL−1, SF was randomly tested to combat P. aeruginosa growth and it stopped P. aeruginosa growth completely up to 72 h but was unable to limit the growth of K. pneumoniae (Fig. 2(a3)). Bare PU could not inhibit the growth of any strain (ESI Fig. 3(a1) and (a2)†). In each strain's growth kinetics curve, three different phases were observed, i.e., lag phase, exponential phase, and a plateau. At 8 mg mL−1 SF, it was capable to inhibit the complete growth of K. pneumoniae (Fig. 2(a4)) as well as the entire growth kinetic curve for four strains. Thus, 8 mg mL−1 concentration was found to be the effective minimum inhibitory concentration to cease the growth of all the studied pathogenic stains up to 72 h. Above 8 mg mL−1, SF was selected to be incorporated into the scaffold matrix.| RMS roughness (μm) | Avg roughness (μm) | Mean ht (μm) | Median ht (μm) | Projected area (μm2) | Surface area (μm2) | Valley (μm) | Peak (μm) | |
|---|---|---|---|---|---|---|---|---|
| PU | 197.6 | 468 | 3.88 | 3.978 | 225 | 1403 | −3.88 | 4 |
| Blended PU/SF | 534 | 425 | 1.389 | 1.357 | 225 | 276.9 | −1.389 | 3.819 |
| Immobilized PU/SF | 464.38 | 377.7 | 1.615 | 1.725 | 100 | 113 | −1.615 | 687 |
| A. mylitta silk fibroin (SF) | Micro-organism | |||
|---|---|---|---|---|
| P. aeruginosa | K. pneumoniae | E. coli | S. aureus | |
| a ‘+’ indicates growth of bacteria (resistant to SF), ‘−’ indicates inhibition of growth of bacteria (sensitive to SF). | ||||
| 4 mg mL−1 | + | + | + | + |
| 4.8 mg mL−1 | + | + | − | + |
| 5.6 mg mL−1 | + | + | − | − |
| 6.4 mg mL−1 | − | + | − | − |
| 8 mg mL−1 | − | − | − | − |
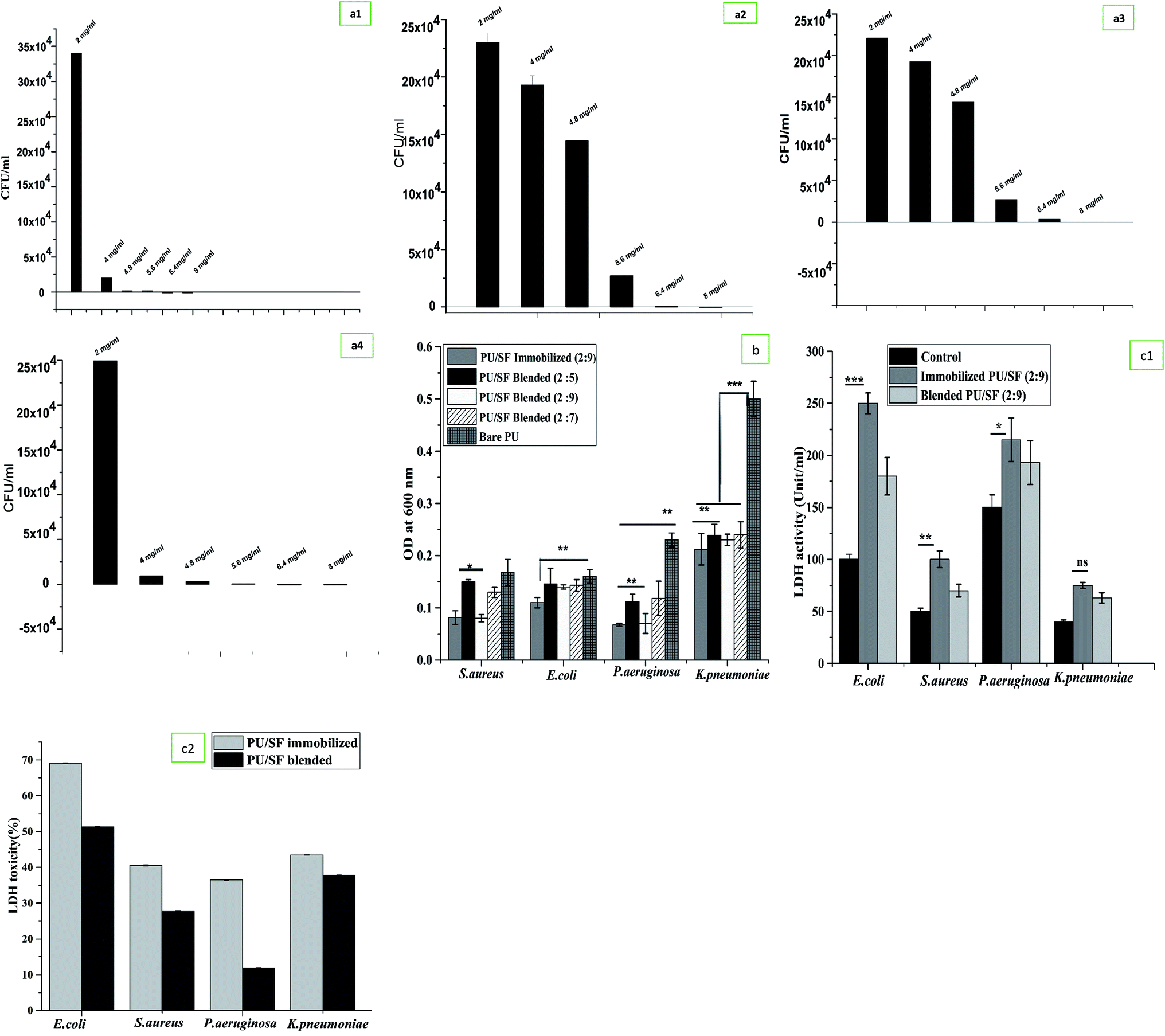 | ||
| Fig. 2 (a) Minimal inhibitory concentrations determination against (a1) E. coli, (a2) P. aeruginosa, (a3) K. pneumoniae, (a4) S. aureus against various concentration of SF. (b) Microbial adherence inhibition to different bacterial strains by different PU/SF matrices. Y-error bars represent standard deviation. (c1) Bacterial LDH release assay upon exposure to blended PU/SF and immobilized PU/SF; Y-error bars represent standard deviation. (c2) LDH cytotoxicity analyses of bacterial upon exposure to blended PU/SF and immobilized PU/SF; Y-error bars represent standard deviation. | ||
Inhibition of microbial adherence was evaluated in a dose-dependent manner of PU/SF (2:5), PU/SF (2:7), and PU/SF (2:9) ratio using S. aureus, E. coli, P. aeruginosa, and K. pneumoniae strains (Fig. 2b). All the absolute concentrations of SF in all the doses of PU/SF, i.e., the weight ratios of 2:5, 2:7, and 2:9 were selected above the MIC (8 mg mL−1). In the control, no treatment was given in the bacterial culture. With the increasing concentration of SF, the microbial load decreased accordingly, resulting in stronger prevention of the adherence of all the strains with ascending concentrations of SF in the matrix. Blended PU/SF (2:9) and immobilized PU/SF (2:9) has almost similar adherence inhibiting efficiency against S. aureus and P. aeruginosa but immobilized PU/SF (2:9) showed greater microbial prevention against E. coli and K. pneumoniae. Microbial adherence inhibition by blended PU/SF (2:9) and immobilized PU/SF (2:9) compared to PU/SF blended (2:5) are statistically significant (p ≤ 0.05) for S. aureus, and statistically significant (p ≤ 0.01) against P. aeruginosa. Preventing the potential of immobilized PU/SF (2:9) is significant (p ≤ 0.01) against E. coli, while K. pneumoniae adherence towards all PU/SF groups is significantly (p ≤ 0.001) lesser than that of K. pneumoniae adhered to the control (bare PU) group.
In the disk diffusion test, the concentration of SF (PU/SF: 2:9) was chosen above the highest MIC value considering all the strains, and it was compared between blended PU/SF (2:9) and immobilized PU/SF (2:9). The zone of inhibition against E. coli, K. pneumoniae, and P. aeruginosa exhibited by the immobilized scaffolds was strikingly more and statistically significant at p ≤ 0.001, p ≤ 0.01, and p ≤ 0.05 level with that of the blended variety (Table 3). Though the zone against S. aureus was not statistically significant, the highest zone was observed for E. coli by the immobilized scaffold. However, whether blended or immobilized, all of them showed satisfactory zones for each strain, indicating that these microbial loads are susceptible to all non-leaching PU/SF matrices, whereas the control plate showed a full bacterial lawn. Likewise, while the LDH activity of blended and immobilized PU/SF (2:9) was compared among four strains, the highest LDH was quantified against E. coli, followed by P. aeruginosa, S. aureus, and K. pneumoniae (Fig. 2(c1)). Compared to the control, LDH release from bacterial cells treated with immobilized PU/SF were statistically significant with that of E. coli (p ≤ 0.001), P. aeruginosa (p ≤ 0.05), and S. aureus (p ≤ 0.01). In the four pathogenic strains, LDH release due to bacterial membrane rupture was greater than immobilized PU/SF compared to that of blended PU/SF, which causes greater antimicrobial efficacy of the immobilized PU/SF over the blended PU/SF. LDH cytotoxicity analyses of bacterial upon exposure to blended PU/SF and immobilized PU/SF is shown in Fig. 2(c2). For all the strains, the percent toxicity was higher due to immobilized PU/SF due to the increased amount of LDH released in the medium from the bacterial cell membrane. The underlying mechanism of microbial growth prevention lies in the prompt chelation of trace elements or metals present in the bacterial cell wall and cell membrane with the SF polypeptide backbone. The pKa value of aspartate, glutamate-like acidic groups of SF polypeptide, lies around 4–4.8, which is less than the neutral pH. The amphoteric nature of SF is accredited to ionizable groups present in the side chains of amino acid residues; these amino acids dissociate at different pH values. Upon dissociation, the available anionic groups bind to oppositely charged moieties, similar to trace metal ions by electrostatic interactions. When, these groups dissociate, adequate negatively charged free ions are released and bind to trace metal cations of the bacterial cell wall and membrane, resulting in the instability of the cell wall and the membrane.55–57
| Discs | Micro-organism | |||
|---|---|---|---|---|
| P. aeruginosa | K. pneumoniae | E. coli | S. aureus | |
| a Mean zone size (cm). | ||||
| PU/SF blended | 1.05 | 0.88 | 1 | 0.9 |
| PU/SF immobilized | 1.5 | 1.32 | 1.85 | 1.2 |
| Bare PU | 4.3 | 4.7 | 3.9 | 5.1 |
3.3 Silk fibroin (SF) immobilization and surface modification of the scaffold
According to the calibration curve, 3.56 mg of 1,6-hexane diamine was estimated to exist on the PU matrix after aminolysis. As depicted in (Fig. 3B), blue ninhydrin reaction product with free amino (–NH2) groups in 1,4-dioxane/isopropanol (v/v, 1/1) solvent has an absorbance maxima at 558 nm. Successful aminolysis is evident as the absorbance increased with increasing 1,6- hexanediamine concentration across a range; in contrast, untreated PU did not show any absorbance under identical measuring conditions.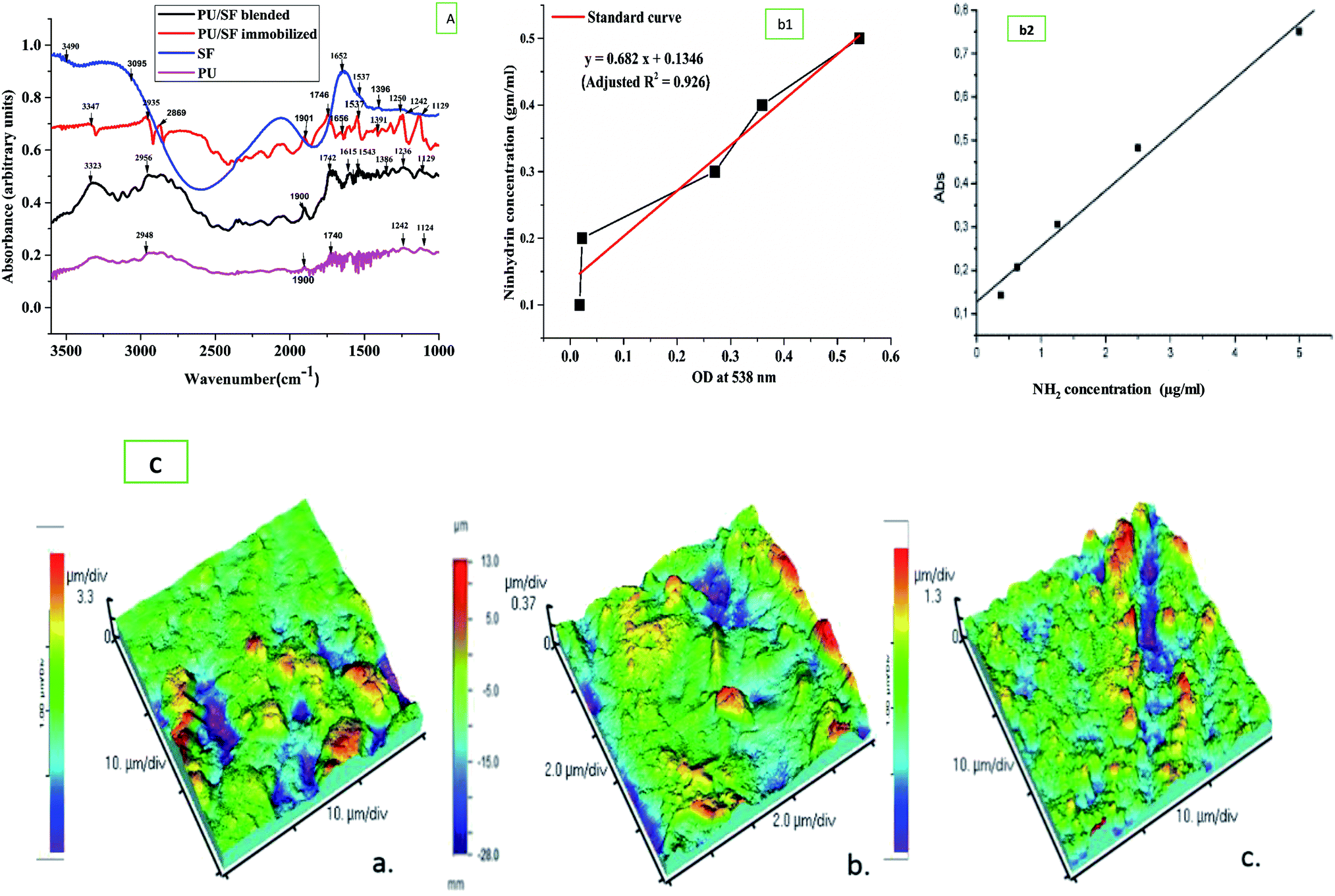 | ||
| Fig. 3 (A) FTIR spectra of IPDI based PU, bended PU/SF, immobilized PU/SF, (B) (b1) The absorbance (at 538 nm) of ninhydrin–NH2 (with 1,6-hexanediamine) reaction products as a function of NH2 concentration (b2). (C) 3D AFM micrographs showing surface roughness of (a) blended PU/SF, (b) immobilized PU/SF, (c) PU scaffold. | ||
Free –NH2 groups were introduced by aminolysis and the presence of amino groups on the scaffold surface was confirmed by ninhydrin estimation. Successful aminolysis was evident from the 1,6-hexane diamine calibration curve (Fig. 3B), which revealed the introduction of 3.863 mg amine groups on the scaffold surface after incubating it in ninhydrin/ethanol solution for 10 min at 37 °C, as calculated from the calibration curve of ninhydrin. This surface exposure not only improved the hydrophilicity, which was measured by respective contact angles, but also provided exposed amino group sites for protein binding with the help of glutaraldehyde as a cross-linking agent to ensure prominent protein grafting on the scaffold surface.58 With this method, any other protein molecules can be immobilized onto the amino group exposed surface. The covalent binding sites of the SF polypeptide sequences to form complexes with amorphous PU backbone are free carboxyl endings of aspartate and glutamate.
3.4 Physicochemical characterization
Fourier transform infrared studies confirmed the blending of PU and SF as the individual components' groups are found in the FTIR peaks. In both the blended and immobilized PU/SF scaffold, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O– stretching of the urethane linkage was seen at 1739 cm−1 (Fig. 3A) and at 2061 cm−1, the characteristic isophorone diisocyanate (-NCO) group was present. At 1680 cm−1, urea CO vibration and at 1606 cm−1, aromatic CC vibration was found.60 These most important IPDI-based PU-associated peaks confirmed its presence in both types of the blended and immobilized scaffold. Silk fibroin has signature peaks of amide I in the regions from 1600 to 1700 cm−1 (CO stretching) and amide II in the region from 1500 to 1600 cm−1 (N–H bending). For non-mulberry SF (Antheraea mylitta), the characteristic peak of amide I was found at 1660 cm−1 and for amide II at 1537 cm−1 due to the N–H bending of the secondary β sheet structure, and 1391 cm−1 for the amide III structure. The peaks at 1537 cm−1 and 1660 cm−1 in the PU/SF matrix corresponded to the β-sheet structure and 2065 cm−1 matched with the urethane linkage, confirming the synthesis of the blend. The blend did not show any alterations in the peaks in the range of 1320–1370 cm−1, indicating the presence of Amide III and 1550-Amide II, while peaks in the range of 1610–1654 cm−1 confirmed amide I (CO stretching). In immobilized PU/SF, two additional strong broad peaks at 2869 cm−1 and 2935 cm−1 were obtained for N–H stretching due to aminolysis with the amine salt, which was absent in blended PU/SF. The presence of these peaks confirmed aminolysis in immobilized PU/SF. The medium peak at 3347 cm−1 in immobilized PU/SF arises due to NH stretching from aliphatic primary amine caused due to aminolysis, whereas the strong peak at 3323 cm−1 in blended PU/SF was obtained due to OH-stretching for an intermolecular bond. No peak was found in blended PU/SF due to aminolysis. The weaker medium vibrations below 3000 cm−1 at 2956 cm−1 (blended PU/SF) and 3089 cm−1 (SF) arises from alkane-type CH bonds derived from the SF protein side chains and the backbone. The broad absorption at 3490 cm−1 (SF) indicates the presence of other functional groups in addition to O–H bonds due to the presence of water.
O– stretching of the urethane linkage was seen at 1739 cm−1 (Fig. 3A) and at 2061 cm−1, the characteristic isophorone diisocyanate (-NCO) group was present. At 1680 cm−1, urea CO vibration and at 1606 cm−1, aromatic CC vibration was found.60 These most important IPDI-based PU-associated peaks confirmed its presence in both types of the blended and immobilized scaffold. Silk fibroin has signature peaks of amide I in the regions from 1600 to 1700 cm−1 (CO stretching) and amide II in the region from 1500 to 1600 cm−1 (N–H bending). For non-mulberry SF (Antheraea mylitta), the characteristic peak of amide I was found at 1660 cm−1 and for amide II at 1537 cm−1 due to the N–H bending of the secondary β sheet structure, and 1391 cm−1 for the amide III structure. The peaks at 1537 cm−1 and 1660 cm−1 in the PU/SF matrix corresponded to the β-sheet structure and 2065 cm−1 matched with the urethane linkage, confirming the synthesis of the blend. The blend did not show any alterations in the peaks in the range of 1320–1370 cm−1, indicating the presence of Amide III and 1550-Amide II, while peaks in the range of 1610–1654 cm−1 confirmed amide I (CO stretching). In immobilized PU/SF, two additional strong broad peaks at 2869 cm−1 and 2935 cm−1 were obtained for N–H stretching due to aminolysis with the amine salt, which was absent in blended PU/SF. The presence of these peaks confirmed aminolysis in immobilized PU/SF. The medium peak at 3347 cm−1 in immobilized PU/SF arises due to NH stretching from aliphatic primary amine caused due to aminolysis, whereas the strong peak at 3323 cm−1 in blended PU/SF was obtained due to OH-stretching for an intermolecular bond. No peak was found in blended PU/SF due to aminolysis. The weaker medium vibrations below 3000 cm−1 at 2956 cm−1 (blended PU/SF) and 3089 cm−1 (SF) arises from alkane-type CH bonds derived from the SF protein side chains and the backbone. The broad absorption at 3490 cm−1 (SF) indicates the presence of other functional groups in addition to O–H bonds due to the presence of water.
Cross-sectional and top views of the SEM images of bare PU scaffolds and blended, immobilized PU/SF matrix exhibited (Fig. 4) interconnected porous architecture. Ideally, the fibroblast and keratinocyte size ranges from 11 to 20 μm.60 The bare PU scaffold had a larger pore size, majorly ranging between 150 and 200 μm (Fig. 4(a1)), which was not suitable for fibroblast or keratinocyte cells. A significant reduction in the pore size was obtained when PU was treated with SF (Fig. 4(b1) and (c1)). Frequency vs. pore size distribution by means of pore width is shown (Fig. 4(a2), (b2) and (c2)), respectively. The pore width size (Fig. 4(b2) and (c2)) obtained was in the range of 22.248 ± 6.818 μm and 16.102 ± 4.802 μm for blended and immobilized PU/SF (2:9) matrix, respectively. As the pore size range of both the blended and immobilized PU/SF is closer to that of the NIH3T3 cells (15 μm), it reflects that dermal fibroblasts could be seeded and intercommunicate between the interconnected pores. Immobilized PU/SF presented rougher and tortuous surface morphology compared to the blended PU/SF variety. It could be attributed to the treatment of 1,6-hexanediamine followed by glutaraldehyde. Therefore, the rougher PU/SF surface is supposed to exhibit greater cellular attachment and proliferation towards the graft. However, the exact estimate of surface roughness has been measured using atomic force microscopy for both the varieties.
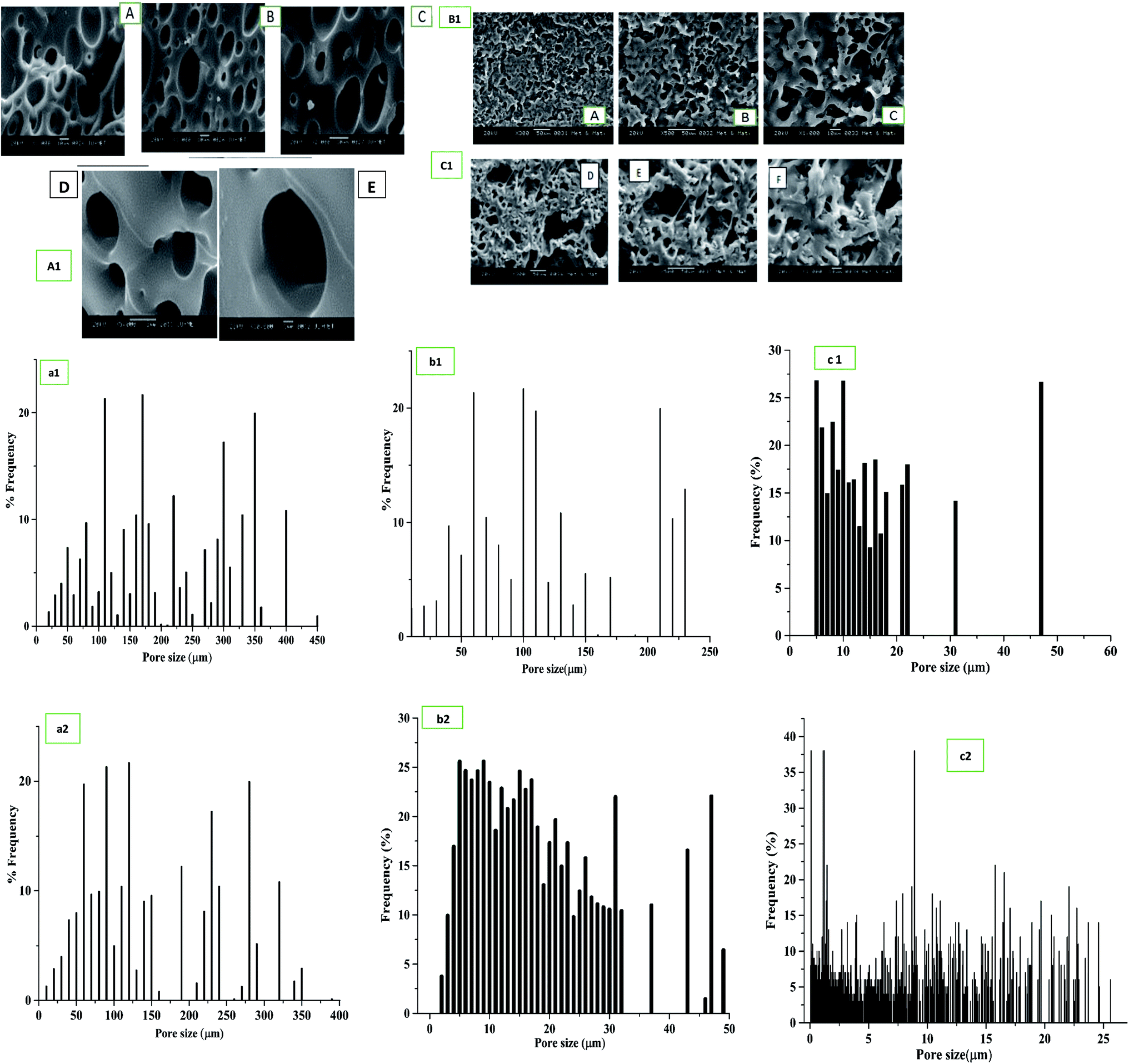 | ||
| Fig. 4 Scaffold morphology in Scanning electron microscopy of (A1) bare PU scaffold 1000× (A & B), 2000 × (C), 5000× (D), 10000× magnification (E); (B1) blended PU/SF [2:9] (A–C) 300×, 500× and 1000× magnification; (C1) immobilized PU/SF [2:9] (D–F) 300×, 500× and 1000× magnification; pore diameter distribution of (a1) bare PU, (b1) blended PU/SF (2:9), and (c1) immobilized PU/SF (2:9). Pore width distribution of (a2) bare PU, (b2) blended PU/SF (2:9), and (c2) immobilized PU/SF (2:9). | ||
Atomic force microscopic studies revealed (Table 1) greater surface roughness of the PU/SF matrix scaffolds due to the presence of peaks and valleys of the surface compared to that of bare PU (Fig. 3C). More the difference between the average height and valleys, higher is the surface roughness. The effect of incorporation of SF into the PU scaffold is reflected in the surface properties. RMS roughness is 197.6 μm, 534 μm, and 464.3 μm for PU, blended PU/SF, and immobilized PU/SF scaffolds, respectively (Table 1). The valleys for PU, blended PU/SF, and immobilized PU/SF are −3.88, −1.615 μm, and −1.389 μm, and the corresponding peaks are 4, 3.819, and 687 μm for PU, blended PU/SF, and immobilized PU/SF, respectively. The difference in the peaks and valleys in immobilized PU/SF was maximum, which reflected the effect of SF incorporation and efficiency of the route of immobilization. Surface troughs and crests contributed to surface roughness, resulting in improved cell attachment and adhesion. As in the blended PU/SF, SF is almost uniformly spread in the inner architecture of PU by covalent bonding, and the functional groups of both SF and PU are bonded at different three-dimensional planes, resulting in more surface irregularities compared to that of immobilized PU/SF. Thus, both RMS and Avg roughness are found to be higher for blended PU/SF.
3.5 Effective dose (ED50) of SF, cell proliferation assay, immunostaining, and DNA quantification
Fibroblast cell proliferation study was initially performed to determine the effective dose (ED50) of SF to be selected for scaffold fabrication (Fig. 5a). Fifty percent NIH3T3 cell population was alive (ED50) at 5.25 mg mL−1 SF concentration, which was lower than the MIC value of P. aeruginosa (8 mg mL−1) and K. pneumoniae (6.4 mg mL−1) but was higher than the MIC values of E. coli (4.8 mg mL−1) and closer to S. aureus (5.6 mg mL−1), respectively. Though the MIC values were higher than the ED50, the chosen 9 mg mL−1 concentration was greater than that for both MIC and ED50. Up to 8.5 mg mL−1, NIH3T3 cell proliferation gradually increased up to 91% and after that, even at 15 mg mL−1, it remained constant. Thus, in all the ratios of PU/SF (2; 5), PU/SF (2:7), PU/SF (2:9), the SF concentrations were kept higher than the ED50 value of SF. Fig. 5b(i)–(viii) depicts the cell proliferation in SF in a concentration-dependent manner at 8 mg mL−1 SF (Fig. 5b(ii) and (vi)) 8.5 mg mL−1 SF (Fig. 5b(iii) and (vii)), and 9 mg mL−1 (Fig. 5b(iv) and (viii)) at 24 h and 48 h, respectively.
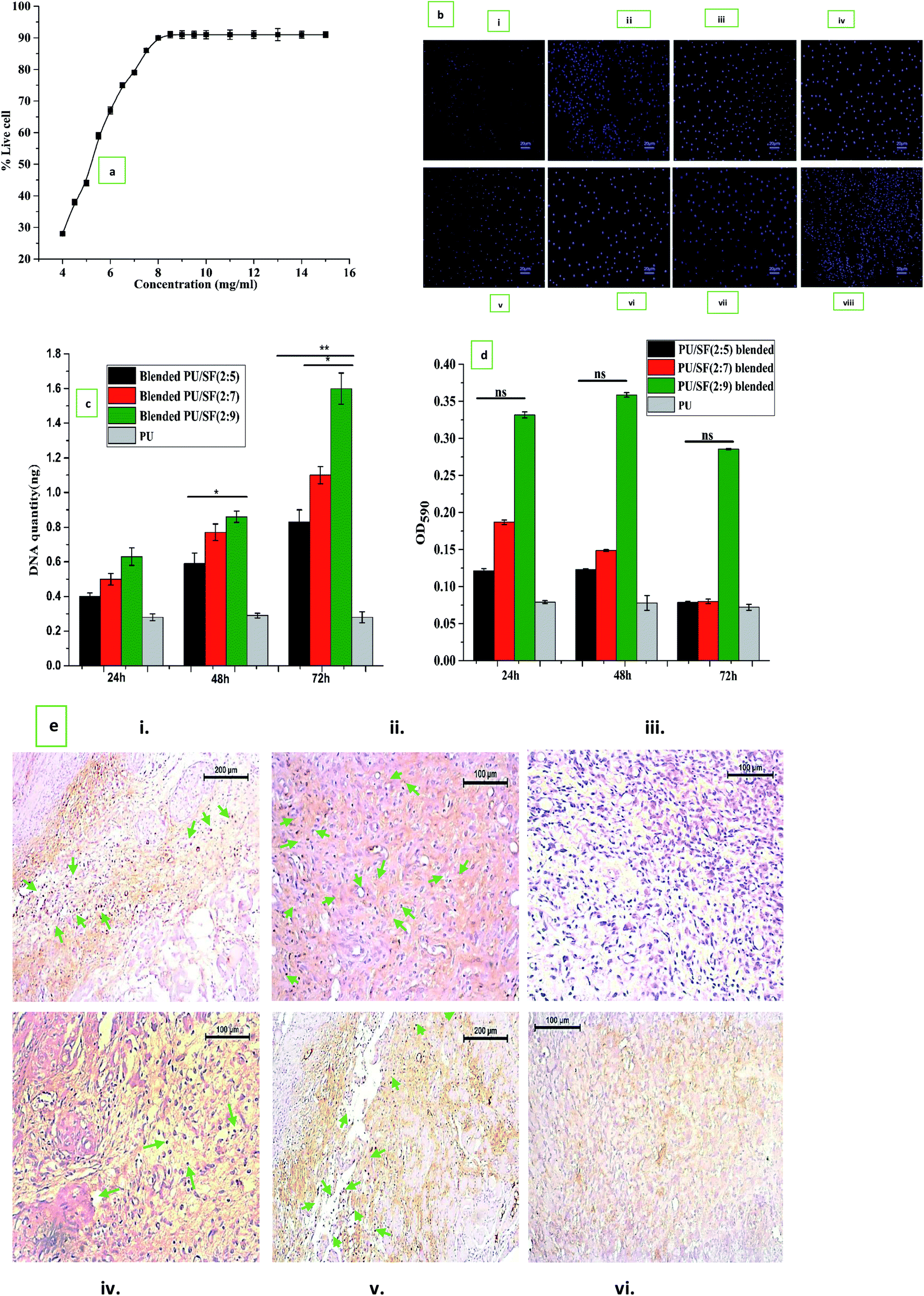 | ||
| Fig. 5 (a) Dose response relationship for calculation of ED50 value of silk fibroin on NIH3T3 cell line. (b) Fluorescence imaging using DAPI for nuclear staining of NIH3T3 cells on TCP at 24 h (i), 48 h (v), cells treated with 8 mg mL−1, 8.5 mg mL−1, 9 mg mL−1 SF concentrations at 24 h (ii–iv) and at 48 h (vi–viii) incubation. (c) In vitro cell viability by MTT assay. Y-error bars represent standard deviation (*p ≤ 0.05, **p ≤ 0.01). (d) DNA quantification study of PU/SF blended and PU/SF immobilized; Y-error bars represent standard deviation (*p ≤ 0.05, **p ≤ 0.01). (e) Ki-67 expression in immunostaining of blended at 7th and 5th days culture on blended PU/SF (i and iv), immobilized PU/SF (ii and v), bare PU treated cells (iii and vi). | ||
The OD595 value obtained in the MTT assay was observed up to 72 h and reflected all the scaffold varieties to be completely non-toxic. The percent cell viability increased with increasing SF concentration, though the differences were insignificant to each other at all the intervals (Fig. 5c). DNA analysis was used to gain more insight into the underlying effect of fibroblast proliferation by dose-dependent PU/SF (Fig. 5d). There was no significant difference in the cell viability assessment results at 24 h between the immobilized PU/SF and blended PU/SF. Doubtfully, the quantified DNA harvested from fibroblasts was the highest for immobilized PU/SF (2:9) at all the intervals. Fibroblasts extracted DNA quantity was directly proportional to the SF concentrations present in PU/SF. Moreover, the obtained DNA quantity by immobilized PU/SF (2:9) was statistically significant compared to the DNA quantity by blended PU/SF (2:5) [p ≤ 0.05], blended PU/SF (2:7), and blended PU/SF (2:9) [p ≤ 0.01] at 72 h, respectively.
Cell proliferation marker Ki-67 was visualized by immunostaining (Fig. 5e) at 5th and 7th days fibroblast cultures on blended PU/SF (Fig. 5e(i) and (iv)) and immobilized PU/SF (Fig. 5e(ii) and (v)) treated NIH3T3 cells. Almost no Ki-67 was visualized in the cells cultured on the bare PU scaffold (Fig. 5e(iii) and (vi)) treated cells. In immobilized PU/SF cultured NIH3T3 cells, more intense Ki-67 marker was visualized compared to cells cultured on blended PU/SF. The visualization of the Ki-67 (marker for cell proliferation) marker in both the types of PU/SF (blended PU/SF and immobilized PU/SF) cultured cells and absence of the Ki-67 marker in the bare PU matrix cultured cells ensured that the PU/SF matrices have regenerative potential, which is credited to the A. mylitta variety SF.
3.6 Measurement of anti-oxidant potential by DPPH scavenging assay and the protective effect assessment by trypan blue dye exclusion assay, LDH activity, and anti-lipid peroxidase (ALP) assay
Three different SF concentrations above ED50 value were subjected to DPPH free radical scavenging activity compared to citric acid taken as the standard (Fig. 6a). All the three chosen concentrations showed greater than 50% DPPH inhibition, which indicates that all the three SF concentrations are above IC50 (3.1 mg mL−1), where 9 mg mL−1 SF scavenged nearly 68% free radicals, while 8.5 mg mL−1 and 8 mg mL−1 SF scavenged 62% and 57% DPPH, respectively, which is directly proportional to the SF concentrations. This finding can be correlated to the LDH release activity of the NIH3T3 cells.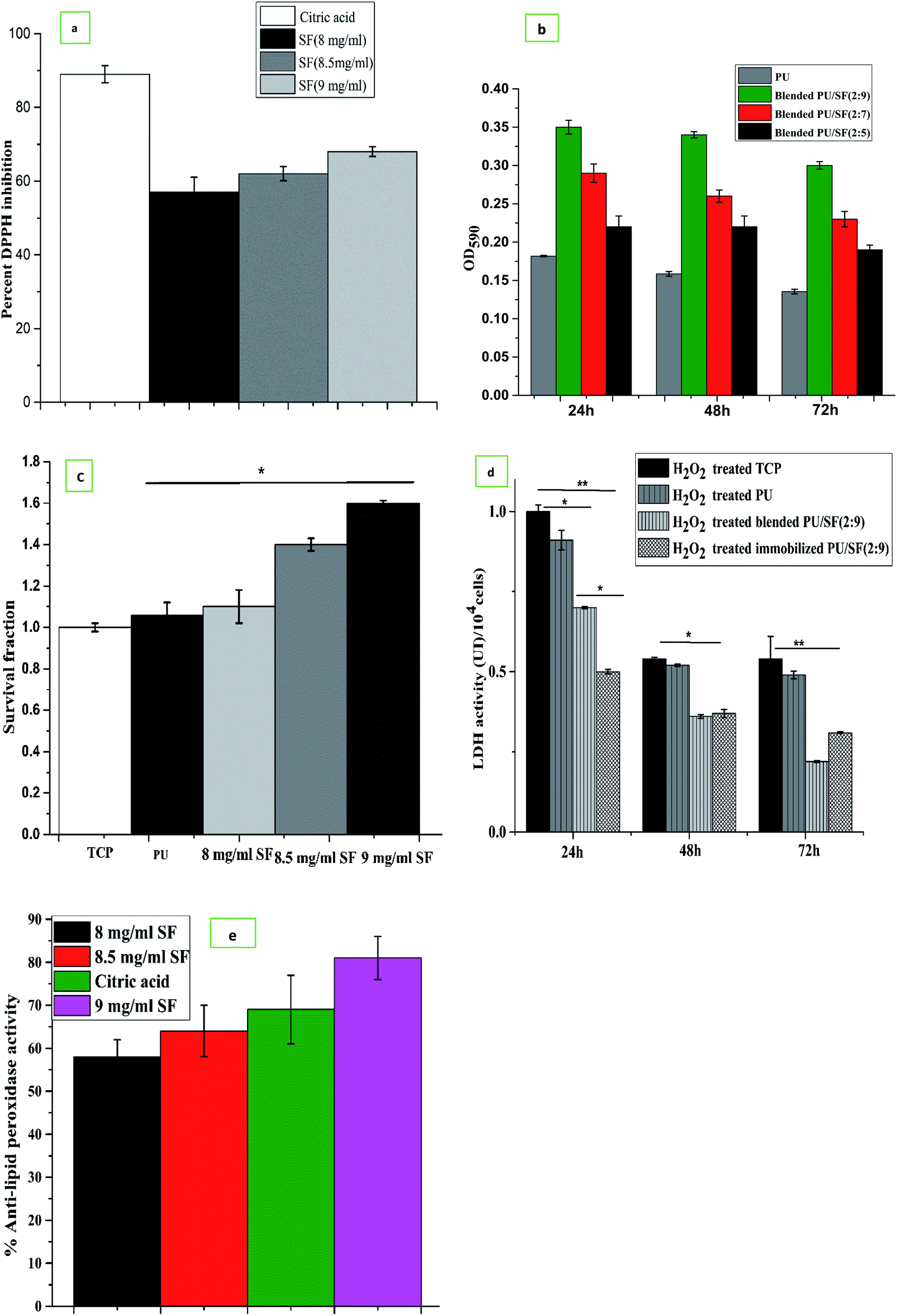 | ||
| Fig. 6 (a) DPPH free radical scavenging by different SF concentrations. (b) Cell viability on PU/SF after H2O2 induced oxidative damage. Y-error bars represent standard deviation (*p ≤ 0.05, **p ≤ 0.01). (c) Cell death was measured by TBE assay after exposure of H2O2 induced oxidative damaged cells treated with different SF concentrations for 24 h. (d) In vitro cytotoxicity study by LDH release assay. (e) Percent alkaline lipid peroxidase activity by different SF concentrations treated NIH3T3 cells on H2O2 induced oxidative damage. | ||
Reactive oxygen species (ROS) are highly unstable free radicals, which are produced by aerobic organisms during cellular metabolism.61 Low level of ROS enhances cell survival (proliferation), whereas elevated ROS oxidizes the intracellular biomolecules, which subsequently leads to cytotoxicity. Hydrogen peroxide (H2O2) is a stable ROS, which reacts with various intracellular targets, including proteins, lipids, and DNA, which would lead to cell death.62 Here, we used H2O2 induced oxidant model to investigate the antioxidant properties of A. mylitta SF. The pretreatment of SF conferred significant protection in few groups against H2O2 induced oxidative stress. In cell viability assessment, the PU/SF (2:9) scaffolds exhibited the highest percent of viable cells (Fig. 6b) after 24 h oxidative damage due to H2O2 treatment, which supports the untreated NIH3T3 cell proliferation on the PU/SF (2:9) scaffold; however, the number of live cells in the MTT assay significantly decreased due to H2O2 induced damage on the same variety of the scaffold.
In order to check the protective role of dose-dependent SF against cellular oxidative stress (induced by overnight NIH3T3 incubation in H2O2), SF treated and untreated (TCP) NIH3T3 cells were subjected to trypan blue dye exclusion assay to quantify the cell viability after going through H2O2-mediated oxidative stress. Correlating with the MTT assay findings and DPPH free radical scavenging activity results, the cell survival fraction was directly proportional to the doses of SF and the viable cell count in the 9 mg mL−1 SF concentration was the highest and statistically significant (p ≤ 0.05) with that of untreated TCP (Fig. 6c). This represents that 9 mg mL−1 SF concentration scavenged the highest amount (62%) of free radicals generated by DPPH, reflecting that 9 mg mL−1 SF is effective enough to eradicate reactive oxygen species (ROS) produced by both hydrogen peroxide and DPPH induced oxidative damage.
To investigate further the effect of dose-dependent A. mylitta silk fibroin on H2O2-induced oxidative stress, the activities of LDH and anti-lipid peroxidase (ALP) were examined. H2O2, one of the major reactive oxygen species, is produced at a high rate as a product of normal aerobic metabolism. Hence, an acceleration of anaerobic metabolic pathway to cope with oxidative stress is reflected by an increase in the LDH activity. The LDH activity of the cells subjected to oxidative stress significantly increased when compared to the normal ones, which indicate loss of membrane integrity.63
LDH release activity was lesser in the case of H2O2 treated cells, which were cultured on blended and immobilized PU/SF matrices (incorporated with 9 mg mL−1 SF concentration) compared to the LDH release activity by H2O2-treated cells cultured on TCP. This can be corroborated with the fact that lesser LDH release is associated with lower cellular damage due to the protective effect of 9 mg mL−1 SF concentration. In the LDH release experiment (Fig. 6d), at all intervals, H2O2 treated cells on the control TCP group released the maximum LDH, which was significant with the LDH released by the immobilized PU/SF treatment at 24 h (p ≤ 0.01), 48 h (p ≤ 0.05), and 72 h (p ≤ 0.01), respectively. This exhibits the treatment of SF decreased cell damage and aided in greater cell survival. The untreated group presented significant (p ≤ 0.05) LDH release with blended PU/SF releasing 0.65 UI LDH/104 cells, whereas the immobilized PU/SF releases 0.5 UI LDH/104 cells at 24 h, which was also statistically significant, reflecting that the SF-treated matrix protects the NIH3T3 cells from natural damage and rupture. It indicates that the immobilized PU/SF allows lesser LDH release due to membrane damage compared to that of the blended PU/SF. Therefore, immobilized PU/SF provides higher protection against cell damage due to oxidative stress. Though LDH release equalizes at 48 h by two types of PU/SF, this finding at 48 h was not statistically significant. The LDH release behavior of the PU/SF matrices acted differently for animal cells and bacterial cells; it ruptured bacterial cell membranes and showed antimicrobial efficacy, whereas it released less LDH from NIH3T3 cell membranes and protected cell membrane damage from oxidative stress. Likewise, anti-lipid peroxidation activity was obtained depending on the concentration of SF. The ALP assay showed (Fig. 6e) 70.2% anti-lipid peroxidation activity by 9 mg mL−1 SF, whereas citric acid showed 69.8% anti-lipid peroxidation activity.
3.7 In vitro pro-inflammatory and anti-inflammatory cytokine profile
The effect of silk fibroin on the production of IL-6 (pg mL−1) due to scratch wound stimulus given to PBMC cultured on the tissue culture plate, bare PU scaffold, blended PU/SF (2:9), and immobilized PU/SF (2:9) scaffold was investigated. In low glucose medium, at initial 24 h, IL-6 release was higher in PU (2550 pg mL−1) and PU/SF immobilized (2:9) (2530 pg mL−1), PU/SF blended (2 : 9) (2305 pg mL−1), TCP (2150 pg mL−1), though the difference is lesser; however, from 48 h to 96 h, both the control (TCP) (3200 pg mL−1 at 96 h) and PU (2750 pg mL−1 at 96 h) kept on increasing, whereas both PU/SF blended (pg mL−1 at 96 h) and immobilized (1350 pg mL−1 at 96 h) decreased at 96 h. In the initial hours after scratching, burst IL-6 release was found by silk-coated PU, which was attenuated after 48 h; in contrast, pristine PU could not attenuate IL-6 response with time like the silk treated PUs, though it exhibited inflammatory property but not in the required controlling manner. Likewise, the pro-inflammatory response of TNF-alpha by bare PU behaved similarly in agreement with the findings of Battiston et al.64 to that of IL-6. TNF-alpha by TCP control kept on increasing from 24 h to 96 h (1600 ng mL−1 at 24 h to 2178 ng mL−1 at 96 h) post scratching in an uncontrolled way and TNF-alpha by bare PU (2270 ng mL−1 at 24 h to 2247 ng mL−1 at 96 h) remained almost the same, whereas TNF-alpha decreased by immobilized PU/SF (2500 ng mL−1 at 24 h to 1750 ng mL−1 at 96 h). TCP control and pristine PU kept on stimulating both the wound associated pro-inflammatory cytokines IL-6 and TNF-alpha. Higher IL-6, IL-10, and TNF-alpha synergistically negated the healing effect, causing delayed fibroblast, keratinocytes repair, and infiltration. SF-coated PU merely increased the amount of TNF-alpha at 48 h and narrowed it down from 72 h to 96 h, indicating gradually lowering pro-inflammatory response at a cellular level after 48 h. It is well known for attaining successful wound healing that the IL-10 level should be lower initially due to the inhibitory effect of anti-inflammatory cytokines (IL-10) on cellular repair.64 TNF alpha/IL-10 ratio should be ideally high at initial hours of a wound and the ratio normally lowers down after a certain period depending on the wound size as IL-10 starts to rise and TNF-alpha falls.65 The contribution of pro-inflammatory cytokines is seen at early hours of inflammation when the anti-inflammatory cytokine IL-10 level ideally should be less as raised IL-10 levels inhibit fibroblast and keratinocytes infiltration, whereas a high level of pro-inflammatory cytokines promotes growth and recruitment of fibroblast and keratinocyte at the beginning (Fig. 7).
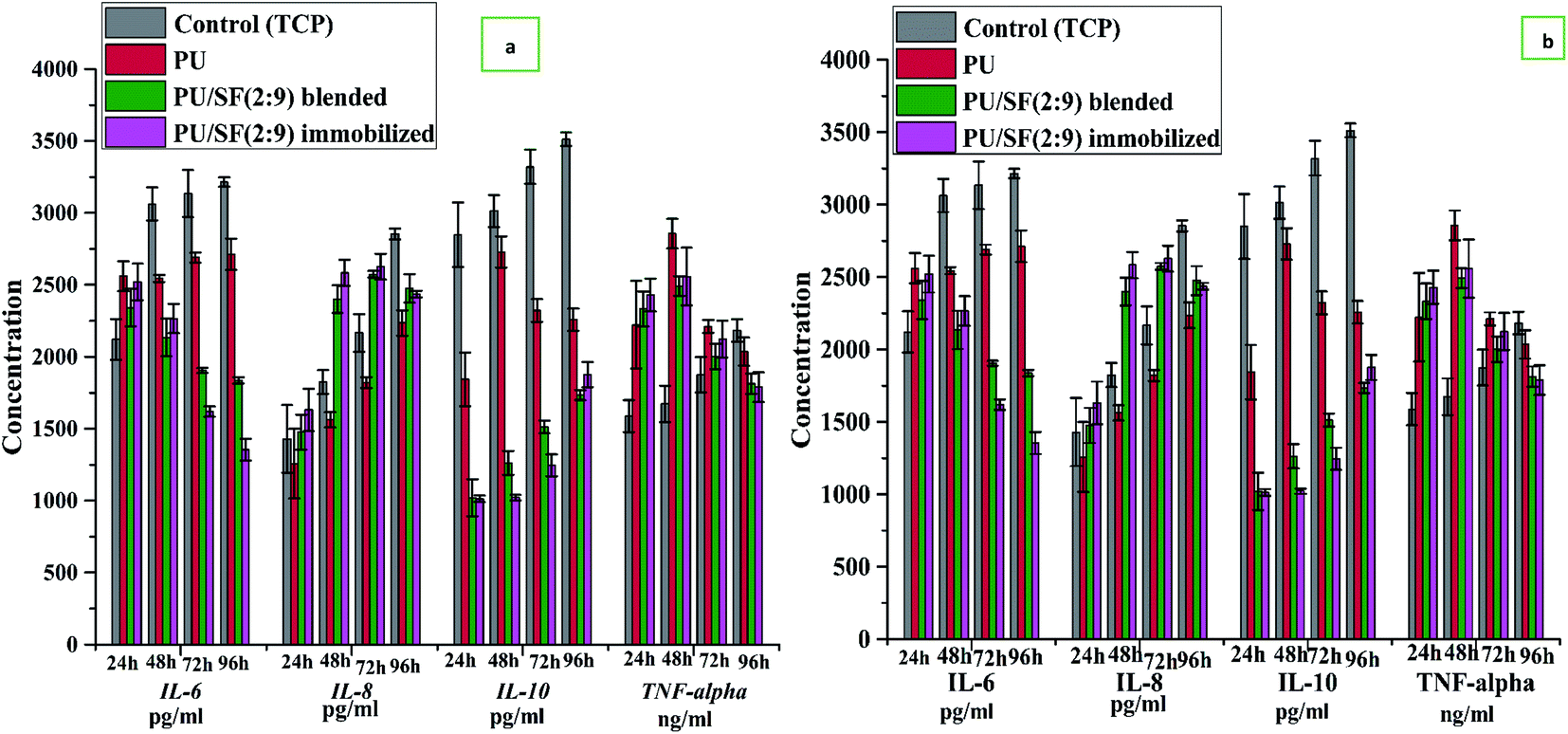 | ||
| Fig. 7 Inflammatory cytokine response upon scratch stimulus on PBMC culture in (a) high glucose RPMI 1640 media, (b) glucose deprived RPMI 1640 media. | ||
In a high glucose medium, the control (TCP) and pristine PU manifested a sequential raise of IL-10 along the entire study period, where the TNF-alpha/IL-10 ratio did not exhibit as high a level as that presented by silk-coated PUs. This is because both TNF-alpha and IL-10 are high at the beginning for bare PU. Since the effect of SF immobilization on PU and the blending of silk with PU triggered a major lowering of IL-10 relative to bare PU over the entire period of 24 h to 96 h, it showed an increase in the TNF-alpha/IL-10 ratio up to 48 h; however, the ratio gradually decreased after 48 h as the elicited IL-10 started to increase and the TNF-alpha sequentially fell by PU/SF. This manifests that PU/SF timely immunomodulated with the initial stimulation of the pro-inflammatory cytokine (TNF-alpha, IL-6) and in a low level of anti-inflammatory cytokine (IL-10 level, 1050 pg mL−1 at 24 h to 1978 pg mL−1 at 96 h in glucose deprived media) to fasten successful scratched PBMC repair and regeneration by PU (1800 pg mL−1 at 24 h to 2250 pg mL−1 at 96 h) and TCP control (2870 at 24 h to 3612 at 96 h). Later on, the influence of TNF alpha dissipates while excess IL-10 stimulation helps to avoid fibroblast scarring due to its inhibitory effect. IL-8 release in high glucose medium depicts initial (24 h) lowered release by PU (1500 pg mL−1) and PU/SF varieties (1675 pg mL−1) but higher for the control TCP (2750 pg mL−1). Although in high glucose media, IL-8 for all groups at 24 h were much elevated relative to that of glucose deprived media, the control group showed the maximum hike in IL-8 level at all time points due to high glucose. However, in low glucose medium, PU and PU/SF exhibited raised IL-8 at 48, 72, and 96 h, favoring the guided healing process in a positive direction. In both high and glucose deprived medium, noticeable IL-6 and IL-10 quantity difference were observed between immobilized and blended PU/SF. Immobilized PU/SF released less IL-6 and IL-10 at all time points in both high and low glucose medium. Firstly, blended and immobilized PU/SF mediated IL-6, IL-8, IL-10, and TNF-alpha profile remained unaffected by glucose in the medium, which interprets that PU/SF directed cytokine responses could escape the effect of glucose, unlike bare PU. Immobilized PU/SF elevated the effect of blended PU/SF by decreasing the IL-6 profile at 72 and 96 h and by decreasing the IL-10 profile at 48 and 72 h. This difference in blended and immobilized PU/SF demonstrated more precise regulation by immobilized PU/SF at desired time intervals of 3–4 days culture, which suppressed the pro-inflammatory cytokines and helped the anti-inflammatory cytokines come into play to accelerate the repair process.
The control (TCP) IL-6 values were not much influenced by different glucose levels over 24 h to 96 h as there was not much difference in IL-6 in low glucose (2000 pg mL−1 at 24 h to 3000 pg mL−1 at 96 h) and high glucose (2500 pg mL−1 at 24 h to 3500 pg mL−1 at 96 h) levels exhibited by TCP. On the other hand, IL-6 elicited by pristine PU was well affected by glucose-deprived media as significant difference was observed at all time points between high and low glucose medium. In glucose-deprived media, PU/SF elicited IL-6 lowered (2500 pg mL−1 at 24 h to 1400 pg mL−1 at 96 h) prominently with respect to high glucose media (2800 at 24 h to 2100 pg mL−1 at 96 h) but the difference in the amount of PU/SF elicited IL-6 between high glucose and glucose-deprived media was much lesser compared to the difference in the amount of pristine PU elicited IL-6 between high (3174 pg mL−1 at 24 h to 3337 pg mL−1 at 96 h) and glucose-deprived media (2100 pg mL−1 at 24 h to 2700 pg mL−1 at 96 h). It indicates that glucose concentration in the culture media could not alter the IL-6 response by PU/SF significantly; thus, PU/SF easily escaped the effect of glucose on IL-6, whereas pristine PU IL-6 response was much affected by glucose in the culture media. In low glucose medium, IL-8 was lower in PU (1243 pg mL−1 at 24 h) and immobilized PU/SF (1490 pg mL−1 at 24 h) but it significantly increased in blended PU/SFs (2494 pg mL−1) and immobilized PU/SFs (2511 pg mL−1) at 96 h, whereas pristine PU increased only upto 2124 pg mL−1 at 96 h. It was earlier reported that the initial lowering of IL-8 level, followed by upregulation, favors the healing process, which is presented by PU/SFs mostly relative to others. The study findings agree with the earlier report65 that uncontrolled high IL-6, TNF-alpha,38 and IL-10 secretion by PU treated cells hinders mitogenic recruitment and interferes in healing, unlike PU/SF treated PBMCs. In PU/SF treated PBMCs, IL-10 was low at the beginning, allowing pro-inflammatory (IL-6, TNF-alpha) activity to prevail and gradually letting anti-inflammatory (IL-8, IL-10) cytokines predominate, coordinated with the downregulation of the pro-inflammatory (IL-6, TNF-alpha) profile to escape excess scarring and facilitate the inflammatory phase of healing.66
4. Conclusion
SF doses above highest the MIC values and ED50 were considered for verifying fibroblast adhesion, as well as the antioxidant and antimicrobial properties. Among the doses, 9 mg mL−1 showed 68% DPPH scavenging activity and 70% ALP activity (close to citric acid (79%)). Based on experimental evidence, 9 mg mL−1 SF was found to be a potential antioxidant and supportive of cell proliferation as it managed to escape the oxidative damage caused by 24 h H2O2 incubation. Using 9 mg mL−1 SF, when blended PU/SF was fabricated, PU/SF (2:9) presented the highest cell growth over PU/SF (2:7) and PU/SF (2:5) in MTT and DNA quantification assay. Thus, only PU/SF (2:9) was fabricated employing immobilization. Immobilized PU/SF (2:9) exhibited greater bacterial zone of inhibition against all microbial strains over blended PU/SF. Immobilized PU/SF (2:9) released more LDH compared to the blended variety, showing the higher potency to disrupt bacterial cell membrane integrity for all the tested strains. Immobilized PU/SF not only exhibited greater potential towards microbial growth inhibition but it also demonstrated higher fibroblast cell retrieval from H2O2 damage, as evidenced from the ALP activity, LDH release, trypan blue dye exclusion, and MTT assay. It could be anticipated that the microbial inhibitory effect, metabolic ROS elimination, and fibroblast re-generative potential can synergistically aid in high wound healing efficiency. Immunomodulation by SF on the shifting pro-inflammatory to anti-inflammatory cytokines would neither cause delayed inflammation nor excess fibroblast scar formation and the immunomodulatory role also escaped the effect of varying glucose concentration microenvironment. In contrast, pristine PU could not escape the effect of glucose on the cytokines profile and lacked precise immunomodulation of fibroblast re generation. The immunomodulatory and antioxidant properties of PU/SF can be utilized in topical dressing, where it would accelerate the sequential cascade of fibroblast, keratinocyte repair, and protect delayed inflammation by favorable cytokines profile along with the scavenging of free radicals, which would protect from cellular oxidative damage.
Author contributions
S. S. conceptualized, designed and performed all the experimental work and prepared the entire manuscript with critical review. S. S., S. G., S. D. have prepared and characterized the samples for experimental work. S. S. performed all the biological experiments. S. G. and S. D. equally contributed to data curation, visualization and data validation for the experimental work. P. B. provided the funding acquisition, supervised and administered the project work including providing the resources, and software. P. M. provided partial resources and supervised the SF extraction, purification and identification experiments and partially provided resources. T. K. M. helped in immunostaining and LDH assay experiment. N. K. J. provided partial resources for a few experiments. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
First author, Sohini Sen is grateful to Council of Scientific and Industrial Research (CSIR) for providing senior research fellowship (File no. 09/096(786)/2013-EMR-I). Authors are grateful to Tathagata Adhikary and Pratik Das, School of Bioscience & Engineering, Jadavpur University for assisting in Fluorescence microscopy and acknowledge Dr Preetam Guha Ray, IIT Kharagpur for providing cell line and DAPI stain.References
- A. Ali, S. Bano, S. S. Poojary, D. Kumar and Y. S. Negi, Colloids Surf., B, 2019, 180, 75–82 CrossRef CAS PubMed.
- N. A. Ayoub, J. E. Garb, R. M. Tinghitella, M. A. Collin and C. Y. Hayashi, PLoS One, 2007, 2, e514 CrossRef PubMed.
- I. Banerjee, D. Mishra, T. Das, S. Maiti and T. K. Maiti, J. Biomater. Sci., Polym. Ed., 2012, 23, 355–373 CrossRef CAS PubMed.
- J. D. Roh, R. Sawh-Martinez, M. P. Brennan, S. M. Jay, L. Devine, D. A. Rao, T. Yi, T. L. Mirensky, A. Nalbandian and B. Udelsman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4669–4674 CrossRef CAS PubMed.
- W. Brodbeck, Y. Nakayama, T. Matsuda, E. Colton, N. Ziats and J. Anderson, Cytokine, 2002, 18, 311–319 CrossRef CAS PubMed.
- B. Kundu and S. C. Kundu, Prog. Polym. Sci., 2010, 35, 1116–1127 CrossRef CAS.
- F. G. Omenetto and D. L. Kaplan, Science, 2010, 329, 528–531 CrossRef CAS PubMed.
- X. Zhang, M. R. Reagan and D. L. Kaplan, Adv. Drug Delivery Rev., 2009, 61, 988–1006 CrossRef CAS PubMed.
- Z. Gong, G. Calkins, E.-c. Cheng, D. Krause and L. E. Niklason, Tissue Eng., Part A, 2009, 15, 319–330 CrossRef CAS PubMed.
- S. Sen, P. Basak, B. P. Sinha, P. Maurye, K. K. Jaiswal, P. Das and T. K. Mandal, Int. J. Biol. Macromol., 2020, 143, 1009–1032 CrossRef CAS PubMed.
- B. Kundu, R. Rajkhowa, S. C. Kundu and X. Wang, Adv. Drug Delivery Rev., 2013, 65, 457–470 CrossRef CAS PubMed.
- S.-i. Inoue, H. Tsuda, T. Tanaka, M. Kobayashi, Y. Magoshi and J. Magoshi, Nano Lett., 2003, 3, 1329–1332 CrossRef CAS.
- B. M. Min, L. Jeong, K. Y. Lee and W. H. Park, Macromol. Biosci., 2006, 6, 285–292 CrossRef CAS PubMed.
- I. D. Prà, P. Petrini, A. Chiarini, S. Bozzini, S. Farè and U. Armato, Tissue Eng., 2003, 9(6), 1113–1121 CrossRef PubMed.
- N. Namviriyachote, V. Lipipun, Y. Akkhawattanangkul, P. Charoonrut and G. C. Ritthidej, Asian J. Pharm. Sci., 2019, 14(1), 63–77 CrossRef PubMed.
- S. Akita, K. Akino, T. Imaizumi, K. Tanaka, K. Anraku and H. Yano, Burns, 2006, 32(4), 447–451 CrossRef PubMed.
- A. Chiarini, P. Petrini, S. Bozzini, I. D. Pra and U. Armato, Biomaterials, 2003, 24(5), 789–799 CrossRef CAS PubMed.
- I. D. Prà, P. Petrini, A. Charini, S. Bozzini, S. Farè and U. Armato, Tissue Eng., 2003, 9, 1113–1121 CrossRef PubMed.
- H. J. Yoo and H. D. Kim, J. Appl. Polym. Sci., 2008, 107, 331 CrossRef CAS.
- S. Sen, B. P. Sinha, T. K. Mandal Sr and P. Basak, TERMIS – Americas Conference & Exhibition, Charlotte, NC, USA, 2017 Search PubMed.
- R. Dash, S. Mukherjee and S. C. Kundu, Int. J. Biol. Macromol., 2006, 38, 255–258 CrossRef CAS PubMed.
- R. Dash, S. K. Ghosh, D. L. Kaplan and S. C. Kundu, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2007, 147, 129–134 CrossRef PubMed.
- J. H. Choi, M. J. Song, S. H. Kim, S. M. Choi and D.-G. Lee, Antimicrob. Agents Chemother., 2003, 12, 3704–3707 CrossRef PubMed.
- B. N. Brown, R. Londono, S. Tottey, L. Zhang, K. A. Kukla and M. T. Wolf, Acta Biomater., 2012, 8, 978–987 CrossRef CAS PubMed.
- M. B. Chan-Park, J. Y. Shen, Y. Cao, Y. Xiong, Y. Liu and S. Rayatpisheh, et al., J. Biomed. Mater. Res., Part A, 2009, 88, 1104–1121 CrossRef PubMed.
- J. Ross, Z. Hong, B. Willenbring, L. Zeng, B. Isenberg, E. H. Lee, M. Reyes, S. A. Keirstead, E. K. Weir and R. T. Tranquillo, J. Clin. Invest., 2006, 116, 3139–3149 CrossRef CAS PubMed.
- T. L. Mirensky, N. Hibino, R. F. Sawh-Martinez, T. Yi, G. Villalona, T. Shinoka and C. K. Breuer, J. Pediatr. Surg., 2010, 45, 1299–1305 CrossRef PubMed.
- N. Kato, S. Sato, A. Yamanaka, H. Yamada, N. Fuwa and M. Nomura, Biosci., Biotechnol., Biochem., 1998, 62, 145–147 CrossRef CAS PubMed.
- J. B. Fan, L. P. Wu, L. S. Chen, X. Y. Mao and F. Z. Ren, J. Food Biochem., 2009, 33, 74–88 CrossRef CAS.
- H.-O. Rennekampff, J. F. Hansbrough, V. Kiessig, C. Doré, M. Sticherling and J. M. Schroder, J. Surg. Res., 2000, 93, 41–54 CrossRef CAS PubMed.
- J. E. McBane, K. G. Battiston, A. Wadhwani, S. Sharifpoor, R. S. Labow and J. P. Santerre, Biomaterials, 2011, 32, 3584–3595 CrossRef CAS PubMed.
- D. G. Seifu, A. Purnama, K. Mequanint and D. Mantovani, Nat. Rev. Cardiol., 2013, 10, 410–421 CrossRef CAS PubMed.
- H. I. Kim, K. Ishihara, S. Lee, J. Seo, H. Y. Kim, D. Suh and M. U. Kim, et al., Biomaterials, 2011, 32, 2241–2247 CrossRef CAS PubMed.
- J. P. Stegemann and R. M. Nerem, Ann. Biomed. Eng., 2003, 31, 391–402 CrossRef PubMed.
- K. G. Battiston, B. Ouyang, R. S. Labow, C. A. Simmons and J. P. Santerre, Acta Biomater., 2014, 10, 1146–1155 CrossRef CAS PubMed.
- M. Mazighi, A. Pelle, W. Gonzalez, E. Mtairag, M. Philippe and D. Henin, et al., Am. J. Physiol.: Heart Circ. Physiol., 2004, 287, H866–H87142 CrossRef CAS PubMed.
- A. A. Thomay, J. M. Daley, E. Sabo, P. J. Worth, L. J. Shelton and M. W. Harty, Am. J. Pathol., 2009, 174, 2129–2136 CrossRef CAS PubMed.
- M. Shinozaki, Y. Okada, A. Kitano, K. Ikeda, S. Saika and M. Shinozaki, Arch. Dermatol. Res., 2009, 301, 531–537 CrossRef CAS PubMed.
- S. Werner and R. Grose, Physiol. Rev., 2003, 83, 835–870 CrossRef CAS PubMed.
- L. M. Nilsson, Z. Sun, J. Nilsson, I. Nordstrom, Y. Chen and J. D. Molkentin, et al., Am. J. Physiol.: Cell Physiol., 2007, 292, C1167–C1178 CrossRef CAS PubMed.
- A. Schepers, D. Eefting, P. I. Bonta, J. M. Grimbergen, M. R. de Vries and V. van Weel, et al., Arterioscler., Thromb., Vasc. Biol., 2006, 26, 2063–2069 CrossRef CAS PubMed.
- W. G. Brodbeck, Y. Nakayama, T. Matsuda, E. Colton, N. P. Ziats and J. M. Anderson, Cytokine, 2002, 18, 311–319 CrossRef CAS PubMed.
- P. Maurye, A. Basu, S. Sen, J. K. Biswas, T. K. Bandyopadhyay and M. Naskar, Biochem. Mol. Biol. Educ., 2018, 46, 566–577 CrossRef CAS PubMed.
- Y. Zhu, C. Gao, T. He and J. Shen, Biomaterials, 2004, 25, 423–430 CrossRef CAS PubMed.
- P. Basak and S. Sen, Adv. Mater. Res., 2012, 584, 474–478 CAS.
- Y. Zhu, C. Gao, X. Liu and J. Shen, Biomacromolecules, 2002, 3, 1312–1319 CrossRef CAS PubMed.
- A. Haldar, S. Sen, D. Banerjee, N. K. Jana and P. Basak, in Biopolymers and Biomaterials, ed. A. Padinjakkara, A. Thankappan, F. G. Souza Jr and S. Thomas, Apple Academic Press, Inc., Waretown, NJ08758 USA, 2018, ch. 15, vol. 1, pp. 254–264 Search PubMed.
- N. Arora, A. Ali, S. Sen, A. Haldar, N. K. Jana and P. Basakin, Biopolymers and Biomaterials, ed. A. Padinjakkara, A. Thankappan, F. G. Souza Jr and S. Thomas, Apple Academic Press, Inc., Waretown, NJ08758 USA, 2018, ch. 16, vol. 1, pp. 267–280 Search PubMed.
- S. Sen, A. Haldar, P. Basak and N. K. Jana, Mater. Today: Proc., 2021, 43(2), 947–953 CAS.
- T. Myginda, M. Stiehlera, A. Baatrupa, H. Lia, X. Zoua, A. Flyvbjerg, M. Kassem and C. Bunger, Biomaterials, 2007, 28, 1036–1047 CrossRef PubMed.
- D. Manna, R. Bhuyan, F. Saikh, S. Ghosh, J. Basak and R. Ghosh, Apoptosis, 2018, 23, 532–553 CrossRef CAS PubMed.
- R. Dash, C. Acharya, P. C. Bindu and S. C. Kundu, BMB Rep., 2008, 3, 236–241 CrossRef PubMed.
- S. Sen, M. H. Chowdhury, B. Mukherjee and P. Basak, Eur. Cells Mater., 2016, 31, 488 Search PubMed.
- S. Sen, H. Pandey, K. K. Jaiswal and P. Basak, in Recent Advances in Biosciences, ed. R. C. Sobti, K. Jaiswal and S. Mishra, Narendra Publishing House, New Delhi, India, 2015, ch. 18, vol. 1, pp. 163–166 Search PubMed.
- J. Kaur, R. Rajkhowa, T. Afrin, T. Tsuzuki and X. Wang, Biopolymers, 2014, 101, 237–245 CrossRef CAS PubMed.
- G. P. Bienert, J. K. Schjoerring and T. P. Jahn, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 994–1003 CrossRef CAS PubMed.
- S. Ennahar, K. Sonomoto and A. Ishizaki, J. Biosci. Bioeng., 1999, 87, 705–716 CrossRef CAS PubMed.
- Q. L. Loh and C. Choong, Tissue Eng., Part B, 2013, 19, 485–502 CrossRef CAS PubMed.
- N. Arora, A. Ali, S. Sen, N. K. Jana and P. Basak, Compos. Interfaces, 2013, 21, 51–58 CrossRef.
- J. Rnjak-Kovacina and A. S. Weiss, Tissue Eng., Part B, 2011, 17, 365 CrossRef CAS PubMed.
- M. M. A. E. L. Salem, M. Shalbaf, N. C. J. Gibbons, B. Chavan, J. M. Thornton and K. U. Schallreuter, FASEB J., 2009, 23, 3790–3807 CrossRef CAS PubMed.
- J. P. Kumar and B. B. Mandal, Free Radicals Biol. Med., 2017, 108, 803–818 CrossRef CAS PubMed.
- N. Hibino, T. Yi, D. R. Duncan, A. Rathore, E. Dean and Y. Naito, et al., FASEB J., 2011, 25, 4253–4263 CrossRef CAS PubMed.
- K. G. Battiston, R. S. Labow and J. P. Santerre, Biomaterials, 2012, 33(33), 8316–8328 CrossRef CAS PubMed.
- B. N. Brown, R. Londono, S. Tottey, L. Zhang, K. A. Kukla, M. T. Wolf, K. A. Daly, J. E. Reing and S. F. Badylak, Acta Biomater., 2012, 8, 978–987 CrossRef CAS PubMed.
- B. N. Brown, B. D. Ratner, S. B. Goodman, S. Amar and S. F. Badylak, Biomaterials, 2012, 33, 3792–3802 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08538c |
| ‡ indicates both the authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |